

Form F-1/A Medlab Clinical Ltd.
 Tweet
Tweet Share
ShareAs filed with the U.S. Securities and Exchange Commission on November 22, 2022.
Registration No. 333-267873
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Washington, D.C. 20549
___________________
Amendment No. 2
to
FORM F-1
REGISTRATION STATEMENT
UNDER THE SECURITIES ACT OF 1933
REGISTRATION STATEMENT
UNDER THE SECURITIES ACT OF 1933
___________________
Medlab Clinical Ltd.
(Exact name of registrant as specified in its charter)
(Exact name of registrant as specified in its charter)
___________________
|
Australia
|
|
2834
|
|
Not Applicable
|
|
(State or other jurisdiction of
incorporation or organization) |
|
(Primary Standard Industrial
Classification Code Number) |
|
(I.R.S. Employer
Identification Number) |
Medlab Clinical Ltd.
Units 5 and 6, 11-13 Lord Street
Units 5 and 6, 11-13 Lord Street
Botany, New South Wales 2019
+ 61 2 8188 0311
(Address, including zip code, and telephone number, including area code, of Registrant's principal executive offices)
___________________
Puglisi & Associates
850 Library Avenue, Suite 204
Newark, Delaware
(302) 738-6680
850 Library Avenue, Suite 204
Newark, Delaware
(302) 738-6680
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
|
Edward S. Horton
|
|
David E. Danovitch, Esq.
|
|
Seward & Kissel LLP
|
|
Joseph Segilia, Esq.
|
|
One Battery Park Plaza
|
|
Caroline J. Lambert, Esq.
|
|
New York, New York 10004
|
|
Sullivan & Worcester LLP
|
|
United States
|
|
1633 Broadway
|
|
(212) 574-1200
|
|
New York, NY 10019
|
|
|
|
United States
|
|
|
|
(212) 660-3000
|
___________________
Approximate date of commencement of proposed sale to the public: As soon as practicable after this
Registration Statement becomes effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the
Securities Act, check the following box: ☐
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the
following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the
Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the
Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
Indicate by check mark whether the registrant is an emerging growth company as defined in Rule 405 of the Securities Act. Emerging growth
company ☒
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant
has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act. ☐
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the
registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act or until the Registration Statement shall become effective on
such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.
The information in this preliminary prospectus is not complete and may be changed. We may not sell these securities until the
registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and we are not soliciting an offer to buy these securities in any jurisdiction where the offer
or sale is not permitted.
Subject to completion, dated November 22, 2022
|
PRELIMINARY PROSPECTUS
|

[•] Ordinary Shares
Pre-Funded Warrants to Purchase
[•] Ordinary Shares and Warrants to Purchase
[•] Ordinary Shares and Warrants to Purchase
[•] Ordinary Shares
___________________
Medlab Clinical Ltd. (“we,” “the Company,” “Medlab,” “us,” or “our”) is offering [•] ordinary shares, no par value (the “Shares” and each a
“Share”), and warrants (“Share Purchase Warrants”) to purchase [•] Shares pursuant to this prospectus. Each whole Share Purchase Warrant is exercisable to purchase one Share at an assumed exercise price of US$ [•], will be exercisable upon issuance
and will expire five years from the date of issuance. The Shares and Share Purchase Warrants will be issued and sold to purchasers in the ratio of one-to-one. This prospectus also relates to the offering of the Shares issuable upon exercise of Share
Purchase Warrants.
We are also offering to certain purchasers whose purchase of Shares in this offering would otherwise result in the purchaser, together with
its affiliates and certain related parties, beneficially owning more than 4.99% (or 9.99% at the election of the purchaser) of our outstanding Shares immediately following the consummation of this offering, the opportunity to purchase, if any such
purchaser so chooses, pre-funded warrants (“Pre-Funded Warrants”) in lieu of Shares that would otherwise result in such purchaser’s beneficial ownership exceeding 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding Shares. Each
Pre-Funded Warrant will be exercisable for one Share. The exercise price of each Pre-Funded Warrant will be US$0.0001 per Share. The Pre-Funded Warrants will be immediately exercisable and may be exercised at any time until all of the Pre-Funded
Warrants are exercised in full. This offering also relates to the Shares issuable upon exercise of any Pre-Funded Warrants sold in this offering.
The Shares and the accompanying Share Purchase Warrants will be sold in units (each, a “Share Unit”) and the Pre-Funded Warrants and the
accompanying Share Purchase Warrants will be sold in units (each, a “Pre-Funded Warrant Unit” and, together with the Share Units, the “Units”), with each Share Unit consisting of one Share and one Share Purchase Warrant to purchase one Share and each
Pre-Funded Warrant Unit consisting of one Pre-Funded Warrant and one Share Purchase Warrant to purchase one Share. For each Pre-Funded Warrant Unit we sell, the number of Share Units we are offering will be decreased on a one-for-one basis. The
Shares and Share Purchase Warrants and the Pre-Funded Warrants and Share Purchase Warrants constituting the Share Units and the Pre-Funded Warrant Units, respectively, will be immediately separable on issuance. Each Share Unit will be sold at a price
of US$[•] per Share Unit. The purchase price of each Pre-Funded Warrant Unit will be equal to the price at which each Share Unit is sold to the public in this offering, minus US$0.0001, and the exercise price of each Pre-Funded Warrant will be
US$0.0001 per Share.
On July 28, 2022, our shareholders approved our board of directors to effect a share consolidation of our outstanding Shares at a specific
ratio within a range from 1-for-1 to 1-for-150, and also granted authorization to our board of directors to determine, in its sole discretion, the specific ratio and timing of such share consolidation. Effective September 1, 2022, our board of
directors has effected such share consolidation at a ratio of 149.846889 (the “Share Consolidation”) in connection with this offering and intended listing of our common shares on the Nasdaq Capital Market (“Nasdaq”).
The offering is being underwritten on a firm commitment basis. We have granted the underwriter the right to purchase up to an additional
[•] Shares and/or Share Purchase Warrants to cover over-allotments, if any, assuming an initial public offering price of US$[•] per Unit. The underwriter can exercise this right at any time within 45 days after the date of this prospectus. In
addition, we will issue to the underwriter warrants to purchase a number of Shares equal to an aggregate of [•]% of the Shares and/or Pre-Funded Warrants sold in the offering. The exercise price of the underwriter’s warrants is equal to [•]% of the
offering price of the Shares and/or Pre-Funded Warrants offered hereby. The underwriter’s warrants are exercisable beginning [•] months from the date of issuance, from time to time, in whole or in part, within five years commencing from the date of
issuance.
Our Shares are listed on the Australian Securities Exchange (“ASX”) under the symbol “MDC.” We have applied to list the Shares on Nasdaq
under the symbol “MDLB”. On , 2022, the last reported sale price of our Shares on the ASX was A$[•] per Share, equivalent to a price of US$[•] per Share, after giving effect to the Australian dollar/U.S. dollar exchange rate of as
of , 2022.
The final offering price per Share in U.S. dollars will be determined through negotiations between us and the underwriter and will be
based, in part, on prevailing market prices of our Shares on the ASX, after taking into account market conditions and other factors. For a discussion of the other factors considered in determining the final offering price per Share, see
“Underwriting.”
|
|
Per Share Unit (1)
|
Per Pre-Funded Warrant Unit
|
Total
|
|||||||||
|
Public offering price
|
$
|
$
|
$
|
|||||||||
|
Underwriting discounts and commissions(2)
|
$
|
$
|
$
|
|||||||||
|
Proceeds to us, before expenses (3)
|
$
|
$
|
$
|
|||||||||
___________________
| (1) |
The public offering price and underwriting discounts and commissions correspond to an assumed public offering price per Share Unit of US$[•] and an assumed public offering
price per Pre-Funded Warrant Unit of US$[•].
|
| (2) |
In addition, we have agreed to reimburse the underwriter for certain expenses. See “Underwriting” on page 132 of this prospectus for additional information.
|
| (3) |
The amount of the offering proceeds to us presented in this table does not give effect to any exercise of the Pre-Funded Warrants or the Share Purchase Warrants being issued in
this offering.
|
Investing in our securities involves a high degree of risk. See the section entitled “Risk Factors” appearing on page 15 of this prospectus
and elsewhere in this prospectus for a discussion of information that should be considered in connection with an investment in our securities.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or
passed upon the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense.
The underwriter expects to deliver the Shares to purchasers on or about , 2022 through the book-entry facilities of The
Depository Trust Company.
___________________
Sole Book-Running Manager
A.G.P.
The date of this prospectus is , 2022.
TABLE OF CONTENTS
Page
|
ABOUT THIS PROSPECTUS
|
1
|
|
PROSPECTUS SUMMARY
|
2
|
|
THE OFFERING
|
13
|
|
RISK FACTORS
|
15
|
|
USE OF PROCEEDS
|
49
|
|
DIVIDEND POLICY
|
50
|
|
CAPITALIZATION
|
51 |
|
DILUTION
|
52
|
|
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
|
53
|
|
BUSINESS
|
64
|
|
MANAGEMENT
|
108
|
|
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
|
113
|
|
PRINCIPAL SHAREHOLDERS
|
114
|
|
DESCRIPTION OF SHARE CAPITAL
|
116
|
|
SHARES ELIGIBLE FOR FUTURE SALE
|
121
|
|
MATERIAL UNITED STATES FEDERAL INCOME AND AUSTRALIAN TAX CONSIDERATIONS
|
122
|
|
ENFORCEMENT OF CIVIL LIABILITIES
|
131
|
|
UNDERWRITING
|
132
|
|
EXPENSES RELATING TO THE OFFERING
|
141
|
|
LEGAL MATTERS
|
142
|
|
EXPERTS
|
142
|
|
WHERE YOU CAN FIND MORE INFORMATION
|
142
|
|
INDEX TO FINANCIAL STATEMENTS
|
F-1
|
ABOUT THIS PROSPECTUS
We are responsible for the information contained in this prospectus and any free writing prospectus we prepare or
authorize. We and the underwriter have not authorized anyone to provide you with different information. We and the underwriter take no responsibility for, and can provide no assurance as to the reliability of, any other information others may give
you. We are not, and the underwriter are not, making an offer to sell these securities in any jurisdiction where the offer or sale is not permitted. You should not assume that the information contained in this prospectus is accurate as of any date
other than its date.
For investors outside the United States: neither we nor the underwriter has done anything that would permit this
offering or possession or distribution of this prospectus or any free writing prospectus in any jurisdiction where action for that purpose is required, other than in the United States. Persons outside the United States who come into possession of
this prospectus or any free writing prospectus must inform themselves about, and observe any restrictions relating to, the offering of the Shares and the distribution of this prospectus and any free writing prospectus outside the United States.
We are incorporated under the laws of Australia, and a majority of our outstanding Shares are owned by non-U.S. residents. Under the rules
of the U.S. Securities and Exchange Commission (“SEC”), we are eligible for treatment as a “foreign private issuer.” As a foreign private issuer, we will not be required to file periodic reports and financial statements with the SEC as frequently or
as promptly as domestic registrants whose securities are registered under the Securities Exchange Act of 1934, as amended (the “Exchange Act”).
Our reporting and functional currency is the Australian dollar, and our financial statements included elsewhere in this prospectus are
presented in Australian dollars. The consolidated financial statements and related notes included elsewhere in this prospectus have been prepared under U.S. Generally Accepted Accounting Principles (“U.S. GAAP”).
All references in this prospectus to “US$” and “U.S. dollars” mean U.S. dollars and all references to “A$” mean Australian dollars, unless
otherwise noted.
This prospectus contains conversions of some Australian dollar amounts into U.S. dollars. Except as otherwise stated in this prospectus,
all conversions from Australian dollars to U.S. dollars are based on the exchange rate of US$ to A$1.00 published by the Reserve Bank of Australia as of , 2022. No representation is made that the Australian dollar amounts referred to in
this prospectus could have been or could be converted into U.S. dollars at such rate.
“Medlab”, the Medlab logo and other trademarks or service marks of Medlab appearing in this prospectus are the property of Medlab or its
subsidiaries. Solely for convenience, the trademarks, service marks and trade names referred to in this prospectus are listed without the ® and ™ symbols, but such references should not be construed as any indicator that their
respective owners will not assert, to the fullest extent under applicable law, their right thereto. All other trademarks, trade names and service marks appearing in this prospectus are the property of their respective owners.
Market and Industry Data
This prospectus contains estimates and information concerning our industry and our business, including estimated market size and projected
growth rates of the markets for our drug candidates. Unless otherwise expressly stated, we obtained this industry, business, market, medical and other information from reports, research surveys, studies and similar data prepared by third parties,
industry, medical and general publications, government data and similar sources.
This information involves a number of assumptions and limitations. We are responsible for all of the disclosure contained in this
prospectus. We have relied on third-party market position, market opportunity and market size data included in this prospectus that we believe are reliable. In addition, projections, assumptions and estimates of our future performance and the
future performance of the industry in which we operate are necessarily subject to a high degree of uncertainty and risk due to a variety of factors, including those described in “Risk Factors.” These and other factors could cause results to differ
materially from those expressed in these publications and reports.
1
PROSPECTUS SUMMARY
This summary highlights selected information contained elsewhere in this prospectus. This summary does not contain all
of the information you should consider before investing in the Shares. You should read this entire prospectus, and the registration statement of which this prospectus is a part, including “Risk Factors,” “Management’s Discussion and Analysis of
Financial Condition and Results of Operations,” and our consolidated financial statements and the related notes included elsewhere in this prospectus, before making an investment decision. Unless otherwise indicated or the context otherwise requires,
“Medlab,” the “Company,” “our company,” “we,” “us” and “our” refer to Medlab Clinical Ltd. and its consolidated subsidiaries, taken as a whole.
Overview
Our legal name is Medlab Clinical Ltd. We are an Australian biotechnology company that formulates innovative solutions and innovative
consumer health care products to provide integrative patient care. Our aim is to disrupt the multi-billion-dollar market of delivery platform technologies for pharmaceutical products that are designed to treat pain and mental health. We were
incorporated in Australia in April 2014, and in 2015 listed on the ASX under the symbol “MDC.” Our corporate headquarters are located in Australia, and we maintain offices in the United States and Europe.
Our mission is to create premier pharmaceutical drugs, therapies and delivery platforms for patients dealing with unmet medical needs, each
of which will comply with applicable regulatory requirements, including the United States Food and Drug Administration (“FDA”), the European Medicines Association (“EMA”) and the Therapeutic Goods Administration of Australia (“TGA”), the medicine and
therapeutic regulatory agency of the Australian Government. We aim to be recognized as a leading specialty drug development company, committed to restoring health and transforming the lives of patients through the development of novel pharmaceutical
products and treatments.
We develop targeted fixed-dose combinations of the NanoCelle® delivery platform and various active pharmaceutical ingredients (“APIs”),
applying proprietary insights to create long-term products that we believe will better meet the medical needs of our patients compared to what is currently available and that we believe will create value for our shareholders. Our focus is on clinical
indications that we believe represent unmet or inadequately addressed medical needs and represent compelling commercial opportunities.
We strongly believe there are considerable unmet needs of patients in our target markets, and that the current drug intervention therapies
that are available can be significantly improved. We believe that we are able to provide these improvements to patients through products that we have and are developing. These products are discussed below.
Though we do have sales from our products, we are primarily in a research and product development phase and do not generate significant
revenue, except from research and development Australian federal grants and pharmaceutical products that we sell through special access pre-registration schemes. Accordingly, we have incurred recurring losses from operations. Our operations have
been funded substantially through capital equity raising. Our focus is on raising capital through equity financings to fund future clinical trials.
We believe the net proceeds of this offering, together with the Australian Research and Development Tax Incentive and our current cash,
will be sufficient to meet our working capital and capital expenditure requirements at least through June 2023.
2
Our Lead Product Candidate: NanoCelle®
NanoCelle®, our core product offering, is a pharmaceutical-delivery platform which is designed to administer pharmaceutical products using
nanoparticles, or small particles ranging between 1 to 100 nanometres in size and undetectable by the human eye. NanoCelle® is an alternative, and, we believe, more effective method of consuming medical products compared to the traditional methods.
NanoCelle® delivery may include oromucosal (oro-buccal/sublingual) sprays, intranasal sprays, dermal and transdermal formulations, topical lotions, oral gel capsules and liquids, and adsorption from solid carriers. NanoCelle® is designed to allow
for vast improvements in patient care, quality of life and side effect reduction. Our primary and commercially available delivery platform, NanoCelle® is now patented in several countries, including Australia, Canada, the European Union Countries(1), Hong Kong, New Zealand, the United States, the United Kingdom and with a pending patent application in Singapore, until 2036. In developing NanoCelle®, we have placed a high research priority on
tolerability, and without the use of harmful toxic products such as heavy metals or gases. To date, NanoCelle® has administered over 350,000 doses of pharmaceutical products to patients through its delivery platform through compassionate use programs
in the United Kingdom and Australia or clinical trials and/or sales of medicines approved by such governments or applicable pharmaceutical regulatory agents. The term “compassionate use” refers to a governmentally-sanctioned expedited pathway for a
patient with an immediately life-threatening condition or serious disease or condition to gain access to an investigational medical product for treatment outside of clinical trials when no comparable or satisfactory alternative therapy options are
available.
(1) “European Union Countries” include Albania, Austria, Belgium, Bulgaria, Croatia, Cyprus, Czech Republic, Denmark, Estonia, Finland, France, Germany,
Greece, Hungary, Ireland, Iceland, Italy, Lithuania, Luxembourg, Latvia, Malta, Monaco, Netherlands, North Macedonia, Norway, Poland, Portugal, Romania, San Marino, Serbia, Slovakia, Slovenia, Spain, Sweden, Switzerland and Turkey.
Our Other Drug Product Candidates, both in-market and in-development
In addition to developing the NanoCelle® delivery method, we have also developed several drug development programs that are specifically
designed to be administered through the NanoCelle® technology. Our NanoCelle® delivery method has produced additional drug candidates which share design and chemistry, manufacturing and controls (“CMC”) processes with our lead candidate,
resulting in tolerability profiles that may be similar to our lead product candidate, as well as regulatory agency familiarity, and a consistent multi-target therapeutic approach.
Several of our products are listed, in contrast to being registered, with the Australian Register of Therapeutic Goods (the “ARTG”). All
medicines registered with the ARTG generally are evaluated for efficacy (that the medicine can do what it says it will) before they may be sold in Australia. However, not all listed medicines are evaluated for efficacy. Our registered medicines, such
as NanaBis™, are higher risk, and because of this, the ARTG fully assesses all registered medicines for safety, quality and efficacy before they go on sale. Registered medicines, which are intended to treat, manage and/or cure one or more
conditions, require investment in clinical evidence with no guarantee of success at TGA. Listed medicines, such as NanoCelle® D3 and NanoCelle® B12, are lower risk and can be purchased off the shelf from pharmacies, health shops, and supermarkets.
Listed products, such as complementary medicines and food supplements, are not exposed to the same risks and do not have to undergo the same rigorous regulatory assessments as registered products but are subject to a random audit after listing.
As such, since our in-market drug products, are lower risk and do not go through pre-market assessment, they are considered ARTG-listed. Both ARTG-listed and ARTG-registered medicines must be manufactured in a licensed or approved facility in accordance with the principles of good manufacturing practice. There is no requirement under the ARTG for our products
to be registered in their present form.
Most therapeutic goods are required to undergo an evaluation for quality, safety and efficacy and be included in the ARTG before they can
be supplied in Australia. In recognition that there are circumstances where patients need access to therapeutic goods that are not included in the ARTG, the TGA manages the Australian Special Access Scheme (the “Special Access Scheme”). The Special
Access Scheme allows registered health practitioners to access therapeutic goods (such as medicines, medical devices or biologicals) that are not included in ARTG for a single patient via approval by the TGA on a named patient basis. Therapeutic
goods that are not included in the ARTG (and are not otherwise exempt from being in the ARTG) are described by us as “unapproved”.
3
In addition to having our products listed and registered with the ARTG, we have also used drug master files in connection with our submissions to
the FDA. Drug master files are submissions to the FDA used to provide confidential, detailed information about facilities, processes, or articles used in the manufacturing, processing, packaging, and storing
of human drug products. The FDA will assess the drug master file with the Company’s overall CMC package and clinical data packages. The drug master file forms part of the CMC package, which is a
significant FDA requirement for a new drug application. The confidential information submitted to the FDA via a drug master file will be reviewed when the FDA reviews the
application that references the drug master file, such as a New Drug Application (“NDA”) or Investigational New Drug (“IND”). To date, the Company has not made NDA or IND submissions. For more information on
drug master files, please see the section entitled “Business—Drug Master Files.”
Our currently available products include the following:
| 1. |
NanoCelle® D3 – Designed to foster immunity and bone health, and currently an ARTG-listed product. This product is currently available through PharmaCare Laboratories Pty Ltd. (“PharmaCare”)
in Australia over-the-counter in pharmacies and through healthcare providers and in New Zealand, operating under Medsafe, as a food supplement;
|
| 2. |
NanoCelle® B12 – Designed to reduce homocysteine levels and support the nervous system, and currently an ARTG-listed product. This product is currently available through PharmaCare in
Australia over-the-counter in pharmacies and through healthcare providers and in New Zealand, operating under Medsafe, as a food supplement;
|
| 3. |
NanaBis™- cannabidiol (“CBD”) and tetrahydrocannabinol (“THC”) with an FDA active pharmaceutical ingredient (“API”) drug master file, and currently an ARTG-registered product. The
intended use of NanaBis™ is for proposed indication of cancer-induced bone pain, with the goal of benefiting larger neuropathic pain populations. This product is available in Australia through the Australian Special Access Scheme
(“Special Access Scheme”) and is scheduled to launch in the United Kingdom through the Named Patient Programme by the end of October 2022, whereby doctors seek government regulatory approval for use for patients on a case-by-case basis.
|
NanaBis™ is pursuing FDA approval first and, if approved by the FDA, then will seek approval by the TGA and the European Medicines Agency
(“EMA”). The NanaBis™ product is currently in Phase II trial studies conducted at an Australian Hospital and met all primary and secondary endpoints. During COVID, the company focused on the CMC and pivoted to 100% synthetics in accordance with
prior FDA guidance.
NanaBis™, to date, has only conducted pharmacokinetic studies on advanced cancer patients and not healthy participants. However, the
Company is expected to include pharmacokinetic studies on a small number of healthy participants in mid-2023. A Phase III trial for NanaBis™, which is expected to include approximately 360 participants from the United States of America, Australia and
the United Kingdom, will plan to use a randomized withdrawal method to demonstrate the analgesic tolerability and exploratory endpoints of NanaBis™ as a monotherapy in cancer patients. The Company expects to complete the Phase III clinical trials for
NanaBis™ in the United States using the net proceeds from this offering, together with the Australian Research and Development Tax Incentive and existing cash.
| 4. |
NanoCBD™- CBD with an FDA API drug master file for proposed indication of occupational stress, with the goal of benefiting mild, chronic pain populations. This product is available
in Australia through the Special Access Scheme and is scheduled to launch in the United Kingdom through the Named Patient Programme by the end of October 2022, whereby doctors seek government regulatory approval for use for patients on a
case-by-case basis. NanoCBD™ is an Australian TGA-lead trial for over-the-counter registered medicines targeting stress. Secondary regulatory agencies include both the Australian TGA and the EMA. NanoCBD™ is expected to
undertake pharmacokinetic and pharmadynamic studies in healthy participants in mid-2023.
|
4
In addition to developing the NanoCelle® delivery method, we have also spearheaded several drug developmental programs that are
specifically designed to be administered through the NanoCelle® technology. Our NanoCelle® delivery method has produced additional drug candidates which share design and CMC processes with our lead candidate, resulting in tolerability
profiles that may be similar to our lead product candidate, as well as regulatory agency familiarity, and a consistent multi-target therapeutic approach. Our products that are currently in the earlier drug development stages include:
| 1. |
MDC2000 - a product targeting major depressive disorders that has been optimized as a single molecule encapsulated in NanoCelle® following clinical studies in 2020 that were closed early due
to the COVID-19 pandemic. Based on this optimization, the program has returned to a pre-clinical stage with clinical returns expected in early 2023; and
|
| 2. |
NanoCelle®-Nucleic Acid is expected to be TGA-focused with the intention to utilize the Company’s NanoCelle® delivery method for RNA COVID-19 vaccines. Nasal adherence studies for a
NanoCelle® insulin have been completed, demonstrating that the studies have met their endpoints. Our next step is to encapsulate siRNA within the NanoCelle® and approach the Australian government for next potential steps. On April 4, 2022,
we entered into a Collaborative Research Agreement with the University of New South Wales and Macquarie University to conduct testing during the pre-clinical stages for our NanoCelle®-Nucleic Acid, a nasal vaccine delivery utilizing nucleic
acid to develop a new vaccine and/or anti-viral technologies. Pursuant to the Collaborative Research Agreement, all intellectual property created or developed under such agreement is owned collectively by all parties to such agreement.
Accordingly, we will not have full ownership over the intellectual property developed under the Collaborative Research Agreement, and any of our products incorporating such intellectual property will be subject to the joint ownership rights
under the agreement.
|
Our Strategy
Our strategy is to expand our market opportunity by gaining United States, United Kingdom, Australian and/or other regulatory approval for
our products. Furthermore, the superior performance that we believe we can achieve through our patented drug delivery platform, NanoCelle®, will be a key differentiator for us in the pharmaceutical space. We are seeking to develop a body of clinical
and real-world evidence and findings to support the benefits of our products.
We intend to take several steps to achieve our goals, including:
| • |
Advancing our novel investigational drug candidates towards approval in the United States and elsewhere;
|
| • |
Developing future clinical products targeting unmet medical needs;
|
| • |
Maintaining a strong intellectual property portfolio in key global markets, including Australia, Canada, the European Union Countries, Hong Kong, New Zealand, the United States, the United
Kingdom and with a pending patent application in Singapore, until 2036; and
|
| • |
Expanding the availability of our products globally and meeting applicable global regulatory and market needs.
|
Our revenue strategy is premised around licensing our intellectual property and assets with large established companies that offer market
expertise. We are seeking to benefit from the health care industry’s search for delivery platforms across all molecules in the pursuit of better patient outlines.
5
For example, over the past year, we have made several key strategic achievements and milestones, including:
| 1. |
Partnered with Purisys LLC (“Purisys”), a United States-based biosynthetic partner for FDA drug master files, for the two cannabinoids, CBD, and THC. It was noted at the time that the
technology required for making a “neat” or 100% dronabinol (the synthetic of THC) was not commercially available. While working with Purisys in Georgia, the Company was able to deliver a 100% dronabinol, as well as a 100% cannabinoid with
relevant documents for an FDA drug master files submission. The Company believes the biosynthetic partnership is a key strategic agreement/milestone.
|
| 2. |
Partnered with a strategic United States-based manufacturing company for (i) chemistry, manufacturing, and controls development for the cannabinoid programs; (ii) manufacturing optimisation
and scale; and (iii) future commercial scale manufacture.
|
| 3. |
Secured patents in several countries for the NanoCelle® delivery platform, including Australia, Canada, the European Union Countries, Hong Kong, New Zealand, the United States, the United
Kingdom and with a pending patent application in Singapore, until 2036 across all territories.
|
| 4. |
Entered agreements with United Kingdom partners for the compassionate use of NanoCBD™ and NanaBis™ to at-risk patients.
|
| 5. |
Entered agreements with Macquarie University and the University of New South Wales for the joint development of NanoCelle®-Nucleic Acid, as a nasal delivery for vaccines.
|
| 6. |
Strengthened our datapoints from our Australian data bank to understand sustainability of the NanaBis™ program over 6 to 12 months and tolerability as it relates to adverse events and in
relation to opioid products.
|
The Company has currently licensed its commercial IP to the following entities:
| 1. |
PharmaCare– Licensed to use Nanocelle® D3 and Nanocelle® B12 in TGA-listed medicines in Australia and New Zealand only for the nutraceuticals.
|
| 2. |
Cultech Ltd.– Licensed to use NRGBiotic™ in TGA-listed medicine and is United Kingdom exclusive and U.S. nonexclusive;
|
| 3. |
YesHealth Sdn Bhd.– Licensed to use several TGA-listed medicines (not including the NanoCelle®) and is Malaysia and Vietnam exclusive and Singapore nonexclusive; and
|
| 4. |
American Nutritional Corporation Inc.– Licensed several TGA-listed medicines inclusive of those NanoCelles® in existing range (Nanocelle® D3 and Nanocelle® B12) and is U.S. exclusive under
their private label called NuScripts.
|
NanoCelle® Technology Transfer Licenses for Manufacturing:
| 1. |
Renaissance Lakewood LLC (“Renaissance”) – a U.S. Contract Development and Manufacturing Organization experienced in FDA and U. S. Drug Enforcement Administration (“DEA”) drugs and
manufacturing;
|
| 2. |
Natural Factors – a Canadian nutraceutical manufacturing facility approved by TGA;
|
| 3. |
Extractas (formerly known as Tasmanian Alkaloids) - an Australian experienced poppy grower and manufacturer for medicines that is approved by TGA; and
|
| 4. |
Universities New South Wales and the Woolcott Institute of Macquarie University – research collaboration partners for the NanoCelle®-Nucleic Acid nasal vaccine program.
|
6
Corporate Information
Our registered office is located at Units 5 and 6, 11-13 Lord Street, Botany, New South Wales, 2019, Australia and our telephone number is
+61 2 8188 0311. Our website address is www.medlab.co. The information on, or accessible through, our website is not part of this prospectus. We have included our website address in this prospectus solely as
an inactive textual reference. All information we file with the SEC is available through the SEC’s Electronic Data Gathering, Analysis and Retrieval system, which may be accessed through the SEC’s website at www.sec.gov.
Implications of Being an Emerging Growth Company
As a company with less than US$1.235 billion in revenue during our last fiscal year, we qualify as an “emerging growth company” as defined
in the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”). As an emerging growth company, we may take advantage of specified reduced disclosure and other requirements that are otherwise applicable generally to public companies. These
provisions include:
| • |
exemption from the auditor attestation requirement of Section 404 of the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”) in the assessment of our internal controls over financial
reporting;
|
| • |
to the extent that we no longer qualify as a foreign private issuer, (i) reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements and (ii)
exemptions from the requirements of holding a non-binding advisory vote on executive compensation, including golden parachute compensation; and
|
| • |
an exemption from compliance with the requirement that the Public Company Accounting Oversight Board has adopted regarding a supplement to the auditor’s report providing
additional information about the audit and the financial statements.
|
We may take advantage of these exemptions until such time that we are no longer an emerging growth company. Accordingly, the information
that we provide shareholders and holders of the Shares may be different than you might obtain from other public companies. We will cease to be an emerging growth company upon the earliest to occur of (i) the last day of the fiscal year in which we
have more than US$1.235 billion in annual revenue; (ii) the last day of the fiscal year in which we qualify as a “large accelerated filer”; (iii) the date on which we have, during the previous three-year period, issued more than US$1.0 billion in
non-convertible debt securities; and (iv) the last day of the fiscal year in which the fifth anniversary of this offering occurs.
Implications of Being a Foreign Private Issuer
We also qualify as a “foreign private issuer” under U.S. securities laws. In our capacity as a foreign private issuer, we are exempt from
certain rules under the Exchange Act that impose certain disclosure obligations and procedural requirements for proxy solicitations under Section 14 of the Exchange Act. In addition, our senior management, the members of our board of directors and
our principal shareholders are exempt from the reporting and “short-swing” profit recovery provisions of Section 16 of the Exchange Act and the rules under the Exchange Act with respect to their purchases and sales of our securities. Moreover, we are
not required to file periodic reports and financial statements with the SEC as frequently or as promptly as U.S. companies whose securities are registered under the Exchange Act. In addition, we are not required to comply with Regulation FD, which
restricts the selective disclosure of material information.
We may take advantage of these exemptions until such time as we are no longer a foreign private issuer. Unless we elect to terminate our
status as a foreign private issuer, we will remain a foreign private issuer until such time that 50% or more of our outstanding voting securities are held by U.S. residents and any of the following three circumstances applies: (i) the majority of the
members of board of directors or our senior management are U.S. citizens or residents; (ii) more than 50% of our assets are located in the United States; or (iii) our business is administered principally in the United States.
7
Risk Factors Summary
Our business is subject to a number of risks of which you should be aware prior to making a decision to invest in our Shares. You should
carefully consider all of the information set forth in this prospectus and, in particular, you should evaluate the specific factors set forth in the section titled “Risk Factors” before deciding whether to invest in our Shares. Among these important
risks are, but not limited to, the following:
| • |
We are subject to going-concern risks.
|
| • |
Escalating global trade tensions, the conflict between Russia and Ukraine, and the adoption or expansion of economic sanctions or trade restrictions could negatively impact us.
|
| • |
The increase in expenses may adversely impact our business if our sources of funding and revenue are insufficient.
|
| • |
We will require additional financing and may be unable to raise sufficient capital, which could have a material adverse impact on our research and development programs or commercialization of
our drug candidates.
|
| • |
We do not own full rights to the intellectual property developed pursuant to our collaborative research agreement with the University of New South Wales and Macquarie University (the
“Collaborative Research Agreement”).
|
| • |
We may find it difficult to enroll patients in our current and any future clinical trials, and patients could discontinue their participation in our current and any future clinical trials,
which could delay or prevent our current and any future clinical trials of our drug candidates and make those trials more expensive to undertake.
|
| • |
Positive results from preclinical studies of our drug candidates are not necessarily predictive of the results of our planned clinical trials of our drug candidates.
|
| • |
Ongoing and future clinical trials of drug candidates may not show sufficient safety and efficacy to obtain requisite regulatory approvals for commercial sale.
|
| • |
If we do not obtain the necessary regulatory approvals, we may be unable to commercialize our drug candidates.
|
| • |
Even if our drug candidates receive regulatory approval, it may still face development and regulatory difficulties that may delay or impair future sales of drug candidates.
|
| • |
Cannabis continues to be a controlled substance under the United States Federal Controlled Substances Act (“CSA”) and our business may result in federal civil or criminal prosecution.
|
| • |
The regulatory approval processes of the FDA, the EMA and other comparable foreign regulatory authorities are lengthy, time-consuming and inherently unpredictable, and if we are ultimately
unable to obtain regulatory approval for our product candidates, our business will be substantially harmed.
|
| • |
We depend on, and will continue to depend on, collaboration and strategic alliances with third partners. To the extent we are able to enter into collaborative arrangements or strategic
alliances, we will be exposed to risks related to those collaborations and alliances.
|
8
| • |
Because we rely on third-party manufacturing and supply partners, our supply of research and development, preclinical and clinical development materials may become limited or interrupted or
may not be of satisfactory quantity or quality.
|
| • |
Future potential sales of our drug candidates may suffer if they are not accepted in the marketplace by physicians, patients and the medical community.
|
| • |
We face competition from entities that may develop drug candidates for our target disease indications, including companies developing novel treatments and technology platforms based on
modalities and technology similar to ours.
|
| • |
If healthcare insurers and other organizations do not pay for our drug candidates or impose limits on reimbursement, our future business may suffer.
|
| • |
We may be exposed to product liability claims which could harm our business.
|
| • |
The continuing COVID-19 pandemic could adversely impact our business, including our non-clinical studies and clinical trials.
|
| • |
The success of our prospective product candidates and future approved products, if any, especially those containing cannabinoids, are subject to a number of constantly evolving state and
federal laws, regulations, and enforcement policies pertaining to cannabis and/or cannabis derivatives.
|
| • |
The approach to the enforcement of cannabis laws may be subject to change, which may create uncertainty for our business.
|
| • |
Research regarding the medical benefits, viability, safety, efficacy, use and social acceptance of cannabis or isolated cannabinoids (such as CBD and THC) remains in early stages.
|
| • |
Our NanaBis™ and NanoCBD™ use of CBD and THC individually and/or in combination, are subject to U.S. and international controlled substance laws and regulations; our
ability to commercialize any product containing these substances will depend, in part, on the ultimate classification of the product under these laws and regulations.
|
| • |
Our drug candidates may be subject to controlled substance laws and regulations. Failure to receive necessary approvals may delay the launch of our drug candidates and failure to comply with
these laws and regulations may adversely affect the results of our business operations.
|
| • |
Our success depends on our ability to protect our intellectual property and our proprietary technology.
|
| • |
Intellectual property rights of third parties could adversely affect our ability to commercialize our drug candidates, such that we could be required to litigate with or obtain licenses from
third parties in order to develop or market our drug candidates.
|
| • |
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies,
and our patent protection could be reduced or eliminated for non-compliance with these requirements.
|
| • |
Intellectual property rights do not address all potential threats to our competitive advantage.
|
9
| • |
We may face difficulties with protecting our intellectual property in certain jurisdictions, which may diminish the value of our intellectual property rights in those jurisdictions.
|
| • |
Price controls may be imposed in markets in which we operate, which may negatively affect our future profitability.
|
| • |
The trading price of the Shares may be volatile, and purchasers of the Shares could incur substantial losses.
|
| • |
The requirements of being a public company may strain our resources and divert management’s attention and if we are unable to maintain effective internal control over financial reporting in
the future, the accuracy and timeliness of our financial reporting may be adversely affected.
|
| • |
We are eligible to be treated as an “emerging growth company” as defined in the JOBS Act, and we cannot be certain if the reduced disclosure requirements applicable to emerging growth
companies will make our Shares less attractive to investors.
|
| • |
There is no public market for the Pre-Funded Warrants or the Share Purchase Warrants being offered by us in this offering.
|
| • |
You will experience immediate and substantial dilution in the net tangible book value of the Shares you purchase in this offering.
|
| • |
Our issuance of additional Shares in connection with financings, acquisitions, investments, or otherwise will dilute all other Shareholders.
|
| • |
As long as we remain subject to the rules of the ASX and Nasdaq, we will be unable to access equity capital without shareholder approval if such equity capital sales would result in an equity
issuance above regulatory thresholds and, consequently, we could be unable to obtain financing sufficient to sustain our business if we are unsuccessful in soliciting requisite shareholder approvals.
|
| • |
The dual listing of our Shares following this offering may negatively impact the liquidity and value of the Shares.
|
| • |
We are subject to risks associated with currency fluctuations, and changes in foreign currency exchange rates could impact our results of operations.
|
| • |
As a foreign private issuer whose shares are listed on Nasdaq, we may follow certain home country corporate governance practices instead of certain Nasdaq requirements.
|
10
Special Note Regarding Forward-Looking Statements
This prospectus contains forward-looking statements about us and our industry that involve substantial risks and uncertainties. All
statements other than statements of historical facts contained in this prospectus, including statements regarding our future results of operations, financial condition, business strategy and plans and objectives of management for future operations,
are forward-looking statements. In some cases, you can identify forward-looking statements because they contain words such as “anticipate,” “believe,” “contemplate,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potential,”
“predict,” “project,” “should,” “target,” “will,” or “would,” or the negative of these words or other similar terms or expressions.
We have based these forward-looking statements largely on our current expectations and projections about future events and trends that we
believe may affect our financial condition, results of operations, business strategy and financial needs. These forward-looking statements are subject to a number of known and unknown risks, uncertainties, other factors and assumptions, including the
risks described in “Risk Factors” and elsewhere in this prospectus, regarding, among other things:
| • |
our product development and business strategy, including the potential size of the markets for our drug candidates and future development and/or expansion of our drug candidates in our
markets;
|
| • |
our current and future research and development activities, including clinical testing and manufacturing and the costs and timing thereof;
|
| • |
our future projections on the amount of our cash necessary to support working capital and capital expenditure requirements;
|
| • |
the impact that the COVID-19 pandemic could have on business operations;
|
| • |
the impact that political instability and international hostilities, including the ongoing aggression between Russia and Ukraine, could have on business operations and agreements with third
parties;
|
| • |
sufficiency of our cash resources;
|
| • |
our ability to commercialize drug candidates and generate product revenues;
|
| • |
our ability to raise additional funding when needed;
|
| • |
any statements concerning anticipated regulatory activities or licensing or collaborative arrangements, including our ability to obtain regulatory clearances;
|
| • |
our research and development and other expenses;
|
| • |
our operations and intellectual property risks;
|
| • |
our ability to remain compliant with the ASX’s and Nasdaq’s continuing listing standards;
|
| • |
any statement of assumptions underlying any of the foregoing; and
|
| • |
other risks and uncertainties, including those listed under “Risk Factors”.
|
These risks are not exhaustive. Other sections of this prospectus may include additional factors that could harm our business and financial
performance. New risk factors may emerge from time to time and it is not possible for our management to predict all risk factors, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of
factors, may cause actual results to differ materially from those contained in, or implied by, any forward-looking statements.
11
You should not rely on forward-looking statements as predictions of future events. We have based the forward-looking statements contained
in this prospectus primarily on our current expectations and projections about future events and trends that we believe may affect our business, financial condition and operating results. We undertake no obligation to update any forward-looking
statements made in this prospectus to reflect events or circumstances after the date of this prospectus or to reflect new information or the occurrence of unanticipated events, except as required by law. We may not actually achieve the plans,
intentions or expectations disclosed in our forward-looking statements, and you should not place undue reliance on our forward-looking statements. Our forward-looking statements do not reflect the potential impact of any future acquisitions, mergers,
dispositions, joint ventures or investments.
In addition, statements that “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. These statements
are based on information available to us as of the date of this prospectus. While we believe that information provides a reasonable basis for these statements, that information may be limited or incomplete. Our statements should not be read to
indicate that we have conducted an exhaustive inquiry into, or review of, all relevant information. These statements are inherently uncertain, and investors are cautioned not to unduly rely on these statements.
You should read this prospectus and the documents that we reference in this prospectus and have filed as exhibits to the registration
statement of which this prospectus is a part with the understanding that our actual future results, levels of activity, performance and achievements may be different from what we expect. We qualify all of our forward-looking statements by these
cautionary statements.
12
THE OFFERING
|
Shares offered by us
|
ordinary shares.
|
|
Pre-Funded Warrants offered by us
|
We are offering to certain purchasers whose purchase of Shares in this offering would otherwise result in the purchaser, together with its affiliates and
certain related parties, beneficially owning more than 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding Shares immediately following the consummation of this offering, the opportunity to purchase, if such purchaser so
chooses, Pre-Funded Warrants in lieu of Shares that would otherwise result in any such purchaser's beneficial ownership exceeding 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding Shares. Each Pre-Funded Warrant will be
exercisable for one Share. The purchase price of each Pre-Funded Warrant will equal the price at which the Shares are being sold to the public in this offering, minus US$0.0001, and the exercise price of each Pre-Funded Warrant will be
US$0.0001 per share. The Pre-Funded Warrants will be exercisable immediately and may be exercised at any time until all of the Pre-Funded Warrants are exercised in full. This offering also relates to the Shares issuable upon exercise of any
Pre-Funded Warrants sold in this offering. For each Pre-Funded Warrant we sell, the number of Shares we are offering will be decreased on a one-for-one basis. For additional information, see "Description of Share Capital —Pre-Funded Warrants
to be Issued as Part of this Offering" on page 118 of this prospectus.
|
|
Share Purchase Warrants offered by us
|
Share Purchase Warrants to purchase an aggregate of [•] Shares. Each Share Purchase Warrant will have an assumed exercise price of US$[•] per share, will
be immediately exercisable and will expire on the [•] anniversary of the original issuance date. This prospectus also relates to the offering of the Shares issuable upon exercise of the Share Purchase Warrants. For additional information, see
"Description of Share Capital — Share Purchase Warrants to be Issued as Part of this Offering" on page 119 of this prospectus.
|
|
Share Units
|
The Shares and accompanying Share Purchase Warrants will be sold in Units, with each Unit consisting of one Share and one Share Purchase Warrant to
purchase one Share. Each Share Unit will be sold at a price of US$ per Unit (assuming a public offering price of US$ per Share). The Share Units will be separable immediately upon issuance.
|
|
Pre-Funded Warrant Units
|
The Pre-Funded Warrants and accompanying Share Purchase Warrants will be sold in units, with each unit consisting of one Pre-Funded Warrant and one Share
Purchase Warrant to purchase one Share. The assumed purchase price of each Pre-Funded Warrant Unit will equal the price at which a Share Unit is being sold to the public in this offering, minus US$0.0001. The Pre-Funded Warrant Units will be
separable immediately upon issuance. For each Pre-Funded Warrant Unit we sell, the number of Share Units we are offering will be decreased on a one-for-one basis.
|
13
|
Over-allotment option
|
We have granted the underwriter a 45-day option to purchase up to additional Shares and/or Share Purchase Warrants at the public
offering price, less underwriting discounts and commissions, on the same terms as set forth in this prospectus.
|
|
Shares to be outstanding after this offering
|
Shares (or Shares if the over-allotment option is exercised in full) (assuming none of the Share Purchase Warrants issued in this offering
are exercised and no sale of any Pre-Funded Warrants).
|
|
Use of proceeds
|
We estimate that the net proceeds from the sale of the Shares that we are selling in this offering will be approximately US$ million (or approximately
US$ million if the underwriter’s option to purchase additional Shares is exercised in full), based upon an assumed initial public offering price of US$ per Share Unit (or US$ per Pre-Funded Warrant Unit), after deducting
underwriting discounts and commissions and estimated offering expenses payable by us.
|
|
|
We intend to use the net proceeds from this offering, together with our existing cash, to further our clinical trials, for working capital and other
general corporate purposes. See “Use of Proceeds” for additional information.
|
|
Risk factors
|
See “Risk Factors” and the other information included in this prospectus for a discussion of the risks you should carefully consider before investing in
the Shares.
|
|
Proposed Nasdaq symbol for the Shares
|
“MDLB”
|
|
No Listing of Warrants
|
We do not intend to apply for listing of the Pre-Funded Warrants or Share Purchase Warrants on any national securities exchange or trading system.
|
|
ASX symbol for the Shares
|
“MDC”
|
The number of Shares that will be outstanding after this offering is based on 2,283,502 Shares outstanding as of November 18, 2022 and
excludes 17,223 Shares issuable upon the exercise of outstanding options as of November 18, 2022, with a weighted-average exercise price of A$ per Share.
14
RISK FACTORS
Investing in the Units involves a high degree of risk. You should consider and read carefully all of the risks and uncertainties described
below, as well as other information included in this prospectus, including our consolidated financial statements and related notes included elsewhere in this prospectus, before making an investment decision. If any of the following risks actually
occur, it could harm our business, prospects, results of operations and financial condition. In such event, the trading price of the Units could decline and you might lose all or part of your investment.
Risks Related to Our Business
We have a history of operating losses and may not achieve or maintain profitability in the future.
We have experienced significant recurring operating losses and negative cash flows from operating activities since inception. For example,
for the years ended June 30, 2022 and 2021, we had net losses of approximately A$7.2 million and approximately A$12.4 million.
We are a clinical stage pharmaceutical development company and the success of our drug candidates is therefore uncertain. We focus on
pharmaceutical solutions for large patient groups with clear, unmet needs, inclusive of non-opioid pain management and mental health. In addition, in efforts to improve patient compliance and associated medication risk profiles, we utilize
proprietary delivery platforms that we believe are more effective for medication offerings.
We expect to continue to incur losses from operations for the foreseeable future and expect the costs of drug development to increase in
the future as more patients are recruited to the clinical trials. In particular, we expect to continue to incur significant losses in the development of our clinical trials and drug candidates. Because of the numerous risks and uncertainties
associated with the development, manufacturing, sales and marketing of our drug candidates, we may experience larger than expected future losses and may never become profitable.
Moreover, there is a substantial risk that we, or our development partners, may not be able to complete the development of our current drug
candidates or develop other pharmaceutical products. It is possible that none of them will be successfully commercialized, which would prevent us from ever achieving profitability.
We are subject to going concern risks.
The Company’s consolidated financial statements have been prepared on a going concern basis under which an entity is considered to be able
to realize its assets and satisfy its liabilities in the ordinary course of business. Our future operations are dependent upon the identification and successful completion of equity or debt financing and the achievement of profitable operations at an
indeterminate time in the future. While we believe that the completion of this offering and the application of the net proceeds as described under “Use of Proceeds” in this prospectus will result in the elimination of the going concern qualification
included in the Company’s consolidated financial statements following this offering, there cannot be any assurances that such qualification will be removed or that the Company will be successful in obtaining necessary financing in the future to
continue as a going concern or achieve profitability. We expect that the Company will need raise additional capital following the completion of this equity offering in order to complete the necessary trials to achieve commercial viability of some or
all of its drug products.
Escalating global trade tensions, the conflict between Russia and Ukraine, and the adoption or expansion of economic
sanctions or trade restrictions could negatively impact us.
On February 24, 2022, Russian troops began a full-scale military invasion of Ukraine. This conflict between Russia and Ukraine has led to
disruption, instability and volatility in global markets and industries that could negatively impact our business, results of operations and financial condition. The conflict has resulted in significant volatility in certain equity, debt and currency
markets, material increases in certain commodity prices, and economic uncertainty. The conflict may escalate and its resolution is unclear. The U.S. government and other governments have imposed severe sanctions against Russia and Russian interests
and Belarus and has threatened additional sanctions and controls. Sanctions and export control laws and regulations are complex, frequently changing, and increasing in number, and they may impose additional legal compliance costs or business risks
associated with our operations. Although the Company does not conduct business directly with companies based in Ukraine, Russia or Belarus, the impact of these measures, as well as potential responses to them by Russia, is currently unknown and they
could adversely affect our business, results of operations and financial condition. Furthermore, this conflict could affect our business by preventing us from entering into agreements with certain counterparties, customers or vendors.
15
The increase in expenses may adversely impact our business if our sources of funding and revenue are insufficient.
We anticipate that as the costs related to the development of our clinical trials will increase, we will require additional funds to
achieve our long-term goals of commercialization and further development of our drug candidates. In addition, we will require funds to pursue regulatory applications, defend intellectual property rights, contract manufacturing capacity, potentially
develop marketing and sales capability and fund operating expenses. We intend to seek such additional funding through public or private financings and/or through licensing of our assets or other arrangements with corporate partners. However, such
financing, licensing opportunities or other arrangements may not be available from any sources on acceptable terms, or at all. Any shortfall in funding could result in us having to curtail or cease our research and development activities, thereby
harming our business, financial condition and results of operations.
In addition, because of the numerous risks and uncertainties associated with the development of our drug candidates, we are unable to
predict the timing or amount of increased expenses, or when, or if, we will be able to achieve or maintain profitability. Our expenses could significantly increase beyond current expectations if the applicable regulatory authorities require further
studies in addition to those currently anticipated. In any case, even if our drug candidates are approved for commercial sale, we anticipate incurring significant costs associated with the commercial launch of such drug candidates and there can be no
guarantee that we will ever generate significant revenues.
We will require additional financing and may be unable to raise sufficient capital, which could have a material adverse
impact on our research and development programs or commercialization of our drug candidates.
We have historically devoted most of our financial resources to research and development, including pre-clinical and clinical development
activities. To date, we have financed a significant amount of our operations through equity financings. The amount of our future net losses will depend, in part, on the rate of our future expenditures and our ability to obtain funding through equity
or debt financings or strategic collaborations. The amount of such future net losses, as well as the possibility of future profitability, will also depend on our success in developing and commercializing products that generate significant revenue.
Our failure to become and remain profitable would depress the value of our Shares and could impair our ability to raise capital, expand our business, maintain our research and development efforts, diversify our product offerings or even continue our
operations.
We anticipate that our expenses will increase substantially for the foreseeable future if, and as, we:
| • |
continue our research and preclinical and clinical development of our drug candidates;
|
| • |
expand the scope of our current proposed clinical studies for our drug candidates;
|
| • |
initiate additional preclinical, clinical or other studies for our drug candidates;
|
| • |
change or add additional manufacturers or suppliers;
|
| • |
seek regulatory and marketing approvals for our drug candidates that successfully complete clinical studies;
|
| • |
seek to identify and validate additional drug candidates;
|
| • |
acquire or in-license other drug candidates and technologies;
|
| • |
maintain, protect and expand our intellectual property portfolio;
|
| • |
attract and retain skilled personnel;
|
| • |
create additional infrastructure to support our operations as a publicly quoted company and our product development and planned future commercialization efforts;
|
| • |
add an internal sales force; and
|
| • |
experience any delays or encounter issues with any of the above.
|
16
Until we are profitable, we will need to obtain additional funding in connection with the further development of our drug candidates. Our
ability to obtain additional financing will be subject to a number of factors, including market conditions, our operating performance and investor sentiment. As such, additional financing may not be available to us when needed, on acceptable terms,
or at all. If we are unable to raise capital when needed or on attractive terms, we could be forced to delay, reduce or eliminate our research and development programs or any future commercialization efforts or obtain funds by entering agreements on
unattractive terms.
Furthermore, any additional equity fundraising in the capital markets may be dilutive for shareholders and any debt-based funding may bind
us to restrictive covenants and curb our operating activities and ability to pay potential future dividends even when profitable. We cannot guarantee that future financing will be available in sufficient amounts or on acceptable terms, if at all. If
we are unable to raise additional capital in sufficient amounts or on acceptable terms, we will be prevented from pursuing research and development efforts. This could harm our business, operating results and financial condition and cause the price
of our Shares to fall.
If we are unable to secure sufficient capital to fund our operations, we may be required to delay, limit, reduce or terminate our product
development or future commercialization efforts or grant rights to third parties to develop and market drug candidates that we would otherwise prefer to develop and market ourselves. For example, additional strategic collaborations could require us
to share commercial rights to our drug candidates with third parties in ways that we do not intend currently or on terms that may not be favorable to us. Moreover, we may also have to relinquish valuable rights to our technologies, future revenue
streams, research programs or drug candidates or grant licenses on terms that may not be favorable to us.
We do not own full rights to the intellectual property developed pursuant to our Collaborative Research Agreement with
the University of New South Wales and Macquarie University.
On April 4, 2022, we entered into a Collaborative Research Agreement with the University of New South Wales and Macquarie University to
conduct testing during the pre-clinical stages for our NanoCelle®-Nucleic Acid, a nasal vaccine delivery utilizing nucleic acid to develop a new vaccine and/or anti-viral technologies. Pursuant to the Collaborative Research Agreement, all
intellectual property created or developed under such agreement is owned collectively by all parties to such agreement. Accordingly, we will not have full ownership over the intellectual property developed under the Collaborative Research Agreement,
and any of our products incorporating such intellectual property will be subject to the joint ownership rights under the agreement. All new intellectual property and commercial returns from the Collaborative Research Agreement will result in a
one-third return for each of the three parties. In addition, any revenues and expenses derived from or allocable to products incorporating the intellectual property developed under the Collaborative Research Agreement may be shared with the other
parties to the Collaborative Research Agreement. We do not believe that our future growth strategy is dependent on the intellectual property developed under the Collaborative Research Agreement, however, we cannot assure you that the Collaborative
Research Agreement will not reduce our potential future revenue or profitability.
We may find it difficult to enroll patients in our current and any future clinical trials, and patients could
discontinue their participation in our current and any future clinical trials, which could delay or prevent our current and any future clinical trials of our drug candidates and make those trials more expensive to undertake.
Identifying and qualifying patients to participate in current and any future clinical trials of our drug candidates is critical to our
success. The timing of our clinical trials depends on the speed at which we can recruit patients to participate in testing our drug candidates. Patients may be unwilling to participate in any future clinical trials because of negative publicity from
adverse events in the biotechnology industry. Patients may be unavailable for other reasons, including competitive clinical trials for similar patient populations, and the timeline for recruiting patients, conducting trials and obtaining regulatory
approval of potential products may be delayed. If we have difficulty enrolling a sufficient number of patients to conduct any future clinical trials as planned, we may need to delay, limit or discontinue those clinical trials. Clinical trial delays
could result in increased costs, slower product development, setbacks in testing the safety and effectiveness of our technology or discontinuation of the clinical trials altogether.
17
Any failure to implement our business strategy could negatively impact our business, financial condition and results of
operations.
The development and commercialization of our drug candidates is subject to many risks, including:
| • |
additional clinical or pre-clinical trials may be required beyond what we currently expect;
|
| • |
regulatory authorities may disagree with our interpretation of data from our preclinical studies and clinical studies or may require that we conduct additional studies;
|
| • |
regulatory authorities may disagree with our proposed design of future clinical trials;
|
| • |
regulatory authorities may delay approval of our drug candidates, thus preventing milestone payments from our collaboration partners;
|
| • |
regulatory authorities may not accept data generated at our clinical study sites;
|
| • |
we may be unable to obtain and maintain regulatory approval of our drug candidate in any jurisdiction;
|
| • |
the prevalence and severity of any side effects of any drug candidate could delay or prevent commercialization, limit the indications for any approved drug candidate, require the establishment
of a risk evaluation and mitigation strategy, or cause an approved drug candidate to be taken off the market;
|
| • |
regulatory authorities may identify deficiencies in manufacturing processes;
|
| • |
regulatory authorities may change their approval policies or adopt new regulations;
|
| • |
the third-party manufacturers we expect to depend on to supply or manufacture our drug candidates may not produce adequate supply;
|
| • |
we may not be able to manufacture our drug candidates at a cost or in quantities necessary to make commercially successful products;
|
| • |
we may not be able to obtain adequate supply of our drug candidates for our clinical trials;
|
| • |
we may experience delays in the commencement of, enrollment of patients in and timing of our clinical trials;
|
| • |
we may not be able to demonstrate that our drug candidates are safe and effective as a treatment for its indications to the satisfaction of regulatory authorities, and we may not be able to
achieve and maintain compliance with all regulatory requirements applicable to our drug candidates;
|
| • |
we may not be able to maintain a continued acceptable safety profile of our products following approval;
|
| • |
we may be unable to establish or maintain collaborations, licensing or other arrangements;
|
| • |
the market may not accept our drug candidates;
|
18
| • |
we may be unable to establish and maintain an effective sales and marketing infrastructure, either through the creation of a commercial infrastructure or through strategic collaborations, and
the effectiveness of our own or any future strategic collaborators’ marketing, sales and distribution strategy and operations will affect our profitability;
|
| • |
we may experience competition from existing products or new products that may emerge;
|
| • |
we and our licensors may be unable to successfully obtain, maintain, defend and enforce intellectual property rights important to protect our drug candidates; and
|
| • |
we may not be able to obtain and maintain coverage and adequate reimbursement from third-party payors.
|
If any of these risks materializes, we could experience significant delays or an inability to successfully develop and commercialize our
drug candidates we or our partners may develop, which would have a material adverse effect on our business, financial condition and results of operations.
Positive results from preclinical studies of our drug candidates are not necessarily predictive of the results of our
planned clinical trials of our drug candidates.
Positive results in preclinical proof of concept and animal studies of our drug candidates may not result in positive results in clinical
trials in humans. Many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in clinical trials after achieving positive results in preclinical development or early-stage clinical trials, and we cannot be
certain that we will not face similar setbacks. These setbacks can be caused by, among other things, preclinical findings made while clinical trials were underway or safety or efficacy observations made in clinical trials, including adverse events.
Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that believed their drug candidates performed satisfactorily in preclinical studies and clinical trials nonetheless failed to
obtain FDA or other regulatory authority approval. If we fail to produce positive results in our clinical trials of our drug candidates, the development timeline and regulatory approval and commercialization prospects for our drug candidates, and,
correspondingly, our business and financial prospects, would be negatively impacted.
Ongoing and future clinical trials of drug candidates may not show sufficient safety and efficacy to obtain requisite
regulatory approvals for commercial sale.
Phase I and Phase II clinical trials are not primarily designed to test the efficacy of a drug candidate but rather to test safety and to
understand the drug candidate’s side effects at various doses and schedules. Furthermore, success in preclinical and early clinical trials does not ensure that later large-scale trials will be successful nor does it predict final results. Acceptable
results in early trials may not be repeated in later trials. Further, Phase III clinical trials may not show sufficient safety or efficacy to obtain regulatory approval for marketing. In addition, clinical results are frequently susceptible to
varying interpretations that may delay, limit or prevent regulatory approvals. Negative or inconclusive results or adverse medical events during a clinical trial could require that the clinical trial be redone or terminated. The length of time
necessary to complete clinical trials and to submit an application for marketing approval by applicable regulatory authorities may also vary significantly based on the type, complexity and novelty of the drug candidate involved, as well as other
factors. If we suffer any significant delays, quality issues, setbacks or negative results in, or termination of, our clinical trials, we may be unable to continue the development of our drug candidates or generate revenue and our business may be
severely harmed.
19
If we do not obtain the necessary regulatory approvals, we may be unable to commercialize our drug candidates.
The clinical development, manufacturing, sales and marketing of our drug candidates are subject to extensive regulation by regulatory
authorities in the United States, the United Kingdom, the European Union, Australia and elsewhere. Despite the substantial time and expense invested in preparation and submission of an application or equivalents in other jurisdictions, regulatory
approval is never guaranteed. The number, size and design of preclinical studies and clinical trials that will be required will vary depending on the product, the disease or condition for which the product is intended to be used and the regulations
and guidance documents applicable to any particular product. Additionally, during the review process and prior to approval, the FDA and/or other regulatory bodies may require additional data, including with respect to whether our products have abuse
potential, which may delay approval and any potential controlled substance scheduling processes. The FDA or other regulators can delay, limit or deny approval of a product for many reasons, including, but not limited to, the fact that regulators may
not approve our or a third-party manufacturer’s processes or facilities or that new laws may be enacted or regulators may change their approval policies or adopt new regulations requiring new or different evidence of safety and efficacy for the
intended use of a product.
Successful results in clinical trials and in the subsequent application for marketing approval are not guaranteed. If we are unable to
obtain regulatory approvals, we will not be able to generate revenue from our drug candidates. Even if we receive regulatory approval for any of our drug candidates, our profitability will depend on our ability to generate revenues from their sale or
the licensing of our technology.
Even if our drug candidates receive regulatory approval, it may still face development and regulatory difficulties that
may delay or impair future sales of drug candidates.
Even if we or our licensing partners receive regulatory approval to sell any drug candidates, the relevant regulatory authorities may,
nevertheless, impose significant restrictions on the indicated uses, manufacturing, labelling, packaging, adverse event reporting, storage, advertising, promotion and record keeping or impose ongoing requirements for post-approval studies. In
addition, regulatory agencies subject a marketed product, its manufacturer and the manufacturer’s facilities to continual review and periodic inspections. Previously unknown problems with the drug candidate, including adverse events of unanticipated
severity or frequency, may result in restrictions on the marketing of the product, and could include withdrawal of the product from the market. In addition, new statutory requirements may be enacted or additional regulations may be enacted that could
prevent or delay regulatory approval of one or more of our drug candidates.
Risks Relating to the Development and Regulatory Approval of Our Product Candidates
We have a limited number of product candidates, all which are still in early clinical or pre-clinical development. If
we do not obtain regulatory approval of one or more of our product candidates, or experience significant delays in doing so, our business will be materially adversely affected.
We currently have no products approved for sale or marketing in any country, and may never be able to obtain regulatory approval for any of our product candidates. As a result, we are not currently
permitted to market any of our product candidates in the United States or in any other country until we obtain regulatory approval from the FDA or regulatory authorities outside the United States. Our product candidates are in early stages of
development and we have not submitted an application, or received marketing approval, for any of our product candidates. Obtaining regulatory approval of our product candidates will depend on many factors, including, but not limited to, the
following:
| • |
successfully completing formulation and process development activities;
|
| • |
completing clinical trials that demonstrate the efficacy and safety of our product candidates;
|
| • |
receiving marketing approval from applicable regulatory authorities;
|
| • |
establishing commercial manufacturing capabilities; and
|
| • |
launching commercial sales, marketing and distribution operations.
|
Many of these factors are wholly or partially beyond our control, including clinical advancement, the regulatory submission process and changes in the
competitive landscape. If we do not achieve one or more of these targets in a timely manner, we could experience significant delays or may be unable to develop our product candidates at all, which may have a material adverse effect on our business
and results of operations.
20
Clinical trials are expensive, time consuming, difficult to design and implement, and involve uncertain outcomes. Results of previous
pre-clinical studies and clinical trials may not be predictive of future results, and the results of our current and planned clinical trials may not satisfy the requirements of the FDA or other regulatory authorities.
Positive or timely results from pre-clinical or early-stage trials do not ensure positive or timely results in late-stage clinical trials or product approval by
the FDA or comparable foreign regulatory authorities. We will be required to demonstrate with substantial evidence through well-controlled clinical trials that our product candidates are safe and effective for use in a diverse population before we
can seek regulatory approvals for their commercialization. Our planned clinical trials may produce negative or inconclusive results, and we or any of our current and future strategic partners may decide, or regulators may require us, to conduct
additional clinical or pre-clinical testing.
Success in pre-clinical studies or early-stage clinical trials does not mean that future clinical trials or registration clinical trials will be successful
because product candidates in later-stage clinical trials may fail to demonstrate sufficient safety and efficacy to the satisfaction of the FDA and foreign regulatory authorities, despite having progressed through pre-clinical studies and initial
clinical trials. Product candidates that have shown promising results in early clinical trials may still suffer significant setbacks in subsequent clinical trials or registration clinical trials. For example, a number of companies in the
biopharmaceutical industry, including those with greater resources and experience than us, have suffered significant setbacks in advanced clinical trials, even after obtaining promising results in earlier clinical trials. Similarly, pre-clinical
interim results of a clinical trial are not necessarily predictive of final results.
If clinical trials for our product candidates are prolonged, delayed or stopped, we may be unable to obtain regulatory
approval and commercialize our product candidates on a timely basis, or at all, which would require us to incur additional costs and delay our receipt of any product revenue.
We may experience delays in our ongoing or future pre-clinical studies or clinical trials, and we do not know whether future pre-clinical studies or clinical trials will begin on time, need to be
redesigned, enroll an adequate number of patients or be completed on schedule, if at all. The commencement or completion of these planned clinical trials could be substantially delayed or prevented by many factors, including, but not limited to:
| • |
discussions with the FDA or other regulatory agencies regarding the scope or design of our clinical trials;
|
| • |
the limited number of, and competition for, suitable sites to conduct our clinical trials, many of which may already be engaged in other clinical trial programs, including some that may be for
the same indication as our product candidates;
|
| • |
any delay or failure to obtain approval or agreement to commence a clinical trial in any of the countries where enrollment is planned;
|
| • |
inability to obtain sufficient funds required for a clinical trial;
|
| • |
clinical holds on, or other regulatory objections to, a new or ongoing clinical trial;
|
| • |
delay or failure to manufacture sufficient supplies of product candidates for our clinical trials;
|
| • |
delay or failure to reach agreement on acceptable clinical trial agreement terms or clinical trial protocols with prospective sites or clinical research organizations (“CROs”), the terms of
which can be subject to extensive negotiation and may vary significantly among different sites or CROs;
|
| • |
delay or failure to obtain institutional review board (“IRB”) approval to conduct a clinical trial at a prospective site;
|
21
| • |
slower than expected rates of patient recruitment and enrollment;
|
| • |
failure of patients to complete the clinical trial;
|
| • |
the inability to enroll a sufficient number of patients in studies to ensure adequate statistical power to detect statistically significant treatment effects;
|
| • |
unforeseen safety issues, including severe or unexpected drug-related adverse effects experienced by patients, including possible deaths;
|
| • |
lack of efficacy during clinical trials;
|
| • |
termination of our clinical trials by one or more clinical trial sites;
|
| • |
inability or unwillingness of patients or clinical investigators to follow our clinical trial protocols;
|
| • |
inability to monitor patients adequately during or after treatment;
|
| • |
clinical study sites failing to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all, deviating from the protocol or dropping out of a
study;
|
| • |
inability to address any non-compliance with regulatory requirements or safety concerns that arise during the course of a clinical trial;
|
| • |
the need to repeat or terminate clinical trials as a result of inconclusive or negative results or unforeseen complications in testing; and
|
| • |
our clinical trials may be suspended or terminated upon a breach or pursuant to the terms of any agreement with, or for any other reason by, current or future strategic partners that have
responsibility for the clinical development of any of our product candidates.
|
Changes in regulatory requirements, policies and guidelines may also occur and we may need to significantly amend clinical trial protocols
to reflect these changes with appropriate regulatory authorities. These changes may require us to renegotiate terms with CROs or resubmit clinical trial protocols to IRBs for re-examination, which may impact the costs, timing or successful completion
of a clinical trial. Our clinical trials may be suspended or terminated at any time by the FDA, other regulatory authorities, the IRB overseeing the clinical trial at issue, any of our clinical trial sites with respect to that site, or us. Any
failure or significant delay in commencing or completing clinical trials for our product candidates may adversely affect our ability to obtain regulatory approval and our commercial prospects and our ability to generate product revenue will be
diminished.
22
Cannabis continues to be a controlled substance under the United States Federal Controlled Substances Act (“CSA”) and
our business may result in federal civil or criminal prosecution.
Though we are not directly engaged in the medical and adult-use cannabis industry, some of our products involve cannabinoids. In the U.S.
local state law may permit such activities however all such activities remain illegal under federal law in the U.S. Investors are cautioned that in the U.S., cannabis is highly regulated at the state level. To our knowledge, there are to date 38
states, the District of Columbia, and four U.S. territories that have legalized medical cannabis in some form, although not all states have fully implemented their legalization programs. To our knowledge, 18 states and the District of Columbia have
legalized cannabis for adult use. Additional states have legalized high-cannabidiol CBD, low THC oils for a limited class of patients. Notwithstanding the permissive regulatory environment of cannabis at the state level, cannabis continues to be
categorized as a Schedule I controlled substance under the CSA. Under United States federal law, a Schedule I drug is considered to have a high potential for abuse, no accepted medical use in the United States, and a lack of accepted safety for the
use of the substance under medical supervision. Federal law prohibits commercial production and sale of all Schedule I controlled substances, and as such, cannabis-related activities, including without limitation, the importation, cultivation,
manufacture, distribution, sale and possession of cannabis remain illegal under U.S. federal law. It is also illegal to aid or abet such activities or to conspire or attempt to engage in such activities. Strict compliance with state and local laws
with respect to cannabis may neither absolve us of liability under U.S. federal law, nor provide a defense to any federal proceeding brought against us. An investor’s contribution to and involvement in such activities may result in federal civil
and/or criminal prosecution, including, but not limited to, forfeiture of his, her or its entire investment, fines and/or imprisonment.
An appropriations rider contained in the fiscal year 2015, 2016, 2017, 2018, 2019, 2020 and 2021 Consolidated Appropriations Acts (formerly
known as the “Rohrabacher-Farr Amendment”; now known as the “Rohrabacher-Blumenauer Amendment” and currently proposed for the next appropriations rider as the “Joyce Amendment”, referred to herein as the “Amendment”) provides budgetary constraints on
the federal government’s ability to interfere with the implementation of state-based medical cannabis laws. The Ninth Circuit Court of Appeals and other courts have interpreted the language to mean that the U.S. Department of Justice (the “DOJ”)
cannot expend funds to prosecute state-law-abiding medical cannabis operators complying strictly with state medical cannabis laws. The Amendment prohibits the federal government from using congressionally appropriated funds to prevent states from
implementing their own medical cannabis laws. The Rohrabacher Amendment was renewed for fiscal year 2021 and again through various spending bills and is effective until September 2022. Continued reauthorization of the Amendment is predicated on
future political developments and cannot be guaranteed. If the Amendment expires, federal prosecutors could prosecute even state-compliant medical cannabis operators for conduct within the five-year statute of limitations. The Amendment does not
protect state legal adult-use cannabis businesses and the DOJ may spend funds to prosecute persons that are operating in accordance with state adult use cannabis laws.
Violations of any federal laws and regulations could result in significant fines, penalties, administrative sanctions, convictions or
settlements arising from civil proceedings conducted by either the federal government or private citizens, or criminal charges and penalties, including, but not limited to, disgorgement of profits, cessation of business activities, divestiture, or
prison time. This could have a material adverse effect on us, including our reputation and ability to conduct business, our holding (directly or indirectly) of medical and adult-use cannabis licenses in the U.S., the listing of our securities on
Nasdaq, our financial position, operating results, profitability or liquidity or the market price of our publicly traded shares. In addition, it is difficult for us to estimate the time or resources that would be needed for the investigation or
defense of any such matters or our final resolution because, in part, the time and resources that may be needed are dependent on the nature and extent of any information requested by the applicable authorities involved, and such time or resources
could be substantial.
23
We may be in violation of anti-money laundering laws and regulations which could impact our ability to obtain banking
services, result in the forfeiture or seizure of our assets and could require us to suspend or cease operations.
We are subject to a variety of laws and regulations in Australia and in the U.S. that involve money laundering, financial recordkeeping and
proceeds of crime, including the Bank Secrecy Act, as amended by Title III of the Uniting and Strengthening America by Providing Appropriate Tools Required to Intercept and Obstruct Terrorism Act of 2001, as amended and the rules and regulations
thereunder, any related or similar rules, regulations or guidelines, issued, administered or enforced by governmental authorities in the U.S. and Canada. Since the cultivation, manufacture, distribution and sale of cannabis remains illegal under the
CSA, banks and other financial institutions providing services to cannabis-related businesses risk violation of federal anti-money laundering statutes (18 U.S.C. §§ 1956 and 1957), the unlicensed money-remitter statute (18 U.S.C. § 1960) and the Bank
Secrecy Act, among other applicable federal statutes. Banks or other financial institutions that provide cannabis businesses with financial services such as a checking account or credit card in violation of the Bank Secrecy Act could be criminally
prosecuted for willful violations of money laundering statutes, in addition to being subject to other criminal, civil, and regulatory enforcement actions. Banks often refuse to provide banking services to businesses involved in the cannabis industry
due to the present state of the laws and regulations governing financial institutions in the U.S. The lack of banking and financial services presents unique and significant challenges to businesses in the cannabis industry. The potential lack of a
secure place in which to deposit and store cash, the inability to pay creditors through the issuance of checks and the inability to secure traditional forms of operational financing, such as lines of credit, are some of the many challenges presented
by the unavailability of traditional banking and financial services. These statutes can impose criminal liability for engaging in certain financial and monetary transactions with the proceeds of a “specified unlawful activity” such as distributing
controlled substances which are illegal under federal law, including cannabis, and for failing to identify or report financial transactions that involve the proceeds of cannabis-related violations of the CSA. We may also be exposed to the foregoing
risks.
As previously introduced, in February 2014, the Financial Crimes Enforcement Network (“FinCEN”) issued a memorandum (the “FinCEN
Memorandum”) providing instructions to banks seeking to provide services to cannabis-related businesses. The FinCEN Memorandum states that in some circumstances, it is permissible for banks to provide services to cannabis-related businesses without
risking prosecution for violation of the Bank Secrecy Act. It refers to supplementary guidance that former Deputy Attorney General James M. Cole issued to federal prosecutors relating to the prosecution of money laundering offenses predicated on
cannabis-related violations of the CSA. Although the FinCEN Memorandum remains in effect today, it is unclear at this time whether the current administration will follow the guidelines of the FinCEN Memorandum. Overall, the DOJ continues to have the
right and power to prosecute crimes committed by banks and financial institutions, such as money laundering and violations of the Bank Secrecy Act, that occur in any state, including in states that have legalized the applicable conduct and the DOJ’s
current enforcement priorities could change for any number of reasons. A change in the DOJ’s enforcement priorities could result in the DOJ prosecuting banks and financial institutions for crimes that previously were not prosecuted. If we do not have
access to a U.S. banking system, its business and operations could be adversely affected.
Other potential violations of federal law resulting from cannabis-related activities include the Racketeer Influenced Corrupt Organizations
Act (“RICO”). RICO is a federal statute providing criminal penalties in addition to a civil cause of action for acts performed as part of an ongoing criminal organization. Under RICO, it is unlawful for any person who has received income derived from
a pattern of racketeering activity to use or invest any of that income in the acquisition of any interest, or the establishment or operation of, any enterprise which is engaged in interstate commerce. RICO also authorizes private parties whose
properties or businesses are harmed by such patterns of racketeering activity to initiate a civil action against the individuals involved. Although RICO suits against the cannabis industry are rare, a few cannabis businesses have been subject to a
civil RICO action. Defending such a case has proven extremely costly, and potentially fatal to a business’ operations.
In the event that any of our operations, or any proceeds thereof, any dividends or distributions therefrom, or any profits or revenues
accruing from such operations in the United States were found to be in violation of money laundering legislation or otherwise, such transactions may be viewed as proceeds of crime under one or more of the statutes noted above or any other applicable
legislation. This could restrict or otherwise jeopardize our ability to declare or pay dividends, effect other distributions and subject us to civil and/or criminal penalties. Furthermore, while there are no current intentions to declare or pay
dividends on our Shares in the foreseeable future, in the event that a determination was made that our proceeds from operations (or any future operations or investments in the United States) could reasonably be shown to constitute proceeds of crime,
we may decide or be required to suspend declaring or paying dividends without advance notice and for an indefinite period of time. We could likewise be required to suspend or cease operations entirely.
24
Uncertainty surrounding existing protection from U.S. federal prosecution may adversely affect our operations and
financial performance.
Pursuant to the Amendment, until such time as it is not renewed or expires of its own accord, the DOJ is prohibited from expending any
funds to prevent states from implementing their own medical cannabis laws. If the Amendment or an equivalent thereof is not successfully included in the next or any subsequent federal omnibus spending bill, the protection which has been afforded
thereby to U.S. medical cannabis businesses in the past would lapse, and such businesses would be subject to a higher risk of prosecution under federal law. Although unlikely, there is a possibility that all amendments may be banned from federal
omnibus spending bills, and if this occurs and the substantive provisions of the Amendment are not included in the base federal omnibus spending bill or other law, these protections will lapse. To the extent the Amendment is included in a continuing
resolution, the protections of the Amendment would lapse if Congress does not reauthorize the resolution or pass another funding measure that includes the Amendment.
The regulatory approval processes of the FDA, the EMA and other comparable foreign regulatory authorities are lengthy,
time-consuming and inherently unpredictable, and if we are ultimately unable to obtain regulatory approval for our product candidates, our business will be substantially harmed.
If we are not eligible to receive or do not seek approvals of our products through the FDA Expanded Access pathway, or a compassionate use
program or a similar medium, we are not permitted to market our product candidates in the United States or the European Union until we receive approval of a NDA from the FDA or a marketing authorization application (“MAA”) from the EMA, respectively,
or in any foreign countries until we receive the requisite approval from such countries. Prior to submitting an NDA to the FDA or an MAA to the EMA for approval of our product candidates, we will need to complete our ongoing preclinical studies, as
well as Phase I, Phase II and Phase III clinical trials. Our drug candidates are in various stages, including some that are still conducting preclinical studies and have not yet commenced our clinical program or human tests. For approval with
regulatory bodies, such as the FDA or TGA, we plan to conduct Phase I, and possibly Phase II, clinical trials in the U.S. or Australia, subject to applicable regulatory approval. Successfully initiating and completing our clinical program and
obtaining approval of an NDA or MAA is a complex, lengthy, expensive and uncertain process, and the FDA or EMA may delay, limit or deny approval of our product candidates for many reasons, including, among others, because:
| • |
we may not be able to demonstrate that our product candidates are safe and effective in treating patients to the satisfaction of the FDA or EMA;
|
| • |
the results of our clinical trials may not meet the level of statistical or clinical significance required by the FDA or EMA for marketing approval;
|
| • |
the FDA or EMA may disagree with the number, design, size, conduct or implementation of our clinical trials;
|
| • |
the FDA or EMA may require that we conduct additional clinical trials;
|
| • |
the FDA or EMA or other applicable foreign regulatory authorities may not approve the formulation, labelling or specifications of our product candidates;
|
| • |
the contract research organizations (“CROs”) and other contractors that we may retain to conduct our clinical trials may take actions outside of our control that materially adversely impact
our clinical trials;
|
| • |
the FDA or EMA may disagree with our interpretation of data from our preclinical studies and clinical trials;
|
25
| • |
the FDA or EMA may not accept data generated at our clinical trial sites or may disagree with us over whether to accept efficacy results from clinical trial sites outside the United States
where the standard of care is potentially different from that in the United States;
|
| • |
if and when our NDAs or MAAs are submitted to the FDA or EMA, as applicable, the regulatory agency may have difficulties scheduling the necessary review meetings in a timely manner, may
recommend against approval of our application or may recommend or require, as a condition of approval, additional preclinical studies or clinical trials, limitations on approved labelling or distribution and use restrictions;
|
| • |
the FDA may require development of a Risk Evaluation and Mitigation Strategy, which would use risk minimization strategies beyond the professional labelling to ensure that the benefits of
certain prescription drugs outweigh their risks, as a condition of approval or post-approval, and the EMA may grant only conditional approval or impose specific obligations as a condition for marketing authorization, or may require us to
conduct post-authorization safety studies;
|
| • |
the FDA, EMA, and the DEA or other applicable foreign regulatory agencies may not approve the manufacturing processes or facilities of third-party manufacturers with which we contract, or DEA
or other applicable foreign regulatory agency quotas may limit the quantities of controlled substances available to our manufacturers; or
|
| • |
the FDA or EMA may change their approval policies or adopt new regulations.
|
Clinical trials conducted in Australia are subject to various regulatory controls to ensure the safety of clinical trial participants. The
TGA regulates the use of therapeutic goods supplied in clinical trials in Australia under the therapeutic goods legislation. Clinical trial sponsors must comply with various import, export, manufacture and supply requirements promulgated by the TGA,
the compliance with which are not always clear and require a significant amount of subjective interpretation on the part of the Australian clinical trial sponsor.
In 2014, the Australian Advisory Council on Medicines Scheduling recommended rescheduling CBD from a prohibited substance to being a
prescription medicine because, according to the Advisory Council on Medicines Scheduling, “there is a low risk of misuse or abuse as CBD does not possess psychoactive properties.” The TGA accepted this recommendation and the decision took effect in
July 2015. From June 2015, CBD has been included under Schedule 4 (S4) Prescription Only Medicine of the Poisons Standard when preparations for therapeutic use contain 2% or less of other cannabinoids found in cannabis.
In February 2016, the Australian Federal Government passed legislation that amended the Narcotic Drugs Act, allowing the supply of suitable
medicinal cannabis products for the management of painful and chronic conditions. This legislation does not relate to the decriminalization of cannabis for general cultivation or recreational use and it does not include the provision of medicinal
grade herbal cannabis, only processed, non-smokable medicinal grade products. Much of the detail remains unclear. For example, the legislation does not specify which products will be covered under the amendment, and it does not specify which
particular conditions or symptoms will be eligible for treatment with cannabis-based products. Before products can be prescribed, they must be registered with the TGA or, in rare circumstances, receive special approval from the TGA. The registration
process requires evidence of testing and efficacy, and it is therefore unlikely Australia will see an TGA registered medicinal cannabis product that GPs can prescribe any time soon.
There are currently few cannabis-based products that are lawfully produced in Australia. The medicinal use of pharmaceutical products
containing cannabinoids is not prohibited, as long as authorization for prescribing is granted from the TGA.
Despite the 2016 legislation discussed above, there are many factors, a significant number of which are beyond our control, that could
jeopardize our ability to obtain regulatory approval to commence our clinical trials in Australia. If we are unable to obtain the necessary regulatory approvals in Australia, we will have to consider alternative locations for our clinical trials,
including the United States, which may be more costly or have stringent regulatory requirements of their own, and which would likely delay aspects of our development plan.
26
We depend on, and will continue to depend on, collaboration and strategic alliances with third partners. To the extent
we are able to enter into collaborative arrangements or strategic alliances, we will be exposed to risks related to those collaborations and alliances.
An important element of our strategy for developing, manufacturing and commercializing our drug candidates is entering into partnerships
and strategic alliances with other pharmaceutical companies or other industry participants.
Any partnerships or alliance we have or may have in the future may be terminated for reasons beyond our control or we may not be able to
negotiate future alliances on acceptable terms, if at all. These arrangements may result in us receiving less revenue than if we sold our products directly, may place the development, sales and marketing of our products outside of our control, may
require us to relinquish important rights or may otherwise be on unfavorable terms. Collaborative arrangements or strategic alliances will also subject us to a number of risks, including the risk that:
| • |
we may not be able to control the amount and timing of resources that our strategic partner/collaborators may devote to the drug candidates;
|
| • |
strategic partner/collaborators may experience financial difficulties;
|
| • |
the failure to successfully collaborate with third parties may delay, prevent or otherwise impair the development or commercialization of our drug candidates or revenue expectations;
|
| • |
products being developed by partners/collaborators may never reach commercial stage resulting in reduced or even no milestone or royalty payments;
|
| • |
business combinations or significant changes in a collaborator’s business strategy may also adversely affect a collaborator’s willingness or ability to complete their obligations under any
arrangement;
|
| • |
a collaborator could independently move forward with a competing product developed either independently or in collaboration with others, including our competitors; and
|
| • |
collaborative arrangements are often terminated or allowed to expire, which would delay the development and may increase the cost of developing drug candidates.
|
Because we rely on third-party manufacturing and supply partners, our supply of research and development, preclinical
and clinical development materials may become limited or interrupted or may not be of satisfactory quantity or quality.
We rely on third-party supply and manufacturing partners to manufacture and supply the materials for our research and development and
preclinical and clinical study supplies. Additionally, we have no “in-house” manufacturing capabilities and are dependent on third parties to manufacture our drug candidates in a cost effective manner. Problems with third-party manufacturers or the
manufacturing process, or inability to scale up manufacturing activities, may result in delayed clinical trials and commercialization of our drug candidates.
There can be no assurance that our supply of research and development, preclinical and clinical development biologics and other materials
will not be limited, interrupted or restricted in certain geographic regions, be of satisfactory quality or continue to be available at acceptable prices. Replacement of a third-party manufacturer could require significant effort, cost and expertise
because there may be a limited number of qualified replacements.
The manufacturing process for a drug candidate is subject to FDA and foreign regulatory authority review. Suppliers and manufacturers must
meet applicable manufacturing requirements and undergo rigorous facility and process validation tests required by regulatory authorities in order to comply with regulatory standards. In the event that any of our suppliers or manufacturers fails to
comply with such requirements or to perform its obligations to us in relation to quality, timing or otherwise, or if our supply of components or other materials becomes limited or interrupted for other reasons, we may be forced to manufacture the
materials ourselves or enter into an agreement with another third party, which would be costly and delay any future clinical trials.
Further, if any third-party provider fails to meet its obligations to manufacture our products, or fails to maintain or achieve
satisfactory regulatory compliance, the development of such substances and the commercialization of any therapies, if approved, could be stopped, delayed or made commercially unviable, less profitable or may result in enforcement actions against us.
27
Our research and development efforts may be jeopardized if we are unable to retain key personnel and cultivate key
academic and scientific collaborations.
Changes in our senior management may be disruptive to our business and may adversely affect our operations. For example, when we have
changes in senior management positions, we may elect to adopt different business strategies or plans. Any new strategies or plans, if adopted, may not be successful and if any new strategies or plans do not produce the desired results, our business
may suffer.
Moreover, competition among biotechnology and pharmaceutical companies for qualified employees is intense and as such we may not be able to
attract and retain personnel critical to our success. Our success depends on our continued ability to attract, retain and motivate highly qualified management, clinical and scientific personnel, manufacturing personnel, sales and marketing personnel
and on our ability to develop and maintain important relationships with clinicians, scientists and leading academic and health institutions. If we fail to identify, attract, retain and motivate these highly skilled personnel, we may be unable to
continue our product development and commercialization activities.
In addition, biotechnology and pharmaceutical industries are subject to rapid and significant technological change. Our drug candidates may
be or become uncompetitive. To remain competitive, we must employ and retain suitably qualified staff that are continuously educated to keep pace with changing technology but may not be in a position to do so.
We may encounter difficulties in managing our growth, which could negatively impact our operations.
As we advance our clinical development programs for drug candidates, seek regulatory approval in the United States, Europe and elsewhere
and increase the number of ongoing product development programs, we anticipate that we will need to increase our product development, scientific and administrative headcount. We will also need to establish commercial capabilities in order to
commercialize any drug candidates that may be approved. Such an evolution may impact our strategic focus and our deployment and allocation of resources.
Our ability to manage our operations and growth effectively depends upon the continual improvement of our procedures, reporting systems and
operational, financial and management controls. We may not be able to implement administrative and operational improvements in an efficient or timely manner and may discover deficiencies in existing systems and controls. If we do not meet these
challenges, we may be unable to execute our business strategies and may be forced to expend more resources than anticipated addressing these issues.
We may acquire additional technology and complementary businesses in the future. Acquisitions involve many risks, any of which could
materially harm our business, including the diversion of management’s attention from core business concerns, failure to effectively exploit acquired technologies, failure to successfully integrate the acquired business or realize expected synergies
or the loss of key employees from either our business or the acquired businesses.
In addition, in order to continue to meet our obligations as a publicly listed company in both Australia and the United States and to
support our anticipated long-term growth, we will need to increase our general and administrative capabilities. Our management, personnel and systems may not be adequate to support this future growth.
If we are unable to successfully manage our growth and the increased complexity of our operations, our business, financial position,
results of operations and prospects may be harmed.
28
Future potential sales of our drug candidates may suffer if they are not accepted in the marketplace by physicians,
patients and the medical community.
There is a risk that our drug candidates may not gain market acceptance among physicians, patients and the medical community, even if they
are approved by the regulatory authorities. The degree of market acceptance of any of our approved drug candidates will depend on a variety of factors, including:
| • |
timing of market introduction, number and clinical profile of competitive products;
|
| • |
our ability to provide acceptable evidence of safety and efficacy and our ability to secure the support of key clinicians and physicians for our drug candidates;
|
| • |
cost-effectiveness compared to existing and new treatments;
|
| • |
availability of coverage, reimbursement and adequate payment from health maintenance organizations and other third-party payers;
|
| • |
prevalence and severity of adverse side effects; and
|
| • |
other advantages over other treatment methods.
|
As controlled substances, the products may generate public controversy. Physicians, patients, payers or the medical community may be
unwilling to accept, use or recommend our drug candidates which would adversely affect our potential revenues and future profitability. Adverse publicity or public perception regarding cannabis to our current or future investigational therapies using
these substances may negatively influence the success of these therapies.
We face competition from entities that may develop drug candidates for our target disease indications, including
companies developing novel treatments and technology platforms based on modalities and technology similar to ours.
The development and commercialization of drug candidates is highly competitive. Multinational pharmaceutical companies and specialized
biotechnology companies could develop drug candidates and processes competitive with our drug candidates. Competitive therapeutic treatments include those that have already been approved and accepted by the medical community, patients and third-party
payers, and any new treatments that enter the market.
There may be a significant number of products that are currently under development, and may become commercially available in the future,
for the treatment of conditions for which we are developing, and may in the future try to develop, drug candidates.
Multinational pharmaceutical companies and specialized biotechnology companies could have significantly greater financial, technical,
manufacturing, marketing, sales and supply resources and experience than we have. If we successfully obtain approval for any drug candidate, we could face competition based on many different factors, including the safety and effectiveness of our drug
candidates, the ease with which our drug candidates can be administered and the extent to which patients accept relatively new routes of administration, the timing and scope of regulatory approvals for these drug candidates, the availability and cost
of manufacturing, marketing and sales capabilities, price, reimbursement coverage and patent position.
Competing products could present superior treatment alternatives, including by being more effective, safer, less expensive or marketed and
sold more effectively than any products we may develop. Competitive products may make any products we develop obsolete or non-competitive before we recover the expense of developing and commercializing our drug candidates. Such competitors could also
recruit our employees, which could negatively impact our level of expertise and our ability to execute our business plan.
29
If healthcare insurers and other organizations do not pay for our drug candidates or impose limits on reimbursement,
our future business may suffer.
Our drug candidates may be rejected by the market due to many factors, including cost. The continuing efforts of governments, insurance
companies and other payers of healthcare costs to contain or reduce healthcare costs may affect our future revenues and profitability. In Australia and certain foreign markets, the pricing of some pharmaceutical products is subject to government
control. We expect initiatives for similar government control to continue in the United States and elsewhere. The adoption of any such legislative or regulatory proposals could harm our business and prospects.
Successful commercialization of our drug candidates will depend in part on the extent to which reimbursement for the cost of our products
and related treatment will be available from government health administration authorities, private health insurers and other organizations. Our drug candidates may not be considered cost-effective and reimbursement may not be available to consumers
or may not be sufficient to allow our products to be marketed on a competitive basis. Third-party payers are increasingly challenging the price of medical products and treatment.
If third-party coverage is not available for our drug candidates the market acceptance of these drug candidates will be reduced.
Cost-control initiatives could decrease the price we might establish for drug candidates, which could result in product revenues lower than anticipated. If the price for our drug candidates decreases or if governmental and other third-party payers do
not provide adequate coverage and reimbursement levels our potential revenue and prospects for profitability will suffer.
We may be exposed to product liability claims which could harm our business.
The testing, marketing and sale of therapeutic products entails an inherent risk of product liability. We rely on a number of third-party
researchers and contractors to produce, collect, and analyze data regarding the safety and efficacy of our drug candidates. We also have quality control and quality assurance in place to mitigate these risks, as well as professional liability and
clinical trial insurance to cover financial damages in the event that human testing is done incorrectly or the data is analyzed incorrectly.
Notwithstanding our control procedures, we may face product liability exposure related to the testing of our drug candidates in human
clinical trials. If any of our drug candidates are approved for sale, we may face exposure to claims by an even greater number of persons than were involved in the clinical trials once marketing, distribution and sales of our drug candidates begin.
Regardless of merit or eventual outcome, liability claims may result in:
| • |
decreased demand for our drug candidates;
|
| • |
injury to our reputation;
|
| • |
withdrawal of clinical trial participants;
|
| • |
costs of related litigation;
|
| • |
substantial monetary awards to patients and others;
|
| • |
loss of revenues; and
|
| • |
the inability to commercialize drug candidates.
|
With respect to product liability claims, we could face additional liability beyond insurance limits if testing mistakes were to endanger
any human subjects. In addition, if a claim is made against us in conjunction with these research testing activities, the market price of our Shares may be negatively affected.
30
Our ability to generate revenues from product sales and achieve profitability will depend on our ability to
successfully commercialize products.
Our ability to generate revenues from product sales and achieve profitability will depend on our ability to successfully commercialize
products, including our current product candidates and other product candidates that we may develop, in-license or acquire in the future. Our ability to generate revenues and achieve profitability also depends on a number of additional factors,
including our ability to:
| • |
successfully complete development activities, including the necessary clinical trials;
|
| • |
complete and submit either Biologics License Applications (“BLAs”) or NDAs to the FDA and obtain U.S. regulatory approval for indications for which there is a commercial market;
|
| • |
complete and submit applications to foreign regulatory authorities;
|
| • |
obtain regulatory approval in territories with viable market sizes;
|
| • |
obtain coverage and adequate reimbursement from third parties, including government and private payors;
|
| • |
set commercially viable prices for our products, if any;
|
| • |
establish and maintain supply and manufacturing relationships with reliable third parties, legally globally compliant manufacturing of bulk drug substances and drug products to maintain that
supply;
|
| • |
develop distribution processes for our product candidates;
|
| • |
develop commercial quantities of our product candidates, if approved, at acceptable cost levels;
|
| • |
obtain additional funding if required to develop and commercialize our product candidates;
|
| • |
develop sales, marketing and distribution capabilities for products we intend to sell;
|
| • |
achieve market acceptance of our products;
|
| • |
attract, hire and retain qualified personnel; and
|
| • |
protect our intellectual property rights.
|
Our revenues for any product candidates for which regulatory approval is obtained will be dependent, in part, upon the size of the markets
in the territories for which it gains regulatory approval, the accepted price for the products, the ability to get reimbursement at any price, and whether we own the commercial rights for that territory. If the number of our addressable disease
patients is not as significant as our estimates, the indication approved by regulatory authorities is narrower than we expect, or the reasonably accepted population for treatment is narrowed by competition, physician choice or treatment guidelines,
we may not generate significant revenues from sales of such products, even if approved. In addition, we anticipate incurring significant costs associated with commercializing any approved product candidates. As a result, even if we generate revenues,
we may not become profitable and may need to obtain additional funding to continue operations. If we fail to become profitable or are unable to sustain profitability on a continuing basis, then we may be unable to continue our operations at planned
levels and may be forced to reduce our operations.
31
The continuing COVID-19 pandemic could adversely impact our business, including our non-clinical studies and clinical
trials.
The outbreak of COVID-19 severely impacted global economic activity and caused significant volatility and negative pressure in financial
markets. The global impact of the outbreak continues to evolve and many countries, including the United States, have, and may in the future, institute quarantines or travel restrictions. The COVID-19 pandemic may continue to have an adverse impact on
global economic growth. COVID-19 or another pandemic could have material and adverse effects on our ability to successfully operate due to, among other factors:
| • |
delays or difficulties in enrolling patients in our clinical trials;
|
| • |
delays or difficulties in clinical site initiation, including difficulties in recruiting clinical site investigators and clinical site staff;
|
| • |
delays or disruptions in non-clinical experiments and IND application-enabling good laboratory practice standard toxicology studies due to unforeseen circumstances at CROs and vendors along
their supply chain;
|
| • |
increased rates of patients withdrawing from our clinical trials following enrolment as a result of contracting COVID-19, being forced to quarantine, or not wanting to attend hospital visits;
|
| • |
diversion of healthcare resources away from the conduct of clinical trials, including the diversion of hospitals serving as our clinical trial sites and hospital staff supporting the conduct
of our clinical trials;
|
| • |
interruption of key clinical trial activities, such as clinical trial site data monitoring, due to limitations on travel imposed or recommended by national, state or local governments,
employers and others or interruption of clinical trial subject visits and study procedures (particularly any procedures that may be deemed non-essential), which may impact the integrity of subject data and clinical study endpoints;
|
| • |
interruption or delays in the operations of the FDA, the EMA, the TGA or other foreign regulatory agencies, which may impact approval timelines;
|
| • |
interruption of, or delays in receiving, supplies of our drug candidates from our contract manufacturing organizations due to staffing shortages, production slowdowns or stoppages and
disruptions in our supply chain or distribution vendors’ ability to ship drug candidates; and
|
| • |
limitations on employee resources that would otherwise be focused on the conduct of our non-clinical studies and clinical trials, including because of sickness of employees or their families,
the desire of employees to avoid contact with large groups of people, an increased reliance on working from home or mass transit disruptions.
|
32
Product shipment delays could have a material adverse effect on our business, results of operations and financial
condition.
The shipment, import and export of our drug candidates will require import and export licenses. In the United States, the FDA, U.S. Customs
and Border Protection, and the DEA; in Canada, the Canada Border Services Agency, Health Canada; in Europe, the EMA and the European Commission; in Australia and New Zealand, the Australian Customs and Border Protection Service, the TGA, the New
Zealand Medicines and Medical Device Safety Authority and the New Zealand Customs Service; and in other countries, similar regulatory authorities, regulate the import and export of pharmaceutical products that contain controlled substances.
Specifically, the import and export processes require the issuance of import and export licenses by the relevant controlled substance authority in both the importing and exporting country.
We may not be granted, or if granted, maintain, such licenses from the authorities in certain countries. Even if we obtain the relevant
licenses, and our drug candidates may be held up or lost in transit, which could cause significant delays and may lead to product batches being stored outside required temperature ranges. Inappropriate storage may damage the product shipment
resulting in delays in clinical trials or, upon commercialization, a partial or total loss of revenue from one or more shipments of API or our drug candidates. A delay in a clinical trial or, upon commercialization, a partial or total loss of revenue
from one or more shipments of API or our drug candidates could have a material adverse effect on our business, results of operations and financial condition.
The success of our prospective product candidates and future approved products, if any, especially those containing
cannabinoids, are subject to a number of constantly evolving state and federal laws, regulations, and enforcement policies pertaining to cannabis and/or cannabis derivatives.
The Agriculture Improvement Act of 2018 (the “2018 Farm Bill”) was signed into law on December 20, 2018. This 2018 Farm Bill expressly
excluded “hemp” from the CSA and the Controlled Substances Import and Export Act’s, as amended, definition of marijuana and, accordingly, declassified substances derived from or containing any part(s) of the cannabis plant containing not more than
0.3% THC on a dry-weight basis from Schedule I. In effect, the 2018 Farm Bill legalized the cultivation and commercial sale of hemp in the United States, subject to applicable state laws and regulations and applicable Federal Food, Drug, and Cosmetic
Act (“FDCA”) provisions, including any implementing regulations, as interpreted, and enforced by the FDA.
Local, state, federal, and international hemp and CBD laws and regulations are broad in scope and subject to evolving interpretations,
which could require us to incur substantial costs associated with compliance requirements. In addition, violations of these laws, or allegations of such violations, could disrupt our business and result in a material adverse effect on our operations.
In addition, it is possible that regulations may be enacted in the future that will be directly applicable to our proposed business regarding cannabinoid production. It is also possible that the federal government will begin strictly enforcing
existing laws, which may limit the legal uses of the hemp plant and its derivatives and extracts, such as cannabinoids. We cannot predict the nature of any future laws, regulations, interpretations, or applications, nor can we determine what effect
additional governmental regulations or administrative policies and procedures, when and if promulgated, could have on our activities in the cannabis industry.
The 2018 Farm Bill did not alter the FDA’s authority to regulate products containing cannabis or cannabis-derived compounds, including
cannabinoids, under the FDCA. Hemp products, including cannabinoids, that qualify as drugs, food, dietary supplements, veterinary products, and cosmetics, for example, are subject to regulation by the FDA. Following passage of the 2018 Farm Bill, the
FDA reaffirmed its enforcement authority and reiterated the requirement that a product containing CBD or other cannabinoid(s) (hemp-derived or otherwise) that is marketed with a claim of therapeutic benefit implicitly or explicitly attributed to, or
based on, the presence of the cannabinoid as an ingredient, or any other health/medical claim, must be approved by the FDA for its intended use(s) before it may be introduced into interstate commerce. Our prospective product candidates are currently
intended for development under an IND and, eventually, approval under an NDA, which will mean that, if approved, we can market such products with claims about their proven medical benefits for the applicable indications for use to the extent
consistent with the product’s NDA.
33
While we believe that the 2018 Farm Bill and analogous state legislation has reduced the amount of DEA oversight of hemp-derived
cannabinoids, this is a rapidly evolving area of U.S. law and substantial uncertainty remains as to the future of federal and state regulation of cannabinoid products. In addition, the FDA has approved only one natural cannabis-based drug product,
which contains only hemp-derived CBD. There can be no assurance that our product candidates containing cannabinoids (as the active drug ingredient(s)) will be similarly approved for commercialization in the United States at any time in the near or
distant future. Any regulations the FDA issues relating to the sale, marketing, and/or other activities involving cannabinoid or certain cannabinoid-containing products could have a material adverse effect on our business, financial condition, and
results of operations.
Given the uncertainty surrounding future state regulations and the continuing barriers that still exist for cannabinoids in certain product
categories due to FDA regulation, it is unknown what impact the removal of hemp from the CSA, and any resulting commercialization of hemp products, may have on our business.
We will be subject to extensive ongoing regulatory obligations and continued regulatory review, which may
result in significant additional expense and we may be subject to penalties if we fail to comply with regulatory requirements or experience unanticipated problems with our products.
Any regulatory approvals that we may receive for our products will require the submission of reports to regulatory authorities
and surveillance to monitor the safety and efficacy of our products, may contain significant limitations related to use restrictions for specified age groups, warnings, precautions or contraindications, and may include burdensome post-approval study
or risk management requirements. In addition, if the FDA, EMA, or comparable foreign regulatory authorities approve our products and the activities associated with their development and commercialization, including its design, testing, manufacture,
safety, efficacy, recordkeeping, labeling, storage, approval, advertising, promotion, sale, distribution, import and export will be subject to comprehensive regulation by the FDA and other regulatory agencies in the United States and by the EMA in
the E.U. and comparable foreign regulatory authorities. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as on-going compliance with current good manufacturing practices (cGMPs)
and GCPs for any clinical trials that we conduct following approval. In addition, manufacturers of drug products and their facilities are subject to continual review and periodic, unannounced inspections by the FDA, EMA, and other regulatory
authorities for compliance with cGMPs.
If we or a regulatory authority discover previously unknown problems with our products, such as adverse events of unanticipated
severity or frequency, or problems with the facilities where our products are manufactured, a regulatory authority may impose restrictions on our products, the manufacturing facility or us, including requiring recall or withdrawal of our products
from the market or suspension of manufacturing, restrictions on our ability to conduct clinical trials, including full or partial clinical holds on ongoing or planned trials, restrictions on the manufacturing process, warning or untitled letters,
civil and criminal penalties, injunctions, product seizures, detentions or import bans, voluntary or mandatory publicity requirements and imposition of restrictions on operations, including costly new manufacturing requirements. The occurrence of any
event or penalty described above may inhibit our ability to commercialize our products and generate revenue and could require us to expend significant time and resources in response and could generate negative publicity.
The FDA’s, EMA’s and other regulatory comparable authorities’ policies may change and additional government regulations may be
enacted that could prevent, limit, delay, increase the cost or risks of obtaining regulatory approval of our product candidates, including if as a result new or more costly or difficult to achieve clinical trial or manufacturing quality requirements
are imposed. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any regulatory approval that we may have
obtained, which would adversely affect our business, prospects and ability to achieve or sustain profitability.
34
The approach to the enforcement of cannabis laws may be subject to change, which may create uncertainty for our
business.
As a result of the conflicting views between state legislatures and the federal government regarding cannabis, investments in, and the
operations of, cannabis businesses in the United States are subject to inconsistent laws and regulations. The so-called “Cole Memo” issued by former Deputy Attorney General James Cole on August 29, 2013 and other Obama-era cannabis policy guidance,
discussed below, provided the framework for managing the tension between federal and state cannabis laws. Subsequently, as discussed below, former Attorney General Jeff Sessions rescinded the Cole Memo and related policy guidance. Although no longer
in effect, these policies, and the enforcement priorities established within, appear to continue to be followed during the Biden Administration and remain critical factors that inform the past and future trend of state-based legalization.
The Cole Memo directed U.S. Attorneys not to prioritize the enforcement of federal cannabis laws against individuals and businesses that
comply with state medical or adult-use cannabis regulatory programs, provided certain enumerated enforcement priorities (such as diversion or sale of cannabis to minors) were not implicated. In addition to general prosecutorial guidance issued by the
DOJ, FinCEN issued the FinCEN Memorandum on February 14, 2014 outlining Bank Secrecy Act-compliant pathways for financial institutions to service state-sanctioned cannabis businesses, which echoed the enforcement priorities outlined in the Cole
Memorandum. On the same day the FinCEN Memorandum was published, the DOJ issued complementary policy guidance directing prosecutors to apply the enforcement priorities of the Cole Memo when determining whether to prosecute individuals or institutions
with crimes related to financial transactions involving the proceeds of cannabis-related activities.
On January 4, 2018, then-Attorney General Jeff Sessions rescinded the Cole Memo and all other related Obama-era DOJ cannabis enforcement
guidance. While the rescission did not change federal law, as the Cole Memo and other DOJ guidance documents were not themselves laws, the rescission removed the DOJ’s formal policy that state-regulated cannabis businesses in compliance with the Cole
Memo guidelines should not be a prosecutorial priority. Notably, former Attorney General Sessions’ rescission of the Cole Memo and the Cole Banking Memorandum has not affected the status of the FinCEN Memorandum issued by the Department of Treasury,
which remains in effect. In addition to his rescission of the Cole Memo, former Attorney General Sessions issued a one-page memorandum known as the “Sessions Memorandum.” The Sessions Memorandum explains the DOJ’s rationale for rescinding all past
DOJ cannabis enforcement guidance, claiming that Obama-era enforcement policies are “unnecessary” due to existing general enforcement guidance adopted in the 1980s, in chapter 9.27.230 of the U.S. Attorney’s Manual (the “USAM”). The USAM enforcement
priorities, like those of the Cole Memo, are based on the use of the federal government’s limited resources and include “law enforcement priorities set by the Attorney General,” the “seriousness” of the alleged crimes, the “deterrent effect of
criminal prosecution,” and “the cumulative impact of particular crimes on the community.” Although the Sessions Memorandum emphasizes that cannabis is a federally illegal Schedule I controlled substance, it does not otherwise instruct U.S. Attorneys
to consider the prosecution of cannabis-related offenses a DOJ priority, and in practice, most U.S. Attorneys have not changed their prosecutorial approach to date. However, due to the lack of specific direction in the Sessions Memorandum as to the
priority federal prosecutors should ascribe to such cannabis activities and the lack of additional guidance since the resignation of former Attorney General Sessions, there can be no assurance that the federal government will not seek to prosecute
cases involving cannabis businesses that are otherwise compliant with state law.
William Barr served as United States Attorney General from February 14, 2019 to December 23, 2020. The DOJ under Mr. Barr did not take a
formal position on federal enforcement of laws relating to cannabis. United States President Biden appointed Merrick Garland to succeed Mr. Barr as the U.S. Attorney General. Though Attorney General Garland has not yet formally reissued the Cole
Memo, he has stated that prosecuting marijuana use is not a priority for the DOJ. Thus, it is still unclear what impact, if any, the Biden administration will have on U.S. federal government enforcement policy on cannabis.
Such potential proceedings could involve significant restrictions being imposed upon us or third parties, while diverting the attention of
key executives. Such proceedings could have a material adverse effect on our business, revenues, operating results and financial condition as well as our reputation and prospects, even if such proceedings were concluded successfully in our favor. In
the extreme case, such proceedings could ultimately involve the criminal prosecution of our key executives, the seizure of our corporate assets, and consequently, our inability to continue our business operations. Strict compliance with state and
local laws with respect to cannabis does not absolve us of potential liability under federal law, nor provide a defense to any federal proceeding which may be brought against us. Any such proceedings brought against us may adversely affect our
operations and financial performance.
35
A demand for cannabis and derivate products may never develop.
The global sale of cannabis and hemp products is a new industry resulting from recent legal and regulatory changes. Although we expect the
demand for licensed cannabis to be in excess of the supply being produced by the licensed producers, there is a risk that such demand does not develop as anticipated. Further, there is a risk that the adoption rate by pharmacies to sell medical
cannabis is lower than expected or that such adoption rate may take longer than anticipated. There is also a risk that the international export market for medicinal cannabis and extracts, such as CBD, Cannabigerol and cannabichromene, will not
materialize as projected or not be commercially viable. Should any of such events materialize, they may have a material adverse effect on our business, results of operations and financial condition.
Research regarding the medical benefits, viability, safety, efficacy, use and social acceptance of cannabis or isolated
cannabinoids (such as CBD and THC) remains in early stages.
There have been relatively few clinical trials on the benefits of cannabis or isolated cannabinoids (such as CBD and THC). Although we
believe that the articles, reports and studies support our beliefs regarding the medical benefits, viability, safety, efficacy, dosing and social acceptance of cannabis, future research and clinical trials may prove such statements to be incorrect,
or could raise concerns regarding, and perceptions relating to, cannabis. Given these risks, uncertainties and assumptions, investors should not place undue reliance on such articles and reports. Future research studies and clinical trials may draw
opposing conclusions to those stated herein or reach negative conclusions related to medical cannabis, which could have a material adverse effect on the demand for our products and could result in a material adverse effect on our business, financial
condition and results of operations or prospects.
Our NanaBis™ and NanoCBD™ use of CBD and THC individually and/or in combination, are subject to
U.S. and international controlled substance laws and regulations; our ability to commercialize any product containing these substances will depend, in part, on the ultimate classification of the product under these laws and regulations.
Our cannabinoids product candidates for treatment of various pain indications use pharmaceutically pure CBD, and THC, which is
synthetically derived. These products are quite distinct from crude herbal “medical marijuana,” and we intend to seek FDA approval for these products in accordance with the customary FDA approval process and based on adequate and well-controlled
clinical studies. However, the active ingredients in our products are defined as controlled substances under the federal CSA. Under the CSA, the DEA may place each drug that has abuse potential into one of five categories. The five categories,
referred to as Schedules I-V, carry different degrees of restriction. Each schedule is associated with a distinct set of controls that affect manufacturers, researchers, healthcare providers, and patients. The controls include registration with the
DEA, labelling and packaging, production quotas, security, recordkeeping, and dispensing. Schedule I is the most restrictive, covering drugs that have “no accepted medical use” in the United States and that have high abuse potential.
If and when any of our product candidates receive FDA approval, the DEA will make a scheduling determination and place the product in a
schedule other than Schedule I in order for it to be prescribed to patients in the United States. Accordingly, our ability to ultimately commercialize the product will depend in part on the ultimate scheduling classification determination by DEA for
our product.
The FDA has stated that it will continue to facilitate the work of companies interested in bringing safe, effective, and quality products
to market, including scientifically based research concerning the medical uses of products derived from marijuana and the FDA has approved synthetic compositions of the active ingredients found in marijuana. However, the use and abuse of controlled
substances is currently subject to political and social pressures from certain constituencies related to their usage which could result in additional difficulty with respect to the approval of cannabinoids as a prescription pharmaceutical. For
example, the FDA or DEA may require us to generate more clinical data about the potential for abuse than that which is currently anticipated, which could increase the cost and/or delay the launch of our product. In addition, DEA scheduling may limit
our ability to achieve market share in the United States due to restricted access and the disinclination of some physicians to prescribe more restrictive scheduled controlled substances. For example, Schedule II drugs may not be refilled without a
new prescription. These factors may limit the commercial viability of cannabinoids in the United States.
36
Our drug candidates may be subject to controlled substance laws and regulations. Failure to receive necessary approvals
may delay the launch of our drug candidates and failure to comply with these laws and regulations may adversely affect the results of our business operations.
Some of our drug candidates contain controlled substances as defined in the CSA. Controlled substances that are pharmaceutical products are
subject to a high degree of regulation under the CSA, which establishes, among other things, certain registration, manufacturing quotas, security, recordkeeping, reporting, import, export and other requirements administered by the DEA. The DEA
classifies controlled substances into five schedules: Schedule I, II, III, IV or V substances. Schedule I substances by definition have a high potential for abuse, have not currently “accepted medical use” in the United States, lack accepted safety
for use under medical supervision, and may not be prescribed, marketed or sold in the United States. Pharmaceutical products approved for use in the United States may be listed as Schedule II, III, IV or V, with Schedule II substances considered to
present the highest potential for abuse or dependence and Schedule V substances the lowest relative risk of abuse among such substances. Schedule I and II drugs are subject to the strictest controls under the CSA, including manufacturing and
procurement quotas, security requirements and criteria for importation. In addition, dispensing of Schedule II drugs is further restricted. For example, they may not be refilled without a new prescription.
As one of our drug candidates classifies as a synthetic cannabinoids pharmaceutical product with psychedelic agents, our drug candidates
are likely to be scheduled as Schedule II or III controlled substance. We will need to identify wholesale distributors with the appropriate DEA registrations and authority to distribute the products to pharmacies and other healthcare providers, and
these distributors would need to obtain Schedule II or III distribution registrations. The failure to obtain, or delay in obtaining, or the loss of any of those registrations could result in increased costs to us. If any of our drug candidates is a
Schedule II drug, pharmacies would have to maintain enhanced security with alarms and monitoring systems, and they must adhere to additional recordkeeping and inventory requirements. This may discourage some pharmacies from carrying the product.
Furthermore, state and federal enforcement actions, regulatory requirements, and legislation intended to reduce prescription drug abuse, such as the requirement that physicians consult a state prescription drug monitoring program, may make physicians
less willing to prescribe, and pharmacies to dispense, Schedule II products.
Individual states in the United States have also established controlled substance laws and regulations. Though state-controlled substance
laws often mirror federal law, because the states are separate jurisdictions, they may separately schedule our drug candidates as well. While some states automatically schedule a drug based on federal action, other states schedule drugs through
rulemaking or a legislative action. State scheduling may delay commercial sale of any product for which we obtain federal regulatory approval and adverse scheduling could have a material adverse effect on the commercial attractiveness of such
product. We or our partners must also obtain separate state registrations, permits or licenses in order to be able to obtain, handle, and distribute controlled substances for clinical trials or commercial sale, and failure to meet applicable
regulatory requirements could lead to enforcement and sanctions by the states in addition to those from the DEA or otherwise arising under federal law.
We intend to contract manufacturers in the United States to produce the drug product for our clinical trials and the API for our drug
candidates. In addition, we may decide to develop, manufacture or commercialize our drug candidates in additional countries. As a result, we will also be subject to controlled substance laws and regulations from the TGA and from other regulatory
agencies in other countries where we develop, manufacture or commercialize our drug candidates in the future. We plan to submit NDAs for our drug candidates to the FDA upon completion of all requisite clinical trials and may require additional DEA
scheduling decisions at such time as well.
37
Risks Related to Intellectual Property
Our success depends on our ability to protect our intellectual property and our proprietary technology.
Our success is to a certain degree also dependent on our ability to obtain and maintain patent protection or where applicable, to
receive/maintain orphan drug designation/status and resulting marketing exclusivity for our drug candidates.
We may be materially adversely affected by our failure or inability to protect our intellectual property rights. Without the granting of
these rights, the ability to pursue damages for infringement would be limited. Similarly, any know-how that is proprietary or particular to our technologies may be subject to risk of disclosure by employees or consultants despite having
confidentiality agreements in place.
Any future success will depend in part on whether we can obtain and maintain patents to protect our own products and technologies; obtain
licenses to the patented technologies of third parties; and operate without infringing on the proprietary rights of third parties. Biotechnology patent matters can involve complex legal and scientific questions, and it is impossible to predict the
outcome of biotechnology and pharmaceutical patent claims. Any of our future patent applications may not be approved, or we may not develop additional products or processes that are patentable. Some countries in which we may sell our drug candidate
or license our intellectual property may fail to protect our intellectual property rights to the same extent as the protection that may be afforded in the United States or Australia. Some legal principles remain unresolved and there has not been a
consistent policy regarding the breadth or interpretation of claims allowed in patents in the United States, the United Kingdom, the European Union, Australia or elsewhere. In addition, the specific content of patents and patent applications that are
necessary to support and interpret patent claims is highly uncertain due to the complex nature of the relevant legal, scientific and factual issues. Changes in either patent laws or in interpretations of patent laws in the United States, the United
Kingdom, the European Union or elsewhere may diminish the value of our intellectual property or narrow the scope of our patent protection. Even if we are able to obtain patents, they may not issue in a form that will provide us with any meaningful
protection, prevent competitors from competing with us or otherwise provide us with any competitive advantage. Our competitors may be able to circumvent our patents by developing similar or alternative technologies or products in a non-infringing
manner. We may also fail to take the required actions or pay the necessary fees to maintain our patents.
Moreover, any of our pending applications may be subject to a third party pre-issuance submission of prior art to the U.S. Patent and
Trademark Office (“USPTO”), the European Patent Office , the Intellectual Property Office in the United Kingdom (“IPO”), IP Australia, the Australian Government agency that administers intellectual property rights and legislation in Australia, and/or
any patents issuing thereon may become involved in opposition, derivation, reexamination, inter parties review, post grant review, interference proceedings or other patent office proceedings or litigation, in the United States or elsewhere,
challenging our patent rights. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, and allow third parties to commercialize our technology or products and compete
directly with us, without payment to us. In addition, if the breadth or strength of protection provided by our patents and patent applications is threatened, it could dissuade companies from collaborating with us to exploit our intellectual property
or develop or commercialize current or future drug candidates.
The issuance of a patent is not conclusive as to the inventorship, scope, validity or enforceability, and our patents may be challenged in
the courts or patent offices in the United States, the European Union, Australia and elsewhere. Such challenges may result in loss of ownership or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could
limit the duration of the patent protection of our technology and products. As a result, our patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
In addition, other companies may attempt to circumvent any regulatory data protection or market exclusivity that we obtain under applicable
legislation, which may require us to allocate significant resources to preventing such circumvention. Such developments could enable other companies to circumvent our intellectual property rights and use our clinical trial data to obtain marketing
authorizations in the European Union, Australia and in other jurisdictions. Such developments may also require us to allocate significant resources to prevent other companies from circumventing or violating our intellectual property rights.
38
Intellectual property rights of third parties could adversely affect our ability to commercialize our drug candidates,
such that we could be required to litigate with or obtain licenses from third parties in order to develop or market our drug candidates.
Our commercial success may depend upon our future ability and the ability of our potential collaborators to develop, manufacture, market
and sell our drug candidates without infringing valid intellectual property rights of third parties.
If a third-party intellectual property right exists it may require the pursuit of litigation or administrative proceedings to nullify or
invalidate the third-party intellectual property right concerned, or entry into a license agreement with the intellectual property right holder, which may not be available on commercially reasonable terms, if at all.
Third-party intellectual property right holders, including our competitors, may bring infringement claims against us. We may not be able to
successfully settle or otherwise resolve such infringement claims. If we are unable to successfully settle future claims or otherwise resolve such claims on terms acceptable to us, we may be required to engage in or continue costly, unpredictable and
time-consuming litigation and may be prevented from, or experience substantial delays in, marketing our drug candidate.
If we fail to settle or otherwise resolve any such dispute, in addition to being forced to pay damages, we or our potential collaborators
may be prohibited from commercializing any drug candidates we may develop that are held to be infringing, for the duration of the patent term. We might, if possible, also be forced to redesign our formulations so that we no longer infringe such
third-party intellectual property rights. Any of these events, even if we were ultimately to prevail, could require us to divert substantial financial and management resources that we would otherwise be able to devote to our business.
Our reliance on third parties requires us to share our trade secrets, which increases the possibility that a competitor
will discover them or that our trade secrets will be misappropriated or disclosed.
Because we collaborate with various organizations and academic institutions on the advancement of our technology and drug candidates, we
may, at times, share trade secrets with them. We seek to protect our proprietary technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, collaborative research agreements, consulting agreements
or other similar agreements with our collaborators, advisors, employees and consultants prior to beginning research or disclosing proprietary information. These agreements typically limit the rights of the third parties to use or disclose our
confidential information, such as trade secrets. Despite these contractual provisions, the need to share trade secrets and other confidential information increases the risk that such trade secrets become known by potential competitors, are
inadvertently incorporated into the technology of others, or are disclosed or used in violation of these agreements. Given that our proprietary position is based, in part, on our know-how and trade secrets, discovery by a third party of our trade
secrets or other unauthorized use or disclosure would impair our intellectual property rights and protections in our drug candidates.
In addition, these agreements typically restrict the ability of our collaborators, advisors, employees and consultants to publish data
potentially relating to our trade secrets. Our academic collaborators typically have rights to publish data, provided that we are notified in advance and may delay publication for a specified time in order to secure our intellectual property rights
arising from the collaboration. In other cases, publication rights are controlled exclusively by us. In other cases, we may share these rights with other parties. Despite our efforts to protect our trade secrets, our competitors may discover our
trade secrets, either through breach of these agreements, independent development or publication of information including our trade secrets in cases where we do not have proprietary or otherwise protected rights at the time of publication.
39
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee
payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and applications are required to be
paid to the USPTO and other governmental patent agencies outside of the United States in several stages over the lifetime of the patents and applications. The USPTO and various corresponding governmental patent agencies outside of the United States
require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process and after a patent has issued. There are situations in which non-compliance can result in abandonment or lapse
of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction.
We may become involved in lawsuits to protect and defend our patents or other intellectual property, which could be
expensive, time consuming and unsuccessful.
Competitors may infringe our patents or other intellectual property and we may inadvertently infringe the patent or intellectual property
of others. To counter infringement or unauthorized use, we may be required to file claims, and any related litigation and/or prosecution of such claims can be expensive and time consuming. Any claims we assert against perceived infringers could
provoke these parties to assert counterclaims against us alleging that we infringe their intellectual property. In addition, in a patent infringement proceeding, a court may decide that a patent of ours is invalid in whole or in part, unenforceable,
or construe the patent’s claims narrowly allowing the other party to commercialize competing products on the grounds that our patents do not cover such products.
Even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims may cause us to incur
significant expenses and could distract our technical and management personnel from their normal responsibilities. Such litigation or proceedings could substantially increase our operating losses and reduce our resources available for development
activities. We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because
of their substantially greater financial resources. The effects of patent litigation or other proceedings could therefore have a material adverse effect on our ability to compete in the marketplace.
Confidentiality and invention assignment agreements with our employees, advisors and consultants may not adequately
prevent disclosure of trade secrets and protect other proprietary information.
We consider proprietary trade secrets and/or confidential know-how and unpatented know-how to be important to our business. We may rely on
trade secrets and/or confidential know-how to protect our technology, especially where patent protection is believed by us to be of limited value. However, trade secrets and/or confidential know-how can be difficult to maintain as confidential.
To protect this type of information against disclosure or appropriation by competitors, our policy is to require our employees, advisors
and consultants to enter into confidentiality and invention assignment agreements with us. However, current or former employees, advisors and consultants may unintentionally or willfully disclose our confidential information to competitors, and
confidentiality and invention assignment agreements may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. Enforcing a claim that a third party obtained illegally and is using trade secrets and/or
confidential know-how is expensive, time consuming and unpredictable. The enforceability of confidentiality and invention assignment agreements may vary from jurisdiction to jurisdiction.
Failure to obtain or maintain trade secrets and/or confidential know-how trade protection could adversely affect our competitive position.
Moreover, our competitors may independently develop substantially equivalent proprietary information and may even apply for patent protection in respect of the same. If successful in obtaining such patent protection, our competitors could limit our
use of our trade secrets and/or confidential know-how.
40
Intellectual property rights do not address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have
limitations, and may not adequately protect our business, or permit us to maintain our competitive advantage. The following examples are illustrative:
| • |
Others may be able to make products that are similar to ours but that are not covered by our intellectual property rights.
|
| • |
Others may independently develop similar or alternative technologies or otherwise circumvent any of our technologies without infringing our intellectual property rights.
|
| • |
We or any of our collaboration partners might not have been the first to conceive and reduce to practice the inventions covered by the patents or patent applications that we own, license or
will own or license.
|
| • |
We or any of our collaboration partners might not have been the first to file patent applications covering certain of the patents or patent applications that we or they own or have obtained a
license, or will own or will have obtained a license.
|
| • |
It is possible that any pending patent applications that we have filed, or will file, will not lead to issued patents.
|
| • |
Issued patents that we own may not provide us with any competitive advantage, or may be held invalid or unenforceable, as a result of legal challenges by our competitors.
|
| • |
Our competitors might conduct research and development activities in countries where we do not have patent rights, or in countries where research and development safe harbor laws exist, and
then use the information learned from such activities to develop competitive products for sale in our major commercial markets.
|
| • |
Ownership of our patents or patent applications may be challenged by third parties.
|
| • |
The patents of third parties or pending or future applications of third parties, if issued, may have an adverse effect on our business.
|
We may face difficulties with protecting our intellectual property in certain jurisdictions, which may diminish the
value of our intellectual property rights in those jurisdictions.
The laws of some jurisdictions do not protect intellectual property rights to the same extent as the laws in the United States and the
European Union, and many companies have encountered significant difficulties in protecting and defending such rights in such jurisdictions. If we or our collaboration partners encounter difficulties in protecting, or are otherwise precluded from
effectively protecting, the intellectual property rights important for our business in such jurisdictions, the value of these rights may be diminished and we may face additional competition from others in those jurisdictions.
Some countries in Europe and China have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third
parties. In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent.
If we are, or any of our licensors is, forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position or commercial advantage may be impaired and our business and results of operations may be
adversely affected.
41
Price controls may be imposed in markets in which we operate, which may negatively affect our future profitability.
In some countries, particularly EU member states, Japan, Australia and Canada, the pricing of prescription drugs is subject to governmental
control. In these countries, pricing negotiations with governmental authorities can take considerable time after receipt of marketing approval for a product. In addition, there can be considerable pressure by governments and other stakeholders on
prices and reimbursement levels, including as part of cost containment measures. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained.
Reference pricing used by various EU member states and parallel distribution, or arbitrage between low-priced and high-priced member states, can further reduce prices. In some countries, we or our collaborators may be required to conduct a clinical
trial or other studies that compare the cost-effectiveness of our drug candidates to other available therapies in order to obtain or maintain reimbursement or pricing approval. Publication of discounts by third-party payors or authorities may lead to
further pressure on the prices or reimbursement levels within the country of publication and other countries. If reimbursement of our drug candidates is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our
business, revenues or profitability could be harmed.
Risks Relating to this Offering
The trading price of the Shares may be volatile, and purchasers of the Shares could incur substantial losses.
The market price of our Shares may be subject to significant fluctuations due to factors specific to us, to changes in analysts’
recommendations and earnings estimates, to potential arbitrage between our Australian-listed Shares and Nasdaq-listed Shares, to changes in exchange rates, or to factors affecting the biopharmaceutical industry or the securities markets in general.
Market fluctuations, as well as general political and economic conditions, such as a recession, interest rate or currency fluctuations, could adversely affect the market price of our securities.
We may experience a material decline in the market price of our shares, regardless of our operating performance. Therefore, a holder of our
Shares may not be able to sell those Shares at or above the price paid by such holder for such Shares. Price declines in our Shares could result from a variety of factors, including many outside our control. These factors include:
| • |
the results of pre-clinical testing and clinical trials by us and our competitors;
|
| • |
unforeseen safety issues or adverse side effects resulting from the clinical trials or the commercial use of our drug candidate;
|
| • |
regulatory actions in respect of any of our drug candidates or the products of any of our competitors;
|
| • |
announcements of the introduction of new products by us or our competitors;
|
| • |
market conditions, including market conditions in the pharmaceutical and biotechnology sectors;
|
| • |
increases in our costs or decreases in our revenues due to unfavorable movements in foreign currency exchange rates;
|
| • |
developments or litigation concerning patents, licenses and other intellectual property rights;
|
| • |
litigation or public concern about the safety of our drug candidates;
|
| • |
changes in recommendations or earnings estimates by securities analysts;
|
| • |
actual and anticipated fluctuations in our quarterly operating results;
|
42
| • |
deviations in our operating results from the estimates of securities analysts;
|
| • |
rumors relating to us or our competitors;
|
| • |
additions or departures of key personnel;
|
| • |
changes in third party reimbursement policies; and
|
| • |
developments concerning current or future strategic alliances or acquisitions.
|
In addition, volatility and low market price of our Shares may adversely impact investors’ interest in our securities. A decline in
investors’ interest may prompt further volatility and decrease in market price.
There has been no prior active market for the Shares in the United States and an active and liquid market for our
securities may fail to develop.
While our Shares have been listed on the ASX prior to this offering, there has been no active public market on a U.S. national securities
exchange for our Shares. Although we have applied for the listing of the Shares on Nasdaq, an active trading market for the Shares may never develop or be sustained following this offering. The initial offering price of the Shares will be determined
through negotiations between us and the underwriter and will be based, in part, on prevailing market prices of our Shares on the ASX, after taking into account market conditions and other factors. This offering price may not be indicative of the
market price of the Shares after this offering. In the absence of an active trading market for the Shares, investors may not be able to sell their Shares at or above the offering price or at the time that they would like to sell.
If we are or become a passive foreign investment company (“PFIC” ), then that would subject our U.S. investors to
adverse tax rules.
Holders of our Shares who are U.S. taxpayers (a U.S. Holder) will be subject to particular income tax rules if we are a passive foreign
investment company (“PFIC”). These rules could result in a reduction in the after-tax return to a U.S. Holder of our Shares and reduce the value of our Shares. For U.S. federal income tax purposes, we will be classified as a PFIC for any taxable year
in which (i) 75% or more of our gross income is passive income, or (ii) at least 50% of the average value of all of our assets for the taxable year produce or are held for the production of passive income. For this purpose, cash is considered to be
an asset that produces passive income.
If we are classified as a PFIC in any year that a U.S. Holder owns Shares, the U.S. Holder will generally continue to be treated as holding
Shares of a PFIC in all subsequent years, notwithstanding that we are not classified as a PFIC in a subsequent year. Dividends received by the U.S. Holder and gains realized from the sale of our Shares would be taxed as ordinary income and subject to
an interest charge. We urge U.S. investors to consult their own tax advisors about the application of the PFIC rules and certain elections that may help to minimize adverse U.S. federal income tax consequences in their particular circumstances.
43
The requirements of being a public company may strain our resources and divert management’s attention and if we are
unable to maintain effective internal control over financial reporting in the future, the accuracy and timeliness of our financial reporting may be adversely affected.
As a U.S. publicly traded company, we will be subject to the reporting requirements of the the Exchange Act, the Sarbanes-Oxley Act, the
Dodd-Frank Act, the listing requirements of Nasdaq and other applicable securities rules and regulations. Compliance with these rules and regulations will increase our legal and financial compliance costs, make some activities more difficult,
time-consuming or costly and increase demand on our systems and resources. The Exchange Act requires, among other things, that we file certain reports with respect to our business and results of operations. The Sarbanes-Oxley Act requires that we
maintain effective disclosure controls and procedures and internal control over financial reporting. In order to maintain and, if required, improve our disclosure controls and procedures and internal control over financial reporting to meet this
standard, significant resources and management oversight are required. As a result, management’s attention may be diverted from other business concerns, which could adversely affect our business and results of operations.
The Sarbanes-Oxley Act requires that we assess the effectiveness of our internal control over financial reporting annually and disclosure
controls and procedures quarterly. If we identify material weaknesses in future periods or we are not able to comply with the requirements of Section 404 in a timely manner, our reported financial results could be restated, we could be subject to
investigations or sanctions by regulatory authorities, which would require additional financial and management resources, and the market price of our Shares could decline.
We are eligible to be treated as an “emerging growth company” as defined in the JOBS Act, and we cannot be certain if
the reduced disclosure requirements applicable to emerging growth companies will make our Shares less attractive to investors.
We are an “emerging growth company” as defined in the JOBS Act. For as long as we continue to be an emerging growth company, we may take
advantage of exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies, including (i) not being required to comply with the auditor attestation requirements of Section 404 of
the Sarbanes-Oxley Act of 2002, (ii) reduced disclosure obligations regarding executive compensation in this registration statement and periodic reports and proxy statements, and (iii) exemptions from the requirements of holding a nonbinding advisory
vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. We could be an emerging growth company for up to five years, although circumstances could cause us to lose that status earlier,
including if the market value of our Shares held by non-affiliates exceeds US$700 million as of July 31, 2022, or if we have total annual gross revenue of US$1.235 billion or more during any fiscal year before that time, in which case we would no
longer be an emerging growth company as of the following October 31. Additionally, if we issue more than US$1.0 billion in non-convertible debt during any three-year period before July 31, 2022, we would cease to be an emerging growth company
immediately. We cannot predict if investors will find our Shares less attractive because we may rely on these exemptions. If some investors find our Shares less attractive as a result, there may be a less active trading market for our Shares, and the
stock price may be more volatile.
Under the JOBS Act, emerging growth companies can also delay adopting new or revised accounting standards until such time as those
standards apply to private companies. We have elected to take advantage of the extended transition period allowed for emerging growth companies for complying with new or revised accounting guidance as allowed by Section 107 of the JOBS Act and
Section 7(a)(2)(B) of the Securities Act.
There is no public market for the Pre-Funded Warrants or the Share Purchase Warrants being offered by us in this
offering.
There is no established public trading market for the Pre-Funded Warrants or the Share Purchase Warrants and we do not expect a market to
develop. In addition, we do not intend to apply to list the Pre-Funded Warrants or the Share Purchase Warrants on any national securities exchange or other nationally recognized trading system, including Nasdaq. Without an active market, the
liquidity of the Pre-Funded Warrants and the Share Purchase Warrants will be limited, which may adversely affect their value.
44
Holders of Pre-Funded Warrants or Share Purchase Warrants purchased in this offering will have no rights as
shareholders until such holders exercise their Pre-Funded Warrants or Share Purchase Warrants and acquire our Shares.
Until holders of Pre-Funded Warrants or Share Purchase Warrants acquire our Shares upon exercise thereof, such holders will have no rights
with respect to our Shares underlying the Pre-Funded Warrants and Share Purchase Warrants. Upon exercise of the Pre-Funded Warrants or Share Purchase Warrants, the holders will be entitled to exercise the rights of a shareholder only as to matters
for which the record date occurs after the exercise date.
The Share Purchase Warrants and Pre-Funded Warrants are speculative in nature.
The Share Purchase Warrants and Pre-Funded Warrants do not confer any rights of Share ownership on its holders, such as voting rights or
the right to receive dividends, but rather merely represent the right to acquire Shares at a fixed price for a limited period of time. Following this offering, the market value of the Share Purchase Warrants and Pre-Funded Warrants, if any, is
uncertain and there can be no assurance that the market value of the Share Purchase Warrants and Pre-Funded Warrants will equal or exceed their imputed offering price. The Share Purchase Warrants and Pre-Funded Warrants will not be listed or quoted
for trading on any market or exchange. There can be no assurance that the market price of the Shares will ever equal or exceed the exercise price of the Share Purchase Warrants and Pre-Funded Warrants, and consequently, it may not ever be profitable
for holders of the Share Purchase Warrants and Pre-Funded Warrants to exercise the Share Purchase Warrants or the Pre-Funded Warrants.
You will experience immediate and substantial dilution in the net tangible book value of the Shares you purchase in
this offering.
The initial public offering price of the Shares is substantially higher than the net tangible book value per Share immediately after this
offering. If you purchase Shares in this offering, you will suffer immediate dilution of US$ per Share, or US$ per Share if the underwriter exercises its option to purchase additional shares in full, representing the difference
between our as adjusted net tangible book value per Share or per Share after giving effect to the sale of Shares in this offering and the initial public offering price of US$ per Share. See “Dilution.”
Our issuance of additional Shares in connection with financings, acquisitions, investments, or otherwise will dilute
all other Shareholders.
We expect to issue additional Shares in the future that may result in dilution to all other Shareholders. We may also raise capital through
equity financings in the future. As part of our business strategy, we may acquire or make investments in complementary companies, products, or technologies and issue equity securities to pay for any such acquisition or investment. While we will be
subject to the constraints of the ASX Listing Rules regarding the percentage of our capital that we are able to issue within a 12-month period (subject to applicable exceptions), any such issuances of additional Shares may cause Shareholders to
experience significant dilution of their ownership interests and the per Share value of our Shares to decline.
45
As long as we remain subject to the rules of the ASX and Nasdaq, we will be unable to access equity capital without
shareholder approval if such equity capital sales would result in an equity issuance above regulatory thresholds and, consequently, we could be unable to obtain financing sufficient to sustain our business if we are unsuccessful in soliciting
requisite shareholder approvals.
Our ability to access equity capital is currently limited by ASX Listing Rule 7.1, which provides that a company must not, subject to
specified exceptions, issue or agree to issue during any consecutive 12-month period any equity securities, or other securities with rights to conversion to equity, if the number of those securities in aggregate would exceed 15% of the number of
ordinary securities on issue at the commencement of that 12-month period, without first obtaining approval from the holders of its ordinary securities. Therefore, if the equity we seek to issue is greater than 15% of the number of Shares on issue at
the time of issuance, we will require shareholder approval via an extraordinary general meeting or annual general meeting prior to the new capital issue, unless an exception applies.
Our equity issuances will be limited by ASX Listing Rule 7.1 as long as we continue to be listed on the ASX and this constraint may prevent
us from raising the full amount of equity capital needed for operations without prior shareholder approval.
Our ability to access equity capital is also limited by Nasdaq rule 5625(d), which provides that shareholder approval is required prior to
a 20% issuance at a price that is less than the minimum price. Nasdaq defines “20% issuance” for purposes of Rule 5635(d) as a private placement transaction involving the sale, issuance, or potential issuance by the issuer of Shares (or securities
convertible into or exercisable for Shares), which, alone or together with sales by officers, directors, or substantial shareholders (persons with more than 5% of the number of securities or voting power outstanding) of the issuer, equals 20% or more
of the Shares or 20% or more of the voting power outstanding before the issuance. To the extent we remain a foreign private issuer at the time of such issuance, this rule may not apply to us.
We have broad discretion in the use of the net proceeds from this offering and may not use them effectively.
We will have broad discretion in the application of the net proceeds that we receive from this offering as well as of our existing cash,
and we may spend or invest these funds in a way with which our shareholders or holders of the Shares disagree. Our failure to apply these funds effectively could harm our business and financial condition. Pending their use, we may invest the net
proceeds from the offering in a manner that does not produce income or that loses value. These investments may not yield a favorable return to our investors.
The dual listing of our Shares following this offering may negatively impact the liquidity and value of the Shares.
Following this offering and after the Shares are listed on Nasdaq, our Shares will continue to be listed on the ASX. We cannot predict the
effect of this dual listing on the value of our Shares. However, the dual listing of our Shares may dilute the liquidity of these securities in one or both markets and may negatively impact the development of an active trading market for the Shares
in the United States. The price of the Shares could also be negatively impacted by trading in our Shares on the ASX.
We are subject to risks associated with currency fluctuations, and changes in foreign currency exchange rates could
impact our results of operations.
Our Shares are quoted in Australian dollars on the ASX and the Shares listed on Nasdaq will be quoted in U.S. dollars. In the past year,
the Australian dollar has generally weakened against the U.S. dollar; however, this trend may not continue and may be reversed. As such, any significant change in the value of the Australian dollar may have a negative effect on the value of the
Shares in U.S. dollars. In addition, if the Australian dollar weakens against the U.S. dollar, then, if we decide to convert our Australian dollars into U.S. dollars for any business purpose, appreciation of the U.S. dollar against the Australian
dollar would have a negative effect on the U.S. dollar amount available to us. To the extent that we need to convert U.S. dollars we receive from this offering into Australian dollars for our operations, appreciation of the Australian dollar against
the U.S. dollar would have a negative effect on the Australian dollar amount we would receive from the conversion.
46
Risks Relating to Our Location in Australia
Australian takeovers laws may discourage takeover offers being made for us or may discourage the acquisition of large
numbers of our shares.
We are incorporated in Australia and are subject to the takeover laws of Australia. Amongst other things, we are subject to the Australian
Corporations Act 2001 (the “Corporations Act”). Subject to a range of exceptions, the Corporations Act prohibits the acquisition of a direct or indirect interest in our issued voting shares if the acquisition of that interest will lead to a person’s
or someone else’s voting power in us increasing from 20% or below to more than 20% or increasing from a starting point that is above 20% and below 90%. Exceptions to the general prohibition include circumstances where the person makes a formal
takeover bid for us, if the person obtains shareholder approval for the acquisition or if the person acquires no more than 3% of the voting power of us in any rolling six-month period. Australian takeovers laws may discourage takeover offers being
made for us or may discourage the acquisition of large numbers of our Shares.
Holders of our Shares may have difficulty in effecting service of process in the United States or enforcing judgments
obtained in the United States.
Holders of our Shares may have difficulties enforcing, in actions brought in courts in jurisdictions located outside the U.S., liabilities
under U.S. securities laws. In particular, if such a holder sought to bring proceedings in Australia based on U.S. securities laws, the Australian court might consider:
| • |
that it did not have jurisdiction; and/or
|
| • |
that it was not an appropriate forum for such proceedings; and/or
|
| • |
that, in the event of a conflict between Australian law and U.S. law (including U.S. securities laws), U.S. law should not be applied; and/or
|
| • |
that the U.S. securities laws were of a public or penal nature and should not be enforced by the Australian court.
|
Holders of our Shares may also have difficulties enforcing in courts outside the U.S. judgments obtained in the U.S. courts against any of
our directors and executive officers or us, including actions under the civil liability provisions of the U.S. securities laws.
As a foreign private issuer whose shares are listed on Nasdaq, we may follow certain home country corporate governance
practices instead of certain Nasdaq requirements.
As a foreign private issuer whose shares are listed on Nasdaq, we are permitted to follow certain home country corporate governance
practices instead of certain requirements of the Nasdaq Marketplace Rules. As an Australian company listed on Nasdaq, we may follow home country practice with regard to, among other things, the composition of the board of directors, director
nomination process, compensation of officers and quorum at shareholders’ meetings. In addition, we may follow Australian law instead of the Nasdaq Marketplace Rules that require that we obtain shareholder approval for certain dilutive events, such as
for the establishment or amendment of certain equity-based compensation plans, an issuance that will result in a change of control of the company, certain transactions other than a public offering involving issuances of a 20% or more interest in the
company and certain acquisitions of the shares or assets of another company. As a foreign private issuer that has elected to follow a home country practice instead of Nasdaq requirements. Accordingly, our shareholders may not be afforded the same
protection as provided under Nasdaq’s corporate governance rules that are applicable to U.S. companies.
47
As a foreign private issuer, we are exempt from a number of rules under the U.S. securities laws and are permitted to
file less information with the SEC than a U.S. company.
We are a foreign private issuer (as defined in the SEC’s rules) and, consequently, we are not subject to all of the disclosure requirements
applicable to public companies organized within the United States. For example, we are exempt from certain rules under the Exchange Act that regulate disclosure obligations and procedural requirements related to the solicitation of proxies under the
Exchange Act. In addition, our senior management and directors are exempt from the reporting and “short-swing” profit recovery provisions of Section 16 of the Exchange Act and related rules with respect to their purchases and sales of our securities.
Moreover, while we currently make periodic filings with respect to our listing on the ASX and expect to file financial reports on an annual and semi-annual basis, we will not be required to file periodic reports and financial statements with the SEC
as frequently or as promptly as U.S. public companies and will not be required to file quarterly reports on Form 10-Q or current reports on Form 8-K under the Exchange Act. In addition, foreign private issuers are not required to file their annual
report on Form 20-F until four months after the end of each fiscal year. Accordingly, there may be less publicly available information concerning our company than there would be if we were not a foreign private issuer.
Any loss of our foreign private issuer status in the future could result in significant additional cost.
While we currently qualify as a foreign private issuer, the determination of foreign private issuer status is made annually on the last
business day of an issuer’s most recently completed second fiscal quarter. In the future, we would lose our foreign private issuer status if we to fail to meet the requirements necessary to maintain our foreign private issuer status as of the
relevant determination date. For example, if 50% or more of our securities are held by U.S. residents and the majority of our the executive offices or directors are United States citizens or residents, we could lose our foreign private issuer status.
U.S. investors may have difficulty enforcing civil liabilities against our company, our directors or members of senior
management and the experts named in this prospectus.
Certain members of our senior management and board of directors named in this prospectus are non-residents of the United States, and a
substantial portion of the assets of such persons are located outside the United States. As a result, it may be impracticable to serve process on such persons in the United States or to enforce judgments obtained in U.S. courts against them based on
civil liability provisions of the securities laws of the United States. Even if you are successful in bringing such an action, there is doubt as to whether Australian courts would enforce certain civil liabilities under U.S. securities laws in
original actions or judgments of U.S. courts based upon these civil liability provisions. In addition, awards of punitive damages in actions brought in the United States or elsewhere may be unenforceable in Australia or elsewhere outside the United
States. An award for monetary damages under U.S. securities laws would be considered punitive if it does not seek to compensate the claimant for loss or damage suffered and is intended to punish the defendant. The enforceability of any judgment in
Australia will depend on the particular facts of the case as well as the laws and treaties in effect at the time. The United States and Australia do not currently have a treaty or statute providing for recognition and enforcement of the judgments of
the other country (other than arbitration awards) in civil and commercial matters.
As a result, our public shareholders may have more difficulty in protecting their interests through actions against us, our management or
our directors than would shareholders of a corporation incorporated in a jurisdiction in the United States. In addition, as a company incorporated in Australia, the provisions of the Corporations Act regulate the circumstances in which shareholder
derivative actions may be commenced which may be different, and in many ways less permissive, than for companies incorporated in the United States.
48
USE OF PROCEEDS
We estimate that the net proceeds from this offering will be approximately US$ million, based upon an assumed initial public offering
price of US$ per Share Unit (or US$ per Pre-Funded Warrant Unit), without deducting underwriting discounts and commissions and estimated offering expenses payable by us and assuming none of the Warrants issued in this offering are exercised.
If the underwriter exercises its over-allotment option to purchase additional Shares and/or Pre-Funded Warrants from us in full, we estimate that the gross proceeds will be approximately US$ million without deducting underwriting discounts and
commissions and estimated offering expenses payable by us.
We expect to complete the Phase III clinical trials for NanaBis™ in the United States using the net proceeds from this offering, together with the Australian Research and
Development Tax Incentive and our existing cash. We expect that approximately US$8 million of the net proceeds will be allocated to NanaBis™ clinical trials in 2023 and completion of product development CMC packages for NanaBis™ and partial
completion of product development CMC packages for NanoCBD™.
However, due to the uncertainties inherent in the product development process, it is difficult to estimate with certainty the exact amounts
of the net proceeds from this offering that may be used for the above purposes. Our management will have broad discretion over the use of the net proceeds from this offering. The amounts and timing of our expenditures will depend upon numerous
factors including the results of our research and development efforts, the timing and success of preclinical studies and any ongoing clinical trials or clinical trials we may commence in the future, the timing of regulatory submissions and the amount
of cash obtained through future collaborations, if any.
We believe opportunities may exist from time to time to expand our current business through acquisitions or in-licensing of complementary
companies, medicines or technologies. While we have no current agreements, commitments or understandings for any specific acquisitions or in-licensing at this time, we may use a portion of the net proceeds for these purposes.
49
DIVIDEND POLICY
We have never declared or paid any dividends on our Shares or any of our securities. We intend to retain any earnings for use in our
business and do not currently intend to pay cash dividends on our Shares. Dividends, if any, on our outstanding Shares will be declared by and subject to the discretion of our board of directors, and subject to Australian law.
Any dividend we declare will be paid to the holders of our Shares, to the extent permitted by applicable law and regulations, less the fees
and expenses payable under the deposit agreement.
50
CAPITALIZATION
The following table sets forth our cash and our capitalization as of , 2022, after giving effect to the Share Consolidation:
| • |
on an actual basis; and
|
| • |
on an as adjusted basis to give effect to the sale of securities offered hereby at the assumed combined offering price of US$ per Share Unit (US$ per Pre-Funded Warrant Unit), without
deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. The assumed offering price may not be the final price of this offering and will be adjusted based on the offering price and other terms
of the offering determined at pricing.
|
You should read this information in conjunction with our consolidated financial statements and the related notes included elsewhere in this
prospectus, the information set forth in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and other financial information contained elsewhere in this prospectus.
51
DILUTION
Investors purchasing Share Units and/or Pre-Funded Warrant Units in this offering will experience immediate and substantial dilution in the
as adjusted net tangible book value of their Shares. Dilution in as adjusted net tangible book value represents the difference between the assumed public offering price per Share and the as adjusted net tangible book value per Share immediately after
the offering.
The historical net tangible book value of our Shares as of June 30, 2022 was US$ or US$ per Share after giving effect to the Share
Consolidation. Historical net tangible book value per Share represents our total tangible assets (total assets less intangible assets) less total liabilities divided by the number of Shares outstanding as of that date after giving effect to the Share
Consolidation.
After giving effect to the sale of Share Units or Pre-Funded Warrant Units in this offering and excluding the Shares issuable upon the
exercise of the Share Purchase Warrants being offered in this offering at the assumed offering price of US$ per Share Unit (US$ per Pre-Funded Warrant Unit), and without deducting estimated underwriting discounts and commissions and estimated
offering expenses payable by us, our net tangible book value as of June 30, 2022 would have been US$ , or US$ per Share. The assumed offering price may not be the final price of this offering and will be adjusted based on the actual offering
price and other terms of the offering determined at pricing. This amount represents an immediate increase in net tangible book value of US$ per share to our existing shareholders and an immediate dilution in net tangible book value of
approximately US$ per share to new investors purchasing our Shares in this offering.
The following table illustrates this dilution on a per Share basis, assuming all Shares outstanding as of , 2022, and after
giving effect to the Share Consolidation:
|
Assumed initial public offering price per Share Unit / Pre-Funded Warrant Unit
|
|
|
|
US$
|
/
|
US$
|
|
Historical net tangible book value per Share as of , 2022
|
|
|
|
US$
|
|
|
|
Increase in net tangible book value per Share attributed to investors purchasing Shares in this offering
|
|
|
|
|
|
|
|
As adjusted net tangible book value per Share after this offering
|
|
|
|
|
|
|
|
Dilution in net tangible book value per Share to investors in this offering
|
|
|
|
US$
|
|
|
If the underwriter exercises its option to purchase additional Shares and/or Share Purchase Warrants, the as adjusted net tangible
book value of our Shares after this offering would be US$ or US$ per share representing an immediate increase in net tangible book value of approximately US$ per share to existing shareholders and an immediate dilution of US$ per
share to the investors in this offering, in each case assuming an initial public offering price of US$ per Share, without deducting the underwriting discount and estimated offering expenses payable by us.
The dilution information above is for illustration purposes only. Our as adjusted net tangible book value following the closing of this
offering will depend on the actual offering price and other terms of this offering determined at pricing. To the extent that outstanding options or warrants are exercised, you may experience further dilution. In addition, we may choose to raise
additional capital due to market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans. To the extent that additional capital is raised through the sale of equity or convertible
debt securities, the issuance of these securities may result in further dilution to our shareholders.
52
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following “Management’s Discussion and Analysis of Financial Condition and Results of Operations” should be read
together with the section titled “Selected Consolidated Financial Data” and our consolidated financial statements and the accompanying notes included elsewhere in this prospectus. This discussion includes both historical information and
forward-looking information based upon current expectations that involve risk, uncertainties, and assumptions. Our actual results may differ materially from management’s expectations as a result of various factors, including, but not limited to,
those discussed in “Risk Factors” and elsewhere in this prospectus.
Overview
We are an Australian biotechnology company that formulates innovative solutions and consumer health care products to provide integrative
patient care. We develop targeted fixed-dose combinations of the NanoCelle® delivery platform and various active pharmaceutical ingredients, applying proprietary insights to create long-term value for our shareholders. Our strategy is to expand our
market opportunity by gaining United States, United Kingdom, Australian and/or other regulatory approval for our products. NanoCelle®, our core product offering, is a pharmaceutical-delivery platform which is designed to administer pharmaceutical
products using nanoparticles, or small particles ranging between 1 to 100 nanometres in size and undetectable by the human eye. NanoCelle® is an alternative, and we believe, more effective method of consuming medical products, compared to the
traditional methods, which include intravenous, intramuscular, subcutaneous, oral (ingested, and sublingual), rectal, inhalation and transdermal. In addition to developing the NanoCelle® delivery method, we have also developed several drug programs
that are specifically designed to be administered through the NanoCelle® technology. Our NanoCelle® delivery method has produced additional drug candidates which share design and CMC processes with our lead candidate, resulting in
tolerability profiles that may be similar to our lead product candidate, as well as regulatory agency familiarity, and a consistent multi-target therapeutic approach.
On October 19, 2021, the Company licensed its Australian nutraceutical business to PharmaCare for cash consideration of A$750,000 (the “Nutraceutical License”). The principal
assets that were licensed comprised registered patents and trademarks, inventory, customer lists, and material contracts. In connection with the licensing deal, the Company sold its nutraceutical inventory to PharmaCare for $1,025,910. Following
the Nutraceutical License, the Company continued to maintain its rights to market and sell its nutraceutical lines of business outside of Australia. The purpose of the Nutraceutical License was to restructure our business to concentrate on
developing and producing pharmaceutical products that are compatible with our NanoCelle® delivery method.
We have experienced net losses in every period since our inception in 2012. We incurred net losses of A$(7,228,814) and A$(12,402,829) for
the years ended June 30, 2022, and 2021, respectively. As of June 30, 2022, we had accumulated losses of A$(59,529,093). We anticipate that we will continue to incur significant losses for the foreseeable future as our operating expenses and
capital expenditures increase substantially due to our continued investment in research and development as well as in sales and marketing activities and as we hire additional employees over the coming years. Furthermore, upon closing of this
offering, we expect to incur additional expenses associated with operating as a U.S. public company, including significant legal, accounting, investor relations and other expenses.
For more information regarding our business and operations, see the section entitled “Business” below.
Components of Operating Results
Sales
Our revenues from continuing operations were A$1,536,366 and A$732,727 for the years ended June 30, 2022, and 2021, respectively.
Currently, the vast majority of our business activity is in Australia. As a result, our revenues are derived mainly from our patient
special access and compassionate use programs for our NanaBis™ and NanoCBD™ products. This is a significant change, as in comparative year sales, a larger proportion was attributed to nutraceuticals. The other revenue we
report in our accounts is the cash rebate from the Australian Federal Government for research and development spend pursuant to the Australian Research and Development Tax Incentive.
53
Cost of Sales
Our cost of sales was A$336,292 and A$16,425 for the years ended June 30, 2022, and 2021, respectively. Most of the cost of sales expenses
consists of the cost of materials and cost of shipments of the materials.
For the year ended June 30, 2022, cost of sales included costs of inventory for nutraceuticals up until November 2021, including any
write-offs and sale of inventory to PharmaCare in connection with the Nutraceutical License.
Research and development expenses
Our research and development expenses were A$1,503,443 and A$2,102,751 for the years ended June 30, 2022, and 2021, respectively.
| • |
Research and development expenses include the following items:
|
| • |
employee-related expenses, such as salaries and any other cash remuneration.
|
| • |
expenses relating to outsourced and contracted services, such as consulting and advisory services; and
|
| • |
costs associated with regulatory compliance.
|
We recognize research and development expenses as we incur them.
Our research and development efforts relating to our products have been financed in part through research and development grants in an
aggregate amount of A$3,688,000 received from the Australian Federal Government, as of June 30, 2022, which provides us with a cash grant each year in an amount based on the total amounts expensed to research and development related costs.
Currently, the annual grants are in an amount equal to approximately 43.5% of qualifying research and development expenses.
The qualifying research and development expenses on which the amount of our annual research and development grants are determined include items related to research and
development that exceed, by a material amount, the Research and Development Expenses reported in our audited financial statements. Expenses included in the calculation of the research and development grant
awards that are not included in our audited financial statements as research and development expenses include contract and consulting fees and expenses, compensation and payroll expenses, including compensation expenses related to the employment of
certain of our senior executive officers and other employees not engaged solely in scientific research, depreciation expenses, as well as product development costs, Special Access Scheme material expenditures and all clinical trial related
activities. The actual amount of qualifying research and development expenses on which the annual grant that we receive is calculated is determined by an outside consultant firm retained by us.
Personnel expenses
Our sales and personnel expenses were A$7,877,497 and A$6,016,686 for the years ended June 30, 2022, and 2021, respectively.
Sales and personnel expenses include the following:
| • |
employee-related expenses, such as salaries and share-based compensation; and
|
| • |
expenses relating to outsourced and contracted services, such as consulting.
|
54
General and administrative expenses
Our general and administrative expenses were A$3,952,633 and A$3,217,213 for the years ended June 30, 2022, and 2021, respectively.
General and administrative expenses consist primarily of personnel costs, including share-based compensation related to directors and
employees, facility costs, patent application and maintenance expenses, and external professional service costs, including legal, accounting, audit, finance, business development, investor relations and human resource services, and other consulting
fees.
We anticipate that our general and administrative expenses will increase in the future as we increase our administrative headcount and
infrastructure to support our continued research and development programs and the potential commercialization of our products. We also anticipate that we will incur increased expenses related to audit, legal, regulatory, and tax-related services
associated with maintaining compliance with Nasdaq and SEC requirements, director and officer insurance premiums, director compensation, and other costs associated with being a public company.
Finance expenses
Our finance expenses were A$101,916 and A$139,065 for the years ended June 30, 2022, and 2021, respectively.
The finance expenses consist primarily of exchange rate differences expenses and interest costs.
Income Taxes
As of June 30, 2022, our net operating loss carry forwards for tax purposes was A$1.642 million. We anticipate that we will continue to
generate losses for the foreseeable future and that we will be able to carry forward these losses for tax purposes indefinitely to future taxable years, subject to meeting certain requirements in the tax loss integrity rules each year. Accordingly,
subject to meeting those requirements, we do not expect to pay taxes in Australia until we have taxable income after the full utilization of our carry forward tax losses.
Results of Operations
Our results of operations have varied in the past and can be expected to vary in the future due to numerous factors. We believe that
period-to-period comparisons of our operating results are not necessarily meaningful and should not be relied upon as indications of future performance.
Below is a summary of our results of operations for the periods indicated:
|
|
Year Ended June 30,
|
|||||||
|
|
2022
|
2021
|
||||||
|
|
(AUD)
|
|||||||
|
Sales
|
$
|
1,536,366
|
$
|
732,727
|
||||
|
Cost of Sales
|
336,292
|
16,425
|
||||||
|
Gross Profit
|
1,200,074
|
716,302
|
||||||
|
Operating expenses:
|
||||||||
|
Personnel expenses
|
7,877,497
|
6,016,686
|
||||||
|
Research and development expenses
|
1,503,443
|
3,217,213
|
||||||
|
General and administrative expenses
|
3,952,633
|
2,102,751
|
||||||
|
Professional fees
|
-
|
1,731,430
|
||||||
|
Depreciation and amortization expenses
|
850,745
|
873,497
|
||||||
|
Loss from operations
|
(12,984,244
|
)
|
(13,225,275
|
)
|
||||
|
Other income and expenses:
|
||||||||
|
Research and development incentive
|
4,621,501
|
2,265,000
|
||||||
|
Gain on forgiveness of debt
|
-
|
-
|
||||||
|
Grant income
|
61,954
|
928,585
|
||||||
|
Legal settlement
|
-
|
500,000
|
||||||
|
Other expenses
|
-
|
-
|
||||||
|
Other income
|
20,655
|
6,454
|
||||||
|
Interest income
|
44,727
|
25,198
|
||||||
|
Loss on impairment of assets
|
-
|
-
|
||||||
|
Loss on foreign currency exchange
|
(51,129
|
)
|
6,120
|
|||||
|
Interest expense
|
(101,916
|
)
|
(139,065
|
)
|
||||
|
Other income and expenses
|
4,595,792
|
3,592,292
|
||||||
|
Income tax
|
-
|
-
|
||||||
|
Loss from continuing operations
|
(8,388,452
|
)
|
(9,632,983
|
)
|
||||
|
Loss from operations of discontinued operations
|
(36,178
|
)
|
(2,769,846
|
)
|
||||
|
Gain from disposition of discontinued operations
|
1,195,816
|
-
|
||||||
|
Foreign currency translation adjustments
|
(5,269
|
)
|
(15,649
|
)
|
||||
|
Total comprehensive loss for the period
|
$
|
(7,234,083
|
)
|
$
|
(12,418,478
|
)
|
||
|
|
||||||||
55
Year Ended June 30, 2022, Compared to Year Ended June 30, 2021
Revenues
Sales increased by A$803,639, or 109%, to A$1,536,366 for the year ended June 30, 2022, compared to A$737,727 for the year ended June 30,
2021. The increase resulted mainly from an improvement in sales of nutraceuticals prior to divestment to PharmaCare.
Cost of sales
Cost of sales increased by A$319,867, or 1947%, to A$336,292 for the year ended June 30, 2022, compared to A$16,425 for the year ended June
30, 2021. The increase resulted mainly from inventory adjustments at year end. For the year ended June 30, 2022, cost of sales included a write-down of inventory and sale of remaining nutraceutical inventory to PharmaCare.
Research and development expenses
Research and development expenses decreased by A$599,308, or 29%, to A$1,503,443 for the year ended June 30, 2022, compared to
A$2,102,751 for the year ended June 30, 2021. The increase resulted mainly from reduced research and development and personnel costs, reduction in payments to third-party CROs and phasing of product development costs into 2023.
General and administrative expenses
General and administrative expenses increased by A$735,420 or 23%, to A$3,952,633 for the year ended June 30, 2022, compared to A$3,217,213
for the year ended June 30, 2021. The increase resulted mainly from corporate-related costs such as public relations and compliance. We expect these expenses to increase in the future due to global inflationary pressures and as a result of becoming
a publicly listed company in the United States.
Other income (expenses)
Other income (expenses) increased by A$1,003,500 or 28%, to A$4,595,792 for the year ended June 30, 2022, compared to A$3,592,292 for the
year ended June 30, 2021. The increase resulted mainly from insurance, post-COVID travel, contractors, and reversal of 2021 provisions, such as doubtful debts.
Discontinued Operations
Total loss from operations of discontinued operations decreased by A$2,733,668 or 99%, to $36,178 for the year ended June 30, 2022,
compared to A$2,769,846 for the year ended June 30, 2021. The decrease resulted mainly from improvement in operational expenditure of nutraceuticals prior to divestment.
Net loss
Net loss decreased by A$5,174,015, or 42%, to A$7,228,814 for the year ended June 30, 2022, compared to A$12,402,829 for the year ended
June 30, 2021. The increase was mainly the result of improvement in operational expenditure outgoings, and reversal of provisions.
56
Impact of COVID-19
The global spread of COVID-19 led many countries to impose stringent limitations on movement, gatherings, transit of passengers and goods
and to close the borders between countries. The responses of governments have notably impacted many economies as well as capital markets worldwide. Our company was able to maintain almost ordinary levels of operations during the period. We were
affected by service providers lack of availability and extended times of delivery from suppliers abroad. Future possible impact of COVID-19 will depend on future developments with the pandemic which are highly uncertain and cannot be predicted,
including new information which may emerge concerning the severity of COVID-19 and the actions by governments around the world to contain COVID-19 or treat its impact, among others.
Going Concern
The Company’s consolidated financial statements are prepared using the U.S. GAAP applicable to a going concern, which contemplates the
realization of assets and liquidation of liabilities in the normal course of business. On June 30, 2022 and 2021, the Company had A$5,191,031 and A$13,434,762 in cash and A$6,748,692 and A$13,866,342 in working capital, respectively. For the years
ended June 30, 2022, and 2021, the Company had a net loss of A$7,228,814 and A$12,402,829, respectively. Continued losses may adversely affect the liquidity of the Company in the future.
In view of the matters described in the preceding paragraph, recoverability of a major portion of the recorded asset amounts shown in the
accompanying consolidated balance sheets is dependent upon continued operations of the Company, which in turn is dependent upon the Company’s ability to raise additional capital, obtain financing and in profitable future operations. The consolidated
financial statements do not include any adjustments relating to the recoverability and classification of recorded asset amounts or amounts and classification of liabilities that might be necessary should the Company be unable to continue as a going
concern.
Liquidity and Capital Resources
Overview
Though we do have sales from our products, we are primarily in research and product development phase and do not generate significant
revenue, except from research and development Australian federal grants special access pre-registration schemes. Accordingly, we have suffered recurring losses from operations. Our operations have been funded substantially through capital equity
raising. Our focus is on raising capital through equity financings to fund future clinical trials.
We believe the net proceeds of this offering, together with our current cash, will be sufficient to meet our working capital and capital expenditure requirements at least
through June 2023. Our future capital requirements will depend on many factors, including:
| • |
the progress and costs of our research and development activities, including for new technologies;
|
| • |
the costs of manufacturing our products, including increasing production capacity;
|
| • |
the costs of filing, prosecuting, enforcing and defending patent claims and other intellectual property rights;
|
| • |
the potential costs of contracting with third parties to provide marketing and distribution services for us or for building such capacities internally; and
|
| • |
the magnitude of our general and administrative expenses.
|
57
Until we can generate significant recurring revenues, profit and cash flow provided by operating activity, we expect to satisfy future cash
needs through debt or equity financings as well as governmental grants and proceeds from exercises of options and warrants. In the event that we require additional financing, we may not be able to raise such financing on terms acceptable to us or at
all. If we are unable to raise additional capital or generate cash flows necessary to expand our operations and invest in continued innovation, we may not be able to compete successfully, which would harm our business, results of operations, and
financial condition.
The table below shows a summary of our cash flows for the periods indicated:
|
|
Year Ended June 30,
|
|||||||
|
|
2022
|
2021
|
||||||
|
|
(AUD)
|
|||||||
|
Cash at beginning of the period
|
13,434,762
|
9,063,044
|
||||||
|
Net cash used in operating activities
|
(9,410,955
|
)
|
(10,349,087
|
)
|
||||
|
Net cash used in investing activities
|
1,744,775
|
(83,304
|
)
|
|||||
|
Net cash provided by financing activities
|
(572,282
|
)
|
14,810,460
|
|||||
|
Net increase in cash and cash equivalents
|
(8,238,462
|
)
|
4,378,069
|
|||||
|
Effects of exchange rate changes on cash
|
(5,269
|
)
|
(6,351
|
)
|
||||
|
Cash at the end of the period
|
5,191,031
|
13,434,762
|
||||||
Year Ended June 30, 2022, Compared to Year Ended June 30, 2021
Net cash provided by (used in) operating activities
Net cash used in operating activities decreased by A$938,132, or 9%, to A$(9,410,955) for the year ended June 30, 2022, compared to
A$(10,349,087) for the year ended June 30, 2021. This increase was mainly due to the impact of the Nutraceutical License in October 2021.
Net cash used in investing activities
Net cash (used in) provided by investing activities increased by A$1,828,079, or 22%, to A$1,744,775 for the year ended June 30, 2022,
compared to A$(83,304) for the year ended June 30, 2021. This increase was mainly due to the Company receiving proceeds from the disposal of continued operations in the amount of $1,775,910.
Net cash used in financing activities
Net cash (used in) provided by financing activities increased by A$15,382,742, or 104%, to A$572,282 for the year ended June 30, 2022,
compared to A$(14,810,460) for the year ended June 30, 2021. This increase was due to a capital raise undertaken in Australia, with ASX, in 2021.
We have incurred losses and cash flow deficits from operations since our inception, resulting in accumulated losses as of June 30, 2022, of
A$59,529,093. We anticipate that we will continue to incur net losses for the foreseeable future. To meet future capital needs, we would need to raise additional capital through equity or debt financing or other strategic transactions. We have
based our estimates on assumptions that may prove to be wrong, and our expenses could prove to be significantly higher than we currently anticipate.
58
Our future capital requirements will depend on many factors, including, but not limited to:
| • |
the progress and costs of our research and development activities;
|
| • |
the costs of development and expansion of our operational infrastructure;
|
| • |
our ability, or that of our collaborators, to achieve development milestones and other events or developments under potential future licensing agreements;
|
| • |
the amount of revenues and contributions we receive under future licensing, collaboration, development and commercialization arrangements with respect to our products;
|
| • |
the costs of filing, prosecuting, enforcing and defending patent claims and other intellectual property rights;
|
| • |
the costs of contracting with third parties to provide sales and marketing capabilities for us or establishing such capabilities ourselves, once our products are developed and ready for
commercialization;
|
| • |
the costs of acquiring or undertaking development and commercialization efforts for any future products;
|
| • |
the costs of our trials for our drug candidates and applications with the relevant regulatory agencies;
|
| • |
the magnitude of our general and administrative expenses; and
|
| • |
any additional costs that we may incur under future in- and out-licensing arrangements relating to our products.
|
Until we can generate significant recurring revenues, we expect to satisfy our future cash needs through capital raising or by
out-licensing and/or co-developing applications of one or more of our product candidates. We cannot be certain that additional funding will be available to us on acceptable terms, if at all. If funds are not available on favourable terms, or at all,
we may be required to delay, reduce the scope of or eliminate research or development efforts or plans for commercialization with respect to our products and make necessary change to our operations to reduce the level of our expenditures in line with
available resources.
We are a development early-stage biotechnology company, and it is not possible for us to predict with any degree of accuracy the outcome of
our research and development efforts. As such, it is not possible for us to predict with any degree of accuracy any significant trends, uncertainties, demands, commitments, or events that are reasonably likely to have a material effect on our net
loss, liquidity or capital resources, or that would cause financial information to not necessarily be indicative of future operating results or financial condition. However, to the extent possible, certain trends, uncertainties, demands, commitments
and events are described herein.
Quantitative and Qualitative Disclosures About Market Risk
We are exposed to a variety of financial risks, which result from our financing, operating, and investing activities. The objective of
financial risk management is to contain, where appropriate, exposures in these financial risks to limit any negative impact on our financial performance and position. Our main financial instruments are our cash and other receivables, trade, and other
payables. The main purpose of these financial instruments is to raise finance for our operations. We actively measure, monitor, and manage our financial risk exposures by various functions pursuant to the segregation of duties and principals. The
risks arising from our financial instruments are mainly credit risk and currency risk. The risk management policies employed by us to manage these risks are discussed below.
59
Interest Rates
We believe the impact of increased interest rates on our Company will be minimal as the Company does not carry financial debt. However, the
increased interest rates may impact the USD, especially as compared to the AUD, thus having a potential impact on our consolidating expenditure. Further, the increase in interest rates may benefit our savings deposits that are held in AUD.
Inflation
The increase in inflation may have an impact on our supply chain, product manufacturing costs and staffing costs. As the U.S. Federal
Reserve interest rates increase at a faster pace than AU Reserve Bank, the Company may be impacted from an exchange rate perspective.
Credit risk
Credit risk arises when a failure by counterparties to discharge their obligations could reduce the amount of future cash inflows from
financial assets on hand at the balance sheet date. We closely monitor the activities of our counterparties and control the access to our intellectual property which enables us to ensure the prompt collection. Our main financial assets are cash as
well as trade receivables and other receivables and represent our maximum exposure to credit risk in connection with our financial assets. Wherever possible and commercially practical, we hold cash with major financial institutions in Australia.
Currency risk
Currency risk is the risk that the value of financial instruments will fluctuate due to changes in foreign exchange rates. Currency risk
arises when future commercial transactions and recognized assets and liabilities are denominated in a currency that is not our functional currency. We are exposed to foreign exchange risk arising from currency exposure resulting primarily from the
majority of our expenses being denominated in Australian dollars, although some of our expenses are U.S. dollar-derived. Our policy is not to enter into any currency hedging transactions.
Concentration of Credit risk
Financial instruments that potentially subject the Company to concentrations of credit risk are cash, accounts receivable and other
receivables arising from its normal business activities. The Company places its cash in what it believes to be credit-worthy financial institutions. The Company controls credit risk related to accounts receivable through credit approvals, credit
limits and monitoring procedures. The Company routinely assesses the financial strength of its customers and, based upon factors surrounding the credit risk, establishes an allowance, if required, for uncollectible accounts and, as a consequence,
believes that its accounts receivable credit risk exposure beyond such allowance is limited.
Liquidity risks
Liquidity risk is the risk that arises when the maturity of assets and the maturity of liabilities do not match. An unmatched position
potentially enhances profitability, but it can also increase the risk of loss. We have procedures to minimize such loss by maintaining sufficient cash and other highly liquid current assets and by having available an adequate amount of committed
credit facilities. As of June 30, 2022, we have a negative working capital.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements.
60
Emerging Growth Company Status
We qualify as an “emerging growth company” as defined in the JOBS Act. An emerging growth company may take advantage of specified reduced
reporting and other burdens that are otherwise applicable generally to public companies. These provisions include:
| • |
a requirement to present only two years of audited financial statements in addition to any required interim financial statements and correspondingly reduced Management’s Discussion and
Analysis of Financial Condition and Results of Operations disclosures;
|
| • |
to the extent that we no longer qualify as a foreign private issuer, (i) reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements and (ii)
exemptions from the requirement to hold a non-binding advisory vote on executive compensation, including golden parachute compensation;
|
| • |
an exemption from the auditor attestation requirement in the assessment of our internal control over financial reporting pursuant to the Sarbanes-Oxley Act of 2002; and
|
| • |
an exemption from compliance with the requirement that the Public Company Accounting Oversight Board has adopted regarding a supplement to the auditor’s report providing additional information
about the audit and the financial statements.
|
Critical Accounting Estimates and Recent Accounting Pronouncements
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the
reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. The most significant assumptions and
estimates relate to the valuation of equity issued for services, valuation of equity associated with convertible debt, the valuation of derivative liabilities, and the valuation of deferred tax assets. Actual results could differ from these
estimates.
Revenue Recognition
The Company recognizes revenue in accordance with Accounting Standards Update 2014-09, “Revenue from contracts with customers,” (Topic
606). Revenue is recognized when a customer obtains control of promised goods or services. In addition, the standard requires disclosure of the nature, amount, timing, and uncertainty of revenue and cash flows arising from contracts with customers.
The amount of revenue that is recorded reflects the consideration that the Company expects to receive in exchange for those goods. The Company applies the following five-step model in order to determine this amount: (i) identification of the promised
goods in the contract; (ii) determination of whether the promised goods are performance obligations, including whether they are distinct in the context of the contract; (iii) measurement of the transaction price, including the constraint on variable
consideration; (iv) allocation of the transaction price to the performance obligations; and (v) recognition of revenue when (or as) the Company satisfies each performance obligation. The Company’s main revenue stream is from product sales. The
Company recognizes as revenues the amount of the transaction price that is allocated to the respective performance obligation when the performance obligation is satisfied or as it is satisfied. Generally, the Company’s performance obligations are
transferred to customers at a point in time, typically upon delivery.
61
Sale of Nutraceuticals
Sale of goods revenue is recognized at the point of sale, which is at the time when the customer’s orders are shipped. Amounts disclosed as
revenue are net of sales returns and trade discounts.
Discounts, promotional and other rebates
The sale of goods revenue is net of any discounts, rebates and any contributions to customers towards promotional activities.
Fair Value Measurements
Accounting Standard Codification (“ASC”) Topic 820, Fair Value Measurements, clarifies the definition of fair value, prescribes methods for
measuring fair value, and establishes a fair value hierarchy to classify the inputs used in measuring fair value as follows:
Level 1: Inputs are unadjusted quoted prices in active markets for identical assets or liabilities available at the measurement date.
Level 2: Inputs are unadjusted quoted prices for similar assets and liabilities in active markets, quoted prices for identical or similar
assets and liabilities in markets that are not active, inputs other than quoted prices that are observable, and inputs derived from or corroborated by observable market data.
Level 3: Inputs are unobservable inputs which reflect the reporting entity’s own assumptions on what assumptions the market participants
would use in pricing the asset or liability based on the best available information.
The estimated fair value of certain financial instruments, including all current liabilities are carried at historical cost basis, which
approximates their fair values because of the short-term nature of these instruments.
Fair Value of Financial Instruments
ASC subtopic 825-10, Financial Instruments requires disclosure of the fair value of certain financial instruments. The carrying value of
cash and cash equivalents, accounts payable and accrued liabilities as reflected in the consolidated balance sheets, approximate fair value because of the short-term maturity of these instruments. All other significant financial assets, financial
liabilities and equity instruments of the Company are either recognized or disclosed in the consolidated financial statements together with other information relevant for making a reasonable assessment of future cash flows, interest rate risk and
credit risk. Where practicable the fair values of financial assets and financial liabilities have been determined and disclosed; otherwise only available information pertinent to fair value has been disclosed.
Cash and Cash Equivalents
The Company considers all highly liquid investments with an original maturity of three months or less to be cash equivalents.
Stock Based Compensation
The Company records stock-based compensation in accordance with the provisions of Financial Accounting Standards Board (“FASB”) ASC Topic
718, “Accounting for Stock Compensation,” which establishes accounting standards for transactions in which an entity exchanges its equity instruments for goods or services. In accordance with guidance provided under ASC Topic 718, the Company
recognizes an expense for the fair value of its stock awards at the time of grant and the fair value of its outstanding stock options as they vest, whether held by employees or others. During the twelve months ended June 30, 2022, and 2021, the
Company recorded A$77,367 and A$619,836, respectively, in stock-based compensation expense.
62
Recent Accounting Pronouncements
The Company has implemented all new accounting pronouncements that are in effect. These pronouncements did not have any material impact on
the consolidated financial statements unless otherwise disclosed, and the Company does not believe that there are any other new accounting pronouncements that have been issued that might have a material impact on its financial position or results of
operations.
Commitments and Contingencies
During the normal course of business, the Company may be exposed to litigation. When the Company becomes aware of potential litigation, it
evaluates the merits of the case in accordance with FASB ASC 450-20-50, Contingencies. The Company evaluates its exposure to the matter, possible legal or settlement strategies and the likelihood of an unfavorable outcome. If the Company determines
that an unfavorable outcome is probable and can be reasonably estimated, it establishes the necessary accruals. As of June 30, 2022, and 2021, the Company is not aware of any contingent liabilities that should be reflected in the consolidated
financial statements.
63
BUSINESS
Overview
Medlab Clinical Ltd. is a company aiming to disrupt the multi-billion-dollar market of delivery platform technologies for pharmaceutical
products that are designed to treat pain and mental health. We were incorporated in Australia in April 2014, and in 2015 listed on the ASX under the symbol “MDC.” Our corporate headquarters are located in Australia, and we maintain offices in the
United States and Europe.
We strongly believe there are considerable unmet needs of patients in our target markets, and that the current drug intervention therapies
that are available can be significantly improved. We believe that we are able to provide these improvements to patients through product that we have and are developing. These products are discussed below.
Our Lead Product Candidate: NanoCelle®
NanoCelle®, our core product offering, is a pharmaceutical-delivery platform which is designed to administer pharmaceutical products using
nanoparticles, or small particles ranging between 1 to 100 nanometers, in size and undetectable by the human eye. NanoCelle® is an alternative, and we believe more effective, method of consuming medical products, compared to traditional methods.
NanoCelle® can administer pharmaceutical products in the form of either an oro-buccal spray, nasal spray, or a topical gel, cream, or spray. NanoCelle® is designed to allow for significant improvements in patient care and quality of life and
significantly reduced side effects. Our primary and only commercially available delivery platform, NanoCelle® is now patented until 2036 in several countries including Australia, Canada, the European Union Countries, Hong Kong, New Zealand, the
United States, the United Kingdom and with a pending patent application in Singapore, until 2036. In developing NanoCelle®, we have placed a high research priority on tolerability, including not using harmful toxic products such as heavy metals or
gases. To date, NanoCelle® has administered over 350,000 doses of pharmaceutical products to patients through its delivery platform through via compassionate use programs in the United Kingdom and Australia or clinical trials and/or sale of approved
medicines, approved by such governments or applicable pharmaceutical regulatory agents. The term “compassionate use” refers to a governmentally-sanctioned expedited pathway for a patient with an immediately life-threatening condition or serious
disease or condition to gain access to an investigational medical product for treatment outside of clinical trials when no comparable or satisfactory alternative therapy options are available.
Our Other Drug Product Candidates, both in-market and in-development
In addition to developing the NanoCelle® delivery method, we have also spearheaded several drug developmental programs that were
specifically designed to be administered through the NanoCelle® technology. Our NanoCelle® delivery method has produced additional drug candidates which share design and CMC processes with our lead candidate, resulting in tolerability
profiles that may be similar to our lead product candidate, as well as regulatory agency familiarity, and a consistent multi-target therapeutic approach. Several of our products are listed, in contrast to being registered, with the ARTG and all
medicines that are sold in Australia must either be registered or listed with the ARTG.
All medicines registered with the ARTG generally are evaluated for efficacy (that the medicine can do what it says it will) before they may
be sold in Australia. However, not all listed medicines are evaluated for efficacy. Our registered medicines, such as NanaBis™, are higher risk and because of this the ARTG fully assesses all registered medicines for safety, quality and
efficacy before they go on sale. Registered medicines, which are intended to treat, manage and/or cure one or more conditions, require investment in clinical evidence with no guarantee of success at TGA. Listed medicines, such as NanoCelle® D3 and
NanoCelle® B12, are lower risk and can be purchased off the shelf from pharmacies, health shops, and supermarkets. Listed products, such as complementary medicines and food supplements, are not exposed to the same risks and do not have to undergo the
same rigorous regulatory assessments as registered products, but are subject to a random audit after listing.
64
As such, since our in-market drug products, are lower risk and do not go through pre-market assessment, they are considered ARTG-listed. Both ARTG-listed and ARTG-registered medicines must be manufactured in a licensed or approved facility in accordance with the principles of good manufacturing practice. There is no requirement under the ARTG for our products
to be registered in their present form.
Though we currently have products available to the public, both listed with the ARTG and administered pursuant to United Kingdom and
Australian compassionate use programs (or their equivalents), our current goal is to continue to expand our suite of products that are able to be administered through our NanoCelle® delivery platform through continued research and
development.
Our mission is to create premier pharmaceutical drugs and therapies and delivery platforms for patients dealing with unmet medical needs,
each of which will comply with applicable regulatory requirements, including the FDA, EMA and TGA. We aim to be recognized as a leading specialty drug development company, committed to restoring health and transforming the lives of patients through
the development of novel pharmaceutical products and treatments.
Below is a summary of our current products that are being sold in Australia and New Zealand and are expected to be launched in the United
Kingdom by the end of October 2022, as well as the potential estimated value in U.S. and other markets:
|
Medlab Drug
Product Name
|
Availability Status
|
Target Market
|
Estimated Size of Target Market
|
Estimated U.S. dollar value of potential demand for product
|
|
NanoCelle® D3
|
Listed with ARTG. This product is available through PharmaCare in Australia over-the-counter in pharmacies and through healthcare providers and in New Zealand, operating under
Medsafe, as a food supplement.
|
Immunity and bone health
|
An estimate of 1 billion people globally have low Vitamin D3 levels
|
US$1.1 billion with potential to reach US$1.6 billion by 2025
|
|
|
|
|
|
|
|
NanoCelle® B12
|
Listed with ARTG. This product is available through PharmaCare in Australia over-the-counter in pharmacies and through healthcare providers and in New Zealand, operating under
Medsafe, as a food supplement.
|
Reduce homocysteine levels and support the nervous system
|
Approximately 15% of global population is deficient in absorption of vitamin B12
|
US$276 million
|
|
|
|
|
|
|
|
NanaBis™
|
Distributable in Australia through the Special Access Scheme and is scheduled to launch in the United Kingdom through the Named Patient Programme by the end of October 2022,
whereby doctors seek government regulatory approval for use for patients on a case-by-case basis.
|
Cancer-induced bone pain
|
100 million in United States
|
US$70 billion
|
|
|
|
|
|
|
|
NanoCBD™
|
Distributable in Australia through the Special Access Scheme and is scheduled to launch in the United Kingdom through the Named Patient Programme by the end of October 2022,
whereby doctors seek government regulatory approval for use for patients on a case-by-case basis.
|
Occupational stress
|
67% of the United States
|
US$17.2 billion
|
65
Below are our drug product candidates that are currently in various stages of regulatory approvals and are not currently listed or approved
drug products or available pursuant to any compassionate use program:
MDC2000 is a product targeting major depressive disorders that has been optimized as a single
molecule encapsulated in NanoCelle® following clinical studies in 2020. The 2020 studies were closed earlier than projected due to the COVID pandemic. Based on this optimization, the program has returned to a pre-clinical stage with clinical returns
expected in early 2023.
NanoCelle® Nucleic Acid: On April 4, 2022, we entered into a Collaborative Research Agreement
with the University of New South Wales and Macquarie University to conduct testing during the pre-clinical stages for our NanoCelle®-Nucleic Acid, a nasal vaccine delivery utilizing nucleic acid to develop a new vaccine and/or anti-viral
technologies. Pursuant to the Collaborative Research Agreement, all intellectual property created or developed under such agreement is owned collectively by all parties to such agreement. Accordingly, we will not have full ownership over the
intellectual property developed under the Collaborative Research Agreement, and any of our products incorporating such intellectual property will be subject to the joint ownership rights under the agreement. Pursuant to our joint collaboration
agreement with Macquarie University and the University of New South Wales, we are involved in the joint development of NanoCelle®-Nucleic Acid, as a nasal delivery for vaccines in pre-clinical stages for a nasal vaccine delivery utilizing nucleic
acid, predictively leading to new vaccine and/or anti-viral technologies. We believe that our nucleic acid-related research can lead to developments in insulin, vaccine and anti-viral products.
Research and Development
Our current focus is to continue expanding our suite of listed drug products, both in-market and in-development, that are compatible with
NanoCelle®, including continually developing our existing products. We currently utilize the Australian Research and Development Tax Incentive, which encourages companies to engage in research and development benefiting Australia, by providing a
refundable tax offset which varies depending on the aggregate annual turnover of certain eligible entities for eligible research and development activities.
The Australian federal government provides a cash rebate each year on the total amount expensed that relates to research and development
costs. This includes personnel costs, product development costs, Special Access Scheme material expenditures and all clinical trial related activities. The cash rebate is 43.5% and is provided as a direct payment back to the Company, while revenue
is less than $A20 million.
The Company engages a research and development consultant to support the Company in the application process.
Research and development incentive income is based on an accrual from the prior year’s actual research and development claim calculation
and is reported at the time of year-end close. Subsequent adjustment to the accounts is made once the closed financial year’s research and development claim is determined by the AusIndustry, the federal tax authority.
Our Strategy
Our mission is to create premier pharmaceutical drugs and therapies and delivery platforms for patients dealing with unmet medical needs,
each of which will be approved by applicable regulatory bodies including the FDA, EMA and TGA. Specifically, we envision the NanoCelle® platforms as a future pinnacle inflexion point in medicine. Drug therapy has played a dramatic role in the
increase in human life expectancy and quality, but contains severe limitations relating to dose control, side effects, diminishing values in patient quality of life and ultimately, real, or self-perceived, diminishing patient compliance. Through our
patented and proprietary drug delivery platform, NanoCelle®, we are committed to formulating on various drug compounds innovative drug-based solutions to today’s most relevant global health priorities, specifically pain and mental health, while
reducing those inherent and existing limitations.
66
We believe that enhanced medicine delivery platforms that are able to control the rate at which a drug is released and the location in the
body where it is released will play a significant role in the administration of pharmaceutical products in the future, and that current delivery methods result in significant and avoidable side effects.
Our strategy is to expand our market opportunity by gaining United States and other regulatory approval for our products. Furthermore, the
superior performance we believe we can achieve through our patented drug delivery platform, NanoCelle® will be a key differentiator in the pharmaceutical space. We are seeking to develop a body of clinical and real-world evidence and findings to
support the benefits of our products.
In March 2021, we raised A$15 million via a two-tranche placement in a private placement offering (the “March 2021 Offering”). The proceeds
from the March 2021 Offering were applied towards funding a pivotal Phase III clinical trial in Australia, the U.S. and the United Kingdom to investigate the analgesic tolerability and exploratory endpoints of our non-opioid analgesic drug candidate
delivered via NanoCelle® for the treatment of cancer-induced bone pain (bone metastases).
On October 19, 2021, the Company licensed its Australian nutraceutical business to PharmaCare for cash consideration of A$750,000. The principal assets that were licensed
comprised registered patents and trademarks, inventory, customer lists, and material contracts. In connection with the licensing deal, the Company sold its nutraceutical inventory to PharmaCare for $1,025,910. Following the Nutraceutical License,
we continued to maintain our rights to market and sell our nutraceutical lines of business outside of Australia. Following the Nutraceutical License, we are able to focus our business on the development and registration of pharmaceutical products
that are compatible with our NanoCelle® delivery platform.
We develop targeted and scientifically-tested fixed-dose combinations of the NanoCelle® delivery platform and various APIs, applying
proprietary insights to create long-term products that we believe will better meet the medical needs of our patients compared to what is currently available and that we believe will create value for our shareholders. Our focus is on clinical
indications that we believe represent unmet or inadequately addressed medical needs.
Over the past year, we have made several key strategic achievements and milestones, including:
| 1. |
Partnered with Purisys, a United States-based biosynthetic partner for FDA drug master files, for the two cannabinoids, CBD, and THC. It was noted at the time that the technology required for
making a “neat” or 100% dronabinol (the synthetic of THC) was not commercially available. While working with Purisys in Georgia, the Company was able to deliver a 100% dronabinol, as well as a 100% cannabinoid with relevant documents for an
FDA drug master files submission. The Company believes the biosynthetic partnership is a key strategic agreement/milestone.
|
| 2. |
Partnered with a strategic United States-based manufacturing company for (1) CMC development for the cannabinoid programs; (2) manufacturing optimization and scale; and (3) future commercial
scale manufacture.
|
| 3. |
Secured patents in several countries, including Australia, Canada, the European Union Countries, Hong Kong, New Zealand, the United States, the United Kingdom and with a pending patent
application in Singapore for the NanoCelle® delivery platform until 2036 across all territories.
|
| 4. |
Entered agreements with United Kingdom and Australian partners for compassionate use of the cannabinoids to at-risk patients.
|
| 5. |
Entered agreements with Macquarie University and University of New South Wales for the joint development of NanoCelle®-Nucleic Acid, as a nasal delivery for vaccines.
|
| 6. |
Strengthened our datapoints from our Australian data bank to understand sustainability of the NanaBis™ program over 6 to 12 months, and tolerability as it relates to adverse events and in
relation to opioid medications.
|
67
The Company has currently licensed its commercial IP to the following:
| 1. |
PharmaCare– Licensed to use Nanocelle® D3 and Nanocelle® B12 in TGA-listed medicines in Australia and New Zealand only for the nutraceuticals.
|
| 2. |
Cultech Ltd.– Licensed to use NRGBiotic™ in TGA-listed medicine and is United Kingdom exclusive and U.S. nonexclusive;
|
| 3. |
YesHealth Sdn Bhd.– Licensed to use several TGA-listed medicines (not including the NanoCelle®) and is Malaysia and Vietnam exclusive and Singapore nonexclusive; and
|
| 4. |
American Nutritional Corporation Inc.– Licensed several TGA-listed medicines inclusive of those NanoCelles® in existing range (Nanocelle® D3 and Nanocelle® B12) and is U.S. exclusive under
their private label called NuScripts.
|
NanoCelle® Technology Transfer Licenses for Manufacturing:
| 1. |
Renaissance – a U.S. Contract Development and Manufacturing Organization experienced in FDA and DEA drugs and manufacturing;
|
| 2. |
Natural Factors – a Canadian nutraceutical manufacturing facility approved by TGA;
|
| 3. |
Extractas (formerly known as Tasmanian Alkaloids) - an Australian experienced poppy grower and manufacturer for medicines that is approved by TGA; and
|
| 4. |
Universities New South Wales and the Woolcott Institute of Macquarie University – research collaboration partners for the NanoCelle®-Nucleic Acid nasal vaccine program.
|
Our Opportunity
The global drug delivery systems market size was valued at US$39.33 billion for 2021. Furthermore, this market is projected to grow from
US$39.33 billion in 2022 to US$71.75 billion by 2029, exhibiting a compound annual growth rate of 9.0% during the forecast period. The global COVID-19 pandemic has been unprecedented and staggering, with the market experiencing
higher-than-anticipated demand across all regions compared to pre-pandemic levels
We intend to license our intellectual property to certain global strategic partners, including larger, established companies in the
biopharma, food and/or cosmetic areas that offer both local and global marketing, sales and distribution capabilities. To maximize our potential return, we are presently engaged in licensing to strategic geographical partners with larger, established
companies in the biopharma, food and/or cosmetic areas, who offer both local and global marketing, sales and distribution capabilities.
68
To achieve our goals, we intend to:
| • |
Advance our novel investigational drug candidates towards approval in the United States and elsewhere. We are pursuing appropriate regulatory
approvals for new medicines, which include approvals from the FDA and may include the EMA and the TGA. NanaBis™ and NanoCBD™ are available in Australia through the Special Access Scheme and are scheduled to launch in the
United Kingdom through the Named Patient Programme by the end of October 2022, whereby doctors seek government regulatory approval for use for patients on a case-by-case basis. We intend to pursue an NDA with the FDA with respect to each of
our drug candidates. If the NDA is approved, NanaBis™ and NanoCBD™ may be marketed in the United States subject to Drug Enforcement Agency scheduling application and approval. Though we cannot ensure approval, other
dronabinol products, such as Marinol in 1985 and Epidiolex in 2018, have obtained approval. Upon such approval, we plan to further pursue marketing approval of the NDA-approved drug(s) in other regions including the European Union, Canada and
Australia.
|
| • |
Develop future clinical products targeting unmet medical needs. We intend to develop and commercialize clinical products that treat unmet
medical conditions.
|
| • |
Maintain a strong intellectual property portfolio. We are working to develop an intellectual property portfolio to support our commercial
objectives. We are monitoring the results of our research and development programs to identify new intellectual property that aligns with those commercial objectives. We intend to take a global approach to our intellectual property strategy
to pursue patent protection in key global markets, including the United States, European Union Countries, Australia, Canada, and the United Kingdom. For more information on our intellectual property portfolio, please see the sections entitled
“Current Commercial IP Licensing” and NanoCelle® Technology Transfer Licenses for Manufacturing.”
|
| • |
Globalize the Company. We intend to globalize the company to meet both regulatory and potential partners’ needs, including articulation of
pricing re-imbursement, clinical optimization of future research design, expanding compassionate use, where appropriate, lecturing at strategic medical, scientific and industry events, as well as increased marketing of our products in the
United States.
|
Our Lead Product Candidate: NanoCelle®
NanoCelle®
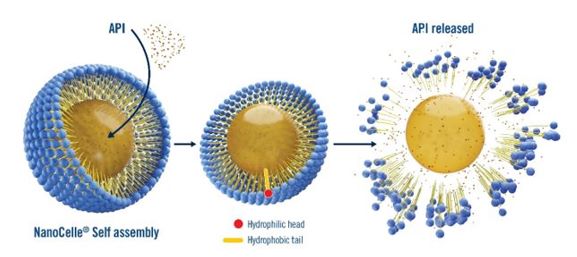
The above illustration provides a nanoscopic view of how NanoCelle® disperses and releases a pharmaceutical product for absorption.
69
NanoCelle® is a clinically trialed, patented delivery technology utilizing standardized nanoparticles and administered in the forms of
either an oro-buccal spray, nasal spray, or a topical gel, cream, or spray. The types of clinical trials that NanoCelle® has conducted includes both disease and healthy population groups with studies conducted for pharmacokinetic,
bioequivalence and analgesic tolerability and exploratory endpoints. NanoCelle® is an alternative, and, we believe, more effective means of consuming medications. For example, NanoCelle® would avoid the requirement of swallowing medication
for patients who temporarily or permanently are unable to ingest medications by traditional oral consumption.
Via the oro-buccal route of administration, NanoCelle® utilizes nanoparticles ranging from 5 nanometers to 90 nanometers to distribute a
compound directly into the blood stream, bypassing first-pass metabolism, a phenomenon in which a drug gets metabolized at a specific location in the body that results in a reduced concentration of the active drug upon reaching its site of action or
the systemic circulation. The NanoCelle® platform allows medicines to be delivered via multiple routes that bypass first-pass metabolism, allowing for quicker absorption and greater drug bioavailability. By bypassing the gastrointestinal
environment, the drug substance is not subject to extensive degradation as found in other ingestible medicines.
We believe NanoCelle® delivers pharmaceutical products, without several harmful side effects inherent in other existing mediums which
traditionally rely on toxic heavy metals or toxins. By changing the drug solubility, we can increase absorption and potentially reduce side effects to the patient. Furthermore, there has been limited evidence of complement activation-related pseudo
allergy (CARPA), a stress reaction in blood triggered by nanomedicines and biologicals, in over 350,000 doses of pharmaceutical products dispensed through NanoCelle®.
NanoCelle® Clinical Trial Work
|
Study
|
Trial Information
|
Status
|
Results
|
|
NanaBis™ Phase II Study
|
• Approval: Australian Ethics
approved;
• Trial Number: Clinical trial
number conferred by TGA;
• Duration: 8 months;
• Type of Study: Phase II study;
• Dosage: Single ascending,
multi-ascending dose study to determine tolerability;
• Number
of Subjects/Patients: 36, advanced cancer with unmanaged pain conducted at Royal North Shore Hospital, Oncology Unit;
• Intervention: Intervention period
30 days;
• Endpoints: All endpoints met;
• Dropouts: 3 (2 deceased due to
unrelated cancer progression; 1 due to severe nausea and vomiting);
• Common
adverse events included mild drowsiness, with four instances of severe drowsiness, similarly with dry mouth, fogginess, nausea, vomiting and fatigue. It is worth noting this patient cohort had advanced cancers with unmanaged pain and as
such were on a number of medications of which are known to produce similar adverse events.
|
Completed in 2020.
|
Published.
|
|
NanaBidial™ (NanaBis™ early variant) Study
|
• Approval: Australian Ethics
approved;
• Trial Number: Clinical trial
number conferred by TGA;
• Duration: 1 week;
• Type of Study: Pharmacokinetic
study
• Dosage: Single dose of 5mg and
single dose of 5mg twice per day;
• Number of Subjects/Patients: 12
healthy participants at Scientia Unit University New South Wales;
• Intervention: Intervention period
3 days;
• Endpoints: All endpoints met;
• Dropouts: 0;
• No adverse events reported.
|
Completed in 2018.
|
Published.
|
70
|
Study
|
Trial Information
|
Status
|
Results
|
|
NanaBis™ Real World Clinical Data Study
|
• Approval: Australian Ethics
approved;
• Trial Number: Clinical trial
number conferred by TGA;
• Type
of Study: Real World Evidence Study– open label, illicit Adverse Events in real world with a look at long term sustainability;
• Dosage: No fixed dose;
• Number
of Subjects/Patients: 1184 participants and growing as approximately 290 Australian doctors, predominantly from the east coast of Australia, are recruiting;
• Endpoints: Ongoing.
• Adverse Events are only accrued
for NanaBis™ with 12% considered non-serious and 1.3% considered serious.
|
Study is ongoing and progressively recruiting.
|
Study is ongoing.
|
|
NanoCelle® B12 Study
|
• Approval: Australian Ethics
approved;
• Trial Number: Clinical trial
number conferred by TGA;
• Duration: 6 months;
• Type of Study: Head-to-head
comparison with single dose pharmacokinetic study;
• Dosage: Single dose against
comparable strength tablets, capsules and emulsions;
• Number of Subjects/Patients: 60
healthy participants conducted in the Company’s lab with Company clinical trial staff;
• Endpoints: NanoCelle® B12 was five
times more absorbable than other oral B12;
• Dropouts: 1 (patient unable to
make follow-up appointment);
• No adverse events reported.
|
Completed in 2018.
|
Published.
|
|
NanoCelle® Atorvastatin Study
|
• Approval: New Zealand Ethics
approved;
• Duration: 4 weeks (including 2
week washout period);
• Type
of Study: Bioequivalence pilot, using Lipitor as the comparator with study cohort cross-overs and was conducted by Zenith New Zealand;
• Dosage: Single dose of 2mg against
20mg tablet;
• Number of Subjects/Patients: 12
healthy participants;
• Endpoints: as bioequivalent as
20mg Lipitor;
• Dropouts: 0;
• No adverse events reported.
|
Completed in 2017.
|
Not Published.
|
71
|
Study
|
Trial Information
|
Status
|
Results
|
|
NanoCelle® D3 Study
|
• Approval: Australian Ethics
approved;
• Trial Number: Clinical trial
number conferred by TGA;
• Duration: 4 weeks;
• Type
of Study: Single dose pharmacokinetic study that was conducted in the Company’s lab with Company clinical trial staff;
• Dosage: Single dose of 1,000 iu;
• Number of Subjects/Patients: 6
healthy participants;
• Endpoints: All endpoints met,
designed to understand the rate of absorption;
• Dropouts: 2 (due to missed
appointment);
• No adverse events reported.
|
Completed in 2018.
|
Not Published.
|
|
NanoCelle® CoQ10 Study
|
• Approval: Australian Ethics
approved;
• Duration: 1 week;
• Type of Study: Single dose
pharmacokinetic study conducted in the Company’s lab with Company clinical trial staff;
• Dosage: Multi-dose of 5mg,
totaling 20mg;
• Number of Subjects/Patients: 6
healthy participants;
• Endpoints: All endpoints met,
designed to understand the rate of absorption and maturation;
• Dropouts: 0;
• No adverse events reported.
|
Completed in 2018.
|
Not Published.
|
72

The above image indicates the difference of solubility between NanoCelle® and the generic versions of the product. The solution on the
left, Atorvastatin Calcium (or Lipitor) in water, versus a NanoCelle® Atorvastatin. The NanoCelle® Atorvastatin is completely solubilized in water, which results in more of the pharmaceutical product being absorbed by the body.
Bioavailability and characteristics of different routes of administration include:
|
ROUTE
|
SPEED
|
BIOAVAILABILITY
|
CHARACTERISITICS
|
|
Intravenous
|
30-60 seconds
|
100%
|
Most rapid
|
|
Intramuscular
|
10-20 minutes
|
75≤100%
|
Large volume may be injected but painful method
|
|
Subcutaneous
|
15-30 minutes
|
75≤100%
|
Smaller volume than IM, may be painful
|
|
Oral – Ingested
|
30-90 minutes
|
5% or more
|
Convenient, first pass metabolism occurs
|
|
Oral - Sublingual
|
3-5 minutes
|
c.35%
|
May avoid first pass metabolism, but may be ingested as well pending the medicine
|
|
Oral - Buccal
|
3-5 minutes
|
30% or more
|
Direct absorption into venous circulation. First pass metabolism is avoided
|
|
Rectal
|
5-30 minutes
|
30<100%
|
Less first pass metabolism than oral route
|
|
Inhalation
|
2-3 minutes
|
5<100%
|
Rapid Onset
|
|
Transdermal
|
Highly varied
|
80≤100%
|
Usually slow absorption, lack of first pass metabolism and prolonged duration of action
|
73
We believe the preferred route of administration is the oro-buccal route, as listed in the above chart, using nanoparticles to infiltrate
the buccal membrane contained in the mouth for systemic response, giving, patient convenience, speed and superior bioavailability. In a 2018 ethics-approved study that was conducted in the Company lab with Company clinical trial staff, NanoCelle®
B12 administration was deemed up to five times more absorbable than other industry-approved oral B12 administrations. The other B12 medicines are ARTG-listed and found in Australian pharmacies in the form of tablets, capsules, emulsions and liposomal
formulations. This head-to-head comparison involved a single dose in 30 subjects. The ethics-approved study was approved by the human ethics research unit of the IRB.
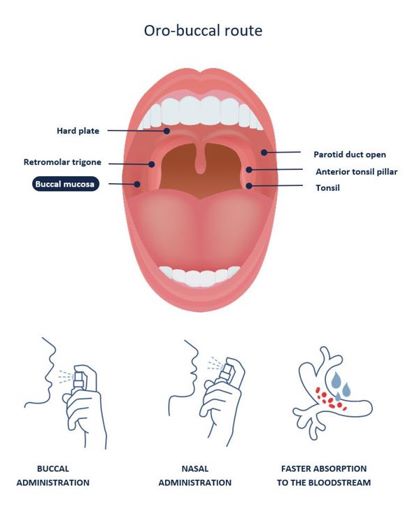
74
NanoCelle® contains a nanoscopic fat soluble core surrounded by a water-soluble outer layer. This structure enables the NanoCelle® solution
to maintain high concentrations of selected nutrients, while delivering nano-particle sized molecules of the compound for absorption across the appropriate membrane. NanoCelle® is designed to increase absorption rate, compared to other dosage forms
such as tablets, capsules, emulsions, or sublingual liposomal spray. The below study demonstrated that when compared to other delivery methods, NanoCelle® exhibited a higher rate of absorption of a pharmaceutical product, with a 28% rate
of absorption.

Our patent portfolio for NanoCelle® includes granted patents spread across multiple jurisdictions, including Australia, Canada, the
European Union Countries, Hong Kong, New Zealand, the United States, the United Kingdom and with a pending patent application in Singapore, until 2036.
75
Our Other Drug Product Candidates, both in-market and in-development
Our drug product candidates in-market that utilize the NanoCelle® platform consist of NanoCelle® B12 and
NanoCelle® D3 while our drug product candidates in-development consist of NanaBis™ , NanoCBD™, MDC2000 and NanoCelle® Nucleic Acid. NanaBis™ and NanoCBD™
are investigational cannabinoid-based medications. MDC2000 and NanoCelle® Nucleic Acid are other investigational products utilizing the NanoCelle® platform.
TGA approved NanoCelle® Products
While we have received TGA approvals for the following specific products utilizing NanoCelle® technology, the Company is aware that this
alone does not guarantee future regulatory approval with other NanoCelle® products.
|
Name of Product
|
Summary
|
Benefits and Highlights
|
|
NanoCelle® Activated B12
|
● B12 as either mecobalamin or cyanocobalamin that is five times more absorbable than other ingestive forms.
● ARTG-listed medicine
|
● Helps reduce homocysteine levels
● Supports nervous system function
● Promotes the mitochondrial energy production cycle
● Assists healthy red blood cell production
● Supports energy production
|
|
NanoCelle® D3
|
● D3 as Cholecalciferol ARTG-listed medicine
|
● Clinically trialed (ACTRN12616001027404)
● Helps prevent dietary vitamin D3 deficiency
● Applied to oral mucosa by passing absorptive barriers in gastrointestinal tract
● Supports healthy immune system function
● Water soluble composition not an emulsion
● Recommended for those with impaired digestive and absorptive capabilities
|
|
NanoCelle® D3+K2
|
● D3 as Cholecalciferol and K2 as Menaquinone
● ARTG-listed medicine
|
● Supports normal process of blood coagulation cascade
● Helps prevent dietary vitamin D deficiency
● Uses menaquinone-7 form of vitamin K2 for greater bioavailability and effect
● Supports healthy immune system function and helps stimulate a healthy immune system response
● Anti-inflammatory and relieves inflammation
● Helps support bone mineralization, bone growth and bone density
|
76
Investigational NanoCelle® Cannabinoid Medications
In additional to our NanoCelle® delivery platforms, we have developed several proprietary cannabinoid-based medications, each of which we
believe can be effectively administered through the NanoCelle®. Both NanaBis™ and NanoCBD™ are unapproved medicines in Australia that are currently undergoing investigation under the Special Access Scheme.
Special Access Scheme
Most therapeutic goods are required to undergo an evaluation for quality, safety and efficacy and be included in the ARTG before they can
be supplied in Australia. In recognition that there are circumstances where patients need access to therapeutic goods that are not included in the ARTG, the TGA manages the Special Access Scheme. In addition, the Company can benefit from patients
access to therapeutic goods through the Special Access Scheme by gaining access to data that may be used by the Company for future marketing and regulatory purposes.
The Special Access Scheme allows registered health practitioners to access therapeutic goods (such as medicines, medical devices or
biologicals) that are not included in ARTG for a single patient via approval by the TGA on a named patient basis. Therapeutic goods that are not included in the ARTG (and are not otherwise exempt from being in the ARTG) are described by us as
“unapproved”.
NanaBis™
We believe the world needs a new, innovative solution towards pain management and reducing the reliance on opioids. We aimed to develop a
drug that treats cancer and non-cancer pain to directly compete against the US$42 billion opioid market. NanaBis™ is a viable, multi-jurisdictional patent-protected non-opioid analgesic, initially designated to treat cancer bone pain.
NanaBis™ consists of a 1:1 mixture of CBD and delta-9- THC in a 1:1 ratio of 9.62 mg/mL CBD to 9.62 mg/mL THC. The formulation is a synthetic, standardized 1:1 blend of CBD and dronabinol, the delta 9 isomer of THC, in a patented submicron
particle delivery platform optimized for buccal delivery.
Pain is classified as a chronic illness and results in loss of economic productivity in the United States of approximately US$3.7 trillion
each year, per the American Action Forum. In 2010, the National Academy of Sciences estimated that more than 100 million American individuals suffered from pain, resulting in estimated costs from health care and missed work time of approximately
US$3.7 trillion per year. Traditionally, pain management has centered around opioid use. Over 81,000 drug overdose deaths occurred in the United States in the 12 months ending in May 2020, the highest number of overdose deaths ever recorded in a
12-month period, according to recent provisional data from the Centers for Disease Control and Prevention.
77
While overdose deaths were already increasing in the months preceding the COVID-19 pandemic, recent statistics suggest an acceleration of overdose deaths during the pandemic.
Synthetic opioids, primarily illicitly manufactured fentanyl, appear to be the primary driver of the increases in overdose deaths. The AMA has embarked on measures to mitigate the opioid overdose epidemic and recognizes that it is imperative to
develop a new and safer alternative solution.
78
79

Since the outbreak of COVID-19 in March 2020, we have been focused on scalable manufacturing and enhancing the CMC package. Presently, we
have two FDA drug master files for the two cannabinoid compounds, THC and CBD, each consisting of 100% pure cannabinoid synthetic. We are preparing for the Phase III clinical trial for NanaBis™, which has an anticipated study completion
of 2024.
The Phase III trial for NanaBis™, which is expected to include 360 participants and to be located in the United States, will
use a randomized-withdrawal method to demonstrate the buccal administration of cannabis as an adjunct to opioid analgesia for the management of intractable pain in patients diagnosed with advanced cancer. The Phase III study will test cancer
patients, aged 18 to 70 in adult males and females. The primary endpoints are to: (i) demonstrate that at the end of the 6-week study period the proportion of responders in the NanaBis™ treated group is significantly greater than the
proportion of responders in the placebo group; and (ii) demonstrate that at the end of the 6-week study period the proportion of responders in the NanaBis™ group is similar to the proportion of responders in the Oxycodone group. The
secondary endpoints are to: (i) demonstrate that at the end of the 6-week study period the HR-QoL scores in the NanaBis™ treated group are significantly greater than in the placebo group and non-inferior to the Oxycodone-treated group;
(ii) demonstrate that NanaBis™ is tolerable; and (iii) demonstrate that half or more of the NanaBis™ treated group preferred further treatment with NanaBis™ in the open label extension study.
An observational study conducted under the Clinical Trial Notification scheme of the ATRG observational study to test the effects of
NanaBis™ has recruited approximately 1100 Australian patients of which 119 patients who have completed six and twelve-month administrations of the drug demonstrated 55% reduction in pain scores, with significant quality improvements in
sleep, mood, and general activities. Of the 119 patients who completed the six and twelve-month courses of NanaBis™, approximately 50% had muscular or neuropathic pain, 32% had soft tissue pain with muscular pain, 11% had visceral pain,
and 7% registered “other categorized pain.” A preliminary analysis of their Brief Pain Inventory scores after six and twelve months showed improvement in mean pain scores from 6.5 at the commencement of the study to 5.5 at six months and 4.7 at
twelve months. We believe the results of the study support tolerability of NanaBis™.
On February 23, 2021, we entered into a Project Scope Document contract with Renaissance whereby Renaissance will manufacture NanaBis™
in accordance with both our instructions and specifications and FDA regulations. On July 9, 2021, we entered into a Master Services Agreement with WEP Clinical Ltd. for the exclusive development and delivery of Named Patient Programs, a type of
compassionate use program, relating to the unlicensed supply of NanaBis™ and NanoCBD™ to patients in the United Kingdom and European Union. This represented our first partnership to supply our cannabinoid medications outside of
our current Australian Special Assess Scheme.
In November 2021, we announced that we received an award of approximately A$12 million from an “Advanced and Overseas Finding” award from
the federal government of Australia’s Research and Development Tax Incentive program for the further development of NanaBis™. We believe that the outcome of the Advance and Overseas Finding will significantly extend the company’s cash
runway, as well as its overall financial performance.
80
Drug Master Files
Drug master files are submissions to the FDA used to provide confidential, detailed information about
facilities, processes, or articles used in the manufacturing, processing, packaging, and storing of human drug products. The FDA will assess the drug master file with the Company’s overall CMC package and clinical data packages. The drug master file forms part of the CMC package, which is a significant FDA requirement for a new drug application. The confidential information submitted to the FDA via a drug master file will be reviewed when the FDA reviews the application that references the drug master file, such as a NDA or IND. To date, the Company has not made NDA or IND
submissions. The Company believes that drug master files are critical as they provide the FDA with necessary information that the Company will use in connection with future drug applications.
Australia’s Research and Development Tax Incentive Program
On October 28, 2021, the Company received acceptance to Australia’s Research and Development Tax Incentive program for overseas research
and development expenditures for the 2020-2021, 2021-2022 and 2022-2023 financial years.
To be eligible for the Australian Research and Development Tax Incentive program and the related overseas expenditure, the Company must
have a significant scientific link to the Australian activities. The Company’s current plan is to use the grant to develop and progress the NanaBis™ product to Phase III clinical trial studies and to develop the product in U.S. markets.
The award that the Company receives from the federal government of Australia’s Research and Development Tax Incentive program for the
further development of NanaBis™ does not have ongoing conditions or requirements apart from the standard eligibility requirements on the cost to be claimed as research and development rebates.
81
NanoCBD™
NanoCBD™ is a cannabinoid-related product aimed at treating stress and stress-like symptoms and is optimally delivered through
our NanoCelle® delivery platform. Our goal is to gain registration for the lawful supply of NanoCBD™ in Australian pharmacies as an over-the-counter medicine. The NanoCBD™ program is currently in the pre-clinical phase and
seeking approval from the TGA primarily with the FDA secondary. NanoCBD™ has been designed to share nearly all the same components used in NanaBis™, inclusive of the drug substance, CBD. NanoCBD™ is a non-TGA product
manufactured in the same FDA facility as NanaBis™ and available to certain approved patients through the TGA compassionate use program via the Special Access Scheme. Its formulation consists of 16.67 mg/mL synthetic CBD as an active
ingredient, in a sub-micron spray applied to the oro-buccal membrane.
Historically, occupational stress was a term used to describe “an undesired factor causing discomfort for healthcare workers,” and stress
within any workplace being mental, physical and/or emotional. Over the last few years, stress has been applied to scenarios beyond the workplace to include loss of income, loss of home, loss of life, and incarceration. The global stress management
treatments market is expected to reach US$20.6 billion by 2024 from US$17.2 billion in 2019 at a compound annual growth rate of 3.7% for the forecast period of 2019 to 2024.
Other Investigational NanoCelle® Programs
MDC2000 (formerly known as NRGBiotic™)
MDC2000 is a novel pharmaceutical program, formerly known as NRGBiotic™, used to produce specific compounds in the
gastrointestinal tract that reduce symptomatology of major depressive disorders and seeking approval from the FDA. We believe mental health continues to be recognized globally as a growing issue. Like opioids, the global mental health problem was
exacerbated by some 25% due to the recent COVID-19 pandemic. Data from the World Health Organization stated that in 2021, the percentage of the global population suffering from depression was 3.8%, and specifically 5.0% of adults. MDC2000 is a
product targeting major depressive disorders that has been optimized as a single molecule encapsulated in NanoCelle® following clinical studies in 2020. The 2020 studies were closed earlier than projected due to the COVID pandemic. Based on this
optimization the program has returned to a pre-clinical stage with clinical returns expected in early 2023.
We believe that the currently available antidepressant therapies do not effectively control or cure depressive symptoms. Furthermore,
refractory major depressive disorder is characterized by recurrent, long-lasting cycles of severe, often suicidal depressive episodes that do not remit using multiple types of antidepressant therapies.
On July 5, 2021, we announced that the Phase IIA trial results for MDC2000, conducted in the University of Technology, Queensland,
fulfilled its objectives and displayed that normalization of major depressive symptoms occurred in eight weeks when MDC2000 was used in conjunction with an anti-depressant medicine. Furthermore, the clinical evidence highlighted a significant
potential for optimization, easing the future regulatory burden and availing the product to a simpler manufacturing requirement.
Our research data found that in approximately 130 Australian patients, a normalization of major depressive symptoms occurred in eight weeks
when used in conjunction with an anti-depressant medicine. This was opposed to a placebo and anti-depressant medicine in a randomized, placebo-controlled, Phase II clinical study that was independently carried out by the Queensland University of
Technology.
82
Given that major depressive disorder (“MDD”) is a leading cause of disability worldwide; only 50% of patients with MDD respond to the first
trial of an antidepressant, and 36.8% of patients achieve remission after a first course of antidepressant treatment. Patients with MDD who respond partially to an adequate trial of antidepressant often need an augmentation agent to further optimize
the response. Our clinical trial with NRGBiotic™ as an adjunct to pharmacotherapy has provided such an augmentation response in patients diagnosed with MDD.
Our goal is for MDC2000 to become a more scalable product that is supportive of an FDA 505(b)(2) pathway.
Nanocelle® Nucleic Acid
On April 4, 2022, we entered into a Collaborative Research Agreement with the University of New South Wales and Macquarie University to conduct testing during the pre-clinical
stages for our NanoCelle®-Nucleic Acid, a nasal vaccine delivery utilizing nucleic acid to develop a new vaccine and/or anti-viral technologies. Pursuant to the Collaborative Research Agreement, all intellectual property created or developed under
such agreement is owned collectively by all parties to such agreement. Accordingly, we will not have full ownership over the intellectual property developed under the Collaborative Research Agreement, and any of our products incorporating such
intellectual property will be subject to the joint ownership rights under the agreement. We plan to combine NanoCelle® with nucleic acid and to administer NanoCelle® by the nose. Our goal for this collaboration is to develop a nasal NanoCelle®
COVID-19 vaccine with optionality to “spin-out” to other vaccines and/or anti-viral agents, effectively reducing the public health burden of administering medicines via injections.
While the program is embryotic, the nasal adherence work for the NanoCelle® platform was conducted using insulin, and the work demonstrated
strong properties for nasal adherence and delivery, thereby showing NanoCelle® was appropriate for nasal delivery.
NanoCelle®-Nucleic Acid is expected to be TGA-focused with the intention to utilize the Company’s NanoCelle delivery method for RNA
COVID-19 vaccines. The program goals are to deliver a validated proof of concept for a NanoCelle® nasal delivery of either an mRNA or SiRNA molecule to the Australian Government by year end 2022. Subsequently, it is expected that next steps and
potential funding would be made known at this time.
NanoCelle® was optimized to improve small molecules, however, the Nanocelle® Nucleic Acid program provides a unique opportunity
to demonstrate the NanoCelle® capabilities in large molecules, ultimately establishing NanoCelle® as a robust and diverse delivery platform for a majority of today’s medicines. Ultimately, the NanoCelle®-Nucleic Acid program will require approvals
from the TGA. Below is a summary of our developmental NanoCelle®-Nucleic Acid-based products:
|
Medlab Product Name
|
Target Market
|
Estimated Size of Target Market
|
Estimated U.S. Dollar Value of Potential Demand for Product
|
|
Nasal Nucleic Acid
|
Vaccines
|
N/A
|
US$ 61.0 billion
|
|
Nasal Nucleic Acid
|
Anti-Viral
|
56% of global marketplace
|
US$ 49.1 billion
|
|
Nasal Nucleic Acid
|
Insulin
|
425 million people globally
|
US$ 21.26 billion
|
Nutraceuticals
Since inception, we developed a portfolio of nutraceuticals, or fortified foods or dietary supplements, that provide health benefits in
addition to their basic nutritional value. Nutraceuticals is globally valued at approximately US$353 billion, with more people turning toward some form of supplementation as a potential preventative to a future disease state.
On October 19, 2021, the Company licensed its Australian nutraceutical business to PharmaCare for cash consideration of A$750,000. The principal assets that were licensed
comprised registered patents and trademarks, inventory, customer lists, and material contracts. In connection with the licensing deal, the Company sold its nutraceutical inventory to PharmaCare for $1,025,910.
83
Government Regulation
The FDA and other regulatory authorities at federal, state and local levels, as well as in foreign countries, extensively regulate, among
other things, the research, development, testing, manufacture, quality control, import, export, safety, effectiveness, labeling, packaging, storage, distribution, record keeping, approval, advertising, promotion, marketing, post-approval monitoring
and post-approval reporting of biologics such as those we are developing. We, along with third-party contractors, will be required to navigate the various preclinical, clinical and commercial approval requirements of the governing regulatory agencies
of the countries in which we wish to conduct studies or seek approval or licensure of our product candidates.
FDA
In the United States, pharmaceutical products are subject to extensive regulation by the FDA pursuant to the Federal Food, Drug and
Cosmetic Act, or FDCA. The FDCA and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution,
post-approval monitoring and reporting, sampling and import and export of pharmaceutical products. Failure to comply with applicable FDA or other requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA
refusal to approve pending applications, clinical holds, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, withdrawal of product from the market, injunctions, fines, civil
penalties and criminal prosecution.
FDA approval is required before any new unapproved drug or dosage form, including a new use of a previously approved drug, can be marketed
in the United States. The process required by the FDA before a new drug may be marketed in the United States generally involves:
| • |
completion of pre-clinical laboratory and animal testing and formulation studies;
|
| • |
submission to the FDA of an IND for human clinical testing which must become effective before human clinical trials may begin in the United States;
|
| • |
approval of clinical protocols and informed consent documents by an independent institutional review board, or IRB, at each clinical trial site before each clinical trial may be initiated;
|
| • |
performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug product for each intended use;
|
| • |
development of a manufacturing process, formulation and packaging processes;
|
| • |
satisfactory completion of an FDA pre-approval inspection of the facility or facilities at which the product is manufactured;
|
| • |
submission to the FDA of an NDA;
|
| • |
potential review by an FDA advisory committee, if applicable; and
|
| • |
FDA review and approval of the NDA.
|
84
Preclinical and Clinical Development
Prior to beginning the first clinical trial with a product candidate, we must submit an IND to the FDA. An IND is a request for
authorization from the FDA to administer an investigational new drug product to humans. The central focus of an IND submission is on the general investigational plan and the protocol or protocols for preclinical studies and clinical trials. The IND
also includes results of animal and in vitro studies assessing the toxicology, pharmacokinetics, pharmacology and pharmacodynamic characteristics of the product, chemistry, manufacturing and controls information, and any available human data or
literature to support the use of the investigational product. An IND must become effective before human clinical trials may begin. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day period,
raises safety concerns or questions about the proposed clinical trial. In such a case, the IND may be placed on clinical hold and the IND sponsor and the FDA must resolve any outstanding concerns or questions before the clinical trial can begin.
Submission of an IND therefore may or may not result in FDA authorization to begin a clinical trial.
In addition to the IND submission process, supervision of human gene transfer trials includes evaluation and assessment by an institutional
biosafety committee, or IBC, a local institutional committee that reviews and oversees research utilizing recombinant or synthetic nucleic acid molecules at that institution. The IBC assesses the safety of the research and identifies any potential
risk to public health or the environment and such review may result in some delay before initiation of a clinical trial.
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified
investigators in accordance with GCPs, which include the requirement that all research subjects provide their informed consent for their participation in any clinical study. Clinical trials are conducted under protocols detailing, among other things,
the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A separate submission to the existing IND must be made for each successive clinical trial conducted during product development
and for any subsequent protocol amendments. Furthermore, an independent IRB for each site proposing to conduct the clinical trial must review and approve the plan for any clinical trial and its informed consent form before the clinical trial begins
at that site, and must monitor the study until completed. Regulatory authorities, the IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health
risk or that the trial is unlikely to meet its stated objectives. Some studies also include oversight by an independent group of qualified experts organized by the clinical study sponsor, known as a data safety monitoring board, which provides
authorization for whether or not a study may move forward at designated check points based on access to certain data from the study and may halt the clinical trial if it determines that there is an unacceptable safety risk for subjects or other
grounds, such as no demonstration of efficacy. There are also requirements governing the reporting of ongoing preclinical studies and clinical trials and clinical study results to public registries.
The preclinical and clinical testing and approval process may take a number of years and the actual time required to obtain approval, if
any, may vary substantially based upon the product. The conduct of the preclinical tests must comply with federal and other regulations and requirements. The results of preclinical testing are submitted to the FDA as part of an IND along with other
information, including information about product chemistry, manufacturing and controls, analytical data, any available clinical data or literature to support the use of the investigational drug in humans and a proposed clinical trial protocol. The
IND may rely in part on information contained in already approved drug applications, where a 505(b)(2) NDA is planned.
85
Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, the parameters to be
used in monitoring safety, and the efficacy criteria to be evaluated. Sponsors of clinical trials generally must register and report, at the NIH-maintained website ClinicalTrials.gov, key parameters of certain clinical trials. For purposes of an NDA
submission and approval, human clinical trials are typically conducted in the following sequential phases, which may overlap or be combined:
| • |
Phase 1: In Phase 1, through the initial introduction of the drug into healthy human subjects or patients, the drug is tested to assess metabolism,
pharmacokinetics, pharmacological actions, side effects associated with increasing doses, and, if possible, early evidence on effectiveness.
|
| • |
Phase 2: Phase 2 usually involves trials in a limited patient population to determine the effectiveness of the drug for a particular indication, dosage
tolerance and optimum dosage and to identify common adverse effects and safety risks.
|
| • |
Phase 3: Phase 3 trials are undertaken to obtain the additional information about clinical efficacy and safety in a larger number of patients,
typically at geographically dispersed clinical trial sites, to permit the FDA to evaluate the overall benefit-risk relationship of the drug and to provide adequate information for the labeling of the drug. In most cases, the FDA requires two
adequate and well controlled clinical trials to confirm the safety and efficacy of the drug. A single Phase 3 trial with other confirmatory evidence may be sufficient in rare instances where the clinical trial is a large multicenter trial
demonstrating internal consistency and a statistically persuasive finding of a clinically meaningful effect on mortality, irreversible morbidity or prevention of a disease with a potentially serious outcome and confirmation of the result in a
second trial would be practically or ethically impossible.
|
After completion of the required clinical testing, an NDA is prepared and submitted to the FDA. FDA approval of the NDA is required before
marketing of the product may begin in the United States. The NDA must include, among other things, the results of all preclinical, clinical and other testing, including negative or ambiguous results as well as positive findings and a compilation of
data relating to the product’s pharmacology, chemistry, manufacture and controls, as well as proposed labeling.
Future changes to some of the conditions established in an approved application, including changes in indications, labeling, or
manufacturing processes or facilities, typically require submission and FDA approval of a new NDA or NDA supplement before the change can be implemented, which may require us to develop additional data or conduct additional pre-clinical studies and
clinical trials.
Section 505(b)(2) NDAs
As an alternative path to FDA approval for modifications to formulations or uses of products previously approved by the FDA, an applicant
may submit an NDA under Section 505(b)(2) of the FDCA. Section 505(b)(2) was enacted as part of the Hatch-Waxman Amendments to the FDCA and enables the applicant to rely, in part, on the FDA’s previous approval of a similar product, or published
literature, in support of its application. Section 505(b)(2) permits the filing of an NDA where at least some of the information required for approval comes from studies not conducted by, or for, the applicant and for which the applicant has not
obtained a right of reference. If the Section 505(b)(2) applicant can establish that reliance on FDA’s previous findings of safety and effectiveness is scientifically appropriate, it may eliminate the need to conduct certain preclinical studies or
clinical trials of the new product. The FDA may also require companies to perform additional studies or measurements, including clinical trials, to support the change from the approved reference drug. The FDA may then approve the new product
candidate for all, or some, of the label indications for which the reference drug has been approved or for any new indication sought by the Section 505(b)(2) applicant.
86
ANDA Approval Process
The Hatch-Waxman Amendments also established an abbreviated FDA approval process for drugs that are shown to be bioequivalent to drugs
previously approved by the FDA through the NDA process. Approval to market and distribute these drugs is obtained by filing an abbreviated new drug application, or ANDA, with the FDA. An ANDA provides for marketing of a drug product that has the same
active ingredients in the same strengths and dosage form as the listed drug and has been shown to be bioequivalent to the listed drug. An ANDA is a comprehensive submission that contains, among other things, data and information pertaining to the
active pharmaceutical ingredient, drug product formulation, specifications and stability of the generic drug, as well as analytical methods, manufacturing process validation data and quality control procedures. ANDAs are termed abbreviated because
they generally do not include preclinical and clinical data to demonstrate safety and effectiveness. Instead, a generic applicant must demonstrate that its product is bioequivalent to the innovator drug. Drugs approved in this way are commonly
referred to as “generic equivalents” to the listed drug and can often be substituted by pharmacists under prescriptions written for the original listed drug.
Orange Book Listing
In seeking approval for a drug through an NDA, including a 505(b)(2) NDA, applicants are required to list with the FDA certain patents
whose claims cover the applicant’s product. Upon approval, each of the patents listed in the application for the drug is then published in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book.
Any applicant who files an ANDA seeking approval of a generic equivalent version of a drug listed in the Orange Book or a Section 505(b)(2) NDA referencing a drug listed in the Orange Book must certify to the FDA that (1) no patent information on the
drug product that is the subject of the application has been submitted to the FDA; (2) such patent has expired; (3) the date on which such patent expires; or (4) such patent is invalid or will not be infringed upon by the manufacture, use or sale of
the drug product for which the application is submitted. This last certification is known as a paragraph IV certification. A notice of the paragraph IV certification must be provided to each owner of the patent that is the subject of the
certification and to the holder of the approved NDA to which the ANDA or Section 505(b)(2) application refers. The applicant may also elect to submit a “section viii” statement certifying that its proposed label does not contain (or carves out) any
language regarding the patented method-of-use rather than certify to a listed method-of-use patent.
DEA Regulation
The active ingredients in our current drug product candidates are listed by the U.S. Drug Enforcement Administration, or DEA, as controlled
substances under the Controlled Substances Act of 1970, or CSA. The CSA and its implementing regulations establish a closed chain of distribution for entities handling controlled substances and impose registration, record-keeping and reporting,
security, storage, procurement, manufacturing, distribution, importation, exportation, labeling, packaging, and other requirements on such entities. The DEA requires individuals or entities that handle controlled substances to comply with these
requirements to ensure legitimate use and prevent diversion of controlled substances to illicit channels of commerce.
The CSA categorizes controlled substances into one of five schedules, Schedule I, II, III, IV or V, depending on the potential for abuse
and physical or psychological dependence. Schedule I substances by definition have a high potential for abuse, have no currently accepted medical use in treatment in the U.S. and lack accepted safety for use under medical supervision. They may not be
marketed or sold for dispensing to patients in the U.S. Pharmaceutical products having a currently accepted medical use and that are otherwise approved for marketing may be listed as Schedule II, III, IV, or V substances, with Schedule II substances
presenting the highest potential for abuse and physical or psychological dependence, and Schedule V substances presenting the lowest relative potential for abuse and dependence. Schedule II substances (as well as substances defined as narcotics in
any Schedule) are subject to the strictest requirements for registration, security, recordkeeping and reporting, and the distribution and dispensing of these substances are highly regulated. For example, all Schedule II drug prescriptions must be
signed by a physician, physically presented to a pharmacist in most situations, unless they are electronically prescribed pursuant to DEA regulations, and may not be refilled. The active ingredients in our product candidates (dexmethylphenidate and
dextroamphetamine) are Schedule II controlled substances and are under various restrictions. Consequently, the procurement, manufacturing, shipping, storage, sales and use of the products, if approved, will be subject to a high degree of regulation.
87
Facilities that manufacture, distribute, import or export controlled substances must register annually with the DEA. The registration is
specific to the particular location, activity and controlled substance schedule. For example, separate registrations are needed for import and manufacturing, and each registration will specify which schedules of controlled substances are authorized.
Similarly, separate registrations are also required for separate facilities.
The DEA inspects manufacturers, distributors, importers, and exporters to review compliance with the CSA and DEA regulations, including
security, record keeping and reporting prior to issuing a controlled substance registration and on a periodic basis. The specific security requirements vary by the type of business activity and the schedule and quantity of controlled substances
handled by the registrant, with the most stringent requirements applying to Schedule I and Schedule II substances. Required security measures include background checks on employees and physical control of inventory through measures such as vaults and
inventory reconciliations. Manufacturers and distributors must also submit regular reports to the DEA of the distribution of Schedule I and II controlled substances, Schedule III narcotic substances, and other designated substances. Records must be
maintained for the handling of all controlled substances, for example, a complete and accurate record of each substance manufactured, received, sold, delivered, or otherwise disposed of. All DEA registrants must also report any controlled substance
thefts or significant losses and must obtain authorization to destroy or dispose of controlled substances. In addition to maintaining an importer and/or exporter registration, importers and exporters of controlled substances must obtain a permit for
every import or export of a Schedule I or II substance and a narcotic substance in Schedule III, IV and V. For all other drugs in Schedule III, IV and V, importers and exporters must submit an import or export declaration.
In addition, a DEA quota system controls and limits the availability and production of controlled substances in Schedule I or II. The DEA
establishes annually an aggregate quota for how much of a controlled substance may be produced in total in the United States based on the DEA’s estimate of the quantity needed to meet legitimate scientific and medicinal needs. The limited aggregate
number of opioids and stimulants that the DEA allows to be produced in the United States each year is allocated among individual companies, which must submit applications annually to the DEA for individual production and procurement quotas. We must
receive an annual quota from the DEA in order to produce or procure our Schedule II substance for use in manufacturing of our product and product candidates. The DEA may adjust aggregate production quotas and individual production and procurement
quotas from time to time during the year, although the DEA has substantial discretion in whether or not to make such adjustments. Distributions of any Schedule I or II controlled substance must also be accompanied by special order forms, with copies
provided to the DEA.
Failure to maintain compliance with applicable DEA requirements, particularly as manifested in loss or diversion or controlled substances,
can result in administrative, civil or criminal enforcement action. The DEA may seek civil penalties, refuse to renew necessary registrations, or initiate administrative proceedings to revoke those registrations. In some circumstances, violations
could lead to criminal prosecution.
The various states and the District of Columbia, also regulate controlled substances and impose similar licensing, recordkeeping, and
reporting requirements on entities that handle controlled substances. Entities must independently comply with the various state requirements in addition to the federal controlled substance requirements.
88
Other Healthcare Laws and Compliance Requirements
Pharmaceutical companies are subject to additional healthcare regulation and enforcement by the federal government and by authorities in
the states and foreign jurisdictions in which they conduct their business. Such laws include, without limitation: the federal Anti-Kickback Statute, the federal False Claims Act, HIPAA and similar foreign, federal and state fraud, abuse and
transparency laws.
The federal Anti-Kickback Statute prohibits, among other things, persons and entities from knowingly and willfully soliciting, receiving,
offering or paying remuneration, to induce, or in return for, either the referral of an individual, or the purchase or recommendation of an item or service for which payment may be made under any federal healthcare program. The term remuneration has
been interpreted broadly to include anything of value, including stock options. The federal Anti-Kickback Statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand, and prescribers and purchasers on the
other. There are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, but they are drawn narrowly and practices that involve remuneration, such as consulting agreements, that may be alleged
to be intended to induce prescribing, purchasing or recommending may be subject to scrutiny if they do not qualify for an exception or safe harbor. Our practices may not in all cases meet all of the criteria for protection under a statutory exception
or regulatory safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the federal Anti-Kickback Statute. Instead, the legality of the
arrangement will be evaluated on a case-by-case basis based on a cumulative review of all of its facts and circumstances. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have
committed a violation.
Civil and criminal false claims laws, including the federal False Claims Act, and civil monetary penalty laws, which can be enforced
through civil whistleblower or qui tam actions, prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment to the federal government, including federal healthcare programs, that are
false or fraudulent. For example, the federal False Claims Act prohibits any person or entity from knowingly presenting, or causing to be presented, a false claim for payment to the federal government or knowingly making, using or causing to be made
or used a false record or statement material to a claim to the federal government. A claim includes “any request or demand” for money or property presented to the U.S. government. Several pharmaceutical and other healthcare companies have been
prosecuted under these laws for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. In addition, the government may assert that a claim resulting from a violation of the
federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act.
HIPAA created additional federal criminal statutes that prohibit, among other things, executing a scheme to defraud any healthcare benefit
program, including private third-party payors, and making false statements relating to healthcare matters. A person or entity does not need to have actual knowledge of the healthcare fraud statute implemented under HIPAA or specific intent to violate
the statute in order to have committed a violation.
The FDCA addresses, among other things, the design, production, labeling, promotion, manufacturing, and testing of drugs, biologics and
medical devices, and prohibits such acts as the introduction into interstate commerce of adulterated or misbranded drugs or devices. The U.S. Public Health Service Act also prohibits the introduction into interstate commerce of unlicensed or
mislabeled biological products.
The U.S. federal Physician Payments Sunshine Act requires certain manufacturers of drugs, devices, biologics and medical supplies for which
payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, to annually report to CMS information related to payments or other transfers of value made to physicians and teaching hospitals, as
well as ownership and investment interests held by physicians and their immediate family members. Beginning in 2022, such reporting obligations will be expanded to include payments and other transfers of value provided in 2021 to certain other
healthcare professionals, including physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists, and certified nurse-midwives.
89
We are also subject to additional similar U.S. state and foreign law equivalents of each of the above federal laws, which, in some cases,
differ from each other in significant ways, and may not have the same effect, thus complicating compliance efforts. If our operations are found to be in violation of any of such laws or any other governmental regulations that apply, we may be subject
to penalties, including, without limitation, civil, criminal and administrative penalties, damages, fines, exclusion from government-funded healthcare programs, such as Medicare and Medicaid or similar programs in other countries or jurisdictions,
integrity oversight and reporting obligations to resolve allegations of non-compliance, disgorgement, individual imprisonment, contractual damages, reputational harm, diminished profits and the curtailment or restructuring of our operations.
Data Privacy and Security
Numerous state, federal and foreign laws, govern the collection, dissemination, use, access to, confidentiality and security of personal
information, including health-related information. In the United States, numerous federal and state laws and regulations, including state data breach notification laws, state health information privacy laws, and federal and state consumer protection
laws and regulations, govern the collection, use, disclosure, and protection of health-related and other personal information could apply to our operations or the operations of our partners. For example, HIPAA, as amended by HITECH, and their
respective implementing regulations, imposes privacy, security and breach notification obligations on certain health care providers, health plans, and health care clearinghouses, known as covered entities, as well as their business associates that
perform certain services that involve creating, receiving, maintaining or transmitting individually identifiable health information for or on behalf of such covered entities. Entities that are found to be in violation of HIPAA may be subject to
significant civil, criminal and administrative fines and penalties and/or additional reporting and oversight obligations if required to enter into a resolution agreement and corrective action plan with HHS to settle allegations of HIPAA
non-compliance. Further, entities that knowingly obtain, use, or disclose individually identifiable health information maintained by a HIPAA covered entity in a manner that is not authorized or permitted by HIPAA may be subject to criminal penalties.
Even when HIPAA does not apply, according to the FTC, violating consumers’ privacy rights or failing to take appropriate steps to keep
consumers’ personal information secure may constitute unfair acts or practices in or affecting commerce in violation of Section 5(a) of the Federal Trade Commission Act.
In addition, certain state and non-U.S. laws, such as the GDPR govern the privacy and security of personal information, including
health-related information, in certain circumstances. Failure to comply with these laws, where applicable, can result in the imposition of significant civil and/or criminal penalties and private litigation. For example, the CCPA, which went into
effect on January 1, 2020, creates new data privacy obligations for covered companies and provides new privacy rights to California residents. In Europe, the GDPR went into effect in May 2018 and introduces strict requirements for processing the
personal data of individuals within the EEA. Further, recent legal developments in Europe have created complexity and compliance uncertainty regarding certain transfers of personal data from the EEA. For example, on July 16, 2020, the CJEU
invalidated the Privacy Shield under which personal data could be transferred from the EEA to United States entities who had self-certified under the Privacy Shield scheme. Moreover, it is uncertain whether the standard contractual clauses will also
be invalidated by the European courts or legislature
Companies that must comply with the GDPR face increased compliance obligations and risk, including more robust regulatory enforcement of
data protection requirements and potential fines for noncompliance of up to €20 million or 4% of the annual global revenues of the noncompliant company, whichever is greater. Additionally, following the United Kingdom’s withdrawal from the European
Union and the EEA, companies have to comply with the GDPR and the GDPR as incorporated into United Kingdom national law, the latter regime having the ability to separately fine up to the greater of £17.5 million or 4% of global turnover. The
relationship between the United Kingdom and the European Union in relation to certain aspects of data protection law remains unclear, for example around how data can lawfully be transferred between each jurisdiction, which exposes us to further
compliance risk.
90
Coverage and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any pharmaceutical product for which we obtain regulatory
approval. Sales of any product, if approved, depend, in part, on the extent to which such product will be covered by third-party payors, such as federal, state, and foreign government healthcare programs, commercial insurance and managed healthcare
organizations, and the level of reimbursement, if any, for such product by third-party payors. Decisions regarding whether to cover any of our product candidates, if approved, the extent of coverage and amount of reimbursement to be provided are made
on a plan-by-plan basis. Further, no uniform policy for coverage and reimbursement exists in the United States, and coverage and reimbursement can differ significantly from payor to payor. Third-party payors often rely upon Medicare coverage policy
and payment limitations in setting their own reimbursement rates, but also have their own methods and approval process apart from Medicare determinations.
As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and
clinical support for the use of our product candidates to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance.
For products administered under the supervision of a physician, obtaining coverage and adequate reimbursement may be particularly difficult
because of the higher prices often associated with such drugs. Additionally, separate reimbursement for the product itself or the treatment or procedure in which the product is used may not be available, which may impact physician utilization. In
addition, companion diagnostic tests require coverage and reimbursement separate and apart from the coverage and reimbursement for their companion pharmaceutical or biological products. Similar challenges to obtaining coverage and reimbursement,
applicable to pharmaceutical or biological products, will apply to companion diagnostics.
In addition, the U.S. government, state legislatures and foreign governments have continued implementing cost-containment programs,
including price controls, restrictions on coverage and reimbursement and requirements for substitution of generic products. Third-party payors are increasingly challenging the prices charged for medical products and services, examining the medical
necessity and reviewing the cost effectiveness of pharmaceutical or biological products, medical devices and medical services, in addition to questioning safety and efficacy. Adoption of price controls and cost-containment measures, and adoption of
more restrictive policies in jurisdictions with existing controls and measures, could further limit sales of any product that receives approval. Decreases in third-party reimbursement for any product or a decision by a third-party not to cover a
product could reduce physician usage and patient demand for the product.
91
Healthcare Reform
The United States and some foreign jurisdictions are considering or have enacted a number of reform proposals to change the healthcare
system. There is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality or expanding access. In the United States, the pharmaceutical industry has been a particular
focus of these efforts and has been significantly affected by federal and state legislative initiatives, including those designed to limit the pricing, coverage, and reimbursement of pharmaceutical and biopharmaceutical products, especially under
government-funded health care programs, and increased governmental control of drug pricing.
The ACA, which was enacted in March 2010, substantially changed the way healthcare is financed by both governmental and private insurers in
the United States, and significantly affected the pharmaceutical industry. The ACA contains a number of provisions of particular import to the pharmaceutical and biotechnology industries, including, but not limited to, those governing enrollment in
federal healthcare programs, a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, and annual fees based on pharmaceutical
companies’ share of sales to federal health care programs. Since its enactment, there have been judicial and Congressional challenges to certain aspects of the ACA, and we expect there will be additional challenges and amendments to the ACA in the
future. For example, the Tax Act was enacted, which, among other things, removes penalties for not complying with ACA’s requirement to carry health insurance, known as the “individual mandate,” effective January 1, 2019. Since the enactment of the
Tax Act, there have been additional amendments to certain provisions of the ACA. In December 2019, the U.S. District Court for the Fifth Circuit upheld a ruling by a Texas U.S. District Court Judge that the ACA is unconstitutional in its entirety
because the “individual mandate” was repealed by Congress as part of the Tax Act. On March 2, 2020, the Supreme Court of the United States granted certiorari to hear the appeal of this decision. While various parties, including the Trump
administration and CMS have stated that the ruling will have no immediate effect, it is unclear how this and other efforts to repeal and replace the ACA will impact the ACA.
Other legislative changes have been proposed and adopted since the ACA was enacted, including automatic aggregate reductions of Medicare
payments to providers of 2% per fiscal year as part of the federal budget sequestration under the Budget Control Act of 2011. These reductions went into effect in April 2013 and, due to subsequent legislative amendments, will remain in effect through
2030 with the exception of a temporary suspension from May 1, 2020 through December 31, 2020, unless additional action is taken by Congress.
Moreover, there has recently been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed
products, which has resulted in several Congressional inquiries and proposed and enacted federal and state measures designed to, among other things, reduce the cost of prescription drugs, bring more transparency to product pricing, review the
relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. For example, in May 2019, CMS adopted a final rule allowing Medicare Advantage Plans the option to use step
therapy for Part B drugs, permitting Medicare Part D plans to apply certain utilization controls to new starts of five of the six protected class drugs, and requiring the Explanation of Benefits for Part D beneficiaries to disclose drug price
increases and lower cost therapeutic alternatives beginning January 1, 2021. In addition, on March 10, 2020, the Trump administration sent “principles” for drug pricing to Congress, calling for legislation that would, among other things, cap Medicare
Part D beneficiary out-of-pocket pharmacy expenses, provide an option to cap Medicare Part D beneficiary monthly out-of-pocket expenses, and place limits on pharmaceutical price increases.
Congress and the Trump administration have each indicated that they will continue to seek new legislative and/or administrative measures to
control drug costs. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on
certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
92
Other Government Regulation Outside of the United States
In addition to regulations in the United States, we are subject to a variety of regulations in other jurisdictions governing, among other
things, research and development, clinical trials, testing, manufacturing, safety, efficacy, quality control, labeling, packaging, storage, record keeping, distribution, reporting, export and import, advertising, marketing and other promotional
practices involving biological products as well as authorization, approval as well as post-approval monitoring and reporting of our products. Because biologically sourced raw materials are subject to unique contamination risks, their use may be
restricted in some countries.
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in foreign
countries prior to the commencement of clinical trials or marketing of the product in those countries. Certain countries outside of the United States have a similar process that requires the submission of a clinical trial application, or a CTA, much
like the IND prior to the commencement of human clinical trials.
The requirements and process governing the conduct of clinical trials, including requirements to conduct additional clinical trials,
product licensing, safety reporting, post-authorization requirements, marketing and promotion, interactions with healthcare professionals, pricing and reimbursement may vary widely from country to country. No action can be taken to market any product
in a country until an appropriate approval application has been approved by the regulatory authorities in that country. The current approval process varies from country to country, and the time spent in gaining approval varies from that required for
FDA approval. In certain countries, the sales price of a product must also be approved. The pricing review period often begins after market approval is granted. Even if a product is approved by a regulatory authority, satisfactory prices may not be
approved for such product, which would make launch of such products commercially unfeasible in such countries.
Regulation in the European Union
Drug and Biologic Development Process
The conduct of clinical trials is currently governed by the EU Clinical Trials Directive 2001/20/EC, or Clinical Trials Directive, and will
be replaced by the EU Clinical Trials Regulation (EU) No. 536/2014, or Clinical Trials Regulation, once the latter comes into effect. The Clinical Trials Regulation introduces a complete overhaul of the existing regulation of clinical trials for
medicinal products in the EU. Currently it is not expected to come into force before December 2021.
Under the current regime, before a clinical trial can be initiated, it must be approved in each EU Member State where there is a site at
which the trial is to be conducted. The approval must be obtained from two separate entities: the National Competent Authority, or NCA, and one or more Ethics Committees. The NCA of the EU Member States in which the clinical trial will be conducted
must authorize the conduct of the trial, and the independent Ethics Committee must grant a positive opinion in relation to the conduct of the clinical trial in the relevant EU Member State before the commencement of the trial. Any substantial changes
to the trial protocol or other information submitted with the clinical trial applications must be submitted to or approved by the relevant NCA and Ethics Committees. Under the current regime all suspected unexpected serious adverse reactions to the
investigated drug that occur during the clinical trial must be reported to the NCA and to the Ethics Committees of the EU Member State where they occur.
A more unified procedure will apply under the new Clinical Trials Regulation. A sponsor will be able to submit a single application for
approval of a clinical trial through a centralized EU clinical trials portal. One national regulatory authority (the reporting EU Member State proposed by the applicant) will take the lead in validating and evaluating the application and the other
regulatory authorities will have limited involvement. If an application is rejected, it may be amended and resubmitted through the EU clinical trials portal. If an approval is issued, the sponsor may start the clinical trial in all concerned Member
States. However, a concerned EU Member State may in limited circumstances declare an “opt-out” from an approval and prevent the clinical trial form being conducted in such Member State. The Clinical Trials Regulation also aims to streamline and
simplify the rules on safety reporting, and introduces enhanced transparency requirements such as mandatory submission of a summary of the clinical trial results to the EU Database.
93
Under both the current regime and the new Clinical Trials Regulation, national laws, regulations, and the applicable Good Clinical Practice
and Good Laboratory Practice standards must also be respected during the conduct of the trials, including the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use, or ICH, guidelines on Good Clinical
Practice, or GCP, and the ethical principles that have their origin in the Declaration of Helsinki.
During the development of a medicinal product, the European Medical Agency, or EMA, and national regulators within the EU provide the
opportunity for dialogue and guidance on the development program, usually in the form of scientific advice. A fee is incurred with each scientific advice procedure. Advice from the EMA is typically provided based on questions concerning, for example,
quality (chemistry, manufacturing and controls testing), nonclinical testing and clinical studies, and pharmacovigilance plans and risk-management programs.
Drug Marketing Authorization
In the European Union, medicinal products, including advanced therapy medicinal products, or ATMPs, are subject to extensive pre- and
post-market regulation by regulatory authorities at both the European Union and national levels. ATMPs comprise gene therapy products, somatic cell therapy products and tissue engineered products, which are genes, cells or tissues that have undergone
substantial manipulation and that are administered to human beings in order to cure, diagnose or prevent diseases or regenerate, repair or replace a human tissue. We anticipate that our gene therapy development products will be regulated as ATMPs in
the European Union under the EU Regulation (EC) No 1394/2007 on advanced therapy medicinal products, or ATMP Regulation. Pursuant to the ATMP Regulation, the Committee on Advanced Therapies, or CAT, is responsible in conjunction with the CHMP for the
evaluation of ATMPs. The CHMP and CAT are also responsible for providing guidelines on ATMPs. These guidelines provide additional guidance on the factors that the EMA will consider in relation to the development and evaluation of ATMPs and include,
among other things, the preclinical studies required to characterize ATMPs; the manufacturing and control information that should be submitted in a marketing authorization application; and post-approval measures required to monitor patients and
evaluate the long term efficacy and potential adverse reactions of ATMPs. Although such guidelines are not legally binding, compliance with them is often necessary to gain and maintain approval for product candidates.
In the European Union and in Iceland, Norway and Liechtenstein (together the European Economic Area, or EEA), after completion of all
required clinical testing, medicinal products may only be placed on the market after a related Marketing Authorization, or MA, has been granted. MAs can be obtained through, amongst others, a centralized procedure, which is compulsory for certain
medicinal products such as ATMPs. The centralized procedure provides for the grant of a single MA by the European Commission, or EC, that is valid for all 27 EU Member States and, after respective national implementing decisions, in the three
additional EEA Member States (Iceland, Norway and Liechtenstein). The centralized procedure is compulsory for certain medicinal products, including medicinal products derived from biotechnological processes, orphan medicinal products, ATMPs and
products with a new active substance indicated for the treatment of AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and viral diseases. It is optional for medicinal products containing a new active substance not yet authorized in the
EEA before May 20, 2004, that constitute significant therapeutic, scientific or technical innovations, or for which the grant of a MA through the centralized procedure would be in the interest of public health at EU level. The timeframe for the
evaluation of an application under the centralized procedure is 210 days, excluding clock stops. Typically, the overall process takes a year or more unless the application is eligible for an accelerated assessment.
All new marketing authorization applications must include a Risk Management Plan, or RMP, describing the risk management system that the
company will put in place and documenting measures to prevent or minimize the risks associated with the product. The regulatory authorities may also impose specific obligations as a condition of the MA. RMPs and Periodic Safety Update Reports, or
PSURs, are routinely available to third parties requesting access, subject to limited redactions.
Additionally, the holder of a marketing authorization for an ATMP must put in place and maintain a system to ensure that each individual
product and its starting and raw materials, including all substances coming into contact with the cells or tissues it may contain, can be traced through the sourcing, manufacturing, packaging, storage, transport and delivery to the relevant
healthcare institution where the product is used.
MAs have an initial duration of five years. The authorization may subsequently be renewed for an unlimited period unless the EC or the
national competent authority grants only a five-year renewal.
94
Data and Market Exclusivity
As in the United States, the European Union also provides opportunities for market and/or data exclusivity. For example, new Chemical
Entities, or NCE, approved in the European Union generally qualify for eight years of data exclusivity and ten years of market exclusivity. Data exclusivity is the period during which another applicant cannot rely on the MA holder’s pharmacological,
toxicological and clinical data in support of another MA for the purposes of submitting an application, obtaining marketing authorization or placing the product on the market. But after eight years, a generic or biosimilar product application may be
submitted and generic companies may rely on the MA holder’s data.
However, even if a generic or biosimilar product is authorized it cannot be placed on the market in the European Union until the expiration
of the 10-year market exclusivity period. An additional non-cumulative one-year period of marketing exclusivity is possible if during the data exclusivity period (the first eight years of the 10-year marketing exclusivity period), the MA holder
obtains an authorization for one or more new therapeutic indications that are deemed to bring a significant clinical benefit compared to existing therapies.
Products may not be granted data exclusivity since there is no guarantee that a product will be considered by the European Union’s
regulatory authorities to include a NCE. Even if a compound is considered to be a NCE and the MA applicant is able to gain the prescribed period of data exclusivity, another company nevertheless could also market another version of the medicinal
product if such company can complete a full marketing authorization application with their own complete database of pharmaceutical tests, preclinical studies and clinical trials and obtain MA of its product.
Orphan Designation and Exclusivity
The criteria for designating an orphan medicinal product in the European Union are similar in principle to those in the United States. The
EMA grants orphan drug designation if the medicinal product is intended for the diagnosis, prevention or treatment of (i) a life-threatening or chronically debilitating condition affecting no more than five in 10,000 persons in the European Union
when the application is made or a life-threatening, seriously debilitating or serious and chronic condition in the European Union and that without the benefits derived from orphan status, would not generate sufficient return in the European Union to
justify investment and (ii) where there exists no satisfactory method of diagnosis, prevention or treatment of such condition authorized in the European Union, or if such a method exists, the product will be of significant benefit to those affected
by the condition. Orphan medicinal products are eligible for financial incentives such as reduction of fees or fee waivers and are, upon grant of a marketing authorization that covers only the therapeutic indication(s) that meet the orphan drug
designation criteria, entitled to ten years of market exclusivity for the approved therapeutic indication. An application for orphan drug designation must be submitted first before an application for marketing authorization of the medicinal product
is submitted. The applicant will receive a fee reduction for the marketing authorization application if the orphan drug designation has been granted, but not if the designation is still pending at the time the marketing authorization is submitted.
Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
During the 10-year period of market exclusivity, with a limited number of exceptions, the regulatory authorities of the EU Member States
and the EMA may not accept applications for marketing authorization, accept an application to extend an existing marketing authorization or grant marketing authorization for other similar medicinal products for the same therapeutic indication. A
similar medicinal product is defined as a medicinal product containing a similar active substance or substances as contained in a currently authorized orphan medicinal product, and which is intended for the same therapeutic indication. An orphan
medicinal product can also obtain an additional two years of market exclusivity for an orphan-designated condition when the results of specific studies are reflected in the Summary of Product Characteristics, or SmPC, addressing the pediatric
population and completed in accordance with a fully compliant Pediatric Investigation Plan, or PIP. No extension to any supplementary protection certificate can be granted on the basis of pediatric studies for orphan indications.
The 10-year market exclusivity may be reduced to six years if, at the end of the fifth year, it is established that the product no longer
meets the criteria for orphan designation, for example, if the product is sufficiently profitable not to justify maintenance of market exclusivity. Additionally, a marketing authorization may be granted to another medicinal product (orphan or not)
for the same or overlapping indication at any time subject to certain requirements.
95
Pediatric Development
In the European Union, companies developing a new medicinal product are obligated to study their product in children and must therefore
agree upon a PIP with the EMA’s Pediatric Committee. The companies must conduct pediatric clinical trials in accordance with that PIP, unless a waiver applies, e.g., because the relevant disease or condition occurs only in adults. The marketing
authorization application for the product must include the results of pediatric clinical trials conducted in accordance with the PIP, unless a waiver applies, or a deferral has been granted, in which case the pediatric clinical trials must be
completed at a later date. Products that are granted a marketing authorization on the basis of the pediatric clinical trials conducted in accordance with the PIP are eligible for a six month extension of the protection under a supplementary
protection certificate (if any is in effect at the time of approval) or, in the case of orphan medicinal products, a two year extension of the orphan market exclusivity. This pediatric reward is subject to specific conditions and is not automatically
available when data in compliance with the PIP are developed and submitted.
PRIME Designation
In March 2016, the EMA launched an initiative to facilitate development of product candidates in indications, often rare, for which few or
no therapies currently exist. The PRIority MEdicines, or PRIME, scheme is intended to encourage drug development in areas of unmet medical need and provides accelerated assessment of products representing substantial innovation reviewed under the
centralized procedure. Products from small- and medium-sized enterprises may qualify for earlier entry into the PRIME scheme than larger companies on the basis of compelling non-clinical data and tolerability data from initial clinical trials. Many
benefits accrue to sponsors of product candidates with PRIME designation, including but not limited to, early and proactive regulatory dialogue with the EMA, frequent discussions on clinical trial designs and other development program elements, and
potentially accelerated marketing authorization application assessment once a dossier has been submitted. Importantly, once a candidate medicine has been selected for the PRIME scheme, a dedicated contact point and rapporteur from the CHMP or from
CAT are appointed facilitating increased understanding of the product at EMA’s Committee level. A kick-off meeting with the CHMP/CAT rapporteur initiates these relationships and includes a team of multidisciplinary experts to provide guidance on the
overall development plan and regulatory strategy. PRIME eligibility does not change the standards for product approval, and there is no assurance that any such designation or eligibility will result in expedited review or approval.
Post-Approval Regulation
Similar to the United States, both MA holders and manufacturers of medicinal products are subject to comprehensive regulatory oversight by
the EMA, the European Commission and/or the competent regulatory authorities of the EU Member States. This oversight applies both before and after grant of manufacturing licenses and marketing authorizations. It includes control of compliance with EU
good manufacturing practices rules, manufacturing authorizations, pharmacovigilance rules and requirements governing advertising, promotion, sale, and distribution, recordkeeping, importing and exporting of medicinal products.
Failure by us or by any of our third-party partners, including suppliers, manufacturers and distributors to comply with EU laws and the
related national laws of individual EU Member States governing the conduct of clinical trials, manufacturing approval, marketing authorization of medicinal products and marketing of such products, both before and after grant of marketing
authorization, statutory health insurance, bribery and anti-corruption or other applicable regulatory requirements may result in administrative, civil or criminal penalties.
96
These penalties could include delays or refusal to authorize the conduct of clinical trials or to grant marketing authorization, product
withdrawals and recalls, product seizures, suspension, withdrawal or variation of the marketing authorization, total or partial suspension of production, distribution, manufacturing or clinical trials, operating restrictions, injunctions, suspension
of licenses, fines and criminal penalties.
The holder of a marketing authorization for a medicinal product must also comply with EU pharmacovigilance legislation and its related
regulations and guidelines, which entail many requirements for conducting pharmacovigilance, or the assessment and monitoring of the safety of medicinal products. These pharmacovigilance rules can impose on holders of MAs the obligation to conduct a
labor intensive collection of data regarding the risks and benefits of marketed medicinal products and to engage in ongoing assessments of those risks and benefits, including the possible requirement to conduct additional clinical studies or
post-authorization safety studies to obtain further information on a medicine’s safety, or to measure the effectiveness of risk-management measures, which may be time consuming and expensive and could impact our profitability. MA holders must
establish and maintain a pharmacovigilance system and appoint an individual qualified person for pharmacovigilance, who is responsible for oversight of that system. Key obligations include expedited reporting of suspected serious adverse reactions
and submission of PSURs in relation to medicinal products for which they hold MAs. The EMA reviews PSURs for medicinal products authorized through the centralized procedure. If the EMA has concerns that the risk benefit profile of a product has
varied, it can adopt an opinion advising that the existing MA for the product be suspended, withdrawn or varied. The agency can advise that the MA holder be obliged to conduct post-authorization Phase 4 safety studies. If the EC agrees with the
opinion, it can adopt a decision varying the existing MA. Failure by the marketing authorization holder to fulfill the obligations for which the EC’s decision provides can undermine the ongoing validity of the MA.
More generally, non-compliance with pharmacovigilance obligations can lead to the variation, suspension or withdrawal of the MA for the
product or imposition of financial penalties or other enforcement measures.
Sales and Marketing Regulations
The advertising and promotion of our products is also subject to EU laws concerning promotion of medicinal products, interactions with
physicians, misleading and comparative advertising and unfair commercial practices. In addition, other national legislation of individual EU Member States may apply to the advertising and promotion of medicinal products and may differ from one
country to another. These laws require that promotional materials and advertising in relation to medicinal products comply with the product’s SmPC as approved by the competent regulatory authorities. The SmPC is the document that provides information
to physicians concerning the safe and effective use of the medicinal product. It forms an intrinsic and integral part of the marketing authorization granted for the medicinal product. Promotion of a medicinal product that does not comply with the
SmPC is considered to constitute off-label promotion. All advertising and promotional activities for the product must be consistent with the approved SmPC and therefore all off-label promotion is prohibited. Direct-to-consumer advertising of
prescription-only medicines is also prohibited in the European Union. Violations of the rules governing the promotion of medicinal products in the European Union could be penalized by administrative measures, fines and imprisonment. These laws may
further limit or restrict the advertising and promotion of our products to the general public and may also impose limitations on its promotional activities with healthcare professionals.
Anti-Corruption Legislation
In the EU, interactions between pharmaceutical companies and physicians are also governed by strict laws, regulations, industry
self-regulation codes of conduct and physicians’ codes of professional conduct both at EU level and in the individual EU Member States. The provision of benefits or advantages to physicians to induce or encourage the prescription, recommendation,
endorsement, purchase, supply, order or use of medicinal products is prohibited in the European Union. The provision of benefits or advantages to physicians is also governed by the national anti-bribery laws of the EU Member States. Violation of
these laws could result in substantial fines and imprisonment.
Payments made to physicians in certain EU Member States also must be publicly disclosed. Moreover, agreements with physicians must often be
the subject of prior notification and approval by the physician’s employer, his/her regulatory professional organization, and/or the competent authorities of the individual EU Member States. These requirements are provided in the national laws,
industry codes, or professional codes of conduct, applicable in the individual EU Member States. Failure to comply with these requirements could result in reputational risk, public reprimands, administrative penalties, fines or imprisonment.
97
Australia
The Therapeutic Goods Administration, or the TGA, and the National Health and Medical Research Council set the GCP requirements for clinical research in Australia, and compliance with these codes is
mandatory. Australia has also adopted international codes, such as those promulgated by the International Council for Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, or the ICH. The ICH guidelines must be
complied with across all fields of clinical research, including those related to pharmaceutical quality, nonclinical and clinical data requirements and trial designs. The basic requirements for preclinical data to support a first-in-human trial
under ICH guidelines are applicable in Australia. Requirements related to adverse event reporting in Australia are similar to those required in other major jurisdictions.
Clinical trials conducted using “unapproved therapeutic goods” in Australia, being those which have not yet been evaluated by the TGA for
quality, safety and efficacy must occur pursuant to either the Clinical Trial Notification Scheme, or the CTN Scheme, or the Clinical Trial Exemption Scheme, or the CTX Scheme. In each case, the trial is supervised by a Human Research Ethics
Committee (“HREC”), an independent review committee set up under guidelines of the Australian National Health and Medical Research Council that ensures the protection of rights, safety and well-being of human subjects involved in a clinical trial. A
HREC does this by reviewing, approving and providing continuing examination of trial protocols and amendments, and of the methods and material to be used in obtaining and documenting informed consent of the trial subjects.
The CTN Scheme broadly involves:
| • |
completion of preclinical laboratory and animal testing;
|
| • |
submission to a HREC, of all material relating to the proposed clinical trial, including the trial protocol;
|
| • |
the institution or organization at which the trial will be conducted, referred to as the “Approving Authority”, giving final approval for the conduct of the trial at the site, having regard to
the advice from the HREC; and
|
| • |
the investigator submitting a ‘Notification of Intent to Conduct a Clinical Trial’ form, or CTN Form, to the TGA. The CTN form must be signed by the sponsor, the principal investigator, the
chairman of the HREC and a person responsible from the Approving Authority. The TGA does not review any data relating to the clinical trial however CTN trials cannot commence until the trial has been notified to the TGA.
|
Under the CTX Scheme:
| • |
a sponsor submits an application to conduct a clinical trial to the TGA for evaluation and comment; and
|
| • |
a sponsor must forward any comments made by the TGA Delegate to the HREC(s) at the sites where the trial will be conducted.
|
A sponsor cannot commence a trial under the CTX Scheme until written advice has been received from the TGA regarding the application and approval for the
conduct of the trial has been obtained from an ethics committee and the institution at which the trial will be conducted.
98
Approval for inclusion in the Australian Register of Therapeutic Goods, or ARTG, is required before a pharmaceutical product may be marketed (or imported,
exported or manufactured) in Australia. In order to obtain registration of the product on the ARTG, it is required that:
| • |
adequate and well-controlled clinical trials demonstrate the quality, safety and efficacy of the therapeutic product;
|
| • |
evidence is compiled which demonstrates that the manufacture of the therapeutic product complies with the principles of cGMP;
|
| • |
manufacturing and clinical data is derived to submit to the Advisory Committee on Prescription Medicines, which makes recommendations to the TGA as to whether or not to grant approval to
include the therapeutic product in the ARTG; and
|
| • |
an ultimate decision is made by the TGA whether to include the therapeutic product in the ARTG.
|
Other Markets
Since the United Kingdom has formally left the European Union on January 31, 2020 and the transition period, during which EU laws continued
to apply to the United Kingdom, has expired on December 31, 2020, EU laws now only apply to the United Kingdom in respect of Northern Ireland as laid out in the Protocol on Ireland and Northern Ireland. The European Union and the United Kingdom have
concluded a trade and cooperation agreement (“TCA”), which was ratified by the United Kingdom Parliament on December 30, 2020. The TCA was applied provisionally as of January 1, 2021 and has entered into force on May 1, 2021.
The TCA includes provisions affecting the life sciences sector (including on customs and tariffs) but areas for further discussion between
the European Union and the United Kingdom remain. In addition, there are some specific provisions concerning pharmaceuticals. These include the mutual recognition of Good Manufacturing Practice (“GMP”),
inspections of manufacturing facilities for medicinal products and GMP documents issued. The TCA does not, however, contain wholesale mutual recognition of United Kingdom and EU pharmaceutical regulations and product standards.
Since January 1, 2021, the EU laws which have been transposed into United Kingdom law through secondary legislation continue to be
applicable as “retained EU law.” As there is no general power to amend these regulations, the United Kingdom government has adopted the Medicines and Medical Devices Act 2021 which seeks to address this regulatory gap through introducing
regulation-making, delegated powers covering the fields of human medicines, clinical trials of human medicines, veterinary medicines and medical devices. The purpose of the act is to enable the existing regulatory frameworks to be updated, with the
powers granted under it only exercisable in relation to four pieces of legislation: the Human Medicines Regulations 2012, the Medicines for Human Use (Clinical Trials) Regulations 2004, the Medicines (Products for Human Use) Regulations 2016 and
limited parts of the Medicines Act 1968 (specifically those parts which make provision related to pharmacies). It is then further restricted to amending or updating only those provisions stated in the act, which include clinical trials.
Specified provisions of the Medicines and Medical Devices Act 2021 entered into force on February 11, 2021 when the legislation formally
became law. The remaining provisions came into effect within two months of February 11, 2021 or will come into effect otherwise as stipulated in subsequent statutory instruments. The new Medicines and Medical Devices Act 2021 supplements the United
Kingdom Medical Devices Regulations 2002 (the “Regulations”), which are based on the EU Medical Devices Directive as amended to reflect the United Kingdom’s post-Brexit regulatory regime. Notably, the United Kingdom Medical Devices Regulations 2002
do not include any of the revisions that have been made by the EU Medical Devices Regulation (EU) 2017/745, which has gained full application in all EU Member States since May 26, 2021. Additionally, the United Kingdom’s Medicines and Healthcare
products Regulatory Agency (MHRA) launched a comprehensive consultation on September 16, 2021 with proposals to amend the regulatory framework for medical devices in the United Kingdom. The stated objectives of the proposals include expansion of the
scope of the Regulations (for example, by expanding the in vitro diagnostic medical device definition to include software and other products, including products without an intended medical purpose but with similar functioning and risk profiles) and
potentially through use of internationally recognized definitions (for example, by excluding products that contain viable biological substances and excluding food), remove trade barriers, further the availability of medical devices and improve the
favorability of the United Kingdom market. The consultation period closes on November 25, 2021 with a view to the new regulations coming into force on July 1, 2023 with appropriate transitional measures.
99
For other countries outside of the European Union, such as countries in Eastern Europe, Latin America or Asia, the requirements governing
the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, again, the clinical trials must be conducted in accordance with GCP and the applicable regulatory requirements and the ethical
principles that have their origin in the Declaration of Helsinki.
If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension of
clinical trials, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Intellectual Property
As of August 2022, we own, or exclusively license through our various wholly owned subsidiaries, nine (9) patent families in connection
with our product offerings. Five (5) of the patent families have granted patents registered in various countries, as detailed below. Six (6) families have active pending application/s under examination. We also rely on trade secrets to protect
certain aspects of our technology.
Patent families with granted patents:
| • |
Transmucosal and Transdermal Delivery Systems – granted in the United States, the European Union Countries (2 patents), Canada, Australia, Hong Kong, New Zealand;
|
| • |
Treatment for Depression and Depressive Disorders – granted in the United States, Australia, New Zealand;
|
| • |
Probiotic Compositions and Uses Thereof for Treatment of Obesity-Related Disorders – granted in Australia (innovation patent);
|
| • |
Probiotic Combinations and Uses Thereof – granted in Australia (standard patent) and Australia (innovation patent);
|
| • |
Compositions and Uses Thereof – granted in Australia (innovation patent).
|
100
Patent Portfolio
|
Product
|
Jurisdiction
|
IP Licensed
|
Patent
|
Type of Patent
|
Status
|
Patent Expiry
|
|||
|
MultiBiotic
|
Australia (innovation)
|
PharmaCare
|
Probiotic compositions and uses thereof for treatment of obesity-related disorders
|
Composition of matter & Use
|
Granted
|
5/11/2023(1)
|
|||
|
NRGBiotic
|
Australia
|
PharmaCare
|
Probiotic combinations and uses thereof
|
Composition of matter & Use
|
Granted
|
5/21/2035
|
|||
|
NRGBiotic
|
Australia (innovation)
|
PharmaCare
|
Probiotic combinations and uses thereof
|
Composition of matter & Use
|
Granted
|
5/21/2023
|
|||
|
Orotate
|
Australia
|
PharmaCare
|
Treatment for depression and depressive disorders
|
Use
|
Granted
|
10/28/2035
|
|||
|
Orotate
|
Canada
|
Cultech Ltd. (non-exclusive for use with a single product (NRGBiotic))
|
Treatment for depression and depressive disorders
|
Use
|
Pending
|
10/28/2035
|
|||
|
Orotate
|
Belgium
France
Germany
Italy
Luxembourg Netherlands
Spain
United Kingdom
|
Cultech Ltd. (exclusive in the United Kingdom and non-exclusive in Belgium, France, Germany, Italy, Luxembourg, Netherlands and Spain for use with a single product (NRGBiotic))
|
Treatment for depression and depressive disorders
|
Use
|
Granted
|
10/28/2035
|
|||
|
Orotate
|
New Zealand
|
PharmaCare
|
Treatment for depression and depressive disorders
|
Use
|
Granted
|
10/28/2035
|
|||
| Orotate |
Singapore |
N/A |
Treatment for depression and depressive disorders |
Use |
Pending |
10/28/2035 | |||
101
|
Product
|
Jurisdiction
|
IP Licensed
|
Patent
|
Type of Patent
|
Status
|
Patent Expiry
|
|
Orotate
|
United States
|
Cultech Ltd. (non-exclusive for use with a single product (NRGBiotic))
|
Treatment for depression and depressive disorders
|
Use
|
Granted
|
5/28/2037
|
|
Orotate
|
Hong Kong
|
N/A
|
Treatment for depression and depressive disorders
|
Use
|
Pending
|
10/28/2035
|
|
Nanocelle
|
Australia
|
PharmaCare (nutraceuticals only)
|
Transmucosal and transdermal delivery systems
|
Composition of matter & Use
|
Granted
|
3/31/2036
|
|
Nanocelle
|
Australia (divisional)
|
PharmaCare (nutraceuticals only)
|
Transmucosal and transdermal delivery systems
|
Composition of matter & Use
|
Granted
|
3/31/2036
|
|
Nanocelle
|
Canada
|
N/A
|
Transmucosal and transdermal delivery systems
|
Composition of matter & Use
|
Granted
|
3/31/2036
|
|
Nanocelle
|
Europe
Albania
Austria
Belgium
Bulgaria
Switzerland
Cyprus
Czech Republic
Germany
Denmark
Estonia
Spain
Finland
France
United Kingdom
Greece
Croatia
Hungary
Ireland
Iceland
Italy
Lithuania
Luxembourg
Latvia
Monaco
North Macedonia
Malta
Netherlands
Norway
Poland
Portugal
Romania
Serbia
Sweden
Slovenia
Slovakia
San Marino
Turkey
|
N/A
|
Transmucosal and transdermal delivery systems
|
Composition of matter & Use
|
Granted
|
3/31/2036
|
102
|
Product
|
Jurisdiction
|
IP Licensed
|
Patent
|
Type of Patent
|
Status
|
Patent Expiry
|
|
Nanocelle
|
Europe (divisional)
Belgium
Switzerland
Czech Republic
Germany
Denmark
Spain
Finland
France
United Kingdom
Greece
Ireland
Italy
Netherlands
Norway
Poland
|
N/A
|
Transmucosal and transdermal delivery systems
|
Composition of matter & Use
|
Granted
|
3/31/2036
|
|
Nanocelle
|
New Zealand
|
N/A
|
Transmucosal and transdermal delivery systems
|
Composition of matter & Use
|
Granted
|
3/31/2036
|
|
Nanocelle
|
New Zealand (divisional)
|
N/A
|
Transmucosal and transdermal delivery systems
|
Composition of matter & Use
|
Pending
|
3/31/2036
|
|
Nanocelle
|
Singapore
|
N/A
|
Transmucosal and transdermal delivery systems
|
Composition of matter & Use
|
Accepted
|
3/31/2036
|
|
Nanocelle
|
United States
|
N/A
|
Transmucosal and transdermal delivery systems
|
Composition of matter & Use
|
Granted
|
3/31/2036
|
|
Nanocelle
|
United States (continuation)
|
N/A
|
Transmucosal and transdermal delivery systems
|
Composition of matter & Use
|
Pending
|
3/31/2036
|
|
Nanocelle
|
Hong Kong
|
N/A
|
Transmucosal and transdermal delivery systems
|
Composition of matter & Use
|
Granted
|
3/31/2036
|
|
Nanocelle
|
Hong Kong (divisional)
|
N/A
|
Transmucosal and transdermal delivery systems
|
Composition of matter & Use
|
Pending
|
3/31/2036
|
103
|
Product
|
Jurisdiction
|
IP Licensed
|
Patent
|
Type of Patent
|
Status
|
Patent Expiry
|
|
Mg Optima + CBD
|
Australia (innovation)
|
N/A
|
Compositions and uses thereof
|
Composition of matter & Use
|
Granted
|
9/13/2028
|
|
Mesothelioma(2)
|
International (PCT)
|
N/A
|
Combination Therapy
|
Use
|
Pending
|
2/22/2042
|
|
API Degradation
|
Australia
|
N/A
|
Protection of plant extracts and compounds from degradation
|
Process
|
Pending
|
5/11/2037
|
|
API Degradation
|
Canada
|
N/A
|
Protection of plant extracts and compounds from degradation
|
Process
|
Pending
|
5/11/2037
|
|
API Degradation
|
Europe
|
N/A
|
Protection of plant extracts and compounds from degradation
|
Process
|
Pending
|
5/11/2037
|
|
API Degradation
|
New Zealand
|
N/A
|
Protection of plant extracts and compounds from degradation
|
Process
|
Pending
|
5/11/2037
|
|
API Degradation
|
Singapore (divisional)
|
N/A
|
Protection of plant extracts and compounds from degradation
|
Process
|
Pending
|
5/11/2037
|
|
API Degradation
|
USA (continuation)
|
N/A
|
Protection of plant extracts and compounds from degradation
|
Process
|
Pending
|
5/11/2037
|
|
API Degradation
|
Hong Kong
|
N/A
|
Protection of plant extracts and compounds from degradation
|
Process
|
Pending
|
5/11/2037
|
(1) The expiration of the Multibiotic patent in Australia on May 11, 2023 will not have an impact on our patent portfolio and business.
(2) The mesothelioma application is still a pending international application under the Patent Cooperation Treaty (PCT), which provides provisional protection for
156 jurisdictions, as contracting states of the PCT. The specific jurisdictions for national/regional phase entry in connection with the pending international application (PCT) will not be decided until August 2023.
104
Quantitative and Qualitative Disclosures about Market Risk
Our cash consists entirely of cash held in interest-bearing accounts with banks in Australia, the U.S. and the United Kingdom. Thus, our
primary exposure to market risk is interest income sensitivity, which is affected by changes in the general level of Australian interest rates. However, a sudden change in market interest rates would not be expected to have a material impact on our
financial condition and/or results of operation.
Employees
As of June 30, 2022, we had 42 employees. Of these employees, 24 were employed in research and development and 18 in general management and
administration. 35 employees were located in Australia, 6 employees were located in the U.S. and one employee was located in the United Kingdom. As at the end of fiscal year June 30, 2021, we had 59 employees.
Each of our employees has entered into an employment agreement with an unlimited term. We may only terminate the employment of any of our
employees in accordance with the relevant employee’s contract of employment.
Our standard contract of employment for full time provides that we can terminate the employment of an employee without notice for serious
misconduct or with between one to six months’ notice without cause (as set out in the relevant employee’s contract of employment).
Property and Facilities
We entered into a five-year lease agreement, ending on May 31, 2024, for a facility at units A5-A6, 11-13 Lord Street, Botany, New South
Wales, 2019, Australia for our medical laboratory, corporate head office and warehouse space associated with our medical laboratory. The rent payments on this facility are A$484,213 annually. We also entered into a five-year lease agreement, ending
in January 2025, to lease a property of 1,376 feet, through our wholly owned U.S. facility, in Rancho Santa Margarita, California. The rent payments on the California property are A$3,027 a month.
Inflation and Seasonality
Management believes inflation has not had a material impact on our operations or financial condition. Management further believes that our
operations are not currently subject to seasonal influences due to our current lack of marketed products. Moreover, the targets of our drug candidates, are not seasonal diseases. Accordingly, once we have marketed products, management does not expect
that our business will be subject to seasonal influences.
Legal Proceedings
To our knowledge, we are not currently a party to any lawsuit, and are not aware of any pending or threatened lawsuit, that, if adversely
determined, would have a material adverse effect on our financial position, results of operations or liquidity. As such, we do not believe that any pending or threatened legal proceedings, taken as a whole, should have any significant impact on our
financial statements. From time to time in the future we may be subject to legal proceedings and claims in the ordinary course of business. While we expect that these claims would be covered by our existing insurance policies, those claims, even if
lacking merit, could result in the expenditure of significant financial and managerial resources.
Manufacturing and Raw Materials
We have no manufacturing capabilities and are dependent on third parties for cost effective manufacture and manufacturing process
development of our drug candidates. Problems with third-party manufacturers or the manufacturing process as such may delay or jeopardize clinical trials and commercialization of our drug candidates.
105
Organizational Structure
Below is a list of our significant subsidiaries, including our ownership percentage, date of formation and jurisdiction. These subsidiaries
were established to allow us to conduct commercial and clinical operations in Europe and the United States and expand our operations in Australia.
|
Subsidiary
|
|
Ownership
|
|
Date of
Formation/Acquisition |
|
Jurisdiction
|
|
Medlab Pty Ltd
|
|
100%
|
|
May 13, 2012
|
|
New South Wales, Australia
|
|
Medlab Clinical U.S., Inc.
|
|
100%
|
|
June 5, 2014
|
|
California, USA
|
|
Medlab Nutraceuticals, Inc. (1)
|
|
60%
|
|
February 4, 2014
|
|
California, USA
|
|
Medlab Research Ltd.
|
|
100%
|
|
August 6, 2013
|
|
London, UK
|
|
Medlab US Operations, Inc.
|
|
100%
|
|
April 11, 2022
|
|
California, USA
|
|
Medlab IP Pty Ltd.
|
|
100%
|
|
August 16, 2013
|
|
New South Wales, Australia
|
|
MDC Europe Ltd.
|
|
100%
|
|
November 26, 2018
|
|
Malta
|
(1) Dr. David Rutolo, an employee of the Company, owns 40% of Medlab Nutraceuticals, Inc.
Competition
Potential competitors to our products are either cannabinoid medicines that are regulatory (FDA) approved for specific purpose or
proprietary drug enhancement and delivery technology platforms. We do not believe that direct competition exists for either our proprietary delivery platform, NanoCelle®, or our lead drug candidate, NanaBis™.
Below are several companies that own FDA-approved cannabinoid products for specific purposes, each of whom we consider a potential
competitor:
| 1. |
Epidiolex® (CBD) which was developed by GW Pharmaceuticals, PLC and since February 2021, is now owned by Jazz Pharmaceuticals plc.
|
| 2. |
Syndros® (dronabinol) which was developed by Insys Therapeutics Inc. and is now owned by Benuvia Therapeutics Inc. (“Benuvia”).
|
| 3. |
Marinol® (dronabinol) which was developed by Solvay Pharmaceuticals B.V., and is now owned by AbbVie Inc.
|
| 4. |
Cesamet® (nabilone) which was developed by Eli Lilly and Company and is now owned by Bausch Health Companies Inc.
|
Epidiolex® was FDA-approved in 2018 and is the first FDA-approved prescription CBD (botanical cannabis-derived CBD) product, used for the
treatment for childhood-onset epilepsy, specifically Lennox-Gastaut syndrome, Dravet syndrome (“DS”), or Tuberous Sclerosis Complex.
106
Syndros®, asynthetic THC – Dronabinol, was FDA-approved in 2016 to treat nausea and vomiting caused by anti-cancer medicine (e.g.,
chemotherapy) in people whose nausea and vomiting have not improved with usual anti-nausea medicines, and loss of appetite (e.g., anorexia) in people with acquired immune deficiency syndrome (“AIDS”) who have lost weight.
| • |
In 2019, Syndros® was acquired by Chilion Group Holdings, (parent company of Benuvia) in a sale of multiple CBD formulations and other products from Insys Therapeutics Inc. for US$12.2
million.
|
| • |
In March 2022, Benuvia Therapeutics Inc. signed an agreement with Pono Capital Corp. to combine their businesses and become a publicly traded company with a combined company pro forma
enterprise value of US$440 million.
|
| • |
Syndros® is currently being examined by the University of Arizona in a pilot program for decreasing the use of opioids in breast cancer subjects with bone metastases (early Phase I clinical
trial).
|
Marinol®, synthetic THC – Dronabinol, was FDA-approved in 1985, and is another registered trade name for dronabinol and like Syndros® is
approved for treating severe nausea and vomiting caused by cancer chemotherapy and loss of appetite contributing to weight loss in people with AIDS.
Cesamet®, synthetic THC – Nabilone, was FDA-approved in 1985 and approved for the treatment of
nausea and vomiting associated with cancer chemotherapy in May 2006, and treats severe nausea and vomiting caused by cancer chemotherapy. Similar to Syndros® and Marinol, Cesamaet is usually given after other medicines to control nausea and vomiting
have been tried without success.
The following companies could be considered competitors to us in the FDA-approved market or clinical trial stage cannabinoid
pharmaceuticals market:
| • |
GW Pharmaceuticals, PLC (now Jazz Pharmaceuticals plc);
|
| • |
Valeant Pharmaceuticals (now Bausch Health Companies Inc.);
|
| • |
AbbVie Inc.;
|
| • |
Benuvia;
|
| • |
Corbus Pharmaceuticals Holdings, Inc.;
|
| • |
Teva Pharmaceutical Industries Ltd.;
|
| • |
Avicanna Inc.;
|
| • |
Solvay Pharmaceuticals B.V.; and
|
| • |
Cannabics Pharmaceuticals Inc.
|
The companies listed above have advanced cannabinoid research programs and in some specific cases, such as GW Pharmaceuticals, PLC (now
Jazz Pharmaceuticals plc), have already demonstrated the ability to progress these programs to FDA-approved products and subsequent commercialization.
Although we do not believe any of these companies are known to be actively progressing cannabinoid products purposed toward managing bone
cancer pain, and therefore do not directly compete with our lead candidate NanaBis™ at this time, prior expertise in drug research and development and experience with the FDA regulatory process for approval of a cannabinoid pharmaceutical
product provides them a theoretical ability to compete with us in the future. Should the University of Arizona pilot program examining Syndros®’s effectiveness in pain management and opioid-sparing for breast cancer and bone metastases deliver
positive results, it is possible that Benuvia Therapeutics (or its parent company, Chilion Group Holdings) would pursue an FDA application for Syndros®, or AbbVie Inc. would pursue an FDA application for Marinol, for each to be used for this purpose.
107
MANAGEMENT
Directors and Senior Management
The following table sets forth our directors and senior management, their age and the positions they held as of , 2022.
|
Name
|
|
Age
|
|
Position
|
|
Mr. Michael Hall
|
|
81
|
|
Chairman
|
|
Dr. Sean Hall
|
|
52
|
|
Chief Executive Officer and Managing Director
|
|
Mr. Drew Townsend
|
|
57
|
|
Director
|
|
Ms. Cheryl Maley
|
|
54
|
|
Director
|
|
Mr. Mohit Gupta
|
|
46
|
|
Director
|
|
Mr. Kerem Kaya
|
|
49
|
|
Chief Financial Officer and Company Secretary
|
Michael Hall has over 35 years of experience working with Australian-based nutrition products and
contributes his insight and experience towards our Company’s goals and product offerings. He has served as our Chairman since January 2013. He last served as the Managing Director of FIT-BioCeuticals Ltd. from January 1999 until January 2010, at
which time the company was sold to a third party. Mr. Hall received a Bachelor of Commerce degree from the University of New South Wales and was a Certified Practicing Accountant (currently retired).
Dr. Sean Hall has over 25 years of experience in health care senior management. He has served as our Chief Executive Officer since July 2012. From July 2008 to July 2012, Dr. Hall was Chief Executive Officer for FIT-BioCeuticals Ltd. and prior to that role was Chief Operating Officer from February 2004
until July 2008. Dr. Hall is also currently a member of World Medical Association, American Academy of Anti-Aging Medicine, and the Special Operations Medical Association. He has an undergraduate degree from
Blue Marble Medical School, an MBA from Academy of Business Management, as well as education in Leadership, Board Governance and continuing medical education from Harvard University.
Drew Townsend has over 35 years of experience in the finance industry as a Chartered Accountant.
He has served as a director of the Company since August 2020. He has worked as an accountant at Hall Chadwick Chartered Accountants since April 1987 and was named a Partner in January 1993. He received a Bachelor of Commerce, majoring in accounting
and finance, from the University of New South Wales, is a member of the Institute of Chartered Secretaries and Accountants and member of the Institute of Internal Auditors.
Cheryl Maley has served a director of the Company since April 2021. She has over 20 years of
pharmaceutical industry experience with roles spanning across sales, marketing, business development, commercial excellence, patient access and general management. Cheryl has worked at Novartis AG since November 2013, and currently serves as the
General Manager of Oncology for Australia and New Zealand. She served as a Director at Commercial Excellence at AbbVie Inc. from January 2012 until October 2013. She worked at Abbot from June 2017 until December 2011, last serving as a Director of
Global Strategic Initiatives Immunology. Cheryl has a Bachelor of Science Degree, a Diploma of Education, a Master of Business Administration and is a graduate of the Australian Institute of Company Directors. She has a passion for innovation and has
completed formal innovation training with What-If Innovation (UK), the Kellogg Institute (USA), Inventium (Australia) and RAW Innovation (Australia).
Mohit Gupta has served as our director since August 2022 and has more than 20 years’ experience in
various roles and geographies, with last 10 years in pharmaceuticals in various commercial capacities based in Australia and Switzerland. Since January 2021, he has served as managing director of MetTAS Pty Ltd, a pharmaceutical company servicing
Australia. From August 2018 until December 2020, he served as Head of Business Development (EU) of Mylan GmbH, and prior was the Head of Business Development from October 2014 until August 2018. From April 2012 until August 2018, he worked in various
business developmental roles at Alphapharm. From April 2011 until March 2012, he served as a project manager at Ingram Micro. Mr. Gupta received a Bachelors in Commerce in Accountancy from Delhi University and an MBA in Finance from Birla Institute
of Management Technology.
108
Kerem Kaya has served as our Chief Financial Officer and Secretary since May 2021 and has over 26
years of experience in pharmaceuticals and chemicals industries, holding positions including Chief Financial Officer, Financial Planning and Analysis Heads and Project Management roles at various companies, both locally and globally. From November
2004 until December 2020, he worked at Novartis AG in numerous positions. He has an undergraduate degree in Business from Western Sydney University and is a Certified Practicing Accountant.
Family Relationships
Michael Hall, our Chairman, is the father of our Chief Executive Officer, Mr. Sean Hall.
Compensation of Directors and Executive Officers
For the fiscal year ended June 30, 2022, we paid aggregate cash compensation of approximately A$1,635,045 to our directors and executive
officers as a group.
Executive Compensation
The following table indicates the remuneration received by our non-executive directors, executive directors, and other key management
personnel during the fiscal year ending June 30, 2022:
|
|
Short-term benefits
|
Post-employment
benefits |
Non-cash
Long-term benefits |
Non-cash
Share-based benefits
|
||||||||||||||||||||||||
|
2022
|
Cash salary and fees
A$
|
Cash in lieu of leave
A$
|
Non-monetary
A$
|
Super-annuation
A$
|
Long service
leave |
Equity settled
(see note b) (2) A$ |
Total
A$
|
|||||||||||||||||||||
|
Non-Executive Directors:
|
||||||||||||||||||||||||||||
|
Michael Hall
|
176,923
|
-
|
-
|
10,192
|
-
|
-
|
187,115
|
|||||||||||||||||||||
|
Drew Townsend
|
66,000
|
-
|
-
|
-
|
-
|
-
|
66,000
|
|||||||||||||||||||||
|
Cheryl Maley
|
66,000
|
-
|
-
|
-
|
-
|
179,805
|
245,805
|
|||||||||||||||||||||
|
Laurence McAllister(1)
|
66,000
|
-
|
-
|
-
|
-
|
-
|
66,000
|
|||||||||||||||||||||
|
|
||||||||||||||||||||||||||||
|
Executive Directors:
|
||||||||||||||||||||||||||||
|
Laurence McAllister
|
554,500
|
-
|
-
|
-
|
-
|
-
|
554,500
|
|||||||||||||||||||||
|
Sean Hall
|
443,346
|
66,826
|
-
|
51,017
|
14,921
|
-
|
576,110
|
|||||||||||||||||||||
|
|
||||||||||||||||||||||||||||
|
Other Key Management Personnel:
|
||||||||||||||||||||||||||||
|
Kerem Kaya
|
262,260
|
-
|
-
|
26,226
|
4,343
|
-
|
292,829
|
|||||||||||||||||||||
|
Luis Vitetta
|
297,003
|
-
|
-
|
29,581
|
11,638
|
-
|
338,222
|
|||||||||||||||||||||
|
David Rutolo
|
173,913
|
-
|
-
|
13,527
|
-
|
-
|
187,440
|
|||||||||||||||||||||
|
Ian Curtinsmith
|
209,808
|
-
|
-
|
20,981
|
3,632
|
-
|
234,421
|
|||||||||||||||||||||
|
|
2,315,753
|
66,826
|
-
|
151,524
|
34,534
|
179,805
|
2,748,442
|
|||||||||||||||||||||
| (1) |
Laurence McAllister was an Executive Director until April 30, 2022 and a Non-Executive Director thereafter until his resignation in August 2022.
|
| (2) |
The amounts included in the share-based remuneration represent the grant date fair value of performance rights and options, amortized on a straight-line basis over the expected vesting period.
|
The total amounts set aside or accrued by the Company or its subsidiaries to provide pension, retirement or similar benefits for the fiscal
year ended June 30, 2022 was A$0.7 million.
109
Corporate Governance
ASX Corporate Governance Principles
In Australia, there are no defined corporate governance structures and practices that must be observed by a company listed on the ASX,
except those entities of a certain size are required to have audit and remuneration committees and trading policies for key management personnel. Instead, the ASX corporate governance council has published the 4th Edition of its Corporate
Governance Principles and Recommendations, which contains what are called the “Recommendations” which articulate eight core principles which are intended to provide a reference point for companies about their corporate governance structures and
practices. Under ASX Listing Rule 4.10.3, companies are required to attach a copy of the Company’s corporate governance statement (which has been approved by the board of directors) and provide a statement in their annual report to shareholders
disclosing the extent to which they have followed the Recommendations in the reporting period and where they have not followed all the Recommendations, identify the Recommendations that have not been followed, and the reasons for not following them
and what (if any) alternative governance practices it adopted in lieu of the recommendations during that period. It is not mandatory to follow the Recommendations. As compliance with the Recommendations would entail excessive costs to us, and in
light of our current size, we do not fully follow the Recommendations because the costs of doing so would outweigh the benefits.
Non-Executive and Independent Directors
Australian law does not require a company to appoint a certain number of independent directors to its board of directors or audit
committee. However, under the Corporate Governance Principles and Recommendations, the ASX corporate governance council recommends, but does not require, that an ASX-listed company have a majority of independent directors on its board of directors
and is chaired by an independent director. Our board of directors has determined that Messrs. Gupta, Townsend and Mrs. Maley qualify as independent directors under the requirements of the ASX.
Our board of directors does not have regularly scheduled meetings at which only independent directors are present. The board of directors
does meet regularly and independent directors are expected to attend all such meetings.
Committees of the Board of Directors
Audit Committee. Nasdaq Marketplace Rules require us to establish an audit committee comprised
of at least three members, each of whom is financially literate and satisfies the respective “independence” requirements of the SEC and Nasdaq and one of whom has accounting or related financial management expertise at senior levels within a company.
Our Audit Committee assists our board of directors in overseeing the accounting and financial reporting processes of our company and audits
of our financial statements, including the integrity of our financial statements, compliance with legal and regulatory requirements, our independent public accountants’ qualifications and independence, and independent public accountants, and such
other duties as may be directed by our board of directors. The Audit Committee is also required to assess risk management.
Our Audit Committee currently consists of two board members, including Drew Townsend and Michael Hall. We believe our Audit Committee
members each satisfy the “independence” requirements of the SEC and Nasdaq Marketplace Rules.
110
Corporate Governance Requirements under Nasdaq listing rules.
As we are incorporated in Australia, we are allowed to follow Australian “home country” corporate governance practices in lieu of the
otherwise applicable Nasdaq corporate governance standards, as long as we disclose each requirement of Rule 5600 that we do not follow and describe the home country practice we follow in lieu of the relevant Nasdaq corporate governance standards. We
intend to take all actions necessary to maintain compliance with applicable corporate governance requirements under the rules adopted by the SEC and listing standards of Nasdaq. We follow Australian corporate governance practices in lieu of the
corporate governance requirements of the Nasdaq Marketplace Rules in respect of:
| • |
Nasdaq requirement under Rule 5605(d) that a compensation committee be constituted — The ASX Listing Rules do not have an express requirement for every issuer listed on ASX to have a
compensation committee. There is a requirement under the ASX Listing Rules for each issuer which is included in the S&P/ASX 300 index at the start of a financial year to have a compensation committee, but we are not currently included on
this list. We expect to rely on an exemption from the requirement to constitute a compensation committee under the Nasdaq listing rules and we seek to claim such exemption.
|
| • |
Nasdaq requirement under Rule 5605(e) that a nominations committee be constituted — The ASX Listing Rules do not have an express requirement that each issuer listed on ASX have a nominations
committee. We expect to rely on an exemption from the requirement to constitute a nominations committee under the Nasdaq listing rules and we seek to claim such exemption.
|
| • |
Nasdaq requirement under Rule 5620(c) that a quorum consist of holders of 33 1/3% of the outstanding Shares — The ASX Listing Rules do not have an express requirement that each issuer listed
on ASX have a quorum of any particular number of the outstanding Shares, but instead allow a listed issuer to establish its own quorum requirements. Our quorum is currently two persons who are entitled to vote. We believe this quorum
requirement is consistent with the requirements of the ASX and is appropriate and typical of generally accepted business practices in Australia.
|
| • |
Nasdaq requirements under Rules 5605(b)(1) and (2) relating to director independence, including the requirements that a majority of the board of directors must be comprised of independent
directors and that independent directors must have regularly scheduled meetings at which only independent directors are present — the Nasdaq and ASX definitions of what constitute an independent director are not identical and the requirements
relating to the roles and obligations of independent directors are not identical. The ASX, unlike Nasdaq, permits an issuer to establish its own materiality threshold for determining whether a transaction between a director and an issuer
affects the director’s status as independent and it does not require that a majority of the issuer’s board of directors be independent, as long as the issuer publicly discloses this fact and the reasons for why it has not chosen to adopt this
corporate governance recommendation. In addition, the ASX does not require that the independent directors have regularly scheduled meetings at which only independent directors are present. We believe that our Board composition is consistent
with the requirements of the ASX and that it is appropriate and typical of generally accepted business practices in Australia.
|
| • |
The requirement that our independent directors meet regularly in executive sessions under Nasdaq Listing Rules. The ASX Listing Rules and the Corporations Act do not require the independent
directors of an Australian company to have such executive sessions.
|
111
| • |
The Nasdaq requirements under Rules 5605(d) that compensation of an issuer’s officers must be determined, or recommended to the Board for determination, either by a majority of the independent
directors, or a compensation committee comprised solely of independent directors, and that director nominees must either be selected, or recommended for the Board’s selection, either by a majority of the independent directors, or a
nominations committee comprised solely of independent directors. The Nasdaq compensation committee requirements are not identical to the ASX remuneration and nomination committee requirements. Issuers listed on the ASX are recommended under
applicable listing standards to establish a remuneration committee consisting of a majority of independent directors and an independent chairperson, or publicly disclose that fact and the reasons for why it has not adopted this recommendation
We do not have a compensation committee.
|
| • |
The requirement prescribed by Nasdaq Listing Rules that issuers obtain shareholder approval prior to the issuance of securities in connection with certain acquisitions, private placements of
securities, or the establishment or amendment of certain share option, purchase or other compensation plans. Applicable Australian law and the ASX Listing Rules differ from Nasdaq requirements, with the ASX Listing Rules providing generally
for prior shareholder approval in numerous circumstances, including (i) issuance of equity securities exceeding 15% (or 25% under certain circumstances) of our issued share capital in any 12-month period (but, in determining the 15% limit,
securities issued under an exception to the rule or with shareholder approval are not counted), (ii) issuance of equity securities to related parties (as defined in the ASX Listing Rules) and (iii) issuances of securities to directors or
their associates under an employee incentive plan.
|
Indemnification of Directors and Officers
Our constitution (the “Constitution”) provides that, we may indemnify a person who is, or has been, a director or an officer of the
Company, to the full extent permissible by law, out of our property against any liability incurred by such person as a director or an officer in defending proceedings, whether civil or criminal, and whatever their outcome.
In addition, our Constitution provides that to the extent permitted by law, we may pay, or agree to pay, a premium in respect of a contract
insuring a person who is or has been a director or an officer of the Company or one of our subsidiaries against any liability:
| • |
incurred by the person in his or her capacity as a director or an officer of the Company or a subsidiary of the Company, and
|
| • |
for costs and expenses incurred by that person in defending proceedings relating to that person acting as a director or an officer of the Company, whether civil or criminal, and whatever their
outcome.
|
We maintain a directors’ and officers’ liability insurance policy. We have established a policy for the indemnification of our directors
and officers against certain liabilities incurred as a director or officer, including costs and expenses associated in successfully defending legal proceedings.
Code of Conduct
We have adopted a Code of Conduct applicable to all of our directors, officers and employees. Our Code of Conduct is available on our
website at www.medlab.co. We post on our website all disclosures that are required by law or the listing standards of Nasdaq concerning any amendments to, or waivers from, any provision of the Code of
Conduct. The reference to our website address does not constitute incorporation by reference of the information contained at or available through our website, and you should not consider it to be a part of, this prospectus.
112
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
In 2020, 2021 and 2022, the Company paid $A11,560, $A13,996 and $A17,032, respectively, in taxation services from Hall Chadwick. Drew
Townsend, an independent director of the Company, is a partner at Hall Chadwick.
In 2020, 2021 and 2022, the Company paid $A57,920, $A109,313 and $A160,897 in employee benefits to Sean Hall, respectively.
113
PRINCIPAL SHAREHOLDERS
The following table presents the beneficial ownership of our Shares based on 2,283,502 Shares outstanding as of November 18, 2022 by each
person known by us to be the beneficial owner of more than 5% of our Shares.
We have determined beneficial ownership in accordance with the rules and regulations of the SEC, and the information is not necessarily
indicative of beneficial ownership for any other purpose. Except as indicated by the footnotes below, we believe, based on information furnished to us, that the persons and entities named in the table below have sole voting and sole investment power
with respect to all shares that they beneficially own.
Applicable percentage ownership before the offering is based on 2,283,502 Shares outstanding as of November 18, 2022. In computing the
number of shares beneficially owned by a person or entity and the percentage ownership of such person or entity, we deemed to be outstanding all shares subject to options and warrants held by the person or entity that are currently exercisable, or
exercisable within 60 days of , 2022. However, except as described above, we did not deem such shares outstanding for the purpose of computing the percentage ownership of any other person or entity.
The address for our directors, executive officers and 5% shareholders is: c/o Medlab Clinical Ltd., Units 5 and 6, 11-13 Lord Street,
Botany, New South Wales 2019.
The following table sets forth information known to us with respect to the beneficial ownership of our ordinary shares as of November 18,
2022 by:
| • |
each of our named executive officers;
|
| • |
each of our directors;
|
| • |
all of our executive officers and directors as a group; and
|
| • |
each person or group of affiliated persons known by us to beneficially own more than 5% of our ordinary shares.
|
|
|
Ordinary Shares Beneficially Owned
prior to the Offering
|
Ordinary Shares Beneficially Owned
after the Offering
|
||||||||||||||
|
Shareholder
|
Number
|
Percentage
|
Number
|
Percentage
|
||||||||||||
|
Named Executive Officers and Directors
|
||||||||||||||||
|
Sean Hall
|
392,375
|
17.18
|
%
|
392,375
|
|
|||||||||||
|
Drew Townsend(1)
|
107,572
|
4.71
|
%
|
107,572
|
|
|||||||||||
|
Michael Hall
|
105,383
|
4.61
|
%
|
105,383
|
|
|||||||||||
|
5% or Greater Shareholders
|
|
|||||||||||||||
|
Farjoy Pty Ltd. (2)
|
205,663
|
9.01
|
%
|
205,663
|
|
|||||||||||
|
All other officers and directors as a group
|
11,667
|
*
|
11,667
|
|
||||||||||||
* Less than 1.0% of the outstanding shares.
(1) Includes (1) 70,000 Shares owned by Realm Group Pty Limited, 37,038 Shares owned by Rolay Pty Ltd, with both entities controlled by Mr. Townsend, and (ii) 534 Shares owned by Mr. Townsend directly.
(2) Farjoy Pty Ltd. is an investment holding company that is beneficially owned by Tim Robertson, QC.
114
On June 23, 2021, we entered into an option agreement with Kerem Kaya, our Chief Financial Officer, to purchase 1,667 Shares. These options
are exercisable from June 25, 2021 until June 24, 2024 at an exercise price of A$31.50 per share.
As of November 18, 2022, we had 5,081 shareholders of record and 5,081 beneficial holders. We are not aware of any arrangements the
operation of which may at a subsequent date result in our change of control.
115
DESCRIPTION OF SHARE CAPITAL
General
As of November 18, 2022, we had 2,283,502 fully paid Shares outstanding with no par value.
Memorandum and Articles of Association
General
Our constituent document is a Constitution. The Constitution is subject to the terms of the ASX Listing Rules and the Corporations Act. The
Constitution may be amended or repealed and replaced by special resolution of shareholders, which is a resolution of which notice has been given and that has been passed by at least 75% of the votes cast by shareholders entitled to vote on the
resolution.
Purposes and Objects
Our Constitution does not provide for or prescribe any specific objects or purposes.
The Powers of the Directors
Under the provision of our Constitution our directors may exercise all the powers of our company except any powers that the Corporations
Act or the Constitution requires the Company to exercise in a general meeting.
Interested Directors
According to our constitution, if a director discloses his or her interest in accordance with the Australian Corporations Act 2001, the
director may (i) contract or make an arrangement with the Company, or a related body corporate of the Company or a body corporate in which the Company is interested, in any matter in any capacity, (ii) be counted in a quorum for a meeting of
directors considering the contract or arrangement, (iii) vote on whether the Company enters into the contract or arrangement, and on any matter that relates to the contract or arrangement, or (iv) sign on behalf of the Company, or witness the
affixing of the common seal of the Company to, any document in respect of the contract or arrangement.
Unless a relevant exception applies, the Corporations Act requires our directors to provide disclosure of certain interests or conflicts of
interests and prohibits directors from voting on matters in which they have a material personal interest and from being present at the meeting while the matter is being considered. In addition, the Corporations Act and the ASX Listing Rules require
shareholder approval of any provision of related party benefits to our directors.
Directors’ compensation
The directors will be paid such remuneration as is from time to time determined by the Company in general meeting, and that remuneration
accrues from day to day. The remuneration may be divided among the directors in such proportion as they from time to time agree and, in default of agreement, equally.
The directors will be reimbursed for all travelling and other expenses properly incurred by them in attending and returning from meetings
of the directors or any committee of the directors or general meetings of the Company or otherwise in connection with the business of the Company. In addition to other remuneration provided in our Constitution, all of our directors are entitled to be
paid by us for travel accommodation and other expenses incurred by the directors in attending general meetings, board meetings, committee meetings or otherwise in connection with our business. Directors may also be paid for extra services or any
special exertions performed on behalf of the Company (subject to the Corporations Act and the ASX Listing Rules).
116
Rights Attached to Our Shares
The concept of authorized share capital no longer exists in Australia and as a result, our authorized share capital is unlimited. All our
outstanding Shares are validly issued, fully paid and non-assessable. The rights attached to our Shares are as follows:
Dividend Rights.
The directors may declare that a dividend be paid to the shareholders according to the shareholders’ pro rata shareholdings and the
directors may fix the amount, the time for payment and the method of payment. No dividend is payable except in accordance with the Corporations Act as amended from time to time and no dividend carries interest as against the Company.
Voting Rights.
Holders of Shares have one vote for each Share held on all matters submitted to a vote of shareholders. Such voting rights may be affected
by the grant of any special voting rights to the holders of a class of shares with preferential rights that may be authorized in the future.
The quorum required for an ordinary meeting of shareholders consists of at least two shareholders present in person, or by proxy, attorney
or representative appointed pursuant to our Constitution. A meeting adjourned for lack of a quorum generally is adjourned to the same day in the following week at the same time and place. At the reconvened meeting, the required quorum consists of any
two members present in person, or by proxy, attorney or representative appointed pursuant to our Constitution. The meeting is dissolved if a quorum is not present within 15 minutes from the time appointed for the meeting.
An ordinary resolution, such as a resolution for the declaration of dividends, requires approval by the holders of a majority of the voting
rights represented at the meeting, in person, by proxy, or by written ballot and voting thereon. Under our Constitution, the Corporations Act and the ASX Listing Rules, certain matters must be passed by way of a special resolution. A special
resolution must be passed by at least 75% of the votes cast by shareholders entitled to vote on the resolution and who vote at the meeting in person. Matters which are not required to be passed by special resolution are required to be passed by
ordinary resolution.
Rights in Our Profits.
Our shareholders have the right to share in our profits distributed as a dividend and any other permitted distribution.
Rights in the Event of Liquidation.
In the event of our liquidation, after satisfaction of liabilities to creditors, our assets will be distributed to the holders of Shares in
proportion to the capital at the commencement of the liquidation paid up or which ought to have been paid up on the shares held by them, respectively. This right may be affected by the grant of preferential dividend or distribution rights to the
holders of a class of shares with preferential rights, such as the right in winding up to payment in cash of the amount then paid up on the share, and any arrears of dividend in respect of that share, in priority to any other class of shares.
Directors may make calls
Our Constitution provides that subject to the terms on which the shares have been issued directors may make calls on a shareholder for
amounts unpaid on shares held by that shareholder, other than monies payable at fixed times under the conditions of allotment.
117
Changing Rights Attached to Shares
According to our Constitution, the rights attached to any class of shares, unless otherwise provided by the terms of the class, may be
varied with either the written consent of the holders of not less than 75% of the issued shares of that class or the sanction of a special resolution passed at a separate general meeting of the shares of that class.
Annual and Extraordinary Meetings
Our directors must convene an annual meeting of shareholders at least once every calendar year. Notice of at least 21 days prior to the
date of the meeting is required. A general meeting may be convened by any director, or shareholders in compliance with the Corporations Act.
Limitations on the Rights to Own Securities in Our Company
Subject to certain limitations on the percentage of shares a person may hold in the Company, neither our Constitution nor the laws of the
Commonwealth of Australia restrict in any way the ownership or voting of shares in the Company.
Changes in Our Capital
Pursuant to the ASX Listing Rules, our directors may in their discretion issue securities to persons who are not related parties of our
company, without the approval of shareholders, if such issue, when aggregated with securities issued by our company during the previous 12-month period would be an amount that would not exceed 15% of our issued capital at the commencement of the
12-month period. Other allotments of securities require approval by an ordinary resolution of shareholders unless an exception applies.
Pre-Funded Warrants to be Issued as Part of this Offering
Duration and Exercise Price
Each Pre-Funded Warrant offered hereby will have an initial exercise price per share equal to US$0.0001. The Pre-Funded Warrants will be
immediately exercisable and may be exercised at any time until the Pre-Funded Warrants are exercised in full. The exercise price and number of Shares issuable upon exercise is subject to appropriate adjustment in the event of stock dividends, stock
splits, reorganizations or similar events affecting our Shares and the exercise price. The Pre-Funded Warrants will be issued separately from the accompanying Share Purchase Warrants and may be transferred separately immediately thereafter.
Exercisability
The Pre-Funded Warrants will be exercisable, at the option of each holder, in whole or in part, by delivering to us a duly executed
exercise notice accompanied by payment in full for the number of Shares purchased upon such exercise (except in the case of a cashless exercise as discussed below). Purchasers of the Pre-Funded Warrants in this offering may elect to deliver their
exercise notice following the pricing of the offering and prior to the issuance of the Pre-Funded Warrants at closing to have their Pre-Funded Warrants exercised immediately upon issuance and receive Shares underlying the Pre-Funded Warrants upon
closing of this offering. A holder (together with its affiliates) may not exercise any portion of the Pre-Funded Warrant to the extent that the holder would own more than 4.99% of the outstanding Shares immediately after exercise, except that upon at
least 61 days’ prior notice from the holder to us, the holder may increase the amount of ownership of outstanding stock after exercising the holder’s Pre-Funded Warrants up to 9.99% of the number Shares outstanding immediately after giving effect to
the exercise, as such percentage ownership is determined in accordance with the terms of the Pre-Funded Warrants. Purchasers of Pre-Funded Warrants in this offering may also elect prior to the issuance of the Pre-Funded Warrants to have the initial
exercise limitation set at 9.99% of our outstanding Shares. No fractional Shares will be issued in connection with the exercise of a Pre-Funded Warrant. In lieu of fractional shares, we will round down to the next whole share.
118
Cashless Exercise
If, at the time a holder exercises its Pre-Funded Warrants, a registration statement registering the issuance of the Shares underlying the
Pre-Funded Warrants under the Securities Act is not then effective or available, then in lieu of making the cash payment otherwise contemplated to be made to us upon such exercise in payment of the aggregate exercise price, the holder may elect
instead to receive upon such exercise (either in whole or in part) the net number of Shares determined according to a formula set forth in the Pre-Funded Warrants.
Transferability
Subject to applicable laws, a Pre-Funded Warrant may be transferred at the option of the holder upon surrender of the Pre-Funded Warrant to
us together with the appropriate instruments of transfer.
Exchange Listing
There is no trading market available for the Pre-Funded Warrants on any securities exchange or nationally recognized trading system. We do
not intend to list the Pre-Funded Warrants on any securities exchange or nationally recognized trading system.
Right as a Stockholder
Except as otherwise provided in the Pre-Funded Warrants or by virtue of such holder’s ownership of our Shares, the holders of the
Pre-Funded Warrants do not have the rights or privileges of holders of our Shares, including any voting rights, until they exercise their Pre-Funded Warrants.
Fundamental Transaction
In the event of a fundamental transaction, as described in the Pre-Funded Warrants and generally including any reorganization,
recapitalization or reclassification of our Shares, the sale, transfer or other disposition of all or substantially all of our properties or assets, our consolidation or merger with or into another person, the acquisition of more than 50% of our
outstanding Shares, or any person or group becoming the beneficial owner of 50% of the voting power represented by our outstanding Shares, the holders of the Pre-Funded Warrants will be entitled to receive upon exercise of the Pre-Funded Warrants the
kind and amount of securities, cash or other property that the holders would have received had they exercised the Pre-Funded Warrants immediately prior to such fundamental transaction.
Share Purchase Warrants to be Issued as Part of this Offering
The following summary of certain terms and provisions of the Share Purchase Warrants that are being offered hereby is not complete and is
subject to, and qualified in its entirety by, the provisions of the Share Purchase Warrants, the form of which is filed as an exhibit to the registration statement of which this prospectus forms a part. Prospective investors should carefully review
the terms and provisions of the form of Share Purchase Warrants for a complete description of the terms and conditions of the Share Purchase Warrants.
Duration and Exercise Price
Each Share Purchase Warrant included in the Units will have an initial exercise price equal to US$ per Share. The Share
Purchase Warrants will be immediately exercisable and will expire on the fifth anniversary of the original issuance date. The exercise price and number of Shares issuable upon exercise is subject to appropriate adjustment in the event of stock
dividends, stock splits, reorganizations or similar events affecting our Shares and the exercise price. The Share Purchase Warrants will be issued separately from the Shares included in the Share Units, or the Pre-Funded Warrants included in the
Pre-Funded Warrant Units, as the case may be. A Share Purchase Warrant to purchase Shares will be included in each Share Unit or Pre-Funded Warrant Unit purchased in this offering.
119
Exercisability
The Share Purchase Warrants will be exercisable, at the option of each holder, in whole or in part, by delivering to us a duly executed
exercise notice accompanied by payment in full for the number of Shares purchased upon such exercise (except in the case of a cashless exercise as discussed below). A holder (together with its affiliates) may not exercise any portion of the Share
Purchase Warrant to the extent that the holder would own more than 4.99% of the outstanding Shares immediately after exercise, except that upon at least 61 days’ prior notice from the holder to us, the holder may increase the amount of ownership of
outstanding Shares after exercising the holder’s Share Purchase Warrants up to 9.99% of the number of Shares outstanding immediately after giving effect to the exercise, as such percentage ownership is determined in accordance with the terms of the
Share Purchase Warrants. Purchasers of Share Purchase Warrants in this offering may also elect prior to the issuance of the Share Purchase Warrants to have the initial exercise limitation set at 9.99% of our outstanding Shares.
Cashless Exercise
If, at the time a holder exercises its Share Purchase Warrants, a registration statement registering the issuance of the Shares underlying
the Share Purchase Warrants under the Securities Act is not then effective or available for the issuance of such Shares, then in lieu of making the cash payment otherwise contemplated to be made to us upon such exercise in payment of the aggregate
exercise price, the holder may elect instead to receive upon such exercise (either in whole or in part) the net number of Shares determined according to a formula set forth in the Share Purchase Warrants.
Fractional Shares
No fractional Shares will be issued upon the exercise of the Share Purchase Warrants. Rather, the number of Shares to be issued will be
rounded to the nearest whole number, or the Company will pay a cash adjustment in respect of the fractional share.
Transferability
Subject to applicable laws, the Share Purchase Warrants may be offered for sale, sold, transferred or assigned without our consent. There
is currently no trading market for the Share Purchase Warrants.
Exchange Listing
There is no trading market available for the Share Purchase Warrants on any securities exchange or nationally recognized trading system. We
do not intend to list the Share Purchase Warrants on any securities exchange or nationally recognized trading system.
Right as a Stockholders
Except as otherwise provided in the Share Purchase Warrants or by virtue of such holder’s ownership of our Shares, the holders of the Share
Purchase Warrants do not have the rights or privileges of holders of our Shares, including any voting rights, until they exercise their Share Purchase Warrants.
Fundamental Transaction
In the event of a fundamental transaction, as described in the Share Purchase Warrants and generally including any reorganization,
recapitalization or reclassification of our Shares, the sale, transfer or other disposition of all or substantially all of our properties or assets, our consolidation or merger with or into another person, the acquisition of more than 50% of our
outstanding Shares, or any person or group becoming the beneficial owner of 50% of the voting power represented by our outstanding Shares, the holders of the Share Purchase Warrants will be entitled to receive upon exercise of the Share Purchase
Warrants the kind and amount of securities, cash or other property that the holders would have received had they exercised the Share Purchase Warrants immediately prior to such fundamental transaction.
120
SHARES ELIGIBLE FOR FUTURE SALE
Future sales of substantial amounts of our Shares in the public market could adversely affect prevailing market prices. Furthermore, since
only a limited number of Shares will be available for sale shortly after the offering because of contractual and legal restrictions on resale described below, sales of substantial amounts of Shares in the public market after the restrictions lapse
could adversely affect the prevailing market price for our Shares as well as our ability to raise equity capital in the future.
Upon completion of this offering, we will have Shares issued and outstanding (or Shares if the underwriter exercises in full its
option to purchase additional Shares).
Of these shares, the Shares sold in this offering (or Shares, if the underwriter exercises in full its option to purchase additional
Shares) will be freely tradable without further restriction or registration under the Securities Act, except that any shares purchased by our affiliates may generally only be sold in compliance with Rule 144, which is described below. The remaining
Shares will be deemed “restricted securities” under the Securities Act. Restricted securities may be sold in the public market only if they are registered under the Securities Act or if they qualify for an exemption from registration under Rules 144
or 701 under the Securities Act, which are discussed below.
Lock-up Agreements
We, and each of our executive officers and directors, will enter into lock-up agreements in connection with this offering under which these
parties will agree not to sell or otherwise transfer their Shares (including securities convertible into or exchangeable for Shares) for a period of ninety (90) days after the date of this prospectus. These lock-up restrictions are subject to certain
exceptions and may be waived by the representatives of the underwriter at any time. As a result of these contractual restrictions, Shares subject to lock-up agreements will not be eligible for sale, including pursuant to Rules 144 or 701 under the
Securities Act as discussed below, until these agreements expire or the restrictions are waived by the representatives of the underwriter.
See “Underwriting” for a more complete description of the lock-up agreements.
Rule 144
In general, Rule 144 provides that once we have been subject to the public company reporting requirements of Section 13 or Section 15(d) of
the Exchange Act for at least 90 days, a person who is not deemed to have been one of our affiliates for purposes of the Securities Act at any time during the 90 days preceding a sale and who has beneficially owned the shares of our Shares proposed
to be sold for at least six months is entitled to sell those shares without complying with the manner of sale, volume limitation, or notice provisions of Rule 144, subject to compliance with the public information requirements of Rule 144. If such a
person has beneficially owned the shares proposed to be sold for at least one year, including the holding period of any prior owner other than our affiliates, then that person would be entitled to sell those shares without complying with any of the
requirements of Rule 144.
In general, Rule 144 provides that our affiliates or persons selling Shares on behalf of our affiliates are entitled to sell upon
expiration of the lock-up agreements described above, within any three-month period, a number of Shares that does not exceed the greater of:
| • |
1% of the number of shares of our Shares then outstanding, which will equal shares immediately after the completion of this offering; or
|
| • |
the average weekly trading volume of our Shares during the four calendar weeks preceding the filing of a notice on Form 144 with respect to that sale.
|
121
MATERIAL UNITED STATES FEDERAL INCOME AND AUSTRALIAN TAX CONSIDERATIONS
The material U.S. federal income tax and Australian tax law consequences of an investment in the Shares is based upon laws and relevant
interpretations thereof in effect as of the date of this prospectus, all of which are subject to change, possibly with retroactive effect. This discussion does not deal with all possible tax consequences relating to an investment in the Shares, such
as the tax consequences under U.S. state, local and other tax laws other than U.S. federal income tax laws and certain Australian tax laws
Material U.S. Federal Income Tax Considerations
In the opinion of Seward & Kissel LLP, our United States counsel, the following discussion describes material U.S. federal income tax
consequences to U.S. Holders (as defined below) of an investment in our Shares and applies only to U.S. Holders that acquire our Shares in exchange for cash in this offering, hold our Shares as capital assets within the meaning of Section 1221 of the
Code (as defined below) and have the U.S. dollar as their functional currency.
This discussion is based on the tax laws of the United States as in effect on the date of this prospectus, including the Internal Revenue
Code of 1986, as amended (the “Code”), and U.S. Treasury regulations in effect or, in some cases, proposed, as of the date of this prospectus, as well as judicial and administrative interpretations thereof available on or before such date. All of the
preceding authorities are subject to change, and any such change could apply retroactively and affect the U.S. federal income tax consequences described below. The statements in this prospectus are not binding on the U.S. Internal Revenue Service
(the “IRS”) or any court. Thus, the Company can provide no assurance that the U.S. federal income tax consequences discussed below will not be challenged by the IRS or will be sustained by a court if challenged by the IRS. Furthermore, this
discussion does not address any estate or gift tax consequences, state, local or non-U.S. tax consequences, or other tax consequences other than U.S. federal income tax consequences.
The following discussion does not describe all the tax consequences that may be relevant to any particular investor or to persons in
special tax situations such as:
| • |
banks and certain other financial institutions;
|
| • |
regulated investment companies;
|
| • |
real estate investment trusts;
|
| • |
insurance companies;
|
| • |
broker-dealers;
|
| • |
traders that elect to mark our Shares to market for U.S. federal income tax purposes;
|
| • |
tax-exempt entities;
|
| • |
persons liable for alternative minimum tax or the Medicare contribution tax on net investment income;
|
| • |
U.S. expatriates;
|
| • |
persons holding our Shares as part of a straddle, hedging, constructive sale, conversion, or integrated transaction;
|
| • |
persons that actually or constructively own 10% or more of the Company’s stock by vote or value;
|
| • |
persons that are resident or ordinarily resident in or have a permanent establishment in a jurisdiction outside the United States;
|
| • |
persons who acquired our Shares pursuant to the exercise of any employee share option or otherwise as compensation; or
|
| • |
persons holding our Shares through partnerships or other pass-through entities or arrangements.
|
PROSPECTIVE PURCHASERS ARE URGED TO CONSULT THEIR TAX ADVISORS ABOUT THE APPLICATION OF THE U.S. FEDERAL TAX RULES TO
THEIR PARTICULAR CIRCUMSTANCES AS WELL AS THE STATE, LOCAL AND NON-U.S. TAX CONSEQUENCES TO THEM OF THE PURCHASE, OWNERSHIP AND DISPOSITION OF OUR SHARES.
122
As used herein, the term “U.S. Holder” means a beneficial owner of our Shares that, for U.S. federal income tax purposes, is or is treated
as:
| • |
an individual who is a citizen or resident of the United States;
|
| • |
a corporation created or organized in or under the laws of the United States, any state thereof or the District of Columbia;
|
| • |
an estate whose income is subject to U.S. federal income taxation regardless of its source; or
|
| • |
a trust that (1) is subject to the supervision of a court within the United States and the control of one or more U.S. persons or (2) has a valid election in effect under applicable U.S.
Treasury regulations to be treated as a U.S. person.
|
The tax treatment of a partner in an entity or arrangement treated as a partnership for U.S. federal income tax purposes that holds our
Shares generally will depend on such partner’s status and the partnership’s activities. Accordingly, a U.S. Holder that is a partner in such a partnership should consult its tax advisor.
Dividends and Other Distributions on Our Shares
Subject to the passive foreign investment company considerations discussed below, the gross amount of distributions made by the Company
with respect to our Shares (including the amount of any non-U.S. taxes withheld therefrom) generally will be includible as dividend income in a U.S. Holder’s gross income in the year received, to the extent such distributions are paid out of the
Company’s current or accumulated earnings and profits, as determined under U.S. federal income tax principles. Because the Company does not maintain its earnings and profits calculations under U.S. federal income tax principles, a U.S. Holder should
expect all cash distributions to be reported as dividends for U.S. federal income tax purposes. Such dividends will not be eligible for the dividends-received deduction allowed to U.S. corporations with respect to dividends received from other U.S.
corporations. Dividends received by non-corporate U.S. Holders may be “qualified dividend income,” which is taxed at the lower applicable capital gains rate, provided that (1) the Company is eligible for the benefits of the tax treaty between the
United States and Australia (the “Treaty”) or our Shares are readily tradable on an established securities market in the United States, (2) the Company is not a passive foreign investment company (as discussed below) for either the taxable year in
which the dividend was paid or the preceding taxable year, (3) the U.S. Holder satisfies specific holding period requirements and (4) the U.S. Holder is not under an obligation to make related payments with respect to positions in substantially
similar or related property. U.S. Holders should consult their tax advisors regarding the availability of the lower rate for dividends paid with respect to our Shares.
The amount of any distribution paid in a foreign currency will be equal to the U.S. dollar value of such currency, translated at the spot
rate of exchange on the date such distribution is received, regardless of whether the payment is in fact converted into U.S. dollars at that time. If the dividend is converted into U.S. dollars on the date of receipt, a U.S. Holder should not be
required to recognize foreign currency gain or loss in respect of the dividend income. A U.S. Holder may have foreign currency gain or loss if the dividend is converted into U.S. dollars after the date of receipt. In general, foreign currency gain or
loss will be treated as U.S.-source ordinary income or loss.
Dividends on our Shares generally will constitute foreign source income for foreign tax credit limitation purposes. Subject to certain
complex conditions and limitations, any Australian taxes withheld on any distributions on our Shares may be eligible for credit against a U.S. Holder’s federal income tax liability or, at such holder’s election, may be eligible as a deduction in
computing such holder’s U.S. federal taxable income. If a refund of the tax withheld is available under the laws of Australia or under the Treaty, the amount of tax withheld that is refundable will not be eligible for such credit against a U.S.
Holder’s U.S. federal income tax liability (and will not qualify for the deduction against U.S. federal taxable income). If the dividends constitute qualified dividend income as discussed above, the amount of the dividend taken into account for
purposes of calculating the foreign tax credit limitation will generally be limited to the gross amount of the dividend, multiplied by the reduced rate applicable to the qualified dividend income, divided by the highest rate of tax normally
applicable to dividends. The limitation on foreign taxes eligible for the credit is calculated separately concerning specific classes of income. For this purpose, dividends distributed by the Company with respect to our Shares will generally
constitute “passive category income”. The rules relating to the determination of the U.S. foreign tax credit are complex, and U.S. Holders should consult their tax advisors regarding the availability of a foreign tax credit in their particular
circumstances and the possibility of claiming an itemized deduction (in lieu of the foreign tax credit) for any foreign taxes paid or withheld.
123
Sale or Other Taxable Disposition of Our Shares
Subject to the passive foreign investment company considerations discussed below, upon a sale or other taxable disposition of our Shares, a
U.S. Holder will recognize capital gain or loss in an amount equal to the difference between the amount realized and the U.S. Holder’s adjusted tax basis in such Shares. A U.S. Holder’s initial tax basis in our Shares generally will equal the cost of
such Shares. Generally, any such gain or loss will be treated as long-term capital gain or loss if the U.S. Holder’s holding period in our Shares exceeds one year. Non-corporate U.S. Holders (including individuals) generally will be subject to U.S.
federal income tax on long-term capital gain at preferential rates. The deductibility of capital losses is subject to significant limitations.
Gain or loss, if any, realized by a U.S. Holder on the sale or other disposition of our Shares generally will be treated as U.S. source
gain or loss for U.S. foreign tax credit limitation purposes. The use of U.S. foreign tax credits relating to any Australian tax imposed upon the sale or other disposition of our Shares may be unavailable or limited. U.S. Holders should consult their
tax advisors regarding the tax consequences if Australian taxes are imposed on or connected with a sale or other disposition of our Shares and their ability to credit any Australian tax against their U.S. federal income tax liability.
Passive Foreign Investment Company Considerations
The Company will be classified as a PFIC for any taxable year if either: (a) at least 75% of its gross income is “passive income” for
purposes of the PFIC rules or (b) at least 50% of the value of its assets (determined on the basis of a quarterly average) is attributable to assets that produce or are held for the production of passive income. For this purpose, passive income
includes interest, dividends and other investment income, with certain exceptions. Cash, cash-equivalents and digital assets generally are passive assets for these purposes. Goodwill is active to the extent attributable to activities that produce or
are intended to produce active income. The PFIC rules also contain a look-through rule whereby the Company will be treated as owning its proportionate share of the gross assets and earning its proportionate share of the gross income of any other
corporation in which it owns, directly or indirectly, 25% or more (by value) of the stock.
Under the PFIC rules, if the Company were considered a PFIC at any time that a U.S. Holder holds our Shares, the Company would continue to
be treated as a PFIC with respect to such investment unless (i) the Company ceases to be a PFIC and (ii) the U.S. Holder made a “deemed sale” election under the PFIC rules.
Based on the current and anticipated composition of the income, assets, and operations of the Company and the expected price of our Shares
in this offering, the Company does not expect to be treated as a PFIC for the current taxable year or in the foreseeable future. However, whether the Company is treated as a PFIC is a factual determination made on an annual basis after the close of
each taxable year. This determination will depend on, among other things, the ownership and the composition of the Company’s income and assets, as well as the relative value of the Company’s assets (which may fluctuate with the Company’s market
capitalization), at the relevant time. Moreover, the application of the PFIC rules to digital assets and transactions related thereto is subject to uncertainty. Among other things, the IRS has issued limited guidance on the treatment of income from
mining digital assets. The IRS or a court may disagree with the Company’s determinations, including how the Company determines the value of the Company’s assets and the percentage of the Company’s assets that are passive assets under the PFIC rules.
Therefore, there can be no assurance that the Company will not be classified as a PFIC for the current taxable year or for any future taxable year.
If the Company is considered a PFIC at any time that a U.S. Holder holds our Shares, any gain recognized by the U.S. Holder on a sale or
other disposition of our Shares, as well as the amount of any “excess distribution” (defined below) received by the U.S. Holder, would be allocated ratably over the U.S. Holder’s holding period for our Shares. The amounts allocated to the taxable
year of the sale or other disposition (or the taxable year of receipt, in the case of an excess distribution) and to any year before the Company became a PFIC would be taxed as ordinary income. The amount allocated to each other taxable year would be
subject to tax at the highest rate in effect for individuals or corporations, as appropriate, for that taxable year, and an interest charge would be imposed. For the purposes of these rules, an excess distribution is the amount by which any
distribution received by a U.S. Holder on our Shares exceeds 125% of the average of the annual distributions on our Shares received during the preceding three years or the U.S. Holder’s holding period, whichever is shorter. Certain elections may be
available that would result in alternative treatments (such as mark-to-market treatment) of our Shares if the Company is considered a PFIC.
If the Company is considered a PFIC, a U.S. Holder will also be subject to annual information reporting requirements. U.S. Holders should
consult their tax advisors about the potential application of the PFIC rules to an investment in our Shares.
124
Information Reporting and Backup Withholding
Dividend payments with respect to our Shares and proceeds from the sale, exchange, or redemption of our Shares may be subject to
information reporting to the IRS and U.S. backup withholding. A U.S. Holder may be eligible for an exemption from backup withholding if the U.S. Holder furnishes a correct taxpayer identification number and makes any other required certification or
is otherwise exempt from backup withholding. U.S. Holders who are required to establish their exempt status may be required to provide such certification on IRS Form W-9. U.S. Holders should consult their tax advisors regarding applying the U.S.
information reporting and backup withholding rules.
Backup withholding is not an additional tax. Amounts withheld as backup withholding may be credited against a U.S. Holder’s U.S. federal
income tax liability, and such U.S. Holder may obtain a refund of any excess amounts withheld under the backup withholding rules by timely filing an appropriate claim for refund with the IRS and furnishing any required information.
Additional Information Reporting Requirements
Certain U.S. Holders who are individuals (and certain entities) that hold an interest in “specified foreign financial assets” (which may
include our Shares) are required to report information relating to such assets, subject to certain exceptions (including an exception for our Shares held in accounts maintained by certain financial institutions). Penalties can apply if U.S. Holders
fail to satisfy such reporting requirements. U.S. Holders should consult their tax advisors regarding the applicability of these requirements to their acquisition and ownership of our Shares.
THE DISCUSSION ABOVE DOES NOT COVER ALL TAX MATTERS THAT MAY BE IMPORTANT TO YOU. EACH PROSPECTIVE PURCHASER SHOULD
CONSULT ITS OWN TAX ADVISOR ABOUT THE TAX CONSEQUENCES OF AN INVESTMENT IN OUR SHARES UNDER ITS CIRCUMSTANCES.
Material Australian Tax Considerations
In the opinion of Pitch Partners, our Australian tax counsel, the following provides a general discussion of the material Australian income
tax, stamp duty, and goods and services tax considerations generally applicable to the acquisition, ownership, and disposal by the owners of the Shares.
This section is based upon existing Australian tax law as of the date of this registration statement, which is subject to change, possibly
retrospectively. This discussion does not address all aspects of Australian tax law, which may be important to particular investors in light of their investment circumstances, such as shares held by investors subject to special tax rules (for
example, financial institutions, insurance companies, or tax-exempt organizations).
It does not purport to address all possible tax situations that may be relevant to a decision to purchase, own, or deposit our Shares. It
is included herein solely for preliminary information purposes and is not intended to be, nor should it be construed to be, legal or tax advice. The Company and their officers, employees, taxation or other advisers do not accept any liability or
responsibility in respect of any statement concerning taxation consequences or the taxation consequences.
Prospective purchasers of our Shares should consult their tax advisers on the applicable tax consequences related to the ownership of our
Shares, based on their particular circumstances. The comments in this section deal only with the Australian taxation implications of the ownership and disposition of Shares if you hold your Shares as investments on a capital account. In addition,
this discussion does not include any non-Australian or state tax considerations, other than stamp duty and goods and services tax.
For this discussion, a holder of our Shares that is not an Australian tax resident and is not carrying on business in Australia at or
through a permanent establishment is referred to as a “Non-Australian Holder”.
Conversely, for the purposes of this discussion, a holder that is an Australian tax resident or is carrying on business in Australia at or
through a permanent establishment is referred to as an “Australian Resident Holder”.
Please be aware that the residence concept used in this section applies for Australian tax assessment purposes only. Any reference in this
section to a tax, duty, levy impost, or other charge or withholding of a similar nature refers to Australia’s tax laws and/or concepts only. Also, please note that a reference to Australian income tax encompasses corporate income tax and personal
income tax generally.
125
Taxation of the Company
As the Company is a fully taxable Australian company, its taxable income is subject to corporate income tax in Australia. All Australian
companies are subject to a corporate income tax rate of 30% unless it is classified as a ‘base rate entity’. Base rate entities are subject to a reduced corporate income tax rate of 25% for the 2021/2022 income year.
Broadly, the Company may qualify as a base rate entity for any particular income year in which (a) the Company’s aggregated turnover for
that income year is less than the aggregated turnover threshold that is applicable for that income year (currently $50 million), and (b) 80% or less of the Company’s assessable income in that income year is base rate entity passive income (“BREPI”).
Broadly, BREPI is corporate distributions and franking credits on these distributions, royalties and rent, interest income (some exceptions apply), gains on qualifying securities, net capital gains, and any amounts included in a company’s assessable
income because the company is a partner in a partnership or a beneficiary of a trust, to the extent it is traceable (either directly or indirectly) to an amount that is otherwise BREPI.
Taxation of Australian Resident Holders
Taxation of Dividends
Dividends paid by the Company on the Shares should be included in the assessable income of an Australian Resident Holder. Australia
operates a dividend imputation system under which dividends may be declared to be “franked” to the extent they are paid out of company profits that have been subject to income tax, are “frankable distributions”, and the company has a sufficient
credit balance in its franking account to frank the dividend.
Individuals and complying superannuation entities
Australian Resident Holders who are individuals (not acting as a trustee) or complying superannuation entities should include the dividend
in their assessable income in the year the dividend is paid, together with any franking credit attached to that dividend.
Subject to the comments concerning ‘Qualified Persons’ below, such Australian Resident Holders should be entitled to a tax offset equal to
the franking credit attached to the dividend. The tax offset can be applied to reduce the tax payable on the investor’s taxable income. Where the tax offset exceeds the tax payable on the investor’s taxable income, the investor should be entitled to
a tax refund equal to the excess.
To the extent that the dividend is unfranked, an Australian individual Shareholder will generally be taxed at their prevailing marginal tax
rate on the dividend received (with no tax offset). Complying Australian superannuation entities will generally be taxed at the prevailing rate for complying superannuation entities on the dividend received (with no tax offset).
126
Companies
Australian Resident Holders that are companies (not acting as a trustee) are also required to include both the dividend and the associated
franking credits (if any) in their assessable income.
Subject to the comments in relation to ‘Qualified Persons’ below, such companies should be entitled to a tax offset up to the amount of the
franking credit attached to the dividend. Likewise, the company should be entitled to a credit in its own franking account to the extent of the franking credits attached to the distribution received. This will allow the Australian Resident Holders
that are companies to pass on the franking credits to their investor(s) on the subsequent payment of franked dividends.
Excess franking credits received by the company shareholder will not give rise to a refund entitlement for a company but may be converted
into carry forward tax losses instead. This is subject to specific rules on how the carry forward tax loss is calculated and utilized in future years. For completeness, this tax loss cannot be carried back under the loss carry back tax offset rules
introduced in the 2020-21 Federal Budget.
Trusts and partnerships
Australian Resident Holders who are trustees (other than trustees of complying superannuation entities, which are dealt with above) or
partnerships are also required to include any dividends and any franking credits in calculating the net income of the trust or partnership. Where a fully franked or partially franked dividend is received, the relevant beneficiary or partner may be
entitled to a tax offset equal to the beneficiary’s or partner’s share of the net income of the trust or partnership subject, in the case of a beneficiary of a trust, to specific tax rules which apply to determine the ability of the beneficiaries of
a trust to claim franking credits attached to franked dividends received by a trust.
To the extent that the dividend is unfranked, an Australian trustee (other than trustees of complying superannuation entities) or
partnership, will be required to include the unfranked dividend in the net income of the trust or partnership. The relevant beneficiary or partner will be taxed at the relevant prevailing tax rate on their share of the net income of the trust or
partnership (with no tax offset).
Qualified Persons
The benefit of franking credits can be denied where an Australian Resident Holder is not a ‘qualified person’ in which
case the Holder will not be able to include an amount for the franking credits in their assessable income and will not be entitled to a tax offset.
Broadly, to be a qualified person, a shareholder must satisfy the holding period rule and, if necessary, the related
payment rule.
These are complex issues and specific advice should be obtained to determine if these requirements have been
satisfied.
127
Capital Gains Tax (“CGT”) Implications
Disposal of Shares
For Australian Resident Holders, who hold their Shares on capital account, the future disposal of Shares will give rise to a CGT event. The
time of the CGT event will generally be the time when the contract for disposal of the shares is entered into. Australian Resident Holders will derive a capital gain on the disposal of their Shares to the extent that the capital proceeds exceed the
cost base of their Shares.
A capital loss will be made where the capital proceeds are less than the cost base of their Shares. Where a capital loss is made, capital
losses can only be offset against capital gains derived in the same or later incomes years. They cannot be offset against ordinary income nor carried back to offset net capital gains arising in earlier income years. Capital losses may be carried
forward to future income years subject to the satisfaction of the relevant loss testing provisions.
Capital Proceeds
The capital proceeds for a disposal to a buyer dealing with the seller at arm’s length should generally include any consideration received
by the Australian Resident Holder in respect to the disposal of their Shares. The capital proceeds will be replaced with the market value of the Shares in certain circumstances, for example where the consideration received did not represent the
market value of the Shares and the transferee and transferor did not deal with each other at arm’s length.
Cost base of Shares
The cost base of a Share will generally include the cost of acquiring the Share, plus any incidental costs of acquisition and disposal
(i.e., brokerage costs and legal fees).
CGT Discount
The CGT discount may apply to Australian Resident Holders that are individuals complying Australian superannuation funds or trusts, who
hold their Shares on capital account and have held, or are taken to have held, their Shares for at least 12 months (not including the date of acquisition or date of disposal) at the time of the disposal of their Shares.
The CGT discount is:
| • |
One-half if the Australian Resident Holder is an individual or trustee: meaning only 50% of the capital gain will be included in the Australian Resident Holder’s assessable income; and
|
| • |
One-third if the Australian Resident Holder is a trustee of a complying superannuation entity: meaning only two-thirds of the capital gain will be included in the Australian Resident Holder’s
assessable income.
|
The CGT discount is not available to Australian Resident Holders that are companies and is not available if the Australian Resident Holder
holds their shares as trading stock or on revenue account.
If an Australian Resident Holder makes a discounted capital gain, any current year and/or carried-forward capital losses will be applied
(subject to certain tax loss integrity rules being satisfied, particularly for companies) to reduce the undiscounted capital gain before the relevant CGT discount is applied (noting the discount is not available for companies). The resulting amount
forms the Australian Resident Holder’s net capital gain for the income year and is included in its assessable income.
The CGT discount rules relating to trusts are complex. Subject to certain requirements being satisfied, the capital gain may flow through
to the beneficiaries in that trust, who will assess the eligibility for the CGT discount in their own right. Accordingly, we recommend trustees seek their own independent advice on how the CGT discount applies to the trust and its beneficiaries.
128
Taxation of Non-Australian Holders
Taxation of Dividends
Non-Australian Holders should not be subject to Australian income tax. However, the Company may be required to
withhold Australian dividend withholding tax on Share dividends paid to Non-Australian Holders.
Australia has a franking system wherein dividends can be franked, and Australian resident shareholders receive a
franking credit which effectively represents the corporate tax paid by the underlying company (i.e., Medlab).
Dividends received by Non-Australian Holders which are franked should not be subject to Australian dividend
withholding tax to the extent of the franking (i.e., if the dividend is fully franked, it should not be subject to Australian dividend withholding tax at all). However, refunds of franking credits are not available to Non-Australian Holders.
Dividends attributable to Conduit Foreign Income
Where the Company declares an unfranked dividend to be conduit foreign income (“CFI”), the Company is not required to withhold withholding tax on such dividends paid to
Non-Australian Holders.
CFI generally includes amounts received by the Company that have been derived from a non-Australian source, for example dividends received
from foreign subsidiaries which are treated as non-assessable non-exempt income for Australian tax purposes.
Unfranked dividends
Dividend withholding tax will be imposed on dividends to the extent they are unfranked and are not declared to be CFI.
Under the provisions of the current Double Taxation Convention between Australia and the United States, the Australian tax withheld on unfranked dividends that are not declared
to be CFI paid by the Company to a resident of the United States who is beneficially entitled to the dividend is in most cases limited to 15% if the Non-Resident Holder qualifies for the benefit of the Double Taxation Convention as a resident of
the United States.
Under the Double Taxation Convention between Australia and the United States, certain qualifying companies that are a Non-Australian Holder
that directly own 10% or more of the voting power in an Australian company (i.e. Medlab) and also qualify for the benefit of the Double Taxation Convention as a resident of the United States (and do not have such benefits limited by the Limitation on
Benefits article of the Double Taxation Convention), may qualify for a lower 5% Australian withholding tax on unfranked dividends that are not declared to be CFI paid by the Company to which they are beneficially entitled.
129
CGT Implications
Disposal of shares
Non-Australian Holders who hold their shares on capital account may qualify for an exemption from Australian capital gains tax on the gain
made on a sale or other disposal of Shares subject to certain integrity rules. By way of guidance, Non-Australian Holders will not be able to qualify for an exemption where:
| • |
they, together with associates, hold 10% or more of our issued capital, at the time of disposal or for a 12-month period during the two years prior to disposal; and
|
| • |
more than 50% of our assets held directly or indirectly, determined by reference to market value, consists of direct or indirect interests in Australian real property (which includes land and
leasehold interests) or Australian mining, quarrying or prospecting rights at the time of disposal.
|
Australian capital gains tax applies to net capital gains at a taxpayer’s marginal tax rates. Net capital gains are calculated after
reduction for capital losses, which may only be offset against capital gains.
The capital gains tax discount is not available to individual (i.e. natural person) or trust Non-Australian Holders on gains in respect of Shares, where they were
Non-Australian Holders during the entire holding period. Where individual or trust Non-Australian Holders were Australian tax residents for some of the holding period and acquired their Shares prior to May 9, 2012, some portion of the capital gain
may be eligible for the capital gains tax discount. Companies are not entitled to a capital gains tax discount.
Broadly, where there is a disposal of certain taxable Australian property, the purchaser will be required to withhold and remit to the
Australian Taxation Office (the “ATO”) 12.5% of the proceeds from the sale. A transaction is excluded from the withholding requirements in certain circumstances, including where the transaction is an on-market transaction conducted on an approved
stock exchange, a securities lending arrangement, or the transaction is conducted using a broker operated crossing system. There may also be an exception to the requirement to withhold where a Non-Australian Holder provides a declaration that their
Shares are not ‘indirect Australian real property interests’. The Non-Australian Holder may be entitled to receive a tax credit for the tax withheld by the purchaser which they may claim in their Australian income tax return.
Shareholders Holding Shares on Revenue Account
As noted above, this summary deals only with the Australian taxation implications of the
ownership and disposition of Shares if you hold your Shares as investments on a capital account.
Some Non-Australian Holders may hold their Shares on revenue rather than capital account – for
example, share traders. Depending on the particular circumstances of such Non-Australian Holders, any gains they make from the sale or other disposal of Shares may be included in their assessable income in Australia. Specific advice should be
obtained.
Dual Residency
If a holder of Shares is a resident of both Australia and the United States under those countries’ domestic taxation laws, that holder may
be subject to tax as an Australian resident. If, however, the holder is determined to be a U.S. resident for the purposes of the Double Taxation Convention between the United States and Australia, the Australian tax would be subject to limitation by
the Double Taxation Convention. Holders should obtain specialist taxation advice in these circumstances.
General Australian Tax Matters
Stamp Duty
No Australian stamp duty is payable on the issue, transfer and/or surrender of the Shares, except where the Company is a “land holder” and the acquirer of the Shares owns or
acquires on an associate inclusive basis 90% or more of the Shares in the Company.
Goods and Services Tax
No Australian Goods and Services Tax (“GST”) will be payable on the supply or acquisition of the Shares to a
Non-Australian Holder. To the extent the Non-Australian Holder pays GST relating to the acquisition or disposal of the Shares, advice should be obtained regarding whether they might be entitled to a credit for the GST paid.
THE DISCUSSION OF THE AUSTRALIAN TAX CONSEQUENCES OF AN INVESTMENT IN OUR SHARES IS BASED UPON LAWS AND RELEVANT
INTERPRETATIONS THEREOF IN EFFECT AS OF THE DATE OF THIS PROSPECTUS, ALL OF WHICH ARE SUBJECT TO CHANGE, POSSIBLY WITH RETROACTIVE EFFECT. EACH PROSPECTIVE INVESTOR IS URGED TO CONSULT ITS OWN TAX ADVISOR.
130
ENFORCEMENT OF CIVIL LIABILITIES
We are a public limited company incorporated under the laws of Australia. Certain of our directors are non-residents of the United States
and substantially all of their assets are located outside the United States. As a result, it may not be possible or practicable for you to:
| • |
effect service of process within the United States upon our non-U.S. resident directors or on us;
|
| • |
enforce in U.S. courts judgments obtained against our non-U.S. resident directors or us in the United States courts in any action, including actions under the civil liability provisions of
U.S. securities laws;
|
| • |
enforce in U.S. courts judgments obtained against our non-U.S. resident directors or us in courts of jurisdictions outside the United States in any action, including actions under the civil
liability provisions of U.S. securities laws; or
|
| • |
bring an original action in an Australian court to enforce liabilities against our non-U.S. resident directors or us based solely upon U.S. securities laws.
|
You may also have difficulties enforcing in courts outside the United States judgments that are obtained in U.S. courts against any of our
non-U.S. resident directors or us, including actions under the civil liability provisions of the U.S. securities laws.
With that noted, there are no treaties between Australia and the United States that would affect the recognition or enforcement of foreign
judgments in Australia. We also note that investors may be able to bring an original action in an Australian court against us to enforce liabilities based in part upon U.S. federal securities laws. The disclosure in this section is not based on the
opinion of counsel.
131
UNDERWRITING
A.G.P./Alliance Global Partners is acting as the sole book-running manager in this offering (the “underwriter”). We intend to enter into an
underwriting agreement with the underwriter. Subject to certain conditions, we have agreed to sell to the underwriter named below and the underwriter named below has agreed to purchase from us, at the respective public offering price less the
underwriting discounts and commissions set forth on the cover page of this prospectus, our Share Units and Pre-Funded Warrant Units listed next to its name in the following table:
|
Underwriter
|
|
Number of
Share Units |
|
|
Number of Pre-
Funded Warrant Units |
|
|
Total
|
|
|||
|
A.G.P./Alliance Global Partners
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
|
|
|
|
|
|
|
|
|
|
|
A copy of the underwriting agreement will be filed as an exhibit to the registration statement of which this prospectus is part.
The underwriter is committed to purchase all the securities we are offering other than those covered by the over-allotment option to
purchase additional securities described below, if they purchase any Share Units or Pre-Funded Warrant Units. The obligations of the underwriter may be terminated upon the occurrence of certain events to be specified in the underwriting agreement.
Furthermore, the underwriter’s obligations will be subject to customary conditions and representations and warranties contained in the underwriting agreement to be entered into by and between the Company and the underwriter, such as receipt by the
underwriter of officers’ certificates and legal opinions.
We have agreed to indemnify the underwriter against specified liabilities, including liabilities under the Securities Act, and to
contribute to payments the underwriter may be required to make in respect thereof.
The underwriter is offering the Share Units and/or Pre-Funded Warrant Units, subject to prior sale, when, as and if issued to and accepted
by them, subject to approval of legal matters by their counsel and other conditions specified in the underwriting agreement. The underwriter reserves the right to withdraw, cancel or modify offers to the public and to reject orders in whole or in
part.
Over-allotment Option to Purchase Additional Securities
Pursuant to the underwriting agreement, we have granted the underwriter an option, exercisable for up to 45 days from the date of this
prospectus, to purchase up to additional Shares and/or Share Purchase Warrants (15% of the Shares and Shares underlying the Pre-Funded Warrants and accompanying Share Purchase Warrants sold in this offering) at
the public offering price set forth on the cover page hereto, less the underwriting discounts and commissions. The underwriter may exercise the option solely to cover over-allotments, if any, made in connection with this offering. If any additional
Shares and accompanying Share Purchase Warrants are purchased pursuant to the over-allotment option, the underwriter will offer these Shares and accompanying Share Purchase Warrants on the same terms as those on which the other securities are being
offered. If this over-allotment option is exercised in full, the total gross proceeds will be approximately US$ million and the total net proceeds, after expenses, to us will be approximately US$ million.
132
Discounts, Commissions and Expense Reimbursement
The following table shows the public offering price, underwriting discount and proceeds, before expenses, to us. The information assumes
either no exercise or full exercise by the underwriter of its over-allotment option to purchase additional securities.
|
|
|
Per
Share Unit |
|
|
Per Pre-
Funded Warrant Unit |
|
|
Total
Without Over- Allotment |
|
|
Total With
Over- Allotment |
|
||||
|
Public offering price
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Underwriting discounts and commissions (7%)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds, before expenses, to us
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The underwriter proposes to offer the Share Units and/or Pre-Funded Warrant Units offered by us to the public at the public offering price
per respective Share Unit and/or Pre-Funded Warrant Unit set forth on the cover of this prospectus. In addition, the underwriter may offer some of the Share Units and/or Pre-Funded Warrant Units to other securities dealers at such price less a
concession of up to US$ per Share Unit and US$ per Pre-Funded Warrant Unit.
If all of the Share Units and/or Pre-Funded Warrant Units offered by us are not sold at the respective public offering prices per Share
Unit and Pre-Funded Warrant Unit, the underwriter may change the offering price per Share Unit and Pre-Funded Warrant Unit and other selling terms by means of a supplement to this prospectus.
We have also agreed to reimburse certain of the underwriter’s accountable expenses not to exceed US$150,000 in the aggregate, and
non-accountable expenses not to exceed 1% of the aggregate gross proceeds of this offering.
We estimate that the total expenses of the offering payable by us, excluding the total underwriting discounts, commissions and underwriter
expense reimbursement, will be approximately US$ million.
Lock-Up Agreements
For a period of ninety (90) days after the date of this prospectus, we have agreed with the underwriter not to (a) offer, pledge, sell,
contract to sell, sell any option or contract to purchase, purchase any option or contract to sell, grant any option, right or warrant to purchase, lend, or otherwise transfer or dispose of, directly or indirectly, any shares of our capital stock or
any securities convertible into or exercisable or exchangeable for shares of our capital stock; (b) file or cause to be filed with the SEC any registration statement relating to the offering of any shares of our capital stock or any securities
convertible into or exercisable or exchangeable for shares of our capital stock; or (c) enter into any swap or other arrangement that transfers to another, in whole or in part, any of the economic consequences of ownership of capital stock of the
Company. In addition, all of our directors, executive officers, and 5% stockholders will enter into lock-up agreements with the underwriter prior to the commencement of this offering pursuant to which each of these persons, for a period of ninety
(90) days from the date of this offering, without the prior written consent of the underwriter, agree not to offer, issue, sell, contract to sell, encumber, grant any option for sale or otherwise dispose of any of our securities.
133
Underwriter’s Warrants
Upon closing of this offering, we will issue to the underwriter a compensation warrant entitling the underwriter or its designees to
purchase our ordinary shares (equal to up to 5.0% of the aggregate number of the Shares and Shares issuable upon exercise of the Pre-Funded Warrants that we issue in this offering), subject to any reductions necessary to comply with the rules and
regulations of the Financial Industry Regulatory Authority, Inc. (“FINRA”). This warrant will be exercisable at any time and from time to time, in whole or in part, during the five-year period commencing from the commencement of sales of the offering
at a price of US$ per Share. The warrant will provide for registration rights for the ordinary shares underlying the warrant, pursuant to FINRA Rule 5110(f)(2)G), including a one-time demand registration right with
a duration of no more than five years from the commencement of sales of the offering and unlimited piggyback rights with a duration of no more than seven years from the commencement of sales of the Offering,
as well as customary anti-dilution provisions. Pursuant to FINRA Rule 5110(g), the underwriter warrants and any shares issued upon exercise of the underwriter warrants shall not be sold, transferred, assigned, pledged, or hypothecated, or be the
subject of any hedging, short sale, derivative, put or call transaction that would result in the effective economic disposition of the securities by any person for a period of 180 days immediately following the date of effectiveness or commencement
of sales of this offering, except the transfer of any security: (i) by operation of law or by reason of our reorganization; (ii) to any FINRA member firm participating in the offering and the officers or partners thereof, if all securities so
transferred remain subject to the lock-up restriction set forth above for the remainder of the time period; (iii) if the aggregate amount of our securities held by the underwriter or related persons do not exceed 1% of the securities being offered;
(iv) that is beneficially owned on a pro rata basis by all equity owners of an investment fund, provided that no participating member manages or otherwise directs investments by the fund and the participating members in the aggregate do not own more
than 10% of the equity in the fund; or (v) the exercise or conversion of any security, if all securities remain subject to the lock-up restriction set forth above for the remainder of the time period.
Electronic Offer, Sale and Distribution of Securities
A prospectus in electronic format may be made available on the websites maintained by the underwriter or selling group members, if any,
participating in this offering and the underwriter participating in this offering may distribute prospectuses electronically. The underwriter may agree to allocate a number of ordinary Share Units and Pre-Funded Warrant Units to underwriter and
selling group members for sale to their online brokerage account holders. Internet distributions will be allocated by the underwriter and selling group members that will make internet distributions on the same basis as other allocations. Other than
the prospectus in electronic format, the information on these websites is not part of, nor incorporated by reference into, this prospectus or the registration statement of which this prospectus forms a part, has not been approved or endorsed by us or
any underwriter in its capacity as underwriter, and should not be relied upon by investors.
The Listing
We have applied to list our ordinary shares on Nasdaq under the symbol “MDLB”. We do not intend to apply for listing of the Pre-Funded
Warrants or the Share Purchase Warrants on any securities exchange or other nationally recognized trading system.
134
Stabilization
In connection with this offering, the underwriter may engage in stabilizing transactions, over-allotment transactions, syndicate-covering
transactions, penalty bids and purchases to cover positions created by short sales. Stabilizing transactions permit bids to purchase shares so long as the stabilizing bids do not exceed a specified maximum and are engaged in for the purpose of
preventing or retarding a decline in the market price of the shares while the offering is in progress.
Over-allotment transactions involve sales by the underwriter of shares in excess of the number of shares the underwriter is obligated to
purchase. This creates a syndicate short position that may be either a covered short position or a naked short position. In a covered short position, the number of shares over-allotted by the underwriter is not greater than the number of shares in
the over-allotment option. In a naked short position, the number of shares involved is greater than the number of shares in the over-allotment option. The underwriter may close out any short position by exercising its option to purchase additional
Shares and accompanying Share Purchase Warrants or Pre-Funded Warrants and accompanying Share Purchase Warrants and/or purchasing Shares and accompanying Share Purchase Warrants or Pre-Funded Warrants and accompanying Share Purchase Warrants in the
open market.
Syndicate covering transactions involve purchases of shares in the open market after the distribution has been completed in order to cover
syndicate short positions. In determining the source of shares to close out the short position, the underwriter will consider, among other things, the price of shares available for purchase in the open market as compared with the price at which they
may purchase shares through exercise of the over-allotment option. If the underwriter sells more shares than could be covered by exercise of the over-allotment option and, therefore, have a naked short position, the position can be closed out only by
buying shares in the open market. A naked short position is more likely to be created if the underwriter is concerned that after pricing there could be downward pressure on the price of the shares in the open market that could adversely affect
investors who purchase in the offering.
Penalty bids permit the underwriter to reclaim a selling concession from a syndicate member when the shares originally sold by that
syndicate member are purchased in stabilizing or syndicate covering transactions to cover syndicate short positions.
These stabilizing transactions, syndicate covering transactions and penalty bids may have the effect of raising or maintaining the market
price of our ordinary shares or preventing or retarding a decline in the market price of our ordinary shares. As a result, the price of our ordinary shares in the open market may be higher than it would otherwise be in the absence of these
transactions. Neither we nor the underwriter make any representation or prediction as to the effect that the transactions described above may have on the price of our ordinary shares. These transactions may be effected on Nasdaq in the
over-the-counter market or otherwise and, if commenced, may be discontinued at any time.
Passive Market Making
In connection with this offering, the underwriter and selling group members may engage in passive market making transactions in our
ordinary shares on Nasdaq in accordance with Rule 103 of Regulation M under the Exchange Act, during a period before the commencement of offers or sales of the shares and extending through the completion of the distribution. A passive market maker
must display its bid at a price not in excess of the highest independent bid of that security. However, if all independent bids are lowered below the passive market maker’s bid, then that bid must then be lowered when specified purchase limits are
exceeded.
135
Certain Relationships
The underwriter and its affiliates have in the past and may in the future provide various investment banking, commercial banking, financial
advisory, brokerage, and other services to us and have and may receive customary fees and expense reimbursement.
The underwriter and its affiliates may, from time to time, engage in transactions with and perform services for us in the ordinary course
of their business for which they may receive customary fees and reimbursement of expenses. In the ordinary course of their various business activities, the underwriter and its affiliates may make or hold a broad array of investments and actively
trade debt and equity securities (or related derivative securities) and financial instruments (including bank loans) for their own accounts and for the accounts of their customers, and such investment and securities activities may involve securities
and/or instruments of our company. The underwriter and its affiliates may also make investment recommendations and/or publish or express independent research views in respect of such securities or instruments and may at any time hold, or recommend to
clients that they acquire, long and/or short positions in such securities and instruments.
Determination of Offering Price
Our ordinary shares are listed on the ASX under the symbol “MDC.” On , 2022, the last reported sale price of our ordinary shares
on the ASX was A$[•] per share, equivalent to a price of US$[•] per share, after giving effect to the Australian dollar/U.S. dollar exchange rate of as of , 2022.
The public offering price of each Share Unit and the exercise price of each Share Purchase Warrant offered by this prospectus will be
determined by negotiations between us and the underwriter. Among the factors considered in determining the public offering price of such securities were:
| • |
our history, capital structure and our business prospects;
|
| • |
the industry in which we operate;
|
| • |
our financial information and historical performance;
|
| • |
the experience and skills of our senior management;
|
| • |
the status and development prospects for our products;
|
| • |
the recent trading and closing bid prices of our ordinary shares listed on the ASX; and
|
| • |
the general condition of the securities markets at the time of this offering.
|
The offering price stated on the cover page of this prospectus should not be considered an indication of the actual value of the securities
sold in this offering. The values of such securities are subject to change as a result of market conditions and other factors.
136
Offer Restrictions Outside the United States
This prospectus does not constitute an offer to sell to, or a solicitation of an offer to buy from, anyone in any country or jurisdiction
(i) in which such an offer or solicitation is not authorized, (ii) in which any person making such offer or solicitation is not qualified to do so or (iii) in which any such offer or solicitation would otherwise be unlawful. No action has been taken
that would, or is intended to, permit a public offer of the securities or possession or distribution of this prospectus or any other offering or publicity material relating to the securities in any country or jurisdiction (other than the United
States) where any such action for that purpose is required. Accordingly, each underwriter has undertaken that it will not, directly or indirectly, offer or sell any securities offered hereby or have in its possession, distribute or publish any
prospectus, form of application, advertisement or other document or information in any country or jurisdiction except under circumstances that will, to the best of its knowledge and belief, result in compliance with any applicable laws and
regulations and all offers and sales of securities by it will be made on the same terms.
European Economic Area
In relation to each member state of the European Economic Area which has implemented the Prospectus Directive (each, a “Relevant Member
State”) an offer to the public of any securities may not be made in that Relevant Member State, except that an offer to the public in that Relevant Member State of any securities may be made at any time under the following exemptions under the
Prospectus Directive, if they have been implemented in that Relevant Member State:
| • |
to legal entities which are qualified investors as defined under the Prospectus Directive;
|
| • |
by the underwriter to fewer than 150, natural or legal persons (other than qualified investors as defined in the Prospectus Directive), as permitted under the Prospectus Directive, subject to
obtaining the prior consent of the representatives of the underwriter for any such offer; or
|
| • |
in any other circumstances falling within Article 3(2) of the Prospectus Directive, provided that no such offer of securities shall result in a requirement for us or any underwriter to publish
a prospectus pursuant to Article 3 of the Prospectus Directive.
|
For the purposes of this provision, (1) the expression an “offer of securities to the public” in relation to any securities in any Relevant
Member State means the communication in any form and by any means of sufficient information on the terms of the offer and any securities to be offered so as to enable an investor to decide to purchase or subscribe for the securities, as the same may
be varied in that Relevant Member State by any measure implementing the Prospectus Directive in that Relevant Member State, (2) the expression “Prospectus Directive” means Directive 2003/71/EC (and amendments thereto, including the 2010 PD Amending
Directive), and includes any relevant implementing measure in each Relevant Member State and (3) the expression “2010 PD Amending Directive” means Directive 2010/73/EU.
United Kingdom
This prospectus has only been communicated or caused to have been communicated and will only be communicated or caused to be communicated
as an invitation or inducement to engage in investment activity (within the meaning of Section 21 of the Financial Services and Markets Act of 2000 (the “FSMA”)) as received in connection with the issue or sale of the securities in circumstances in
which Section 21(1) of the FSMA does not apply to us. All applicable provisions of the FSMA will be complied with in respect to anything done in relation to the securities in, from or otherwise involving the United Kingdom.
137
Canada
The securities may be sold only to purchasers purchasing, or deemed to be purchasing, as principal that are accredited investors, as
defined in National Instrument 45-106 Prospectus Exemptions or subsection 73.3(1) of the Securities Act (Ontario), and are permitted clients, as defined in National Instrument 31-103 Registration Requirements, Exemptions and Ongoing Registrant
Obligations. Any resale of the securities must be made in accordance with an exemption from, or in a transaction not subject to, the prospectus requirements of applicable securities laws.
Securities legislation in certain provinces or territories of Canada may provide a purchaser with remedies for rescission or damages if
this prospectus (including any amendment thereto) contains a misrepresentation, provided that the remedies for rescission or damages are exercised by the purchaser within the time limit prescribed by the securities legislation of the purchaser’s
province or territory. The purchaser should refer to any applicable provisions of the securities legislation of the purchaser’s province or territory for particulars of these rights or consult with a legal advisor.
Pursuant to section 3A.3 of National Instrument 33-105 Underwriting Conflicts (NI 33-105), the underwriter is not required to comply with
the disclosure requirements of NI 33-105 regarding underwriter conflicts of interest in connection with this offering.
Australia
No placement document, prospectus, product disclosure statement or other disclosure document has been lodged with the Australian Securities
and Investments Commission in relation to the offering. This prospectus does not constitute a prospectus, product disclosure statement or other disclosure document under the Corporations Act, and does not purport to include the information required
for a prospectus, product disclosure statement or other disclosure document under the Corporations Act.
Any offer in Australia of the securities may only be made to persons, or the Exempt Investors, who are “sophisticated investors” (within
the meaning of section 708(8) of the Corporations Act), “professional investors” (within the meaning of section 708(11) of the Corporations Act) or otherwise pursuant to one or more exemptions contained in section 708 of the Corporations Act so that
it is lawful to offer the securities without disclosure to investors under Chapter 6D of the Corporations Act.
The securities applied for by Exempt Investors in Australia must not be offered for sale in Australia in the period of 12 months after the
date of allotment under the offering, except in circumstances where disclosure to investors under Chapter 6D of the Corporations Act would not be required pursuant to an exemption under section 708 of the Corporations Act or otherwise or where the
offer is pursuant to a disclosure document which complies with Chapter 6D of the Corporations Act. Any person acquiring securities must observe such Australian on-sale restrictions.
This prospectus contains general information only and does not take account of the investment objectives, financial situation or needs of
any particular person. It does not contain any securities recommendations or financial product advice. Before making an investment decision, investors need to consider whether the information in this prospectus is appropriate to their needs,
objectives and circumstances and, if necessary, seek expert advice on those matters.
138
Switzerland
The securities may not be publicly offered in Switzerland and will not be listed on the SIX Swiss Exchange (SIX”) or on any other stock
exchange or regulated trading facility in Switzerland. This document has been prepared without regard to the disclosure standards for issuance prospectuses under art. 652a or art. 1156 of the Swiss Code of Obligations or the disclosure standards for
listing prospectuses under art. 27 ff. of the SIX Listing Rules or the listing rules of any other stock exchange or regulated trading facility in Switzerland. Neither this document nor any other offering or marketing material relating to the
securities or the offering may be publicly distributed or otherwise made publicly available in Switzerland.
Neither this document nor any other offering or marketing material relating to the offering, or the securities have been or will be filed
with or approved by any Swiss regulatory authority. This document will not be filed with, and the offer of securities will not be supervised by, the Swiss Financial Market Supervisory Authority , and the offer of securities has not been and will not
be authorized under the Swiss Federal Act on Collective Investment Schemes (“CISA”). The investor protection afforded to acquirers of interests in collective investment schemes under the CISA does not extend to acquirers of securities.
Dubai International Financial Centre
This prospectus relates to an exempt offer (“Exempt Offer”) in accordance with the Offered Securities Rules of the Dubai Financial Services
Authority (“DFSA”). This prospectus is intended for distribution only to persons of a type specified in the Offered Securities Rules of the DFSA. It must not be delivered to, or relied on by, any other person. The DFSA has no responsibility for
reviewing or verifying any documents relating to Exempt Offers. The DFSA has not approved this prospectus nor taken steps to verify the information set forth herein and has no responsibility for the prospectus. The securities to which this prospectus
relates may be illiquid and/or subject to restrictions on their resale. Prospective purchasers of the securities offered should conduct their own due diligence on the securities. If you do not understand the contents of this prospectus, you should
consult an authorized financial advisor.
Hong Kong
The securities have not been offered or sold and will not be offered or sold in Hong Kong, by means of any document, other than (a) to
“professional investors” as defined in the Securities and Futures Ordinance (Cap. 571) of Hong Kong and any rules made under that Ordinance or (b) in other circumstances which do not result in the document being a “prospectus” as defined in the
Companies Ordinance (Cap. 32) of Hong Kong or which do not constitute an offer to the public within the meaning of that Ordinance. No advertisement, invitation or document relating to the securities has been or may be issued or has been or may be in
the possession of any person for the purposes of issue, whether in Hong Kong or elsewhere, which is directed at, or the contents of which are likely to be accessed or read by, the public of Hong Kong (except if permitted to do so under the securities
laws of Hong Kong) other than with respect to securities which are or are intended to be disposed of only to persons outside Hong Kong or only to “professional investors” as defined in the Securities and Futures Ordinance and any rules made under
that Ordinance.
Japan
The securities have not been and will not be registered under the Financial Instruments and Exchange Law of Japan (Law No. 25 of 1948, as
amended) and, accordingly, will not be offered or sold, directly or indirectly, in Japan, or for the benefit of any Japanese Person or to others for re-offering or resale, directly or indirectly, in Japan or to any Japanese Person, except in
compliance with all applicable laws, regulations and ministerial guidelines promulgated by relevant Japanese governmental or regulatory authorities in effect at the relevant time. For the purposes of this paragraph, “Japanese Person” shall mean any
person resident in Japan, including any corporation or other entity organized under the laws of Japan.
139
Israel
THIS DOCUMENT DOES NOT CONSTITUTE A PROSPECTUS UNDER THE ISRAELI SECURITIES LAW, 5728-1968, AND HAS NOT BEEN FILED WITH OR APPROVED BY THE
ISRAEL SECURITIES AUTHORITY. IN ISRAEL, THIS PROSPECTUS MAY BE DISTRIBUTED ONLY TO, AND IS DIRECTED ONLY AT, INVESTORS LISTED IN THE FIRST ADDENDUM, OR THE ADDENDUM, TO THE ISRAELI SECURITIES LAW, CONSISTING PRIMARILY OF JOINT INVESTMENT IN TRUST
FUNDS; PROVIDENT FUNDS; INSURANCE COMPANIES; BANKS; PORTFOLIO MANAGERS, INVESTMENT ADVISORS, MEMBERS OF THE TEL AVIV STOCK EXCHANGE LTD., UNDERWRITERS, EACH PURCHASING FOR THEIR OWN ACCOUNT; VENTURE CAPITAL FUNDS; ENTITIES WITH EQUITY IN EXCESS OF
NIS 50 MILLION AND “QUALIFIED INDIVIDUALS,” EACH AS DEFINED IN THE ADDENDUM (AS IT MAY BE AMENDED FROM TIME TO TIME), COLLECTIVELY REFERRED TO AS QUALIFIED INVESTORS. QUALIFIED INVESTORS SHALL BE REQUIRED TO SUBMIT WRITTEN CONFIRMATION THAT THEY FALL
WITHIN THE SCOPE OF THE ADDENDUM.
140
EXPENSES RELATING TO THE OFFERING
The following table sets forth the costs and expenses, other than underwriting discounts and commissions, payable in connection with the
sale of Shares in the offering. All amounts are estimated except the SEC registration fee, the FINRA filing fee, and the Nasdaq initial listing fee. Except as otherwise noted, all the expenses below will be paid by us.
|
Expense
|
|
Amount
|
|
|
|
SEC registration fee
|
|
|
US$
|
|
|
FINRA filing fee
|
|
|
*
|
|
|
Nasdaq initial listing fee
|
|
|
5,000
|
|
|
Legal fees and expenses
|
|
|
*
|
|
|
Accounting fees and expenses
|
|
|
*
|
|
|
Printing expenses
|
|
|
*
|
|
|
Miscellaneous fees and expenses
|
|
|
*
|
|
|
Total
|
|
|
US$*
|
|
___________________
* To be provided by amendment.
141
LEGAL MATTERS
The validity of the Shares and certain other matters of Australian law will be passed upon for us by McCabes Lawyers Pty Ltd. Certain
matters as to U.S. federal law and New York state law will be passed upon for us by Seward & Kissel LLP. Legal counsels to the underwriter in connection with this offering are Sullivan & Worcester LLP with respect to U.S. federal law and
Baker & McKenzie with respect to Australian law.
EXPERTS
The consolidated financial statements of Medlab Clinical Ltd. as of June 30, 2022, and 2021, and for the years appearing in the prospectus
have been audited by Accell Audit & Compliance, P.A. (“Accell”), independent registered public accounting firm, as set forth in their report thereon, relating to the consolidated financial statements of the Company, appearing elsewhere in the
prospectus, and are included in reliance on the report of such firm given upon their authority as experts in accounting and auditing.
The offices of Accell are located at 4806 W Gandy Blvd, Tampa, FL 33611.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC a Registration Statement on Form F-1 under the Securities Act with respect to the Shares offered in this
prospectus. This prospectus, which forms a part of the Registration Statement, does not contain all of the information included in the Registration Statement. Certain information is omitted and you should refer to the Registration Statement and its
exhibits for that information. With respect to references made in this prospectus to any contract or other document of Medlab, such references are not necessarily complete and you should refer to the exhibits attached to the Registration Statement
for copies of the actual contract or document.
Upon the closing of this offering, we will become subject to periodic reporting and other informational requirements of the Exchange Act as
applicable to foreign private issuers. Accordingly, we will be required to file reports, including annual reports on Form 20-F, periodic reports and other information, with the SEC.
We are allowed four months after the end of our fiscal year to file our annual report with the SEC, and we are not required to disclose
certain detailed information regarding executive compensation that is required from U.S. domestic issuers. Also, as a foreign private issuer, we are exempt from the rules of the Exchange Act prescribing the furnishing of proxy statements to
shareholders, and the members of our board of directors, our senior management and our principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act.
The SEC maintains a website at www.sec.gov that contains reports, proxy and information
statements and other information regarding registrants that file electronically with the SEC. You also can inspect our registration statement, as well as any other information we file with or furnish to the SEC, on this website. This reference to the
SEC’s website is an inactive textual reference only and is not a hyperlink.
We expect to make our annual reports and other information filed with or furnished to the SEC available, free of charge, through our
website at www.medlab.co as soon as reasonably practicable after those reports and other information are filed with or furnished to the SEC. The information contained on, or that can be accessed through, our
website is not part of, and is not incorporated into, this prospectus.
142
MEDLAB CLINICAL LIMITED
FOR THE YEARS ENDED JUNE 30, 2022 AND 2021
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED JUNE 30, 2022 AND 2021
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
|
Consolidated Financial Statements
|
|
|
Report of Independent Registered Public Accounting Firm
|
F-2
|
|
Consolidated Balance Sheets at June 30, 2022 and 2021
|
F-3
|
|
Consolidated Statements of Operations and Comprehensive Loss for the years ended June 30, 2022 and 2021
|
F-4
|
|
Consolidated Statements of Stockholders’ Equity for the years ended June 30, 2022 and 2021
|
F-5
|
|
Consolidated Statements of Cash Flows for the years ended June 30, 2022 and 2021
|
F-6
|
|
Notes to Consolidated Financial Statements
|
F-7
|
F-1

REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and
Stockholders of Medlab Clinical Limited
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Medlab Clinical Limited (the Company) as of June 30, 2022 and 2021, and the related
consolidated statements of operations and comprehensive loss, stockholders’ equity and cash flows for the years then ended, and the related notes (collectively referred to as the financial statements). In our opinion, the consolidated financial
statements present fairly, in all material respects, the financial position of the Company as of June 30, 2022 and 2021, and the results of its operations and its cash flows for the two years then ended, in conformity with accounting principles
generally accepted in the United States of America.
Substantial Doubt about the Company’s Ability to Continue as a Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 3, the
Company has incurred net losses and does not have enough working capital to meet its needs for the next twelve months. These factors, and the need for additional financing in order for the Company to meet its business plans, raise substantial doubt
about the Company’s ability to continue as a going concern. Our opinion is not modified with respect to that matter.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial
statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal
securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable
assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part
of our audits, we are required to obtain an understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we
express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and
performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and
significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinions.

We have served as the Company’s auditor since 2021. Tampa, Florida
September 19, 2022
3001 N. Rocky Point Dr. East, Suite 200 ☐ Tampa, Florida 33607 ☐ +1.813.367.3527
F-2
|
MEDLAB CLINICAL LIMITED
|
||||||||
|
CONSOLIDATED BALANCE SHEETS
|
||||||||
|
|
As of June 30, 2022
|
As of June 30, 2021
|
||||||
|
Assets
|
||||||||
|
Current assets
|
||||||||
|
Cash and cash equivalents
|
$
|
5,191,031
|
$
|
13,434,762
|
||||
|
Accounts receivables, net
|
78,982
|
55,893
|
||||||
|
Research and development tax credit incentive receivable
|
3,545,243
|
2,306,662
|
||||||
|
Inventories, net
|
80,107
|
8,375
|
||||||
|
Deposits and prepaids
|
346,636
|
243,720
|
||||||
|
Assets of discontinued operations
|
-
|
2,030,064
|
||||||
|
Total current assets
|
9,241,999
|
18,079,476
|
||||||
|
|
||||||||
|
Property and equipment, net
|
344,306
|
483,316
|
||||||
|
Right of use assets, net
|
1,030,085
|
1,600,978
|
||||||
|
Other assets
|
709,208
|
482,536
|
||||||
|
Total Assets
|
$
|
11,325,598
|
$
|
20,646,306
|
||||
|
|
||||||||
|
Liabilities and Stockholders' Equity
|
||||||||
|
Current liabilities
|
||||||||
|
Accounts payables
|
$
|
530,959
|
$
|
659,031
|
||||
|
Accrued liabilities
|
931,560
|
674,122
|
||||||
|
Accrued employee benefits, current
|
463,119
|
412,668
|
||||||
|
Current maturities of long-term debt
|
-
|
67,834
|
||||||
|
Lease liabilities, current
|
547,467
|
638,066
|
||||||
|
Liabilities of discontinued operations
|
-
|
1,761,413
|
||||||
|
Total current liabilities
|
2,473,105
|
4,213,134
|
||||||
|
|
||||||||
|
Contingent lease termination liability
|
305,422
|
305,422
|
||||||
|
Accrued employee benefits, non-current
|
264,177
|
189,732
|
||||||
|
Lease liabilities, non-current
|
533,757
|
989,176
|
||||||
|
Liabilities of discontinued operations, non-current
|
-
|
42,989
|
||||||
|
Total Liabilities
|
3,576,461
|
5,740,453
|
||||||
|
|
||||||||
|
Commitments and contingencies (Note 12)
|
||||||||
|
|
||||||||
|
Stockholders' Equity
|
||||||||
|
Common stock, no par value; unlimited shares authorized; 2,283,502 shares issued and outstanding
|
67,508,316
|
67,430,949
|
||||||
|
Other comprehensive income
|
101,840
|
80,120
|
||||||
|
Accumulated deficit
|
(59,529,093
|
)
|
(52,366,660
|
)
|
||||
|
Controlling interest
|
8,081,063
|
15,144,409
|
||||||
|
Non controlling interest
|
(331,926
|
)
|
(238,556
|
)
|
||||
|
Total Stockholders' Equity
|
7,749,137
|
14,905,853
|
||||||
|
Total Liabilities and Stockholders' Equity
|
$
|
11,325,598
|
$
|
20,646,306
|
||||
|
|
||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-3
|
MEDLAB CLINICAL LIMITED
|
|
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
|
|
|
For the years ended
|
|||||||
|
|
June 30,
|
|||||||
|
|
2022
|
2021
|
||||||
|
Sale of goods (net discounts)
|
$
|
1,536,366
|
$
|
732,727
|
||||
|
Cost of sales
|
336,292
|
16,425
|
||||||
|
Gross profit
|
1,200,074
|
716,302
|
||||||
|
|
||||||||
|
Operating expenses:
|
||||||||
|
Personnel expenses
|
7,877,497
|
6,016,686
|
||||||
|
General and administrative expenses
|
3,952,633
|
3,217,213
|
||||||
|
Research and development expenses
|
1,503,443
|
2,102,751
|
||||||
|
Professional fees
|
-
|
1,731,430
|
||||||
|
Depreciation and amortization expenses
|
850,745
|
873,497
|
||||||
|
Total operating expenses
|
14,184,318
|
13,941,577
|
||||||
|
|
||||||||
|
Loss from operations
|
(12,984,244
|
)
|
(13,225,275
|
)
|
||||
|
|
||||||||
|
Other income (expense):
|
||||||||
|
Research and development incentive
|
4,621,501
|
2,265,000
|
||||||
|
Grant income
|
61,954
|
928,585
|
||||||
|
Legal settlement
|
-
|
500,000
|
||||||
|
Other income
|
20,655
|
6,454
|
||||||
|
Interest income
|
44,727
|
25,198
|
||||||
|
Loss on foreign currency exchange
|
(51,129
|
)
|
6,120
|
|||||
|
Interest expense
|
(101,916
|
)
|
(139,065
|
)
|
||||
|
Total other income (expense)
|
4,595,792
|
3,592,292
|
||||||
|
|
||||||||
|
Net loss from continuing operations
|
(8,388,452
|
)
|
(9,632,983
|
)
|
||||
|
|
||||||||
|
Discontinued operations:
|
||||||||
|
Loss from operations of discontinued operations
|
(36,178
|
)
|
(2,769,846
|
)
|
||||
|
Gain from disposition of discontinued operations
|
1,195,816
|
-
|
||||||
|
|
1,159,638
|
(2,769,846
|
)
|
|||||
|
|
||||||||
|
Net loss
|
(7,228,814
|
)
|
(12,402,829
|
)
|
||||
|
Foreign currency translation adjustments
|
(5,269
|
)
|
(15,649
|
)
|
||||
|
Comprehensive loss
|
$
|
(7,234,083
|
)
|
$
|
(12,418,478
|
)
|
||
|
|
||||||||
|
Net loss attributable:
|
||||||||
|
Non-controlling interest
|
(66,381
|
)
|
(79,124
|
)
|
||||
|
Common stockholders
|
(7,162,433
|
)
|
(12,323,705
|
)
|
||||
|
|
(7,228,814
|
)
|
(12,402,829
|
)
|
||||
|
|
||||||||
|
Comprehensive loss attributable:
|
||||||||
|
Non-controlling interest
|
(93,370
|
)
|
(96,698
|
)
|
||||
|
Common stockholders
|
(7,140,713
|
)
|
(12,321,780
|
)
|
||||
|
|
(7,234,083
|
)
|
(12,418,478
|
)
|
||||
|
|
||||||||
|
Basic and diluted earnings per share on net loss from continuing operations
|
$
|
(3.67
|
)
|
$
|
(4.90
|
)
|
||
|
Basic and diluted earnings per share on net loss
|
$
|
(3.17
|
)
|
$
|
(6.31
|
)
|
||
|
Weighted average shares outstanding - basic and diluted
|
2,283,502
|
1,964,038
|
||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-4
|
MEDLAB CLINICAL LIMITED
|
||||||||||||||||||||||||
|
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY
|
||||||||||||||||||||||||
|
FOR THE YEARS ENDED JUNE 30, 2022 AND 2021
|
||||||||||||||||||||||||
|
|
||||||||||||||||||||||||
|
|
Common Stock
|
Other Comprehensive
|
Accumulated
|
Non-Controlling
|
||||||||||||||||||||
|
|
Income
|
Deficit
|
Interest
|
Total
|
||||||||||||||||||||
|
|
Shares
|
Amount
|
||||||||||||||||||||||
|
Balance June 30, 2020
|
1,797,036
|
$
|
51,361,909
|
$
|
78,195
|
$
|
(40,042,955
|
)
|
$
|
(141,858
|
)
|
$
|
11,255,291
|
|||||||||||
|
|
||||||||||||||||||||||||
|
Sale of common stock, net
|
486,466
|
15,449,204
|
-
|
-
|
-
|
15,449,204
|
||||||||||||||||||
|
|
||||||||||||||||||||||||
|
Stock based compensation (stock options)
|
-
|
619,836
|
-
|
-
|
-
|
619,836
|
||||||||||||||||||
|
|
||||||||||||||||||||||||
|
Foreign currency translation adjustment
|
-
|
-
|
1,925
|
-
|
(17,574
|
)
|
(15,649
|
)
|
||||||||||||||||
|
|
||||||||||||||||||||||||
|
Net loss
|
-
|
-
|
-
|
(12,323,705
|
)
|
(79,124
|
)
|
(12,402,829
|
)
|
|||||||||||||||
|
|
||||||||||||||||||||||||
|
Balance June 30, 2021
|
2,283,502
|
67,430,949
|
80,120
|
(52,366,660
|
)
|
(238,556
|
)
|
14,905,853
|
||||||||||||||||
|
|
||||||||||||||||||||||||
|
Stock based compensation (stock options)
|
-
|
77,367
|
-
|
-
|
-
|
77,367
|
||||||||||||||||||
|
|
||||||||||||||||||||||||
|
Foreign currency translation adjustment
|
-
|
-
|
21,720
|
-
|
(26,989
|
)
|
(5,269
|
)
|
||||||||||||||||
|
|
||||||||||||||||||||||||
|
Net loss
|
-
|
-
|
-
|
(7,162,433
|
)
|
(66,381
|
)
|
(7,228,814
|
)
|
|||||||||||||||
|
|
||||||||||||||||||||||||
|
Balance June 30, 2022
|
2,283,502
|
$
|
67,508,316
|
$
|
101,840
|
$
|
(59,529,093
|
)
|
$
|
(331,926
|
)
|
$
|
7,749,137
|
|||||||||||
|
|
||||||||||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements
F-5
|
MEDLAB CLINICAL LIMITED
|
||||||||
|
CONSOLIDATED STATEMENTS OF CASH FLOWS
|
||||||||
|
|
||||||||
|
|
For the years ended
|
|||||||
|
|
June 30,
|
|||||||
|
|
2022
|
2021
|
||||||
|
Cash Flows from Operating Activities
|
||||||||
|
Net loss
|
$
|
(7,228,814
|
)
|
$
|
(12,402,829
|
)
|
||
|
|
||||||||
|
Adjustments to reconcile net loss to net
|
||||||||
|
cash from operating activities:
|
||||||||
|
Depreciation and amortization
|
850,745
|
873,497
|
||||||
|
Stock based compensation
|
77,367
|
619,836
|
||||||
|
Gain from disposition of discontinued operations
|
(1,195,816
|
)
|
-
|
|||||
|
Accretion of earn-out
|
(24,819
|
)
|
-
|
|||||
|
Changes in operating assets & liabilities
|
||||||||
|
Accounts receivable
|
(23,089
|
)
|
(260,968
|
)
|
||||
|
Research and development tax incentive credit receivable
|
(1,238,581
|
)
|
284,066
|
|||||
|
Inventories, net
|
(71,732
|
)
|
677,599
|
|||||
|
Deposits and prepaids
|
(102,916
|
)
|
13,052
|
|||||
|
Other assets
|
(706,997
|
)
|
404
|
|||||
|
Accounts payable
|
(128,072
|
)
|
(346,496
|
)
|
||||
|
Accrued liabilities
|
256,873
|
119,776
|
||||||
|
Accrued employee benefits
|
124,896
|
72,976
|
||||||
|
Net cash from operating activities
|
(9,410,955
|
)
|
(10,349,087
|
)
|
||||
|
|
||||||||
|
Cash Flows from Investing Activities
|
||||||||
|
Purchase of property and equipment
|
(31,135
|
)
|
(83,304
|
)
|
||||
|
Proceeds from disposal of discontinued operations
|
1,775,910
|
-
|
||||||
|
Net cash from investing activities
|
1,744,775
|
(83,304
|
)
|
|||||
|
|
||||||||
|
Cash Flows from Financing Activities
|
||||||||
|
Sale of common stock
|
15,449,204
|
|||||||
|
Payments on long-term debt
|
(67,834
|
)
|
(26,387
|
)
|
||||
|
Repayment of lease liabilities
|
(504,448
|
)
|
(612,357
|
)
|
||||
|
Net cash from financing activities
|
(572,282
|
)
|
14,810,460
|
|||||
|
|
||||||||
|
Foreign currency adjustment
|
(5,269
|
)
|
(6,351
|
)
|
||||
|
|
||||||||
|
Increase in Cash and cash equivalents
|
(8,238,462
|
)
|
4,378,069
|
|||||
|
|
||||||||
|
Cash and cash equivalents at beginning of period
|
13,434,762
|
9,063,044
|
||||||
|
|
||||||||
|
Cash and cash equivalents at end of period
|
$
|
5,191,031
|
$
|
13,434,762
|
||||
|
|
||||||||
|
Supplemental Cash Flow Information
|
||||||||
|
Cash paid for interest
|
$
|
-
|
$
|
139,065
|
||||
|
Cash paid for income taxes
|
$
|
-
|
$
|
-
|
||||
|
|
||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-6
MEDLAB CLINICAL LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Nature of operations
Medlab Clinical Limited (“MDC” or the “Company”) is an Australian biotechnology medical research and development
company positioned within the nutraceutical products, pharmaceutical research, and drug delivery industries.
Through its pharmaceutical division, MDC engages in the development of nutraceutical products, drug delivery systems and bio-therapeutics
for chronic health issues including kidney disease, pre-diabetes/obesity, depression, musculoskeletal muscle loss associated with aging, and non-opioid pain management.
MDC’s nutraceuticals product division comprises of 21 formulations and provides nutraceuticals for anti-inflammatory,
cardiovascular support, endocrine support, gastrointestinal support, immunological support, sports performance, urogenital support, and weight management, as well as for men, pregnancy, seniors, teenagers, and women support.
MDC principal activities include:
| • |
The development of NanaBis™ via clinical trials;
|
| • |
The use of NanaBis™ and other cannabis-related products through the Australian Special Access Scheme;
|
| • |
The continued development of its proprietary delivery platform, NanoCelle®; and
|
| • |
The development and commercialisation of its nutraceutical range of products.
|
Since its inception, a priority focus of MDC has been the development and distribution of its patented drug delivery product, NanoCelle®, and the continued research, development, and commercialisation of cannabis-based products, with its principal product being NanaBis™.
The Company has the following seven subsidiaries:
| • |
Medlab Pty Ltd
|
| • |
Medlab Clinical US Inc
|
| • |
Medlab IP Pty Ltd
|
| • |
Medlab Research Pty Ltd
|
| • |
Medlab Nutraceuticals Inc
|
| • |
Medlab Research Ltd
|
| • |
MDC Europe Ltd
|
F-7
2. Summary of significant accounting policies
Basis of Presentation and Principles of Consolidation
The consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States of America (“U.S.
GAAP”). The consolidated financial statements, which include the accounts of the Company, and its wholly-owned subsidiaries, Medlab Pty Ltd, Medlab Clinical US, Inc., Medlab IP Pty Ltd, Medlab Research Pty Ltd, Medlab Nutraceuticals Inc., Medlab
Research Ltd, and MDC Europe Ltd, are prepared in conformity with U.S. GAAP. All significant intercompany balances and transactions have been eliminated. The consolidated financial statements and related disclosures have been prepared pursuant to the
rules and regulations of the Securities and Exchange Commission (“SEC”). The consolidated financial statements have been prepared using the accrual basis of accounting in accordance with accounting principles generally accepted in the U.S. GAAP and
presented in Australian dollars. The fiscal year end is June 30.
Non-controlling interest represents the 13% third-party interest in Medlab Nutraceuticals Inc.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentrations of credit risk are cash, accounts receivable and other receivables arising from
its normal business activities. The Company places its cash in what it believes to be credit-worthy financial institutions. The Company controls credit risk related to accounts receivable through credit approvals, credit limits and monitoring
procedures. The Company routinely assesses the financial strength of its customers and, based upon factors surrounding the credit risk, establishes an allowance, if required, for uncollectible accounts and, as a consequence, believes that its
accounts receivable credit risk exposure beyond such allowance is limited.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of
assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. The most significant assumptions and estimates relate to
the valuation of equity issued for services, valuation of equity associated with convertible debt, the valuation of derivative liabilities, and the valuation of deferred tax assets. Actual results could differ
from these estimates.
Revenue Recognition
The Company recognizes revenue in accordance with Accounting Standards Update 2014-09, “Revenue from contracts with customers,” Accounting Standard Codification (“ASC”) Topic 606, Revenue Recognition. Revenue is recognized when a customer obtains control of promised goods or services. In addition, the standard requires disclosure of the nature, amount,
timing, and uncertainty of revenue and cash flows arising from contracts with customers. The amount of revenue that is recorded reflects the consideration that the Company expects to receive in exchange for those goods. The Company applies the
following five-step model in order to determine this amount: (i) identification of the promised goods in the contract; (ii) determination of whether the promised goods are performance obligations, including whether they are distinct in the context of
the contract; (iii) measurement of the transaction price, including the constraint on variable consideration; (iv) allocation of the transaction price to the performance obligations; and (v) recognition of revenue when (or as) the Company satisfies
each performance obligation. The Company’s main revenue stream is from product sales. Revenue is recognized when the performance obligation is satisfied. The performance obligation is satisfied at the point of sale, which is the time when the
customer’s orders are shipped.
Sale of nutraceuticals
The Company’s nutraceutical line of business consists primarily of powders, capsules and NanoCelle® sprays. The sprays are administered to end users through
the Company’s NanoCelle® technology, which is a pharmaceutical-delivery platform which is designed to administer pharmaceutical products using nanoparticles.
These products are sold through the Company’s wholesale distribution network to pharmacy and healthcare specialists. The Company has wholesale agreements with
its customers. The selling price is determined based on competitive wholesale pricing. The products are warehoused by the Company and shipped directly to the Customer. The Company recognizes revenue at the point of sale, which is at the time when the
customer’s orders are shipped. Amounts disclosed as revenue are net of sales returns and trade discounts.
The Company licensed its nutraceutical line of business on October 19, 2021. See Note 7.
F-8
Sale of pharmaceuticals
The Company’s pharmaceutical line of business consists of NanaBis™ and NanoCBD™. These products are also administered to end users through the Company’s
NanoCelle® technology.
These products are sold to doctors and pharmacies on a case-by-case basis through the Company’s compassionate use program. The prices are set based on a
case-by-case basis based on compassionate use. The average selling price is A$100 per unit. The Company recognizes revenue at the point of sale, which is at the time when the customer’s orders are shipped. Amounts disclosed as revenue are net of
sales returns and trade discounts.
Discounts, promotional and other rebates
The sale of goods revenue is net of any discounts, rebates and any contributions to customers towards promotional activities.
For the years ended June 30, 2022 and 2021, the Company had A$350,692 and A$849,568 in discounts, rebates and promotion allowances that were netted against
the sale of products.
Fair Value Measurements
ASC Topic 820, Fair Value Measurements, clarifies the definition of fair value, prescribes methods for measuring
fair value, and establishes a fair value hierarchy to classify the inputs used in measuring fair value as follows:
Level 1: Inputs are unadjusted quoted prices in active markets for identical assets or liabilities available at the measurement date.
Level 2: Inputs are unadjusted quoted prices for similar assets and liabilities in active markets, quoted prices for identical or similar
assets and liabilities in markets that are not active, inputs other than quoted prices that are observable, and inputs derived from or corroborated by observable market data.
Level 3: Inputs are unobservable inputs which reflect the reporting entity's own assumptions on what assumptions the market participants
would use in pricing the asset or liability based on the best available information.
The estimated fair value of certain financial instruments, including all current liabilities are carried at historical cost basis, which approximates their
fair values because of the short-term nature of these instruments.
Fair Value of Financial Instruments
ASC Subtopic 825-10, Financial Instruments requires disclosure of the fair value of certain financial instruments.
The carrying value of cash and cash equivalents, accounts payable and accrued liabilities as reflected in the consolidated balance sheets, approximate fair value because of the short-term maturity of these instruments. All other significant financial
assets, financial liabilities and equity instruments of the Company are either recognized or disclosed in the consolidated financial statements together with other information relevant for making a reasonable assessment of future cash flows, interest
rate risk and credit risk. Where practicable the fair values of financial assets and financial liabilities have been determined and disclosed; otherwise only available information pertinent to fair value has been disclosed.
Cash and Cash Equivalents
The Company considers all highly liquid investments with an original maturity of three months or less to be cash equivalents.
Stock Based Compensation
The Company records stock-based compensation in accordance with the provisions of ASC Topic 718, “Accounting for Stock
Compensation,” which establishes accounting standards for transactions in which an entity exchanges its equity instruments for goods or services. In accordance with guidance provided under ASC Topic 718, the Company recognizes an expense for
the fair value of its stock awards at the time of grant and the fair value of its outstanding stock options as they vest, whether held by employees or others. During the years ended June 30, 2022 and 2021, the Company recorded A$77,367 and A$619,836,
respectively, in stock-based compensation expense.
F-9
Advertising and Promotion
The Company follows the policy of charging the costs of advertising, marketing, and public relations to expense as incurred. The Company has A$186,778 and
A$770,694 in advertising expenses, which is included under general and administrative expenses in the statement of operations, for the years ended June 30, 2022 and 2021, respectively.
Income Taxes
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences
attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss, capital loss and tax credit carryforwards. Deferred tax assets and liabilities are
measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized
in income in the period that includes the enactment date.
The Company recognizes the effect of income tax positions only if those positions are more likely than not of being sustained. Recognized income tax positions
are measured at the largest amount that is greater than 50% likely of being realized. Changes in recognition or measurement are reflected in the period in which the change in judgment occurs. The Company records
interest and penalties related to unrecognized tax benefits as a component of general and administrative expenses. The Company’s federal tax return and any state tax returns are not currently under examination.
The Company has adopted ASC Subtopic 740-10, Accounting for Income Taxes, which requires an asset and liability approach to financial accounting and reporting
for income taxes. Deferred income tax assets and liabilities are computed annually from differences between the financial statement and tax basis of assets and liabilities that will result in taxable or deductible amounts in the future based on
enacted tax laws and rates applicable to the periods in which the differences are expected to affect taxable income. Valuation allowances are established when necessary to reduce deferred tax assets to the amount expected to be realized.
Inventory
Raw materials, work in progress and finished goods are stated at the lower of cost and net realizable value on a 'first in
first out' basis. Cost comprises of direct materials and delivery costs, direct labor, import duties and other taxes, and an appropriate proportion of variable and fixed overhead expenditure based on normal operating capacity. Costs of purchased
inventory are determined after deducting rebates and discounts received or receivable.
The provision for impairment of inventories requires a degree of estimation and judgment. The level of the provision is assessed by taking into account the
recent sales experience, the ageing of inventories and other factors that affect inventory obsolescence. During the years ended June 30, 2022 and 2021, the Company had A$0 and A$80,001 recorded in inventory reserves.
Inventory in transit is stated at the lower of cost and net realizable value. Cost comprises purchase and delivery costs, net of rebates and discounts
received or receivable.
Net realizable value is the estimated selling price in the ordinary course of business less the estimated costs of completion and the estimated costs
necessary to make the sale.
Accounts Receivable
The Company has applied the simplified approach to measuring expected credit losses, which uses a lifetime expected loss allowance. To measure the expected
credit losses, trade receivables have been grouped based on days overdue. Account balances deemed to be uncollectible are charged to bad debt expense and included in the allowance after all means of collection have been exhausted and the potential
for recovery is considered remote. As of June 30, 2022 and 2021, the allowance for doubtful accounts amounted to A$10,000 and A$675,000, respectively.
Property and Equipment
Property and equipment are recorded at historical cost. Depreciation is computed on the straight-line method over the estimated useful lives of the respective
assets. The Company capitalizes asset amounts A$1,000 or greater.
F-10
Net Loss Per Common Share
The Company computes loss per common share, in accordance with ASC Topic 260, Earnings Per Share, which requires dual
presentation of basic and diluted earnings per share. Basic income or loss per common share is computed by dividing net income or loss by the weighted average number of common shares outstanding during the period. Diluted income or loss per common
share is computed by dividing net income or loss by the weighted average number of common shares outstanding, plus the issuance of common shares, if dilutive, that could result from the exercise of outstanding stock options and warrants. No potential
dilutive common shares are included in the computation of any diluted per share amount when a loss is reported.
Segments
The Company determined its reporting units in accordance with ASC 280, “Segment Reporting” (“ASC 280”). Management
evaluates a reporting unit by first identifying its’ operating segments under ASC 280. The Company then evaluates each operating segment to determine if it includes one or more components that constitute a business. If there are components within an
operating segment that meet the definition of a business, the Company evaluates those components to determine if they must be aggregated into one or more reporting units. If applicable, when determining if it is appropriate to aggregate different
operating segments, the Company determines if the segments are economically similar and, if so, the operating segments are aggregated.
Management has determined that the Company has two consolidated operating segments: nutraceutical and pharmaceutical. The Company’s reporting segment reflects
the manner in which its chief operating decision maker reviews results and allocates resources. The Company’s reporting segment meets the definition of an operating segment and does not include the aggregation of multiple operating segments.
Leases
The Company accounts for leases in accordance with ASC Topic 842, Leases, applying the package of practical
expedients to leases that commenced before the effective date whereby the Company elected to not reassess the following: (i) whether any expired or existing contracts contain leases and; (ii) initial direct costs for any existing leases. For
contracts entered into on or after the effective date, at the inception of a contract the Company assessed whether the contract is, or contains, a lease. The Company’s assessment is based on: (1) whether the contract involves the use of a distinct
identified asset, (2) whether the Company obtains the right to substantially all the economic benefit from the use of the asset throughout the period, and (3) whether it has the right to direct the use of the asset. The Company will allocate the
consideration in the contract to each lease component based on its relative stand-alone price to determine the lease payments.
Operating lease right of use assets represents the right to use the leased asset for the lease term and operating lease liabilities are recognized based on
the present value of the future minimum lease payments over the lease term at commencement date. As most leases do not provide an implicit rate, the Company uses an incremental borrowing rate based on the information available at the adoption date in
determining the present value of future payments. Lease expense for minimum lease payments is amortized on a straight-line basis over the lease term and is presented on the consolidated statements of operations.
As permitted under the guidance, the Company has made an accounting policy election not to apply the recognition provisions of the new guidance to short term
leases (leases with a lease term of twelve months or less that do not include an option to purchase the underlying asset that the lessee is reasonably certain to exercise); instead, the Company will recognize the lease payments for short term leases
on a straight-line basis over the lease term.
F-11
Subsequent Events
In accordance with ASC 855, Subsequent Events, the Company evaluated subsequent events through the date of this
report; the date the consolidated financial statements were available for issue.
Recent Accounting Pronouncements
The Company has implemented all new accounting pronouncements that are in effect. These pronouncements did not have any material impact on the consolidated
financial statements unless otherwise disclosed, and the Company does not believe that there are any other new accounting pronouncements that have been issued that might have a material impact on its financial position or results of operations.
3. Going
Concern
The Company's consolidated financial statements are prepared using the U.S. GAAP applicable to a going concern, which contemplates the realization of assets
and liquidation of liabilities in the normal course of business. At June 30, 2022 and 2021, the Company had A$5,191,031 and A$13,434,762 in cash and A$6,748,692 and A$13,866,342 in working capital, respectively. For the years ended June 30, 2022 and
2021, the Company had a net loss of A$7,228,814 and A$12,402,829, respectively. Continued losses may adversely affect the liquidity of the Company in the future.
In view of the matters described in the preceding paragraph, recoverability of a major portion of the recorded asset amounts shown in the accompanying
consolidated balance sheets is dependent upon continued operations of the Company, which in turn is dependent upon the Company's ability to raise additional capital, obtain financing and to succeed in its future operations. The consolidated financial
statements do not include any adjustments relating to the recoverability and classification of recorded asset amounts or amounts and classification of liabilities that might be necessary should the Company be unable to continue as a going concern.
4. Research
and Development Tax Incentive Credit Receivable
The balance of research and development tax incentive credit receivable at June 30, 2022 and 2021 was A$3,545,243 and A$2,311,457, respectively. Other
receivables represent amounts due from government agencies for the Research and Development Tax Incentive (Australia) and indirect tax for which there is no expected credit loss. Other receivables are recognized at amortized costs, less any expected
credit losses. The Company expects to collect these receivables within the next fiscal year.
5. Inventory
|
|
As of
|
As of
|
||||||
|
|
June 30, 2022
|
June 30, 2021
|
||||||
|
|
||||||||
|
Raw materials - at cost
|
$
|
56,774
|
$
|
253,754
|
||||
|
Finished goods - at cost
|
23,333
|
618,618
|
||||||
|
Inventories, gross
|
80,107
|
872,372
|
||||||
|
|
||||||||
|
Inventory reserves
|
-
|
(80,001
|
)
|
|||||
|
|
||||||||
|
Inventory, net
|
$
|
80,107
|
$
|
792,371
|
||||
During the year ended June 30, 2022, the Company wrote off its inventory reserve balance of A$80,001. The Company did not record an inventory reserve balance
as of June 30, 2022.
F-12
6. Property
and equipment
Property and equipment consisted of the following at June 30, 2022 and 2021:
|
|
|
|
June 30,
|
|
|
June 30,
|
|
||
|
Useful Life
|
|
2022
|
|
|
2021
|
|
|||
|
|
|
|
|
|
|
|
|
||
|
Leasehold improvements
|
3-15 Years
|
|
$
|
429,102
|
|
|
$
|
429,102
|
|
|
Equipment
|
3-13 Years
|
|
|
611,227
|
|
|
|
607,432
|
|
|
Office furniture and equipment
|
3-10 Years
|
|
|
480,377
|
|
|
|
628,115
|
|
|
|
|
|
|
1,520,706
|
|
|
|
1,664,649
|
|
|
Less: Accumulated depreciation
|
|
|
|
(1,176,400
|
)
|
|
|
(1,181,333
|
)
|
|
Property, plant and equipment - net
|
|
|
$
|
344,306
|
|
|
$
|
483,316
|
|
Depreciation expense was A$192,916 and A$188,498 for the years ended June 30, 2022 and 2021, respectively.
7. Discontinued
operations
On October 19, 2021, the Company licensed its Australian nutraceutical business to PharmaCare Pty Ltd (“PharmaCare”) for cash consideration of A$750,000. As part of the agreement, PharmaCare will
have use of the nutraceutical brand’s registered patents and trademarks, inventory, customer lists, and material contracts. In connection with the transaction, the Company licensed its nutraceutical inventory to PharmaCare for $1,025,910.
In terms of the sale agreement, the Company is entitled to receive an earn-out of the greater of A$250,000 or 5% of net sales for each of the two successive years following completion. At the
time of the sale, the fair value of the minimum additional cash consideration was determined to be A$445,816 and has been recognized as a deferred consideration receivable. Subsequent to recognition, the deferred consideration receivable is
accounted for at amortized cost and at June 30, 2022, the receivable increased to A$470,635 as a result of the unwinding of the discount. The fair value of any contingent consideration above the minimum annual earn-out of A$250,000 has been
assessed to have a nil fair value. The Company has applied the guidance of ASC 606 to conclude that this transaction constitutes a sale of intellectual property. The deferred consideration receivable consists of the
two future guaranteed payments of $250,000, discounted to present value using an implied rate of 8%. The Company estimates that any additional contingent consideration, above the $250,000 per year, is unlikely; however, if additional
consideration is received, it will be recognized in the period received.
The operating results for PharmaCare have been presented in the accompanying consolidated statement of operations for the years ended June 30, 2022 and 2021
as discontinued operations and are summarized below:
|
|
Years Ended June 30,
|
|||||||
|
|
2022
|
2021
|
||||||
|
Sale of goods (net discounts)
|
$
|
2,521,307
|
$
|
3,666,685
|
||||
|
Cost of sales
|
2,027,305
|
2,923,369
|
||||||
|
Gross profit
|
494,002
|
743,316
|
||||||
|
Operating expenses
|
908,346
|
3,507,042
|
||||||
|
Loss from operations
|
(414,344
|
)
|
(2,763,726
|
)
|
||||
|
Other income (expenses)
|
378,166
|
(6,120
|
)
|
|||||
|
|
$
|
(36,178
|
)
|
$
|
(2,769,846
|
)
|
||
|
|
||||||||
F-13
The operating results for PharmaCare Pty Ltd have been presented in the accompanying consolidated statement of operations for the period from July 1, 2021 to
October 19, 2021 as discontinued operations and are summarized below:
|
From July 1, 2021
|
||||
|
to October 19, 2021
|
||||
|
Sale of goods (net discounts)
|
$
|
2,521,307
|
||
|
Cost of sales
|
2,027,305
|
|||
|
Gross profit
|
494,002
|
|||
|
Operating expenses
|
908,346
|
|||
|
Loss from operations
|
(414,344
|
)
|
||
|
Other income (expenses)
|
378,166
|
|||
|
|
$
|
(36,178
|
)
|
|
The assets and liabilities of the discontinued operations at June 30, 2022 and June 30, 2021 are summarized below:
|
Years Ended June 30,
|
||||||||
|
2022
|
2021
|
|||||||
|
Accounts receivables, net
|
$
|
-
|
$
|
993,369
|
||||
|
Inventories, net
|
-
|
783,996
|
||||||
|
Deposits and prepaids
|
-
|
252,699
|
||||||
|
Total assets
|
$
|
-
|
$
|
2,030,064
|
||||
|
|
||||||||
|
Accounts payables
|
$
|
-
|
$
|
957,127
|
||||
|
Accrued liabilities
|
-
|
700,525
|
||||||
|
Accrued employee benefits, current
|
-
|
103,761
|
||||||
|
Total current liabilities
|
-
|
1,761,413
|
||||||
|
Accrued employee benefits, non-current
|
-
|
42,989
|
||||||
|
Total liabilities
|
$
|
-
|
$
|
1,804,402
|
||||
The assets and liabilities of the discontinued operations at October 19, 2021 are summarized below:
|
A s of
|
||||
|
October 19, 2021
|
||||
|
Inventories, net
|
$
|
1,025,910
|
||
|
Total assets
|
$
|
1,025,910
|
||
|
|
||||
|
Total liabilities
|
$
|
-
|
||
F-14
Calculation of Loss on Disposition of Discontinued Operations
|
|
October 19,
|
|
||
|
|
2021
|
|
||
|
Sale consideration
|
|
$
|
2,221,726
|
|
|
Net assets of discontinued operations
|
|
|
(1,025,910
|
)
|
|
Gain from disposition of discontinued operations
|
|
$
|
1,195,816
|
|
8. Notes payable
As of June 30, 2022 and 2021, the balance of the notes payable was A$0 and A$67,834. The notes payable consists of an insurance premium funding facility which
was established with a 12-month term and with an interest rate of 8%. The amount was paid in full during the year ended June 30, 2022.
9. Contingent lease termination liability
In connection with the facilities lease agreement, the Company is required to pay any necessary costs for the space to be returned to its condition prior to
the Company occupying the space. As a result, the Company has estimated these make good costs at A$305,422. This amount is held in non-current liabilities on the consolidated balance sheet.
10. Operating lease right-of-use assets and operating lease liabilities
The Company leases office space in three locations: 8,008 square feet of office space located in Alexandria, New South Wales (“Alexandria”); 14,354 square
feet of office space located in Botany, New South Wales (“Botany”); and 1,376 square feet of office space located in Rancho Santa Margarita, CA 92688 (“USA”).
Operating lease right-of-use assets and liabilities are recognized at the present value of the future lease payments at the lease commencement date. The
interest rate used to determine the present value is the Company’s incremental borrowing rate, estimated to be 4%, as the interest rate implicit in most of its leases is not readily determinable. Operating lease expense is recognized on a
straight-line basis over the lease term. During the years ended June 30, 2022 and 2021, the Company recorded A$374,768 and A$185,814 in rent expense, which is included under general and administrative expenses in the statement of operations.
Right-of- use assets are summarized below:
|
|
As of June 30, 2022
|
|||||||||||||||
|
|
Alexandria
|
Botany
|
USA
|
Total
|
||||||||||||
|
Office Lease
|
$
|
541,434
|
$
|
2,308,211
|
$
|
105,445
|
$
|
2,955,090
|
||||||||
|
Less accumulated amortization
|
(499,882
|
)
|
(1,410,478
|
)
|
(14,645
|
)
|
(1,925,005
|
)
|
||||||||
|
Right-of-use, net
|
$
|
41,552
|
$
|
897,733
|
$
|
90,800
|
$
|
1,030,085
|
||||||||
|
|
||||||||||||||||
|
|
As of June 30, 2021
|
|||||||||||||||
|
|
Alexandria
|
Botany
|
USA
|
Total
|
||||||||||||
|
Office Lease
|
$
|
541,434
|
$
|
2,308,211
|
$
|
90,159
|
$
|
2,939,804
|
||||||||
|
Less accumulated amortization
|
(332,421
|
)
|
(936,765
|
)
|
(69,640
|
)
|
(1,338,826
|
)
|
||||||||
|
Right-of-use, net
|
$
|
209,013
|
$
|
1,371,446
|
$
|
20,519
|
$
|
1,600,978
|
||||||||
|
|
||||||||||||||||
F-15
Operating lease liabilities are summarized below:
|
|
As of June 30, 2022
|
|||||||||||||||
|
|
Alexandria
|
Botany
|
USA
|
Total
|
||||||||||||
|
Office Lease
|
$
|
45,112
|
$
|
944,063
|
$
|
92,049
|
$
|
1,081,224
|
||||||||
|
Less: current portion
|
(45,112
|
)
|
(465,577
|
)
|
(36,778
|
)
|
(547,467
|
)
|
||||||||
|
Long term portion
|
$
|
-
|
$
|
478,486
|
$
|
55,271
|
$
|
533,757
|
||||||||
|
|
||||||||||||||||
|
|
As of June 30, 2021
|
|
||||||||||||||
|
|
Alexandria
|
|
Botany
|
|
USA
|
|
|
Total
|
|
|||||||
|
Office Lease
|
|
$
|
220,399
|
|
|
$
|
1,384,887
|
|
|
$
|
21,956
|
|
|
$
|
1,627,242
|
|
|
Less: current portion
|
|
|
(175,302
|
)
|
|
|
(440,823
|
)
|
|
|
(21,941
|
)
|
|
|
(638,066
|
)
|
|
Long term portion
|
|
$
|
45,097
|
|
|
$
|
944,064
|
|
|
$
|
15
|
|
|
$
|
989,176
|
|
Future minimum lease payments are summarized below:
|
|
Alexandria
|
Botany
|
USA
|
Total
|
||||||||||||
|
Year ending June 30, 2023
|
$
|
45,274
|
$
|
503,582
|
$
|
36,778
|
$
|
585,634
|
||||||||
|
Year ending June 30, 2024
|
-
|
478,486
|
37,881
|
516,367
|
||||||||||||
|
Year ending June 30, 2025
|
-
|
-
|
22,479
|
22,479
|
||||||||||||
|
Total future minimum lease payments
|
45,274
|
982,068
|
97,138
|
1,124,480
|
||||||||||||
|
Less imputed interest
|
(162
|
)
|
(38,005
|
)
|
(5,089
|
)
|
(43,256
|
)
|
||||||||
|
PV of Payments
|
$
|
45,112
|
$
|
944,063
|
$
|
92,049
|
$
|
1,081,224
|
||||||||
11. Equity
Employee share option plan
An employee share option plan has been established by the Company and approved by stockholders at a general meeting, whereby the Company may, at the
discretion of the board of directors, grant options over common stock in the Company to certain staff of the Company. The options are issued for nil consideration and are granted in accordance with performance guidelines established by the Nomination
and Remuneration Committee. No options have been issued under this employee share option plan as of June 30, 2022(1).
| • |
On November 12, 2020, the Company granted 12,000,000 unlisted options to the following Directors for nil consideration:
|
| • |
Michael Hall - 2,000,000 options exercisable at 20 cents per share
|
| • |
Drew Townsend - 2,000,000 options exercisable at 20 cents per share
|
| • |
Sean Hall - 4,000,000 options exercisable at 20 cents per share
|
| • |
Laurence McAllister - 4,000,000 options exercisable at 18 cents per share
|
(1) The options described above do not reflect the reverse stock split.
F-16
The grant of options is designed to incentivize the Directors by participating in the future growth and prosperity of the Company through share ownership and
in recognition made to the Company by the Directors and their ongoing responsibility. The options vested on January 31, 2021 and expire on October 31, 2022. The value of the options at grant date was A$580,000.
On June 25, 2021, the Company granted 833,333 and 250,000 unlisted options to the Investor Relations Consultant and the Chief Financial Officer respectively.
The options are exercisable at 21 cents per share. The options vest on grant date and expire on June 24, 2024. The value of the options at grant date was A$39,836.
On October 18, 2021, the Company granted 1,500,000 options to a non-executive director. The options are exercisable at 21 cents per share. The options vest on
grant date and expire on October 18, 2024. The value of the options at grant date was A$77,367.
The Company selected the Black-Scholes-Merton valuation technique to calculate the grant date fair values for the stock options because it believes that this
technique is reflective of all the inputs that market participants would likely consider in transactions involving warrants. The inputs include the strike price, underlying price, term to expiration, historical volatility, and risk-free interest
rate. The Company recognizes forfeitures as they occur.
The Company valuated the options using the Black-Scholes Model using inputs as detailed below:
|
|
||||||||||||
|
|
October
18, 2021
|
June
25, 2021
|
November
20, 2020
|
|||||||||
|
Underlying price
|
$
|
A0.16
|
$
|
A0.15
|
$
|
A0.185
|
||||||
|
Contractual strike price
|
$
|
A0.21
|
$
|
A0.21
|
$
|
A0.18 - A$0.20
|
||||||
|
Expected term
|
3 Years
|
3 Years
|
2 Years
|
|||||||||
|
Market volatility:
|
||||||||||||
|
Equivalent Historical Volatility
|
60.00
|
%
|
60.00
|
%
|
50.00
|
%
|
||||||
|
Interest rate
|
0.68
|
%
|
0.22
|
%
|
0.10
|
%
|
||||||
Stock option activity for the two year period ended June 30, 2022 is summarized as follows:
|
|
Weighted
|
|||||||
|
|
Average
|
|||||||
|
|
Shares
|
Exercise Price
|
||||||
|
Options outstanding June 30, 2020
|
1,541,725
|
$
|
0.30
|
|||||
|
Options granted
|
13,083,333
|
$
|
0.19
|
|||||
|
Options exercised
|
-
|
-
|
||||||
|
Options cancelled
|
(1,541,725
|
)
|
$
|
0.30
|
||||
|
Options outstanding at June 30, 2021
|
13,083,333
|
$
|
0.19
|
|||||
|
Options granted
|
1,500,000
|
$
|
0.21
|
|||||
|
Options exercised
|
-
|
-
|
||||||
|
Options cancelled
|
-
|
$
|
-
|
|||||
|
Options outstanding at June 30, 2022
|
14,583,333
|
$
|
0.20
|
|||||
|
Options exercisable at June 30, 2022
|
14,583,333
|
$
|
0.20
|
|||||
During the years ended June 30, 2022 and 2021, the Company recorded A$77,367 and A$619,836 in stock-based compensation expense, respectively. At June 30,
2022, the weighted average remaining life of the stock options is 0.66 years. The aggregate weighted average intrinsic value as of June 30, 2022 was A$7.00. The weighted average grant date fair values of the options issued during the years ended June
30, 2022 and 2021 was A$77,367 and A$292,190, respectively.
F-17
12. Commitments
and contingencies
During the normal course of business, the Company may be exposed to litigation. When the Company becomes aware of potential litigation, it evaluates the
merits of the case in accordance with ASC Subtopic 450-20-50, Contingencies. The Company evaluates its exposure to the matter, possible legal or settlement strategies and the likelihood of an unfavorable
outcome. If the Company determines that an unfavorable outcome is probable and can be reasonably estimated, it establishes the necessary accruals. As of June 30, 2022 and 2021, the Company is not aware of any contingent liabilities that should be
reflected in the consolidated financial statements. During the year ended June 30, 2021, the Company paid a legal settlement in the amount of A$500,000 related to a dispute with RNA for nutraceuticals business.
13. Segment
reporting
Identification of reportable operating segments
The Company has two operating segments based on pharmaceutical research and nutraceutical sales. These operating segments are based on the internal reports
that are reviewed and used by the board of directors (who are identified as the chief operating decision makers ) in assessing performance and in determining the allocation of resources. There is no aggregation of operating segments.
The principal products and services of each of these operating segments are as follows:
| • |
Nutraceutical: The supply of Medlab’s self-branded nutraceutical range, predominantly in Australia
|
| • |
Pharmaceutical: Various research activities (depression and oncology) and the supply of cannabis-based medicines
|
As mentioned in Note 7, the Company licensed its Australian nutraceutical business to PharmaCare on October 19, 2021, which included the sale of its
nutraceutical inventory for $1,025,910. As of June 30, 2022, the Company has assets under its nutraceutical division, which solely relate to the deferred consideration receivable ($470,635) established as part of the licensing agreement.
Major customers
During the years ended June 30, 2022 and 2021 approximately 49% and 47% of the Company’s external revenue was derived from sales to two major distributors in
the nutraceuticals business.
|
Years Ended June 30, 2022
|
||||||||
|
Continuing Operations
|
Discontinued Operations
|
|||||||
|
Revenue:
|
Pharmaceutical research
|
Nutraceutical
|
||||||
|
Segment revenue
|
$
|
1,536,366
|
$
|
2,521,307
|
||||
|
Years Ended June 30, 2021
|
||||||||
|
Continuing Operations
|
Discontinued Operations
|
|||||||
|
Revenue:
|
Pharmaceutical research
|
Nutraceutical
|
||||||
|
Segment revenue
|
$
|
732,727
|
$
|
3,666,685
|
||||
|
Years Ended June 30, 2022
|
||||||||
|
Continuing Operations
|
Discontinued Operations
|
|||||||
|
|
Pharmaceutical research
|
Nutraceutical
|
||||||
|
(Loss) gain from operations
|
$
|
(12,984,244
|
)
|
$
|
1,159,638
|
|||
|
Years Ended June 30, 2021
|
||||||||
|
Continuing Operations
|
Discontinued Operations
|
|||||||
|
|
Pharmaceutical research
|
Nutraceutical
|
||||||
|
(Loss) gain from operations
|
$
|
(13,225,275
|
)
|
$
|
(2,769,846
|
)
|
||
F-18
A reconciliation of the Company's consolidated segment operating income to consolidated earnings before income taxes is as follows:
|
Years Ended
|
||||||||
|
|
June 30,
|
|||||||
|
|
2022
|
2021
|
||||||
|
Loss from operations
|
$
|
(12,984,244
|
)
|
$
|
(13,225,275
|
)
|
||
|
Other income – Research and development incentive
|
4,621,501
|
2,265,000
|
||||||
|
Grant income
|
61,954
|
928,585
|
||||||
|
Legal settlement
|
-
|
500,000
|
||||||
|
Other income
|
20,655
|
6,454
|
||||||
|
Interest income
|
44,727
|
25,198
|
||||||
|
Loss on foreign currency exchange
|
(51,129
|
)
|
6,120
|
|||||
|
Interest expense
|
(101,916
|
)
|
(139,065
|
)
|
||||
|
Loss from continuing operations before income taxes
|
(8,388,452
|
)
|
(9,632,983
|
)
|
||||
|
Loss from operations of discontinued operations
|
(36,178
|
)
|
(2,769,846
|
)
|
||||
|
Gain from disposition of discontinued operations
|
1,195,816
|
-
|
||||||
|
Income tax (expense) benefit
|
-
|
-
|
||||||
|
Net loss
|
$
|
(7,228,814
|
)
|
$
|
(12,402,829
|
)
|
||
|
Years Ended June 30, 2022
|
||||||||||||||||||||
|
Continuing Operations
|
Discontinued Operations
|
Total
|
||||||||||||||||||
|
|
Pharmaceutical research
|
Nutraceutical - Continuing
|
Non-operating corporate
|
Nutraceutical - Discontinued
|
Assets
|
|||||||||||||||
|
Total Assets
|
$
|
5,221,996
|
$
|
470,635
|
$
|
5,673,972
|
$
|
-
|
$
|
11,366,603
|
||||||||||
|
Years Ended June 30, 2021
|
||||||||||||||||||||
|
Continuing Operations
|
Discontinued Operations
|
Total
|
||||||||||||||||||
|
|
Pharmaceutical research
|
Nutraceutical - Continuing
|
Non-operating corporate
|
Nutraceutical - Discontinued
|
Assets
|
|||||||||||||||
|
Total Assets
|
$
|
1,038,041
|
$
|
3,660,903
|
$
|
13,917,298
|
$
|
2,030,064
|
$
|
20,646,306
|
||||||||||
14. Related party transactions
The following transactions occurred with related parties:
|
|
June 30,
|
June 30,
|
||||||
|
|
2022
|
2021
|
||||||
|
Director fees
|
||||||||
|
Accrued
|
$
|
481,784
|
$
|
421,260
|
||||
|
Expenses
|
75,000
|
75,000
|
||||||
15. Income taxes
The Company adopted the provisions of uncertain tax positions as addressed in ASC Subtopic 740-10-65-1. As a result of the implementation of ASC Subtopic
740-10-65-1, the Company recognized no increase in the liability for unrecognized tax benefits. As of June 30, 2022 the Company had net operating loss carry forwards of approximately $1,642,153 that may be available to reduce future years’ taxable
income in varying amounts through 2030. Future tax benefits which may arise as a result of these losses have not been recognized in these financial statements, as their realization is determined not likely to occur and accordingly, the Company has
recorded a valuation allowance for the deferred tax asset relating to these tax loss carry-forwards.
The valuation allowance at June 30, 2022 and 2021 was $1,642,153 and $2,206,251, respectively. The net decrease in valuation allowance during the year ended
June 30, 2022 was $564,098. In assessing the realizability of deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred income tax assets will not be realized.
The components of the net deferred tax asset (liability) at June 30, 2022 and, 2021 and the statutory tax rate, the effective tax rate and the elected amount
of the valuation allowance are indicated below:
|
|
||||||||
|
|
June 30,
|
June 30,
|
||||||
|
|
2022
|
2021
|
||||||
|
|
||||||||
|
Net operating loss
|
$
|
1,879,345
|
$
|
3,224,736
|
||||
|
Non-deductible:
|
||||||||
|
Non-deductible research and development expense
|
(543,522
|
)
|
(1,641,847
|
)
|
||||
|
Entertainment expenses
|
(2,298
|
)
|
(5,217
|
)
|
||||
|
Donations
|
(1,170
|
)
|
(1,907
|
)
|
||||
|
Cash flow boost receipts
|
195
|
26,000
|
||||||
|
Difference in overseas tax rates
|
9,205
|
15,586
|
||||||
|
Research and development incentive receivable
|
300,398
|
588,900
|
||||||
|
Deferred tax asset
|
1,642,153
|
2,206,251
|
||||||
|
Less: valuation allowance
|
(1,642,153
|
)
|
(2,206,251
|
)
|
||||
|
Net deferred tax asset (liability)
|
$
|
-
|
$
|
-
|
||||
F-19
Income tax benefit resulting from applying statutory rates in jurisdictions in which the Company is taxed (Australia, United Kingdom and the United States)
differs from the income tax provision (benefit) in the Company’s consolidated financial statements. The following table reflects the reconciliation for the years ended June 30, 2022 and 2021:
|
|
Years Ended June 30,
|
|||||||
|
|
2022
|
2021
|
||||||
|
Benefit at federal and statutory rate
|
(26
|
)%
|
(26
|
)%
|
||||
|
Change in valuation allowance
|
26
|
%
|
26
|
%
|
||||
|
Effective tax rate
|
0
|
%
|
0
|
%
|
||||
The Company periodically evaluates the likelihood of the realization of deferred tax assets, and adjusts the carrying amount of the deferred tax assets by the
valuation allowance to the extent the future realization of the deferred tax assets is not judged to be more likely than not. The Company considers many factors when assessing the likelihood of future realization of its deferred tax assets, including
its recent cumulative earnings experience by taxing jurisdiction, expectations of future taxable income or loss, the carryforward periods available to the Company for tax reporting purposes, and other relevant factors.
Future changes in the unrecognized tax benefit will have no impact on the effective tax rate due to the existence of the valuation allowance. The Company
estimates that the unrecognized tax benefit will not change significantly within the next twelve months. The Company will continue to classify income tax penalties and interest as part of general and administrative expense in its consolidated
statements of operations. There were no interest or penalties accrued as of June 30, 2022 and 2021.
16. Subsequent
Events
On July 28, 2022, the Company had a shareholder meeting to approve the following:
| • |
Share consolidation of every 150 shares into 1 share; and
|
| • |
The issuance of up to 4,000,000 new securities in connection with a U.S. Nasdaq initial public offering.
|
F-20

[•] Ordinary Shares
Pre-Funded Warrants to Purchase
Pre-Funded Warrants to Purchase
[•] Ordinary Shares and Warrants to Purchase
[•] Ordinary Shares
[•] Ordinary Shares
________________________
A.G.P.
________________________
Until , 2022 (25 days after the commencement of this offering), all dealers that buy, sell or trade our
ordinary shares, whether or not participating in this offering, may be required to deliver a prospectus. This delivery requirement is in addition to the obligation of dealers to deliver a prospectus when acting as underwriters and with respect to
their unsold allotments or subscriptions.
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 6. Indemnification of Directors and Officers
Australian law. Australian law provides that a company or a related body corporate of the
company may provide for indemnification of officers and directors, except to the extent of any of the following liabilities incurred as an officer or director of the company:
| • |
a liability owed to the company or a related body corporate of the company;
|
| • |
a liability for a pecuniary penalty order made under section 1317G or a compensation order under section 961M, 1317H, 1317HA, 1317HB 1317HC or 1317HE of the Corporations Act;
|
| • |
a liability that is owed to someone other than the company or a related body corporate of the company and did not arise out of conduct in good faith; or
|
| • |
legal costs incurred in defending an action for a liability incurred as an officer or auditor of the company if the costs are incurred;
|
| • |
in defending or resisting proceedings in which the person is found to have a liability for which they cannot be indemnified as set out above;
|
| • |
in defending or resisting criminal proceedings in which the person is found guilty;
|
| • |
in defending or resisting proceedings brought by the Australian Securities and Investments Commission or a liquidator for a court order if the grounds for making the order are found by the
court to have been established (except costs incurred in responding to actions taken by the Australian Securities and Investments Commission or a liquidator as part of an investigation before commencing proceedings for a court order); or
|
| • |
in connection with proceedings for relief to the person under the Australian Corporations Act 2001 in which the court denies the relief.
|
Constitution. Our Constitution provides that, except to the extent prohibited by the law and the
Corporations Act and, to the extent that the officer is not otherwise indemnified by us pursuant to an indemnity, we indemnify every person who is or has been an officer of the Company against any liability or claim (other than legal costs that are
unreasonable) incurred by that person as an officer. This includes any liability or claim incurred by that person in their capacity as an officer of a subsidiary of the Company where the Company requested that person to accept that appointment.
SEC Position. Insofar as indemnification for liabilities arising under the Securities Act may be
permitted to directors, officers or persons controlling us pursuant to the foregoing provisions, we have been informed that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is therefore
unenforceable.
Item 7. Recent Sales of Unregistered Securities
On December 23, 2019, we raised a total of A$5 million in a private placement offering to certain institutional and accredited investors
for the sale of 17,857,143 Shares at A$0.28 per Share.
On June 19, 2020, we raised a total of A$5 million in a private placement offering to certain institutional and accredited investors for
the sale of 35,984,020 Shares at A$0.15 per Share.
On March 25, 2021 and April 30, 2021, we raised a total of A$15 million in a private placement offering to certain institutional and
accredited investors for the sale of 62,500,000 Shares at A$0.24 per Share.
II-1
Item 8. Exhibits and Financial Statement Schedules
|
Exhibits:
|
|
|
|
|
|
Exhibit No.
|
Description
|
|
1.1
|
|
|
3.1
|
|
|
3.2
|
|
|
4.1
|
|
|
4.2
|
|
|
4.3
|
|
| 4.4 |
|
|
5.1
|
|
|
5.2
|
|
|
8.1
|
|
|
8.2
|
|
|
10.1
|
|
|
14.1
|
|
|
21.1
|
|
|
23.1
|
|
|
23.2
|
Consent of McCabes Lawyers Pty Ltd (included in Exhibit 5.1) **
|
|
23.3
|
Consent of Seward & Kissel LLP (included in Exhibit 5.2)*
|
|
23.4
|
Consent of Pitcher Partners (included in Exhibit 8.1) **
|
|
23.5
|
Consent of Seward & Kissel LLP (included in Exhibit 8.2)*
|
|
24.1
|
|
|
107
|
* Previously filed.
** Filed herewith.
Schedules:
None
II-2
Item 9. Undertakings.
The undersigned Registrant hereby undertakes to provide to the underwriter at the closing specified in the underwriting agreement,
certificates in such denominations and registered in such names as required by the underwriter to permit prompt delivery to each purchaser.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons
of the Registrant pursuant to the foregoing provisions, or otherwise, the Registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and
is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the Registrant of expenses incurred or paid by a director, officer or controlling person of the Registrant in the
successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the Registrant will, unless in the opinion of its counsel the matter has been
settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
The undersigned Registrant hereby undertakes that:
| (1) |
For purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this Registration Statement in reliance upon
Rule 430A and contained in a form of prospectus filed by the Registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this Registration Statement as of the time it was declared effective.
|
| (2) |
For the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration
statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
|
II-3
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, as amended, the registrant certifies that it has reasonable grounds to believe
that it meets all of the requirements for filing on Form F-1 and has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized in New South Wales, Australia on November 22, 2022.
|
|
|
Medlab Clinical Ltd.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
By:
|
/s/ Sean Hall
|
|
|
|
Name:
|
Sean Hall
|
|
|
|
Title:
|
Chief Executive Officer and Managing Director
|
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints each of Edward S. Horton and
Michael Indelicato his true and lawful attorney-in-fact and agent, with full powers of substitution and re-substitution, for him or her and in his or her name, place and stead, in any and all capacities, to sign any or all amendments (including
post-effective amendments) to this Registration Statement, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorney-in-fact and agent full
power and authority to do and perform each and every act and thing requisite and necessary to be done, as fully for all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorney-in-fact and
agent, or his substitute, may lawfully do or cause to be done by virtue thereof.
Pursuant to the requirements of the Securities Act of 1933, as amended, this registration statement has been signed by the following
persons on behalf of the registrant and in the capacities and on the dates indicated:
|
Signature
|
|
Title
|
|
Date
|
|
/s/ Sean Hall
|
|
Chief Executive Officer and Managing Director
|
|
November 22, 2022
|
|
Sean Hall
|
|
(Principal Executive Officer)
|
|
|
|
|
|
|
|
|
|
/s/ Karem Kaya
|
|
Chief Financial Officer and Company Secretary
|
|
November 22, 2022
|
|
Karem Kaya
|
|
(Principal Financial and Accounting Officer)
|
|
|
|
|
|
|
|
|
|
/s/ Michael Hall
|
|
Director
|
|
November 22, 2022
|
|
Michael Hall
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Drew Townsend
|
|
Director
|
|
November 22, 2022
|
|
Drew Townsend
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Mohit Gupta
|
|
Director
|
|
November 22, 2022
|
|
Mohit Gupta
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Cheryl Maley
|
|
Director
|
|
November 22, 2022
|
|
Cheryl Maley
|
|
|
|
|
II-4
Signature of Authorized U.S. Representative of the Registrant
Pursuant to the requirements of the Securities Act of 1933, as amended, the undersigned, the duly authorized representative in the United
States of Medlab Clinical Ltd., has signed the Registration Statement on November 22, 2022.
|
|
By:
|
/s/ Donald J. Puglisi
|
|
|
|
|
Name:
|
Donald J. Puglisi
|
|
|
|
|
Title:
|
Managing Director
|
|
|
II-5
ATTACHMENTS / EXHIBITS
Serious News for Serious Traders! Try StreetInsider.com Premium Free!
You May Also Be Interested In
- Tiziana Life Sciences (TLSA) Reports Positive 3-Month Neuroimaging Scores in Multiple Sclerosis Patients Receiving Intranasal Foralumab
- Tiziana Life Sciences (TLSA) Announces Study Results from Intranasal Anti-CD3 Foralumab in Multiple Sclerosis Patients with PIRA Highlighted in Neurology Today
- Psyence Biomedical (PBM) Announces Phase IIb Clinical Trial Listing on the Australian New Zealand Clinical Trials Registry
Create E-mail Alert Related Categories
SEC FilingsRelated Entities
F1Sign up for StreetInsider Free!
Receive full access to all new and archived articles, unlimited portfolio tracking, e-mail alerts, custom newswires and RSS feeds - and more!
