

Form S-1 Hoth Therapeutics, Inc.
 Tweet
Tweet Share
ShareAs filed with the Securities and Exchange Commission on August 30, 2019
Registration No. 333-
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
Hoth Therapeutics, Inc.
(Exact name of Registrant as specified in its charter)
| Nevada | 2834 | 82-1553794 | ||
| (State or other jurisdiction of incorporation or organization) |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification Number) |
1 Rockefeller Plaza, Suite 1039
New York, New York 10020
(646)
756-2997
(Address, including zip code, and telephone number, including area code, of Registrant’s principal executive offices)
Robb Knie
Chief Executive Officer
1 Rockefeller Plaza, Suite 1039
New York, New York 10020
(646) 756-2997
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
Richard A. Friedman, Esq. Nazia Khan, Esq. Sheppard,
Mullin, Richter & Hampton LLP |
Approximate date of commencement of proposed sale to the public:
As soon as practicable after the effective date of this registration statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933 check the following box: ☒
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☐ | |
| Non-accelerated filer | ☒ | Smaller reporting company | ☒ | |
| Emerging growth company | ☒ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided to Section 7(a)(2)(B) of the Securities Act. ☐
CALCULATION OF REGISTRATION FEE
| Proposed | Proposed | |||||||||||||||
| Maximum | Maximum | |||||||||||||||
| Amount | Offering | Aggregate | Amount of | |||||||||||||
| Title of Each Class of Securities to be Registered (1) | to be Registered | Price Per Share (2) | Offering Price (2) | Registration Fee | ||||||||||||
| Common Stock, par value $0.0001 per share | 407,424 | $ | 5.12 | $ | 2,086,011 | $ | 252.82 | |||||||||
| Common Stock, par value $0.0001 per share (3) | 921,579 | $ | 5.12 | $ | 4,718,485 | $ | 571.88 | |||||||||
| Total | 1,329,003 | $ | 6,804,496 | $ | 824.70 | |||||||||||
| (1) | The shares of our common stock being registered hereunder are being registered for sale by the selling security holders named in the prospectus. Under Rule 416 of the Securities Act of 1933, as amended, the shares being registered include such indeterminate number of shares of common stock as may be issuable with respect to the shares being registered in this registration statement as a result of any stock splits, stock dividends or other similar event. |
| (2) | The proposed maximum offering price per share and the proposed maximum aggregate offering price have been estimated solely for the purpose of calculating the amount of the registration fee in accordance with Rule 457(c) under the Securities Act of 1933, as amended, using the average of the high and low prices as reported on The Nasdaq Capital Market on August 28, 2019. |
| (3) | Represents shares of common stock issuable upon exercise of outstanding warrants to purchase shares of common stock offered by the selling stockholders. |
The registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the registration statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.
The information in this prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the Securities and Exchange Commission is declared effective. This prospectus is not an offer to sell nor does it seek an offer to buy these securities in any state where the offer or sale is not permitted
PRELIMINARY PROSPECTUS
SUBJECT TO COMPLETION, DATED AUGUST 30, 2019

Hoth Therapeutics Inc.
1,329,003 Shares of Common Stock
This prospectus relates to the resale by certain selling stockholders of Hoth Therapeutics, Inc., a Nevada corporation (the “Company”) identified in this prospectus of up to 1,329,003 shares (the “Resale Shares”) of our common stock, par value $0.0001 per share, including 407,424 outstanding shares of common stock and 921,579 shares of common stock issuable upon exercise of outstanding warrants. All of the Resale Shares were purchased from the Company in private placement transactions and are being offered for resale by the selling stockholders only.
The Resale Shares may be sold by the selling stockholders to or through underwriters or dealers, directly to purchasers or through agents designated from time to time. For additional information regarding the methods of sale you should refer to the section entitled “Plan of Distribution” in this Prospectus.
The prices at which the selling stockholders may sell the Resale Shares will be determined by the prevailing market price for shares of the Company’s common stock or in privately negotiated transactions. We will not receive any proceeds from the sale of the Resale Shares by the selling stockholders; provided, however, we will receive the proceeds from any cash exercise of warrants.
We will bear all costs relating to the registration of the Resale Shares, other than any selling stockholders legal or accounting costs or commissions.
Our common stock is presently listed on The Nasdaq Capital Market under the symbol “HOTH.” The closing price of our common stock on August 29, 2019, as reported on The Nasdaq Capital Market was $5.50 per share.
Investing in our common stock involves a high degree of risk. See the section entitled “Risk Factors” beginning on page 7 of this prospectus and elsewhere in this prospectus for a discussion of information that should be considered in connection with an investment in our common stock.
We may amend or supplement this prospectus from time to time by filing amendments or supplements as required. You should read the entire prospectus and any amendments or supplements carefully before you make your investment decision.
We qualify as an “emerging growth company” as defined in the Jumpstart our Business Startups Act of 2012 (“JOBS Act”). For more information, see the sub-section titled “Implications of Being an Emerging Growth Company.”
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense.
The date of this prospectus is_________, 2019.
INCORPORATION OF CERTAIN INFORMATION BY REFERENCE
The Securities and Exchange Commission (the “SEC”) allows us to “incorporate by reference” information that we file with them. Incorporation by reference allows us to disclose important information to you by referring you to those other documents. The information incorporated by reference is an important part of this prospectus, and information that we file later with the SEC will automatically update and supersede this information. We filed a registration statement on Form S-1, of which this prospectus forms a part, under the Securities Act of 1933, as amended (the “Securities Act”), with the SEC with respect to the securities being offered pursuant to this prospectus. This prospectus omits certain information contained in the registration statement, as permitted by the SEC. You should refer to the registration statement, including the exhibits, for further information about us and the securities being offered pursuant to this prospectus. Statements in this prospectus regarding the provisions of certain documents filed with, or incorporated by reference in, the registration statement are not necessarily complete and each statement is qualified in all respects by that reference. Copies of all or any part of the registration statement, including the documents incorporated by reference or the exhibits, may be obtained upon payment of the prescribed rates at the offices of the SEC listed above in “Where You Can Find More Information.” We are incorporating by reference the documents listed below, which we have already filed with the SEC, and all documents subsequently filed by us pursuant to Sections 13(a), 13(c), 14 or 15(d) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), except as to any portion of any future report or document that is not deemed filed under such provisions:
| 1. | The Company’s Annual Report on Form 10-K for the year ended December 31, 2018, filed with the SEC on April 1, 2019; |
| 2. | The Company’s Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2019 and June 30, 2019 filed with the SEC on May 15, 2019 and August 14, 2019, respectively; |
| 3. | The Company’s Current Reports on Form 8-K (other than Current Reports furnished under Item 2.02 or Item 7.01 of Form 8-K and exhibits filed on such form that are related to such items) filed with the SEC on February 20, 2019, March 7, 2019, April 29, 2019, August 21, 2019 and August 23, 2019; and |
| 4. | The description of the Company’s common stock contained in the registration statement on Form 8-A filed with the SEC on February 6, 2019, including any amendment or report filed for the purpose of updating that description. |
Any statement contained in this prospectus or in a document incorporated or deemed to be incorporated by reference into this prospectus will be deemed to be modified or superseded to the extent that a statement contained in this prospectus or any subsequently filed document that is deemed to be incorporated by reference into this prospectus modifies or supersedes the statement.
You may request, and we will provide you with, a copy of these filings, at no cost, by calling us at (646) 756-2997 or by writing to us at the following address:
Hoth Therapeutics, Inc.
1 Rockefeller Plaza, Suite 1039
New York, New York 10020
Attn.: Secretary
TABLE OF CONTENTS
ABOUT THIS PROSPECTUS
We have not, and the underwriters have not, authorized anyone to provide any information or to make any representations other than those contained in this prospectus or in any free writing prospectus prepared by or on behalf of us or to which we have referred you. We take no responsibility for, and can provide no assurance as to the reliability of, any other information that others may give to you. The information contained in this prospectus is accurate only as of the date of this prospectus, regardless of the time of delivery of this prospectus or any sale of our common stock.
You should rely only on the information contained in this prospectus. No dealer, salesperson or other person is authorized to give information that is not contained in this prospectus. This prospectus is not an offer to sell nor is it seeking an offer to buy these securities in any jurisdiction where the offer or sale is not permitted. The information in this prospectus is accurate only as of the date of this prospectus, regardless of the time of delivery of this prospectus or of any sale of these securities.
i
The following summary highlights selected information contained elsewhere in this prospectus and is qualified in its entirety by the more detailed information and financial statements included elsewhere in this prospectus. It does not contain all the information that may be important to you and your investment decision. You should carefully read this entire prospectus, including the matters set forth under “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and our financial statements and related notes included elsewhere in this prospectus. In this prospectus, unless context requires otherwise, references to “we,” “us,” “our,” “Hoth” or “the Company” refer to Hoth Therapeutics, Inc.
Overview
We are a development stage biopharmaceutical company incorporated in May 2017 focused on targeted therapeutics for patients suffering from conditions such as atopic dermatitis, also known as eczema.
Our primary asset is a sublicense agreement with Chelexa Biosciences, Inc. (“Chelexa”) pursuant to which Chelexa has granted us an exclusive sublicense to use its BioLexa Platform (as defined herein), a proprietary, patented, drug compound platform developed at the University of Cincinnati. The license enables us to develop the platform for any indications in humans. Our initial focus will be on the treatment of eczema through the application of a topical cream. Although our initial focus will be on the treatment of eczema, we intend to develop a second topical cream which, upon application, is intended to reduce post-procedure infections, accelerate healing and improve clinical outcomes for patients undergoing aesthetic dermatology procedures. The BioLexa Platform combines a U.S. Food and Drug Administration (“FDA”) approved zinc chelator with one or more approved antibiotics in a topical dosage form to address unchecked eczema flare-ups by preventing the formation of infectious biofilms and the resulting clogging of sweat ducts which trigger symptoms. To our knowledge, it is the first product candidate intended to prevent the symptom triggering flare-ups rather than simply treating symptoms when they occur.
On May 26, 2017, we entered into a sublicense agreement with Chelexa, as amended on August 22, 2018 and August 29, 2018, pursuant to which Chelexa granted us an exclusive sublicense to make, use, have made, import, offer for sale, and sell products based upon or involving the use of (i) topical compositions comprising a zinc chelator and gentamicin and (ii) zinc chelators to inhibit biofilm formation (the “BioLexa Platform” or “BioLexa”), which rights were originally granted to Chelexa pursuant to an exclusive license agreement with the University of Cincinnati. In addition, Chelexa granted us the right to issue exclusive and nonexclusive sublicenses (with the right to further sublicense to third parties) to make, use, have made, import, offer for sale, and sell products based upon the BioLexa Platform.
We intend to use the BioLexa Platform to develop two different topical cream products: (i) a product to treat eczema and (ii) a product that reduces post-procedure infections, accelerates healing and improves clinical outcomes for patients undergoing aesthetic dermatology procedures. Our initial focus will be on eczema. Eczema is a disease that results in inflammation of the skin and is characterized by rash, red skin, and itchiness. Eczema is also referred to as atopic dermatitis (“AD”). According to the National Eczema Association, eczema affects approximately 32 million Americans and, at $300 per annum per patient, represents an approximate $9.5 billion market in the U.S. alone.
BioLexa’s formulation is a new topical dosage form “repurposing” the antibiotic, enabling it to be developed for use in patients following a special regulatory pathway codified in Section 505(b)(2) of the FDA rules. Section 505(b)(2) of the Federal Food, Drug and Cosmetic Act was enacted to enable sponsors to seek New Drug Application (“NDA”) approval for novel repurposed drugs without the need for such sponsors to undertake time consuming and expensive pre-clinical safety studies and Phase 1 safety studies. Proceeding under this regulatory pathway, we will be able to rely upon all of the publicly available safety and toxicology data with respect to gentamicin and zinc chelator in our FDA submissions. We will be required to conduct a Phase 2 study to show the safety of the combination in humans and after such Phase 2 study will be required to proceed to Phase 3 pivotal clinical trials. We believe that this path will dramatically reduce the required clinical development effort, costs and risks as compared to what would be required of us if we were required to conduct pre-clinical safety, toxicology and animal studies together with Phase 1 human safety trials required for new chemical entities which are not eligible to be reviewed pursuant to the Section 505(b)(2) regulatory pathway. We estimate that by using the Section 505(b)(2) regulatory pathway, that the clinical development process may be five to six years shorter than is required for a new chemical entity, and the FDA approval process may be six to nine months shorter than the typical eighteen month period, which we believe may result in lower development costs and shorter development time. As of the date of this prospectus, we have not submitted an NDA to the FDA. In September 2018, we attended the first of a planned series of meetings with the FDA to review the requirements for submission and activation of an investigational new drug application (“IND”) with respect to the BioLexa Platform for use in eczema. In preparation for such pre-IND meeting, we prepared and presented to the FDA our proposed Phase 2 clinical trial plan for the treatment of eczema in patients over the age of one year old. As part of our pre-IND meeting, the FDA provided us with general guidance with respect to specific animal studies, dosing schedules and suggested human safety studies before we commence clinical trials in pediatric or adult patients. We are currently investigating multiple potential venues for conducting such trial both in and outstand of the U.S. We have engaged Camargo Pharmaceutical Services, LLC (“Camargo”) to assist us with the FDA process required for Section 505(b)(2) applications and with the evaluation of potential clinical trial venues for the proof of concept study should we determine to undertake such study. Specifically, Camargo has provided and will continue to provide advice and guidance relative to the IND preparation phase for the BioLexa platform. Camargo will assist us with the refinement of our non-clinical, clinical, clinical pharmacology and biopharmaceutics strategy incorporating the preliminary feedback we received from the FDA during our pre-IND meeting.
-1-
We intend to conduct our first Phase 1 study in healthy adults with an immediate transition to a randomized, vehicle controlled Phase 1b trial in adolescent eczema patients comparing BioLexa to the base vehicle. This Phase 1b trial is intended to examine both safety and efficacy. We will assess the formulation of Ca-DTPA and Gentamicin 0.1% in our proprietary topical lotion delivered by a metered pump system. We will also assess the ability of BioLexa to clear harmful staph aureus bacterial from the skin of atopic dermatitis patients.
Following our Phase 1b trial, we intend to conduct up to two Phase 2 trials in atopic dermatitis patients comparing BioLexa to the base vehicle. Subject numbers and allocation will be informed by the results of the Phase 1b trial. We expect the clinical program to be completed, subject to receipt of funding by us, by the end of 2020 or early 2021 with an NDA submission targeted for mid to late 2021.
In addition, we conducted an initial pilot study on the efficacy of BioLexa to accelerate diabetic wound healing and intend to conduct additional studies with respect to the regenerative effects of the BioLexa Platform in the context of chronic diabetic ulcers, with and without substantial bacterial burden.
We believe that the key elements for our market success include:
| ● | the proprietary formulation of two FDA-approved drugs to treat bacterial proliferation reduces development time and costs by giving us the ability to rely on safety and efficacy data from the two approved drugs; |
| ● | our proprietary formulation is not a topical corticosteroid, and may not be subject to the same FDA black box warning issues as most commonly prescribed treatments currently in use; and |
| ● | recent peer-reviewed publications highlight that staph-induced biofilms are the root cause of flare-ups in eczema. Our BioLexa product candidate has been demonstrated to prevent the formation of these biofilms with the promise of delaying or completely arresting flare-ups, rather than merely treating symptoms of a flare-up already underway. |
In addition to our sublicense agreement with Chelexa, we entered into an exclusive license agreement with the University of Cincinnati for a patented, novel genetic marker for food allergies. The genetic marker licensed by us from the University of Cincinnati (i) may be used to identify at risk infants in predicting food allergies, including peanut and milk allergies, (ii) may be used to identify a person’s predisposition to an allergic reaction, thereby avoiding such reaction and (iii) may also determine an individual’s propensity to develop AD, such as eczema. We intend to utilize the genetic marker for purposes of determining an individual’s propensity to develop eczema as well as to identify and treat allergies in at-risk infants.
-2-
We also entered into an exclusive sublicense agreement (the “Sublicense Agreement”) with Zylö Therapeutics, Inc. (“Zylö”) pursuant to which Zylö granted us an exclusive sublicense to certain patent rights and technology to, among other things, develop, make and sell the certain licensed products and to practice certain licensed technology in the United States and Canada for all therapeutic uses related to lupus in human beings.
Our Product Pipeline
The following table summarizes the BioLexa expected product development pipeline.
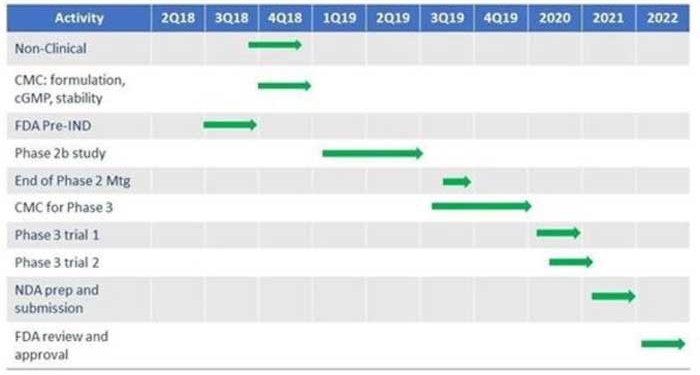
There is currently no active IND for our product candidate in the United States.
Risks Associated with Our Business
Our business is subject to a number of risks of which you should be aware before making a decision to invest in our common shares. These risks are discussed more fully in the “Risk Factors” section of this prospectus. These risks include the following:
| ● | We were formed in May 2017 and have a limited operating history. Since inception, we have incurred losses and expect to continue to operate at a net loss for at least the next several years as we commence our research and development efforts, conduct clinical trials and develop manufacturing, sales, marketing and distribution capabilities. Our net losses for the year ended December 31, 2018 and for the period from May 16, 2017 (inception) through December 31, 2017 were $2,495,525 and $2,015,481, respectively. Our net losses for the three and six months ended June 30, 2019 were $1,267,586 and $2,096,951, respectively. |
| ● | We have generated no revenue from commercial sales to date and our future profitability is uncertain. |
| ● | If we fail to obtain the capital necessary to fund our operations, we will be unable to continue or complete our product development and you will likely lose your entire investment. |
-3-
| ● | We may expand our business through the acquisition of rights to new drug candidates that could disrupt our business, harm our financial condition and may also dilute current shareholders’ ownership interests in our Company. |
| ● | Our management team lacks experience in the pharmaceutical field. |
| ● | The marketing approval process of the FDA is lengthy, time consuming and inherently unpredictable, and if we ultimately are unable to obtain marketing approval for the product candidates we intend to develop, our business will be substantially harmed. |
| ● | There is no guarantee that the FDA will grant NDA approval of our future products, and failure to obtain necessary clearances or approvals for our future products would adversely affect our ability to grow our business. |
| ● | There can be no assurance that the data generated from our clinical trials will be acceptable to FDA. |
| ● | Even if our products are approved by regulatory authorities, if we or our suppliers fail to comply with ongoing FDA regulation or if we experience unanticipated problems with our products, these products could be subject to restrictions or withdrawal from the market. |
| ● | Our products will face significant competition in the markets for such products, and if they are unable to compete successfully, our business will suffer. |
| ● | Our business depends upon securing and protecting critical intellectual property. |
| ● | We may undertake international operations, which will subject us to risks inherent with operations outside of the United States. |
| ● | Market and economic conditions may negatively impact our business, financial condition and share price. |
| ● | Future sales and issuances of our securities could result in additional dilution of the percentage ownership of our shareholders and could cause our share price to fall. |
Corporate Information
We were incorporated as a Nevada corporation on May 16, 2017. Our principal executive offices are located at 1 Rockefeller Plaza, Suite 1039, New York, New York 10020 and our telephone number is (646) 756-2997. Our website address is www.hoththerapeutics.com. The information contained on our website is not incorporated by reference into this prospectus, and you should not consider any information contained on, or that can be accessed through, our website as part of this prospectus or in deciding whether to purchase our common shares.
Implications of Being an Emerging Growth Company
We qualify as an emerging growth company as defined in the JOBS Act because we had less than $1.07 billion in revenues during our last fiscal year. As an emerging growth company, we expect to take advantage of reduced reporting requirements that are otherwise applicable to public companies. These provisions include, but are not limited to:
| ● | being permitted to present only two years of audited financial statements, in addition to any required unaudited interim financial statements, with correspondingly reduced “Management’s Discussion and Analysis of Financial Condition and Results of Operations” disclosure in this prospectus; |
| ● | not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, as amended (“Sarbanes-Oxley”); |
-4-
| ● | reduced disclosure obligations regarding executive compensation in our periodic reports, proxy statements and registration statements; and |
| ● | exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. |
We may use these provisions until the last day of our fiscal year following the fifth anniversary of the completion of our initial public offering. However, if certain events occur prior to the end of such five-year period, including if we become a “large accelerated filer,” our annual gross revenues exceed $1.07 billion or we issue more than $1.0 billion of non-convertible debt in any three-year period, we will cease to be an emerging growth company prior to the end of such five-year period.
As an emerging growth company, we intend to take advantage of an extended transition period for complying with new or revised accounting standards as permitted by the JOBS Act. To the extent that we continue to qualify as a “smaller reporting company,” as such term is defined in Rule 12b-2 under the Securities Exchange Act of 1934, after we cease to qualify as an emerging growth company, certain of the exemptions available to us as an emerging growth company may continue to be available to us as a smaller reporting company, including: (i) not being required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes Oxley Act; (ii) scaled executive compensation disclosures; and (iii) the requirement to provide only two years of audited financial statements, instead of three years.
August 2019 Private Placement
On August 16, 2019 (the “Closing Date”), we sold an aggregate of $2,037,120 of units (the “Units”), each Unit consisting of (i) one share of the Company’s common stock and (ii) a warrant to purchase one-half share pursuant to a private placement offering (the “August 2019 Private Placement”) at a purchase price of $5.00 per Unit. Each warrant is exercisable for a period of two years beginning six months from the Closing Date at an exercise price of $8.00 per whole share, subject to adjustment.
In connection with the August 2019 Private Placement, we agreed to (i) pay Laidlaw and Company (UK) Ltd., a U.S. registered broker-dealer (“Laidlaw”) a cash commission of 10% of the gross proceeds raised from investors in the August 2019 Private Placement introduced by them, and (ii) issued to Laidlaw, warrants to purchase that number of shares of common stock equal to 10% of the number of shares of common stock sold to investors (including shares of common stock issuable upon exercise of the warrants) in the August 2019 Private Placement introduced by them, with a term of five years from the closing date of the August 2019 Private Placement, and an exercise price of $5.00 per share (the “Placement Agent Warrants”).
As a result of the foregoing, we paid Laidlaw an aggregate commission of approximately $0.2 million and issued Laidlaw Placement Agent Warrants to purchase up to an aggregate of 61,113 shares of our common stock in connection with the August 2019 Private Placement. We also paid Laidlaw a one time fee of $50,000 and reimbursed the Laidlaw for approximately $0.4 million of expenses incurred in connection with the August 2019 Private Placement. In the event that the investors exercise their warrants, we will also be required to pay Laidlaw a cash fee of 5% of the gross proceeds received from the exercise of such warrants.
-5-
THE OFFERING
| Common stock offered by selling stockholders: | 1,329,003 shares of common stock including 407,424 outstanding shares of common stock and 921,579 shares of common stock issuable upon exercise of the warrants. | |
| Offering price: | Market price or privately negotiated prices. | |
| Common stock outstanding after the offering: | 10,077,068 | |
| Use of proceeds: | We will not receive any proceeds from the sale of the Resale Shares by the selling stockholders; however, we will receive the proceeds from any cash exercise of warrants. | |
| Risk factors: | An investment in our securities involves a high degree of risk and could result in a loss of your entire investment. Prior to making an investment decision, you should carefully consider all of the information in this prospectus and, in particular, you should evaluate the risk factors set forth under the caption “Risk Factors” beginning on page 7. | |
| Nasdaq Capital Market Symbol: | HOTH |
The number of shares of common stock to be outstanding immediately after this offering is based on 10,077,068 shares of common stock outstanding as of August 27, 2019 and excludes:
| ● | 717,870 shares of common stock issuable upon exercise of warrants with an exercise price of $1.00 per share; |
| ● | 203,709 shares of common stock issuable upon exercise of warrants with an exercise price of $8.00 per share; |
| ● | 50,000 shares of common stock issuable upon exercise of warrants with a weighted average exercise price of $7.00 per share; and |
| ● | 830,683 shares of common stock reserved for future issuance under our 2018 Equity Incentive Plan. |
-6-
Any investment in our common stock involves a high degree of risk. Before deciding whether to purchase our common stock, investors should carefully consider the risks described below together with the “Risk Factors” described in our most recent Annual Report on Form 10-K, which are incorporated herein by reference, as may be amended, supplemented or superseded from time to time by other reports we file with the SEC. Our business, financial condition, operating results and prospects are subject to the following material risks as well as those material risks incorporated by reference. Additional risks and uncertainties not presently foreseeable to us may also impair our business operations. If any of the following risks actually occurs, our business, financial condition or operating results could be materially adversely affected. In such case, the trading price of our common stock could decline, and our stockholders may lose all or part of their investment in the shares of our common stock.
Risks Related to Our Financial Position and Need for Capital
We have generated no revenue from commercial sales to date and our future profitability is uncertain.
We were incorporated in May 2017 and have a limited operating history and our business is subject to all of the risks inherent in the establishment of a new business enterprise. Our likelihood of success must be considered in light of the problems, expenses, difficulties, complications and delays frequently encountered in connection with development and expansion of a new business enterprise. Since inception, we have incurred losses and expect to continue to operate at a net loss for at least the next several years as we commence our research and development efforts, conduct clinical trials and develop manufacturing, sales, marketing and distribution capabilities. Our net losses for the year ended December 31, 2018 and for the period from May 16, 2017 (inception) through December 31, 2017 were $2,495,525 and $2,015,481, respectively, and our accumulated deficit as of December 31, 2018 and 2017 was $4,511,006 and $2,015,481, respectively. Our net losses for the three and six months ended June 30, 2019 were $1,267,586 and $2,096,951, respectively, and our accumulated deficit as of June 30, 2019 was $6,607,957. There can be no assurance that the products under development by us will be approved for sale in the U.S. or elsewhere. Furthermore, there can be no assurance that if such products are approved they will be successfully commercialized, and the extent of our future losses and the timing of our profitability are highly uncertain. If we are unable to achieve profitability, we may be unable to continue our operations.
If we fail to obtain the capital necessary to fund our operations, we will be unable to continue or complete our product development and you will likely lose your entire investment.
We will need to continue to seek capital from time to time to continue development of our lead drug candidate beyond the initial Phase 2 clinical trial and to acquire and develop other product candidates. Our first product is not expected to be commercialized until at least 2022 and we cannot provide any assurances that any revenues it may generate in the future will be sufficient to fund our ongoing operations. We believe that we will need to raise substantial additional capital to fund our continuing operations and the development and commercialization of our product candidate.
Our business or operations may change in a manner that would consume available funds more rapidly than anticipated and substantial additional funding may be required to maintain operations, fund expansion, develop new or enhanced products, acquire complementary products, business or technologies or otherwise respond to competitive pressures and opportunities, such as a change in the regulatory environment or a change in preferred eczema treatment modalities. In addition, we may need to accelerate the growth of our sales capabilities and distribution beyond what is currently envisioned, and this would require additional capital. However, we may not be able to secure funding when we need it or on favorable terms. We may not be able to raise sufficient funds to commercialize the product candidates we intend to develop.
If we cannot raise adequate funds to satisfy our capital requirements, we will have to delay, scale back or eliminate our research and development activities, clinical studies or future operations. We may also be required to obtain funds through arrangements with collaborators, which arrangements may require us to relinquish rights to certain technologies or products that we otherwise would not consider relinquishing, including rights to future product candidates or certain major geographic markets. This could result in sharing revenues which we might otherwise retain for ourselves. Any of these actions may harm our business, financial condition and results of operations.
-7-
The amount of capital we may need depends on many factors, including the progress, timing and scope of our product development programs; the progress, timing and scope of our preclinical studies and clinical trials; the time and cost necessary to obtain regulatory approvals; the time and cost necessary to further develop manufacturing processes and arrange for contract manufacturing; our ability to enter into and maintain collaborative, licensing and other commercial relationships; and our partners’ commitment of time and resources to the development and commercialization of our products.
Even if we can raise additional funding, we may be required to do so on terms that are dilutive to you.
The capital markets have been unpredictable in the recent past for unprofitable companies such as ours. In addition, it is generally difficult for early stage companies to raise capital under current market conditions. The amount of capital that a company such as ours is able to raise often depends on variables that are beyond our control. As a result, we may not be able to secure financing on terms attractive to us, or at all. If we are able to consummate a financing arrangement, the amount raised may not be sufficient to meet our future needs. If adequate funds are not available on acceptable terms, or at all, our business, including our results of operations, financial condition and our continued viability will be materially adversely affected.
Risks Related to Product Development, Regulatory Approval, Manufacturing and Commercialization
We depend upon the success of the BioLexa Platform, which has not yet demonstrated efficacy in Phase 2 clinical trials and the genetic marker for food allergies which is in the pre-clinical stage, the use of which we licensed from the University of Cincinnati. If we are unable to generate revenues from the BioLexa Platform or the genetic marker, our ability to create stockholder value will be limited.
We intend to conduct an initial Phase 2 study for our lead product candidate, the BioLexa Platform, which is a new topical dosage form “repurposing” the antibiotic, enabling it to be developed for use in patients following a special regulatory pathway codified in Section 505(b)(2) of the FDA rules. In addition, the genetic marker for food allergies which we licensed from the University of Cincinnati is in the pre-clinical stage. We do not generate revenues from any approved drug products and have no other product candidates in development. We may not be successful in obtaining acceptance from the regulatory authorities to start our clinical trials. If we do not obtain such acceptance, the time in which we expect to commence clinical programs for any product candidate will be extended and such extension will increase our expenses and increase our need for additional capital. Moreover, there is no guarantee that our clinical trials will be successful or that we will continue clinical development in support of an approval from the regulatory authorities for any indication. We note that most drug candidates never reach the clinical stage and even those that do commence clinical development have only a small chance of successfully completing clinical development and gaining regulatory approval. Therefore, our business currently depends entirely on the successful development, regulatory approval and commercialization of our product candidates, which may never occur.
Members of our management team lack experience in the pharmaceutical field.
Members of our management team lack experience in the pharmaceutical field. This lack of experience may impair our ability to commercialize our pharmaceutical products and attain profitability. We will need to hire or engage managerial personnel with relevant experience in the pharmaceutical field; however, there can be no assurance that such personnel will be available to us or, that once engaged, will be retained by us. Failure to establish and maintain an effective management team with experience in the pharmaceutical field and commercialization of pharmaceuticals products would have a material adverse effect on our business and results of operations.
-8-
The marketing approval process of the FDA is lengthy, time consuming and inherently unpredictable, and if were ultimately are unable to obtain marketing approval for the product candidates we intend to develop, our business will be substantially harmed.
None of the product candidates we intend to develop have gained marketing approval in the U.S. and we cannot guarantee that we will ever have marketable products. Our business is substantially dependent on our ability to complete the development of, obtain marketing approval for, and successfully commercialize our product candidates in a timely manner. We cannot commercialize our product candidates in the United States without first obtaining approval from the FDA to market each product candidate. Our product candidates could fail to receive marketing approval for many reasons, including among others:
| ● | the FDA may disagree with the design or implementation of our clinical trials; |
| ● | the FDA could determine that we cannot rely on Section 505(b)(2) for any or all of our product candidates; and |
| ● | the FDA may determine that we have identified the wrong reference listed drug or drugs or that approval of our Section 505(b)(2) application for any of our product candidates is blocked by patent or non-patent exclusivity of the reference listed drug or drugs. |
In addition, the process of seeking regulatory clearance or approval to market the product candidates we intend to develop is expensive and time consuming and, notwithstanding the effort and expense incurred, clearance or approval is never guaranteed. If we are not successful in obtaining timely clearance or approval of our product candidates from the FDA, we may never be able to generate significant revenue and may be forced to cease operations. The NDA process is costly, lengthy and uncertain. Any NDA application filed by the Company will have to be supported by extensive data, including, but not limited to, technical, preclinical, clinical trial, manufacturing and labeling data, to demonstrate to the FDA’s satisfaction the safety and efficacy of the product for its intended use.
Obtaining clearances or approvals from the FDA and from the regulatory agencies in other countries is an expensive and time consuming process and is uncertain as to outcome. The FDA and other agencies could ask us to supplement our submissions, collect non-clinical data, conduct additional clinical trials or engage in other time-consuming actions, or it could simply deny our applications. In addition, even if we obtain an NDA approval or pre-market approvals in other countries, the approval could be revoked or other restrictions imposed if post-market data demonstrates safety issues or lack of effectiveness. We cannot predict with certainty how, or when, the FDA will act. If we are unable to obtain the necessary regulatory approvals, our financial condition and cash flow may be adversely affected, and our ability to grow domestically and internationally may be limited. Additionally, even if cleared or approved, the Company’s products may not be approved for the specific indications that are most necessary or desirable for successful commercialization or profitability.
We may encounter substantial delays in completing our clinical studies which in turn will require additional costs, or we may fail to demonstrate adequate safety and efficacy to the satisfaction of applicable regulatory authorities.
It is impossible to predict if or when any of our product candidates, will prove safe or effective in humans or will receive regulatory approval. Before obtaining marketing approval from regulatory authorities for the sale of our product candidates, we must conduct extensive clinical studies to demonstrate the safety and efficacy of the product candidates in humans. Clinical testing is expensive, time-consuming and uncertain as to outcome. We cannot guarantee that any clinical studies will be conducted as planned or completed on schedule, if at all. A failure of one or more clinical studies can occur at any stage of testing. Events that may prevent successful or timely completion of clinical development include:
| ● | delays in reaching, or failing to reach, a consensus with regulatory agencies on study design; | |
| ● | delays in reaching, or failing to reach, agreement on acceptable terms with a sufficient number of prospective contract research organizations (“CROs”) and clinical study sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; | |
| ● | delays in obtaining required IRB or Ethics Committee (“EC”) approval at each clinical study site; | |
| ● | delays in recruiting a sufficient number of suitable patients to participate in our clinical studies; | |
| ● | imposition of a clinical hold by regulatory agencies, after an inspection of our clinical study operations or study sites; | |
| ● | failure by our CROs, other third parties or us to adhere to clinical study, regulatory or legal requirements; |
-9-
| ● | failure to perform in accordance with the FDA’s GCPs or applicable regulatory guidelines in other countries; | |
| ● | delays in the testing, validation, manufacturing and delivery of sufficient quantities of our product candidates to the clinical sites; | |
| ● | delays in having patients complete participation in a study or return for post-treatment follow-up; | |
| ● | clinical study sites or patients dropping out of a study; | |
| ● | delay or failure to address any patient safety concerns that arise during the course of a trial; | |
| ● | unanticipated costs or increases in costs of clinical trials of our product candidates; | |
| ● | occurrence of serious adverse events associated with the product candidate that are viewed to outweigh its potential benefits; or | |
| ● | changes in regulatory requirements and guidance that require amending or submitting new clinical protocols. |
We could also encounter delays if a clinical trial is suspended or terminated by us, by the IRBs or ECs of the institutions in which such trials are being conducted, by an independent Safety Review Board (“SRB”) for such trial or by the FDA, EMA, or other regulatory authorities. Such authorities may suspend or terminate a clinical trial due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA, EMA, or other regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a drug, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial.
Any inability to successfully complete preclinical and clinical development could result in additional costs to us or impair our ability to generate revenues from product sales, regulatory and commercialization milestones and royalties. In addition, if we make manufacturing or formulation changes to our product candidates, we may need to conduct additional studies to bridge our modified product candidates to earlier versions.
Clinical study delays could also shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do, which could impair our ability to successfully commercialize our product candidates. In addition, any delays in completing our clinical trials will increase our costs, slow down our product candidate development and approval process and jeopardize our ability to commence product sales and generate revenues. Any of these occurrences may significantly harm our business, financial condition and prospects. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates.
The outcome of preclinical studies and early clinical trials may not be predictive of the success of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. Further, preclinical and clinical data are often susceptible to various interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain marketing approval. If the results of our clinical studies are inconclusive or if there are safety concerns or adverse events associated with our other product candidates, we may:
| ● | be delayed in obtaining marketing approval for our product candidates, if approved at all; |
| ● | obtain approval for indications or patient populations that are not as broad as intended or desired; | |
| ● | obtain approval with labeling that includes significant use or distribution restrictions or safety warnings; | |
| ● | be required to change the way the product is administered; |
-10-
| ● | be required to perform additional clinical studies to support approval or be subject to additional post-marketing testing requirements; | |
| ● | have regulatory authorities withdraw their approval of a product or impose restrictions on its distribution in the form of a modified risk evaluation and mitigation strategy; | |
| ● | be sued; or | |
| ● | experience damage to our reputation. |
Additionally, our product candidates could potentially cause other adverse events that have not yet been predicted. The inclusion of ill patients in our clinical studies may result in deaths or other adverse medical events due to other therapies or medications that such patients may be using. As described above, any of these events could prevent us from achieving or maintaining market acceptance of our product candidates and impair our ability to commercialize our products.
If we are not able to obtain any required regulatory approvals for our product candidates, we will not be able to commercialize our product candidates and our ability to generate revenue will be limited.
We must successfully complete clinical trials for our product candidates before we can apply for marketing approval. Even if we complete our clinical trials, it does not assure marketing approval. Our preclinical trials may be unsuccessful, which would materially harm our business. Even if our initial preclinical trials are successful, we are required to conduct clinical trials to establish our product candidates’ safety and efficacy, before a marketing application (NDA or Biologics License Application, or BLA, or their foreign equivalents) can be filed with the FDA, the European Medicines Agency (“EMA”), or comparable foreign regulatory authorities for marketing approval of our product candidates.
Clinical testing is expensive, is difficult to design and implement, can take many years to complete and is uncertain as to outcome. Success in early phases of pre-clinical and clinical trials does not ensure that later clinical trials will be successful, and interim results of a clinical trial do not necessarily predict final results. A failure of one or more of our clinical trials can occur at any stage of testing. We may experience numerous unforeseen events during, or as a result of, the clinical trial process that could delay or prevent our ability to receive regulatory approval or commercialize our product candidates. The research, testing, manufacturing, labeling, packaging, storage, approval, sale, marketing, advertising and promotion, pricing, export, import and distribution of drug products are subject to extensive regulation by the FDA, EMA, and other regulatory authorities in the United States, European Union, and other countries, where regulations differ from country to country. We are not permitted to market our product candidates as prescription pharmaceutical products in the United States until we receive approval of an NDA from the FDA, or in any foreign countries until we receive the requisite approval from such countries. In the United States, the FDA generally requires the completion of clinical trials of each drug to establish its safety and efficacy and extensive pharmaceutical development to ensure its quality before an NDA is approved. Regulatory authorities in other jurisdictions impose similar requirements. Of the large number of drugs in development, only a small percentage result in the submission of an NDA to the FDA or other regulatory authorities and even fewer are eventually approved for commercialization. We have not submitted an NDA to the FDA or comparable applications to other regulatory authorities. If our development efforts for our product candidates, including regulatory approval, are not successful for their planned indications, or if adequate demand for our product candidates is not generated, our business will be materially adversely affected.
Our success depends on the receipt of regulatory approval and the issuance of such regulatory approvals is uncertain and subject to a number of risks, including the following:
| ● | the results of nonclinical or toxicology studies may not support the filing of an IND or foreign equivalent for our eczema product candidate; | |
| ● | the FDA, EMA, or comparable foreign regulatory authorities or IRBs or ECs may disagree with the design or implementation of our clinical trials; |
-11-
| ● | we may not be able to provide acceptable evidence of our product candidates’ safety and efficacy; | |
| ● | the results of our clinical trials may not be satisfactory or may not meet the level of statistical or clinical significance required by the FDA, EMA, or other regulatory agencies for marketing approval; | |
| ● | the dosing of our product candidates in a particular clinical trial may not be at an optimal level; | |
| ● | patients in our clinical trials may suffer adverse effects for reasons that may or may not be related to our product candidates; | |
| ● | the data collected from clinical trials may not be sufficient to support the submission of an NDA, BLA or other marketing application or to obtain regulatory approval in the United States or elsewhere; | |
| ● | the requirement for additional studies, including a second phase 3 study for the PRV-031 program in T1D; | |
| ● | the FDA, EMA, or comparable foreign regulatory authorities may fail to approve the manufacturing processes or facilities of third-party manufacturers with which we contract for clinical and commercial supplies; | |
| ● | the approval policies or regulations of the FDA, EMA, or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval; | |
| ● | the FDA, EMA, or comparable foreign regulatory authorities may disagree on the design or implementation of our clinical trials, including the methodology used in our studies, our chosen endpoints, our statistical analysis, or our proposed product indication; | |
| ● | our failure to demonstrate to the satisfaction of the FDA, EMA, or comparable regulatory authorities that a product candidate is safe and effective for its proposed indication; | |
| ● | we may fail to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks; | |
| ● | immunogenicity might affect a product candidate efficacy and/or safety; | |
| ● | the FDA, EMA, or comparable foreign regulatory authorities may disagree with our interpretation of data from nonclinical studies or clinical trials; | |
| ● | data collected from clinical trials of our product candidates may be insufficient to support the submission and filing of a marketing application or to obtain marketing approval. For example, the FDA may require additional studies to show that our product candidates are safe or effective; | |
| ● | we may fail to obtain approval of the manufacturing processes or facilities of third-party manufacturers with whom we contract for clinical and commercial supplies; | |
| ● | there may be changes in the approval policies or regulations that render our nonclinical and clinical data insufficient for approval; or | |
| ● | the FDA, EMA or comparable foreign regulatory authority may require more information, including additional nonclinical or clinical data to support approval, which may delay or prevent approval and our commercialization plans, or we may decide to abandon the development program. |
Failure to obtain regulatory approval for our product candidates for the foregoing, or any other reasons, will prevent us from commercializing our product candidates, and our ability to generate revenue will be materially impaired. We cannot guarantee that regulators will agree with our assessment of the results of the clinical trials we intend to conduct in the future or that such trials will be successful. The FDA, EMA and other regulators have substantial discretion in the approval process and may refuse to accept any application or may decide that our data is insufficient for approval and require additional clinical trials, or pre-clinical or other studies. In addition, varying interpretations of the data obtained from pre-clinical and clinical testing could delay, limit or prevent regulatory approval of our product candidates.
-12-
We have not submitted an IND or received regulatory approval to commence clinical trials for our product candidates in any jurisdiction. We have only limited experience in filing the applications necessary to gain regulatory approvals and expect to rely on consultants and third party CROs with expertise in this area to assist us in this process. Securing regulatory approvals to market a product requires the submission of pre-clinical, clinical, and/or pharmacokinetic data, information about product manufacturing processes and inspection of facilities and supporting information to the appropriate regulatory authorities for each therapeutic indication to establish a product candidate’s safety and efficacy for each indication. Our product candidates may prove to have undesirable or unintended side effects, toxicities or other characteristics that may preclude us from obtaining regulatory approval or prevent or limit commercial use with respect to one or all intended indications.
The process of obtaining regulatory approvals is expensive, often takes many years, if approval is obtained at all, and can vary substantially based upon, among other things, the type, complexity and novelty of the product candidates involved, the jurisdiction in which regulatory approval is sought and the substantial discretion of the regulatory authorities. Changes in regulatory approval policies during the development period, changes in or the enactment of additional statutes or regulations, or changes in regulatory review for a submitted product application may cause delays in the approval or rejection of an application. Regulatory approval obtained in one jurisdiction does not necessarily mean that a product candidate will receive regulatory approval in all jurisdictions in which we may seek approval, but the failure to obtain approval in one jurisdiction may negatively impact our ability to seek approval in a different jurisdiction. Failure to obtain regulatory marketing approval for our product candidates in any indication will prevent us from commercializing the product candidate, and our ability to generate revenue will be materially impaired.
If we are unable to submit an application for approval under Section 505(b)(2) of the FDCA or if we are required to generate additional data related to safety and efficacy in order to obtain approval under Section 505(b)(2), we may be unable to meet our anticipated development and commercialization timelines.
Our current strategy for seeking marketing authorization in the United States for our product candidates relies primarily on Section 505(b)(2) of the FDCA which permits use of a marketing application, referred to as a 505(b)(2) application, where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use. The FDA interprets this to mean that an applicant may rely for approval on such data as that found in published literature or the FDA’s finding of safety or effectiveness, or both, of a previously approved drug product owned by a third party. There is no assurance that the FDA would find third-party data relied upon by us in a 505(b)(2) application sufficient or adequate to support approval and may require us to generate additional data to support the safety and efficacy of our intended product candidates. Consequently, we may need to conduct substantial new research and development activities beyond those we currently plan to conduct. Such additional new research and development activities would be costly and time consuming and there is no assurance that such data generated from such additional activities would be sufficient to obtain approval.
If the data to be relied upon in a 505(b)(2) application is related to drug products previously approved by the FDA and covered by patents that are listed in the FDA’s Orange Book, we would be required to submit with our 505(b)(2) application a Paragraph IV Certification in which we must certify that we do not infringe the listed patents or that such patents are invalid or unenforceable, and provide notice to the patent owner or the holder of the approved NDA. The patent owner or NDA holder would have 45 days from receipt of the notification of our Paragraph IV Certification to initiate a patent infringement action against us. If an infringement action is initiated, the approval of our NDA would be subject to a stay of up to 30 months or more while we defend against such a suit. Approval of our product candidates under Section 505(b)(2) may therefore be delayed until patent exclusivity expires or until we successfully challenge the applicability of those patents to our product candidates. Alternatively, we may elect to generate sufficient clinical data so that we would no longer need to rely on third-party data, which would be costly and time consuming and there would be no assurance that such data generated from such additional activities would be sufficient to obtain approval.
-13-
We may not be able to obtain shortened review of our applications, and the FDA may not agree that our product candidates qualify for marketing approval. If we are required to generate additional data to support approval, we may be unable to meet anticipated or reasonable development and commercialization timelines, may be unable to generate the additional data at a reasonable cost, or at all, and may be unable to obtain marketing approval of our product candidates. If the FDA changes its interpretation of Section 505(b)(2) allowing reliance on data in a previously approved drug application owned by a third party, or there is a change in the law affecting Section 505(b)(2), this could delay or even prevent the FDA from approving any Section 505(b)(2) application that we submit.
Modifications to our products may require new NDA approvals.
Once a particular product receives FDA approval or clearance, expanded uses or uses in new indications of our products may require additional human clinical trials and new regulatory approvals or clearances, including additional IND and NDA submissions and premarket approvals before we can begin clinical development, and/or prior to marketing and sales. If the FDA requires new clearances or approvals for a particular use or indication, we may be required to conduct additional clinical studies, which would require additional expenditures and harm our operating results. If the products are already being used for these new indications, we may also be subject to significant enforcement actions. Conducting clinical trials and obtaining clearances and approvals can be a time consuming process, and delays in obtaining required future clearances or approvals could adversely affect our ability to introduce new or enhanced products in a timely manner, which in turn would harm our future growth.
Conducting successful clinical studies may require the enrollment of large numbers of patients, and suitable patients may be difficult to identify and recruit.
Patient enrollment in clinical trials and completion of patient participation and follow-up depends on many factors, including the size of the patient population; the nature of the trial protocol; the attractiveness of, or the discomforts and risks associated with, the treatments received by enrolled subjects; the availability of appropriate clinical trial investigators; support staff; and proximity of patients to clinical sites and ability to comply with the eligibility and exclusion criteria for participation in the clinical trial and patient compliance. For example, patients may be discouraged from enrolling in our clinical trials if the trial protocol requires them to undergo extensive post-treatment procedures or follow-up to assess the safety and effectiveness of our products or if they determine that the treatments received under the trial protocols are not attractive or involve unacceptable risks or discomforts. Patients may also not participate in our clinical trials if they choose to participate in contemporaneous clinical trials of competitive products.
Additional delays to the completion of clinical studies may result from modifications being made to the protocol during the clinical trial, if such modifications are warranted and/or required by the occurrences in the given trial.
Each modification to the protocol during a clinical trial has to be submitted to the FDA. This could result in the delay or halt of a clinical trial while the modification is evaluated. In addition, depending on the quantity and nature of the changes made, the FDA could take the position that the data generated by the clinical trial is not poolable because the same protocol was not used throughout the trial. This might require the enrollment of additional subjects, which could result in the extension of the clinical trial and the FDA delaying clearance or approval of a product. Any such delay could have a material adverse effect on our business and results of operations.
There can be no assurance that the data generated from our clinical trials using modified protocols will be acceptable to FDA.
There can be no assurance that the data generated using modified protocols will be acceptable to the FDA or that if future modifications during the trial are necessary, that any such modifications will be acceptable to the FDA. If the FDA believes that its prior approval is required for a particular modification, it can delay or halt a clinical trial while it evaluates additional information regarding the change.
Serious injury or death resulting from a failure of one of our drug candidates during current or future clinical trials could also result in the FDA delaying our clinical trials or denying or delaying clearance or approval of a product.
-14-
Even though an adverse event may not be the result of the failure of our drug candidate, the FDA or an IRB could delay or halt a clinical trial for an indefinite period of time while an adverse event is reviewed, and likely would do so in the event of multiple such events.
Any delay or termination of our current or future clinical trials as a result of the risks summarized above, including delays in obtaining or maintaining required approvals from IRBs, delays in patient enrollment, the failure of patients to continue to participate in a clinical trial, and delays or termination of clinical trials as a result of protocol modifications or adverse events during the trials, may cause an increase in costs and delays in the filing of any product submissions with the FDA, delay the approval and commercialization of our products or result in the failure of the clinical trial, which could adversely affect our business, operating results and prospects.
If the third parties on which we rely to conduct our clinical trials and to assist us with preclinical development do not perform as contractually required or expected, we may not be able to obtain regulatory approval for or commercialize our products.
We do not have the ability to independently conduct our pre-clinical and clinical trials for our products and we must rely on third parties, such as CROs, medical institutions, clinical investigators and contract laboratories to conduct such trials. If these third parties do not successfully carry out their contractual duties or regulatory obligations, meet expected deadlines or need to be replaced, or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our pre-clinical development activities or clinical trials may be extended, delayed, suspended or terminated, and we may not be able to obtain regulatory approval for, or successfully commercialize, our products on a timely basis, if at all. Furthermore, our third-party clinical trial investigators may be delayed in conducting our clinical trials for reasons outside of their control. The occurrence of any of the foregoing may adversely affect our business, operating results and prospects.
The future results of our current or future clinical trials may not support our product candidate claims or may result in the discovery of unexpected adverse side effects.
Even if our clinical trials are completed as planned, we cannot be certain that their results will support our drug candidate claims or that the FDA or foreign authorities will agree with our conclusions regarding them. Success in preclinical studies and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the later trials will replicate the results of prior trials and preclinical studies. The clinical trial process may fail to demonstrate that our drug candidates are safe and effective for the proposed indicated uses. If the FDA concludes that the clinical trials for BioLexa, or any other product for which we might seek clearance, has failed to demonstrate safety and effectiveness, we would not receive FDA clearance to market that product in the United States for the indications sought.
In addition, such an outcome could cause us to abandon the product candidate and might delay development of others. Any delay or termination of our clinical trials will delay the filing of any product submissions with the FDA and, ultimately, our ability to commercialize our product candidates and generate revenues. It is also possible that patients enrolled in clinical trials will experience adverse side effects that are not currently part of the product candidate’s profile. In addition, our clinical trials for BioLexa involve a relatively small patient population. Because of the small sample size, our results may not be indicative of future results.
BioLexa and future products may never achieve market acceptance.
BioLexa and future products that we may develop may never gain market acceptance among physicians, patients and the medical community. The degree of market acceptance of any of our products will depend on a number of factors, including the actual and perceived effectiveness and reliability of our products; the results of any long-term clinical trials relating to use of our products; the availability, relative cost and perceived advantages and disadvantages of alternative technologies; the degree to which treatments using our products are approved for reimbursement by public and private insurers; the willingness of patients to pay out of pocket in the absence of government or third-party coverage; the strength of our marketing and distribution infrastructure; the level of education and awareness among physicians and hospitals concerning our products; and prevalence and severity of any side effects. Failure of BioLexa or any of our other products to significantly penetrate current or new markets would negatively impact our business, financial condition and results of operations.
-15-
To be commercially successful, physicians must be persuaded that using our products for treatment of eczema are effective alternatives to existing therapies and treatments.
We believe that physicians will not widely adopt our products unless they determine, based on experience, clinical data, and published peer-reviewed journal articles, that the use of our products provides an effective alternative to other means of treating eczema. Patient studies or clinical experience may indicate that treatment with our products does not provide patients with sufficient benefits in quality of life. We believe that recommendations and support for the use of our products from influential physicians will be essential for widespread market acceptance. Our products are still in development and it is premature to attempt to gain support from physicians at this time. We can provide no assurance that such support will ever be obtained. If our products do not receive such support from these physicians and from long-term data, physicians may not use or continue to use, and hospitals may not purchase or continue to purchase, our products.
Even if our products are approved by regulatory authorities, if we or our suppliers fail to comply with ongoing FDA regulation or if we experience unanticipated problems with our products, these products could be subject to restrictions or withdrawal from the market.
Any product for which we obtain clearance or approval, and the manufacturing processes, reporting requirements, post-approval clinical data and promotional activities for such product, will be subject to continued regulatory review, oversight and periodic inspections by the FDA. In particular, we and our suppliers are required to comply with FDA’s Quality System Regulations, or QSR, and International Standards Organization, or ISO, regulations for the manufacture of our products and other regulations which cover the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging, storage and shipping of any product for which we obtain clearance or approval. Regulatory bodies, such as the FDA, enforce these regulations through periodic inspections. The failure by us or one of our suppliers to comply with applicable statutes and regulations administered by the FDA and other regulatory bodies, or the failure to timely and adequately respond to any adverse inspectional observations or product safety issues, could result in, among other things, enforcement actions by the FDA.
If any of these actions were to occur it would harm our reputation and cause our product sales and profitability to suffer and may prevent us from generating revenue. Furthermore, our key component suppliers may not currently be or may not continue to be in compliance with all applicable regulatory requirements which could result in our failure to produce our products on a timely basis and in the required quantities, if at all.
Even if regulatory clearance or approval of a product is granted, such clearance or approval may be subject to limitations on the intended uses for which the product may be marketed and reduce the potential to successfully commercialize the product and generate revenue from the product. If the FDA determines that the product promotional materials, labeling, training or other marketing or educational activities constitute promotion of an unapproved use, it could request that we or our commercialization partners cease or modify our training or promotional materials or subject us to regulatory enforcement actions. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider such training or other promotional materials to constitute promotion of an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement.
In addition, we may be required to conduct costly post-market testing and surveillance to monitor the safety or effectiveness of our products, and we must comply with adverse event and pharmacovigilance reporting requirements, including the reporting of adverse events which occur in connection with, and whether or not directly related to, our products. Later discovery of previously unknown problems with our products, including unanticipated adverse events or adverse events of unanticipated severity or frequency, manufacturing problems, or failure to comply with regulatory requirements, may result in changes to labeling, restrictions on such products or manufacturing processes, withdrawal of the products from the market, voluntary or mandatory recalls, a requirement to recall, replace or refund the cost of any product we manufacture or distribute, fines, suspension of regulatory approvals, product seizures, injunctions or the imposition of civil or criminal penalties which would adversely affect our business, operating results and prospects.
-16-
Our revenue stream will depend upon third-party reimbursement.
The commercial success of our products in both domestic and international markets will be substantially dependent on whether third-party coverage and reimbursement is available for patients that use our products. However, the availability of insurance coverage and reimbursement for newly approved eczema therapies is uncertain, and therefore, third-party coverage may be particularly difficult to obtain even if our products are approved by the FDA as safe and efficacious. Patients using existing approved therapies are generally reimbursed all or part of the product cost by Medicare or other third-party payors. Medicare, Medicaid, health maintenance organizations and other third-party payors are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement of new drugs, and, as a result, they may not cover or provide adequate payment for these products. Submission of applications for reimbursement approval generally does not occur prior to the filing of an NDA for that product and may not be granted for as long as many months after NDA approval. In order to obtain reimbursement arrangements for these products, we or our commercialization partners may have to agree to a net sales price lower than the net sales price we might charge in other sales channels. The continuing efforts of government and third-party payors to contain or reduce the costs of healthcare may limit our revenue. Initial dependence on the commercial success of our products may make our revenues particularly susceptible to any cost containment or reduction efforts.
We will need to make additions to senior management in order to successfully execute our business plan.
The Company will need to identify and recruit prospective executives with proven experience in the biopharmaceutical industry, specifically candidates who have managed and completed FDA-required submissions and clinical trials concerning new products. Robb Knie, the acting Chief Executive Officer, is one of the founders and has agreed to serve in that capacity in the interim. Although his primary background involves electronics and technology, he has experience in venture-level investments and early stage capital formation for emerging growth companies. The Company has entered into an employment agreement with Mr. Knie which includes various provisions that may result in significant financial and severance obligations to the Company. Our inability to recruit and retain executives with proven experience in the biopharmaceutical industry could delay or negatively affect our ability to execute on our business plan, which would have a material adverse effect on our financial condition and results of operation.
Current and future legislation may increase the difficulty and cost for us to obtain marketing approval of and commercialize our product candidates and affect the prices we may obtain for such product candidates.
In the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval for our product candidates, restrict or regulate post-approval activities and affect our ability to profitably sell our product candidates. Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. We do not know whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of our product candidates, if any, may be. In addition, increased scrutiny by the U.S. Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements.
In the United States, the Medicare Modernization Act (“MMA”) changed the way Medicare covers and pays for pharmaceutical products. The legislation expanded Medicare coverage for drug purchases by the elderly and introduced a new reimbursement methodology based on average sales prices for drugs. In addition, this legislation authorized Medicare Part D prescription drug plans to use formularies where they can limit the number of drugs that will be covered in any therapeutic class. As a result of this legislation and the expansion of federal coverage of drug products, we expect that there will be additional pressure to contain and reduce costs. These cost reduction initiatives and other provisions of this legislation could decrease the coverage and price that we receive for our product candidates and could seriously harm our business. While the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates, and any reduction in reimbursement that results from the MMA may result in a similar reduction in payments from private payors.
-17-
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act of 2010 (collectively, the “Health Care Reform Law”) is a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. The Health Care Reform Law revised the definition of “average manufacturer price” for reporting purposes, which could increase the amount of Medicaid drug rebates to states. Further, the law imposed a significant annual fee on companies that manufacture or import branded prescription drug products.
The Health Care Reform Law remains subject to legislative efforts to repeal, modify or delay the implementation of the law. However, if the Health Care Reform Law is repealed or modified, or if implementation of certain aspects of the Health Care Reform Law are delayed, such repeal, modification or delay may materially adversely impact our business, strategies, prospects, operating results or financial condition. We are unable to predict the full impact of any repeal, modification or delay in the implementation of the Health Care Reform Law on us at this time. Due to the substantial regulatory changes that will need to be implemented by the Centers for Medicare & Medicaid Services and others, and the numerous processes required to implement these reforms, we cannot predict which healthcare initiatives will be implemented at the federal or state level, the timing of any such reforms, or the effect such reforms or any other future legislation or regulation will have on our business.
In addition, other legislative changes have been proposed and adopted in the United States since the Health Care Reform Law was enacted. We expect that additional federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, and in turn could significantly reduce the projected value of certain development projects and reduce or eliminate our profitability.
We are dependent on third parties for manufacturing and marketing of our proposed product candidates. If we are not able to secure favorable arrangements with such third parties, our business and financial condition could be harmed.
We will not manufacture any of our proposed product candidates for commercial sale nor do we have the resources necessary to do so. In addition, we currently do not have the capability to market our drug products ourselves. In addition to our internal sales force efforts, we intend to contract with specialized manufacturing companies to manufacture our proposed product candidates and partner with larger pharmaceutical companies for commercialization of our products. In connection with our efforts to commercialize our proposed product candidates, we will seek to secure favorable arrangements with third parties to distribute, promote, market and sell our proposed product candidates. If our internal sales force is unable to successfully distribute, market and promote our product candidates and we are not able to secure favorable commercial terms or arrangements with third parties for the distribution, marketing, promotion and sales of our proposed product candidates, we may have to retain promotional and marketing rights and seek to develop the commercial resources necessary to promote or co-promote or co-market certain or all of our proposed drug candidates to the appropriate channels of distribution in order to reach the specific medical market that we are targeting. We may not be able to enter into any partnering arrangements on this or any other basis. If we are not able to secure favorable partnering arrangements, or are unable to develop the appropriate resources necessary for the commercialization of our proposed product candidates, our business and financial condition could be harmed. In addition, we will have to hire additional employees or consultants, since our current employees have limited experience in these areas. Sufficient employees with relevant skills may not be available to us. Any increase in the number of our employees would increase our expense level, and could have an adverse effect on our financial position.
In addition, we, or our potential commercial partners, may not successfully introduce our proposed product candidates or such candidates may not achieve acceptance by patients, health care providers and insurance companies. Further, it is possible that we may not be able to secure arrangements to manufacture, market, distribute, promote and sell our proposed product candidates at favorable commercial terms that would permit us to make a profit. To the extent that corporate partners conduct clinical trials, we may not be able to control the design and conduct of these clinical trials.
-18-
We may have conflicts with our partners that could delay or prevent the development or commercialization of our product candidates.
We may have conflicts with our partners, such as conflicts concerning the interpretation of preclinical or clinical data, the achievement of milestones, the interpretation of contractual obligations, payments for services, development obligations or the ownership of intellectual property developed during our collaboration. If any conflicts arise with any of our partners, such partner may act in a manner that is adverse to our best interests. Any such disagreement could result in one or more of the following, each of which could delay or prevent the development or commercialization of our product candidates, and in turn prevent us from generating revenues: unwillingness on the part of a partner to pay us milestone payments or royalties we believe are due to us under a collaboration; uncertainty regarding ownership of intellectual property rights arising from our collaborative activities, which could prevent us from entering into additional collaborations; unwillingness by the partner to cooperate in the development or manufacture of the product, including providing us with product data or materials; unwillingness on the part of a partner to keep us informed regarding the progress of its development and commercialization activities or to permit public disclosure of the results of those activities; initiating of litigation or alternative dispute resolution options by either party to resolve the dispute; or attempts by either party to terminate the agreement.
Even if we receive regulatory approval for any of our product candidates, we may not be able to successfully commercialize the product and the revenue that we generate from its sales, if any, may be limited.
If approved for marketing, the commercial success of our product candidates will depend upon each product’s acceptance by the medical community, including physicians, patients and health care payors. The degree of market acceptance for any of our product candidates will depend on a number of factors, including:
| ● | demonstration of clinical safety and efficacy; | |
| ● | relative convenience, dosing burden and ease of administration; | |
| ● | the prevalence and severity of any adverse effects; | |
| ● | the willingness of physicians to prescribe our product candidates, and the target patient population to try new therapies; | |
| ● | efficacy of our product candidates compared to competing products; | |
| ● | the introduction of any new products that may in the future become available targeting indications for which our product candidates may be approved; | |
| ● | new procedures or therapies that may reduce the incidences of any of the indications in which our product candidates may show utility; | |
| ● | pricing and cost-effectiveness; | |
| ● | the inclusion or omission of our product candidates in applicable therapeutic and vaccine guidelines; | |
| ● | the effectiveness of our own or any future collaborators’ sales and marketing strategies; | |
| ● | limitations or warnings contained in approved labeling from regulatory authorities; | |
| ● | our ability to obtain and maintain sufficient third-party coverage or reimbursement from government health care programs, including Medicare and Medicaid, private health insurers and other third-party payors or to receive the necessary pricing approvals from government bodies regulating the pricing and usage of therapeutics; and | |
| ● | the willingness of patients to pay out-of-pocket in the absence of third-party coverage or reimbursement or government pricing approvals. |
-19-
If any of our product candidates are approved, but do not achieve an adequate level of acceptance by physicians, health care payors, and patients, we may not generate sufficient revenue and we may not be able to achieve or sustain profitability. Our efforts to educate the medical community and third-party payors on the benefits of our product candidates may require significant resources and may never be successful.
In addition, even if we obtain regulatory approvals, the timing or scope of any approvals may prohibit or reduce our ability to commercialize our product candidates successfully. For example, if the approval process takes too long, we may miss market opportunities and give other companies the ability to develop competing products or establish market dominance. Any regulatory approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render our product candidates not commercially viable. For example, regulatory authorities may approve any of our product candidates for fewer or more limited indications than we request, may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve any of our product candidates with a label that does not include the labeling claims necessary or desirable for the successful commercialization for that indication. Further, the FDA or comparable foreign regulatory authorities may place conditions on approvals or require risk management plans or a REMS to assure the safe use of the drug. If the FDA concludes a REMS is needed, the sponsor of the NDA must submit a proposed REMS; the FDA will not approve the NDA without an approved REMS, if required. A REMS could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA may also require a REMS for an approved product when new safety information emerges. Any of these limitations on approval or marketing could restrict the commercial promotion, distribution, prescription or dispensing of our product candidates. Moreover, product approvals may be withdrawn for non-compliance with regulatory standards or if problems occur following the initial marketing of the product. Any of the foregoing scenarios could materially harm the commercial success of our product candidates.
Upon commercialization of our products, we may be dependent on third parties to market, distribute and sell our products.
Our ability to receive revenues may be dependent upon the sales and marketing efforts of any future co-marketing partners and third-party distributors. At this time, we have not entered into an agreement with any commercialization partner and only plan to do so after the successful completion of Phase 2 clinical trials and prior to commercialization. If we fail to reach an agreement with any commercialization partner, or upon reaching such an agreement that partner fails to sell a large volume of our products, it may have a negative impact on our business, financial condition and results of operations.
Our products will face significant competition in the markets for such products, and if they are unable to compete successfully, our business will suffer.
Our product candidates face, and will continue to face, intense competition from large pharmaceutical companies, as well as academic and research institutions. We compete in an industry that is characterized by: (i) rapid technological change, (ii) evolving industry standards, (iii) emerging competition and (iv) new product introductions. Our competitors have existing products and technologies that will compete with our products and technologies and may develop and commercialize additional products and technologies that will compete with our products and technologies. Because several competing companies and institutions have greater financial resources than us, they may be able to: (i) provide broader services and product lines, (ii) make greater investments in research and development and (iii) carry on larger research and development initiatives. Our competitors also have greater development capabilities than we do and have substantially greater experience in undertaking preclinical and clinical testing of products, obtaining regulatory approvals, and manufacturing and marketing pharmaceutical products. They also have greater name recognition and better access to customers than us. Our chief competitors include companies such as Pfizer Inc. and Sanofi S.A.
-20-
Adverse events involving our products may lead the FDA to delay or deny clearance for our products or result in product recalls that could harm our reputation, business and financial results.
Once a product receives FDA clearance or approval, the agency has the authority to require the recall of commercialized products in the event of adverse side effects, material deficiencies or defects in design or manufacture. The authority to require a recall must be based on an FDA finding that there is a reasonable probability that the product would cause serious injury or death. Manufacturers may, under their own initiative, recall a product if any material deficiency in a product is found. A government-mandated or voluntary recall by us or one of our distributors could occur as a result of adverse side effects, impurities or other product contamination, manufacturing errors, design or labeling defects or other deficiencies and issues. Recalls of any of our products would divert managerial and financial resources and have an adverse effect on our financial condition and results of operations. The FDA requires that certain classifications of recalls be reported to FDA within ten working days after the recall is initiated. Companies are required to maintain certain records of recalls, even if they are not reportable to the FDA. We may initiate voluntary recalls involving our products in the future that we determine do not require notification of the FDA. If the FDA disagrees with our determinations, they could require us to report those actions as recalls. A future recall announcement could harm our reputation with customers and negatively affect our sales. In addition, the FDA could take enforcement action for failing to report the recalls when they were conducted.
If we fail to comply with healthcare regulations, we could face substantial enforcement actions, including civil and criminal penalties and our business, operations and financial condition could be adversely affected.
We could be subject to healthcare fraud and abuse laws and patient privacy laws of both the federal government and the states in which we conduct our business. The laws include:
| ● | the federal healthcare program anti-kickback law, which prohibits, among other things, persons from soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual, for an item or service or the purchasing or ordering of a good or service, for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs; | |
| ● | federal false claims laws which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payers that are false or fraudulent, and which may apply to entities like us which provide coding and billing information to customers; | |
| ● | the federal Health Insurance Portability and Accountability Act of 1996, which prohibits executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters and which also imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information; | |
| ● | the FDCA which among other things, strictly regulates drug manufacturing and product marketing, prohibits manufacturers from marketing drug products for off-label use and regulates the distribution of drug samples; and | |
| ● | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payer, including commercial insurers, and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by federal laws, thus complicating compliance efforts. |
If our operations are found to be in violation of any of the laws described above or any governmental regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines and the curtailment or restructuring of our operations. Any penalties, damages, fines, curtailment or restructuring of our operations could adversely affect our ability to operate our business and our financial results. Although compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, the risks cannot be entirely eliminated. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert management’s attention from the operation of our business. Moreover, achieving and sustaining compliance with applicable federal and state privacy, security and fraud laws may prove costly.
-21-
If third-party contract manufacturers upon whom we rely to formulate and manufacture our product candidates do not perform, fail to manufacture according to our specifications or fail to comply with strict regulations, our preclinical studies or clinical trials could be adversely affected and the development of our product candidates could be delayed or terminated or we could incur significant additional expenses.
We do not own or operate any manufacturing facilities. We intend to rely on third-party contractors, at least for the foreseeable future, to formulate and manufacture these preclinical and clinical materials. Our reliance on third-party contract manufacturers exposes us to a number of risks, any of which could delay or prevent the completion of our preclinical studies or clinical trials, or the regulatory approval or commercialization of our product candidates, result in higher costs, or deprive us of potential product revenues. Some of these risks include:
| ● | our third-party contractors failing to develop an acceptable formulation to support later-stage clinical trials for, or the commercialization of, our product candidates; | |
| ● | our contract manufacturers failing to manufacture our product candidate according to their own standards, our specifications, the FDA’s cGMP requirements, or otherwise manufacturing material that we or the FDA may deem to be unsuitable in our clinical trials; | |
| ● | our contract manufacturers being unable to increase the scale of, increase the capacity for, or reformulate the form of our product candidates. We may experience a shortage in supply, or the cost to manufacture our products may increase to the point where it adversely affects the cost of our product candidates. We cannot assure you that our contract manufacturers will be able to manufacture our product candidates at a suitable scale, or we will be able to find alternative manufacturers acceptable to us that can do so; | |
| ● | our contract manufacturers placing a priority on the manufacture of their own products, or other customers’ products; | |
| ● | our contract manufacturers failing to perform as agreed or not remain in the contract manufacturing business; and | |
| ● | our contract manufacturers’ plants being closed as a result of regulatory sanctions or a natural disaster. |
Manufacturers of pharmaceutical products are subject to ongoing periodic inspections by the FDA, the U.S. Drug Enforcement Administration and corresponding state and foreign agencies to ensure strict compliance with FDA-mandated cGMPs, other government regulations and corresponding foreign standards. While we are obligated to audit their performance, we do not have control over our third-party contract manufacturers’ compliance with these regulations and standards. Failure by our third-party manufacturers, or us, to comply with applicable regulations could result in sanctions being imposed on us or the drug manufacturer from the production of other third-party products. These sanctions may include fines, injunctions, civil penalties, failure of the government to grant pre-market approval of drugs, delays, suspension or withdrawal of approvals, seizures or recalls of product, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect our business.
In the event that we need to change our third-party contract manufacturers, our preclinical studies, clinical trials or the commercialization of our product candidate could be delayed, adversely affected or terminated, or such a change may result in significantly higher costs.
Various steps in the manufacture of our product candidate may need to be sole-sourced. In accordance with cGMPs, changing manufacturers may require the re-validation of manufacturing processes and procedures, and may require further preclinical studies or clinical trials to show comparability between the materials produced by different manufacturers. Changing our current or future contract manufacturers may be difficult for us and could be costly, which could result in our inability to manufacture our product candidates for an extended period of time and therefore a delay in the development of our product candidates. Further, in order to maintain our development time lines in the event of a change in our third-party contract manufacturer, we may incur significantly higher costs to manufacture our product candidates.
-22-
Healthcare Reform in the United States.
In the United States, there have been, and continue to be, a number of legislative and regulatory changes and proposed changes to the healthcare system that could affect the future results of pharmaceutical manufactures’ operations. In particular, there have been and continue to be a number of initiatives at the federal and state levels that seek to reduce healthcare costs. Most recently, the Patient Protection and Affordable Care Act (“PPACA”) was enacted in March 2010, which includes measures to significantly change the way healthcare is financed by both governmental and private insurers. Among the provisions of the PPACA of greatest importance to the pharmaceutical and biotechnology industry are the following:
| ● | an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare programs; | |
| ● | implementation of the federal physician payment transparency requirements, sometimes referred to as the “Physician Payments Sunshine Act”; | |
| ● | a licensure framework for follow-on biologic products; | |
| ● | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; | |
| ● | establishment of a Center for Medicare Innovation at the Centers for Medicare & Medicaid Services to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending; | |
| ● | an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program, to 23.1% and 13% of the average manufacturer price for most branded and generic drugs, respectively and capped the total rebate amount for innovator drugs at 100% of the Average Manufacturer Price; | |
| ● | a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for certain drugs and biologics, including our product candidates, that are inhaled, infused, instilled, implanted or injected; | |
| ● | extension of manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; | |
| ● | expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for individuals with income at or below 133% of the federal poverty level, thereby potentially increasing manufacturers’ Medicaid rebate liability; | |
| ● | a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; and | |
| ● | expansion of the entities eligible for discounts under the Public Health program. |
-23-
Some of the provisions of the PPACA have yet to be implemented, and there have been legal and political challenges to certain aspects of the PPACA. Since January 2017, President Trump has signed two executive orders and other directives designed to delay, circumvent, or loosen certain requirements mandated by the PPACA. Concurrently, Congress has considered legislation that would repeal or repeal and replace all or part of the PPACA. While Congress has not passed repeal legislation, the Tax Cuts and Jobs Act of 2017 includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the PPACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate”. Congress may consider other legislation to repeal or replace elements of the PPACA.
Many of the details regarding the implementation of the PPACA are yet to be determined, and at this time, the full effect that the PPACA would have on a pharmaceutical manufacturer remains unclear. In particular, there is uncertainty surrounding the applicability of the biosimilars provisions under the PPACA. The FDA has issued several guidance documents, but no implementing regulations, on biosimilars. A number of biosimilar applications have been approved over the past few years. The regulations that are ultimately promulgated and their implementation are likely to have considerable impact on the way pharmaceutical manufacturers conduct their business and may require changes to current strategies. A biosimilar is a biological product that is highly similar to an approved drug notwithstanding minor differences in clinically inactive components, and for which there are no clinically meaningful differences between the biological product and the approved drug in terms of the safety, purity, and potency of the product.
Individual states have become increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access, and marketing cost disclosure and transparency measures, and to encourage importation from other countries and bulk purchasing. Legally mandated price controls on payment amounts by third-party payors or other restrictions could harm a pharmaceutical manufacturer’s business, results of operations, financial condition and prospects. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs. This could reduce ultimate demand for certain products or put pressure product pricing, which could negatively affect a pharmaceutical manufacturer’s business, results of operations, financial condition and prospects.
In addition, given recent federal and state government initiatives directed at lowering the total cost of healthcare, Congress and state legislatures will likely continue to focus on healthcare reform, the cost of prescription drugs and biologics and the reform of the Medicare and Medicaid programs. While no one cannot predict the full outcome of any such legislation, it may result in decreased reimbursement for drugs and biologics, which may further exacerbate industry-wide pressure to reduce prescription drug prices. This could harm a pharmaceutical manufacturer’s ability to generate revenue. Increases in importation or re-importation of pharmaceutical products from foreign countries into the United States could put competitive pressure on a pharmaceutical manufacturer’s ability to profitably price products, which, in turn, could adversely affect business, results of operations, financial condition and prospects. A pharmaceutical manufacturer might elect not to seek approval for or market products in foreign jurisdictions in order to minimize the risk of re-importation, which could also reduce the revenue generated from product sales. It is also possible that other legislative proposals having similar effects will be adopted.
Furthermore, regulatory authorities’ assessment of the data and results required to demonstrate safety and efficacy can change over time and can be affected by many factors, such as the emergence of new information, including on other products, changing policies and agency funding, staffing and leadership. We cannot be sure whether future changes to the regulatory environment will be favorable or unfavorable to our business prospects. For example, average review times at the FDA for marketing approval applications can be affected by a variety of factors, including budget and funding levels and statutory, regulatory and policy changes.
Inadequate funding for the FDA, the SEC and other government agencies could hinder their ability to hire and retain key leadership and other personnel, prevent new products and services from being developed or commercialized in a timely manner, affect whether government agencies promptly pay amounts awarded under grants from such agencies, or otherwise prevent those agencies from performing normal business functions on which the operation of our business may rely, which could negatively impact our business.
The ability of the FDA to review and approve new drugs and medical devices can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory, and policy changes. Average review times at the FDA have fluctuated in recent years as a result. In addition, government funding of the SEC and other government agencies on which our operations may rely, including those that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable.
-24-
Disruptions at the FDA and other agencies may also slow the time necessary for new drugs and medical devices to be reviewed and/or approved by necessary government agencies as well as affect whether we receive timely payment of amounts awarded to us under grants and contracts with government agencies which would adversely affect our business. For example, over the last several years, including from December 22, 2018 until January 25, 2019, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA and the SEC, have had to furlough critical FDA, SEC and other government employees and stop critical activities. If a prolonged government shutdown occurs, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business. Further, in our operations as a public company, future government shutdowns could impact our ability to access the public markets and obtain necessary capital in order to properly capitalize and continue our operations.
Security threats to our information technology infrastructure and/or our physical buildings could expose us to liability and damage our reputation and business.
It is essential to our business strategy that our technology and network infrastructure and our physical buildings remain secure and are perceived by our customers and corporate partners to be secure. Despite security measures, however, any network infrastructure may be vulnerable to cyber-attacks by hackers and other security threats. We may face cyber-attacks that attempt to penetrate our network security, sabotage or otherwise disable our research, products and services, misappropriate our or our customers’ and partners’ proprietary information, which may include personally identifiable information, or cause interruptions of our internal systems and services. Despite security measures, we also cannot guarantee security of our physical buildings. Physical building penetration or any cyber-attacks could negatively affect our reputation, damage our network infrastructure and our ability to deploy our products and services, harm our relationship with customers and partners that are affected, and expose us to financial liability.
Additionally, there are a number of state, federal and international laws protecting the privacy and security of health information and personal data. For example, the HIPAA imposes limitations on the use and disclosure of an individual’s healthcare information by healthcare providers, healthcare clearinghouses, and health insurance plans, or, collectively, covered entities, and also grants individuals rights with respect to their health information. HIPAA also imposes compliance obligations and corresponding penalties for non-compliance on individuals and entities that provide services to healthcare providers and other covered entities. As part of the American Recovery and Reinvestment Act of 2009 (“ARRA”) the privacy and security provisions of HIPAA were amended. ARRA also made significant increases in the penalties for improper use or disclosure of an individual’s health information under HIPAA and extended enforcement authority to state attorneys general. As amended by ARRA and subsequently by the final omnibus rule adopted in 2013, HIPAA also imposes notification requirements on covered entities in the event that certain health information has been inappropriately accessed or disclosed, notification requirements to individuals, federal regulators, and in some cases, notification to local and national media. Notification is not required under HIPAA if the health information that is improperly used or disclosed is deemed secured in accordance with encryption or other standards developed by the U.S. Department of Health and Human Services. Most states have laws requiring notification of affected individuals and/or state regulators in the event of a breach of personal information, which is a broader class of information than the health information protected by HIPAA. Many state laws impose significant data security requirements, such as encryption or mandatory contractual terms, to ensure ongoing protection of personal information. Activities outside of the U.S. implicate local and national data protection standards, impose additional compliance requirements and generate additional risks of enforcement for non-compliance. We may be required to expend significant capital and other resources to ensure ongoing compliance with applicable privacy and data security laws, to protect against security breaches and hackers or to alleviate problems caused by such breaches.
-25-
Risks Relating to Our Intellectual Property Rights
We rely on an exclusive sublicense granted to us by Chelexa with respect to the BioLexa Platform, an exclusive license granted to us by the University of Cincinnati with respect to a genetic marker for food allergies and an exclusive sublicense granted to us by Zylö in connection with the development of a treatment for patients suffering from Cutaneous Lupus Erythematosus (“CLE”), and if Chelexa, the University of Cincinnati and/or Zylö do not adequately defend such licenses, our business may be harmed.
Our primary asset is a sublicense agreement with Chelexa pursuant to which Chelexa has granted us an exclusive sublicense to use its BioLexa Platform, a proprietary, patented, drug compound platform developed at the University of Cincinnati. The license enables us to develop the platform for any indications in humans. In addition, we entered into an exclusive license agreement with the University of Cincinnati with respect to a patented, novel genetic marker for food allergies. We also entered into the Sublicense Agreement with Zylö in connection with the development of a treatment for patients suffering from CLE including patents with respect thereto developed by Albert Einstein College of Medicine. We rely on the Chelexa, the University of Cincinnati, Zylö and Albert Einstein College of Medicine to maintain the patents with respect to the BioLexa Platform, the genetic marker and CLE and otherwise protect the intellectual property covered by our exclusive license and sublicenses. We have limited control over the activities of Chelexa, the University of Cincinnati, Zylö and Albert Einstein College of Medicine or over any other intellectual property that may be related to the BioLexa Platform, the genetic marker or CLE. For example, we cannot be certain that activities by Chelexa, the University of Cincinnati, Zylö or Albert Einstein College of Medicine have been or will be conducted in compliance with applicable laws and regulations. We may have no control or input over whether, and in what manner, the University of Cincinnati and/or Albert Einstein College of Medicine may enforce or defend the patents against a third-party. The University of Cincinnati and/or Albert Einstein College of Medicine may enforce or defend the patent less vigorously than if we had enforced or defended the patents ourselves. Further, the University of Cincinnati and/or Albert Einstein College of Medicine may not necessarily seek enforcement in scenarios in which we would feel that enforcement was in our best interests. For example, the University of Cincinnati and/or Albert Einstein College of Medicine may not enforce the patents against a competitor of ours who is not a direct competitor of the University of Cincinnati or Albert Einstein College of Medicine, as applicable. If our in-licensed intellectual property is found to be invalid or unenforceable, then the University of Cincinnati and/or Albert Einstein College of Medicine may not be able to enforce the patents against a competitor of ours. If we fail to meet our obligations under the sublicense agreement with Chelexa or Chelexa fails to meet its obligations under its license agreement with the University of Cincinnati, then the University of Cincinnati may terminate the license agreement with Chelexa thereby terminating our sublicense agreement with Chelexa, and we will be unable to conduct our business. Similarly, if we fail to meet our obligations under the sublicense agreement with Zylö or Zylö fails to meet its obligations under its license agreement with Albert Einstein College of Medicine, then Albert Einstein College of Medicine may terminate the license agreement with Zylö thereby terminating our sublicense agreement with Zylö, and we will be unable to conduct our business with respect to the development of treatment for patients suffering from CLE. n addition, if we fail to meet our obligations under the license agreement with the University of Cincinnati, then the University of Cincinnati may terminate the license agreement, and we will be unable to continue to use the genetic marker in conducing our business. Although we may choose to terminate our sublicense agreement with Chelexa or Zylö or our license agreement with the University of Cincinnati, doing so would allow a third party to seek and obtain an exclusive license to the BioLexa Platform, the genetic marker and the patents relating to CLE. If a third party obtains an exclusive license to intellectual property with respect to the BioLexa Platform, the genetic marker or CLE formerly licensed to us, then the third party may seek to enforce the intellectual property against us which may have a material adverse effect on our business.
We are dependent upon our sublicense agreement with Chelexa with respect to the BioLexa Platform and Zylö with respect the development of a treatment for patients suffering from CLE; however, we have no control over the license agreement between Chelexa and the University of Cincinnati or the license agreement between Zylö and Albert Einstein College of Medicine.
Our sublicense agreements with Chelexa and Zylö are subject to many risks and uncertainties. Although we are dependent upon our sublicense agreement with Chelexa with respect to the BioLexa Platform and Zylö with respect the development of a treatment for patients suffering from CLE, we have no control over the license agreement between Chelexa and the University of Cincinnati pursuant to which the University of Cincinnati licensed the BioLexa Platform to Chelexa or the license agreement between Zylö and Albert Einstein College of Medicine pursuant to which Albert Einstein College of Medicine licensed certain patent rights relating to CLE to Zylö. In the event that Chelexa is unable to fulfill its obligations to the University of Cincinnati pursuant to the terms of its license agreement, the University of Cincinnati may terminate the license thereby voiding our sublicense. Similarly, in the event that Zylö is unable to fulfill its obligations to Albert Einstein College of Medicine pursuant to the terms of its license agreement, Albert Einstein College of Medicine may terminate the license thereby voiding our sublicense. In the event that either the license agreement between Chelexa and the University of Cincinnati or the license agreement between Zylö and Albert Einstein College of Medicine is terminated, there may be a material adverse effect upon our business.
-26-
Our business depends upon securing and protecting critical intellectual property.
Although we do not own and only license intellectual property, to the extent we develop intellectual property, our commercial success will depend in part on obtaining and maintaining patent, trade secret, copyright and trademark protection of our technologies in the United States and other jurisdictions as well as successfully enforcing and defending such intellectual property rights against third-party challenges. We will only be able to protect our intellectual property from unauthorized use by third parties to the extent that valid and enforceable intellectual property protection, such as patents or trade secrets, cover them. In particular, we place considerable emphasis on obtaining patent and trade secret protection for significant new technologies, products and processes. Furthermore, the degree of future protection of our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. Moreover, the degree of future protection of our proprietary rights is uncertain for products that are currently in the early stages of development because we cannot predict which of these products will ultimately reach the commercial market or whether the commercial versions of these products will incorporate proprietary technologies.
Patent positions in our industry are highly uncertain and involves complex legal and factual questions.
Patent positions in our industry are highly uncertain and involves complex legal and factual questions. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in our patents or in third-party patents. For example, we or our licensors might not have been the first to make the inventions covered by our pending patent applications and issued patents, as applicable; we or our licensors might not have been the first to file patent applications for these inventions; others may independently develop similar or alternative technologies or duplicate any of our technologies; it is possible that none of our pending patent applications or the pending patent applications of our licensors will result in issued patents; our issued patents and issued patents of our licensors may not provide a basis for commercially viable technologies, or may not provide us with any competitive advantages, or may be challenged and invalidated by third parties; and, we may not develop additional proprietary technologies that are patentable.
As a result, our owned and licensed patents may not be valid and we may not be able to obtain and enforce patents and to maintain trade secret protection for the full commercial extent of our technology. The extent to which we are unable to do so could materially harm our business.
We or our licensors have applied for and will continue to apply for patents for certain products. Such applications may not result in the issuance of any patents, and any patents now held or that may be issued may not provide us with adequate protection from competition. Furthermore, it is possible that patents issued or licensed to us may be challenged successfully. In that event, if we have a preferred competitive position because of such patents, any preferred position held by us would be lost. If we are unable to secure or to continue to maintain a preferred position, we could become subject to competition from the sale of generic products. Failure to receive, inability to protect, or expiration of our patents for medical use, manufacture, conjugation and labeling of BioLexa, the product platform that we license from Chelexa, or subsequent related filings, would adversely affect our business and operations.
Patents issued or licensed to us may be infringed by the products or processes of others. The cost of enforcing our patent rights against infringers, if such enforcement is required, could be significant, and we do not currently have the financial resources to fund such litigation. Further, such litigation can go on for years and the time demands could interfere with our normal operations. There has been substantial litigation and other proceedings regarding patent and other intellectual property rights in the pharmaceutical industry. We may become a party to patent litigation and other proceedings. The cost to us of any patent litigation, even if resolved in our favor, could be substantial. Some of our competitors may be able to sustain the costs of such litigation more effectively than we can because of their substantially greater financial resources. Litigation may also absorb significant management time.
-27-
Unpatented trade secrets, improvements, confidential know-how and continuing technological innovation are important to our scientific and commercial success. Although we attempt to and will continue to attempt to protect our proprietary information through reliance on trade secret laws and the use of confidentiality agreements with our corporate partners, collaborators, employees and consultants and other appropriate means, these measures may not effectively prevent disclosure of our proprietary information, and, in any event, others may develop independently, or obtain access to, the same or similar information.
The patent rights for our primary product are licensed to us by third parties. If we fail to comply with the terms of these license agreements, our rights to those patents may be terminated, and we will be unable to conduct our business.
If we are found to be infringing on patents or trade secrets owned by others, we may be forced to cease or alter our product development efforts, obtain a license to continue the development or sale of our products, and/or pay damages.
Our manufacturing processes and potential products may violate proprietary rights of patents that have been or may be granted to competitors, universities or others, or the trade secrets of those persons and entities. As the pharmaceutical industry expands and more patents are issued, the risk increases that our processes and potential products may give rise to claims that they infringe the patents or trade secrets of others. These other persons could bring legal actions against us claiming damages and seeking to enjoin clinical testing, manufacturing and marketing of the affected product or process. If any of these actions are successful, in addition to any potential liability for damages, we could be required to obtain a license in order to continue to conduct clinical tests, manufacture or market the affected product or use the affected process. Required licenses may not be available on acceptable terms, if at all, and the results of litigation are uncertain. If we become involved in litigation or other proceedings, it could consume a substantial portion of our financial resources and the efforts of our personnel.
Our ability to protect and enforce any patents we may obtain does not guaranty that we will secure the right to commercialize such patents.
A patent is a limited monopoly right conferred upon an inventor, and his successors in title, in return for the making and disclosing of a new and non-obvious invention. This monopoly is of limited duration but, while in force, allows the patent holder to prevent others from making and/or using his invention. While a patent gives the holder this right to exclude others, it is not a license to commercialize the invention, where other permissions may be required for permissible commercialization to occur. For example, a drug cannot be marketed without the appropriate authorization from the FDA, regardless of the existence of a patent covering the product. Further, the invention, even if patented itself, cannot be commercialized if it infringes the valid patent rights of another party.
We rely on confidentiality agreements to protect our trade secrets. If these agreements are breached by our employees or other parties, our trade secrets may become known to our competitors.
We rely on trade secrets which we seek to protect through confidentiality agreements with our employees and other parties. If these agreements are breached, our competitors may obtain and use our trade secrets to gain a competitive advantage over us. We may not have any remedies against our competitors and any remedies that may be available to us may not be adequate to protect our business or compensate us for the damaging disclosure. In addition, we may have to expend resources to protect our interests from possible infringement by others.
Related Risks to the Company
We may expand our business through the acquisition of rights to new drug candidates that could disrupt our business, harm our financial condition and may also dilute current shareholders’ ownership interests in our Company.
Our business strategy includes expanding our products and capabilities, and we may seek acquisitions of drug candidates or technologies to do so. Acquisitions involve numerous risks, including substantial cash expenditures; potentially dilutive issuance of equity securities; incurrence of debt and contingent liabilities, some of which may be difficult or impossible to identify at the time of acquisition; difficulties in assimilating the acquired technologies or the operations of the acquired companies; diverting our management’s attention away from other business concerns; risks of entering markets in which we have limited or no direct experience; and the potential loss of our key employees or key employees of the acquired companies.
-28-
We cannot assure you that any acquisition will result in short-term or long-term benefits to us. We may misjudge the value or worth of an acquired product, company or business. In addition, our future success would depend in part on our ability to manage the rapid growth associated with acquisitions. We cannot assure you that we will be able to make the combination of our business with that of acquired products, businesses or companies work or be successful. Furthermore, the development or expansion of our business or any acquired products, business or companies may require a substantial capital investment by us. We may not have these necessary funds or they might not be available to us on acceptable terms or at all. We may also seek to raise funds by selling shares of our preferred or common stock, which could dilute each current shareholder’s ownership interest in the Company.
We may undertake international operations, which will subject us to risks inherent with operations outside of the United States.
Although we do not have any foreign operations at this time, we intend to seek to obtain market clearances in foreign markets that we deem to generate significant opportunities. However, even with the cooperation of a commercialization partner, conducting drug development in foreign countries involves inherent risks, including, but not limited to: difficulties in staffing, funding and managing foreign operations; unexpected changes in regulatory requirements; export restrictions; tariffs and other trade barriers; difficulties in protecting, acquiring, enforcing and litigating intellectual property rights; fluctuations in currency exchange rates; and potentially adverse tax consequences.
If we were to experience any of the difficulties listed above, or any other difficulties, any international development activities and our overall financial condition may suffer and cause us to reduce or discontinue our international development and registration efforts.
We may not be successful in hiring and retaining key employees, including executive officers.
Our future operations and successes depend in large part upon the strength of our management team. We rely heavily on the continued service of Robb Knie, our President and Chief Executive Officer. Accordingly, if Mr. Knie terminates his employment with us, such a departure may have a material adverse effect on our business, and our future success depends on our ability to identify, attract, hire or engage, retain and motivate other well-qualified financial, managerial, technical, clinical and regulatory personnel. There can be no assurance that these professionals will be available in the market, or that we will be able to retain existing professionals or to meet or to continue to meet their compensation requirements. Furthermore, the cost base in relation to such compensation, which may include equity compensation, may increase significantly, which could have a material adverse effect on us. Failure to establish and maintain an effective management team and work force could adversely affect our ability to operate, grow and manage our business.
Managing our growth as we expand operations may strain our resources.
We expect to grow rapidly in order to support additional, larger, and potentially international, pivotal clinical trials of our drug candidates, which will place a significant strain on our financial, managerial and operational resources. In order to achieve and manage growth effectively, we must continue to improve and expand our operational and financial management capabilities. Moreover, we will need to increase staffing and to train, motivate and manage our employees. All of these activities will increase our expenses and may require us to raise additional capital sooner than expected. Failure to manage growth effectively could harm our business, financial condition or results of operations.
-29-
If a product liability claim is successfully brought against us for uninsured liabilities, or such claim exceeds our insurance coverage, we could be forced to pay substantial damage awards that could materially harm our business.
The use of any of our existing or future product candidates in clinical trials and the sale of any approved pharmaceutical products may expose us to significant product liability claims. We currently do not have product liability insurance coverage but we intend to obtain such insurance. Such insurance coverage may not protect us against any or all of the product liability claims that may be brought against us in the future. We may not be able to acquire or maintain adequate product liability insurance coverage at a commercially reasonable cost or in sufficient amounts or scope to protect us against potential losses. In the event a product liability claim is brought against us, we may be required to pay legal and other expenses to defend the claim, as well as uncovered damage awards resulting from a claim brought successfully against us. In the event our product candidate is approved for sale by the FDA and commercialized, we may need to substantially increase the amount of our product liability coverage. Defending any product liability claim or claims could require us to expend significant financial and managerial resources, which could have an adverse effect on our business.
Risks Related to Our Common Stock
The price of our common stock may fluctuate substantially.
You should consider an investment in our common stock to be risky, and you should invest in our common stock only if you can withstand a significant loss and wide fluctuations in the market value of your investment. Some factors that may cause the market price of our common stock to fluctuate, in addition to the other risks mentioned in this “Risk Factors” section and elsewhere in this Annual Report on Form 10-K, are:
| ● | sale of our common stock by our shareholders, executives, and directors; | |
| ● | volatility and limitations in trading volumes of our shares of common stock; | |
| ● | our ability to obtain financings to conduct and complete research and development activities including, but not limited to, our clinical trials, and other business activities; | |
| ● | the timing and success of introductions of new products by us or our competitors or any other change in the competitive dynamics of our industry, including consolidation among competitors; | |
| ● | our ability to attract new customers; | |
| ● | our ability to secure resources and the necessary personnel to conduct clinical trials on our desired schedule; | |
| ● | commencement, enrollment or results of our clinical trials for our product candidates or any future clinical trials we may conduct; | |
| ● | changes in the development status of our product candidates; | |
| ● | any delays or adverse developments or perceived adverse developments with respect to the FDA’s review of our planned preclinical and clinical trials; | |
| ● | any delay in our submission for studies or product approvals or adverse regulatory decisions, including failure to receive regulatory approval for our product candidates; | |
| ● | unanticipated safety concerns related to the use of our product candidates; | |
| ● | changes in our capital structure or dividend policy, future issuances of securities, sales of large blocks of common stock by our shareholders; | |
| ● | our cash position; | |
| ● | announcements and events surrounding financing efforts, including debt and equity securities; |
-30-
| ● | our inability to enter into new markets or develop new products; | |
| ● | reputational issues; | |
| ● | announcements of acquisitions, partnerships, collaborations, joint ventures, new products, capital commitments, or other events by us or our competitors; | |
| ● | changes in general economic, political and market conditions in or any of the regions in which we conduct our business; | |
| ● | changes in industry conditions or perceptions; | |
| ● | analyst research reports, recommendation and changes in recommendations, price targets, and withdrawals of coverage; | |
| ● | departures and additions of key personnel; |
| ● | disputes and litigations related to intellectual properties, proprietary rights, and contractual obligations; | |
| ● | changes in applicable laws, rules, regulations, or accounting practices and other dynamics; and | |
| ● | other events or factors, many of which may be out of our control. |
In addition, if the market for stocks in our industry or industries related to our industry, or the stock market in general, experiences a loss of investor confidence, the trading price of our common stock could decline for reasons unrelated to our business, financial condition and results of operations. If any of the foregoing occurs, it could cause our stock price to fall and may expose us to lawsuits that, even if unsuccessful, could be costly to defend and a distraction to management.
We may acquire other companies or technologies, which could divert our management’s attention, result in dilution to our stockholders and otherwise disrupt our operations and adversely affect our operating results.
We may in the future seek to acquire or invest in businesses, applications and services or technologies that we believe could complement or expand our services, enhance our technical capabilities or otherwise offer growth opportunities. The pursuit of potential acquisitions may divert the attention of management and cause us to incur various expenses in identifying, investigating and pursuing suitable acquisitions, whether or not they are consummated.
In addition, we do not have any experience in acquiring other businesses. If we acquire additional businesses, we may not be able to integrate the acquired personnel, operations and technologies successfully, or effectively manage the combined business following the acquisition. We also may not achieve the anticipated benefits from the acquired business due to a number of factors, including:
| ● | inability to integrate or benefit from acquired technologies or services in a profitable manner; | |
| ● | unanticipated costs or liabilities associated with the acquisition; | |
| ● | difficulty integrating the accounting systems, operations and personnel of the acquired business; | |
| ● | difficulties and additional expenses associated with supporting legacy products and hosting infrastructure of the acquired business; | |
| ● | difficulty converting the customers of the acquired business onto our platform and contract terms, including disparities in the revenue, licensing, support or professional services model of the acquired company; |
-31-
| ● | diversion of management’s attention from other business concerns; | |
| ● | adverse effects to our existing business relationships with business partners and customers as a result of the acquisition; | |
| ● | the potential loss of key employees; | |
| ● | use of resources that are needed in other parts of our business; and | |
| ● | use of substantial portions of our available cash to consummate the acquisition. |
In addition, a significant portion of the purchase price of companies we acquire may be allocated to acquired goodwill and other intangible assets, which must be assessed for impairment at least annually. In the future, if our acquisitions do not yield expected returns, we may be required to take charges to our operating results based on this impairment assessment process, which could adversely affect our results of operations.
Acquisitions could also result in dilutive issuances of equity securities or the incurrence of debt, which could adversely affect our operating results. In addition, if an acquired business fails to meet our expectations, our operating results, business and financial position may suffer.
Market and economic conditions may negatively impact our business, financial condition and share price.
Concerns over inflation, energy costs, geopolitical issues, the U.S. mortgage market and a declining real estate market, unstable global credit markets and financial conditions, and volatile oil prices have led to periods of significant economic instability, diminished liquidity and credit availability, declines in consumer confidence and discretionary spending, diminished expectations for the global economy and expectations of slower global economic growth going forward, increased unemployment rates, and increased credit defaults in recent years. Our general business strategy may be adversely affected by any such economic downturns, volatile business environments and continued unstable or unpredictable economic and market conditions. If these conditions continue to deteriorate or do not improve, it may make any necessary debt or equity financing more difficult to complete, more costly, and more dilutive. Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our growth strategy, financial performance, and share price and could require us to delay or abandon development or commercialization plans.
If securities or industry analysts do not publish research or reports, or publish unfavorable research or reports about our business, our stock price and trading volume may decline.
The trading market for our common stock will rely in part on the research and reports that industry or financial analysts publish about us, our business, our markets and our competitors. We do not control these analysts. If securities analysts do not cover our common stock, the lack of research coverage may adversely affect the market price of our common stock. Furthermore, if one or more of the analysts who do cover us downgrade our stock or if those analysts issue other unfavorable commentary about us or our business, our stock price would likely decline. If one or more of these analysts cease coverage of us or fails to regularly publish reports on us, we could lose visibility in the market and interest in our stock could decrease, which in turn could cause our stock price or trading volume to decline and may also impair our ability to expand our business with existing customers and attract new customers.
Because certain of our shareholders control a significant number of shares of our common stock, they may have effective control over actions requiring shareholder approval.
As of August 27, 2019, our directors and executive officers and their respective affiliates, beneficially own approximately 34.44% of our outstanding shares of common stock. As a result, these shareholders acting together, would have the ability to control the outcome of matters submitted to our shareholders for approval, including the election of directors and any merger, consolidation or sale of all or substantially all of our assets. In addition, these shareholders, acting together, would have the ability to control the management and affairs of our Company. Accordingly, this concentration of ownership might harm the market price of our common stock by:
| ● | delaying, deferring or preventing a change in corporate control; | |
| ● | impeding a merger, consolidation, takeover or other business combination involving us; or | |
| ● | discouraging a potential acquirer from making a tender offer or otherwise attempting to obtain control of us. |
-32-
Future sales and issuances of our securities could result in additional dilution of the percentage ownership of our shareholders and could cause our share price to fall.
We expect that significant additional capital will be needed in the future to continue our planned operations, including research and development, increased marketing, hiring new personnel, commercializing our products, and continuing activities as an operating public company. To the extent we raise additional capital by issuing equity securities, our shareholders may experience substantial dilution. We may sell common stock, convertible securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time. If we sell common stock, convertible securities or other equity securities in more than one transaction, investors may be materially diluted by subsequent sales. Such sales may also result in material dilution to our existing shareholders, and new investors could gain rights superior to our existing shareholders.
We do not intend to pay cash dividends on our shares of common stock so any returns will be limited to the value of our shares.
We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future. Any return to shareholders will therefore be limited to the increase, if any, of our share price.
We are an “emerging growth company” and will be able to avail ourselves of reduced disclosure requirements applicable to emerging growth companies, which could make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the JOBS Act, and we intend to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” including not being required to comply with the auditor attestation requirements of Section 404(b) of Sarbanes-Oxley, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. In addition, pursuant to Section 107 of the JOBS Act, as an “emerging growth company” we intend to take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile. We may take advantage of these reporting exemptions until we are no longer an “emerging growth company.” We will remain an “emerging growth company” until the earliest of (i) the last day of the fiscal year in which we have total annual gross revenues of $1.07 billion or more; (ii) the last day of our fiscal year following the fifth anniversary of the date of our initial public offering; (iii) the date on which we have issued more than $1 billion in nonconvertible debt during the previous three years; or (iv) the date on which we are deemed to be a large accelerated filer under the rules of the SEC.
We may be at risk of securities class action litigation.
We may be at risk of securities class action litigation. In the past, biotechnology and pharmaceutical companies have experienced significant stock price volatility, particularly when associated with binary events such as clinical trials and product approvals. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business and results in a decline in the market price of our common stock.
-33-
Financial reporting obligations of being a public company in the United States are expensive and time-consuming, and our management will be required to devote substantial time to compliance matters.
As a publicly traded company we incur significant legal, accounting and other expenses. The obligations of being a public company in the United States require significant expenditures and places significant demands on our management and other personnel, including costs resulting from public company reporting obligations under the Exchange Act and the rules and regulations regarding corporate governance practices, including those under Sarbanes-Oxley, the Dodd-Frank Wall Street Reform and Consumer Protection Act, and the listing requirements of The Nasdaq Capital Market. These rules require the establishment and maintenance of effective disclosure and financial controls and procedures, internal control over financial reporting and changes in corporate governance practices, among many other complex rules that are often difficult to implement, monitor and maintain compliance with. Moreover, despite recent reforms made possible by the JOBS Act, the reporting requirements, rules, and regulations will make some activities more time-consuming and costly, particularly after we are no longer an “emerging growth company.” In addition, we expect these rules and regulations to make it more difficult and more expensive for us to obtain director and officer liability insurance. Our management and other personnel will need to devote a substantial amount of time to ensure that we comply with all of these requirements and to keep pace with new regulations, otherwise we may fall out of compliance and risk becoming subject to litigation or being delisted, among other potential problems.
If we fail to comply with the rules under Sarbanes-Oxley related to internal controls and procedures in the future, or, if we discover material weaknesses and other deficiencies in our internal controls over financial reporting, our stock price could decline significantly and raising capital could be more difficult.
Section 404 of Sarbanes-Oxley requires annual management assessments of the effectiveness of our internal controls over financial reporting. If we fail to comply with the rules under Sarbanes-Oxley related to disclosure controls and procedures in the future, or, if we discover material weaknesses and other deficiencies in our internal controls over financial reporting, our stock price could decline significantly and raising capital could be more difficult. If material weaknesses or significant deficiencies are discovered or if we otherwise fail to achieve and maintain the adequacy of our internal controls, we may not be able to ensure that we can conclude on an ongoing basis that we have effective internal controls over financial reporting in accordance with Section 404 of Sarbanes-Oxley. Moreover, effective internal controls are necessary for us to produce reliable financial reports and are important to helping prevent financial fraud. If we cannot provide reliable financial reports or prevent fraud, our business and operating results could be harmed, investors could lose confidence in our reported financial information, and the trading price of our common stock could drop significantly.
Comprehensive tax reform bills could adversely affect our business and financial condition.
The U.S. government recently enacted comprehensive federal income tax legislation that includes significant changes to the taxation of business entities. These changes include, among others, a permanent reduction to the corporate income tax rate. Notwithstanding the reduction in the corporate income tax rate, the overall impact of this tax reform is uncertain, and our business and financial condition could be adversely affected. We urge our shareholders to consult with their legal and tax advisors with respect to any such legislation and the potential tax consequences of investing in our common stock.
Our Articles of Incorporation, as amended (“Articles of Incorporation”) our Amended and Restated Bylaws, and Nevada law may have anti-takeover effects that could discourage, delay or prevent a change in control, which may cause our stock price to decline.
Our Articles of Incorporation, Amended and Restated Bylaws, and Nevada law could make it more difficult for a third party to acquire us, even if closing such a transaction would be beneficial to our stockholders. We are authorized to issue up to 10,000,000 shares of preferred stock, none of which are outstanding as of August 27, 2019. This preferred stock may be issued in one or more series, the terms of which may be determined at the time of issuance by our board of directors without further action by shareholders. The terms of any series of preferred stock may include voting rights (including the right to vote as a series on particular matters), preferences as to dividend, liquidation, conversion and redemption rights and sinking fund provisions. As of August 27, 2019, 5,000,000 shares of our preferred stock have been designated as Series A Preferred Stock of which 3,102,480 shares of Series A Preferred Stock which were previously issued were converted into common stock at the time of our initial public offering (“IPO”) and 1,897,520 shares of Series A Preferred Stock remain authorized. The issuance of any preferred stock could materially adversely affect the rights of the holders of our common stock, and therefore, reduce the value of our common stock. In particular, specific rights granted to future holders of preferred stock could be used to restrict our ability to merge with, or sell our assets to, a third party and thereby preserve control by the present management.
-34-
Provisions of our Articles of Incorporation, our Amended and Restated Bylaws and Nevada law also could have the effect of discouraging potential acquisition proposals or making a tender offer or delaying or preventing a change in control, including changes a shareholder might consider favorable. Such provisions may also prevent or frustrate attempts by our shareholders to replace or remove our management. In particular, the Articles of Incorporation, our Amended and Restated Bylaws and Nevada law, as applicable, among other things:
| ● | provide the board of directors with the ability to alter the Amended and Restated Bylaws without shareholder approval; | |
| ● | place limitations on the removal of directors; | |
| ● | establish advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted upon at shareholder meetings; and | |
| ● | provide that vacancies on the board of directors may be filled by a majority of directors in office, although less than a quorum. |
Our Amended and Restated Bylaws provide that the Eighth Judicial District Court of Clark County, Nevada will be the sole and exclusive forum for certain disputes which could limit stockholders’ ability to obtain a favorable judicial forum for disputes with the Company or its directors, officers, employees or agents.
Our Amended and Restated Bylaws provide that unless the Company consents in writing to the selection of an alternative forum, the Eighth Judicial District Court of Clark County, Nevada shall be the sole and exclusive forum for state law claims with respect to: (i) any derivative action or proceeding brought in the name or right of the Company or on its behalf, (ii) any action asserting a claim for breach of any fiduciary duty owed by any director, officer, employee or agent of the Company to the Company or the Company’s stockholders, (iii) any action arising or asserting a claim arising pursuant to any provision of Nevada Revised Statutes Chapters 78 or 92A or any provision of the Company’s Articles of Incorporation or Amended and Restated Bylaws or (iv) any action asserting a claim governed by the internal affairs doctrine, including, without limitation, any action to interpret, apply, enforce or determine the validity of the Company’s Articles of Incorporation or Amended and Restated Bylaws. This exclusive forum provision would not apply to suits brought to enforce any liability or duty created by the Securities Act or the Exchange Act or any other claim for which the federal courts have exclusive jurisdiction. To the extent that any such claims may be based upon federal law claims, Section 27 of the Exchange Act creates exclusive federal jurisdiction over all suits brought to enforce any duty or liability created by the Exchange Act or the rules and regulations thereunder. Furthermore, Section 22 of the Securities Act creates concurrent jurisdiction for federal and state courts over all suits brought to enforce any duty or liability created by the Securities Act or the rules and regulations thereunder.
This choice of forum provision may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with the Company or its directors, officers, other employees or agents, which may discourage such lawsuits against the Company and its directors, officers, other employees and agents. Alternatively, if a court were to find the choice of forum provision contained in our Amended and Restated Bylaws to be inapplicable or unenforceable in an action, the Company may incur additional costs associated with resolving such action in other jurisdictions, which could have a material adverse effect on the Company’s business, results of operations, and financial condition.
-35-
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements that involve risks and uncertainties. You should not place undue reliance on these forward-looking statements. All statements other than statements of historical facts contained in this prospectus are forward-looking statements. The forward-looking statements in this prospectus are only predictions. We have based these forward-looking statements largely on our current expectations and projections about future events and financial trends that we believe may affect our business, financial condition and results of operations. In some cases, you can identify these forward-looking statements by terms such as “anticipate,” “believe,” “continue,” “could,” “depends,” “estimate,” “expects,” “intend,” “may,” “ongoing,” “plan,” “potential,” “predict,” “project,” “should,” “will,” “would” or the negative of those terms or other similar expressions, although not all forward-looking statements contain those words. We have based these forward-looking statements on our current expectations and projections about future events and trends that we believe may affect our financial condition, results of operations, strategy, short-term and long-term business operations and objectives and financial needs. These forward-looking statements include, but are not limited to, statements concerning the following:
| ● | our business strategies; |
| ● | the timing of regulatory submissions; |
| ● | our ability to obtain and maintain regulatory approval of our existing product candidates and any other product candidates we may develop, and the labeling under any approval we may obtain; |
| ● | risks relating to the timing and costs of clinical trials, the timing and costs of other expenses; |
| ● | risks related to market acceptance of products; |
| ● | intellectual property risks; |
| ● | risks associated with our reliance on third party organizations; |
| ● | our competitive position; |
| ● | our industry environment; |
| ● | our anticipated financial and operating results, including anticipated sources of revenues; |
| ● | assumptions regarding the size of the available market, benefits of our products, product pricing, timing of product launches; |
| ● | management’s expectation with respect to future acquisitions; |
| ● | statements regarding our goals, intensions, plans and expectations, including the introduction of new products and markets; and |
| ● | our cash needs and financing plans. |
These forward-looking statements are subject to a number of risks, uncertainties and assumptions, including those described in “Risk Factors.” Moreover, we operate in a very competitive and rapidly changing environment. New risks emerge from time to time. It is not possible for our management to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. In light of these risks, uncertainties and assumptions, the forward-looking events and circumstances discussed in this prospectus may not occur and actual results could differ materially and adversely from those anticipated or implied in the forward-looking statements.
You should not rely upon forward-looking statements as predictions of future events. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee that the future results, levels of activity, performance or events and circumstances reflected in the forward-looking statements will be achieved or occur. Moreover, except as required by law, neither we nor any other person assumes responsibility for the accuracy and completeness of the forward-looking statements. We undertake no obligation to update publicly any forward-looking statements for any reason after the date of this prospectus to conform these statements to actual results or to changes in our expectations.
You should read this prospectus and the documents that we reference in this prospectus and have filed with the SEC as exhibits to the registration statement of which this prospectus is a part with the understanding that our actual future results, levels of activity, performance and events and circumstances may be materially different from what we expect.
-36-
This prospectus contains estimates and other statistical data made by independent parties and by us relating to market size and growth and other data about our industry. We obtained the industry and market data in this prospectus from our own research as well as from industry and general publications, surveys and studies conducted by third parties. This data involves a number of assumptions and limitations and contains projections and estimates of the future performance of the industries in which we operate that are subject to a high degree of uncertainty, including those discussed in “Risk Factors.” We caution you not to give undue weight to such projections, assumptions and estimates. Further, industry and general publications, studies and surveys generally state that they have been obtained from sources believed to be reliable, although they do not guarantee the accuracy or completeness of such information. While we believe that these publications, studies and surveys are reliable, we have not independently verified the data contained in them. In addition, while we believe that the results and estimates from our internal research are reliable, such results and estimates have not been verified by any independent source.
The selling stockholders will receive all of the proceeds from the sale of the Resale Shares offered by them pursuant to this prospectus. We will not receive any proceeds from the sale of the Resale Shares by the selling stockholders covered by this prospectus. If the warrants are exercised for cash, 5% of the proceeds will be paid to Laidlaw and the balance of such proceeds will be used by the Company for working capital.
MARKET FOR OUR COMMON STOCK AND RELATED STOCKHOLDER MATTERS
Market Information
On February 15, 2019, our common stock began trading on The Nasdaq Capital Market under the symbol “HOTH”. Prior to that time, there was no public market for our common stock.
Stockholders
As of August 27, 2019, there were 116 stockholders of record of our common stock. The actual number of holders of our common stock is greater than this number of record holders, and includes stockholders who are beneficial owners, but whose shares are held in street name by brokers or held by other nominees. This number of holders of record also does not include stockholders whose shares may be held in trust by other entities.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table summarizes information about our equity compensation plans as of December 31, 2018.
| Plan Category | Number of securities to be issued upon exercise of outstanding options, warrants and rights (1) | Weighted average exercise price of outstanding options, warrants and rights | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (1)) (2) | |||||||||
| Equity compensation plans approved by security holder | 135,987 | $ | 0.25 | 864,013 | ||||||||
| Equity compensation plans not approved by security holder | - | - | - | |||||||||
| 135,987 | 864,013 | |||||||||||
-37-
We have never declared or paid any cash dividends on our capital stock. We intend to retain future earnings, if any, to finance the operation and expansion of our business and do not anticipate paying any cash dividends in the foreseeable future. Any future determination related to our dividend policy will be made at the discretion of our board of directors after considering our financial condition, results of operations, capital requirements, business prospects and other factors our board of directors deems relevant.
From October 2017 until February 2018, we conducted a private placement (the “Private Placement Offering”) of our securities pursuant to which we issued an aggregate of 3,102,480 shares of Series A Preferred Stock and warrants (the “PPM Warrants”) to purchase an aggregate of 991,367 shares of common stock (including warrants to purchase up to 215,747 shares of common stock issued to the placement agent). As of August 27, 2019, PPM Warrants to purchase 717,870 shares (the “PPM Warrant Shares”) of common stock are outstanding.
On August 16, 2019, we conducted the August 2019 Private Placement pursuant to which we issued an aggregate of 407,424 shares of our common stock (the “Private Placement Shares”) and warrants (the “Private Placement Warrants” and together with the PPM Warrants, the “Warrants”) to purchase up to 203,709 shares (the “Private Placement Warrant Shares” and together with the PPM Warrant Shares, the “Warrant Shares”) of our common stock.
This prospectus relates to the resale from time to time by the selling security holders identified herein of up to an aggregate of 1,329,003 Resale Shares consisting of 407,424 Private Placement Shares and 921,579 Warrant Shares.
The transactions by which the selling stockholders acquired their securities from us were exempt under the registration provisions of the Securities Act.
The Resale Shares referred to above are being registered to permit public sales of the Resale Shares, and the selling stockholders may offer the shares for resale from time to time pursuant to this prospectus. The selling stockholders may also sell, transfer or otherwise dispose of all or a portion of their shares in transactions exempt from the registration requirements of the Securities Act or pursuant to another effective registration statement covering those shares.
The table below sets forth certain information regarding the selling stockholders and the Resale Shares offered in this prospectus. The selling stockholders have had no material relationship with us within the past three years other than as described in the footnotes to the table below or as a result of their acquisition of our shares or other securities.
-38-
Beneficial ownership is determined in accordance with the rules of the SEC. The selling stockholder’s percentage of ownership of our outstanding shares in the table below is based upon 10,077,068 shares of common stock outstanding as of August 27, 2019.
| Name of Selling Stockholder | Number of Shares of Common Stock Beneficially Owned Before this Offering (1) |
Percentage of Common Stock Beneficially Owned Before this Offering |
Shares of Common Stock Offered in this Offering |
Shares of Common Stock Beneficially Owned After this Offering (2) |
Percentage of Common Stock Beneficially Owned After this Offering (2) | |||||||||||||||
| Michael Miller | 16,112 | (3) | * | 2,500 | 10,000 | |||||||||||||||
| Kevin J. Poor | 937,500 | (4) | 9.13 | % | 187,500 | 750,000 | 7.44 | % | ||||||||||||
| Michael C. Fox Revocable Trust DTD 05/05/05 Michael C. Fox TTEE (5) | 31,250 | (6) | * | 6,250 | 25,000 | * | ||||||||||||||
| Phillip Todd Herndon | 348,750 | (7) | 3.44 | % | 69,750 | 279,000 | 2.77 | % | ||||||||||||
| Matthew Eitner (8) | 768,750 | (9) | 7.63 | % | 3,750 | 765,000 | 7.59 | % | ||||||||||||
| Thomas G. Hoffman | 62,500 | (10) | * | 12,500 | 50,000 | * | ||||||||||||||
| Jimmy R. Hasley | 25,000 | (11) | * | 5,000 | 20,000 | * | ||||||||||||||
| Jonathan Plotkin | 12,500 | (12) | * | 2,500 | 10,000 | * | ||||||||||||||
| Ron D. Craig | 62,500 | (13) | * | 12,500 | 50,000 | * | ||||||||||||||
| Gary J. Mabie & Janelle L. Mabie JTWROS | 47,500 | (14) | * | 9,500 | 38,000 | * | ||||||||||||||
| Roger Conan | 124,975 | (15) | 1.24 | % | 24,995 | 99,980 | * | |||||||||||||
| Ronald A. Soicher | 43,750 | (16) | * | 8,750 | 35,000 | * | ||||||||||||||
| Brian E. Jones & Peggy A. Jones JTWROS | 125,000 | (17) | 1.24 | % | 25,000 | 100,000 | * | |||||||||||||
| George T. Hawes | 138,582 |
(18) | 1.37 | % | 25,000 | 113,582 | 1.13 |
% | ||||||||||||
| Rippee Mineral Management LLC (19) | 23,750 | (20) | * | 4,750 | 19,000 | * | ||||||||||||||
| Satheesh Kumar & Anju Satheesh JTWROS | 61,250 | (21) | * | 36,250 | 25,000 | * | ||||||||||||||
| Craig M. Cancienne Jr. | 31,250 | (22) | * | 6,250 | 25,000 | * | ||||||||||||||
| Jonathan Smith | 15,500 | (23) | * | 5,500 | 10,000 | * | ||||||||||||||
| David S. Rippee | 31,250 | (24) | * | 1,500 | 29,750(111) | * | ||||||||||||||
| Jean Marc Henry & Patricia Henry JTWROS | 62,500 | (25) | * | 12,500 | 50,000 | * | ||||||||||||||
| Michael L. Turner | 25,000 | (26) | * | 5,000 | 20,000 | * | ||||||||||||||
| The William D. Woodford & Deborah N. Woodford Revocable Living Trust 01-15-2013 (27) | 15,625 | (28) | * | 3,125 | 12,500 | * | ||||||||||||||
| Thomas S. Murray | 18,750 | (29) | * | 3,750 | 15,000 | * | ||||||||||||||
| Lexecon Pension Trust (30) | 125,000 | (31) | 1.24 | % | 25,000 | 100,000 | * | |||||||||||||
| Andy Horan | 125,000 | (32) | 1.24 | % | 25,000 | 100,000 | * | |||||||||||||
| Marvin S. Rosen | 62,500 | (33) | * | 12,500 | 50,000 | * | ||||||||||||||
| Georgios E. Doutis | 25,000 | (34) | * | 5,000 | 20,000 | * | ||||||||||||||
| Joel Cherande | 125,000 | (35) | 1.24 | % | 25,000 | 100,000 | * | |||||||||||||
| Craig William Bannister | 25,000 | (36) | * | 5,000 | 20,000 | * | ||||||||||||||
-39-
| Name of Selling Stockholder | Number of Shares of Common Stock Beneficially Owned Before this Offering (1) |
Percentage of Common Stock Beneficially Owned Before this Offering |
Shares of Common Stock Offered in this Offering |
Shares of Common Stock Beneficially Owned After this Offering (2) |
Percentage of Common Stock Beneficially Owned After this Offering (2) | |||||||||||||||
| Joy D. Bushwell | 31,250 | (37) | * | 6,250 | 25,000 | * | ||||||||||||||
| Stefaan Verhelst & Katharine Demeestere JTWROS | 80,500 | (38) | * | 30,500 | 50,000 | * | ||||||||||||||
| Graeme Farr | 23,125 | (39) | * | 4,625 | 18,500 | * | ||||||||||||||
| Tim M. Lowney | 12,500 | (40) | * | 2,500 | 10,000 | * | ||||||||||||||
| Donald F. McDonald Jr. | 31,250 | (41) | * | 6,250 | 25,000 | * | ||||||||||||||
| William A. Narduzzi | 31,250 | (42) | * | 6,250 | 25,000 | * | ||||||||||||||
| Dennis J. Steul | 31,250 | (43) | * | 6,250 | 25,000 | * | ||||||||||||||
| Ashok Suppiah | 31,250 | (44) | * | 6,250 | 25,000 | * | ||||||||||||||
| Daniel P. Wikel | 77,500 | (45) | * | 27,500 | 50,000 | * | ||||||||||||||
| Kenneth G. Williamson | 31,250 | (46) | * | 6,250 | 25,000 | * | ||||||||||||||
| Simon C. Guscott | 28,125 | (47) | * | 5,625 | 22,500 | * | ||||||||||||||
| John Van Parys | 38,450 | (48) | * | 13,450 | 25,000 | * | ||||||||||||||
| Edward J. Reagan | 38,750 | (49) | * | 13,750 | 25,000 | * | ||||||||||||||
| Donald E. Garlikov | 375,000 | (50) | 3.69 | % | 75,000 | 300,000 | 2.98 | % | ||||||||||||
| Adam Baker | 70,000 | (51) | * | 14,000 | 56,000 | * | ||||||||||||||
| Pedro Luis Planas & Maria Mercedes Tapia Sasot JTWROS | 12,500 | (52) | * | 2,500 | 10,000 | * | ||||||||||||||
| Duerr Capital LLC (53) | 27,000 | (54) | * | 27,000 | 0 | 0 | % | |||||||||||||
| David J. Hallett | 37,500 | (55) | * | 37,500 | 0 | 0 | % | |||||||||||||
| Surendranath Kavuri | 29,998 | (56) | * | 29,998 | 0 | 0 | % | |||||||||||||
| George A. Parmer & Barbara Parmer JTWROS | 13,950 | (57) | * | 13,950 | 0 | 0 | % | |||||||||||||
| George A. Parmer | 99,150 | (58) | * | 61,500 | 37,650 (112) | * | ||||||||||||||
| Carlos De Serpa Pimentel | 15,000 | (59) | * | 15,000 | 0 | 0 | % | |||||||||||||
| Garth Saunders | 11,100 | (60) | * | 11,100 | 0 | 0 | % | |||||||||||||
| Gregory Alexander | 7,524 | (61) | * | 7,524 | 0 | 0 | % | |||||||||||||
| Scott L. Byer | 7,500 | (62) | * | 7,500 | 0 | 0 | % | |||||||||||||
| Zachary J. Inman | 7,500 | (63) | * | 7,500 | 0 | 0 | % | |||||||||||||
-40-
| Name of Selling Stockholder | Number of Shares of Common Stock Beneficially Owned Before this Offering (1) |
Percentage of Common Stock Beneficially Owned Before this Offering |
Shares of Common Stock Offered in this Offering |
Shares of Common Stock Beneficially Owned After this Offering (2) |
Percentage of Common Stock Beneficially Owned After this Offering (2) | |||||||||||||||
| David Patterson | 7,500 | (64) | * | 7,500 | 0 | 0 | % | |||||||||||||
| Philip Robert Kimbrough 5x5 Trust, Philip R. Kimbrough TTEE (65) | 7,524 | (66) | * | 7,524 | 0 | 0 | % | |||||||||||||
| David Scott | 14,998 | (67) | * | 14,998 | 0 | 0 | % | |||||||||||||
| Robert W. Biederman | 4,500 | (68) | * | 4,500 | 0 | 0 | % | |||||||||||||
| Kevin Carnahan and Laurie B. Carnahan Co TTEES Carnahan Trust (69) | 14,998 | (70) | * | 14,998 | 0 | 0 | % | |||||||||||||
| The Laskowski Revocable Trust DTD 9/20/2018 Jan Laskowski TTEE (71) | 10,500 | (72) | * | 9,000 | 1,500 | * | ||||||||||||||
| Michael Lippitt | 4,050 | (73) | * | 4,050 | 0 | 0 | % | |||||||||||||
| Richard Pitt | 8,985 | (74) | * | 8,985 | 0 | 0 | % | |||||||||||||
| Martin Rost | 7,500 | (75) | * | 7,500 | 0 | 0 | % | |||||||||||||
| Edward Bluestone | 7,500 | (76) | * | 7,500 | 0 | 0 | % | |||||||||||||
| Reed Family Trust DTD 06/24/1999 Clayton A. Reed and Stephanie S. Reed TTEES (77) | 15,000 | (78) | * | 15,000 | 0 | 0 | % | |||||||||||||
| Kaizer Wood | 4,500 | (79) | * | 4,500 | 0 | 0 | % | |||||||||||||
| Dirk A. Dent | 10,800 | (80) | * | 10,800 | 0 | 0 | % | |||||||||||||
| Darrel Welling | 5,400 | (81) | * | 5,400 | 0 | 0 | % | |||||||||||||
| John A. Whiting | 4,500 | (82) | * | 4,500 | 0 | 0 | % | |||||||||||||
| Samuel Bruce Patterson | 7,500 | (83) | * | 7,500 | 0 | 0 | % | |||||||||||||
| Peter James Wealleans | 14,998 | (84) | * | 14,998 | 0 | 0 | % | |||||||||||||
| Clayton Wise | 3,600 | (85) | * | 3,600 | 0 | 0 | % | |||||||||||||
| Jon Luchford | 6,300 | (86) | * | 6,300 | 0 | 0 | % | |||||||||||||
| Sean A. Cavanaugh | 3,600 | (87) | * | 3,600 | 0 | 0 | % | |||||||||||||
| Steven and Louise Feller JTWROS | 5,998 | (88) | * | 5,998 | 0 | 0 | % | |||||||||||||
| Johnson Revocable Trust DTD Feb 13 2000 Todd Johnson and Luann Johnson TTEES (89) | 7,500 | (90) | * | 7,500 | 0 | 0 | % | |||||||||||||
-41-
| Name of Selling Stockholder | Number of Shares of Common Stock Beneficially Owned Before this Offering (1) |
Percentage of Common Stock Beneficially Owned Before this Offering |
Shares of Common Stock Offered in this Offering |
Shares of Common Stock Beneficially Owned After this Offering (2) |
Percentage of Common Stock Beneficially Owned After this Offering (2) | |||||||||||||||
| Michael C. Bellard | 6,000 | (91) | * | 6,000 | 0 | 0 | % | |||||||||||||
| Pieter Deventer Van Wijk | 29,998 | (92) | * | 29,998 | 0 | 0 | % | |||||||||||||
| Richard Levine | 9,000 | (93) | * | 9,000 | 0 | 0 | % | |||||||||||||
| Michael B. Carroll and Sheila J. Carroll JTWROS | 26,115 | (94) | * | 15,000 | 11,115 | * | ||||||||||||||
| Hochman Family, LLLP (95) | 9,000 | (96) | * | 9,000 | 0 | 0 | % | |||||||||||||
| Derry Management Company Inc (97) | 4,800 | (98) | * | 4,800 | 0 | 0 | % | |||||||||||||
| Fine Line Homes LP (99) | 5,400 | (100) | * | 5,400 | 0 | 0 | % | |||||||||||||
| Residential Warranty Company LLC (Health Reserve) (101) | 6,300 | (102) | * | 6,300 | 0 | 0 | % | |||||||||||||
| Residential Warranty Company LLC (Kimball Hill) (103) | 7,200 | (104) | * | 7,200 | 0 | 0 | % | |||||||||||||
| Gunnar Stolze | 9,000 | (105) | * | 9,000 | 0 | 0 | % | |||||||||||||
| Christopher A. Corcoran | 7,500 | (106) | * | 7,500 | 0 | 0 | % | |||||||||||||
| Michael K. Barber and Julia K. Barber JTWROS | 7,500 | (107) | * | 7,500 | 0 | 0 | % | |||||||||||||
| Portofino Ventures LP (108) | 4,500 | (109) | * | 4,500 | 0 | 0 | % | |||||||||||||
| Ram V. Seetharam and Rani J. Seetharam JTWROS | 10,800 | (110) | * | 10,800 | 0 | 0 | % | |||||||||||||
| TOTAL | 1,329,003 | |||||||||||||||||||
* Represents less than 1%.
(1) Under applicable SEC rules, a person is deemed to beneficially own securities which the person has the right to acquire within 60 days through the exercise of any option or warrant or through the conversion of a convertible security. Also under applicable SEC rules, a person is deemed to be the “beneficial owner” of a security with regard to which the person directly or indirectly, has or shares (a) voting power, which includes the power to vote or direct the voting of the security, or (b) investment power, which includes the power to dispose, or direct the disposition, of the security, in each case, irrespective of the person’s economic interest in the security. To our knowledge, subject to community property laws where applicable, each person named in the table has sole voting and investment power with respect to the shares of common stock shown as beneficially owned by such selling stockholder, except as otherwise indicated in the footnotes to the table.
(2) Represents the amount of shares that will be held by the selling stockholder after completion of this offering based on the assumptions that (a) all Resale Shares registered for sale by the registration statement of which this prospectus is part will be sold and (b) no other shares of our common stock are acquired or sold by the selling stockholder prior to completion of this offering. However, each selling stockholder may sell all, some or none of the Resale Shares offered pursuant to this prospectus and may sell other shares of our common stock that they may own pursuant to another registration statement under the Securities Act or sell some or all of their shares pursuant to an exemption from the registration provisions of the Securities Act, including under Rule 144.
-42-
(3) Includes (i) 12,408 shares of common stock and (ii) 3,704 Warrant Shares.
(4) Includes (i) 750,000 shares of common stock and (ii) 187,500 Warrant Shares.
(5) Michael Fox is the Trustee of the Michael C. Fox Revocable Trust DTD 05/05/05 Michael C. Fox TTEE and in such capacity has
voting and dispositive power over the securities held by such trust.
(6) Includes (i) 25,000 shares of common stock and (ii) 6,250 Warrant Shares.
(7) Includes (i) 279,000 shares of common stock and (ii) 69,750 Warrant Shares.
(8) Matthew Eitner is the Chief Executive Officer and Head of Capital Markets of Laidlaw & Company (UK) Ltd. Laidlaw acted as our placement agent in the August 2019 Private Placement and as the sole book-running manager in our IPO.
(9) Includes (i) 765,000 shares of common stock and (ii) 3,750 Warrant Shares.
(10) Includes (i) 50,000 shares of common stock and (ii) 12,500 Warrant Shares.
(11) Includes (i) 20,000 shares of common stock and (ii) 5,000 Warrant Shares.
(12) Includes (i) 10,000 shares of common stock and (ii) 2,500 Warrant Shares.
(13) Includes (i) 50,000 shares of common stock and (ii) 12,500 Warrant Shares.
(14) Includes (i) 38,000 shares of common stock and (ii) 9,500 Warrant Shares.
(15) Includes (i) 99,980 shares of common stock and (ii) 24,995 Warrant Shares.
(16) Includes (i) 35,000 shares of common stock and (ii) 8,750 Warrant Shares.
(17) Includes (i) 100,000 shares of common stock and (ii) 25,000 Warrant Shares.
(18) Includes (i) 100,000 shares of common stock and (ii) 25,000 Warrant Shares.
(19) David Rippee is the Manager of Rippee Mineral Management LLC (“Rippee Mineral”) and in such capacity has voting and dispositive power over the securities held by such entity.
(20) Includes (i) 19,000 shares of common stock and (ii) 4,750 Warrant Shares.
(21) Includes (i) 45,000 shares of common stock and (ii) 16,250 Warrant Shares.
(22) Includes (i) 25,000 shares of common stock and (ii) 6,250 Warrant Shares.
(23) Includes (i) 12,000 shares of common stock and (ii) 3,500 Warrant Shares.
(24) Includes (i) 6,000 shares of common stock owned by David S. Rippee, (ii) 1,500 Warrant Shares owned by David S. Rippee, (iii) 19,000 shares of common stock owned by Rippee Mineral and (iv) 4,750 Warrant Shares owned by Rippee Mineral. David Rippee is the Manager of Rippee Mineral and in such capacity has voting and dispositive power over the securities held by such entity.
-43-
(25) Includes (i) 50,000 shares of common stock and (ii) 12,500 Warrant Shares.
(26) Includes (i) 20,000 shares of common stock and (ii) 5,000 Warrant Shares.
(27) William D. Woodford and Deborah N. Woodford are Trustees of The William D. Woodford & Deborah N. Woodford Revocable Living Trust 01-15-2013 and in such capacity have voting and dispositive power over the securities held by such trust.
(28) Includes (i) 12,500 shares of common stock and (ii) 3,125 Warrant Shares.
(29) Includes (i) 15,000 shares of common stock and (ii) 3,750 Warrant Shares.
(30) William Bishop is the Trustee of the Lexecon Pension Trust and in such capacity has voting and dispositive power over the securities held by such trust.
(31) Includes (i) 100,000 shares of common stock and (ii) 25,000 Warrant Shares.
(32) Includes (i) 100,000 shares of common stock and (ii) 25,000 Warrant Shares.
(33) Includes (i) 50,000 shares of common stock and (ii) 12,500 Warrant Shares.
(34) Includes (i) 20,000 shares of common stock and (ii) 5,000 Warrant Shares.
(35) Includes (i) 100,000 shares of common stock and (ii) 25,000 Warrant Shares.
(36) Includes (i) 20,000 shares of common stock and (ii) 5,000 Warrant Shares.
(37) Includes (i) 25,000 shares of common stock and (ii) 6,250 Warrant Shares.
(38) Includes (i) 62,000 shares of common stock and (ii) 18,500 Warrant Shares.
(39) Includes (i)18,500 shares of common stock and (ii) 4,625 Warrant Shares.
(40) Includes (i) 10,000 shares of common stock and (ii) 2,500 Warrant Shares.
(41) Includes (i) 25,000 shares of common stock and (ii) 6,250 Warrant Shares.
(42) Includes (i) 25,000 shares of common stock and (ii) 6,250 Warrant Shares.
(43) Includes (i) 25,000 shares of common stock and (ii) 6,250 Warrant Shares.
(44) Includes (i) 25,000 shares of common stock and (ii) 6,250 Warrant Shares.
(45) Includes (i) 60,000 shares of common stock and (ii) 17,500 Warrant Shares.
(46) Includes (i) 25,000 shares of common stock and (ii) 6,250 Warrant Shares.
(47) Includes (i) 22,500 shares of common stock and (ii) 5,625 Warrant Shares.
(48) Includes (i) 29,800 shares of common stock and (ii) 8,650 Warrant Shares.
(49) Includes (i) 30,000 shares of common stock and (ii) 8,750 Warrant Shares.
(50) Includes (i) 300,000 shares of common stock and (ii) 75,000 Warrant Shares.
(51) Includes (i) 56,000 shares of common stock and (ii) 14,000 Warrant Shares.
-44-
(52) Includes (i) 10,000 shares of common stock and (ii) 2,500 Warrant Shares.
(53) Peter Duerr is the Managing Member of Duerr Capital LLC and in such capacity has voting and dispositive power over the securities held by such entity.
(54) Includes (i) 18,000 shares of common stock and (ii) 9,000 Warrant Shares.
(55) Includes (i) 25,000 shares of common stock and (ii) 12,500 Warrant Shares.
(56) Includes (i) 19,999 shares of common stock and (ii) 9,999 Warrant Shares.
(57) Includes (i) 9,300 shares of common stock and (ii) 4,650 Warrant Shares.
(58) Includes (i) 41,000 shares of common stock owned George A. Parmer, (ii) 20,500 Warrant Shares owned by George A. Parmer, (iii) 9,300 shares of common stock owned by George A. Parmer & Barbara Parmer JTWROS, (iv) 4,650 Warrant Shares owned by George A. Parmer & Barbara Parmer JTWROS, (v) 3,200 shares of common stock owned by Derry Management Company Inc, (vi) 1,600 Warrant Shares owned by Derry Management Company Inc, (vii) 3,600 shares of common stock owned by Fine Line Homes LP, (viii) 1,800 Warrant Shares owned by Fine Line Homes LP, (ix) 4,200 shares of common stock owned by Residential Warranty Company LLC (Health Reserve), (x) 2,100 Warrant Shares owned by Residential Warranty Company LLC (Health Reserve), (xi) 4,800 shares of common stock owned by Residential Warranty Company LLC (Kimball Hill), and (xii) 2,400 Warrant Shares owned by Residential Warranty Company LLC (Kimball Hill). George A. Parmer is the President of Derry Management Company Inc, the President of Fine Line Homes LP, the Sole Member of Residential Warranty Company LLC (Health Reserve) and the President of Residential Warranty Company LLC (Kimball Hill) and in such capacities has voting and dispositive power over the securities held by such entity.
(59) Includes (i) 10,000 shares of common stock and (ii) 5,000 Warrant Shares.
(60) Includes (i) 7,400 shares of common stock and (ii) 3,700 Warrant Shares.
(61) Includes (i) 5,016 shares of common stock and (ii) 2,508 Warrant Shares.
(62) Includes (i) 5,000 shares of common stock and (ii) 2,500 Warrant Shares.
(63) Includes (i) 5,000 shares of common stock and (ii) 2,500 Warrant Shares.
(64) Includes (i) 5,000 shares of common stock and (ii) 2,500 Warrant Shares.
(65) Philip R. Kimbrough is the Trustee of the Philip Robert Kimbrough 5x5 Trust, Philip R. Kimbrough TTEE and in such capacity has voting and dispositive power over the securities held by such trust.
(66) Includes (i) 5,016 shares of common stock and (ii) 2,508 Warrant Shares.
(67) Includes (i) 9,999 shares of common stock and (ii) 4,999 Warrant Shares.
(68) Includes (i) 3,000 shares of common stock and (ii) 1,500 Warrant Shares.
(69) Kevin Carnahan and Laurie B. Carnahan are the Trustees of the Kevin Carnahan and Laurie B. Carnahan Co TTEES Carnahan Trust and in such capacity have voting and dispositive power over the securities held by such trust.
(70) Includes (i) 9,999 shares of common stock and (ii) 4,999 Warrant Shares.
(71) Jan Laskowski is the Trustee of The Laskowski Revocable Trust DTD 9/20/2018 Jan Laskowski TTEE and in such capacity has voting and dispositive power over the securities held by such trust.
-45-
(72) Includes (I) 7,500 shares of common stock and (ii) 3,000 Warrant Shares.
(73) Includes (i) 2,700 shares of common stock and (ii) 1,350 Warrant Shares.
(74) Includes (i) 5,990 shares of common stock and (ii) 2,995 Warrant Shares.
(75) Includes (i) 5,000 shares of common stock and (ii) 2,500 Warrant Shares.
(76) Includes (i) 5,000 shares of common stock and (ii) 2,500 Warrant Shares.
(77) Clayton A. Reed and Stephanie Reed are the Trustees of the Reed Family Trust DTD 06/24/1999 Clayton A. Reed and Stephanie S. Reed TTEES and in such capacity have voting and dispositive power over the securities held by such trust.
(78) Includes (i) 10,000 shares of common stock and (ii) 5,000 Warrant Shares.
(79) Includes (i) 3,000 shares of common stock and (ii) 1,500 Warrant Shares.
(80) Includes (i) 7,200 shares of common stock and (ii) 3,600 Warrant Shares.
(81) Includes (i) 3,600 shares of common stock and (ii) 1,800 Warrant Shares.
(82) Includes (i) 3,000 shares of common stock and (ii) 1,500 Warrant Shares.
(83) Includes (i) 5,000 shares of common stock and (ii) 2,500 Warrant Shares.
(84) Includes (i) 9,999 shares of common stock and (ii) 4,999 Warrant Shares.
(85) Includes (i) 2,400 shares of common stock and (ii) 1,200 Warrant Shares.
(86) Includes (i) 4,200 shares of common stock and (ii) 2,100 Warrant Shares.
(87) Includes (i) 2,400 shares of common stock and (ii) 1,200 Warrant Shares.
(88) Includes (i) 3,999 shares of common stock and (ii) 1,999 Warrant Shares.
(89) Todd Johnson and Luann Johnson are the are the Trustees of the Johnson Revocable Trust DTD Feb 13 2000 Todd Johnson and Luann Johnson TTEES and in such capacity have voting and dispositive power over the securities held by such trust.
(90) Includes (i) 5,000 shares of common stock and (ii) 2,500 Warrant Shares.
(91) Includes (i) 4,000 shares of common stock and (ii) 2,000 Warrant Shares.
(92) Includes (i) 19,999 shares of common stock and (ii) 9,999 Warrant Shares.
(93) Includes (i) 6,000 shares of common stock and (ii) 3,000 Warrant Shares.
(94) Includes (i) 21,115 shares of common stock and (ii) 5,000 Warrant Shares.
(95) Lawrence D. Hochman Declaration of Trust DTD 8/9/2001, Amended and Restated 9/25/2007 (the “Hochman Trust”) holds the voting and dispositive power over the securities held by Hochman Family, LLLP. Lawrence D. Hochman is the Trustee of the Hochman Trust and in such capacity has voting and dispositive power over the securities held by such trust.
(96) Includes (i) 6,000 shares of common stock and (ii) 3,000 Warrant Shares.
-46-
(97) George A. Parmer is the President of Derry Management Company Inc and in such capacity has voting and dispositive power over the securities held by such entity.
(98) Includes (i) 3,200 shares of common stock and (ii) 1,600 Warrant Shares.
(99) George A. Parmer is the President of Fine Line Homes LP and in such capacity has voting and dispositive power over the securities held by such entity.
(100) Includes (i) 3,600 shares of common stock and (ii) 1,800 Warrant Shares.
(101) George A. Parmer is the Sole Member of Residential Warranty Company LLC (Health Reserve) and in such capacity has voting and dispositive power over the securities held by such entity.
(102) Includes (i) 4,200 shares of common stock and (ii) 2,100 Warrant Shares.
(103) George A. Parmer is the President of Residential Warranty Company LLC (Kimball Hill) and in such capacity has voting and dispositive power over the securities held by such entity.
(104) Includes (i) 4,800 shares of common stock and (ii) 2,400 Warrant Shares.
(105) Includes (i) 6,000 shares of common stock and (ii) 3,000 Warrant Shares.
(106) Includes (i) 5,000 shares of common stock and (ii) 2,500 Warrant Shares.
(107) Includes (i) 5,000 shares of common stock and (ii) 2,500 Warrant Shares.
(108) Portofino Management Inc holds the voting and dispositive power over the securities held by Portofino Ventures LP. Michael Knudsen is the President of Portofino Management Inc and in such capacity holds voting and dispositive power over the securities held by such entity.
(109) Includes (i) 1,500 shares of common stock and (ii) 3,000 Warrant Shares.
(110) Includes (i) 7,200 shares of common stock and (ii) 3,600 Warrant Shares.
(111) Includes (i) 6,000 shares of common stock owned by David S. Rippee, (ii) 19,000 shares of common stock owned by Rippee Mineral and (iii) 4,750 Warrant Shares owned by Rippee Mineral.
(112) Includes (i) 9,300 shares of common stock owned by George A. Parmer & Barbara Parmer JTWROS, (ii) 4,650 Warrant Shares owned by George A. Parmer & Barbara Parmer JTWROS, (iii) 3,200 shares of common stock owned by Derry Management Company Inc, (iv) 1,600 Warrant Shares owned by Derry Management Company Inc, (v) 3,600 shares of common stock owned by Fine Line Homes LP, (vi) 1,800 Warrant Shares owned by Fine Line Homes LP, (vii) 4,200 shares of common stock owned by Residential Warranty Company LLC (Health Reserve), (viii) 2,100 Warrant Shares owned by Residential Warranty Company LLC (Health Reserve), (ix) 4,800 shares of common stock owned by owned by Residential Warranty Company LLC (Kimball Hill) and (x) 2,400 Warrant Shares owned by Residential Warranty Company LLC (Kimball Hill).
-47-
Each selling stockholder of the securities and any of their pledgees, assignees and successors-in-interest may, from time to time, sell any or all of their securities covered hereby on The Nasdaq Capital Market or any other stock exchange, market or trading facility on which the securities are traded or in private transactions. These sales may be at fixed or negotiated prices. A selling stockholder may use any one or more of the following methods when selling securities:
| ● | ordinary brokerage transactions and transactions in which the broker-dealer solicits purchasers; | |
| ● | block trades in which the broker-dealer will attempt to sell the securities as agent but may position and resell a portion of the block as principal to facilitate the transaction; | |
| ● | purchases by a broker-dealer as principal and resale by the broker-dealer for its account; | |
| ● | an exchange distribution in accordance with the rules of the applicable exchange; | |
| ● | privately negotiated transactions; | |
| ● | settlement of short sales entered into after the effective date of the registration statement of which this prospectus is a part; | |
| ● | in transactions through broker-dealers that agree with the selling stockholders to sell a specified number of such securities at a stipulated price per security; | |
| ● | through the writing or settlement of options or other hedging transactions, whether through an options exchange or otherwise; | |
| ● | a combination of any such methods of sale; or | |
| ● | any other method permitted pursuant to applicable law. |
The selling stockholders may also sell securities under Rule 144 under the Securities Act, if available, rather than under this prospectus. Broker-dealers engaged by the selling stockholders may arrange for other brokers-dealers to participate in sales. Broker-dealers may receive commissions or discounts from the selling stockholders (or, if any broker-dealer acts as agent for the purchaser of securities, from the purchaser) in amounts to be negotiated, but, except as set forth in a supplement to this prospectus, in the case of an agency transaction not in excess of a customary brokerage commission in compliance with FINRA Rule 2440; and in the case of a principal transaction a markup or markdown in compliance with FINRA IM-2440.
In connection with the sale of the securities or interests therein, the selling stockholders may enter into hedging transactions with broker-dealers or other financial institutions, which may in turn engage in short sales of the securities in the course of hedging the positions they assume. The selling stockholders may also sell securities short and deliver these securities to close out their short positions, or loan or pledge the securities to broker-dealers that in turn may sell these securities. The selling stockholders may also enter into option or other transactions with broker-dealers or other financial institutions or create one or more derivative securities which require the delivery to such broker-dealer or other financial institution of securities offered by this prospectus, which securities such broker-dealer or other financial institution may resell pursuant to this prospectus (as supplemented or amended to reflect such transaction).
-48-
The selling stockholders and any broker-dealers or agents that are involved in selling the securities may be deemed to be “underwriters” within the meaning of the Securities Act in connection with such sales. In such event, any commissions received by such broker-dealers or agents and any profit on the resale of the securities purchased by them may be deemed to be underwriting commissions or discounts under the Securities Act. Each selling stockholder has informed the Company that it does not have any written or oral agreement or understanding, directly or indirectly, with any person to distribute the securities. In no event shall any broker-dealer receive fees, commissions and markups which, in the aggregate, would exceed 8%.
The Company is required to pay certain fees and expenses incurred by the Company incident to the registration of the securities. The Company has agreed to indemnify the selling stockholders against certain losses, claims, damages and liabilities, including liabilities under the Securities Act.
Because selling stockholders may be deemed to be “underwriters” within the meaning of the Securities Act, they will be subject to the prospectus delivery requirements of the Securities Act including Rule 172 thereunder. In addition, any securities covered by this prospectus which qualify for sale pursuant to Rule 144 under the Securities Act may be sold under Rule 144 rather than under this prospectus. The selling stockholders have advised us that there is no underwriter or coordinating broker acting in connection with the proposed sale of the resale securities by the selling stockholders.
We agreed to keep this prospectus effective until the earliest of (i) one year from the date the Registration Statement is declared effective by the SEC, (ii) the date on which the securities may be resold by the selling stockholders without registration and without regard to any volume or manner-of-sale limitations by reason of Rule 144, without the requirement for the Company to be in compliance with the current public information under Rule 144 under the Securities Act or any other rule of similar effect or (iii) the date on which all of the securities have been sold pursuant to this prospectus or Rule 144 under the Securities Act or any other rule of similar effect. The Resale Shares will be sold only through registered or licensed brokers or dealers if required under applicable state securities laws. In addition, in certain states, the Resale Shares covered hereby may not be sold unless they have been registered or qualified for sale in the applicable state or an exemption from the registration or qualification requirement is available and is complied with.
Under applicable rules and regulations under the Exchange Act, any person engaged in the distribution of the Resale Shares may not simultaneously engage in market making activities with respect to the common stock for the applicable restricted period, as defined in Regulation M, prior to the commencement of the distribution. In addition, the selling stockholders will be subject to applicable provisions of the Exchange Act and the rules and regulations thereunder, including Regulation M, which may limit the timing of purchases and sales of securities of the common stock by the selling stockholders or any other person. We will make copies of this prospectus available to the selling stockholders and have informed them of the need to deliver a copy of this prospectus to each purchaser at or prior to the time of the sale (including by compliance with Rule 172 under the Securities Act).
The following description of the Company’s capital stock and provisions of its Articles of Incorporation and Amended and Restated Bylaws are summaries and are qualified by reference to the Company’s Articles of Incorporation and Amended and Restated Bylaws.
General
Our authorized capital stock will consists of 75,000,000 shares of common stock, par value $0.0001 per share, and 5,000,000 shares of preferred stock, par value $0.0001 per share, of which no shares of preferred stock are designated or issued.
As of August 27, 2019, 10,077,068 shares of our common stock were outstanding and held by 116 stockholders of record. The actual number of holders of our common stock is greater than this number of record holders, and includes stockholders who are beneficial owners, but whose shares are held in street name by brokers or held by other nominees. This number of holders of record also does not include stockholders whose shares may be held in trust by other entities.
-49-
Common Stock
Each share of our common stock entitles the holder to receive notice of and to attend all meetings of our stockholders with the entitlement to one vote. Holders of common stock are entitled, subject to the rights, privileges, restrictions and conditions attaching to any other class of shares ranking in priority to the common stock, to receive any dividend declared by the board of directors. If the Company is voluntarily or involuntarily liquidated, dissolved or wound-up, the holders of common stock will be entitled to receive, after distribution in full of the preferential amounts, if any, all of the remaining assets available for distribution ratably in proportion to the number of shares of common stock held by them. Holders of common stock have no redemption or conversion rights. The rights, preferences and privileges of holders of shares of common stock are subject to, and may be adversely affected by, the rights of the holders of shares of any series of preferred stock that we may designate and issue in the future.
Preferred Stock
Our board of directors is authorized, subject to limitations prescribed by Nevada law, to issue up to 10,000,000 shares of our preferred stock in one or more series, to establish from time to time the number of shares to be included in each series, and to fix the designation, powers, preferences, and rights of the shares of each series and any of its qualifications, limitations or restrictions, in each case without further vote or action by our shareholders. Our board of directors can also increase or decrease the number of shares of any series of preferred stock, but not below the number of shares of that series then outstanding, without any further vote or action by our shareholders. Our board of directors may authorize the issuance of preferred stock with voting or conversion rights that could adversely affect the voting power or other rights of the holders of our common stock. The issuance of preferred stock, while providing flexibility in connection with possible acquisitions and other corporate purposes, could, among other things, have the effect of delaying, deferring or preventing a change in control of our company and might adversely affect the market price of our common stock and the voting and other rights of the holders of our common stock. We have no current plan to issue any shares of preferred stock.
Warrants
From October 2017 until February 2018, we conducted a private placement of our securities pursuant to which we issued the PPM Warrants. As of August 27, 2019, PPM Warrants to purchase up to 717,870 shares of our common stock were issued and outstanding. The PPM Warrants are exercisable for seven years from the date of issuance at an exercise price of $1.00, subject to adjustment for stock dividends, stock splits, pro rata distributions and upon the occurrence of fundamental transactions. If at any time following the issuance date of the PPM Warrants there is no registration statement registering for resale the shares of common stock issuable upon exercise of the PPM Warrants, the PPM Warrants may be exercised on a cashless basis. The PPM Warrants contain an ownership limitation such that the holder may not exercise the warrant to the extent that such exercise would result in the holder’s beneficial ownership being in excess of 4.99% of the Company’s issued and outstanding common stock together with all shares owned by the holder and its affiliates, which beneficial ownership limitation may be increased by the holder up to, but not exceeding, 9.99% of the Company’s issued and outstanding common stock.
On February 20, 2019, we issued Laidlaw & Company (UK) Ltd. a five-year warrant to purchase up to 50,000 shares of our common stock at an exercise price of $7.00 per share in connection with our IPO. The warrants provide for one demand registration of the shares of common stock underlying the warrants at our expense commencing 180 days after the effective date of our IPO and for a period of no more than five years from the effective date of our IPO or the commencement of sales of shares in our IPO. In addition, the warrants provide for unlimited “piggyback” registration rights at our expense with respect to the underlying shares of common stock for a period of not more than seven years from the effective date of our IPO or the commencement of sales of shares in our IPO.
On August 16, 2019, we closed the August 2019 Private Offering pursuant to which we issued the Private Placement Warrants. As of August 27, 2019, Private Placement Warrants to purchase up to 203,709 shares of our common stock were issued and outstanding. The Private Placement Warrants are exercisable for a period of two years beginning six months from the Closing Date at an exercise price of $8.00 per whole share, subject to adjustment. The Company is prohibited from effecting an exercise of the Private Placement Warrants to the extent that, as a result of such exercise, the holder together with the holder’s affiliates, would beneficially own more than 4.99% of the number of shares of common stock outstanding immediately after giving effect to the issuance of shares of common stock upon exercise of the Private Placement Warrants, which beneficial ownership limitation may be increased by the holder up to, but not exceeding, 9.99%.
-50-
On August 16, 2019, we issued Laidlaw & Company (UK) Ltd. a five-year warrant to purchase up to 61,113 shares of our common stock at an exercise price of $5.00 per share in connection with the August 2019 Private Placement. The Placement Agent Warrants provide for unlimited “piggyback” registration rights at our expense with respect to the underlying shares of common stock during the term of the warrant.
Equity Awards
As of August 27, 2019, we issued 170,011 shares of common stock pursuant to our 2018 Equity Incentive Plan (the “2018 Plan”) and had no outstanding options to purchase shares of our common stock or other equity awards. Pursuant to the 2018 Plan, 1,000,000 shares of our common stock will be reserved for issuance of equity awards under such plan; provided, however, the 2018 Plan contains an “evergreen” provision which allows for an annual increase in the Share Limit and the ISO Limit on the first day of each fiscal year commencing on January 1, 2019. The annual Share Limit and ISO Limit increase shall be equal to the lowest of: 250,000 shares of common stock; 5% of the number of shares of our common stock outstanding as of such date; and an amount determined by the Committee.
Registration and Piggyback Rights
Spherix Incorporated
On June 30, 2017, we entered into a registration rights agreement (the “Spherix RRA”) with Spherix Incorporated (“Spherix”) pursuant to which we agreed, among other things, that we will file with the SEC a registration statement on Form S-1 under the Securities Act that covers the resale of 1,700,000 shares of common stock issued to Spherix pursuant to a securities purchase agreement between us and Spherix and any securities issued or issuable upon any stock split, dividend or other distribution, recapitalization or similar event with respect to the foregoing (the “Spherix Registrable Securities”).
Pursuant to the Spherix RRA, we are obligated to use our best efforts to have the registration statement declared effective by the SEC as soon as practicable after it is filed with the SEC, but in no event later than the applicable Effectiveness Date. “Effectiveness Date” means with respect to the initial registration statement required to be filed pursuant to the Spherix RRA, the 18 month anniversary of the closing date of the transactions contemplated by the securities purchase agreement and, with respect to any additional registration statements which may be required pursuant to the Spherix RRA, the earliest practical date on which we are permitted to go effective on such additional registration statement; provided, however, that, in the event we are notified by the SEC that one or more of the above registration statements will not be reviewed or is no longer subject to further review and comments, the Effectiveness Date as to such registration statement shall be the fifth trading day following the date on which we are so notified if such date precedes the dates otherwise required above. In addition, pursuant to the terms of the Spherix RRA, without the consent of Spherix, neither we nor any of our security holders may include our securities in any registration statements other than the Spherix Registrable Securities. Furthermore, subject to certain exemptions, if at any time during the Effectiveness Period there is not an effective registration statement covering all of the Spherix Registrable Securities and we shall determine to prepare and file with the SEC a registration statement relating to an offering for our own account or the account of others under the Securities Act of any of our equity securities, then we shall deliver to Spherix a written notice of such determination and, if within 15 days after the date of the delivery of such notice, Spherix notifies us in writing, we must include in such registration statement all or any part of such Spherix Registrable Securities requested to be registered by Spherix.
-51-
Series A Preferred Stockholders
From October 2017 until February 2018, we conducted Private Placement Offering pursuant to which we issued an aggregate of 3,102,480 shares of Series A Preferred Stock, which were converted into an aggregate of 3,102,480 shares of common stock, and Warrants to purchase an aggregate of 775,620 shares of common stock. Investors in the Private Placement Offering received demand registration rights and piggyback registration rights with respect to the shares of common stock issuable upon conversion of the Series A Preferred Stock and exercise of the Warrants, which rights commence 90 days after the date on which our common stock first becomes publicly traded, or May 16, 2019 (the “Start Date”). In the event such demand registration rights and/or piggyback registration rights are exercised and a registration statement is not filed with the SEC including all the registrable securities no later than 90 days following the Start Date and/or the registration statement is not declared effective by the SEC within 180 days following the Start Date (or is declared effective but can no longer be used to sell registrable securities), we must pay to each holder of registrable securities 1% percent of such holder’s purchase price of the units sold in the private placement for each 30 day period (pro rata for shorter periods) until such registration statement is filed with the SEC and/or declared effective or is able to be used by the holders of registrable securities as the case may be. Notwithstanding the foregoing, in no event shall any payment of liquidated damages exceed 6% of a holder’s purchase price of such holder’s unregistered registrable securities.
August 2019 Private Placement
On August 16, 2019, we entered into a registration rights agreement with the investors in the August 2019 Private Placement pursuant to which we are required to prepare and file with the SEC a registration statement covering the Private Placement Shares and the Warrant Shares (collectively, the “Registrable Securities”) on or prior to the date that is 14 calendar days following the Closing Date (the “Filing Date”). We are required to use our reasonable best efforts to cause the registration statement covering the Registrable Securities to be declared effective as promptly as practicable after the filing thereof, but in any event no later the 120th calendar day following the Filing Date (the “Private Placement Effectiveness Date”). If we fail to file the registration statement by the Filing Date or fail to have such registration statement declared effective by the Private Placement Effectiveness Date (the date on which such failure occurs, the “Event Date”), then on each such Event Date and on each 30 calendar day anniversary of each such Event Date until the applicable failure is cured, we shall pay to each investor, in cash, a fee (the “Fee”) equal to 1% of the aggregate purchase price paid by such investor; provided, however, that we shall not be required to pay any Fees if the applicable failure occurred at such time that the Registrable Securities are eligible for resale pursuant to Rule 144 promulgated under the Securities Act; and provided, further, that we shall not be obligated to pay any such Fees if we are unable to fulfill our registration obligations as a result of rules, regulations, positions or releases issued or actions taken by the SEC pursuant to its authority with respect to Rule 415 under the Securities Act. The maximum aggregate Fees payable to any investor shall not exceed 6% of the aggregate amount invested by such investor.
Anti-Takeover Provisions our Amended and Restated Bylaws
Board of Directors Vacancies
Our Amended and Restated Bylaws authorize only our board of directors to fill vacant directorships. In addition, the number of directors constituting our board of directors may be set only by resolution of the majority of the incumbent directors.
Special Meeting of Shareholders
Our Amended and Restated Bylaws provide that special meetings of our shareholders may be called by the president of the Company, the board of directors or a committee of the board of directors that has been duly designated by the board of directors and whose powers and authority include the power to call such meetings.
Advance Notice Requirements for Shareholder Proposals and Director Nominations
Our Amended and Restated Bylaws provide that shareholders seeking to bring business before our annual meeting of shareholders, or to nominate candidates for election as directors at our annual meeting of shareholders, must provide timely notice of their intent in writing. To be timely, a shareholder’s notice must be delivered to the secretary at our principal executive offices not later than the close of business on the 90th day nor earlier than the close of business on the 120th day prior to the first anniversary of the preceding year’s annual meeting; provided, however, that in the event the date of the annual meeting is not within 25 days before or after such anniversary date, notice by the shareholder to be timely must be so delivered not later than the close of business on the 10th day following the day on which such notice of the date of annual meeting was mailed or public disclosure of the date of the annual meeting was made, whichever occurs first. These provisions may preclude our shareholders from bringing matters before our annual meeting of shareholders or from making nominations for directors at our annual meeting of shareholders.
-52-
Exclusive Forum
Our Amended and Restated Bylaws provide that unless the Company consents in writing to the selection of an alternative forum, the Eighth Judicial District Court of Clark County, Nevada shall be the sole and exclusive forum for state law claims with respect to: (i) any derivative action or proceeding brought in the name or right of the Company or on its behalf, (ii) any action asserting a claim for breach of any fiduciary duty owed by any director, officer, employee or agent of the Company to the Company or the Company’s stockholders, (iii) any action arising or asserting a claim arising pursuant to any provision of Nevada Revised Statutes Chapters 78 or 92A or any provision of the Company’s Articles of Incorporation or Amended and Restated Bylaws or (iv) any action asserting a claim governed by the internal affairs doctrine, including, without limitation, any action to interpret, apply, enforce or determine the validity of the Company’s Articles of Incorporation or Amended and Restated Bylaws. This exclusive forum provision would not apply to suits brought to enforce any liability or duty created by the Securities Act or the Exchange Act or any other claim for which the federal courts have exclusive jurisdiction. To the extent that any such claims may be based upon federal law claims, Section 27 of the Exchange Act creates exclusive federal jurisdiction over all suits brought to enforce any duty or liability created by the Exchange Act or the rules and regulations thereunder. Furthermore, Section 22 of the Securities Act creates concurrent jurisdiction for federal and state courts over all suits brought to enforce any duty or liability created by the Securities Act or the rules and regulations thereunder. The enforceability of similar exclusive forum provisions in other corporations’ bylaws has been challenged in legal proceedings, and it is possible that a court could rule that this provision in our Amended and Restated Bylaws is inapplicable or unenforceable.
Transfer Agent and Registrar
Our transfer agent and registrar is Continental Stock Transfer & Trust Company whose address is 1 State Street, 30th Floor, New York , NY 10004.
Listing
Our common stock is listed on The Nasdaq Capital Market under the symbol “HOTH.”
-53-
Overview
We are a biopharmaceutical company focused on targeted therapeutics for patients suffering from conditions such as atopic dermatitis, also known as eczema. We were incorporated in Nevada in May 2017 and have a limited operating history.
Our primary asset is a sublicense agreement with Chelexa Biosciences, Inc. pursuant to which Chelexa has granted us an exclusive sublicense to make, use, have made, import, offer for sale, and sell products based upon or involving the use of (i) topical compositions comprising a zinc chelator and gentamicin and (ii) zinc chelators to inhibit biofilm formation, which rights were originally granted to Chelexa pursuant to an exclusive license agreement with the University of Cincinnati. In addition, Chelexa granted us the right to issue exclusive and nonexclusive sublicenses (with the right to further sublicense to third parties) to make, use, have made, import, offer for sale, and sell products based upon the BioLexa Platform.
The license enables us to develop the platform for any indications in humans. Our initial focus will be on the treatment of eczema through the application of a topical cream. Although our initial focus will be on the treatment of eczema, we intend to develop a second topical cream which, upon application, is intended to reduce post-procedure infections, accelerate healing and improve clinical outcomes for patients undergoing aesthetic dermatology procedures. In addition, we conducted an initial pilot study on the efficacy of BioLexa to accelerate diabetic wound healing and intend to conduct additional studies with respect to the regenerative effects of the BioLexa Platform in the context of chronic diabetic ulcers, with and without substantial bacterial burden. The BioLexa Platform combines an FDA approved zinc chelator with one or more approved antibiotics in a topical dosage form to address unchecked eczema flare-ups by preventing the formation of infectious biofilms and the resulting clogging of sweat ducts which trigger symptoms. It is the first product candidate intended to prevent the symptom triggering flare-ups rather than simply treating symptoms when they occur.
We intend to initially use the BioLexa Platform to develop two different topical cream products: (i) a product to treat eczema and (ii) a product that reduces post-procedure infections, accelerates healing and improves clinical outcomes for patients undergoing aesthetic dermatology procedures.
-54-
BioLexa Biofilm Platform
The BioLexa Platform is a proprietary, patented, drug compound platform for the treatment of eczema. It combines an FDA-approved zinc chelator with one or more approved antibiotics in a topical dosage form to address unchecked eczema flare-ups by preventing the formation of infectious biofilms and the resulting clogging of sweat ducts.
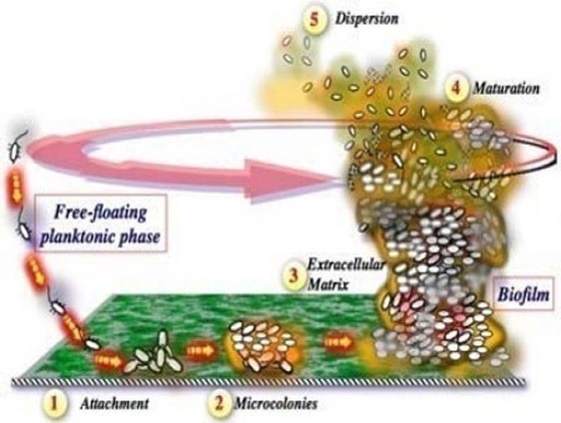
BIOFILMS IN INFECTIONS, DR TV RAO, MD https://www.slideshare.net/doctorrao/biofilms-2172226
The technology is based on scientific research into the pathogenesis of bacterial biofilm formation conducted by Andrew B. Herr, PhD at the University of Cincinnati. Dr. Herr’s work indicated that staph-biofilm formation requires the presence of zinc in the cellular environment. If the zinc is removed, the biofilms’ formation is inhibited, rendering the bacteria susceptible to immune defenses and antibiotic therapy.
Dr. Herr conducted multiple in-vitro experiments, or experiments conducted in a controlled environment outside of a living organism, in his laboratory demonstrating that chelation of zinc can prevent staph bacteria from forming colonies which in turn enables the creation of staph-biofilm. Prevention of the formation of colonies leaves the bacteria in their planktonic, or single cell state and susceptible to host immune defenses, as well as antibiotic therapy.
Dr. Herr’s in-vitro work demonstrating that zinc is an enabler for staph-biofilm formation led to the design and implementation of a series of in-vivo experiments, or experiments conducted using living organisms, specially, pigs, which experiments were conducted at the University of Miami and intended to demonstrate that the combination of zinc removal, or chelation, and broad spectrum antibiotic therapy was far more effective than either approach on its own.
The in-vivo porcine deep partial thickness wound study was undertaken to determine the effects of an antimicrobial agent on the proliferation of 106 Staphylococcus aureus (MRSA USA 300).
Swine were chosen for the in-vivo study due to the morphological and biochemical similarity between porcine and human skin. Two young female white Yorkshire/landrace specific pathogen-free pigs weighing 35-40 kg were kept in-house for at least one week prior to initiating the study, and were studied under the same protocol with approximately two weeks separating the two studies. Skin was prepared by washing with a non-antibiotic soap (Neutrogena) and sterile water. The area was blotted dry with sterile gauze. Forty-four rectangular wounds per animal (88 total wounds) measuring 10mm x 7mm x 0.5mm deep were made in the paravertebral and thoracic area with a specialized electrokeratome. The wounds were separated from one another by approximately 15mm of unwounded skin. Four wounds (four per each treatment group) were randomly assigned to each treatment group (n=11), inoculated with 106 Staphylococcus aureus (MRSA USA 300) and then treated once per day for two days.
-55-
The BioLexa Platform was formulated as a topical cream made up of Glyceryl Stearate/PEG-100 Stearate, Lanolin Alcohol, Cetyl Alcohol, Mineral Oil, Sorbitol 70% Solution, Purified Water and the active components, Gentamicin and Ca-DTPA. Gentamicin 0.1% cream (1-gram cream contains 1 mg of Gentamicin base), a broad-spectrum antibiotic exhibiting bactericidal activity against both gram-positive and gram-negative bacteria, is FDA cleared for both internal (not oral) and external application and provides a highly effective topical treatment in primary and secondary bacterial infections of the skin. Ca-DTPA, at the concentrations used, is treated as an excipient and has also received FDA clearance to be safe and effective for internal usage to increase the rates of elimination of heavy metals.
The concentration of Gentamicin 0.1% was kept constant in the study, since that is the FDA-cleared topical cream concentration. Ca-DTPA concentration was varied with the goal of achieving an optimal dose-response antimicrobial effect. Results revealed that the combination of both Gentamicin and Ca-DTPA is greater than the results achieved by Gentamicin alone or Ca-DTPA alone. In addition, no new chemical entities were formed within this formulation.
The data tables below highlight these results.
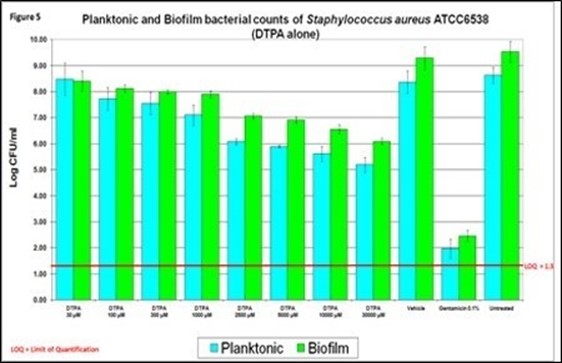
Miller School of Medicine, of the University of Miami and University of Cincinnati - Determination of the effects of a novel antimicrobial agent used in conjunction with Gentamicin on Staphylococcus aureus using a porcine model: preliminary evaluations Jose Valdes, Joel Gil, Andrew Herr, Andrew Harding and Stephen Davis
-56-

Miller School of Medicine, of the University of Miami and University of Cincinnati - Determination of the effects of a novel antimicrobial agent used in conjunction with Gentamicin on Staphylococcus aureus using a porcine model: preliminary evaluations Jose Valdes, Joel Gil, Andrew Herr, Andrew Harding and Stephen Davis
The BioLexa Platform has achieved positive results in its initial clinical studies conducted at the University of Miami. BioLexa’s formulation is a new topical dosage form “repurposing” the antibiotic, enabling it to be developed for use in patients following a special regulatory pathway codified in Section 505(b)(2) of the FDA rules. Section 505(b)(2) of the Federal Food, Drug and Cosmetic Act (“FDCA”) was enacted to enable sponsors to seek NDA approval for novel repurposed drugs without the need for such sponsors to undertake time consuming and expensive pre-clinical safety studies and Phase 1 safety studies. Proceeding under this regulatory pathway, we will be able to rely upon all of the publicly available safety and toxicology data with respect to gentamicin and zinc chelator in our FDA submissions. We will be required to conduct a Phase 2 study to show the safety of the combination in humans and after such Phase 2 study will be required to proceed to Phase 3 pivotal clinical trials. We believe that this path will dramatically reduce the required clinical development effort, costs and risks as compared to what would be required of us if we were required to conduct pre-clinical safety, toxicology and animal studies together with Phase 1 human safety trials required for new chemical entities which are not eligible to be reviewed pursuant to the Section 505(b)(2) regulatory pathway. We estimate that by using the Section 505(b)(2) regulatory pathway, that the clinical development process may be five to six years shorter than is required for a new chemical entity, and the FDA approval process may be six to nine months shorter than the typical eighteen month period, which we believe may result in lower development costs and shorter development time. As of the date hereof, we have not submitted an NDA to the FDA. In September 2018, we attended the first of a planned series of meetings with the FDA to review the requirements for submission and activation of an IND with respect to the BioLexa Platform for use in eczema. In preparation for such pre-IND meeting, we prepared and presented to the FDA our proposed Phase 2 clinical trial plan for the treatment of eczema in patients over the age of one year old. As part of our pre-IND meeting, the FDA provided us with general guidance with respect to specific animal studies, dosing schedules and suggested human safety studies before we commence clinical trials in pediatric or adult patients. We are currently investigating multiple potential venues for conducting such trial both in and outstand of the U.S. We have engaged Camargo Pharmaceutical Services, LLC to assist us with the FDA process required for Section 505(b)(2) applications and with the evaluation of potential clinical trial venues for the proof of concept study should we determine to undertake such study. Specifically, Camargo has provided and will continue to provide advice and guidance relative to the IND preparation phase for the BioLexa Platform. Camargo will assist us with the refinement of our non-clinical, clinical, clinical pharmacology and biopharmaceutics strategy incorporating the preliminary feedback we received from the FDA during our pre-IND meeting.
-57-
Sublicense with Chelexa Biosciences, Inc.
On May 26, 2017, we entered into a sublicense agreement with Chelexa, as amended on August 22, 2018 and August 29, 2018, pursuant to which Chelexa granted us an exclusive worldwide sublicense to make, use, have made, import, offer for sale, and sell products based upon or involving the use of the BioLexa Platform, which rights were originally granted to Chelexa pursuant to an exclusive license agreement with the University of Cincinnati. In addition, Chelexa granted us the right to issue exclusive and nonexclusive sublicenses (with the right to further sublicense to third parties) to make, use, have made, import, offer for sale, and sell the products based upon the BioLexa Platform.
In May 2017, we paid $300,000 to Chelexa pursuant to the sublicense agreement. In addition, we issued Chelexa 250,000 shares of our common stock, which was 10% of our fully-diluted equity at May 26, 2017, and Chelexa had the right to receive such number of additional shares of common stock required to maintain its 10% interest in our fully-diluted equity until such time we raised a minimum of $3,000,000 (the “Preemptive Right”). As of the date hereof, we have issued Chelexa an aggregate of 476,943 additional shares of common stock in accordance with the Preemptive Right. However, the Company has raised more than $3,000,000 and therefore the Preemptive Right has been terminated. Furthermore, pursuant to the sublicense agreement, Chelexa has the right to participate (the “Chelexa Participation Right”) in certain equity issuances made by us for purposes of raising capital based upon its pro-rata share to enable Chelexa to retain 10% of our fully-diluted equity until such time as we consummate an initial public offering pursuant to which we receive aggregate gross proceeds of not less than $5,000,000. However, since we consummated an initial public offering pursuant to which we received aggregate gross proceeds of $7,000,000, the Chelexa Participation Right has been terminated.
The Chelexa agreement requires us to use our best commercial efforts to develop, produce and commercialize the BioLexa products on a global basis. It further provides for the payment by us of all development and commercialization expenses along with sales-based royalties at percentages which range from mid to high single digits, with high sales volumes being subject to lower royalty rates, and total milestone payments of $3.5 million. Industry standard performance obligations for us are provided for in the Chelexa agreement with remedies for breach of such obligations. The sublicense agreement will continue until the later of April 16, 2034 and the last to expire patent, unless earlier terminated pursuant to the terms of the agreement. We, in our sole discretion, have the first right of refusal to renew the term. In addition, at any time after one year from the effective date of the sublicense agreement, Chelexa may, at its sole option, terminate or render the license granted to us nonexclusive if, in Chelexa’s judgment, our progress reports do not demonstrate that we have used our best commercial efforts to develop and seek regulatory approval of BioLexa and/or we are engaged in manufacturing, marketing or sublicensing activity which is reasonably expected to ensure that BioLexa is available to the public.
License with the University of Cincinnati
On May 18, 2018, we entered into an exclusive license agreement with the University of Cincinnati for a patented, novel genetic marker for food allergies. The genetic marker licensed by us from the University of Cincinnati (i) is used to identify at risk infants in predicting food allergies, including peanut and milk allergies, (ii) may be used to identify a person’s predisposition to an allergic reaction, thereby avoiding such reaction and (iii) may also determine an individual’s propensity to develop atopic dermatitis, such as eczema. We intend to utilize the genetic marker for purposes of determining an individual’s propensity to develop eczema as well as to identify and treat allergies in at-risk infants.
Pursuant to the terms of the license agreement, we agreed to pay the University of Cincinnati a one-time initial fee within 30 days of the date of the agreement in addition to an annual license fee. In addition, we agreed to pay the University of Cincinnati a yearly minimum annual royalty and certain milestone payments upon successful proof of concept of determining an individual’s propensity to food allergy and within 30 days of a marketing approval in the U.S. The license agreement will continue until the latter of the date upon which a valid claim pursuant to the terms of the license agreement expires or 10 years after the first commercial sale or until earlier terminated pursuant to the terms of the license agreement.
-58-
Sublicense Agreement with Zylö Therapeutics, Inc.+
On August 19, 2019 (the “Zylö Effective Date”), we entered into the Sublicense Agreement with Zylö Therapeutics, Inc. pursuant to which Zylö granted us an exclusive sublicense to the Licensed Patent Rights (as defined in the Sublicense Agreement) and the Licensed Technology (as defined in the Sublicense Agreement) to, among other things, develop, make and sell the Licensed Products (as defined in the Sublicense Agreement) and to practice the Licensed Technology in the United States and Canada for any and all uses within the Field. “Field” means all therapeutic uses related to lupus in human beings, subject to the Field Expansion Rights (as defined in the Sublicense Agreement). The term of the Sublicense Agreement shall commence on the Effective Date and shall continue until the latest of (i) ten years from the date of First Commercial Sale (as defined in the Sublicense Agreement) of the Licensed Product in such country and (ii) expiration of the last to expire Valid Claim (as defined in the Sublicense Agreement) of the Licensed Patent Rights that would be infringed by the composition, use or sale of such Licensed Product in such country. Pursuant to the terms of the Sublicense Agreement, we shall establish, with Zylö, a joint development committee to plan, review, coordinate and oversee our development activities with respect to the Licensed Products in the Field. Pursuant to the Sublicense Agreement, we shall pay Zylö (i) an upfront license fee of $50,000 (less the $10,000 we previously paid); (ii) sales-based royalties at percentages which range from mid to high single digits, with low sales volumes being subject to lower royalty rates; and total milestone payments of up to $13.5 million. In addition, within 45 days of our next equity financing pursuant to which we receive gross proceeds of at least $1 million, we shall purchase equity securities of Zylö in an amount equal to $60,000.
Product Development and Pipeline
We intend to conduct our first Phase 1 study in healthy adults with an immediate transition to a randomized, vehicle controlled Phase 1b trial in adolescent eczema patients comparing BioLexa to the base vehicle. This Phase 1b trial is intended to examine both safety and efficacy. We will assess the formulation of Ca-DTPA and Gentamicin 0.1% in our proprietary topical lotion delivered by a metered pump system. We will also assess the ability of BioLexa to clear harmful staph aureus bacterial from the skin of atopic dermatitis patients.
Following our Phase 1b trial, we intend to conduct up to two Phase 2 trials in atopic dermatitis patients comparing BioLexa to the base vehicle. Subject numbers and allocation will be informed by the results of the Phase 1b trial. We expect the clinical program to be completed, subject to receipt of funding by us, by the end of 2020 or early 2021 with an NDA submission targeted for mid to late 2021. There is currently no active IND for our product candidate in the United States.
-59-
The following table summarizes the BioLexa expected product development pipeline.

Although our initial focus will be on the treatment of eczema, we intend to develop a second topical cream which, upon application, is intended to reduce post-procedure infections, accelerate healing and improve clinical outcomes for patients undergoing aesthetic dermatology procedures. In addition, we conducted an initial pilot study on the efficacy of BioLexa to accelerate diabetic wound healing and intend to conduct additional studies with respect to the regenerative effects of the BioLexa Platform in the context of chronic diabetic ulcers, with and without substantial bacterial burden.
Eczema and Atopic Dermatitis
Eczema is also referred to as atopic dermatitis. According to the National Eczema Association, eczema affects over 32 million Americans alone. Eczema affects 10-20% of children with 60% of cases occurring within a child’s first year and 85% before the age of five.
There is no cure for eczema, but, in most cases, it is manageable. The word eczema comes from a Greek word that means to effervesce or bubble or boil over.
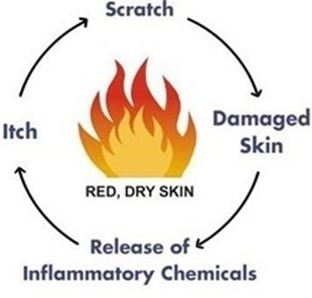
http://www.easeeczema.org/erc/symptoms_of_eczema.htm
-60-
Symptoms
The main symptom of eczema is an inflamed, itchy red rash. It can appear all over the body. Many people have it on their elbows or behind their knees. Babies often have eczema on the face, especially the cheeks and chin. They can also have it on the scalp, trunk (chest and back), and outer arms and legs. Children and adults tend to have eczema on the neck, wrists, and ankles, and in areas that bend, like the inner elbow and knee. People with eczema are usually diagnosed with it when they are babies or young children. Eczema symptoms often become less severe as children grow into adults. For some people, eczema continues into adulthood. Less often, it can start in adulthood. The rash of eczema is different for each person. It may even look different or affect different parts of the body from time to time. It can be mild, moderate or severe. Generally, people with eczema suffer from dry, sensitive skin. Eczema is also known for its intense itch. The itch may be so bad that patients scratch their skin until it bleeds, which can make the rash even worse, leading to increased inflammation and itching. This is called the itch-scratch cycle.
Signs and Symptoms of Eczema
| ● | Dry, sensitive skin | |
| ● | Intense itching | |
| ● | Red, inflamed skin | |
| ● | Recurring rash | |
| ● | Scaly areas | |
| ● | Rough, leathery patches | |
| ● | Oozing or crusting | |
| ● | Areas of swelling | |
| ● | Dark colored patches of skin |
Current Treatments
According to the National Eczema Association, people utilize many treatments for eczema to relieve the itch, including over-the-counter remedies and prescription medications. In addition, some people utilize alternative eczema treatments, such as herbal remedies. However, a study referenced by the National Eczema Association found that the majority of people with eczema are likely not satisfied with the effectiveness of their medications. The most common complaints in the study included that the subjects’ medications:
| ● | Do not work; | |
| ● | Are messy to use; | |
| ● | Are too expensive; and | |
| ● | Cause side effects. |
There can be no assurances that, if approved, the BioLexa Platform will not be subject to the similar complaints set forth above about its use. Until clinical data is available, there can be no assurances that the BioLexa Platform will not have side effects.
In addition to over-the-counter moisturizers, topical steroids are an important part of the treatment plan for most people with eczema. When eczema flares up, applying cream, lotion or ointment containing a steroid will reduce inflammation, ease soreness and irritation, reduce itching and relieve the need to scratch, allowing the skin to heal and recover.
-61-
Steroids are naturally-occurring substances that are produced in our bodies to regulate growth and immune function. There are many kinds of steroids, including “anabolic steroids” such as testosterone, “female hormones” such as estrogen (both produced in the gonads) and corticosteroids such as cortisol, which is produced by the adrenal glands. Corticosteroids are the type of steroid used for the treatment of eczema. Corticosteroids have many functions in the body, including effective control of inflammation. Corticosteroids reduce inflammation by temporarily altering the function of several types of cells and chemicals in the skin.
According to the National Eczema Association, there are many serious risks associated with the chronic use of topical steroids. Thinning of the skin (skin atrophy) is a well-recognized, possible side effect. This is especially true when potent topical corticosteroids are applied too frequently and for a prolonged period of time without a break. Early skin thinning can disappear if the topical corticosteroid use is discontinued, and, while uncommon, prolonged use can cause permanent stretch marks (striae), usually on the upper inner thighs, under the arms and in the elbow and knee creases.
Many patients with undertreated eczema have the opposite of skin thinning, and develop thickening, and sometimes darkening of the skin (changes known as lichenification). This is the skin’s response to rubbing and scratching.
Frequent and prolonged application of a topical corticosteroid to the eyelids can cause glaucoma and even cataracts. Topical corticosteroids can occasionally cause tiny pink bumps and acne, especially when used on the face and around the mouth. On the body, greasy corticosteroid ointments sometimes cause redness around hair follicles, sometimes with a pus bump centered in the follicle (folliculitis). When corticosteroids are applied to large body surface areas, enough may be absorbed to inhibit the body’s own production of cortisol, a condition known as “adrenal suppression.” The risk of adrenal suppression is highest with high potency (Class 1-2) corticosteroids. Infants and young children have a higher ratio of body surface area compared to their weight, so they are more susceptible to topical corticosteroid absorption. Moreover, if a child is given oral corticosteroids in large doses or over a long term, prolonged adrenal suppression can be associated with growth suppression and weakened immune responses.
Alternatives Today
The risks and side effects of prolonged steroid use are driving patients, physicians and the pharmaceutical industry to find safe and effective alternatives. Based upon data from the National Eczema Association our competitors include, but are not limited to, the following:
| Competitor Drug | Types of Therapies in the Market | |
| Eucrisa | Topical - non steroid | |
| Vanos Cream | Topical - Corticosteroid | |
| Aristocort A Cream | Topical - Corticosteroid | |
| Topicort Cream | Topical - Corticosteroid | |
| Temorate E* Emollient | Emollient | |
| Theraplex | Emollient | |
| Mustela | Emollient | |
| Dupixant | Shot |
What is common to all of the above candidates is that they are focused on treating or suppressing symptoms rather than causally preventing or delaying flare-ups.
-62-
The graphic below shows the numerous causes of flare-ups in eczema.

https://infodiseases.com/the-causes-symptoms-and-treatments-of-eczema.html
Our product development pipeline is focused on preventing flare-ups caused by staph biofilms. The fundamental difference between the product candidate we intend to develop and those in the table above is that ours are intended to prevent eczema flare-ups rather than merely treat symptoms of a flare-up already underway.
Preventing Eczema Flare-Ups By Stopping Biofilms
It is well known that the skin of eczema patients is colonized with Staphylococcus aureus (S. Aureus) and this organism has been shown to exist in both dry skin as well as areas of severe dermatitis. It is well known that S. Aureus bacteria are programmed by nature to form micro-colonies as a means of self-preservation. Once formed, these colonies secrete a polysaccharide matrix “shield” enabling the bacteria to grow unfettered by the host immune system or external antibiotic therapy. These shielded bacteria are referred to as “biofilm.” In eczema, biofilms are known to clog sweat ducts, triggering flare-ups. Eczema severity has been directly correlated to the degree of S. Aureus colonization and therapy generally fails to improve symptoms in the presence of high S. Aureuscounts.
Biofilms are implicated in 80% of all human infections. Once formed, bacterial biofilms resist the host immune system and antibiotics. Biofilms may require 1,000 times the antibiotic dose required to kill single bacteria, rendering biofilms virtually nontreatable once formed. Despite these realities, existing technology focuses on treatment rather than prevention.
Competition
The current competition in the eczema therapeutics market consists of conventional forms of therapy such as topical corticosteroids, topical immunomodulators and emollients as the most prominent therapies. Among all the available treatment options, topical corticosteroids hold a majority share and dominate the market. Topical corticosteroids, such as Vanos Cream, Aristorcort A Cream and Topicort Cream are available in various strengths (mild, moderate, potent and very potent) and formulations (ointment, cream, lotion and others), so that they can be used according to the severity of eczema. Calceurin inhibitors (Protopic (tacrolimus) and Elidel (pimecrolimus)) showed higher efficacy in comparison to corticosteroids and these products were widely used after their respective launches. However, in 2005, the FDA issued black box warnings for the calceurin inhibitors (Protopic and Elidel), and this resulted in declining sales of these products. Emollients, such as Theraplex, Mustela and Temorate E* Emollient, have good efficacy as well as good safety. They hydrate, moisturize and repair the skin. These products do not offer first line treatment, but they are useful as maintenance therapy in eczema patients.
-63-
Market Opportunity
We believe we have a two-fold competitive advantage over our competition. First, currently available eczema treatment options focus on treating or suppressing symptoms rather than causally preventing or delaying flare-ups. Recent peer-reviewed publications highlight that staph-induced biofilms are the root cause of flare-ups in eczema. Our BioLexa product candidate has been demonstrated to prevent the formation of these biofilms with the promise of delaying or completely arresting flare-ups, rather than merely treating symptoms of a flare-up already underway. Second, long-term use of corticosteroids, can have harmful side effects. Because the BioLexa Platform does not use steroids, our treatment avoids these harmful side effects and gives us another advantage over our competition.
Commercialization
Our business success with BioLexa depends not only on the successful development and approval of the product but also on its commercialization. At present, our plan anticipates us making the investments necessary to build an in-house marketing and sales capability for the U.S. market for BioLexa. As BioLexa makes its way through clinical development in the U.S., we intend to approach pharmaceutical and biotechnology companies outside the U.S. to negotiate and enter into strategic partnerships that will enable development and commercialization of BioLexa outside the U.S., where we believe the market opportunity is larger than that of the U.S. albeit far more complex to reach. We have no operations outside the U.S., nor are we planning to have any non-U.S. operations.
Manufacturing and Supply
We do not have any manufacturing capability and therefore have engaged Particle Sciences, Inc. (“Particle Sciences”), a company with over 20 years of experience formulating and producing topical therapeutics under current good manufacturing practice requirements (“cGMP”) regulations, to formulate and manufacture the BioLexa product candidate in accordance with cGMP requirements. Although we have not entered into a master service agreement with Particle Sciences, Particle Sciences is charged with, among other things, the following pursuant to the terms of a quote provided to us by Particle Sciences:
| ● | Optimizing the formulation of the BioLexa product candidate for ease of production and analysis; | |
| ● | Producing and packaging the required doses of the BioLexa product candidate for all clinical testing under cGMP conditions; and | |
| ● | Evaluating the shelf life of the BioLexa product candidate employing industry standard stability testing techniques and protocol. |
In addition to the foregoing, Particle Sciences is required to identify and source the two raw materials, Ca-DTPA and Gentamicin, used to produce the BioLexa product candidate. Both DTPA and Gentamicin are available from multiple suppliers in the U.S., Europe and Asia, and the Company anticipates that such raw materials will be readily available to the Company. Particle Sciences is required to vet and engage potential suppliers of the raw materials. Although the Company is engaged in negotiations with suppliers of the raw materials, the Company has not yet entered into any agreements for the supply of such raw materials. The additional components in the BioLexa formulation are all listed in the United States Pharmacopeia and are readily available from multiple U.S. sources who routinely supply similar materials to the pharmaceutical and cosmetic industries.
Intellectual Property Portfolio
We believe that market exclusivity derived from our licensed intellectual property, the Hatch-Waxman provisions applicable to products approved under 505(b)(2) and possible data protection rights will present barriers to entry and are keys to our success.
Our goal is to obtain, maintain and enforce patent protection for our products, formulations, processes, methods and other proprietary technologies, preserve our trade secrets, and operate without infringing on the proprietary rights of other parties, both in the U.S. and in other countries. Our policy is to actively seek the broadest intellectual property protection possible for our products, proprietary information and proprietary technology through a combination of contractual arrangements and patents, both in the U.S. and elsewhere in the world. In addition, we intend to actively pursue product life-cycle management initiatives to extend our market exclusivity.
-64-
We intend to cement our market exclusivity in conjunction with our formulation-development partners through additional patents based on the pharmaceutical and clinical characteristics of our drug in the proprietary formulation and through the introduction of line extensions such as combination drugs and new formulations.
In addition to any granted patents, our products will be eligible for market exclusivity to run concurrently with the term of the patent for three and a half years in the U.S. per the Hatch-Waxman Act and pediatric exclusivity guideline and up to ten years of market exclusivity in the E.U. which includes eight years of data exclusivity and two years of market exclusivity from the date of the NDA or the European equivalent referred to as Marketing Authorization Application, or MAA.
BioLexa, our biofilm-prevention technology, is covered by U.S. Patent No. 9,821,063, which was issued on November 21, 2017 and expires in 2033, and has issued patents in the E.U. and Spain expiring in 2028. Patent applications covering multiple formulations and methods of use for the BioLexa Platform are presently pending in the U.S., Europe and Canada which, if issued, will expire in 2033.
Government Regulation
Governmental authorities in the U.S. and other countries extensively regulate the research, development, testing, manufacture, labeling, promotion, advertising, distribution and marketing of pharmaceutical products such as those being developed by us. In the U.S., the FDA regulates such products under the FDCA and implements related regulations. Failure to comply with applicable FDA requirements, both before and after approval, may subject us to administrative and judicial sanctions, such as a delay in approving or refusal by the FDA to approve pending applications, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions and/or criminal prosecution.
U.S. Food and Drug Administration Regulation
United States Drug Development
In the United States, the FDA regulates drugs, medical devices and combinations of drugs and devices, or combination products, under the FDCA and its implementing regulations. Drugs are also subject to other federal, state and local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. These sanctions could include, among other actions, the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, untitled or warning letters, requests for voluntary product recalls or withdrawals from the market, product seizures, total or partial suspension of production or distribution injunctions, fines, refusals of government contracts, restitution, disgorgement, or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
The process required by the FDA before a drug may be marketed in the United States generally involves the following:
| ● | completion of extensive pre-clinical laboratory tests, animal studies and formulation studies in accordance with applicable regulations, including the FDA’s Good Laboratory Practice regulations; |
| ● | submission to the FDA of an IND, which must become effective before human clinical trials may begin; |
| ● | performance of adequate and well-controlled human clinical trials in accordance with an applicable IND and other clinical study related regulations, sometimes referred to as good clinical practices, or GCPs, to establish the safety and efficacy of the proposed drug for its proposed indication; |
-65-
| ● | submission to the FDA of an NDA; |
| ● | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the product, or components thereof, are produced to assess compliance with the FDA’s cGMP requirements; |
| ● | potential FDA audit of the clinical trial sites that generated the data in support of the NDA; and |
| ● | FDA review and approval of the NDA prior to any commercial marketing or sale. |
Once a pharmaceutical product candidate is identified for development, it enters the pre-clinical testing stage. Pre-clinical tests include laboratory evaluations of product chemistry, toxicity, formulation and stability, as well as animal studies. An IND sponsor must submit the results of the pre-clinical tests, together with manufacturing information, analytical data and any available clinical data or literature, to the FDA as part of the IND. The sponsor must also include a protocol detailing, among other things, the objectives of the initial clinical trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated if the initial clinical trial lends itself to an efficacy evaluation. Some pre-clinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions related to a proposed clinical trial and places the trial on a clinical hold within that 30-day period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Clinical holds also may be imposed by the FDA at any time before or during clinical trials due to safety concerns or non-compliance, and may be imposed on all drug products within a certain class of drugs. The FDA also can impose partial clinical holds, for example, prohibiting the initiation of clinical trials of a certain duration or for a certain dose.
All clinical trials must be conducted under the supervision of one or more qualified investigators in accordance with GCP regulations. These regulations include the requirement that all research subjects provide informed consent in writing before their participation in any clinical trial. Further, an Institutional Review Board (“IRB”) must review and approve the plan for any clinical trial before it commences at any institution, and the IRB must conduct continuing review and reapprove the study at least annually. An IRB considers, among other things, whether the risks to individuals participating in the clinical trial are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the information regarding the clinical trial and the consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed.
Each new clinical protocol and any amendments to the protocol must be submitted for FDA review, and to the IRBs for approval. Protocols detail, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| ● | Phase 1. The product is initially introduced into a small number of healthy human subjects or patients and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion and, if possible, to gain early evidence on effectiveness. In the case of some products for severe or life-threatening diseases, especially when the product is suspected or known to be unavoidably toxic, the initial human testing may be conducted in patients. |
| ● | Phase 2. Involves clinical trials in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage and schedule. |
| ● | Phase 3. Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit relationship of the product and provide an adequate basis for product labeling. |
-66-
Post-approval trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These studies are used to gain additional experience from the treatment of patients in the intended therapeutic indication. In certain instances, the FDA may mandate the performance of Phase 4 trials. Companies that conduct certain clinical trials also are required to register them and post the results of completed clinical trials on a government-sponsored database, such as ClinicalTrials.gov in the United States, within certain timeframes. Failure to do so can result in fines, adverse publicity and civil and criminal sanctions.
Progress reports detailing the results of the clinical trials, among other information, must be submitted at least annually to the FDA, and written IND safety reports must be submitted to the FDA and the investigators for serious and unexpected adverse events, findings from other studies that suggest a significant risk to humans exposed to the product, findings from animal or in vitro testing that suggest a significant risk to human subjects, and any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, if at all. The FDA or the clinical trial sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the product has been associated with unexpected serious harm to patients. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee. This group provides authorization for whether a trial may move forward at designated check points based on access to certain data from the study. The clinical trial sponsor may also suspend or terminate a clinical trial based on evolving business objectives and/or competitive climate.
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the product and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
NDA and FDA Review Process
The results of product development, pre-clinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the drug, proposed labeling and other relevant information, are submitted to the FDA as part of an NDA for a new drug, requesting approval to market the product. The submission of an NDA is subject to the payment of a substantial user fee, and the sponsor of an approved NDA is also subject to an annual program user fee; although a waiver of such fee may be obtained under certain limited circumstances. For example, the agency will waive the application fee for the first human drug application that a small business or its affiliate submits for review.
The FDA reviews all NDAs submitted before it accepts them for filing and may request additional information rather than accepting an NDA for filing. The FDA typically makes a decision on accepting an NDA for filing within 60 days of receipt. The decision to accept the NDA for filing means that the FDA has made a threshold determination that the application is sufficiently complete to permit a substantive review. Under the goals and policies agreed to by the FDA under the Prescription Drug User Fee Act (“PDUFA”), the FDA’s goal to complete its substantive review of a standard NDA and respond to the applicant is ten months from the receipt of the NDA. The FDA does not always meet its PDUFA goal dates, and the review process is often significantly extended by FDA requests for additional information or clarification and may go through multiple review cycles.
-67-
After the NDA submission is accepted for filing, the FDA reviews the NDA to determine, among other things, whether the proposed product is safe and effective for its intended use, and whether the product is being manufactured in accordance with cGMPs to assure and preserve the product’s identity, strength, quality and purity. The FDA may refer applications for novel drug products or drug products which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. The FDA will likely re-analyze the clinical trial data, which could result in extensive discussions between the FDA and us during the review process. The review and evaluation of an NDA by the FDA is extensive and time consuming and may take longer than originally planned to complete, and we may not receive a timely approval, if at all.
Before approving an NDA, the FDA will conduct a pre-approval inspection of the manufacturing facilities for the new product to determine whether they comply with cGMPs. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. In addition, before approving an NDA, the FDA may also audit data from clinical trials to ensure compliance with GCP requirements. After the FDA evaluates the application, manufacturing process and manufacturing facilities, it may issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete and the application will not be approved in its present form. A Complete Response Letter usually describes all the specific deficiencies in the NDA identified by the FDA. The Complete Response Letter may require additional clinical data and/or an additional pivotal Phase 3 clinical trial(s), and/or other significant and time-consuming requirements related to clinical trials, nonclinical studies or manufacturing. If a Complete Response Letter is issued, the applicant may either resubmit the NDA, addressing all the deficiencies identified in the letter, or withdraw the application. Even if such data and information are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive, and the FDA may interpret data differently than we interpret the same data.
There is no assurance that the FDA will ultimately approve a product for marketing in the United States, and we may encounter significant difficulties or costs during the review process. If a product receives marketing approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling or may condition the approval of the NDA on other changes to the proposed labeling, development of adequate controls and specifications, or a commitment to conduct post-market testing or clinical trials and surveillance to monitor the effects of approved products. For example, the FDA may require Phase 4 clinical trials to further assess drug safety and effectiveness and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized. The FDA may also place other conditions on approvals, including the requirement for a risk evaluation and mitigation strategy (“REMS”), to assure the safe use of the drug. If the FDA concludes a REMS is needed, the sponsor of the NDA must submit a proposed REMS; the FDA will not approve the NDA without an approved REMS, if required. A REMS could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. Any of these limitations on approval or marketing could restrict the commercial promotion, distribution, prescription or dispensing of products. Product approvals may be withdrawn for non-compliance with regulatory requirements or if problems occur following initial marketing.
Section 505(b)(2) Regulatory Approval Pathway
Section 505(b)(2) of the FDCA provides an alternate regulatory pathway for approval of a new drug by allowing the FDA to rely on data not developed by the applicant. Specifically, Section 505(b)(2) permits the submission of an NDA where one or more of the investigations relied upon by the applicant for approval was not conducted by or for the applicant and for which the applicant has not obtained a right of reference. The applicant may rely upon published literature and/or the FDA’s findings of safety and effectiveness for an approved drug already on the market. Approval or submission of a 505(b)(2) application, like those for abbreviated new drugs (“ANDAs”), may be delayed because of patent and/or exclusivity rights that apply to the previously approved drug.
-68-
A 505(b)(2) application may be submitted for a new chemical entity, or NCE, when some part of the data necessary for approval is derived from studies not conducted by or for the applicant and when the applicant has not obtained a right of reference. Such data are typically derived from published studies, rather than FDA’s previous findings of safety and effectiveness of a previously approved drug. For changes to a previously approved drug however, an applicant may rely on the FDA’s finding of safety and effectiveness of the approved drug, coupled with information needed to support the change from the approved drug, such as new studies conducted by the applicant or published data. When based on an approved drug, the 505(b)(2) drug may be approved for all of the indications permitted for the approved drug, as well as any other indication supported by additional data.
Section 505(b)(2) applications also may be entitled to marketing exclusivity if supported by appropriate data and information. As discussed in more detail below, three-year new data exclusivity may be granted to the 505(b)(2) application if one or more clinical investigations conducted in support of the application, other than bioavailability/bioequivalence studies, were essential to the approval and conducted or sponsored by the applicant. Five years of marketing exclusivity may be granted if the application is for an NCE, and pediatric exclusivity is likewise available.
Orange Book Listing and Paragraph IV Certification
For NDA submissions, including those under Section 505(b)(2), applicants are required to list with the FDA certain patents with claims that cover the applicant’s product. Upon approval, each of the patents listed in the application is published in Approved Drug Products with Therapeutic Equivalence Evaluations, commonly referred to as the Orange Book. Any applicant who subsequently files an ANDA or 505(b)(2) NDA that references a drug listed in the Orange Book must certify to the FDA that (1) no patent information on the drug product that is the subject of the application has been submitted to the FDA; (2) such patent has expired; (3) the date on which such patent expires; or (4) such patent is invalid or will not be infringed upon by the manufacture, use or sale of the drug product for which the application is submitted. This last certification is known as a Paragraph IV Certification.
If an applicant has provided a Paragraph IV Certification to the FDA, the applicant must also send notice of the Paragraph IV Certification to the holder of the NDA for the approved drug and the patent owner once the application has been accepted for filing by the FDA. The NDA holder or patent owner may then initiate a patent infringement lawsuit in response to notice of the Paragraph IV Certification. The filing of a patent infringement lawsuit within 45 days of the receipt of a Paragraph IV Certification prevents the FDA from approving the ANDA or 505(b)(2) application until the earlier of 30 months from the date of the lawsuit, the applicant’s successful defense of the suit, or expiration of the patent.
Reimbursement
Potential sales of any of our product candidates, if approved, will depend, at least in part, on the extent to which such products will be covered by third-party payors, such as government health care programs, commercial insurance and managed healthcare organizations. These third-party payors are increasingly limiting coverage and/or reducing reimbursements for medical products and services. A third-party payor’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Further, one payor’s determination to provide coverage for a drug product does not assure that other payors will also provide coverage for the drug product. In addition, the U.S. government, state legislatures and foreign governments have continued implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our future revenues and results of operations. Decreases in third-party reimbursement or a decision by a third-party payor to not cover a product candidate, if approved, or any future approved products could reduce physician usage of our products, and have a material adverse effect on our sales, results of operations and financial condition.
In the United States, the Medicare Part D program provides a voluntary outpatient drug benefit to Medicare beneficiaries for certain products. We do not know whether our product candidates, if approved, will be eligible for coverage under Medicare Part D, but individual Medicare Part D plans offer coverage subject to various factors such as those described above. Furthermore, private payors often follow Medicare coverage policies and payment limitations in setting their own coverage policies.
-69-
Pediatric Exclusivity and Pediatric Use
The Pediatric Research Equity Act, or PREA, requires a sponsor to conduct pediatric studies for most drugs and biologics, for a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration. Under PREA, original NDAs, biologics license applications and supplements thereto, must contain a pediatric assessment unless the sponsor has received a deferral or waiver. Unless otherwise required by regulation, PREA does not apply to any drug for an indication for which an orphan drug designation has been granted. The required assessment must assess the safety and effectiveness of the product for the claimed indications in all relevant pediatric subpopulations and support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The sponsor or FDA may request a deferral of pediatric studies for some or all of the pediatric subpopulations. A deferral may be granted for several reasons, including a finding that the drug or biologic is ready for approval for use in adults before pediatric studies are complete or that additional safety or effectiveness data needs to be collected before the pediatric studies begin.
Pediatric exclusivity is another type of non-patent marketing exclusivity in the United States and, if granted, provides for the attachment of an additional six months of marketing protection to the term of any existing regulatory exclusivity, including the non-patent and orphan exclusivity. This six-month exclusivity may be granted if an NDA sponsor submits pediatric data that fairly respond to a written request from the FDA for such data. The data does not need to show the product to be effective in the pediatric population studied; rather, if the clinical trial is deemed to fairly respond to the FDA’s request, the additional protection is granted. If reports of requested pediatric studies are submitted to and accepted by the FDA within the statutory time limits, whatever statutory or regulatory periods of exclusivity or patent protection cover the product are extended by six months.
Healthcare Laws and Regulations
Sales of our product candidates, if approved, or any other future product candidate will be subject to healthcare regulation and enforcement by the federal government and the states and foreign governments in which we might conduct our business. The healthcare laws and regulations that may affect our ability to operate include the following:
| ● | The federal Anti-Kickback Statute makes it illegal for any person or entity to knowingly and willfully, directly or indirectly, solicit, receive, offer, or pay any remuneration that is in exchange for or to induce the referral of business, including the purchase, order, lease of any good, facility, item or service for which payment may be made under a federal healthcare program, such as Medicare or Medicaid. The term “remuneration” has been broadly interpreted to include anything of value |
| ● | Federal false claims and false statement laws, including the federal civil False Claims Act, prohibits, among other things, any person or entity from knowingly presenting, or causing to be presented, for payment to, or approval by, federal programs, including Medicare and Medicaid, claims for items or services, including drugs, that are false or fraudulent. |
| ● | Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) created additional federal criminal statutes that prohibit among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors or making any false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. |
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 and their implementing regulations, impose obligations on certain types of individuals and entities regarding the electronic exchange of information in common healthcare transactions, as well as standards relating to the privacy and security of individually identifiable health information. |
| ● | The federal Physician Payments Sunshine Act requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, to report annually to the Centers for Medicare & Medicaid Services information related to payments or other transfers of value made to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. |
-70-
Also, many states have similar laws and regulations, such as anti-kickback and false claims laws that may be broader in scope and may apply regardless of payor, in addition to items and services reimbursed under Medicaid and other state programs. Additionally, we may be subject to state laws that require pharmaceutical companies to comply with the federal government’s and/or pharmaceutical industry’s voluntary compliance guidelines, state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures, as well as state and foreign laws governing the privacy and security of health information, many of which differ from each other in significant ways and often are not preempted by HIPAA.
Additionally, to the extent that our product is sold in a foreign country, we may be subject to similar foreign laws.
Employees
As of August 27, 2019, we employed a total of 2 full-time employees, 1 employee consultant, and 1 part-time employee. We are not a party to any collective bargaining agreements. We believe that we maintain good relations with our employees.
Our Corporate Information
We were incorporated as a Nevada corporation on May 16, 2017. Our principal executive offices are located at 1 Rockefeller Plaza, Suite 1039, New York, New York 10020 and our telephone number is (646) 756-2997.
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion of our financial condition and results of operations in conjunction with financial statements and notes thereto, as well as the “Risk Factors” and “Description of Business” sections included elsewhere in this prospectus. The following discussion contains forward-looking statements that reflect our plans, estimates and beliefs. Our actual results could differ materially from those discussed in the forward-looking statements. Factors that could cause or contribute to these differences include those discussed below and elsewhere in this prospectus, particularly in “Risk Factors”.
Overview
We are a biopharmaceutical company formed in May 2017 focused on targeted therapeutics for patients suffering from conditions such as atopic dermatitis, also known as eczema.
Our primary asset is a sublicense agreement with Chelexa Biosciences, Inc. (“Chelexa”) pursuant to which Chelexa has granted us an exclusive sublicense to use its BioLexa Platform (as defined herein), a proprietary, patented, drug compound platform developed at the University of Cincinnati. The license enables us to develop the platform for any indications in humans. Our initial focus will be on the treatment of eczema through the application of a topical cream. Although our initial focus will be on the treatment of eczema, we intend to develop a second topical cream which, upon application, is intended to reduce post-procedure infections, accelerate healing and improve clinical outcomes for patients undergoing aesthetic dermatology procedures. The BioLexa Platform combines a U.S. Food and Drug Administration (“FDA”) approved zinc chelator with one or more approved antibiotics in a topical dosage form to address unchecked eczema flare-ups by preventing the formation of infectious biofilms and the resulting clogging of sweat ducts which trigger symptoms. It is the first product candidate intended to prevent the symptom triggering flare-ups rather than simply treating symptoms when they occur.
-71-
On May 26, 2017, we entered into a sublicense agreement with Chelexa, as amended on August 22, 2018 and August 29, 2018, pursuant to which Chelexa granted us an exclusive sublicense to make, use, have made, import, offer for sale, and sell products based upon or involving the use of (i) topical compositions comprising a zinc chelator and gentamicin and (ii) zinc chelators to inhibit biofilm formation (the “BioLexa Platform” or “BioLexa”), which rights were originally granted to Chelexa pursuant to an exclusive license agreement with the University of Cincinnati. In addition, Chelexa granted us the right to issue exclusive and nonexclusive sublicenses (with the right to further sublicense to third parties) to make, use, have made, import, offer for sale, and sell products based upon the BioLexa Platform.
We intend to initially use the BioLexa Platform to develop two different topical cream products: (i) a product to treat eczema and (ii) a product that reduces post-procedure infections, accelerates healing and improves clinical outcomes for patients undergoing aesthetic dermatology procedures. Eczema is a disease that results in inflammation of the skin and is characterized by rash, red skin, and itchiness. Eczema is also referred to as atopic dermatitis. We are concentrating our effort and resources to develop the BioLexa Platform, utilizing our novel formulation and approach for these two markets.
The BioLexa Platform has achieved positive results in its initial clinical studies conducted at the University of Miami. BioLexa’s formulation is a new topical dosage form “repurposing” the antibiotic, enabling it to be developed for use in patients following a special regulatory pathway codified in Section 505(b)(2) of the FDA rules. Section 505(b)(2) of the U.S. Federal Food, Drug and Cosmetic Act was enacted to enable sponsors to seek New Drug Application (“NDA”) approval for novel repurposed drugs without the need for such sponsors to undertake time consuming and expensive pre-clinical safety studies and Phase 1 safety studies. Proceeding under this regulatory pathway, we will be able to rely upon all of the publicly available safety and toxicology data with respect to gentamicin and zinc chelator in our FDA submissions. We will be required to conduct a Phase 2 study to show the safety of the combination in humans and after such Phase 2 study will be required to proceed to Phase 3 pivotal clinical trials. We believe that this path will dramatically reduce the required clinical development effort, costs and risks as compared to what would be required of us if we were required to conduct pre-clinical safety, toxicology and animal studies together with Phase 1 human safety trials required for new chemical entities which are not eligible to be reviewed pursuant to the Section 505(b)(2) regulatory pathway. We estimate that by using the Section 505(b)(2) regulatory pathway, that the clinical development process may be five to six years shorter than is required for a new chemical entity, and the FDA approval process may be six to nine months shorter than the typical eighteen month period, which we believe may result in lower development costs and shorter development time. As of the date hereof, we have not submitted an NDA to the FDA. In September 2018, we attended the first of a planned series of meetings with the FDA to review the requirements for submission and activation of an investigational new drug application (“IND”) with respect to the BioLexa Platform for use in eczema. In preparation for such pre-IND meeting, we prepared and presented to the FDA our proposed Phase 2 clinical trial plan for the treatment of eczema in patients over the age of one year old. As part of our pre-IND meeting, the FDA provided us with general guidance with respect to specific animal studies, dosing schedules and suggested human safety studies before we commence clinical trials in pediatric or adult patients. We are currently investigating multiple potential venues for conducting such trial both in and outstand of the U.S. We have engaged Camargo Pharmaceutical Services, LLC (“Camargo”) to assist us with the FDA process required for Section 505(b)(2) applications and with the evaluation of potential clinical trial venues for the proof of concept study should we determine to undertake such study. Specifically, Camargo has provided and will continue to provide advice and guidance relative to the IND preparation phase for the BioLexa Platform. Camargo will assist us with the refinement of our non-clinical, clinical, clinical pharmacology and biopharmaceutics strategy incorporating the preliminary feedback we received from the FDA during our pre-IND meeting.
We intend to conduct our first Phase 1 study in healthy adults with an immediate transition to a randomized, vehicle controlled Phase 1b trial in adolescent eczema patients comparing BioLexa to the base vehicle. This Phase 1b trial is intended to examine both safety and efficacy. We will assess the formulation of Ca-DTPA and Gentamicin 0.1% in our proprietary topical lotion delivered by a metered pump system. We will also assess the ability of BioLexa to clear harmful staph aureus bacterial from the skin of atopic dermatitis patients.
Following our Phase 1b trial, we intend to conduct up to two Phase 2 trials in atopic dermatitis patients comparing BioLexa to the base vehicle. Subject numbers and allocation will be informed by the results of the Phase 1b trial. We expect the clinical program to be completed, subject to receipt of funding by us, by the end of 2020 or early 2021 with an NDA submission targeted for mid to late 2021.
-72-
In addition, we conducted an initial pilot study on the efficacy of BioLexa to accelerate diabetic wound healing and intend to conduct additional studies with respect to the regenerative effects of the BioLexa Platform in the context of chronic diabetic ulcers, with and without substantial bacterial burden.
We believe that the key elements for our market success include:
| ● | the proprietary formulation of two FDA-approved drugs to treat bacterial proliferation reduces development time and costs by giving us the ability to rely on safety and efficacy data from the two approved drugs; |
| ● | our proprietary formulation is not a topical corticosteroid, and may not be subject to the same FDA black box warning issues as most commonly prescribed treatments currently in use; and |
| ● | a recent peer-reviewed publication titled “Staphylococcal Bacteria May Cause Eczema, Study Reveals”, published by Dr. Herbert B. Allen, highlights that staph-induced biofilms are the root cause of flare-ups in eczema. Our BioLexa product candidate has been demonstrated to prevent the formation of these biofilms with the promise of delaying or completely arresting flare-ups, rather than merely treating symptoms of a flare-up already underway. |
In addition to our sublicense agreement with Chelexa, we entered into an exclusive license agreement with the University of Cincinnati for a patented, novel genetic marker for food allergies. The genetic marker licensed by us from the University of Cincinnati (i) may be used to identify at risk infants in predicting food allergies, including peanut and milk allergies, (ii) may be used to identify a person’s predisposition to an allergic reaction, thereby avoiding such reaction and (iii) may also determine an individual’s propensity to develop atopic dermatitis, such as eczema. We intend to utilize the genetic marker for purposes of determining an individual’s propensity to develop eczema as well as to identify and treat allergies in at-risk infants.
In order to generate revenue from our product candidates, we will need to sell our product candidates either through distribution partnerships or through our own sales efforts. Prior to selling our product candidates, we will need to receive FDA approval of our NDA for each indication that we intend to treat. The first indication we are seeking approval for is the BioLexa Platform for treating eczema. We intend to submit our NDA for such indication by the end of 2021 with approval of such NDA anticipated to be in 2022; however, no assurances can be given that we will receive approval of the NDA in a timely manner, if at all.
Results of Operations
Comparison of the Three Months Ended June 30, 2019 and 2018
Operating Costs and Expenses
Research and Development Expenses
For the three months ended June 30, 2019, research and development expenses were approximately $0.4 million, of which $10,000 was related to a term sheet entered into between the Company and Zylö Therapeutics Inc. (the “Zylö Term Sheet”) and approximately $0.4 million was related to other research and development expenses.
During the three months ended June 30, 2018, research and development expenses were approximately $0.2 million, of which approximately $32,000 was related to the acquisition of a license from University of Cincinnati and approximately $0.2 million was related to other research and development expenses.
We expect our research and development activities to increase as we develop our existing product candidate and potentially acquire new product candidates, reflecting increasing costs associated with the following:
| ● | employee-related expenses, which include salaries and benefits, and rent expenses; |
-73-
| ● | license fees and milestone payments related to in-licensed products and technology; |
| ● | expenses incurred under agreements with contract research organizations, investigative sites and consultants that conduct our clinical trials and a substantial portion of our preclinical activities; |
| ● | the cost of acquiring and manufacturing clinical trial materials; and |
| ● | costs associated with non-clinical activities, and regulatory approvals. |
Compensation, Professional Fees, Rent and Other (“General and Administrative Expenses”)
For the three months ended June 30, 2019, general and administrative expenses were approximately $0.9 million, which primarily consisted of approximately $0.1 million related to payroll expenses and stock-based compensation, $0.5 million for professional fees and $0.2 million for other expenses.
During the three months ended June 30, 2018, general and administrative expenses were approximately $0.5 million, which primarily consisted of approximately $0.2 million related to payroll expenses and approximately $0.2 million for professional fees.
We anticipate that our general and administrative expenses will increase in future periods, reflecting continued and increasing costs associated with:
| ● | support of our research and development activities; |
| ● | stock compensation granted to key employees and non-employees; |
| ● | support of business development activities; and |
| ● | increased professional fees and other costs associated with the regulatory requirements and increased compliance associated with being a public reporting company. |
Comparison of the Six Months Ended June 30, 2019 and 2018
Operating Costs and Expenses
Research and Development Expenses
For the six months ended June 30, 2019, research and development expenses were approximately $0.5 million, of which $10,000 was related to the Zylö Term Sheet, an aggregate of $10,000 was related to a license acquired from the University of Maryland and Isoprene Pharmaceuticals Inc., and approximately $0.4 million was related to other research and development expenses.
During the six months ended June 30, 2018, research and development expenses were approximately $0.4 million, of which $32,000 was related to the acquisition of a license from University of Cincinnati and $0.1 million was related to the acquisition of a license from Chelexa Biosciences, Inc. Pursuant to the terms of the sublicense agreement, we issued Chelexa 0.2 million shares of our common stock valued at $0.1 million. In addition, we incurred approximately $0.3 million of expense related to other research and development expenses.
Compensation, Professional Fees, Rent and Other
For the six months ended June 30, 2019, general and administrative expenses were approximately $1.6 million, which primarily consisted of approximately $0.5 million related to payroll expenses and stock-based compensation and approximately $0.8 million for professional fees.
During the six months ended June 30, 2018, general and administrative expenses were $0.9 million, which primarily consisted of $0.3 million related to payroll expenses and $0.4 million for professional fees.
-74-
Results of Operations
Components of Our Results of Operations for the Year Ended December 31, 2018
Operating Costs and Expenses
Research and Development Expenses
For the year ended December 31, 2018, research and development expenses were approximately $1.0 million, of which approximately $0.1 million was related to license acquired, $0.1 million was related to the issuance of 213,166 shares of our common stock pursuant to the sublicense agreement with Chelexa and $0.8 million was related to other research and development expenses.
We expect our research and development activities to increase as we develop our existing product candidate and potentially acquire new product candidates, reflecting increasing costs associated with the following:
| ● | employee-related expenses, which include salaries and benefits, and rent expenses; |
| ● | license fees and milestone payments related to in-licensed products and technology; |
| ● | expenses incurred under agreements with contract research organizations, investigative sites and consultants that conduct our clinical trials and a substantial portion of our preclinical activities; |
| ● | the cost of acquiring and manufacturing clinical trial materials; and |
| ● | costs associated with non-clinical activities, and regulatory approvals. |
General and Administrative Expenses
For the year ended December 31, 2018, general and administrative expenses were approximately $1.5 million, which primarily consisted of approximately $0.4 million related to payroll expenses, approximately $0.1 million related to the issuance of 145,970 shares of our common stock to two employees and two directors and approximately $0.7 million for professional fees.
We anticipate that our general and administrative expenses will increase in future periods, reflecting continued and increasing costs associated with:
| ● | support of our research and development activities; |
| ● | stock compensation granted to key employees and non-employees; |
| ● | support of business development activities; and |
| ● | increased professional fees and other costs associated with the regulatory requirements and increased compliance associated with being a public reporting company. |
Components of Our Results of Operations for the Period from May 16, 2017 (Inception) through December 31, 2017
Operating Costs and Expenses
Research and Development Expenses
For the period from May 16, 2017 (inception) through December 31, 2017, research and development expenses were approximately $0.6 million, of which approximately $0.5 million was related to the acquisition of our sublicense with Chelexa, to use the BioLexa Platform, a proprietary, patented, drug compound platform developed at the University of Cincinnati. Such amount includes an upfront cash fee of $0.3 million and $0.2 million associated with the value of 513,777 shares of our common stock issued. Additionally, we incurred approximately $67,000 of expense related to other research and development expenses.
-75-
We expect our research and development activities to significantly increase as we develop our existing product candidate and potentially acquire new product candidates, reflecting increasing costs associated with the following:
| ● | employee-related expenses, which include salaries and benefits, and rent expenses; | |
| ● | license fees and milestone payments related to in-licensed products and technology; | |
| ● | expenses incurred under agreements with contract research organizations, investigative sites and consultants that conduct our clinical trials and a substantial portion of our preclinical activities; | |
| ● | the cost of acquiring and manufacturing clinical trial materials; and | |
| ● | costs associated with non-clinical activities and regulatory approvals. |
General and Administrative Expenses
For the period from May 16, 2017 (inception) through December 31, 2017, general and administrative expenses were $1.3 million, which primarily consisted of $1.0 million related to the issuance of 2,430,000 shares of restricted stock to employees and non-employees and $0.2 million related to professional fees.
We anticipate that our general and administrative expenses will increase in future periods, reflecting continued and increasing costs associated with:
| ● | support of our expanded research and development activities; | |
| ● | stock compensation granted to key employees and non-employees; | |
| ● | support of business development activities; and | |
| ● | increased professional fees and other costs associated with the regulatory requirements and increased compliance associated with being a public reporting company. |
Liquidity and Capital Resources
We have incurred substantial operating losses since inception, and expect to continue to incur significant operating losses for the foreseeable future and may never become profitable. As of June 30, 2019, we had approximately $4.1 million in cash and an accumulated deficit of approximately $6.6 million.
We have funded our operations from proceeds from the sale of equity and debt securities. We will require significant additional capital to make the investments we need to execute our longer-term business plan. Our ability to successfully raise sufficient funds through the sale of debt or equity securities when needed is subject to many risks and uncertainties and, even if we are successful, future equity issuances would result in dilution to its existing stockholders and any future debt securities may contain covenants that limit our operations or ability to enter into certain transactions.
The proceeds from our initial public offering (the “IPO”) and the current cash are sufficient to fund operations for at least the next 12 months; however, we will need to raise significant additional capital to continue to fund our operations and the clinical trials for BioLexa. We may seek to sell common stock, preferred stock or convertible debt securities, enter into a credit facility or another form of third-party funding or seek other debt financing. In addition, we may seek to raise cash through collaborative agreements or from government grants. The sale of equity and convertible debt securities may result in dilution to our stockholders and certain of those securities may have rights senior to those of our common shares. If we raise additional funds through the issuance of preferred stock, convertible debt securities or other debt financing, these securities or other debt could contain covenants that would restrict our operations. Any other third-party funding arrangement could require us to relinquish valuable rights.
-76-
The source, timing and availability of any future financing will depend principally upon market conditions, and, more specifically, on the progress of our clinical development program. Funding may not be available when needed, at all, or on terms acceptable to us. Lack of necessary funds may require us to, among other things, delay, scale back or eliminate expenses including some or all of our planned development, including our clinical trials.
Cash Flows from Operating Activities
For the six months ended June 30, 2019, net cash used in operations was $1.8 million, which primarily resulted from a net loss of $2.1 million.
For the six months ended June 30, 2018, net cash used in operations was $0.9 million, which primarily resulted from a net loss of $1.3 million, partially offset by $0.1 million of non-cash research and development expense related to a license acquisition.
Cash Flows from Investing Activities
For the six months ended June 30, 2019, net cash used in investing activities was $20,000, which was related to the purchase of research and development licenses.
For the six months ended June 30, 2018, there were no investing activities.
Cash Flows from Financing Activities
For the six months ended June 30, 2019, net cash provided by financing activities was $5.8 million, including $0.2 million restricted cash, from the net proceeds of the IPO. The $0.2 million restricted cash has been deposited into a third-party escrow account in order to provide a source of funding for certain indemnification obligations the Company has pursuant to its Qualified Independent Underwriter Engagement Agreement.
On February 20, 2019, we closed the IPO pursuant to which we issued 1,250,000 shares of our common stock for net proceeds of approximately $5.8 million, after deducting underwriting discounts and commissions and offering expenses.
For the six months ended June 30, 2018, net cash provided by financing activities was $1.2 million, which reflects the net proceeds from investors in consideration for the issuance of 13.77 units of our securities.
Off-Balance Sheet Arrangements; Commitments and Contractual Obligations
As of June 30, 2019, we did not have any off-balance sheet arrangements as defined in Item 303(a)(4)(ii) of Regulation S-K and did not have any commitments or contractual obligations.
-77-
The following table sets forth the name, age and positions of our executive officers and directors.
| NAME | AGE | POSITION | ||
| Robb Knie | 50 | President, Chief Executive Officer and Director | ||
| David Briones | 43 | Chief Financial Officer | ||
| Vadim Mats | 34 | Director | ||
| Kenneth Rice | 65 | Director | ||
| Anthony Hayes | 51 | Director | ||
| David B. Sarnoff | 51 | Director |
The business background and certain other information about our directors and executive officers is set forth below:
Robb Knie
Robb Knie has served as President and Chief Executive Officer and as a director of the Company since May 2017 and served as our principal financial and accounting officer from June 2018 until March 2019. Mr. Knie served as the President of Lifeline Industries Inc. since its inception in 1995. From 2002 to 2010 he was a Semiconductor Analyst for PAW Partners. From 1993 until 1995, Mr. Knie served as Northeast Regional Manager of American Express Financial Advisors. Mr. Knie served as a board member of Inventergy Global, Inc. (NASDAQ: INVT) from December 2013 until October 2014. We believe that Mr. Knie is qualified to serve as a director because of his business and leadership experience and experience as a board member of public companies.
David Briones
David Briones served as Chief Financial Officer of the Company since March 5, 2019 and has over nineteen years of public accounting and executive level experience. He consults with various public companies in financial reporting, internal control development and evaluation, budgeting and forecasting. Since October 2010, Mr. Briones has served as the managing member and founder of Brio Financial Group, LLC, a financial reporting consulting firm. In addition, since August 2013, Mr. Briones has served as Chief Financial Officer of Petro River Oil Corp., an independent energy company focused on the exploration and development of conventional oil and gas assets. Mr. Briones has also served as interim Chief Financial Officer of AdiTx Therapeutics, Inc., a pre-clinical stage, life sciences company with a mission to prolong life and enhance life quality of transplanted patients, since January 2018. From October 2017 to May 2018, Mr. Briones served as the Chief Financial Officer of Bitzumi, Inc., a Bitcoin exchange and marketplace. Prior to founding Brio Financial Group, LLC, Mr. Briones was an auditor with Bartolomei Pucciarelli, LLC in Lawrenceville, New Jersey and PricewaterhouseCoopers LLP in New York, New York. Mr. Briones received a BS in accounting from Fairfield University.
Vadim Mats
Vadim Mats has served as a director of the Company since May 2017. Mr. Mats currently is the Chief Financial Officer and Chief Operating Officer of Grand Private Equity. Mr. Mats consults with multiple companies in a range of industries on all aspects of finance, accounting, tax and operations. From June 2010 to December 2016, Mr. Mats was Chief Financial Officer of Whalehaven Capital. Mr. Mats also served as the Assistant Controller at Eton Park Capital Management, LP, a multi-strategy fund, from July 2007 to December 2009. From June 2006 to July 2007, Mr. Mats was a Senior Fund Accountant at The Bank of New York Mellon (NYSE: BK), where he was responsible for over fifteen funds. From 2011 until March 2017, Mr. Mats served as Director and Chair of the Audit Committee of Wizard World Inc. (OTCQB: WIZD). Mr. Mats holds a Master of Science degree in accounting and finance and a Bachelor’s degree in Business Administration specializing in finance and investments from the Zicklin School of Business at Bernard Baruch College. Further, Mr. Mats is a CAIA© Charterholder and a Certified Public Accountant in the State of New York. We believe that Mr. Mats is qualified to serve as a director because of his experience as a board member of a public company and his knowledge with respect to finance, accounting, tax, and operations matters.
-78-
Kenneth Rice
Kenneth Rice has served as a director of the Company since May 2017. Mr. Rice served as the President and Chief Financial Officer of LikeMinds, Inc., an affiliate of Alseres Pharmaceuticals, Inc. (“Alseres”) from 2016 through March 2019. From 2005 through March 2019, Mr. Rice served as the Executive Vice President, Chief Financial Officer and in-house counsel to Alseres. In addition, since 2012, Mr. Rice has served as Executive Chairman of Chelexa. From August 1999 through March 2001, Mr. Rice served as Vice President and Chief Financial Officer of MacroChem Corporation, a publicly-traded drug delivery company. Mr. Rice received his BSBA from Babson College, his MBA from Babson College, his Juris Doctorate from Suffolk University Law School and his LLM from Boston University Law School. We believe that Mr. Rice is qualified to serve as a director because of his over 25 years of experience in operations, finance, marketing and sales and business development in both private and public life science companies.
Anthony Hayes
Anthony Hayes has served as a director of the Company since June 2017 and as Chief Executive Officer and director of Spherix Incorporated (NASDAQ: SPEX) since September 2013. In addition, Mr. Hayes has served as the Chief Executive Officer of North South since March 2013. Mr. Hayes was the fund manager of JaNSOME IP Management LLC and JaNSOME Patent Fund LP from August 2012 to August 2013, both of which he co-founded. Mr. Hayes was the founder and Managing Member of Atwater Partners of Texas LLC from March 2010 to August 2012 and a partner at Nelson Mullins Riley & Scarborough LLP from May 1999 to March 2010. Mr. Hayes received his Juris Doctorate from Tulane University School of Law and his B.A. in economics from Mary Washington College. We believe that Mr. Hayes is qualified to serve as a director because of his experience as CEO of Spherix Incorporated and North South.
David B. Sarnoff
David Sarnoff has served as a director of the Company since August 2018. Since June 2015, Mr. Sarnoff has served as the founder and Principal of Sarnoff Group, LLC. From October 2003 until June 2015, Mr. Sarnoff served as the co-founder and Principal of Morandi, Taub & Sarnoff LLC, and from July 1998 until October 2003 he served as a Legal Recruiter for Schneider Legal Search, Inc. From August 1994 until July 1998, Mr. Sarnoff served as a litigation associate attorney at Wachtel Missry LLP (formerly known as Gold & Wachtel LLP). Since July 2018, Mr. Sarnoff has served as a member of the advisory committee of the New Jersey Association of School Resource Officers. From January 2015 until January 2018, Mr. Sarnoff served as board President of Fort Lee Board of Education and served as a board member from January 2013 through January 2019. Mr. Sarnoff received his juris doctor from Rutgers University School of Law and his bachelor of arts from Hofstra University. Mr. Sarnoff is admitted to the New York and New Jersey (retired status) state bars. Mr. Sarnoff is qualified to serve as a director because of his legal experience as well as his extensive experience in executive leadership and business development.
Family Relationships
There are no family relationships among any of our executive officers or directors.
Arrangements between Officers and Directors
To our knowledge, there is no arrangement or understanding between any of our officers and any other person, including directors, pursuant to which the officer was selected to serve as an officer.
Involvement in Certain Legal Proceedings
We are not aware of any of our directors or officers being involved in any legal proceedings in the past ten years relating to any matters in bankruptcy, insolvency, criminal proceedings (other than traffic and other minor offenses), or being subject to any of the items set forth under Item 401(f) of Regulation S-K.
-79-
Committees of Our Board of Directors
Our board of directors directs the management of our business and affairs, as provided by Nevada law, and conducts its business through meetings of the board of directors and its standing committees. We have a standing audit committee, compensation committee and nominating and corporate governance committee. In addition, from time to time, special committees may be established under the direction of the board of directors when necessary to address specific issues.
Our board of directors has determined that all of the members of the audit committee, the compensation committee and the nominating and corporate governance committee are independent as defined under the applicable rules of The Nasdaq Capital Market, including, in the case of all of the members of our audit committee, the independence requirements contemplated by Rule 10A-3 under the Exchange Act. In making such determination, the board of directors considered the relationships that each director has with our Company and all other facts and circumstances that the board of directors deemed relevant in determining director independence, including the beneficial ownership of our capital stock by each director.
Audit Committee
Our audit committee will be responsible for, among other things:
| ● | approving and retaining the independent registered public accounting firm to conduct the annual audit of our financial statements; | |
| ● | reviewing the proposed scope and results of the audit; | |
| ● | reviewing and pre-approval of audit and non-audit fees and services; | |
| ● | reviewing accounting and financial controls with the independent registered public accounting firm and our financial and accounting staff; | |
| ● | reviewing and approving transactions between us and our directors, officers and affiliates; | |
| ● | establishing procedures for complaints received by us regarding accounting matters; | |
| ● | overseeing internal audit functions, if any; and | |
| ● | preparing the report of the audit committee that the rules of the Securities and Exchange Commission require to be included in our annual meeting proxy statement. |
Our audit committee consists of Anthony Hayes, Vadim Mats and David Sarnoff, with Anthony Hayes serving as chair. Each member of our audit committee meets the financial literacy requirements of the Nasdaq rules. In addition, our board of directors has determined that Anthony Hayes qualifies as an “audit committee financial expert,” as such term is defined in Item 407(d)(5) of Regulation S-K.
Our board of directors adopted a written charter for the audit committee, which is available on our principal corporate website at www.hoththerapeutics.com.
Compensation Committee
Our compensation committee is responsible for, among other things:
| ● | reviewing and recommending the compensation arrangements for management, including the compensation for our president and chief executive officer; | |
| ● | establishing and reviewing general compensation policies with the objective to attract and retain superior talent, to reward individual performance and to achieve our financial goals; | |
| ● | administering our stock incentive plans; and | |
| ● | preparing the report of the compensation committee that the rules of the Securities and Exchange Commission require to be included in our annual meeting proxy statement. |
Our compensation committee consists of Anthony Hayes, Vadim Mats and David Sarnoff, with Anthony Hayes serving as chair.
Our board of directors adopted a written charter for the compensation committee, which is available on our principal corporate website at www.hoththerapeutics.com.
-80-
Nominating and Governance Committee
Our nominating and governance committee is responsible for, among other things:
| ● | identifying and nominating members of the board of directors; | |
| ● | developing and recommending to the board of directors a set of corporate governance principles applicable to our Company; and | |
| ● | overseeing the evaluation of our board of directors. |
Our nominating and corporate governance committee consists of Vadim Mats, Anthony Hayes and David Sarnoff, with Vadim Mats serving as chair.
Our board of directors adopted a written charter for the nominating and corporate governance committee, which is available on our principal corporate website at www.hoththerapeutics.com.
Scientific Advisory Board
In July 2017, the board of directors formed a Scientific Advisory Board (formerly known as the Technology Advisory Board). The members of such board are as follows: (i) Kenneth Rice as Chairman, (ii) Dr. Richard Granstein, Dr. Gurjit Hershey, and Adam Friedman as Medical Doctor members and (iii) Dr. Andrew Herr and Dr. Stefanie Johns as Non-Medical Doctor members.
Summary Compensation Table
The following table sets forth the compensation paid or accrued during the fiscal year ended December 31, 2018 and 2017 to:
| ● | Robb Knie, Chief Executive Officer |
| Name and Principal Position | Year | Salary ($) |
Bonus ($) |
Stock
awards ($) |
Option
awards ($) |
Nonequity
incentive plan compensation ($) |
Nonqualified deferred compensation earnings ($) | All
other compensation ($) |
Total ($) |
|||||||||||||||||||||||||
| Robb Knie, | 2018 | $ | 250,000 | — | $ | 58,170 | — | — | — | — | $ | 308,170 | ||||||||||||||||||||||
| Chief Executive Officer, President and Director | 2017 | $ | 145,833 | — | — | — | — | — | — | $ | 145,833 | |||||||||||||||||||||||
Outstanding Equity Awards at December 31, 2018
There were no outstanding equity awards held by our executive officers as of December 31, 2018.
-81-
Non-Employee Director Compensation
The following table presents the total compensation for each person who served as a non-employee member of our board of directors and received compensation for such service during the fiscal year ended December 31, 2018. Other than as set forth in the table and described more fully below, we did not pay any compensation, make any equity awards or non-equity awards to, or pay any other compensation to any of the non-employee members of our board of directors in 2018.
| Name | Fees
earned or paid in cash ($) | Stock awards ($) | Option
awards ($) | Non-equity
incentive plan compensation ($) | Nonqualified
deferred compensation earnings ($) | All
other compensation ($) | Total ($) | |||||||||||||||||||||
| Vadim Mats | $ | 36,000 | $ | 12,500 | — | — | — | — | $ | 48,500 | ||||||||||||||||||
| Kenneth Rice | $ | 40,000 | $ | 12,500 | — | — | — | — | $ | 52,500 | ||||||||||||||||||
| Anthony Hayes | $ | 42,000 | $ | 12,500 | — | — | — | — | $ | 54,500 | ||||||||||||||||||
| David Sarnoff | $ | 12,228 | $ | 3,470 | — | — | — | — | $ | 15,698 | ||||||||||||||||||
Non-Employee Director Compensation Policy
Our directors will receive $30,000 cash compensation per year for their service on the board of directors, as well as reimbursement for out-of-pocket expenses with respect to such directors’ attendance at meetings of the board of directors of the Company. Committee chairs will receive an additional $6,000 cash compensation per year for their added services in such roles. In addition, Anthony Hayes, Vadim Mats and Kenneth Rice have each received a stock grant of 12,500 shares of common stock.
Employment Agreements
Robb Knie Employment Agreement
On February 20, 2019, the Company entered into an amended and restated employment agreement (the “Employment Agreement”) with Robb Knie, the Company’s Chief Executive Officer in connection with the IPO. The term of the Employment Agreement will continue for a period of one year from the date of execution and automatically renews for successive one year periods at the end of each term until either party delivers written notice of their intent not to review at least six months prior to the expiration of the then effective term. Mr. Knie’s base salary was increased to $350,000 per year upon completion of the IPO. Mr. Knie is eligible to receive an annual bonus of up to $100,000 per year at the discretion of the compensation committee of the Company. Mr. Knie is also entitled to participate in any and all Benefit Plans (as defined in the Employment Agreement), from time to time, in effect for senior executives, along with vacation, sick and holiday pay in accordance with the Company’s policies established and in effect from time to time.
The Employment Agreement may be terminated upon (i) Mr. Knie’s death, (ii) Mr. Knie’s Total Disability (as defined in the Employment Agreement), (iii) expiration of the term if either party has provided a timely non-renewal notice, (iv) at Mr. Knie’s option (A) upon 90 days prior written notice; provided, however, Mr. Knie may terminate the Employment Agreement by providing written notice at any time within 40 days of the consummation of a Change in Control Transaction (as defined in the Employment Agreement) or (B) for Good Reason (as defined in the Employment Agreement); or (v) at the Company’s option (A) for Cause (as defined in the Employment Agreement) or (B) upon 90 days prior written notice without Cause (as defined in the Employment Agreement).
Upon the termination of Mr. Knie’s employment for any reason, whether by Mr. Knie or by the Company, Mr. Knie shall be paid accrued but unpaid compensation and vacation pay through the date of termination and any other benefits accrued to him under any Benefit Plans (as defined in the Employment Agreement) outstanding at the date of termination and the reimbursement of expenses incurred on or prior to such date (the “Severance Package”). In addition to the Severance Package, upon Mr. Knie’s termination for death or Total Disability (as defined in the Employment Agreement), Mr. Knie or his estate or beneficiaries, as applicable, shall receive (i) 12 months base salary at the then current rate and (ii) payment on a pro-rated basis of any annual bonus or other payments earned in connection with any bonus plan to which the Mr. Knie was a participant as of the date of death or Total Disability. Upon Mr. Knie’s termination for Good Reason (as defined in the Employment Agreement), without Cause (as defined in the Employment Agreement) or Mr. Knie’s termination upon 90 days prior written notice to the Company or notice to the Company within 40 days of the consummation of a Change in Control Transaction (as defined in the Employment Agreement), in addition to the Severance Package, Mr. Knie shall receive (i) 12 months base salary at the then current rate, (ii) payment on a pro-rated basis of any annual bonus or other payments earned in connection with any bonus plan to which the Mr. Knie was a participant as of the date of termination and (iii) any equity grants to Mr. Knie shall be immediately vested upon termination. The Employment Agreement also contains covenants prohibiting Mr. Knie from disclosing confidential information with respect to the Company.
-82-
David Briones Employment Agreement
On March 6, 2019 (the “Effective Date”), the Company entered into an employment agreement with David Briones pursuant to which Mr. Briones will serve as Chief Financial Officer of the Company (the “Employment Agreement”) and will receive (i) a base salary of $60,000 per year which may be increased from time to time at the discretion of the Company, (ii) will be eligible to receive an annual bonus of up to $30,000 per year at the discretion of the compensation committee of the Company which bonus may be paid in shares of common stock of the Company at the sole discretion of Mr. Briones and (iii) received an option to purchase up to 50,000 shares of the Company’s common stock at an exercise price of $5.88 per share which option vested in full upon grant. Mr. Briones will also be entitled to vacation, sick and holiday pay in accordance with the Company’s policies established and in effect from time to time. The term of the Employment Agreement will continue for a period of one year from the Effective Date and automatically renews for successive one year periods at the end of each term unless either party delivers written notice of their intent not to renew at least six months prior to the expiration of the then effective term.
The Employment Agreement may be terminated (A) by the Company (i) with or without Cause (as defined in the Employment Agreement) (if terminated without Cause, the Company must provide sixty days prior written notice) (ii) upon Mr. Briones’ death or (iii) upon Mr. Briones’ Disability (as defined in the Employment Agreement) or (B) by Mr. Briones for any reason upon sixty days prior written notice. Upon the termination of Mr. Briones’ employment for any of the foregoing reasons, Mr. Briones will be paid (i) his then base salary accrued up to and including the date of termination, (ii) unreimbursed expenses and (iii) any accrued benefits under any Company plans. The Employment Agreement also contains covenants prohibiting Mr. Briones from disclosing confidential information with respect to the Company.
-83-
SECURITY OWNERSHIP OF BENEFICIAL OWNERS AND MANAGEMENT
The following table sets forth certain information regarding beneficial ownership of shares of our common stock as of August 27, 2019 by (i) each person known to beneficially own more than 5% of our outstanding common stock, (ii) each of our directors, (iii) each of our named executive officers and (iv) all of our directors and executive officers as a group. Except as otherwise indicated, the persons named in the table below have sole voting and investment power with respect to all shares beneficially owned, subject to community property laws, where applicable.
Beneficial Owner(1) | Shares of Common Stock Beneficially Owned | Percentage(2) | ||||||||
| Directors and Executive Officers: | ||||||||||
| Robb Knie | 808,170 | 8.02 | % | |||||||
| David Briones | 50,000 | (3) | * | |||||||
| Vadim Mats | 37,500 | * | ||||||||
| Kenneth Rice (4) | 776,944 | (5) | 7.71 | % | ||||||
| Anthony Hayes (6) | 1,805,000 | (7) | 17.91 | % | ||||||
| David Sarnoff | 10,410 | (8) | * | |||||||
| All Executive Officers and Directors as a Group (6 persons) | 3,488,024 | 34.44 | % | |||||||
| 5% or Greater Stockholders: | ||||||||||
| Spherix Incorporated (6) One Rockefeller Plaza New York, NY 10020 | 1,700,000 | 16.87 | % | |||||||
| Chelexa Biosciences, Inc. (4) P.O. Box 7122 Lowell, MA 01852 | 726,944 | 7.21 | % | |||||||
James Ahern 521 Fifth Avenue, 12th Floor New York, NY 10175 | 750,000 | 7.44 | % | |||||||
Matthew Eitner 521 Fifth Avenue, 12th Floor New York, NY 10175 | 768,750 | (9) | 7.63 | % | ||||||
| Kevin Poor 750 Beulahs Lane Idaho Falls, ID 83401 | 937,500 | (10) | 9.13 | % | ||||||
| * | Represents beneficial ownership of less than 1%. |
| (1) | The address of each person is c/o Hoth Therapeutics, Inc., 1 Rockefeller Plaza, Suite 1039, New York, New York 10020 unless otherwise indicated herein. |
| (2) | The calculation in this column is based upon 10,077,068 shares of common stock outstanding on August 27, 2019. Beneficial ownership is determined in accordance with the rules of the SEC and generally includes voting or investment power with respect to the subject securities. Shares of common stock that are currently exercisable or convertible within 60 days of August 27, 2019 are deemed to be beneficially owned by the person holding such securities for the purpose of computing the percentage beneficial ownership of such person, but are not treated as outstanding for the purpose of computing the percentage beneficial ownership of any other person. |
| (3) | Includes options to purchase up to 50,000 shares of the Company’s common stock. |
| (4) | Kenneth Rice is the Executive Chairman of Chelexa and in such capacity has voting and dispositive power over the securities held by such entity. |
| (5) | Includes (i) 50,000 shares of common stock held by Kenneth Rice and (ii) 726,944 shares of common stock held by Chelexa. |
| (6) | Anthony Hayes is the Chief Executive Officer and a member of the board of directors of Spherix Incorporated and in such capacity has voting and dispositive power over the securities held by such entity. |
| (7) | Includes (i) 105,000 shares of common stock held by Anthony Hayes and (ii) 1,700,000 shares of common stock held by Spherix Incorporated. |
| (8) | Includes 10,410 shares of common stock. Excludes 14,590 shares of common stock which vest in equal installments over a 21 month period. |
| (9) | Includes (i) 765,000 shares of common stock and (ii) 3,750 shares of common stock underlying warrants to purchase common stock. |
| (10) | Includes (i) 750,000 shares of common stock and (ii) 187,500 shares of common stock underlying warrants to purchase common stock. |
-84-
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
Since our inception on May 16, 2017, except as set forth below, we have not been a participant to any transactions in which the amount involved exceeded or will exceed the lesser of $120,000 or one percent of our total assets at the end of our last completed fiscal year, and in which any of our directors, executive officers holders of more than 5% of our capital stock, promotor or certain control person or any member of their immediate family had or will have a direct or indirect material interest.
Laidlaw
On May 16, 2017, Matthew Eitner and James Ahern, the Chief Executive Officer and Head of Capital Markets of Laidlaw, respectively, each purchased 750,000 shares of our common stock for an aggregate purchase price of $3,000. In addition, they each loaned us an aggregate of $102,000, which loans, together with a $48,000 original issue discount, have since been repaid. Further, on October 25, 2017, Matthew Eitner purchased 15,000 shares of Series A Preferred Stock and warrants to purchase up to 3,750 shares of common stock pursuant to a private placement of the Company’s securities. Upon the consummation of our IPO, Matthew Eitner’s 15,000 shares of Series A Preferred Stock were automatically converted into 15,000 shares of the Company’s common stock.
On July 6, 2017, we entered into an engagement agreement with Laidlaw. We agreed to pay Laidlaw a fee in the amount of 10% of the gross proceeds of the private placement of our securities received from investors at the closing of such offering, which, in the aggregate, amounted to $310,248, as well as a non-accountable expense reimbursement equal to 2% of the gross proceeds received from investors at the closing of such offering, which, in the aggregate, amounted to $62,050. In addition, Laidlaw received seven year warrants to purchase 215,747 shares of our common stock at an exercise price of $1.00 per share. Furthermore, Laidlaw was paid an activation fee of $50,000 at the initial closing of the offering.
On February 14, 2019, we entered into an underwriting agreement with Laidlaw pursuant to which we paid Laidlaw a fee in the amount of 7% of the gross proceeds of the IPO, or $490,000. We also reimbursed Laidlaw for certain out-of-pocket expenses, including the fees and disbursements of their counsel, up to an aggregate of $200,000. In addition, Laidlaw received five-year warrants to purchase 50,000 shares of our common stock at an exercise price of $7.00 per share.
Chelexa
On May 26, 2017, we entered into a sublicense agreement with Chelexa, as amended on August 22, 2018 and August 29, 2018. Kenneth Rice, a member of our board of directors is the Executive Chairman of Chelexa. Pursuant to the terms of the sublicense agreement, Chelexa granted us an exclusive worldwide sublicense to use the BioLexa Platform, a proprietary, patented, drug compound platform developed at the University of Cincinnati. Furthermore, pursuant to the terms of the sublicense agreement, we will pay Chelexa up to an aggregate of $3.8 million, of which $300,000 has been paid to date. Such amount consists of total milestone payments of $3.5 million in addition to payments by us of certain licensing fees and all development and commercialization expenses. In addition, we will also be required to pay sales-based royalties at percentages which range from mid to high single digits, with high sales volumes being subject to lower royalty rates. We also issued Chelexa 250,000 shares of our common stock, which was 10% of our fully-diluted equity at May 26, 2017, and Chelexa had the right to receive such number of additional shares of common stock required to maintain its 10% interest in our fully-diluted equity until such time that we raised a minimum of $3,000,000. As of the date hereof, we have issued Chelexa an aggregate of 476,943 additional shares of common stock pursuant to the Preemptive Right. We have raised more than $3,000,000 and therefore the Preemptive Right has been terminated. Furthermore, pursuant to the sublicense agreement, Chelexa has the right to participate in certain equity issuances made by us for purposes of raising capital based upon its pro-rata share to enable Chelexa to retain 10% of our fully-diluted equity until such time as we consummate an initial public offering pursuant to which we receive aggregate gross proceeds of not less than $5,000,000. However, since we consummated the IPO pursuant to which we received aggregate gross proceeds of $7,000,000, the Chelexa Participation Right has been terminated. The sublicense agreement shall terminate on the later of April 16, 2034 or the last to expire patent in the Patent Rights (as defined in the sublicense agreement) (the “Sublicense Term”). We have the right of first refusal, in our sole discretion, to renew the Sublicense Term. We may terminate the sublicense agreement at any time upon twelve months prior notice. In the event we are in default of any of our material obligations under the sublicense agreement, Chelexa may, at its option upon 90 days prior written notice, terminate the sublicense agreement if we do not cure such default prior to the expiration of such 90 day period. In addition, at any time after May 26, 2018, Chelexa may, at its sole discretion, terminate or render the license non-exclusive if, in Chelexa’s judgment the Progress Reports (as defined in the sublicense agreement) furnished by us does not demonstrate that we used our best commercial efforts to develop and seek regulatory approval for the BioLexa Platform in the Territory (as defined in the sublicense agreement) and in the Field (as defined in the sublicense agreement) and /or is engaged in manufacturing, marketing or sublicensing activity which is reasonably expected to keep the BioLexa Platform reasonably available to the public. The sublicense agreement will automatically terminate upon the expiration of the UC License (as defined in the sublicense agreement).
-85-
Spherix
On June 30, 2017, we entered into a securities purchase agreement with Spherix Incorporated pursuant to which we sold 1,700,000 shares of our common stock for gross proceeds of $675,000. Anthony Hayes, a member of our board of directors is the Chief Executive Officer and member of the board of directors of Spherix.
In connection with the sale of the shares of common stock, on June 30, 2017, we entered into the Spherix RRA pursuant to which we agreed, among other things, that we will file with the SEC a registration statement on Form S-1 under the Securities Act that covers the resale of 1,700,000 shares of common stock issued to Spherix pursuant to a securities purchase agreement between us and Spherix and any securities issued or issuable upon any stock split, dividend or other distribution, recapitalization or similar event with respect to the foregoing. Pursuant to the Spherix RRA, we are obligated to use our best efforts to have the registration statement declared effective by the SEC as soon as practicable after it is filed with the SEC, but in no event later than the applicable Effectiveness Date. “Effectiveness Date” means with respect to the initial registration statement required to be filed pursuant to the Spherix RRA, the 18 month anniversary of the closing date of the transactions contemplated by the securities purchase agreement and, with respect to any additional registration statements which may be required pursuant to the Spherix RRA, the earliest practical date on which we are permitted to go effective on such additional registration statement; provided, however, that, in the event we are notified by the SEC that one or more of the above registration statements will not be reviewed or is no longer subject to further review and comments, the Effectiveness Date as to such registration statement shall be the fifth trading day following the date on which we are so notified if such date precedes the dates otherwise required above. In addition, pursuant to the terms of the Spherix RRA, without the consent of Spherix, neither we nor any of our security holders may include our securities in any registration statements other than the Spherix Registrable Securities. Furthermore, subject to certain exemptions, if at any time during the Effectiveness Period there is not an effective registration statement covering all of the Spherix Registrable Securities and we shall determine to prepare and file with the SEC a registration statement relating to an offering for our own account or the account of others under the Securities Act of any of our equity securities, then we shall deliver to Spherix a written notice of such determination and, if within 15 days after the date of the delivery of such notice, Spherix notifies us in writing, we must include in such registration statement all or any part of such Spherix Registrable Securities requested to be registered by Spherix.
In addition, we entered into a lock-up agreement with Spherix pursuant to which Spherix and its affiliates have agreed to not take certain actions, including exercising their registration rights, until the 36 month anniversary of the IPO.
Unless otherwise indicated, Sheppard, Mullin, Richter & Hampton LLP, New York, New York, will pass upon the validity of the shares of our common stock to be sold in this offering.
Our balance sheets as of December 31, 2018 and 2017, and the related statements of operations, changes in stockholders’ equity and cash flows for the year ended December 31, 2018 and for the period from May 16, 2017 (inception) through December 31, 2017 incorporated by reference in this prospectus and the registration statement, of which it forms a part, have been audited by WithumSmith+Brown, PC, independent registered public accounting firm, as set forth in their report thereon incorporated by reference herein, and are included in reliance on such report given on the authority of such firm as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC a registration statement on Form S-1 under the Securities Act with respect to the Resale Shares being offered by this prospectus. This prospectus does not contain all of the information in the registration statement of which this prospectus is a part and the exhibits to such registration statement. For further information with respect to us the Resale Shares by this prospectus, we refer you to the registration statement of which this prospectus is a part and the exhibits to such registration statement. Statements contained in this prospectus as to the contents of any contract or any other document are not necessarily complete, and in each instance, we refer you to the copy of the contract or other document incorporated by reference or filed as an exhibit to the registration statement of which this prospectus is a part. Each of these statements is qualified in all respects by this reference.
You may read and copy the registration statement of which this prospectus is a part, as well as our reports, proxy statements and other information, at the SEC’s Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549. Please call the SEC at 1-800-SEC-0330 for more information about the operation of the Public Reference Room. The SEC maintains an Internet site that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC, including Hoth Therapeutics, Inc. The SEC’s Internet site can be found at http://www.sec.gov. You may also request a copy of these filings, at no cost, by writing us at Hoth Therapeutics, Inc., 1 Rockefeller Plaza, Suite 1039, New York, NY 10020 or telephoning us at (646) 756-2997.
We are subject to the information and reporting requirements of the Exchange Act, and, in accordance with this law, file periodic reports, proxy statements and other information with the SEC. These periodic reports, proxy statements and other information are available for inspection and copying at the SEC’s public reference facilities and the website of the SEC referred to above. We also maintain a website at www.hoththerapeutics.com. You may access these materials free of charge as soon as reasonably practicable after they are electronically filed with, or furnished to, the SEC. Information contained on our website is not a part of this prospectus and the inclusion of our website address in this prospectus is an inactive textual reference only.
-86-
1,329,003 Shares of Common Stock
PROSPECTUS
, 2019
PART II- INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution.
The following table sets forth the costs and expenses payable by the Company in connection with the issuance and distribution of the securities being registered hereunder. All amounts are estimates except the SEC registration fee.
| SEC registration fees | $ | 825 | ||
| Printing expenses | $ | 10,000 | ||
| Accounting fees and expenses | $ | 7,500 | ||
| Legal fees and expenses | $ | 25,000 | ||
| Miscellaneous | $ | 1,675 | ||
| Total | $ | 45,000 |
Item 14. Indemnification of Directors and Officers.
Section 78.7502(1) of the Nevada Revised Statutes provides that a corporation may indemnify any person who was or is a party, or is threatened to be made a party, to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (except in an action brought by or on behalf of the corporation) if that person is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation or enterprise, against expenses, including attorneys’ fees, judgments, fines and amounts paid in settlement actually and reasonably incurred by that person in connection with such action, suit or proceeding, if that person acted in good faith and in a manner which that person reasonably believed to be in, or not opposed to, the best interests of the corporation, and, with respect to any criminal action or proceedings, had no reasonable cause to believe his conduct was unlawful. The termination of any action, suit or proceeding by judgment, order, settlement, conviction or upon a plea of nolo contendere or its equivalent, alone, does not create a presumption that the person did not act in good faith and in a manner which the person reasonably believed to be in, or not opposed to, the best interests of the corporation, and that, with respect to any criminal action or proceeding, the person had reasonable cause to believe his action was unlawful.
Section 78.7502(2) of the Nevada Revised Statutes provides that a corporation may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action or suit brought by or on behalf of the corporation to procure a judgment in its favor because the person acted in any of the capacities set forth above, against expenses, including amounts paid in settlement and attorneys’ fees, actually and reasonably incurred by that person in connection with the defense or settlement of such action or suit, if the person acted in accordance with the standard set forth above, except that no indemnification may be made in respect of any claim, issue or matter as to which such person shall have been adjudged by a court of competent jurisdiction after exhaustion of all appeals therefrom to be liable to the corporation or for amounts paid in settlement to the corporation unless and only to the extent that the court in which such action or suit was brought or other court of competent jurisdiction determines that, in view of all the circumstances of the case, such person is fairly and reasonably entitled to indemnity for such expenses as the court deems proper.
Section 78.7502(3) of the Nevada Revised Statutes further provides that, to the extent a director or officer of a corporation has been successful on the merits or otherwise in the defense of any action, suit or proceeding referred to in subsections 1 and 2 thereof, or in the defense of any claim, issue or matter therein, that person shall be indemnified by the corporation against expenses (including attorneys’ fees) actually and reasonably incurred by that person in connection therewith.
Section 78.751 of the Nevada Revised Statutes provides that unless indemnification is ordered by a court, the determination to provide indemnification must be made by the stockholders, by a majority vote of a quorum of the board of directors who were not parties to the action, suit or proceeding, or in specified circumstances by independent legal counsel in a written opinion. In addition, the articles of incorporation, bylaws or an agreement made by the corporation may provide for the payment of the expenses of a director or officer of the expenses of defending an action as incurred upon receipt of an undertaking to repay the amount if it is ultimately determined by a court of competent jurisdiction that the person is not entitled to indemnification. Section 78.751 of the Nevada Revised Statutes further provides that the indemnification provided for therein shall not be deemed exclusive of any other rights to which the indemnified party may be entitled and that the scope of indemnification shall continue as to directors, officers, employees or agents who have ceased to hold such positions, and to their heirs, executors and administrators.
II-1
Section 78.752 of the Nevada Revised Statutes provides that a corporation may purchase and maintain insurance on behalf of a director, officer, employee or agent of the corporation against any liability asserted against him or incurred by him in any such capacity or arising out of his status as such whether or not the corporation would have the authority to indemnify him against such liabilities and expenses.
Articles of Incorporation
Our Articles of Incorporation provide that the Company shall, to the fullest extent permitted by the provisions of Section 78.751 of the Nevada Revised Statutes, indemnify any and all persons whom it shall have the power to indemnify under such section.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the Company pursuant to the foregoing provisions, or otherwise, we have been advised that, in the opinion of the SEC, such indemnification is against public policy as expressed in such Act and is, therefore, unenforceable.
Item 15. Recent Sales of Unregistered Securities.
On May 16, 2017, Robb Knie, Chief Executive Officer of the Company and Matthew Eitner and James Ahern, the Chief Executive Officer and Head of Capital Markets of Laidlaw, respectively, each purchased 750,000 founders’ shares of the Company for an aggregate purchase price of $3,000.
From May, 2017 until February, 2018, the Company issued an aggregate of 726,943 shares of the Company’s common stock pursuant to the terms of a license agreement.
On June 30, 2017, the Company issued 1,700,000 shares of common stock to one investor for gross proceeds of $675,000.
In July 2017, the Company issued an aggregate of 155,000 shares of the Company’s common stock to three consultants for services rendered.
From July 2017 until August 2017, the Company issued an aggregate of 37,500 shares of the Company’s common stock to members of the Company’s Scientific Advisory Board for services rendered.
From October 25, 2017 until February 15, 2018, the Company issued an aggregate of 3,102,480 shares of its Series A Preferred Stock and warrants to purchase up to 775,620 shares of its common stock to accredited investors in the Private Offering for gross proceeds of $3,102,480. In February 2019, the 3,102,480 shares of Series A Preferred Stock were converted into an aggregate of 3,102,480 shares of the Company’s common stock. In addition, in June 2019, warrants to purchase 57,750 shares of the Company’s common stock were exercised on a cashless basis for an aggregate of 47,605 shares of the Company’s common stock.
From October 2017 until December 2017, the Company issued warrants to purchase up to 215,747 of the Company’s common stock to Laidlaw in connection with the Private Offering. In April 2019, such warrants were exercised on a cashless basis for an aggregate of 176,272 shares of the Company’s common stock.
In December 2017, the Company issued 25,000 shares of the Company’s common stock to an employee for services rendered.
II-2
In March 2018, the Company issued 12,500 shares of the Company’s common stock to a member of the Company’s Scientific Advisory Board for services rendered.
In May 2018, the Company issued an aggregate of 130,000 shares of the Company’s common stock pursuant to the 2018 Plan to employees and directors for services rendered.
In August 2018, the Company issued 12,500 shares of the Company’s common stock to a member of the Company’s Scientific Advisory Board for services rendered.
From August to December 2018, the Company issued 3,471 shares of the Company’s common stock to a member of the Company’s Board for services rendered.
From January 2019 until August 2019, the Company issued 4,858 shares of the Company’s common stock to a member of the Company’s Board for services rendered.
On February 20, 2019, the Company issued Laidlaw a warrant to purchase up to 50,000 shares of common stock for services rendered in connection with the Company’s initial public offering.
On April 17, 2019, the Company entered into a Master Service Agreement (the “MSA”) with a consultant (the “Consultant”). In consideration for services provided by the Consultant, the Company issued the Consultant a two year warrant to purchase up to 50,000 shares of the Company’s common stock at an exercise price of $0.01 per share (the “Consultant Warrant”). On May 22, 2019, the Company and Consultant agreed to terminate the MSA and number of shares of the Company’s common stock issuable upon exercise of the Consultant Warrant was reduced to 16,333. In June 2019, the Company issued 16,333 shares of common stock upon exercise of the Consultant Warrant.
On August 16, 2019 the Company sold the Units to accredited investors in the August 2019 Private Placement for aggregate gross proceeds of $2,037,120. Each Unit consists of (i) one share of the Company’s common stock and (ii) a warrant to purchase one-half share of the Company’s common stock.
On August 16, 2019, the Company issued Laidlaw a warrant to purchase up to 61,113 shares of common stock for services rendered in connection with the August 2019 Private Placement.
The foregoing offers, sales and issuances were exempt from registration under Section 4(a)(2) of the Securities Act and/or Rule 506 of Regulation D thereunder.
II-3
Item 16. Exhibits and Financial Statement Schedules.
| (a) | Exhibits. |
II-4
| * | Filed herewith. |
| + | Indicates a management contract or any compensatory plan, contract or arrangement. |
| # | Confidential treatment has been requested to a portion of this exhibit, and such confidential portion has been deleted and filed separately with the SEC. |
| ## | Pursuant to Item 601(b)(10) of Regulation S-K, certain confidential portions of this exhibit were omitted by means of marking such portions with an asterisk because the identified confidential portions (i) are not material and (ii) would be competitively harmful if publicly disclosed. |
II-5
ITEM 17. UNDERTAKINGS.
The undersigned registrant hereby undertakes:
| (1) | To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement: |
| (i) | To include any prospectus required by section 10(a)(3) of the Securities Act; | |
| (ii) | To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20% change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement. | |
| (iii) | To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement; |
| (2) | That, for the purpose of determining any liability under the Securities Act, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
| (3) | To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering. |
| (4) | That, for the purpose of determining liability under the Securities Act of 1933 to any purchaser, if the registrant is subject to Rule 430C, each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use. |
| (5) | That, for the purpose of determining liability of the registrant under the Securities Act to any purchaser in the initial distribution of the securities, the undersigned registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser: |
| (i) | Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424; |
| (ii) | Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant; |
| (iii) | The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and |
II-6
| (iv) | Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser. |
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the SEC such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
The undersigned registrant hereby undertakes to provide to the underwriter at the closing specified in the underwriting agreements certificates in such denominations and registered in such names as required by the underwriter to permit prompt delivery to each purchaser.
The undersigned registrant hereby undertakes that:
| (1) | For purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b) (1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective. |
| (2) | For the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
II-7
Pursuant to the requirements of the Securities Act of 1933, as amended, the registrant has duly caused this Registration Statement on Form S-1 to be signed on its behalf by the undersigned, thereunto duly authorized in the City of New York, State of New York, on the 30th day of August, 2019.
| HOTH THERAPEUTICS, INC. | ||
| By: | /s/ Robb Knie | |
| Robb Knie | ||
| Chief Executive Officer | ||
SIGNATURES AND POWER OF ATTORNEY
Each of the undersigned officers and directors of Hoth Therapeutics, Inc. hereby constitutes and appoints Robb Knie the individual’s true and lawful attorneys-in-fact and agents, each with full power of substitution and resubstitution, for the person and in his or her name, place and stead, in any and all capacities, to sign this registration statement of Hoth Therapeutics, Inc. on Form S-1, and any other registration statement relating to the same offering (including any registration statement, or amendment thereto, that is to become effective upon filing pursuant to Rule 462(b) under the Securities Act of 1933, as amended), and any and all amendments thereto (including post-effective amendments to the registration statement), and to file the same, with all exhibits thereto, and all other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorney-in-fact and agent, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorney-in-fact and agent, or his substitute, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Act of 1933, as amended, this Registration Statement has been signed by the following persons in the capacities held and on the dates indicated.
| Signature | Title | Date | ||
| /s/ Robb Knie | Chief Executive Officer and Director | August 30, 2019 | ||
| Robb Knie | (Principal Executive Officer) | |||
| /s/ David Briones | Chief Financial Officer | August 30, 2019 | ||
| David Briones | (Principal Financial and Accounting Officer) | |||
| /s/ Vadim Mats | Director | August 30, 2019 | ||
| Vadim Mats | ||||
| /s/ Kenneth Rice | Director | August 30, 2019 | ||
| Kenneth Rice | ||||
| /s/ Anthony Hayes | Director | August 30, 2019 | ||
| Anthony Hayes | ||||
| /s/ David B. Sarnoff | Director | August 30, 2019 | ||
| David B. Sarnoff |
II-8
Exhibit 5.1
 |
Sheppard, Mullin, Richter & Hampton LLP 30 Rockefeller Plaza New York, New York 10112-0015 212.653.8700 main 212.653.8701 fax www.sheppardmullin.com |
August 30, 2019
VIA ELECTRONIC MAIL
Hoth Therapeutics, Inc.
1 Rockefeller Plaza, Suite 1039
New York, New York 10020
Re: Registration Statement on Form S-1
Ladies and Gentlemen:
We have acted as counsel to Hoth Therapeutics, Inc., a Nevada corporation (the “Company”), in connection with the issuance of this opinion which relates to a Registration Statement on Form S-1 (the “Registration Statement”) filed by the Company with the Securities and Exchange Commission (the “SEC”) under the Securities Act of 1933, as amended (the “Securities Act”). The Registration Statement covers the resale of 1,329,003 shares of the Company’s common stock, $0.0001 par value per share (“Common Stock”), including an aggregate of 407,424 shares of Common Stock (the “Shares”) and an aggregate of 921,579 shares of Common Stock (the “Warrant Shares”) issuable upon exercise of outstanding warrants to purchase shares of Common Stock (the “Warrants”) issued to the selling stockholders.
This opinion letter is being delivered in accordance with the requirements of Item 601(b)(5)(i) of Regulation S-K under the Securities Act, and no opinion is expressed herein as to any matter pertaining to the contents of the Registration Statement.
In connection with the issuance of this opinion letter, we have examined originals or copies, certified or otherwise identified to our satisfaction, of:
a. the Registration Statement, including the prospectus contained therein and all exhibits thereto;
b. a specimen certificate representing the Shares;
c. the Warrants;
d. the Articles of Incorporation of the Company, as amended and presently in effect (the “Charter”);
e. the Amended and Restated Bylaws of the Company, as presently in effect (the “Bylaws”); and
f. certain resolutions adopted by the Board of Directors of the Company relating to the issuance of the shares of Common Stock being registered pursuant to the Registration Statement.
We have also examined originals or copies, certified or otherwise identified to our satisfaction, of such records of the Company and such agreements, certificates and receipts of public officials, certificates of officers or other representatives of the Company and others, and such other documents as we have deemed necessary or appropriate as a basis for the opinions stated below.
In our examination, we have assumed the genuineness of all signatures, including endorsements, the legal capacity and competency of all natural persons, the authenticity of all documents submitted to us as originals, the conformity to original documents of all documents submitted to us as facsimile, electronic, certified or photostatic copies, and the authenticity of the originals of such copies. As to any facts relevant to the opinions stated herein that we did not independently establish or verify, we have relied upon statements and representations of officers and other representatives of the Company and others and of public officials.
Page 2
For the purposes of this opinion letter, we have assumed that at the time of issuance of each Warrant Share, the Charter, the Bylaws and the Warrants, as applicable, will not have been modified or amended and will be in full force and effect. In addition, it is understood that this opinion is to be used only in connection with the offer and sale of the securities being registered while the Registration Statement is effective under the Securities Act.
Based upon the foregoing and subject to the qualifications and assumptions stated herein, we are of the following opinions:
1. the Shares have been duly authorized by all requisite corporate action on the part of the Company under the Nevada Revised Statutes (the “NRS”) and are validly issued, fully paid and non-assessable.
2. the Warrant Shares have been duly authorized by all requisite corporate action on the part of the Company under the NRS and, when the Warrant Shares are delivered and paid for in accordance with the terms of the Warrant and when evidence of the issuance thereof is duly recorded in the Company’s books and records, the Warrants Shares will be validly issued, fully paid and nonassessable.
The opinion which we render herein is limited to those matters governed by the NRS as of the date hereof. Our opinion expressed herein is as of the date hereof, and we assume no obligation to revise or supplement the opinion rendered herein should the above-referenced laws be changed by legislative or regulatory action, judicial decision or otherwise. We express no opinion as to whether, or the extent to which, the laws of any particular jurisdiction apply to the subject matter hereof.
We hereby consent to the filing of this opinion as an exhibit to the Registration Statement. We also hereby consent to the reference to our firm under the heading “Legal Matters” in the Registration Statement. In giving this consent, we do not thereby admit that we are within the category of persons whose consent is required under Section 7 of the Securities Act or the General Rules and Regulations under the Securities Act.
This opinion letter is rendered as of the date first written above and we disclaim any obligation to advise you of facts, circumstances, events or developments which hereafter may be brought to our attention and which may alter, affect or modify the opinion expressed herein. Our opinion is expressly limited to the matters set forth above and we render no opinion, whether by implication or otherwise, as to any other matters relating to the Company, the Shares, the Warrant Shares, or any other agreements or transactions that may be related thereto or contemplated thereby. We are expressing no opinion as to any obligations that parties other than the Company may have under or in respect of the Shares, the Warrant Shares or as to the effect that their performance of such obligations may have upon any of the matters referred to above. No opinion may be implied or inferred beyond the opinion expressly stated above.
Very truly yours,
/s/ Sheppard, Mullin, Richter & Hampton LLP
SHEPPARD, MULLIN, RICHTER & HAMPTON LLP
Exhibit 23.1
CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
We hereby consent to the incorporation by reference in this Registration Statement on Form S-1, of our report dated March 29, 2019, relating to the balance sheets of Hoth Therapeutics, Inc. as of December 31, 2018 and 2017, and the related statements of operations, changes in stockholders’ equity and cash flows for the year ended December 31, 2018 and for the period from May 16, 2017 (inception) through December 31, 2017, and to the reference to our Firm under the caption “Experts” in the Prospectus.
| /s/ WithumSmith+Brown, PC | |
|
New York, New York |
|
| August 30, 2019 |
Serious News for Serious Traders! Try StreetInsider.com Premium Free!
You May Also Be Interested In
- VinFast Auto (VFS) Appoints Tham Chee Soon to its Board, Ngan Wan Sing Winston Resigns
- Parsons (PSN) Increasing Production of U.S. Air Force’s RADBO System
- Cyngn Inc (CYN) Announces Pricing of $5.0 Million Firm Commitment Public Offering of Common Stock
Create E-mail Alert Related Categories
SEC FilingsRelated Entities
S1Sign up for StreetInsider Free!
Receive full access to all new and archived articles, unlimited portfolio tracking, e-mail alerts, custom newswires and RSS feeds - and more!
