

Form 10-K Jaguar Health, Inc. For: Dec 31
 Tweet
Tweet Share
ShareUNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
Form
(Mark One)
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
For the fiscal year ended | |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
For the transition period from to
COMMISSION FILE NO.
(Exact name of registrant as specified in its charter)
(State or other jurisdiction of | (I.R.S. Employer |
incorporation or organization) | Identification No.) |
(Address of principal executive offices)
Registrant’s telephone number, including area code:
(
SECURITIES REGISTERED PURSUANT TO SECTION 12(b) OF THE ACT:
Title of each class |
| Trading Symbol(s) |
| Name of each exchange on which registered |
SECURITIES REGISTERED PURSUANT TO SECTION 12(g) OF THE ACT: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ◻
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ◻
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ⌧
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer ◻ | Accelerated filer ◻ | Smaller reporting company | |
Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ◻
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes
As of June 30, 2021, the aggregate market value of the registrant’s common stock held by non-affiliates was approximately $
The number of shares of the registrant’s common stock outstanding as of March 10, 2022 was
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the proxy statement for the registrant’s 2022 Annual Meeting of Stockholders, or Proxy Statement, to be filed within 120 days of the end of the fiscal year ended December 31, 2021 are incorporated by reference in Part III hereof. Except with respect to information specifically incorporated by reference in this Form 10-K, the Proxy Statement is not deemed to be filed as a part hereof.
TABLE OF CONTENTS
i
PART I
Forward-looking statements
This Form 10-K contains forward-looking statements within the meaning of Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act. All statements other than statements of historical facts contained in this Form 10-K, including statements regarding our future results of operations and financial position, business strategy, prospective products, product approvals, research and development costs, timing of receipt of clinical trial, field study and other study data, and likelihood of success, commercialization plans and timing, other plans and objectives of management for future operations, and future results of current and anticipated products are forward-looking statements. These statements involve known and unknown risks, uncertainties and other important factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements.
In some cases, you can identify forward-looking statements by terms such as “may,” “will,” “should,” “expect,” “plan,” “aim,” “anticipate,” “could,” “intend,” “target,” “project,” “contemplate,” “believe,” “estimate,” “predict,” “potential” or “continue” or the negative of these terms or other similar expressions. The forward-looking statements in this Form 10-K are only predictions. We have based these forward-looking statements largely on our current expectations and projections about future events and financial trends that we believe may affect our business, financial condition and results of operations. These forward-looking statements speak only as of the date of this Form 10-K and are subject to a number of risks, uncertainties and assumptions described under the sections in this Form 10-K titled “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and elsewhere in this Form 10-K. Forward-looking statements are subject to inherent risks and uncertainties, some of which cannot be predicted or quantified and some of which are beyond our control. The events and circumstances reflected in our forward-looking statements may not be achieved or occur and actual results could differ materially from those projected in the forward-looking statements. Moreover, we operate in a dynamic industry and economy. New risk factors and uncertainties may emerge from time to time, and it is not possible for management to predict all risk factors and uncertainties that we may face. Except as required by applicable law, we do not plan to publicly update or revise any forward-looking statements contained herein, whether as a result of any new information, future events, changed circumstances or otherwise.
Jaguar Health, our logo, Napo Pharmaceuticals, Napo Therapeutics, Mytesi, Equilevia, Canalevia, Canalevia-CA1, Canalevia-CA2, and Neonorm are our trademarks that are used in this Form 10 K. This Form 10 K also includes trademarks, tradenames and service marks that are the property of other organizations. Solely for convenience, trademarks and tradenames referred to in this Form 10 K appear without the ©, ® or ™ symbols, but those references are not intended to indicate that we will not assert, to the fullest extent under applicable law, our rights or that the applicable owner will not assert its rights, to these trademarks and tradenames.
ITEM 1. BUSINESS
BUSINESS
Jaguar Health, Inc. (“Jaguar” or the “Company”) is a commercial stage pharmaceuticals company focused on developing novel, plant-based, non-opioid, and sustainably derived prescription medicines for people and animals with GI distress, including chronic, debilitating diarrhea. Jaguar Animal Health is a tradename of Jaguar Health. Our wholly owned subsidiary, Napo Pharmaceuticals, Inc. (“Napo”), focuses on developing and commercializing proprietary plant-based human pharmaceuticals for the global marketplace from plants or plant products used traditionally in rainforest areas. Napo’s marketed drug Mytesi (crofelemer 125 mg delayed-release tablets) is a first-in-class oral botanical drug product approved by the U.S. Food and Drug Administration (“FDA”) for the symptomatic relief of noninfectious diarrhea in adults with HIV/AIDS on antiretroviral therapy. To date, this is the only oral plant-based botanical prescription medicine approved under the FDA’s Botanical Guidance. Jaguar Animal Health’s Canalevia-CA1 (crofelemer delayed-release tablets) drug is the first and only oral plant-based prescription product that is FDA conditionally approved to treat chemotherapy-induced diarrhea (“CID”) in dogs. Canalevia-CA1
1
is a canine-specific formulation of crofelemer. Napo Therapeutics S.p.A., Napo’s majority owned Italian subsidiary, focuses on expanding crofelemer access in Europe.
Jaguar, formerly known as Jaguar Animal Health, Inc., was founded in San Francisco, California as a Delaware corporation on June 6, 2013 (inception). The Company was a majority-owned subsidiary of Napo until the close of the Company's initial public offering on May 18, 2015. The Company was formed to develop and commercialize first-in-class prescription and non-prescription products for companion and production animals and horses. The Company's first non-prescription commercial products, Neonorm Calf and Neonorm Foal, were launched in 2014 and 2016, respectively.
On July 31, 2017, Jaguar completed a merger with Napo pursuant to the Agreement and Plan of Merger dated March 31, 2017, by and among Jaguar, Napo, Napo Acquisition Corporation (“Merger Sub”), and Napo's representative (the “Merger Agreement”). In accordance with the terms of the Merger Agreement, upon the completion of the merger, Merger Sub merged with and into Napo, with Napo surviving as the wholly-owned subsidiary (the “Merger” or “Napo Merger”). Immediately following the Merger, Jaguar changed its name from “Jaguar Animal Health, Inc.” to “Jaguar Health, Inc.” Napo now operates as a wholly-owned subsidiary of Jaguar focused on human health including the ongoing development of crofelemer and commercialization of Mytesi.
On March 15, 2021, Jaguar established Napo EU S.p.A (which changed its name in January 2022 to Napo Therapeutics S.p.A. “Napo Therapeutics”) based in Milan, Italy as a subsidiary of Napo. Napo Therapeutics’ mission is to provide access to crofelemer in Europe to address significant rare/orphan disease indications, including, initially, two key orphan target indications: Short bowel syndrome with intestinal failure (“SBS-IF”), and congenital diarrheal disorders (“CDD”). On November 3, 2021, Napo Therapeutics merged with Dragon SPAC S.p.A. (“Dragon SPAC”).
On December 13, 2021, the European Medicines Agency (“EMA”) granted orphan-drug designation (“ODD”) for crofelemer for short bowel syndrome (“SBS”) indication in the European Union following review of the ODD application Napo submitted to the EMA in September 2021. Following this decision from the EMA, Napo Therapeutics is initiating efforts to commence clinical development of crofelemer in both adult and pediatric SBS patients in support of the company’s key focus on leveraging the EMA’s accelerated conditional marketing authorization pathway in Europe for these rare diseases. Napo Therapeutics has also agreed to support an investigator-initiated trial (“IIT”) which will provide proof of concept (“POC”) support for potential expanded access programs for crofelemer for patients with CDD and/or SBS patients with intestinal failure (“IF”). The expanded access program will be initiated following completion of this study and upon publication of POC results, potentially in 2023. Crofelemer previously received ODD in the U.S. from the FDA for SBS. SBS affects approximately 10,000 to 20,000 people in the U.S., according to the Crohn's & Colitis Foundation, and it is estimated that the population of SBS patients in Europe is approximately the same size. Despite limited treatment options, the global SBS market exceeded $568 million in 2019 and is expected to reach $4.6 billion by 2027, according to a report by Vision Research Reports.
On December 21, 2021, we received conditional approval from the FDA to market Canalevia-CA1 (crofelemer delayed-release tablets), our oral plant-based prescription drug and the only drug for the treatment of CID in dogs. We expect Canalevia-CA1 to be available to multiple leading veterinary distributors in the U.S. in the second quarter of 2022 after we complete a post-approval update to the chemistry, manufacturing and controls (“CMC”) related to crofelemer. This update will align with the CMC requirements related to crofelemer used as the active ingredient in Mytesi.
On January 4, 2022, we announced the launch of Canalevia-CA1 (crofelemer delayed-released tablets), which is being commercialized as a prescription drug product under the Company’s Jaguar Animal Health tradename. Canalevia-CA1 is a tablet that is given orally and can be prescribed for home treatment of CID. Canalevia-CA1 is a canine-specific formulation of crofelemer that is conditionally approved by the FDA under application number 141-552. Conditional approval allows for commercialization of the product while Jaguar Animal Health continues to collect the substantial evidence of effectiveness required for a full approval. We have received Minor Use in a Major Species (“MUMS”) designation from the FDA for Canalevia-CA1 to treat CID in dogs. FDA has established a "small number" threshold for minor use in each of the seven major species covered by the MUMS act. The small number threshold is currently 70,000 for dogs, representing the largest number of dogs that can be affected by a disease or
2
condition over the course of a year and still have the use qualify as a minor use. We expect Canalevia-CA1 to be available to multiple leading veterinary distributors in the U.S. in the second quarter of 2022 after we complete a post-conditional-approval update to the CMC related to crofelemer. In the field of animal health, we are continuing limited activities related to developing and commercializing first in class gastrointestinal products for dogs, dairy calves, foals, and high value horses.
Most of the activities of the Company are focused on the commercialization of Mytesi and Canalevia-CA1 and the ongoing clinical development of crofelemer for the prophylaxis of diarrhea in adult patients receiving targeted cancer therapy. In June 2021, Napo Pharmaceuticals recruited Dr. Darlene Horton, as the Chief Medical Officer (“CMO”) of Napo Pharmaceuticals to support the ongoing clinical development activities for prescription products for human health. Dr. Horton is a 25-year veteran of the biotech and pharmaceutical industry. Prior to joining Napo, she led clinical development and regulatory strategy as CMO at Coherus Biosciences, Itero Biopharmaceuticals, and SMC Biotechnology. As Head of Clinical and Medical Affairs at Scios, she led the clinical development program that led to the approval of Natrecor® and was on the senior executive team when Scios was acquired by JNJ for $2.4B. At JNJ, she co-led (with strategic marketing) the cardiovascular therapeutic area when JNJ in-licensed and began developing the blockbuster drug Xarelto®. She also served as CEO at Nile Therapeutics and TulangCo Inc. Dr. Horton completed her Pediatric Cardiology fellowship and Pediatrics Residency at UCSF. She holds M.D. and B.S. in Microbiology degrees from the University of Florida.
We believe Jaguar is poised to realize a number of synergistic, value adding benefits—an expanded pipeline of potential blockbuster human follow on indications of crofelemer, and a second generation anti secretory agent—upon which to build global partnerships. Jaguar, through Napo, holds global unencumbered rights for crofelemer, Mytesi, and Canalevia-CA1. Additionally, several of the drug product opportunities in Jaguar’s crofelemer pipeline are backed by Phase 2 and proof of concept evidence from human clinical trials.
Crofelemer is a novel, first in class anti secretory agent which has a normalizing effect on electrolyte and fluid balance while acting locally in the gut, and this mechanism of action has the potential to benefit multiple disorders that cause gastrointestinal distress, including diarrhea and abdominal discomfort. Crofelemer is also in development for possible follow on indications, including prophylaxis for cancer therapy related diarrhea (“CTD”); for rare disease indications for symptomatic treatment of infants and children with CDD and for adult and pediatric patients with SBS-IF. As mentioned above, crofelemer has received ODD for SBS in the US and in the European Union (“EU”). Furthermore, the drug is being evaluated for management of diarrhea and abdominal discomfort in inflammatory bowel disease (“IBD”); diarrhea-predominant irritable bowel syndrome (“IBS-D”); and for idiopathic/functional diarrhea. A second generation proprietary anti secretory agent, NP-300 (lechlemer), is undergoing preclinical development for symptomatic relief and treatment of diarrhea in patients with acute infection from cholera.
Napo has a direct sales force of 8 sales representatives and a national sales director covering U.S. geographies with the highest commercial potential. In 2019, we hired Ian Wendt, an industry veteran with a broad range of experience that includes commercializing supportive care and HIV treatments, as Vice President of Commercial Strategy. He was promoted to Chief Commercial Officer in 2020. With support provided by concomitant marketing, promotional activities, patient education programs and peer education initiatives described below, we expect continued growth in the number of patients treated with Mytesi. Mr. Wendt will lead disease state educational initiatives that will pave the way for crofelemer’s final development for the CTD market and our commercial role for this next important, potential indication for Mytesi. Additionally, he is leading commercialization activities for Canalevia-CA1 in the U.S. veterinary market.
A key component of our marketing strategies for Mytesi in 2021 was focused on the transition of Mytesi distribution to a closed network of specialty pharmacies rather than to wholesalers that resell the product to retail pharmacies. This transition was intended to help remove access barriers for patients receiving Mytesi and includes services such as a higher level of support for prior authorizations, appeals, adherence counseling, and home delivery options. While patients often visit retail pharmacies for short-term or uncomplicated medical needs, specialty pharmacies focus on serving patients with complex and chronic medical conditions like HIV. The transition to a closed network of specialty pharmacies is expected to result in a meaningful reduction in Mytesi distribution costs and
3
prepare our U.S. commercial distribution network for future indication expansion of crofelemer to other populations of patients with complex medical needs. However, when we made the full transition on September 3, 2021, to selling to specialty pharmacies, it resulted in an underrepresentation of actual Mytesi utilization, as wholesalers depleted their inventory during the transition process.
The goal of Napo’s sales team is to deliver a frequent and consistent selling message to targeted, high volume prescribers of HIV antiretroviral therapies (“ART”) and to gastroenterologists who see large numbers of HIV patients. In 2017 we released the results of a survey of 350 people living with HIV and AIDS regarding the topic of “Talking to Your Doctor About Symptoms.” The survey results show that diarrhea remains prevalent in those living with HIV/AIDS, with 27% of respondents reporting that they currently have diarrhea, and 56% reporting that they have had diarrhea in the past. Additionally, the results of a 2017 Napo sponsored survey of 271 U.S. board certified gastroenterologists indicate that the number one GI complaint for people living with HIV/AIDS is diarrhea, and 93% of U.S. gastroenterologists see patients with HIV/AIDS in their practice.
Key to the success of our sales representatives in growing Mytesi sales is segmenting and targeting the right health care providers—those HIV treaters who are high prescribers of ART and those gastrointestinal doctors who see large populations of people living with HIV/AIDS. The target list of prescribers for our sales reps includes a pool of approximately 1,300 high volume ART prescribing HIV specialists, and gastroenterologists who see the largest number of people living with HIV/AIDs, and we’ve strategically focused our sales force in the US geographies with the highest potential, including San Francisco, Sacramento, Seattle, southern California, Arizona, Nevada, Florida, New York City/Long Island, Connecticut, New Jersey, Pennsylvania, Maryland, Kansas, Texas, Missouri, Chicago, Michigan, Atlanta, Louisiana, DC, Virginia, North Carolina, South Carolina, Indianapolis, and Ohio.
Medical education presentations led by health care providers (“HCPs”) participating in the Napo Speakers Bureau—a group that includes HIV/AIDS treaters, infectious disease specialists, gastroenterologists, colorectal surgeons, nurse practitioners, doctors of pharmacology, and physician assistants—focus on the prevalence and pathophysiology of gastrointestinal consequences of HIV infection and on the latest treatment options for HIV related diarrhea. Presentations given by patient advocate members provide information to HIV/AIDS patients about the prevalence of diarrhea in people living with HIV/AIDS (“PLWHA”) and offer guidance about talking to HCPs regarding diarrhea related concerns.
With the introduction of newer antiretroviral (“ARV”) drug therapies, there has been a reduction in the severity of ARV induced diarrhea. However, a significant portion of this patient population still suffers from diarrhea caused by HIV enteropathy, which is due to the direct and indirect effects of HIV on the intestinal mucosa. Chronic diarrhea remains a significant complaint of PLWHA, particularly those who are older and have lived with the virus in their gut for 10+ years. According to data from the U.S. Centers for Disease Control and Prevention, currently more than 70% of people living with HIV are over age 50 and have lived with HIV for more than 10 years.
Napo is on many AIDS Drug Assistance Program (“ADAP”) formularies. ADAPs provide lifesaving HIV treatments to low income, uninsured, and underinsured individuals living with HIV/AIDS in all 50 states and the territories. The ADAP program provides Mytesi free of charge to patients who qualify and copay support for some patients who have insurance coverage. Based on data from healthcare research firm Decision Resource Group, approximately 86% of ADAP eligible US lives now have access to Mytesi, which is now on the ADAP formularies for 30 states, including the four programs with the largest enrollment.
In May 2020, Napo launched a program to educate insurance companies about the benefits of Mytesi and negotiate better access to Mytesi for commercially insured patients. We believed that our enhanced Mytesi market access strategy engaged select payors in contracting discussions with the objective of removing barriers for patients in order to allow them to more easily start on – and stay on – Mytesi. This initiative represented a commercial opportunity to employ a strategic mechanism that was well-established in the U.S. pharmaceutical industry to help patients access Mytesi.
4
Napo expanded the NapoCares Patient Support Program for Mytesi in April 2020 as part of the Company's enhanced market access strategy. The expansion meaningfully increased co-pay support for commercially insured patients, which also includes allowing the co-pay amount to remain the same whether a patient fills a 30-day or a 90-day prescription of Mytesi. The expansion also increased the income ceiling from two times the Federal poverty limit to five times the Federal poverty limit for our patient assistance program, which will allow more low-income patients to receive Mytesi at no cost. The co-pay program and patient assistance program are components of a comprehensive suite of patient support services Napo rolled out in the second quarter of 2020 with the support of AssistRx, a specialty therapy initiation and patient support company.
On August 2021, Napo signed a license agreement with Napo EU S.p.A. to study, develop, manufacture, and commercialize Napo’s plant-based crofelemer and lechlemer drug product candidates in the European Union (excluding Russia) and in specified non-EU countries in Europe for specific indications, which rights and obligations were assumed by the combined company formed by the merger of Napo Therapeutics with Dragon SPAC (the combined company uses the Napo Therapeutics name). Per the terms of the license agreement, Napo will receive payment of up to $10 million as the initial license fee (to be paid in two installments, the first of which has already been received) as the initial license fee and is eligible to receive additional payments related to milestones, royalties, and product transfers.
In November 2021, Napo Therapeutics appointed Mr. Massimo Mineo, a veteran of the European pharmaceutical industry for 20+ years, as general manager (the equivalent in Europe of chief executive). He has significant experience in the field of orphan-drug development and commercialization activities and his leadership is expected to be instrumental in the ongoing development and commercialization activities for crofelemer for the planned SBS-IF indication in the EU. Mr. Mineo is responsible for the strategy, planning, direction, and implementation of all Napo Therapeutics’ commercial, operational, and product development activities within Europe, with success defined by bringing crofelemer to market for key initial target indications, beginning with SBS-IF and CDD.
In November 2021, Napo Therapeutics announced the appointment of Annabella Amatulli as chief regulatory officer. A recognized expert in global regulatory affairs, Ms. Amatulli is responsible for both high-level strategic planning and hands-on support for Napo Therapeutics’ development programs and licensed products from a regulatory perspective and serves as the primary liaison between Napo Therapeutics and European health authorities.
In January 2022, Napo Therapeutics announced the appointment of Martire Particco, MD, a physician with 30+ years of experience in Europe’s pharmaceutical industry and in clinical practice, as its CMO for the EU-related development activities of Napo Therapeutics. Dr. Particco possesses in-depth experience in the field of orphan and rare diseases, having been involved in the clinical development and launch of Pfizer’s pulmonary hypertension indication for sildenafil and the clinical development of Kerdion Biopharma’s ligneous conjunctivitis indication for plasminogen, with direct experience with patients and experts treating these rare pathologies.
In February 2022, Napo announced the completion of a third-party, investigator-initiated preclinical enterocyte (intestinal cell) in vitro study to evaluate the effects of crofelemer on cells with certain genetic defects that result in specific forms of CDD. Jaguar believes that these study results will support certain requests received from the Office of Orphan Products Development at the U.S. FDA in response to the ODD application Napo filed with the FDA for crofelemer for CDD in infants and children. The data from this study will support the rare disease business model that Napo Therapeutics is pursuing in Europe under its exclusive license for crofelemer from Jaguar and Napo. CDD patients have intestinal failure and morbidity resulting in a failure to thrive due to malabsorption of nutrients and need parenteral nutrition. We believe the novel mechanism of action of crofelemer may have considerable potential to manage the severe secretory loss of electrolytes and fluid resulting in dehydration. There are currently no therapies for CDD except parenteral nutrition. Thus, crofelemer may reduce the associated morbidity and mortality of CDD and lessen the need for total parenteral nutrition (“TPN”).
5
Napo has actively ensured that its intellectual property (“IP”) filings in support of the development of crofelemer for various proposed indications are protected appropriately. The IP portfolio for crofelemer includes the relief and treatment of HIV-associated diarrhea and chemotherapy-induced diarrhea as well as planned indications for inflammatory diarrhea, IBS-D, CID and SBS, with all indications, Napo prioritizes IP protection. Napo currently holds approximately 143 patents, the majority of which do not expire until 2027 - 2031, and approximately 43 patents pending.
In October 2020, Napo initiated its pivotal Phase 3 clinical trial of crofelemer (Mytesi) for prophylaxis of diarrhea in adult cancer patients receiving targeted therapy (OnTarget study). The Phase 3 OnTarget clinical trial is a 24-week (two 12-week stages), randomized, placebo-controlled, double-blind study to evaluate the safety and efficacy of crofelemer in the prophylaxis of diarrhea in adult cancer patients with solid tumors receiving targeted cancer therapy-containing treatment regimens. Patients will be randomized to receive either crofelemer or matching placebo treatment that will start concurrently with the initiation of targeted cancer therapy regimen. The primary endpoint will be assessed at the end of the initial (Stage I) 12-week double-blind placebo-controlled primary treatment phase after the last patient has completed 12 weeks of treatment. After completing the Stage I treatment phase, the subjects will have the option to remain on their assigned treatment arm and reconsent to enter into the Stage II 12-week extension phase. The safety and efficacy of orally administered crofelemer will be evaluated for the prophylaxis of diarrhea in adult cancer patients receiving targeted cancer therapies with or without standard chemotherapy regimens. The assessment of the frequency of diarrhea will be measured by the average number of weekly loose and/or watery stools for the active (crofelemer) or placebo arms over 12-week Stage I treatment period.
A significant proportion of patients undergoing cancer therapy experience diarrhea. Novel “targeted cancer therapy” agents, such as epidermal growth factor receptor (“EGFR”) antibodies and tyrosine kinase inhibitors (“TKIs”), with or without cycle chemotherapy agents, may activate intestinal chloride ion channel-mediated secretory pathways leading to increased electrolyte and fluid content in the gut lumen, which results in passage of loose/watery stools, i.e., secretory diarrhea. With increased approval of several novel targeted therapies, it is estimated that 13.6% of cancer patients in 2020 were eligible for targeted therapies with or without standard chemotherapy regimens, according to a paper published in April 2021 in the journal Annals of Oncology1. According to the National Cancer Institute, in 2020, 1,806,590 new cases of cancer were diagnosed and nearly 250,000 of these newly diagnosed patients could be eligible for available targeted therapies.
Due to the chronic dosing and toxicity associated with targeted therapies, many cancer patients on targeted therapy require drug holidays or dose reductions in their therapy, including those due to diarrhea. By improving stool consistency and reducing the frequency of watery stools, crofelemer is expected to provide improved adherence to the therapeutic dosing of any targeted therapies, potentially leading to better clinical outcomes. We have learned from business development discussions with cancer drug manufacturers that the adoption and continued use of targeted cancer therapies is directly related to the ability of patients to tolerate these therapies—highlighting the importance of supportive care drugs like crofelemer to help manage cancer treatment-related diarrhea in this patient population.
As previously announced, an abstract regarding patient outcomes associated with cancer-related diarrhea (“CRD”) by Napo and Napo's collaborators was accepted for poster presentation at the American Society of Clinical Oncology (ASCO®) Annual Meeting, which was held virtually from June 4-8, 2021. This study found that patients with CRD were 40% more likely to discontinue the chemotherapy or targeted therapy than patients without CRD. The persistence of index cancer therapy and time to switch were also lower for patients with CRD. Strategies to control CRD and continue cancer therapy are urgently needed2.
In addition, two CRD-related abstracts from Napo and its collaborators were accepted for online publication at ASCO. One of these studies found that patients with CRD used significantly more resources, including outpatient services, ED visits, and hospitalizations. Effective prevention of CRD remains an unmet strategy to reduce the overall
1A. Haslam, M.S. Kim, V. Prasad, Updated estimates of eligibility for and response to genome-targeted oncology drugs among US cancer patients, 2006-2020
2Pablo C. Okhuysen, M.D., Lee Schwartzberg, M.D., FACP, Eric Roeland, M.D., FAAHPM, The impact of cancer-related diarrhea on changes in cancer therapy patterns: Real world evidence
6
cost of cancer care3. Findings from the other study indicated that patients with CRD had nearly 2.9 times higher all-cause total cost than patients without CRD after adjusting for covariates. Prevention of CRD may result in a significant reduction in cancer-treatment cost4.
As previously announced in June 2020, the effects of crofelemer (Mytesi) were evaluated in reducing diarrhea associated with the irreversible pan-HER TKI neratinib in female dogs. The data were presented at the American Association for Cancer Research (“AACR”) Annual Meeting II in 2020. The results from the dog study provide further scientific support for the evaluation of crofelemer in providing symptomatic relief of watery diarrhea in patients receiving a targeted cancer therapy drug like neratinib with or without cycle chemotherapy, without the use of antimotility drugs like loperamide, in future clinical studies.
The dog study was conducted without the prophylaxis or concomitant use of loperamide and demonstrated that crofelemer caused an approximate 30% reduction in the incidence and severity of diarrhea associated with daily oral administration of neratinib, which was statistically significant, within the 28-day period. Crofelemer also demonstrated significant improvement in the proportion of responder dogs, and there was a trend for fewer neratinib dose reductions in both crofelemer treatment groups when compared to the control group.
The results and finding from a clinical, third-party investigator-initiated Phase 2 study (called HALT-D) evaluated the effectiveness of crofelemer for reduction of diarrhea in HER2 positive breast cancer patients receiving trastuzumab, pertuzumab, and chemotherapy agents such as docetaxel or pacilitaxel with or without carboplatin. The investigators of the HALT-D study, sponsored by Georgetown University and funded by Genentech, a member of the Roche Group, were presented at the San Antonio Breast Cancer Symposium on December 10, 2021. It has been reported that these pertuzumab-containing regimens cause diarrhea in up to 80% of breast cancer patients and approximately 8 to 12% of patients reach grade 3, which often requires hospitalization. No antidiarrheal medications are currently approved that specifically target the underlying mechanism of CID associated with pertuzumab-containing regimens. Recent studies have shown that EGFR inhibitors cause increased chloride secretion into the lumen of the gut and Crofelemer through its unique and novel mechanism of reducing the chloride-secretory actions of the cystic fibrosis transmembrane conductance regulator (“CFTR”) and calcium-activated chloride channels (CaCC), was considered to be mechanistically- and physiologically-appropriate for reducing the loss of electrolyte and fluid in breast cancer patients receiving this regimen.
The Principal Investigator (“PI”), Paula Pohlmann, MD, PhD, Associated Professor at the University of Texas MD Anderson Cancer Center and formerly from Georgetown University, reported the results of the HALT-D study. This clinical study evaluated 51 breast cancer patients that were eligible to receive at least three cycles of pertuzumab-containing regimen with chemotherapy that were randomly assigned to either crofelemer in cycles 1 and 2 or the control group, in which patients received standard of care. Breakthrough anti-diarrheal medicines (“BAM”) were permitted but not given prophylactically. Findings showed that the primary endpoint, the incidence of diarrhea for at least two consecutive days, was not statistically different for the two groups. However, crofelemer patients demonstrated significantly better outcomes compared to control group patients across a number of key secondary endpoints including reductions in the incidence and severity of diarrhea in cycle 2 based on Investigator and Patient Reported Outcomes (“PRO”) (see Jaguar Health's November 19, 2021 press release). In the presentation, additional findings were reported that showed that CID occurred significantly lesser (by 23%) in the crofelemer group during cycle 1 and crofelemer patients were 1.8 times more likely than control patients to have their diarrhea resolved. These data provide concordance to the planned primary endpoint in Napo’s ongoing phase 3 OnTarget study.
Dr. Pohlmann commented that the HALT-D study showed benefits of crofelemer across a range of important diarrhea-related measures, including its incidence, severity and probability of resolving and that the lack of difference in the primary endpoint was because about 70% of the patients in both groups would have at least two consecutive days of diarrhea regardless of cycle or CID treatment group. The incidence of two consecutive days of diarrhea is
3Lee Schwartzberg, M.D., FACP, Eric Roeland, M.D., FAAHPM, Pablo C. Okhuysen, M.D., Characterizing unplanned resource utilization associated with cancer-related diarrhea
4Eric Roeland, M.D., FAAHPM, Pablo C. Okhuysen, M.D., Lee Schwartzberg, M.D., FACP, Healthcare utilization and costs associated with cancer-related diarrhea
7
typical in cancer patient experiences from receiving chemotherapy regimens and is thus not clinically relevant as it does not differentiate the severity nor duration of CID among treatment groups. Dr. Pohlmann also commented that the secondary endpoints provided a more relevant assessment of CID, which may guide future studies that address this significant comorbidity in cancer patients receiving such regimens.
Napo is also continuing to conduct preclinical and formulation activities to support the evaluation of its second generation, plant-based oral prescription drug product, NP-300 (lechlemer), for its planned evaluation in the symptomatic treatment of diarrhea associated with acute infections including that from Vibrio cholerae. Cholera produces a devastating loss of electrolytes and fluid in patients and without appropriate reduction in loss of fluid and electrolytes, patients experience significant hospitalization and mortality. Lechlemer provides the opportunity to treat cholera patients in combination with oral rehydration salts (“ORS”) and the recommended guidelines from the World Health Organization (“WHO”) for the use of appropriate antibiotics to reduce the burden of the pathogen. Appropriate preclinical toxicity studies and formulation development activities are ongoing to support the conduct of clinical studies with lechlemer.
As previously mentioned, Napo completed 7-day and 28-day preclinical toxicology and safety studies in rats and dogs with lechlemer (NP-300) following repeated daily oral dosing. Napo received partial support for preclinical services from the National Institute of Allergy and Infectious Diseases (“NIAID”) of the National Institutes of Health, and Napo is grateful for their partial support of lechlemer’s development.
Napo is currently conducting additional, prerequisite IND-enabling toxicity studies and also developing appropriate oral solid dosage forms to allow the clinical evaluation of lechlemer. The Company plans to provide appropriate regulatory documents by end of the second quarter of 2022 to support the initiation of first-in-human (“FIH”) evaluation in the third quarter of 2022.
Cholera is an acute diarrheal illness caused by infection of the intestine with the bacterium Vibrio cholerae. According to the Centers for Disease Control and Prevention of the U.S. Department of Health & Human Services, an estimated 3 5 million cholera cases and more than 100,000 cholera-related deaths occur each year around the world. The infection is often mild or without symptoms but can sometimes be severe. Approximately one in 10 of infected persons will have severe disease characterized by profuse watery diarrhea, vomiting, and leg cramps. In these people, rapid loss of body fluids leads to dehydration and shock. Without treatment, death can occur within hours. The largest cholera outbreak in recorded history recently occurred in Yemen. According to Oxfam, the number of cholera cases in Yemen in 2019 was the second largest ever recorded in a country in a single year, surpassed only by the numbers in Yemen in 2017. According to the Brookings Institution, cholera continues to spread in Yemen, with 180,000 new cases reported in the first eight months of 2020.
We expect that lechlemer will be significantly less expensive and would support development efforts to receive a tropical disease priority review voucher from the FDA for an indication for the symptomatic treatment of diarrhea from acute infections such as cholera. Priority review vouchers are granted by the FDA as an incentive to develop treatments for neglected diseases and rare pediatric diseases. Priority review vouchers are transferable and, in past transactions by other companies, have sold for prices ranging from $60 million to $350 million. Additionally, we believe lechlemer may provide a long-term pipeline opportunity as a second-generation anti-secretory agent for multiple gastrointestinal diseases—especially in resource-constrained countries.
Napo received advice from the Division of Anti-Infectives at the U.S. FDA in September 2021 as part of the pre-investigational New Drug Application (“Pre-IND”) discussion. Napo plans to include the advice in its plans for the Phase 1 clinical trial in healthy volunteers, currently planned the second half of 2022. The NP-300 (lechlemer) program is paired with funding from a promissory note related to the potential future sale of a possible TDPRV.
As previously announced, the Company also launched the Entheogen Therapeutics (“ETI”) initiative to support the discovery and development of novel, natural medicines derived from psychoactive and psychedelic plant compounds for treatment of mood disorders, neuro-degenerative diseases, addiction, and other mental health disorders. The initiative is initially focused on plants with the potential to treat depression and leverages Napo’s proprietary library of approximately 2,300 plants with medicinal properties. According to statistics available from the
8
National Institute of Mental Health Disorders, part of the National Institutes of Health, approximately 9.5% of American adults ages 18 and over will suffer from a depressive illness (major depression, bipolar disorder, or dysthymia) each year.
Field research collaborations have been conducted in the past by members of the scientific strategy team (“SST”) of Jaguar’s predecessor company Shaman Pharmaceuticals, who are also members of the ETI SST, yielded possible applications for a compound called alstonine. Alstonine is derived from a plant used by traditional healers in Nigeria, and has demonstrated a potential novel mechanism of action for the treatment of difficult to manage conditions such as schizophrenia.
The ETI SST consists of leading and globally renowned ethnobotanists, physicians, and pharmacologists as well as experts in the fields of natural product chemistry and neuropharmacology. We believe the wealth of expertise, experience, and commitment of the ETI SST—comprised of multiple members of the original SST that contributed to development of Jaguar's proprietary library of approximately 2,300 plants—will play an instrumental role in advancing our shared initial goal of identifying plants in our library that may have the potential to treat mood disorders and neurodegenerative diseases, such as Alzheimer's disease, Parkinson's disease, and amyotrophic sclerosis. Mood disorders and neurodegenerative diseases affect hundreds of millions of people around the globe and represent classic unmet medical needs.
While Napo and Jaguar remain steadfastly focused on the commercial success of Mytesi and Canalevia-CA1, respectively, and on the development of potential crofelemer follow-on indications in the area of gastrointestinal treatments, the Company believes the same competencies and multi-disciplinary scientific strategy that led to the development of crofelemer will support collaborative efforts with potential partners to develop novel first-in-class prescription medicines derived from psychoactive plants.
Our management team has significant experience in gastrointestinal product development for both humans and animals. Napo was founded 32 years ago to perform drug discovery and development by leveraging the knowledge of traditional healers working in rainforest areas. Ten members of the Jaguar and Napo team have been together for more than 15 years. Dr. Steven King, our chief sustainable supply, ethnobotanical research and intellectual property officer, and Lisa Conte, our founder, president and CEO, have worked together for more than 30 years. We have buttressed the early founding team with the expertise and experiences of team members like Dr. Darlene Horton and Dr. Karen Brunke to support the continued development and commercialization activities of the Napo and Jaguar family. We have assembled an impressive group of scientific advisory board (SAB) members that work closely with the Chair of Jaguar’s Scientific Advisory Board, Dr. Pravin Chaturvedi, who also serves as the Chief Scientific Officer (“CSO”) of Jaguar. Together, these dedicated personnel successfully transformed crofelemer, which is extracted from trees growing in the rainforest, to Mytesi and Canalevia-CA1, which are natural, sustainably harvested, FDA-approved drugs.
As announced in February 2020, the American Botanical Council named Napo the recipient of the 2019 Varro E. Tyler Commercial Investment in Phytomedicinal Research Award in recognition of Napo’s ongoing commitment to the sustainable development and production of natural therapeutic preparations. Specifically, this award acknowledges the successful development and approval of crofelemer, which is derived from the medicinal Croton lechleri tree in the Amazon rainforest. Previous recipients of this award include Jaguar’s partner, Italy based Indena S.p.A., one of the world’s largest producers of clinically-tested botanical extracts for the food, dietary supplement, cosmetic, and pharmaceutical markets.
Pipeline within a product—crofelemer
Crofelemer is currently being evaluated for the prophylaxis of CTD in adult patients receiving targeted therapy. As announced in October 2020, Napo has initiated its pivotal Phase 3 clinical trial of crofelemer (Mytesi) for prophylaxis of diarrhea in adult cancer patients receiving targeted therapy (OnTarget study). A significant proportion of patients undergoing cancer therapy experience diarrhea. Novel targeted cancer therapy agents, such as epidermal growth factor receptor antibodies and tyrosine kinase inhibitors, with or without cycle chemotherapy agents, may
9
activate intestinal chloride secretory pathways leading to increased chloride secretion into the gut lumen, coupled with significant loss of water that would result in secretory diarrhea.
According to data appearing in “Treatment Guidelines for CID” (chemotherapy induced diarrhea) in the April 2004 issue of Gastroenterology and Endoscopy News, diarrhea is the most common adverse event reported in chemotherapy patients. Approved third-party supportive care products for chemotherapy induced nausea and vomiting (“CINV”) include Sustol, Aloxi, Akynzeo and Sancuso. According to a report published by Allied Market Research, the global CINV market was valued at $1.66 billion in 2015 and is estimated to reach $2.66 billion by 2022.
Diarrhea has been reported as the most common side effect of the recently approved CDK 4/6 inhibitor abemaciclib and the pan HER TKI neratinib, with occurrence ranging from 86% to >95% and grade 3 over 40%, in published studies. Diarrhea in this patient population has the potential to cause dehydration, potential infections, and non-adherence to treatment. A novel antidiarrheal like crofelemer may hold promise for treating secretory diarrhea—and therefore also support long term cancer treatment adherence—in this population.
Napo has previously received orphan drug designation from the FDA for adult and/or pediatric SBS. The Orphan Drug Act provides for granting special status to a drug or biological product to treat a rare disease or condition upon request of a sponsor. Orphan drug designation qualifies the sponsor of the drug for various development incentives, including extended exclusivity, tax credits for qualified clinical testing, and relief of filing fees. As mentioned above, Napo Therapeutics has licensed the rights for the orphan and rare diseases associated with SBS and CDD with IF.
Napo Therapeutics expects that an IIT in pediatric patients with CDD will study crofelemer in the second half of 2022. This study will be initiated in the Middle East/North Africa (“MENA”) region with sites that treat infants and children with CDD and SBS with IF.
CDD is a group of rare, chronic intestinal channel diseases, with onset in early infancy, that are characterized by severe, lifelong diarrhea and a lifelong need for nutritional intake either parenterally or with a feeding tube. CDD is related to specific genetic defects inherited as autosomal recessive traits. The incidence of CDD is prevalent in regions where consanguineous marriage (related by blood) is part of the culture. CDD is directly associated with serious secondary conditions including dehydration, metabolic acidosis, and failure to thrive, prompting the need for immediate therapy to prevent death and limit lifelong disability. A recent preclinical study shows that crofelemer reduces the chloride secretion in intestinal cells derived from patients with CDD and these preclinical results provide additional support and rationale for the use of crofelemer in treating patients with CDD and/or SBS with IF.
As previously announced (in 2019), a clinical research study sponsored by The University of Texas Health Science Center at Houston (“UTHealth”) is being supported by Napo. This study evaluates the safety and effectiveness of crofelemer for treatment of chronic idiopathic diarrhea in patients. Chronic idiopathic diarrhea is a common complaint of patients presenting to family practitioners and internists and is one of the most common reasons for referral to gastroenterologists. It is estimated that the prevalence of chronic idiopathic diarrhea in developed countries (including the U.S.) is approximately 3-5%. It has a significant negative effect on health-related quality of life and causes a high economic burden on patients and society. The American Gastroenterological Association Burden of Illness study (2012) showed that the estimated annual direct and indirect costs associated with chronic idiopathic diarrhea is up to $524 million per year and $136 million per year, respectively. The principal investigator for this study is Dr. Brooks D. Cash, MD, AGAF, FACG, FACP, FASGE, Chief – Division of Gastroenterology, Hepatology and Nutrition, Sterling Professor of Medicine, McGovern Medical School at UTHealth, Co-Director, Ertan Digestive Disease Center at Memorial Hermann-Texas Medical Center. The Study is titled Yield of Diagnostic Tests and Management of Crofelemer for Chronic Idiopathic Diarrhea in Non-HIV Patients.
10
Crofelemer is also being evaluated in another investigator-initiated trial for the management of functional diarrhea in non-HIV patients. The principal investigator for this clinical study is Dr. Anthony Lembo, Professor, Gastroenterology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA. This clinical study is a randomized double-blind, placebo-controlled study in adult subjects with functional diarrhea. Eligible patients will have functional diarrhea defined by Rome IV criteria as >25% loose watery stools and <25% hard/lumpy stools. The study plans to randomize 80 patients and the subjects will be randomized 1:1 for 4 weeks to either the placebo or crofelemer 125 mg delayed-release tablets (Mytesi) arm, administered twice daily for 4 weeks. Following the four-week placebo-controlled period, all subjects will receive Mytesi for an additional four weeks in an open label extension phase. The safety and tolerability of crofelemer and the clinical response during the placebo-controlled period will be evaluated in this study. Subjects will be allowed to use limited amounts of an antimotility drug (loperamide) during the placebo-controlled and open-label extension phase to manage uncontrolled diarrhea. However, no more than 11 doses of 2 mg loperamide will be permitted during any given week per subject.
Jaguar’s and Napo’s portfolio development strategy involves meeting with Key Opinion Leaders (“KOLs”) to identify indications that are potentially high value because they address important medical needs that are significantly or globally unmet, obtain input on protocol practicality and protocol generation, and then strategically sequencing indication development priorities, second generation product pipeline development, and partnering goals on a global basis.
Mytesi is the only antidiarrheal drug that has been approved by the US FDA for the treatment of chronic, noninfectious diarrhea in adult HIV/AIDS patients receiving ART. This approval was on the basis of the drug’s safety and efficacy in reducing the number of weekly and daily watery stools in patients and improvement of stool consistency, from unformed to formed stools, over a 24-week treatment period.
Unlike other available diarrhea treatments, crofelemer does not act by inhibiting intestinal motility. It has minimal oral absorption and does not have any clinically significant food or drug interactions, thereby allowing patients to maintain their appropriate dosing of treatment to suppress their viral load and maintain adequate CD4 levels in PLWHA. Crofelemer is also the only approved antidiarrheal drug that is approved for chronic use. Moreover, it is not an opioid, like other traditionally used treatments, thus avoiding both the acute side effect of constipation and the potential for abuse.
Napo’s Scientific Advisory Board has focused primarily on physician education, and community and global awareness regarding the importance and availability of solutions for neglected comorbidities, such as the first in class anti secretory mechanism of action of Mytesi for its currently approved indication.
There are significant barriers to entry for Mytesi (crofelemer). Through Napo, we hold an extensive global patent portfolio. At the present time, we hold approximately 143 issued worldwide patents, with coverage in many cases that extends until 2031. These issued patents cover multiple indications, including HIV AIDS diarrhea, irritable bowel syndrome (“IBS”), IBD, manufacturing, enteric protection from gastric juices, among others. We also have approximately 43 pending patent applications worldwide in the human health areas that are being prosecuted.
Mytesi is the first oral drug approved under the FDA’s Botanical Guidance, which provides another barrier to entry from potential generic competition. The FDA requires that the manufacturer of crofelemer use a validated proprietary bioassay to release the drug substance and drug product of Mytesi. While most generic products are fashioned to meet chemical release specifications that are in the public domain, the specifics of this assay are not publicly available. There is no pathway by which a generic product can be developed for a drug approved under botanical guidance. In addition, Mytesi is minimally absorbed systemically, so the classic approach of creating a generic drug by matching pharmacokinetic blood levels is not possible. A generic player would have to conduct costly and risky clinical trials.
While Jaguar’s commercial and development efforts have evolved to focus primarily on Mytesi and human pipeline indications since its merger with Napo, the Company has commenced launch initiatives related to Canalevia-CA1, our drug product which received conditional approval in December 2021 for treatment of CID in dogs. CID in dogs is typically caused by the same mechanism of action as in humans, and hence the work in dogs serves as a
11
preclinical proof of concept for the diarrhea in humans that is related to targeted cancer therapy. CID is an interesting model for human medical need and is being pursued as a prescription indication for animal health. We believe there is an important unmet medical need for the treatment of CID in dogs. Certain cancer treatment agents provided to dogs are human drugs or have the same mechanism of action as human cancer drugs, and these agents and mechanisms of action often have meaningful rates of diarrhea in humans as well.
As previously announced, Jaguar has received MUMS designation status from the FDA for Canalevia-CA1 for the indication of CID in dogs. MUMS designation is modeled on the orphan drug designation for human drug development and offers possible financial incentives to encourage MUMS drug development, such as the availability of grants to help with the cost of developing the MUMS drug.
For Jaguar Animal Health’s second proposed indication for Canalevia, exercise induced diarrhea (“EID”) in dogs, the Company is leveraging the use of many of the same major technical sections that have been submitted in support of the Company's application for Canalevia-CA1 for the indication of CID in dogs. Conditional approval of Canalevia for EID, under the name Canalevia-CA2, is expected in the fourth quarter of 2022.
Crofelemer is extracted and purified from the Croton lechleri tree, which we sustainably harvest and manage through programs that we have been developing over the past 30 years. This process has involved working with local and indigenous communities to plant trees, obtaining permits for export, and creating a supply network that is robust and reliable.
Our team continues to have relationships with partners that we began working with in the 1990s. Additionally, through the establishment of a nonprofit called the Healing Forest Conservancy, our team has created a long term mechanism for benefit sharing that recognizes the intellectual contribution of Indigenous populations. This program is intended to contribute to the continued strength and effectiveness of the valued and strategically important relationships we have carefully cultivated over the past 30+ years.
Product Pipeline
In addition to our Mytesi (crofelemer) product that is approved by the U.S. FDA for the symptomatic relief of noninfectious diarrhea in adults with HIV/AIDS on antiretroviral therapy, we are also developing a pipeline of prescription drug product candidates to address unmet needs in gastrointestinal health through Napo. Mytesi (crofelemer) is a novel, first in class anti secretory agent which has a basic normalizing effect locally in the gut and lumen, and this mechanism of action has the potential to benefit multiple disorders. Clinical trials demonstrated that nearly 80% of Mytesi users experienced an improvement in their diarrhea over a four week period. At week 20 of the pivotal trial, over half the patients had no watery stools, or a 100% decrease, and 83% had at least a 50% decrease in watery stools. Our Mytesi pipeline currently includes prescription drug product candidates for four follow on indications, several of which are backed by Phase 2 evidence from completed Phase 2 trials. In addition, a second generation proprietary anti secretory agent, lechlemer, is in development for cholera-related diarrhea.
12
Napo Prescription Drug Product Candidates
Product Candidates | Indication | Completed Milestones | Current Phase of | Anticipated Near-Term | |
Mytesi | CTD | • Initiated pivotal Phase 3 clinical trial in October 2020 • Findings of Phase 2 HALT-D study presented in Q4 2021 | Phase 3 | • Enrollment in Phase 3 trial ongoing | |
Novel lyophilized crofelemer product | Rare disease indications (SBS & CDD) | • Orphan drug designation for SBS granted by FDA and EMA | Phase 2 | • Initiate Phase 2 proof of concept (POC) study in SBS and/or CDD in 2022 | |
Mytesi | IBS-D | • Two Phase 2 studies completed | Phase 2 | • Potential business development opportunities | |
Mytesi | Chronic idiopathic diarrhea | • Initiated clinical study at The University of Texas Health Science Center at Houston (“UTH”) | Phase 2 | • Top line results of UTH trial expected in 2022 | |
Mytesi | Functional diarrhea | • Initiated clinical study at Beth Israel Deaconess Medical Center, Harvard Medical School, Boston | Phase 2 | • Enrollment ongoing | |
Mytesi | IBD | • Safety evidence from other chronic diarrhea indications | Phase 2 | • KOL discussions ongoing | |
NP-300 (lechlemer) | Second-generation antidiarrheal drug for infectious diarrhea including from cholera | • Ongoing IND-enabling toxicology studies and formulation development | Pre IND | • Initiate Phase 1 trial in the second half of 2022 | |
*Clinical trials are funding dependent
Estimated Size of Mytesi Target Markets
We believe the medical need for Mytesi is significant, compelling, and unmet, and that doctors are looking for a drug product with a mechanism of action that is distinct from the options currently available to resolve diarrhea. A growing percentage of HIV patients have lived with the virus in their gut for 10+ years, often causing gut enteropathy and chronic or chronic episodic diarrhea. According to data from the U.S. Centers for Disease Control and Prevention, by 2020 more than 70% of Americans with HIV are expected to be 50 and older and have lived with HIV for more than 10 years (1).
13
Market | Competition | Market Size/Potential | |
HIV-related Diarrhea | None | We estimate the U.S. market revenue potential for Mytesi to be approximately $50 million in gross annual sales | |
CTD | None | An estimated 650,000 U.S. cancer patients receive chemotherapy in an outpatient oncology clinic(2). Comparable supportive care (i.e., CINV) product sales of ~$620 million in 2013(3). Global CINV market projected to reach a valuation of $2.7 billion by 2022(4) | |
SBS/CDD | 1 | Financial benefits of Orphan Drug Designation. The global SBS market exceeded $568 million in 2019 and is expected to reach $4.6 billion by 2027, according to a report by Vision Research Reports(5) | |
IBS-D | 3 | Most IBS products have an estimated revenue potential of greater than $1.0 billion(6) | |
IBD | None | Estimated 1,171,000 Americans have IBD(7) | |
Infectious Diarrhea from Cholera | None | In recent transactions by other companies, priority review vouchers have sold for $67 million to $350 million(8) | |
(1) | HIV Among People Aged 50 and Older (https://www.cdc.gov/hiv/group/age/olderamericans/index.html) |
(2) | Centers for Disease Control and Prevention. Preventing Infections in Cancer Patients: Information for Health Care Providers (cdc.gov/cancer/prevent infections/providers.htm) |
(3) | Heron Therapeutics, Inc. Form 10 K for the fiscal year ended December 31, 2016 |
(4) | Report published by Allied Market Research, titled, "Chemotherapy-induced Nausea and Vomiting (CINV) Market-Global Opportunity Analysis and Industry Forecast, 2014-2022” (https://www.prnewswire.com/news-releases/chemotherapy-induced-nausea-and-vomiting-cinv-market-expected-to-reach-2659-million-by-2022-611755395.html) |
(5) | Short Bowel Syndrome Market – Global Industry Analysis, Size, Share, Trends, Revenue, Forecast 2020 to 2027 (https://www.mynewsdesk.com/us/medical-technology-news/pressreleases/short-bowel-syndrome-market-global-industry-analysis-size-share-trends-revenue-forecast-2020-to-2027-3069433) |
(6) | Merrill Lynch forecasts peak US sales of roughly $1.5 bn for Ironwood’s Linzess (https://247wallst.com/healthcare-business/2015/04/27/key-analyst-sees-nearly-30-upside-in-ironwood/); Rodman & Renshaw estimate peak annual sales of Synergy Pharmaceuticals’ Trulance at $2.3 bn in 2021 (https://www.benzinga.com/analyst-ratings/analyst-color/17/04/9304883/what-synergys-new-patents-mean-for-its-commercial-prospe) |
(7) | Kappelman, M. et al. Recent Trends in the Prevalence of Crohn’s Disease and Ulcerative Colitis in a Commercially Insured US Population. Dig Dis Sci. 2013 Feb; 58(2): 519 525 |
(8) | In Aug. 2015, AbbVie Inc. bought a priority review voucher from United Therapeutics Corp for $350 million (https://www.wsj.com/articles/united-therapeutics-sells-priority-review-voucher-to-abbvie-for-350-million-1439981104 ). In July 2014, BioMarin announced that it had sold a priority review voucher to Sanofi and Regeneron for $67.5 million. (https://investors.biomarin.com/2014-07-30-BioMarin-Sells-Priority-Review-Voucher-for-67-5-Million). |
14
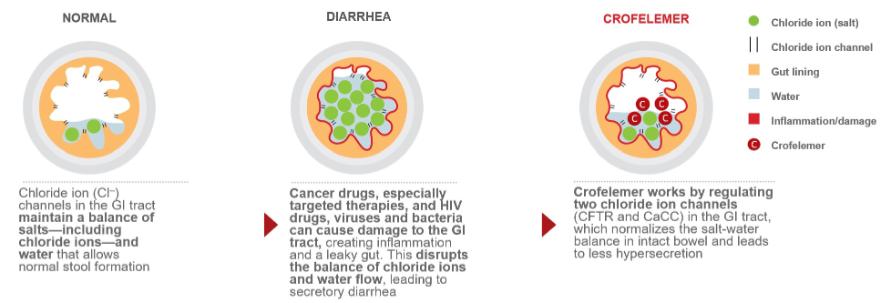
The following diagram illustrates the mechanism of action of our human and animal gastrointestinal drug products and drug product candidates, which normalize chloride and water flow and transit time of fluids within the intestinal lumen.
Business Strategy
Our goal is to become a leading pharmaceutical company with first in class, sustainably derived products that address significant unmet gastrointestinal medical needs globally. To accomplish this goal, we plan to:
Expand Mytesi by leveraging our significant gastrointestinal product knowledge, experience and intellectual property portfolio
Mytesi is a novel, first in class anti secretory antidiarrheal agent which has a basic normalizing effect locally on the gut, and this mechanism of action has the potential to benefit multiple gastrointestinal disorders. Our Mytesi (crofelemer) product is approved by the U.S. FDA for the symptomatic relief of noninfectious diarrhea in adults with HIV/AIDS on antiretroviral therapy. Jaguar, through Napo, holds global unencumbered rights for Mytesi. Mytesi is in development for multiple possible follow on indications, including diarrhea related to targeted cancer therapy; orphan drug indications for infants and children with congenital diarrheal disorders and short bowel syndrome; inflammatory bowel disease; irritable bowel syndrome; and for idiopathic/functional diarrhea. In addition, a second generation proprietary anti secretory agent is in development for cholera.
Our management team collectively has extensive experience in the development of prescription drugs. This experience covers all aspects of product development, including discovery, preclinical and clinical development, GMP manufacturing, regulatory affairs, and commercialization. Key members of this team successfully developed Mytesi.
Maintain commercial capabilities in Mytesi sales and marketing efforts
Napo’s direct sales organization is comprised of Mytesi field sales representatives strategically positioned to cover U.S. geographies with the highest potential. With support provided by concomitant marketing, promotional activities, patient empowerment programs, including an integrated social digital campaign, and medical education initiatives described below, we expect a proportional response in the number of patients treated with Mytesi.
Leverage our relationships with Scientific Advisory Board (SAB) members for crofelemer commercialization and development in follow on indications
The Company has retained 10 SAB members that have extensive clinical experience in HIV, CTD, IBD, SBS, and CDD. In addition, the Company engages key opinion leaders (KOLs) for specific turnkey needs.
15
Establish partnerships to support moving pipeline indications to pivotal clinical trials
Jaguar is actively pursuing the development of a robust pipeline of potential follow on indications for crofelemer, and the Company’s goal is to establish partnerships to support moving pipeline indications to pivotal clinical trials.
Strategically sequence the development of follow on indications of Mytesi and seek geographically focused licensing opportunities
As announced September 24, 2018, Jaguar and Knight Therapeutics Inc. (“Knight”) entered into a Distribution, License and Supply Agreement that grants Knight the exclusive right to commercialize Mytesi and related products in Canada and Israel. The License Agreement has a term of 15 years (with automatic renewals) and provides Knight with an exclusive right to commercialize current and future Jaguar human health products (including crofelemer, Lechlemer, and any product containing a proanthocyanidin or with an anti-secretory mechanism) in Canada and Israel. Knight forfeited its right of first negotiation for expansion to Latin America. Under the License Agreement, Knight is responsible for applying for and obtaining necessary regulatory approvals in the territory of Canada and Israel, as well as marketing, sales and distribution of the licensed products. Knight will pay a transfer price for all licensed products, and upon achievement of certain regulatory and sales milestones, the Company may receive payments from Knight in an aggregate amount of up to approximately $18 million payable throughout the initial 15-year term of the agreement. The Company did not have any license revenues since the execution of this agreement.
Although it is possible that we may enter into additional corporate partnering relationships related to Mytesi, our intention would be to retain all or co commercialization and promotional rights in the U.S., so that we do not become primarily a royalty collecting organization, and we are opposed to entering into any Mytesi partnering relationship that would require splitting indications. We are seeking to put limited geographically focused partnerships in place in the near term, while also considering possibilities for a worldwide partnership with a leading global entity (excluding the U.S. exclusive commercial rights) in the field of gastrointestinal care and cancer in the long term.
Reduce risks relating to product development
Risk reduction is a key focus of our product development planning. Mytesi is approved for chronic indication, providing us the ability to leverage this corresponding safety data when seeking approval for planned follow on indications that are also chronic or chronic episodic indications. In an effort to reduce risk further, we have implemented the following approach: first, we meet with key opinion leaders, typically at medical conferences. Next, we confirm unmet medical needs with these key opinion leaders and discuss the practicality of patient enrollment and trial implementation. We then generate protocols to discuss with the FDA, seeking, when possible, special protocol assessments. Our goal is to have de risked the program as much as we believe we possibly can, by the time we start devoting significant funds to a clinical trial, in particular the regulatory pathway. We believe this approach will lead to better long-term outcomes for our products in development.
We will continue to seek partnerships outside the United States for the above indications while focusing on development and commercial access in the United States directly. We are also focused on investigating (lechlemer) for various gastrointestinal indications. Lechlemer is a proprietary Jaguar pharmaceutical product, a standardized botanical extract distinct from crofelemer, also sustainably derived from the Croton lechleri tree.
We believe lechlemer, which has a similar mechanism of action as crofelemer and is significantly less costly to produce, may support efforts to receive a priority review voucher from the U.S. FDA for symptomatic treatment of diarrhea from cholera infection. Priority review vouchers are granted by the FDA to drug developers for tropical disease indications (TDPRV) as an incentive to develop treatments for neglected diseases and rare pediatric diseases. Additionally, we believe lechlemer represents a long-term pipeline opportunity as a second generation anti secretory agent, on a global basis, for multiple gastrointestinal diseases—especially in resource constrained countries where the cost of goods is a factor, in part, because requirements often exist in such regions for drug prices to decrease annually.
16
The Company has previously presented Phase 2 data on crofelemer for the treatment of diarrhea in cholera patients from a study in Bangladesh. Napo plans to follow a similar clinical study design to support the development of lechlemer (NP-300) for a cholera related indication.
Napo is conducting IND-enabling preclinical toxicology studies and developing novel NP-300 formulations that will support the initiation of clinical studies in 2022.
Our portfolio development strategy is based on identifying indications that are potentially high value because they address important medical needs that are significantly or globally unmet, and then strategically sequencing indication development priorities, second generation product pipeline development, and partnering goals on a global basis.
Our technology for proprietary gastrointestinal disease products is central to the product pipelines of both human and veterinary indications. Crofelemer is also the API in Canalevia-CA1, our prescription drug product recently conditionally approved by the FDA and launched for CID in dogs, and also expected to be approved and launched, under the name Canalevia-CA2, for EID in dogs in the fourth quarter of 2022.
Napo Therapeutics Provides New Opportunities to Treat Orphan Indications Like Short Bowel Syndrome
Jaguar is strategically pursuing multiple important shots on goal for its drug development pipeline: Crofelemer for CTD, led by Napo, and crofelemer for SBS and CDD, led by Napo Therapeutics. Jaguar’s exclusive license agreement with Napo Therapeutics provides a perpetual, royalty-bearing license for Europe (excluding Russia), and includes traditional terms such as up-front fees, milestone payments, royalties on sales in Europe, and a supply agreement, and rights to utilized all data Napo Therapeutics generates for Jaguar development and approval activities globally.
Competition
There are several significantly larger pharmaceutical companies competing with us in the gastrointestinal segment.
Diarrhea in adult patients living with HIV/AIDs. We are not aware of any other FDA approved drugs for the symptomatic relief of diarrhea in HIV/AIDs patients. HIV/AIDs diarrhea patients may also use loperamide or Lomotil but these medications affect motility which can result in rebound diarrhea and are not indicated for chronic use. Other agents’ patients may use include over the counter anti diarrheal remedies such as Mylanta or Kaopectate.
Cancer therapy related diarrhea. We are not aware of any FDA approved drugs specifically indicated for cancer therapy related diarrhea, including chemotherapy related diarrhea. A recent Phase 2b trial of elsiglutide for the treatment of chemotherapy induced diarrhea in colorectal cancer patients did not meet statistical significance. Opioids and over the counter drugs are commonly used to treat chemotherapy induced diarrhea, but these drugs affect motility. Certain tyrosine kinase inhibitor chemotherapy agents have diarrhea as a significant side effect. For example, FDA guidance suggests diarrhea prophylaxis prior to initiating adjuvant therapy with neratinib.
Short Bowel Syndrome and Congenital Diarrheal Disorders. We are not aware of any FDA approved drugs specifically indicated for congenital diarrheal disorders. In the U.S., Takeda Pharmaceuticals’ GATTEX® (teduglutide) is indicated for the treatment of adults and pediatric patients 1 year of age and older with short bowel syndrome who are dependent on parenteral support, and Zorbtive® is a recombinant human growth hormone indicated for the treatment of short bowel syndrome in adult patients receiving specialized nutritional support.
Diarrhea predominant irritable bowel syndrome. Two drugs were approved in 2015 for the treatment of diarrhea predominant irritable bowel syndrome, Allergan plc’s Virbezi and Xifaxan, which is marketed by Valeant Pharmaceuticals International. Also, Lotronex was approved by the FDA in 2000 but was withdrawn from the market and later reintroduced in 2002 under a Risk Management Program. With the exception of Lotronex, the sponsors of
17
Verbezi and Xifaxan employ extensive media and print promotion for the commercialization of these products. We are seeking a partner to further the clinical development and commercialization of crofelemer for IBS-D. There are currently numerous trials ongoing for IBS-D.
Inflammatory Bowel Disorders. We are not aware of any FDA approved drugs specifically indicated as an anti-secretory agent for use to address IBD.
Infectious Diarrhea from Cholera. We are not aware of any FDA approved drugs specifically indicated as an anti-secretory agent for use to address the devastating dehydration in cholera patients.
Manufacturing
The plant material used to manufacture is crude plant latex (“CPL”) extracted and purified from Croton lechleri, a widespread and naturally regenerating tree in the rainforest that is managed as part of sustainable harvesting programs. The tree is found in several South American countries and has been the focus of long term sustainable harvesting research and development work. Napo’s collaborating suppliers obtain CPL and arrange for the shipment of CPL to Napo’s third party contract manufacturer.
Napo’s third party contract manufacturer, India based Glenmark Life Sciences Ltd. (“Glenmark”), a research driven, global, integrated pharmaceutical company, is Napo’s manufacturer of crofelemer, the active pharmaceutical ingredient in Mytesi. Glenmark processes CPL into crofelemer utilizing a proprietary manufacturing process. The processing occurs at an FDA approved Glenmark facility. Additionally, Napo is establishing a second processing site, which will be operated by Indena S.p.A. (“Indena”), a Milan, Italy based contract manufacturer dedicated to the identification, development and production of high quality active principles derived from plants, for use in the pharmaceutical, health food and personal care industries. Indena has completed the required technology transfer and feasibility and preparing for validation activities to support commercial scale manufacturing.
We have contracts in place with all the manufacturers and third-party testing labs required to manufacture Mytesi, Canalevia-CA1, and lechlemer. We are finalizing a master service agreement with Indena for the manufacture of crofelemer. We are evaluating alternate drug substance and drug product manufacturers to establish redundancy for DP manufacturing.
Proprietary Library of Medicinal Plants
We possess a proprietary library of more than 2,300 medicinal plants.
Intellectual Property
Trademarks
We plan to market all of our products under a trademark or trademarks we select, and we will own all rights, title and interest, including all goodwill, associated with such trademarks. Mytesi is a registered trademark owned by Napo. Jaguar Animal Health is a trademark owned by Jaguar.
License Agreements
Patent Portfolio
Napo
Napo owns a portfolio of patents and patent applications covering formulations of and methods of treatment with proanthocyanidin polymers isolated from Croton spp. or Calophyllum spp., including Mytesi (crofelemer). The patent family associated with International Patent publication WO1998/16111 relates to enteric protected formulations
18
of proanthocyanidin polymers isolated from Croton spp. or Calophyllum spp., including crofelemer, and methods of treating watery diarrhea using these enteric protected formulations. There is one U.S. patent in force in this family, US 7,341,744, which has a term until at least June 23, 2019, which term has been extended under 35 U.S.C. 156 by 1,075 days. Based upon the June 23, 2019 expiration date, the expiration date for crofelemer is June 2, 2022, to account for the regulatory delay in obtaining human marketing approval for crofelemer.
Napo additionally owns a family of patents arising from International Patent Application Publication WO2012/058664 that cover methods of treating HIV associated diarrhea and HAART associated diarrhea with proanthocyanidin polymers isolated from Croton spp. or Calophyllum spp., including crofelemer. In the U.S., there are two issued patents, US 8,962,680 and US 9,585,868, both of which expire October 31, 2031. Outside the U.S., patent protection for methods of treating HIV associated diarrhea has been obtained in Australia, Europe, Hong Kong, Japan, Kenya, Mexico, Russia, Ukraine, South Africa, and Zimbabwe, with expiration dates of October 31, 2031, and applications are pending in Brazil, Hong Kong, and China. Napo also has patent families related to methods of treating diarrhea predominant irritable bowel syndrome, methods of treating constipation predominant irritable bowel syndrome, and methods of treating inflammatory bowel disease, familial adenomatous polyposis and colon cancer, with proanthocyanidin polymers isolated from Croton spp. or Calophyllum spp., including crofelemer. In particular, for diarrhea predominant irritable bowel syndrome, Napo has two issued U.S. patents, US 8,846,113 and US 9,980,938, which expire on February 9, 2027, as well as issued patents in Australia, Canada, Europe, Gulf States, Hong Kong, Japan, South Korea, Mexico, New Zealand, Singapore, and Taiwan and pending applications in Bangladesh, Bolivia, Chile, Paraguay, Thailand, and Venezuela, all of which are estimated to expire April 30, 2027; for constipation predominant irritable bowel syndrome, Napo has three issued U.S. patents, with terms to at least April 30, 2027, patents in Australia, Canada, Europe, Hong Kong, Mexico, New Zealand, and Singapore, all of which are estimated to expire April 30, 2027; and for inflammatory bowel disease, familial adenomatous polyposis and/or colon cancer, Napo has two issued U.S. patents, US 8,852,649 and US 9,987,250 with terms until at least January 4, 2028, as well as issued patents in Australia, Hong Kong, and Europe and Canada, which have estimated expiration dates of April 30, 2027. Napo has a pending U.S. non provisional application for the treatment of CID with crofelemer filed on March 9, 2018, as well as International and Taiwanese applications, and two International Patent Applications on other human indications including for the treatments of short bowel syndrome and congenital diarrhea disorder filed on May 31, 2018, with pending national phase applications in the United States, Australia, Canada, China, Europe, Israel, Jordan, Japan and the Gulf States.
For methods of manufacturing proanthocyanidin polymers isolated from Croton spp. or Calophyllum spp., including crofelemer, Napo owns issued patents in India, South Africa, and Eurasia with terms at least until August 26, 2029. Napo also owns issued patents in Brazil, India, Russia, and South Africa and pending applications in Argentina and Venezuela that also cover methods of manufacturing proanthocyanidin polymers isolated from Croton spp. or Calophyllum spp., including crofelemer, with terms at least until January 17, 2032. Lastly, Napo owns two U.S. patents covering a formulation of NP 500 (nordihydroguiaretic acid (“NDGA”)) and its use in treating a metabolic disorder that have terms until April 23, 2031.
Napo grants license to Napo Therapeutics S.p.A. (formerly named Napo EU S.p.A.)
On August 2021, Napo signed a license agreement with Napo EU S.p.A. to study, develop, manufacture, and commercialize Napo’s plant-based crofelemer and lechlemer drug product candidates in the European Union (excluding Russia) and in specified non-EU countries in Europe for specific indications, which rights and obligations were assumed by the combined company formed by the merger of Napo Therapeutics with Dragon SPAC (the combined company uses the Napo Therapeutics name). The license agreement grants Napo Thera the rights for SBS-IF, HIV-related diarrhea, and the symptomatic relief and treatment of IF-related diarrhea in patients with congenital disorders. The license agreement grants Napo Thera the right to study Per the terms of the license agreement, Napo will receive payment of up to $10 million as the initial license fee (to be paid in two installments, the first of which has already been received) as the initial license fee and is eligible to receive additional payments related to milestones, royalties, and product transfers. The license fees will be eliminated in the consolidated financial statements.
19
Government Regulation
The FDA and comparable regulatory authorities in state and local jurisdictions and in other countries impose substantial and burdensome requirements upon companies involved in the clinical development, manufacture, marketing and distribution of prescription drugs such as those Napo is that Jaguar and its subsidiaries are commercializing and/or developing. These agencies and other federal, state and local entities regulate, among other things, the research and development, testing, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping, approval, advertising and promotion, distribution, post approval monitoring and reporting, sampling and export and import of Napo’s drug product candidates. To comply with the regulatory requirements in each of the jurisdictions in which Napo is seeking to market and subsequently sell its prescription products, Napo has established processes and resources to provide oversight of the development, approval processes and launch of its products and to position those products in order to gain market share
U.S. Government Regulation
In the United States, the FDA approves and regulates drugs under the Federal Food, Drug, and Cosmetic Act (“FDCA”), and it’s implementing regulations.
The process of obtaining regulatory approvals and the subsequent compliance with applicable federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to a variety of administrative or judicial sanctions, such as the FDA’s refusal to approve pending New Drug Applications (“NDAs”), withdrawal of an approval, imposition of a clinical hold, issuance of warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties.
The process required by the FDA before a human or animal health prescription drug may be marketed in the United States generally involves the following:
| ● | completion of pre-clinical laboratory tests, animal studies and formulation studies in compliance with the FDA’s good laboratory practice, or good laboratory practices (“GLPs”) regulations; |
| ● | submission to the FDA of an investigational new drug application (“IND”) for human clinical trials, or an investigational new animal drug (“INAD”) for animal health studies; |
| ● | approval by an institutional review board (“IRB”) for human trials, and appropriate animal care and use committees for animal health studies. Multiple sites may necessitate the involvement of multiple IRBs and submissions for human health products; |
| ● | performance of adequate and well controlled human clinical trials in accordance with good clinical practices (“GCPs”), requirements to establish the safety and efficacy of the proposed drug product for each indication; |
| ● | submission to the FDA of an NDA for marketing approval of human prescription drugs; and a new animal drug application (“NADA”) for marketing authorization of animal health products; |
| ● | satisfactory completion of FDA advisory committees review, if applicable; |
| ● | satisfactory completion of an FDA pre-approval inspection (“PAI”) of the manufacturing facility or facilities at which the product is produced to assess compliance with current good manufacturing practices (“cGMPs”), requirements and to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; and |
20
| ● | FDA review and approval of the NDA or NADA. |
Pre-clinical Studies
Pre-clinical studies include laboratory evaluation of the drug product’s chemistry, toxicity and formulation, as well as animal studies to assess potential safety and effectiveness. An IND sponsor must submit the results of the pre-clinical tests, together with manufacturing information, analytical data and any available clinical data or literature, among other things, to the FDA as part of an IND. Some pre-clinical testing may continue even after the IND is submitted. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to one or more proposed clinical trials and places the clinical trial on a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND may not result in the FDA allowing clinical trials to commence.
Clinical Trials for Human Prescription Drugs
Clinical trials involve the administration of the investigational new drug to human subjects pursuant to a clinical protocol, under the supervision of qualified investigators in accordance with GCPs requirements, which include the requirement that all research subjects provide their informed consent in writing for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives or endpoints of the trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA under the IND. In addition, an IRB at each institution participating in the clinical trial must review and approve the plan for any clinical trial before it commences at that institution. Information about certain clinical trials must be submitted within specific timeframes to the National Institutes of Health, or NIH, for public dissemination on their www.clinicaltrials.gov website.
Human clinical trials are typically conducted in three sequential phases, which may overlap or be combined:
• | Phase 1: The drug is initially introduced into healthy human subjects or patients with the target disease or condition and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an early indication of its effectiveness depending on the endpoints set forth in the protocol. |
• | Phase 2: The drug is administered to a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. |
• | Phase 3: The drug is administered to an expanded patient population, generally at geographically dispersed clinical trial sites, in well-controlled clinical trials to generate enough data to statistically evaluate the efficacy and safety of the product for approval, to establish the overall risk-benefit profile of the product, and to provide adequate information for the labeling of the product. |
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if serious adverse events occur. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, or at all. Furthermore, the FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients.
Special Protocol Assessment for Human Health Prescription Drugs
The special protocol assessment, or SPA, process is designed to facilitate the FDA’s review and approval of drugs by allowing the FDA to evaluate issues related to the adequacy of certain clinical trials, including Phase 3
21
clinical trials that can form the primary basis for a drug product’s efficacy claim in an NDA. Upon specific request by a clinical trial sponsor, the FDA will evaluate the protocol and respond to a sponsor’s questions regarding, among other things, primary efficacy endpoints, trial conduct and data analysis, within 45 days of receipt of the request.
The FDA ultimately assesses whether the protocol design and planned analysis of the trial are acceptable to support regulatory approval of the product candidate with respect to effectiveness of the indication studied. All agreements and disagreements between the FDA and the sponsor regarding a SPA must be clearly documented in a SPA letter or the minutes of a meeting between the sponsor and the FDA.
Even if the FDA agrees to the design, execution and analyses proposed in protocols reviewed under the SPA process, the FDA may revoke or alter its agreement under the following circumstances:
• | public health concerns emerge that were unrecognized at the time of the protocol assessment; |
• | the director of the review division determines that a substantial scientific issue essential to determining safety or efficacy has been identified after testing has begun; |
• | a sponsor fails to follow a protocol that was agreed upon with the FDA; or |
• | the relevant data, assumptions, or information provided by the sponsor in a request for SPA change, are found to be false statements or misstatements, or are found to omit relevant facts. |
A documented SPA may be modified, and such modification will be deemed binding on the FDA review division, except under the circumstances described above, if FDA and the sponsor agree in writing to modify the protocol and such modification is intended to improve the study.
Marketing Approval for Human Prescription Drugs
Assuming successful completion of the required clinical testing, the results of the pre-clinical studies and clinical trials, together with detailed information relating to the product’s chemistry, manufacture, controls and proposed labeling, among other things, are submitted to the FDA as part of an NDA requesting approval to market the product for one or more indications. In most cases, the submission of an NDA is subject to a substantial application user fee. Under the Prescription Drug User Fee Act (“PDUFA”), guidelines that are currently in effect, the FDA has a goal of ten months from the date of “filing” of a standard NDA for a new molecular entity to review and act on the submission. This review typically takes twelve months from the date the NDA is submitted to the FDA because the FDA has approximately two months to make a “filing” decision.
In addition, under the Pediatric Research Equity Act of 2003, as amended and reauthorized, certain NDAs or supplements to an NDA must contain data that are adequate to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations; this would include information which supports dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults or full or partial waivers from the pediatric data requirements.
The FDA may also require submission of a risk evaluation and mitigation strategy, or REMS, plan to ensure that the benefits of the drug outweigh its risks. The REMS plan could include medication guides, physician communication plans, assessment plans, and/or elements to assure safe use, such as restricted distribution methods, patient registries, or other risk minimization tools.
The FDA may also require other information as part of the NDA filing, such as an environmental impact statement. The FDA can waive some or delay compliance with some of these requirements.
The FDA conducts a preliminary review of all NDAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the application must be
22
resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews an NDA to determine, among other things, whether the drug is safe and effective and whether the facility in which it is manufactured, processed, packaged or held meets standards designed to assure the product’s continued safety, quality and purity.
The FDA may refer an application for a novel drug to an advisory committee. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMPs requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA may inspect one or more clinical trial sites to assure compliance with GCPs requirements.
After evaluating the NDA and all related information, including the advisory committee recommendation, if any, and inspection reports regarding the manufacturing facilities and clinical trial sites, the FDA may issue an approval letter, or, in some cases, a complete response letter. A complete response letter must contain a statement of specific items that prevent the FDA from approving the application and will also contain conditions that must be met in order to secure final approval of the NDA and may require additional clinical or pre-clinical testing in order for FDA to reconsider the application. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If and when those conditions have been met to the FDA’s satisfaction, the FDA will typically issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications.
Even if the FDA approves a product, it may limit the approved indications for use the of product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies, Phase 4 clinical trials, be conducted to further assess a drug’s safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution and use restrictions or other risk management mechanisms under a REMS, which can materially affect the potential market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-marketing studies or surveillance programs. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes, and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Post-Approval Requirements for Human Prescription Drugs
Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims are subject to prior FDA review and approval. New secondary indications must be supported by clinical trials or data. There also are continuing, annual user fee requirements for any marketed products and the establishments at which such products are manufactured, as well as new application fees for supplemental applications with clinical data.
The FDA may impose a number of post-approval requirements as a condition of approval of an NDA. For example, the FDA may require post-marketing testing, including Phase 4 clinical trials, and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization.
23
In addition, drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies, and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with cGMPs requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMPs requirements and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMPs compliance.
Once approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in mandatory revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, include, but are not limited to:
| ● | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
| ● | fines, warning letters or holds on post-approval clinical trials; |
| ● | refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product approvals; |
| ● | product seizure or detention, or refusal to permit the import or export of products; or |
| ● | injunctions or the imposition of civil or criminal penalties. |
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability. In addition, the FDA regulates the purity and or consistency of the approved product. Products, if deemed adulterated, can lead to serious consequences as set forth above as well as civil and criminal penalties.
Animal Health Prescription Drugs
Under the Federal Food, Drug, and Cosmetic Act (the “Act”), the term "drug" means articles recognized in the official United States Pharmacopoeia, official Homeopathic Pharmacopoeia of the United States, or official National Formulary; articles intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in man or other animals; and articles other than food intended to affect the structure or any function of the body of man or other animals. It also includes articles intended for use as a component of a drug.
Once a product is determined to be a drug for animal use, the next step is to determine whether or not it is a new animal drug. The Act defines a new animal drug (in part) as any drug intended for use for animals other than man, the composition of which is not generally recognized, among experts qualified by scientific training and experience, as safe and effective for use under the conditions prescribed, recommended, or suggested in its labeling. By virtue of Supreme Court interpretations of the necessary basis for general recognition, there are, for all practical purposes, no animal drugs which are not also new animal drugs.
24
Under the Act, a new animal drug may not be legally introduced into interstate commerce unless it is the subject of either:
| ● | an approved NADA or abbreviated new animal drug application (ANADA) under section 512 of the Act; |
| ● | a conditional approval under section 571 of the Act; |
| ● | a listing on the Legally Marketed Unapproved New Animal Drug Index for Minor Species (the Index) under section 572 of the Act; |
| ● | an emergency use authorization (“EUA”) under section 564 of the Act (an EUA may only be issued under very limited circumstances, more information regarding EUAs is available at this webpage: Emergency Use Authorization) ; or |
| ● | an investigational exemption under section 512(j) of the Act. |
Three Regulatory Pathways in the U.S. to Legal Marketing Status for Animal Health Drugs
Approval
An approved animal drug has gone through the New Animal Drug Application (NADA) process, or for an approved generic animal drug, the Abbreviated New Animal Drug Application (ANADA) process. If the information in the application meets the requirements for approval, FDA approves the animal drug. FDA’s approval means the drug is safe and effective when it is used according to the label. FDA’s approval also ensures that the drug’s strength, quality, and purity are consistent from batch to batch, and that the drug’s labeling is truthful, complete, and not misleading.
Conditional Approval
Conditional approval is only available for some animal drugs for use in a minor species or in a major species under special circumstances. A conditionally approved animal drug has gone through FDA's drug approval process except the drug has not yet met the effectiveness standard for full approval. FDA’s conditional approval means that when used according to the label, the drug is safe and has a “reasonable expectation of effectiveness.” FDA's conditional approval also means that the drug is properly manufactured.
The conditional approval is valid for one year. The drug company can ask FDA to renew the conditional approval annually for up to four more years, for a total of five years of conditional approval. During the 5-year period, the drug company can legally sell the animal drug while collecting the remaining effectiveness data. After collecting the remaining effectiveness data, the company submits an application to FDA for full approval. The agency reviews the application and, if appropriate, fully approves the drug.
Indexing
An indexed animal drug is a drug on FDA’s Index of Legally Marketed Unapproved New Animal Drugs for Minor Species, referred to simply as “the Index.” As the name says, a drug listed on the Index is unapproved but has legal marketing status. It can be legally sold for a specific use in certain minor species. Indexing is allowed for drugs for:
| ● | Non-food-producing minor species, such as pet birds, hamsters, and ornamental fish. These animals are typically not eaten by people or by other animals that produce food for people to eat; and |
| ● | An early non-food life stage of a food-producing minor species, such as oyster spat (immature oysters). Because people do not generally eat oyster spat, a drug to treat a disease in spat can be indexed, but a drug to treat a disease in adult oysters, which people commonly eat, cannot be indexed. |
25
Indexing a drug is quite different from the drug approval process. Indexing relies heavily on a panel of qualified experts outside FDA. The experts review the drug’s safety in the specific minor species and the drug’s effectiveness for the intended use. All experts on the panel must agree that, when used according to the label, the drug’s benefits outweigh the risks to the treated animal. If FDA agrees with the panel, the agency adds the drug to the Index.
Animal Health Business Regulations
The development, approval and sale of animal health products are governed by the laws and regulations of each country in which we intend to seek approval, where necessary, to market and subsequently sell our prescription drug and non-drug products. To comply with these regulatory requirements, we have established processes and resources to provide oversight of the development, approval processes and launch of our products and to position those products in order to gain market share in each respective market.
Certain U.S. federal regulatory agencies are charged with oversight and regulatory authority of animal health products in the United States. These agencies, depending on the product and its intended use, may include the FDA, the USDA and the Environmental Protection Agency. The approval of prescription drugs intended for animal use is regulated by the FDA’s CVM. In addition, the Drug Enforcement Administration regulates animal therapeutics that are classified as controlled substances. In addition, the Federal Trade Commission may, in the case of non-drug products, regulate the marketing and advertising claims being made.
Labeling
The FDA plays a significant role in regulating the labeling, advertising and promotion of animal drugs. This is also true of regulatory agencies in the EU and other territories. In addition, advertising and promotion of animal health products is controlled by regulations in many countries. These rules generally restrict advertising and promotion to those claims and uses that have been reviewed and approved by the applicable agency. We will conduct a review of advertising and promotional material for compliance with the local and regional requirements in the markets where we sell our product candidates.
EMA Regulation of Human Prescription Drugs
Napo and Napo Therapeutics intend to leverage the orphan medicines marketing authorization incentives from the EMA for the short bowel syndrome and congenital diarrheal disorders indications for crofelemer for the licensed territories in the European Union. EMA has developed a regulatory procedure for sponsor eligibility for incentives available for drugs with ODD for the appropriate patient populations in an expedited manner. The EMA is responsible for scientific evaluation of centralized marketing authorization applications (“MAA”). Once granted by the European Commission, the centralized MAA is valid in all EU member states, Iceland, Norway and Liechtenstein.
Centralized authorization procedure
Under the centralized authorization procedure, pharmaceutical companies submit a single marketing authorization application to EMA. This allows the marketing-authorization holder to market the medicine and make it available to patients and healthcare professionals throughout the EU on the basis of a single marketing authorization.
EMA's Committee for Medicinal products for Human Use (“CHMP”) or Committee for Medicinal products for Veterinary Use (“CVMP”) carry out a scientific assessment of the application and give a recommendation on whether the medicine should be marketed or not. However, under EU law EMA has no authority to actually permit marketing in the different EU countries. The European Commission is the authorizing body for all centrally authorized product, who takes a legally binding decision based on EMA's recommendation. This decision is issued within 67 days of receipt of EMA’s recommendation.
Once granted by the European Commission, the centralized marketing authorization is valid in all EU Member States as well as in the European Economic Area (“EEA”) countries Iceland, Liechtenstein and Norway. Commission decisions are published in the Community Register of medicinal products for human use.
26
Centralized Procedure is mandatory for certain types of products, such as biotechnology medicinal products, orphan medicinal products, and medicinal products containing a new active substance indicated for the treatment of AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and viral diseases.
Conditional marketing authorization
The EMA supports the development of medicines that address unmet medical needs. In the interest of public health, applicants may be granted a conditional marketing authorization for such medicines on less comprehensive clinical data than normally required, where the benefit of immediate availability of the medicine outweighs the risk inherent in the fact that additional data are still required.
Medicines for human use are eligible if they are intended for treating, preventing or diagnosing seriously debilitating or life-threatening diseases. This includes orphan medicines. Its use is also intended for a public health emergency (e.g. a pandemic). For these medicines, less comprehensive pharmaceutical and non-clinical data may also be accepted. The legal basis is Article 14-a of Regulation (EC) No 726/2004. The provisions for granting a conditional marketing authorization are further elaborated in Regulation (EC) No 507/2006.
Criteria and conditions
EMA's CHMP may grant a conditional marketing authorization for a medicine if it finds that all of the following criteria are met:
| ● | the benefit-risk balance of the medicine is positive; |
| ● | it is likely that the applicant will be able to provide comprehensive data post-authorization; |
| ● | the medicine fulfils an unmet medical need; |
| ● | the benefit of the medicine's immediate availability to patients is greater than the risk inherent in the fact that additional data are still required. |
Conditional marketing authorizations are valid for one year and can be renewed annually. Once a conditional marketing authorization has been granted, the marketing authorization holder must fulfil specific obligations within defined timelines. These obligations could include completing ongoing or new studies or collecting additional data to confirm the medicine's benefit-risk balance remains positive. EMA publishes the conditions of the marketing authorization in the medicine's European public assessment report.
The marketing authorization can be converted into a standard marketing authorization (no longer subject to specific obligations) once the marketing authorization holder fulfils the obligations imposed and the complete data confirm that the medicine's benefits continue to outweigh its risks. Initially, this is valid for 5 years. It can then be renewed for unlimited validity.
As for any medicine, if new data show that the medicine’s benefits no longer outweigh its risks, EMA can take regulatory action, such as suspending or revoking the marketing authorization. EMA can also take regulatory action if the company does not comply with the imposed obligations.
Despite earlier approval, it guarantees that the medicine meets rigorous EU standards for safety, efficacy and quality and that comprehensive data is still generated post-approval. It offers a robust post-authorization regulatory framework based on legally binding obligations, safeguards and controls.
These include:
| ● | full prescribing information and package leaflet with detailed instructions for safe use and conditions for storage; |
| ● | a robust risk-management and safety monitoring plan; |
27
| ● | manufacturing controls including official batch controls for vaccines, as required; |
| ● | legally binding post-approval obligations (i.e. conditions) for the marketing authorization holder and a clear legal framework for the evaluation of emerging efficacy and safety data; |
| ● | a pediatric investigation plan. |
Guidance for applicants for conditional marketing authorization
EMA advises applicants to discuss their development plans with the Agency via scientific advice or protocol assistance early in the development process. Involving health technology assessment bodies early is also encouraged, which is possible via EMA's parallel consultations procedure. The applicant should indicate a request for conditional marketing authorization in their notification of intention to submit a marketing authorization application. They should submit this 6 to 7 month before submitting the application. EMA also encourages applicants to further discuss their plans with EMA as part of a pre-submission meeting. For products deemed suitable for a conditional marketing authorization, EMA encourages applicants to also consider requesting accelerated assessment.
Applicants should include a formal request for a conditional marketing authorization in their marketing authorization application. The CHMP will assess this request together with the application. Guideline on the scientific application and the practical arrangements necessary to implement Regulation (EC) No 507/2006 on the conditional marketing authorization for medicinal products for human use falling within the scope of Regulation (EC) No 726/2004.
Distinction from authorization under exceptional circumstances
EMA may also grant a marketing authorization in absence of comprehensive data under exceptional circumstances. Unlike conditional marketing authorization, where marketing approval is granted in the likelihood that the sponsor will provide such data within an agreed timeframe, EMA can grant authorization under exceptional circumstances when comprehensive data cannot be obtained even after authorization. This authorization route normally does not lead to a standard marketing authorization.
Orphan drug development incentives from EMA
Protocol assistance
The Agency provides a form of scientific advice specifically for orphan medicines called protocol assistance. This allows sponsors to get answers to their questions on the types of studies needed to demonstrate the medicine's quality, benefits and risks, and information on the significant benefit of the medicine. Protocol assistance is available at a reduced charge for designated orphan medicines, linked to a fee-reduction scale that depends on the status of the sponsor. There is no restriction on the number of times a sponsor can request protocol assistance.
The Agency encourages sponsors to consider coordinating the timing of protocol assistance from the Agency with request for scientific advice from the United States Food and Drug Administration (FDA). Parallel scientific advice with the FDA is available.
Access to the centralized authorization procedure
All designated orphan medicines are assessed for marketing authorization centrally in the European Union. This allows companies to make a single application to the European Medicines Agency, resulting in a single opinion and a single decision from the European Commission, valid in all EU Member States. Sponsors may also have access via orphan designation to conditional approval, which is conducted under the centralized procedure.
Ten years of market exclusivity
Authorized orphan medicines benefit from ten years of protection from market competition with similar medicines with similar indications once they are approved. This period of protection is extended by two years for
28
medicines that also have complied with an agreed pediatric investigation plan granted at the time of review of the orphan medicine designation.
Additional incentives for micro, small and medium-sized enterprises (“SMEs”)
Companies classified as SMEs benefit from further incentives when developing medicines with orphan designation. These include administrative and procedural assistance from the Agency's SME office and fee reductions.
Fee reductions
Companies applying for designated orphan medicines pay reduced fees for regulatory activities. This includes reduced fees for protocol assistance, marketing-authorization applications, inspections before authorization, applications for changes to marketing authorizations made after approval, and reduced annual fees. Fee reductions are revised each year in relation to the budget available.
EMA Grants
The Agency does not offer research grants for sponsors of orphan medicines, but funding is available from the European Commission and other sources:
| ● | Horizon 2020, the EU Framework Programme for Research and Innovation; |
| ● | E-Rare, a transnational project for research programs on rare diseases. |
| ● | Grants are also available for sponsors considering research in the United States or Japan: |
| ● | United States: Food and Drug Administration: Orphan products grants program |
| ● | Japan: National Institute of Biomedical Innovation: Services to promote development of medicinal products for rare diseases |
Incentives in Member States
Details on incentives available for designated orphan medicines in EU Member States are available in the European Commission's Inventory of Union and Member State incentives to support research into, and the development and availability of, orphan medicinal products.
Activities after orphan designation
Orphan designation makes the sponsor eligible for a number of orphan incentives. Sponsors need to comply with various activities that take place after a designation has been granted. Sponsors should submit all post-designation activities, including annual reports. For information and guidance on using IRIS, see the IRIS homepage. Sponsors must submit an annual report on development to the Agency summarizing the status of development of the medicine.
Sponsors of medicines with orphan designation should also remember to apply for a pediatric investigation plan (“PIP”), deferral or waiver at the appropriate time, as specified in the Pediatric regulation.
Sponsors also need to submit an application for maintenance of the orphan designation at the time of marketing authorization, in order to be eligible for the ten-year market exclusivity incentive.
A valid and completed PIP could make the sponsor eligible for the two-year marketing exclusivity extension to the ten-year marketing exclusivity which is granted at the time of review of the orphan medicinal designation. Transfers of orphan designation from one sponsor to another are possible. Transfers are free of charge. Sponsors can also request removal of an orphan designation.
29
EMA Compassionate Use Program
Compassionate use is a treatment option that allows the use of an unauthorized medicine. Under strict conditions, products in development can be made available to groups of patients who have a disease with no satisfactory authorized therapies and who cannot enter clinical trials. The EMA provides recommendations through the Committee for Medicinal Products for Human Use (“CHMP”), but these do not create a legal framework. Compassionate use programs are coordinated and implemented by Member States, which set their own rules and procedures.
Established by Article 83 of Regulation (EC) No 726/2004, this tool is designed to:
| ● | facilitate and improve access to compassionate use programs by patients in the EU; |
| ● | favor a common approach regarding the conditions of use, the conditions for distribution and the patients targeted for the compassionate use of unauthorized new medicines; |
| ● | increase transparency between Member States in terms of treatment availability. |
These programs are only put in place if the medicine is expected to help patients with life-threatening, long-lasting or seriously debilitating illnesses, which cannot be treated satisfactorily with any currently authorized medicine. The medicine must be undergoing clinical trials or have entered the marketing-authorization application process and while early studies will generally have been completed, its safety profile and dosage guidelines may not be fully established.
How to request an opinion for Compassionate Use
National competent authorities can ask EMA for an opinion on how to administer, distribute and use certain medicines for compassionate use. The CHMP also identifies which patients would benefit, and Member States should take note of these recommendations when making decisions.
Manufacturers and marketing-authorization applicants should not contact EMA to request an opinion, but they may wish to inform the Agency of applications underway at national level. National competent authorities will inform the Agency if they are making a product available to a group of patients for compassionate use.
Comparison to individual basis treatment (Named Patient Program)
Compassionate use should not be confused with 'named-patient basis' treatments, which see doctors obtain medicines directly from manufacturers before authorization. This is done on an individual basis under the direct responsibility of the doctor, and the Agency does not need to be informed.
In general, medicines that are not yet authorized are first made available through clinical trials and patients should always be considered for inclusion in trials before being offered compassionate use programs.
Compassionate use recommendations
EMA's recommendations cover how a medicine should be used in compassionate use programs across the EU, and the type of patient who may benefit from treatment. EMA does not update its recommendations after a medicine receives marketing authorization, as all relevant information on the medicine's use is available in its European public assessment report (“EPAR”). However, compassionate use programs may continue in certain Member States until the medicine becomes available on the market.
Rewards and incentives for pediatric medicines
Several rewards and incentives for the development of pediatric medicines for children are available in the EU. Medicines authorized across the EU with the results of studies from a pediatric investigation plan included in the
30
product information are eligible for an extension of their supplementary protection certificate by six months. This is the case even when the studies' results are negative.
For orphan medicines, the incentive is an additional two years of market exclusivity.
Scientific advice and protocol assistance at the Agency are free of charge for questions relating to the development of pediatric medicines. Medicines developed specifically for children that are already authorized but are not protected by a patent or supplementary protection certificate are eligible for a pediatric-use marketing authorization (“PUMA”). If a PUMA is granted, the product will benefit from 10 years of market protection as an incentive. The above can be complemented by other incentives to support the research, development and availability of medicinal products for pediatric use.
Market exclusivity: Orphan medicines
Orphan medicines benefit from ten years of market exclusivity once they receive a marketing authorization in the EU. This measure is intended to encourage the development of medicines for rare diseases, by protecting them from competition from similar medicines with similar indications, which cannot be marketed during the exclusivity period. Market exclusivity is an orphan incentive awarded by the European Commission to a specific clinical indication with an orphan designation.
Each indication with an orphan designation confers ten years' market exclusivity for the particular indication. A medicine that has multiple orphan designations for different conditions will benefit from separate market exclusivity periods pertaining to its different orphan designations.
To benefit from market exclusivity, a medicine must maintain its orphan designation at the time of marketing authorization.
Extension of market exclusivity period
The market exclusivity period is extended by two additional years for an orphan-designated condition when the results of specific studies are reflected in the summary of product characteristics (“SmPC”) addressing the pediatric population and completed in accordance with a fully compliant PIP.
The European Commission grants the extension based on a positive compliance check from the Pediatric Committee and opinion from the Committee for Medicinal Products for Human Use (“CHMP”), and includes this information in the Community register of orphan medicinal products.
Review of market exclusivity period
Article 8(2) of the Orphan Regulation establishes the possibility for Member States to request that the market exclusivity be reduced from ten to six years, under certain circumstances.
Expiry of market exclusivity
When the period of market exclusivity for an indication ends, the orphan designation for that indication expires and the European Commission removes it from the Community register of orphan medicinal products.
Once all of the orphan designations associated with an approved medicine have expired or been withdrawn by the sponsor, the medicine ceases to be classified as an orphan medicine and no longer benefits from the orphan incentives.
European Union new chemical entity exclusivity
In the EU, new chemical entities, sometimes referred to as new active substances, qualify for eight years of data exclusivity upon marketing authorization and an additional two years of market exclusivity. This data exclusivity, if granted, prevents regulatory authorities in the European Union from referencing the innovator’s data to assess a
31
generic application for eight years, after which a generic marketing authorization application can be submitted, and the innovator’s data may be referenced, but not approved for two years. The overall ten-year period will be extended to a maximum of 11 years if, during the first eight years of those ten years, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies.
European Union orphan designation and exclusivity
In the EU, the EMA’s Committee for Orphan Medicinal Products grants orphan drug designation to promote the development of products that are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than 5 in 10,000 persons in the European Union Community and for which no satisfactory method of diagnosis, prevention, or treatment has been authorized (or the product would be a significant benefit to those affected). Additionally, designation is granted for products intended for the diagnosis, prevention, or treatment of a life-threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the drug in the European Union would be sufficient to justify the necessary investment in developing the medicinal product.
In the European Union, orphan drug designation entitles a party to financial incentives such as reduction of fees or fee waivers and ten years of market exclusivity is granted following medicinal product approval. This period may be reduced to six years if the orphan drug designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity. Orphan drug designation must be requested before submitting an application for marketing approval. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
European Union Pediatric Plan
In the EEA, marketing authorization applications for new medicinal products not authorized have to include the results of studies conducted in the pediatric population, in compliance with a pediatric investigation plan, or PIP, agreed with the EMA’s Pediatric Committee (“PDCO”). The PIP sets out the timing and measures proposed to generate data to support a pediatric indication of the drug for which marketing authorization is being sought. The PDCO can grant a deferral of the obligation to implement some or all of the measures of the PIP until there are sufficient data to demonstrate the efficacy and safety of the product in adults. Further, the obligation to provide pediatric clinical trial data can be waived by the PDCO when these data is not needed or appropriate because the product is likely to be ineffective or unsafe in children, the disease or condition for which the product is intended occurs only in adult populations, or when the product does not represent a significant therapeutic benefit over existing treatments for pediatric patients. Once the marketing authorization is obtained in all Member States of the EU and study results are included in the product information, even when negative, the product is eligible for six months’ supplementary protection certificate extension. For orphan drug designated medicinal products, the 10 year period of market exclusivity is extended to 12 years.
Clinical Trials Regulation in Europe
In the EU, pursuant to the currently applicable Clinical Trials Directive 2001/20/EC and the Directive 2005/28/EC on GCP, a system for the approval of clinical trials in the EU has been implemented through national legislation of the EU member states. Under this system, an applicant must obtain approval from the national competent authority of an EU member state in which the clinical trial is to be conducted, or in multiple member states if the clinical trial is to be conducted in a number of member states. Furthermore, the applicant may only start a clinical trial at a specific study site after the independent ethics committee for each site has issued a favorable opinion. The clinical trial application must be accompanied by an investigational medicinal product dossier with supporting information prescribed by Directive 2001/20/EC and Directive 2005/28/EC and corresponding national laws of the individual EU member states and further detailed in applicable guidance documents. In April 2014, the EU adopted a new Clinical Trials Regulation (EU) No 536/2014, which is set to replace the current Clinical Trials Directive 2001/20/EC. It is anticipated that the new Clinical Trials Regulation (EU) No 536/2014 may come into effect in late 2021with a three-year transition period for some types of clinical trials. It will overhaul the current system of
32
approvals for clinical trials in the EU. Specifically, the new regulation, which will be directly applicable in all EU member states, aims at simplifying and streamlining the approval of clinical trials in the EU. For instance, the new Clinical Trials Regulation provides for a streamlined application procedure via a single-entry point and strictly defined deadlines for the assessment of clinical trial applications
Other U.S. Healthcare Laws
In addition to FDA restrictions on marketing of pharmaceutical and biological products, other U.S. federal and state healthcare regulatory laws restrict business practices in the pharmaceutical industry, which include, but are not limited to, state and federal anti-kickback, false claims, data privacy and security and physician payment and drug pricing transparency laws.
The U.S. federal Anti-Kickback Statute prohibits, among other things, any person or entity from knowingly and willfully offering, paying, soliciting, receiving or providing any remuneration, directly or indirectly, overtly or covertly, to induce or in return for purchasing, leasing, ordering, or arranging for or recommending the purchase, lease, or order of any good, facility, item or service reimbursable, in whole or in part, under Medicare, Medicaid or other federal healthcare programs. The term “remuneration” has been broadly interpreted to include anything of value. The Anti-Kickback Statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers, formulary managers and beneficiaries on the other. Although there are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, the exceptions and safe harbors are drawn narrowly. Practices that involve remuneration that may be alleged to be intended to induce prescribing, purchases, or recommendations may be subject to scrutiny if they do not meet the requirements of a statutory or regulatory exception or safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the U.S. federal Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case by case basis based on a cumulative review of all its facts and circumstances. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the statute has been violated.
Additionally, the intent standard under the U.S. federal Anti-Kickback Statute was amended by the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively, the ACA, to a stricter standard such that a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. In addition, the ACA codified case law that a claim including items or services resulting from a violation of the U.S. federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. The majority of states also have Anti-Kickback laws, which establish similar prohibitions and, in some cases, may apply to items or services reimbursed by any third-party payor, including commercial insurers.
The federal false claims and civil monetary penalties laws, including the civil False Claims Act, prohibit any person or entity from, among other things, knowingly presenting, or causing to be presented, a false, fictitious or fraudulent claim for payment to, or approval by, the federal government, knowingly making, using, or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government, or from knowingly making a false statement to avoid, decrease or conceal an obligation to pay money to the U.S. federal government. A claim includes “any request or demand” for money or property presented to the U.S. government. Actions under the civil False Claims Act may be brought by the Attorney General or as a qui tam action by a private individual in the name of the government. Violations of the civil False Claims Act can result in very significant monetary penalties and treble damages. Several pharmaceutical and other healthcare companies have been prosecuted under these laws for, among other things, allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. Other companies have been prosecuted for causing false claims to be submitted because of the companies’ marketing of products for unapproved, or off label, uses. Companies also have been prosecuted for allegedly violating the Anti-Kickback Statute and False Claims Act as a result of impermissible arrangements between companies and healthcare practitioners or as a result of the provision of remuneration by the companies to the healthcare practitioners. In addition, the civil monetary penalties statute imposes penalties against any person who is determined to have presented or caused to be presented a claim to a federal health
33
program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent. Many states also have similar fraud and abuse statutes or regulations that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor.
Violations of fraud and abuse laws, including federal and state Anti-Kickback and false claims laws, may be punishable by criminal and civil sanctions, including fines and civil monetary penalties, the possibility of exclusion from federal healthcare programs (including Medicare and Medicaid), disgorgement and corporate integrity agreements, which impose, among other things, rigorous operational and monitoring requirements on companies. Similar sanctions and penalties, as well as imprisonment, also can be imposed upon executive officers and employees of such companies. Given the significant size of actual and potential settlements, it is expected that the government authorities will continue to devote substantial resources to investigating healthcare providers’ and manufacturers’ compliance with applicable fraud and abuse laws.
The federal Health Insurance Portability and Accountability Act (“HIPAA”) of 1996 created additional federal criminal statutes that prohibit, among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the U.S. federal Anti-Kickback Statute, the ACA broadened the reach of certain criminal healthcare fraud statutes created under HIPAA by amending the intent requirement such that a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
In addition, there has been a recent trend of increased federal and state regulation of payments made to physicians and certain other healthcare providers. The ACA imposed, among other things, new annual reporting requirements through the Physician Payments Sunshine Act for covered manufacturers for certain payments and “transfers of value” provided to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Failure to submit timely, accurately and completely the required information for all payments, transfers of value and ownership or investment interests may result in civil monetary penalties of up to an aggregate of $150,000 per year and up to an aggregate of $1 million per year for “knowing failures.” Covered manufacturers must submit reports by the 90th day of each subsequent calendar year. In addition, certain states require the implementation of compliance programs and compliance with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, impose restrictions on marketing practices and/or tracking and reporting of gifts, compensation and other remuneration or items of value provided to physicians and other healthcare professionals and entities.
Napo may also be subject to data privacy and security regulation by both the federal government and the states in which Napo conducts its business. HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act (“HITECH”), and their respective implementing regulations, including the Final HIPAA Omnibus Rule, published on January 25, 2013, impose specified requirements relating to the privacy, security and transmission of individually identifiable health information held by covered entities and their business associates. Among other things, HITECH made HIPAA’s security standards directly applicable to “business associates,” defined as independent contractors or agents of covered entities that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same requirements, thus complicating compliance efforts.
34
Coverage and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any pharmaceutical products for which Napo obtains regulatory approval. In the United States and markets in other countries, patients who are prescribed treatments for their conditions and providers performing the prescribed services generally rely on third-party payors to reimburse all or part of the associated healthcare costs. Patients are unlikely to use Napo’s products unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of Napo’s products. Sales of any products for which Napo receives regulatory approval for commercial sale will, therefore depend, in part, on the availability of coverage and adequate reimbursement from third party payors. Third party payors include government authorities, managed care plans, private health insurers and other organizations. In the United States, the process for determining whether a third party payor will provide coverage for a pharmaceutical or biological product typically is separate from the process for setting the price of such product or for establishing the reimbursement rate that the payor will pay for the product once coverage is approved. Third party payors may limit coverage to specific products on an approved list, also known as a formulary, which might not include all of the FDA approved products for a particular indication. A decision by a third party payor not to cover Napo’s product candidates could reduce physician utilization of Napo’s products once approved and have a material adverse effect on Napo’s sales, results of operations and financial condition. Moreover, a third party payor’s decision to provide coverage for a pharmaceutical or biological product does not imply that an adequate reimbursement rate will be approved. Adequate third party reimbursement may not be available to enable Napo to maintain price levels sufficient to realize an appropriate return on Napo’s investment in product development. Additionally, coverage and reimbursement for products can differ significantly from payor to payor. One third party payor’s decision to cover a particular medical product or service does not ensure that other payors will also provide coverage for the medical product or service, or will provide coverage at an adequate reimbursement rate. As a result, the coverage determination process will require Napo to provide scientific and clinical support for the use of Napo’s products to each payor separately and will be a time consuming process.
In the EEA, governments influence the price of products through their pricing and reimbursement rules and control of national health care systems that fund a large part of the cost of those products to consumers. Some jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed to by the government. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare the cost effectiveness of a particular product candidate to currently available therapies. Other member states allow companies to fix their own prices for medicines, but monitor and control company profits. The downward pressure on health care costs in general, particularly prescription products, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in some countries, cross border imports from low priced markets exert commercial pressure on pricing within a country.
The containment of healthcare costs has become a priority of federal, state and foreign governments, and the prices of pharmaceutical or biological products have been a focus in this effort. Third party payors are increasingly challenging the prices charged for medical products and services, examining the medical necessity and reviewing the cost effectiveness of pharmaceutical or biological products, medical devices and medical services, in addition to questioning safety and efficacy. If these third party payors do not consider Napo’s products to be cost effective compared to other available therapies, they may not cover Napo’s products after FDA approval or, if they do, the level of payment may not be sufficient to allow Napo to sell its products at a profit.
Healthcare Reform
A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Government authorities and other third party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medical products. For example, in March 2010, the ACA was enacted, which, among other things, increased the minimum Medicaid rebates owed by most manufacturers under the Medicaid Drug Rebate Program; introduced a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected; extended the Medicaid Drug Rebate Program to the utilization of prescriptions of individuals enrolled in Medicaid managed care plans; imposed mandatory discounts for
35
certain Medicare Part D beneficiaries as a condition for manufacturers’ outpatient drugs covered under Medicare Part D; subjected drug manufacturers to new annual fees based on pharmaceutical companies’ share of sales to federal healthcare programs; created a new Patient Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; creation of the Independent Payment Advisory Board, once empaneled, will have authority to recommend certain changes to the Medicare program that could result in reduced payments for prescription drugs; and establishment of a Center for Medicare Innovation at the CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending. Since its enactment, the U.S. federal government has delayed or suspended the implementation of certain provisions of the ACA.
Napo expects that the ACA, as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and lower reimbursement and additional downward pressure on the price that Napo receives for any approved product. Any reduction in reimbursement from Medicare or other government-funded programs may result in a similar reduction in payments from private payors. Moreover, recently there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products. The implementation of cost containment measures or other healthcare reforms may prevent Napo from being able to generate revenue, attain profitability or commercialize Napo’s drugs.
Additionally, on August 2, 2011, the Budget Control Act of 2011 created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This included aggregate reductions of Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013, and due to subsequent legislative amendments to the statute, will stay in effect through 2025 unless additional action is taken by Congress. On January 2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several types of providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. More recently, there has been heightened governmental scrutiny recently over the manner in which manufacturers set prices for their marketed products, which have resulted in several recent Congressional inquiries and proposed bills designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for pharmaceutical products.
Napo expects that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for Napo’s products once approved or additional pricing pressures.
Other Regulatory Considerations
We believe regulatory rules relating to human food safety, food additives, or drug residues in food will not apply to the products we currently are developing because our animal prescription drug product candidates are not intended for use in production animals, with the exception of horses, which qualify as food animals in Europe and Canada; and our nonprescription products are not regulated by section 201(g) of the Federal Food, Drug, and Cosmetic Act, which the FDA is authorized to administer.
We do not believe that our animal nonprescription products are currently subject to regulation in the United States. The CVM only regulates those animal supplements that fall within the FDA’s definition of an animal drug, food or feed additive. The Federal Food Drug and Cosmetic Act defines food as “articles used for food or drink for man or other animals and articles used as components of any such article.” Animal foods are not subject to pre-market approval and are designed to provide a nutritive purpose to the animals that receive them. Feed additives are defined as those articles that are added to an animal’s feed or water, as illustrated by the guidance documents. Our nonprescription products are not added to food, are not ingredients in food, nor are they added to any animal’s drinking water. There is no intent to make our nonprescription products a component of an animal food, either directly
36
or indirectly. We do not believe that our nonprescription products fit the definition of an animal drug, food or food additive and therefore are not regulated by the FDA at this time.
In addition to the foregoing, we may be subject to state, federal and foreign healthcare and/or veterinary medicine laws, including but not limited to anti-kickback laws, as we may from time to time enter consulting and other financial arrangements with veterinarians, who may prescribe or recommend our products. If our financial relationships with veterinarians are found to be in violation of such laws that apply to us, we may be subject to penalties.
Legal Proceedings
From time to time, we may become involved in litigation relating to claims arising from the ordinary course of business. Other than as set forth in “Item 3. LEGAL PROCEEDINGS”, there are currently no claims or actions pending against us, the ultimate disposition of which could have a material adverse effect on our results of operations, financial condition or cash flows.
Corporate Information
We were incorporated in the State of Delaware on June 6, 2013. Our principal executive office is located at 200 Pine Street, Suite 400, San Francisco, CA 94104 for human health prescription drugs and the telephone number is (415) 371 8300. We have an additional office at 200 Pine Street, Suite 600, San Francisco, CA 94104 for Jaguar Animal Health. Our website for the corporation is https://jaguar.health. The information contained on, or that can be accessed through, our website is not part of this annual report. Our voting common stock is listed on the NASDAQ Capital Market and trades under the symbol “JAGX.” On July 31, 2017, we completed the acquisition of Napo pursuant to the Agreement and Plan of Merger, dated March 31, 2017, by and among the Company, Napo, Napo Acquisition Corporation, and Napo’s representative (the “Merger”).
Employees
As of December 31, 2021, we had 52 employees. Ten employees hold M.D., D.V.M and/or Ph.D. degrees. Twenty-one of our employees are engaged in research and development activities and 17 employees are engaged in sales and marketing. We have two employees within Napo Therapeutics in Italy. None of our employees are represented by labor unions or covered by collective bargaining agreements.
Description of Properties
Our corporate headquarters are located in San Francisco, California, where we currently lease 10,526 rentable square feet of office space from M & E, LLC.
ITEM 1A. RISK FACTORS
The business, financial condition and operating results of the Company may be affected by a number of factors, whether currently known or unknown, including but not limited to those described below. Anyone or more of such factors could directly or indirectly cause the Company’s actual results of operations and financial condition to vary materially from past or anticipated future results of operations and financial condition. Any of these factors, in whole or in part, could materially and adversely affect the Company’s business, financial condition, results of operations and stock price. The following information should be read in conjunction with Part II, Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and the consolidated financial statements and related notes in Part II, Item 8, “Financial Statements and Supplementary Data” of this Annual Report.
37
Risk Factor Summary
The following is a summary of the principal risks that could adversely affect our business, operations and financial results.
Risks Related to Our Business
| ● | We have a limited operating history, expect to incur further losses as we grow and may be unable to achieve or sustain profitability. |
| ● | We expect to incur significant additional costs as we continue commercialization efforts for current prescription drug candidates or other product candidates, and undertake the clinical trials necessary to obtain any necessary regulatory approvals, which will increase our losses. |
| ● | We will need to raise substantial additional capital in the future in the event that we conduct clinical trials for new indications and we may be unable to raise such funds when needed and on acceptable terms, which would force us to delay, limit, reduce or terminate one or more of our product development programs. |
| ● | We are substantially dependent on the success of Mytesi, our current lead prescription drug product, and Canalevia-CA1, our conditionally approved prescription drug product for CID in dogs, and Canalevia-CA2, our candidate for EID in dogs. We cannot be certain that necessary approvals will be received for planned Mytesi, Canalevia-CA1 or Canalevia-CA2 follow-on indications or that these product candidates will be successfully commercialized, either by us or any of our partners. |
| ● | If we are not successful in identifying, licensing, developing and commercializing additional product candidates and products, our ability to expand our business and achieve our strategic objectives could be impaired. |
| ● | Mytesi faces significant competition from other pharmaceutical companies, both for its currently approved indication and for planned follow-on indications, and our operating results will suffer if we fail to compete effectively. |
| ● | We may be unable to obtain, or obtain on a timely basis, regulatory approval for our existing or future human or animal prescription drug product candidates under applicable regulatory requirements, which would harm our operating results. |
| ● | The results of our earlier studies of Mytesi may not be predictive of the results in any future clinical trials and species-specific formulation studies, respectively, and we may not be successful in our efforts to develop or commercialize line extensions of Mytesi. |
| ● | Development of prescription drug products is inherently expensive, time-consuming and uncertain, and any delay or discontinuance of our current or future pivotal trials would harm our business and prospects. |
| ● | We will partially rely on third parties to conduct our development activities. If these third parties do not successfully carry out their contractual duties, we may be unable to obtain regulatory approvals or commercialize our current or future human or animal product candidates on a timely basis, or at all. |
| ● | Even if we obtain regulatory approval for planned follow-on indications of crofelemer, Canalevia or our other product candidates, they may never achieve market acceptance. Further, even if we are successful in the ongoing commercialization of Mytesi and Canalevia, we may not achieve commercial success. |
| ● | Human and animal gastrointestinal health products are subject to unanticipated post-approval safety or efficacy concerns, which may harm our business and reputation. |
| ● | Future federal and state legislation may result in increased exposure to product liability claims, which could result in substantial losses. |
| ● | If we fail to retain current members of our senior management, or to identify, attract, integrate and retain additional key personnel, our business will be harmed. |
| ● | We are dependent on two suppliers for the raw material used to produce the active pharmaceutical ingredient in Mytesi and Canalevia. The termination of either of these contracts would result in a disruption to product development and our business will be harmed. |
| ● | We are dependent upon third-party contract manufacturers, both for the supply of the active pharmaceutical ingredient in Mytesi and Canalevia-CA1, as well as for the supply of finished products for commercialization. |
| ● | If we are unable to establish sales capabilities on our own or through third parties, we may not be able to market and sell our current or future human products and product candidates, if approved, and generate |
38
| product or other revenue. |
| ● | We will need to increase the size of our organization and may not successfully manage such growth. |
| ● | Canalevia-CA1 and, our animal health prescription drug product candidates, if approved may be marketed in the United States only in the target animals and for the indications for which they are approved, and if we want to expand the approved animals or indications, it will need to obtain additional approvals, which may not be granted. |
| ● | The misuse or extra-label use of Mytesi, Canalevia, and our human or animal prescription drug product candidates if approved by regulatory authorities, may harm our reputation or result in financial or other damages. |
| ● | We may be unable to obtain, or obtain on a timely basis, a renewal of conditional approval for Canalevia-CA1, or to eventually obtain full regulatory approval of Canalevia-CA1, which would harm our operating results. |
| ● | We may not maintain the benefits associated with MUMS designation, including market exclusivity. |
| ● | The market for our human and animal products, and the gastrointestinal health market as a whole, is uncertain and may be smaller than we anticipate, which could lead to lower revenue and harm our operating results. |
| ● | Insurance coverage for Mytesi for its current approved indication could decrease or end, or Mytesi might not receive insurance coverage for any approved follow-on indications, which could lead to lower revenue and harm our operating results. |
| ● | We may engage in future acquisitions that increase our capital requirements, dilute our stockholders, cause us to incur debt or assume contingent liabilities and subject us to other risks. |
| ● | Certain of the countries in which we plan to commercialize our products in the future are developing countries, some of which have potentially unstable political and economic climates. |
| ● | Fluctuations in the exchange rate of foreign currencies could result in currency transactions losses. |
| ● | Laws and regulations governing global trade compliance could adversely impact our business. |
| ● | There are other gastrointestinal-focused human pharmaceutical companies, and we face competition in the marketplaces in which we operate or plan to operate. |
| ● | Our obligations to Streeterville are secured by a security interest in all of Napo’s lechlemer assets, so if we default on those obligations, Streeterville could foreclose on our assets. |
| ● | Our royalty interests require us to make minimum royalty payments, even if we do not sell a sufficient amount of products to cover the amount of such payments, which may strain our cash resources. |
| ● | Failure in our information technology systems, including by cyber-attacks or other data security incidents, could significantly disrupt our operations. |
| ● | The novel coronavirus global pandemic could adversely impact our business, including our supply chain, clinical trials and commercialization of Mytesi and Canalevia. |
| ● | Long-term remote work arrangements may adversely affect our business. |
| ● | Substantially all of our revenue for recent periods has been received from three customers. |
| ● | We are subject to state laws in California that require gender and diversity quotas for boards of directors of public companies headquartered in California. |
Risks Related to Our Intellectual Property
| ● | We cannot be certain that our patent strategy will be effective to protect against competition |
| ● | Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements. |
| ● | Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, which would be costly, time-consuming and, if successfully asserted against us, delay or prevent the development and commercialization of our current or future products and product candidates. |
| ● | Our proprietary position depends upon the botanical guidance of our drug approval and patents that are formulation or method-of-use patents, which do not prevent a competitor from using the same human or animal drug for another use. |
| ● | We may be involved in lawsuits to protect or enforce our patents, which could be expensive, time- |
39
| consuming and unsuccessful, and third parties may challenge the validity or enforceability of our patents and they may be successful. |
| ● | If we are unable to prevent disclosure of our trade secrets or other confidential information to third parties, our competitive position may be impaired. |
| ● | Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our products. |
| ● | We may not be able to protect our intellectual property rights throughout the world, which could impair our business. |
| ● | Our business could be harmed if we fail to obtain certain registered trademarks in the United States or in other countries. |
| ● | We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties. |
| ● | Even if we receive any of the required regulatory approvals for our current or future prescription drug product candidates and non-prescription products, we will be subject to ongoing obligations and continued regulatory review, which may result in significant additional expense. |
| ● | Any of our current or future prescription drug product candidates or non-prescription products may cause or contribute to adverse medical events that we would be required to report to regulatory authorities and, if we fail to do so, we could be subject to sanctions that would harm our business. |
| ● | Legislative or regulatory reforms with respect to animal health may make it more difficult and costly for us to obtain regulatory clearance or approval of any of our current or future product candidates and to produce, market, and distribute our products after clearance or approval is obtained. |
| ● | We believe that our non-prescription products are not subject to regulation by regulatory agencies in the United States, but there is a risk that regulatory bodies may disagree with our interpretation, or may redefine the scope of their regulatory reach in the future, which would result in additional expense and could delay or prevent the commercialization of these products. |
| ● | Even if we receive the required regulatory approvals for our current or future prescription drug product candidates and non-prescription products, we will be subject to ongoing obligations and continued regulatory review, which may result in significant additional expense |
Risks Related to Our Common Stock
| ● | Our failure to meet the continued listing requirements of The Nasdaq Capital Market could result in a delisting of our common stock. |
| ● | If we issue all shares available for issuance pursuant to the ATM Agreement, we will have no shares of common stock available for new securities issuances, which may restrict us from accessing additional capital through the sale of new securities. |
| ● | If our shares become subject to the penny stock rules, it would become more difficult to trade our shares. |
| ● | The price of our common stock could be subject to volatility related or unrelated to our operations, and purchasers of our common stock could incur substantial losses. |
| ● | A possible “short squeeze” due to a sudden increase in demand of our common stock that largely exceeds supply may lead to further price volatility in our common stock. |
| ● | You may not be able to resell our common stock when you wish to sell them or at a price that you consider attractive or satisfactory. |
| ● | If securities or industry analysts do not publish research or reports about our company, or if they issue adverse or misleading opinions regarding us or our stock, our stock price and trading volume could decline. |
| ● | You may be diluted by conversions of outstanding shares of non-voting common stock, exercises of outstanding options and warrants and issuances of securities pursuant to our ATM Agreement. |
| ● | Provisions in our charter documents and under Delaware law could discourage a takeover that stockholders may consider favorable and may lead to entrenchment of management. |
| ● | Our amended and restated bylaws designate the Court of Chancery of the State of Delaware as the sole and exclusive forum for certain actions and proceedings that may be initiated by our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or other employees. |
| ● | We do not intend to pay dividends on our common stock, and your ability to achieve a return on your |
40
| investment will depend on appreciation in the market price of our common stock. |
| ● | The requirements of being a public company, including compliance with the reporting requirements of the Exchange Act and the requirements of the Sarbanes-Oxley Act, may strain our resources, increase our costs and distract management, and we may be unable to comply with these requirements in a timely or cost-effective manner. |
| ● | We are a smaller reporting company and the reduced reporting requirements applicable to smaller reporting companies may make our common stock less attractive to investors. |
Risks Related to Our Business
We have a limited operating history, expect to incur further losses as we grow and may be unable to achieve or sustain profitability.
Since the consummation of our merger with Napo Pharmaceuticals Inc. in 2017, our operations have been primarily focused on research, development and the ongoing commercialization of our lead prescription drug product, Mytesi, which is approved by the U.S. FDA for the symptomatic relief of noninfectious diarrhea in adults with HIV/AIDS on antiretroviral therapy. As a result, we have limited meaningful historical operations upon which to evaluate our business and prospects and have not yet demonstrated an ability to broadly commercialize any of our human health products beyond Mytesi for HIV-related diarrhea or animal health products, obtain any required marketing approval for any of our animal prescription drug product candidates or successfully overcome the risks and uncertainties frequently encountered by companies in emerging fields such as the animal health industry or the gastrointestinal health industry in general. Our revenues to date have been insufficient to offset our expenses, and we expect to continue to incur significant research and development and other expenses. Our net loss and comprehensive loss for the years ended December 31, 2021 and 2020 was $52.6 million and $33.8 million, respectively. As of December 31, 2021, we had total stockholders’ equity of $11.9 million. We expect to continue to incur losses for the foreseeable future, which will increase significantly from historical levels as we expand our product development activities, seek necessary approvals for our human and veterinary drug product candidates, conduct species-specific formulation studies for our non-prescription products and increase commercialization activities. Even if we succeed in developing and broadly commercializing one or more of our products or product candidates, we expect to continue to incur losses for the foreseeable future, and we may never become profitable. If we fail to achieve or maintain profitability, then we may be unable to continue our operations at planned levels and be forced to reduce or cease operations.
As more fully discussed in Note 1 to our consolidated financial statements, we believe there is substantial doubt about our ability to continue as a going concern as we do not currently have sufficient cash resources to fund our operations through March 11, 2023, or one year from the filing date of our Form 10-K. Our financial statements do not include any adjustments that may result from the outcome of this uncertainty. If we are unable to continue as a viable entity, our stockholders may lose their investment.
We expect to incur significant additional costs as we continue commercialization efforts for current prescription drug candidates or other product candidates, and undertake the clinical trials necessary to obtain any necessary regulatory approvals, which will increase our losses.
Napo commenced sales of Mytesi for adults with HIV/AIDS on antiretroviral therapy in September 2016. Jaguar launched Canalevia-CA1 for chemotherapy-induced diarrhea in dogs in December 2021. We will need to continue to invest in developing our internal and third-party sales and distribution network and outreach efforts to key opinion leaders in the gastrointestinal health industry, including physicians and veterinarians as applicable.
We are actively identifying additional products for development and commercialization, and will continue to expend substantial resources for the foreseeable future to develop Mytesi, lechlemer and Canalevia-CA1. These expenditures will include costs associated with:
| ● | identifying additional potential prescription drug product candidates and non-prescription products; |
41
| ● | formulation studies; |
| ● | conducting pilot, pivotal and toxicology studies; |
| ● | completing other research and development activities; |
| ● | payments to technology licensors; |
| ● | maintaining our intellectual property; |
| ● | obtaining necessary regulatory approvals; |
| ● | establishing commercial supply capabilities; and |
| ● | sales, marketing and distribution of our commercialized products. |
We also may incur unanticipated costs in connection with developing and commercializing our products. Because the outcome of our development activities and commercialization efforts is inherently uncertain, the actual amounts necessary to successfully complete the development and commercialization of our current or future products and product candidates may be greater than we anticipate.
Because we anticipate incurring significant costs for the foreseeable future, if we are not successful in broadly commercializing any of our current or future products or product candidates or raising additional funding to pursue our research and development efforts, we may never realize the benefit of our development efforts and our business may be harmed.
We will need to raise substantial additional capital in the future in the event that we conduct clinical trials for new indications and we may be unable to raise such funds when needed and on acceptable terms, which would force us to delay, limit, reduce or terminate one or more of our product development programs.
We are forecasting continued losses and negative cash flows as we continue to fund our operating and marketing activities and research and development programs, and to complete the development of all the current products in our pipeline, or any additional products we may identify. We will need to seek additional funds through public or private equity or debt financings or other sources such as strategic collaborations. Any such financings or collaborations may result in dilution to our stockholders, the imposition of debt covenants and repayment obligations or other restrictions that may harm our business or the value of our common stock. We may also seek from time to time to raise additional capital based upon favorable market conditions or strategic considerations such as potential acquisitions or potential license arrangements.
Our future capital requirements depend on many factors, including, but not limited to:
| ● | the scope, progress, results and costs of researching and developing our current and future prescription drug product candidates and non-prescription products; |
| ● | the timing of, and the costs involved in, obtaining any regulatory approvals for our current and any future products; |
| ● | the number and characteristics of the products we pursue; |
| ● | the cost of manufacturing our current and future products and any products we successfully commercialize; |
| ● | the cost of commercialization activities for Mytesi and Canalevia, if approved, including sales, |
42
| marketing and distribution costs; |
| ● | the expenses needed to attract and retain skilled personnel; |
| ● | the costs associated with being a public company; |
| ● | our ability to establish and maintain strategic collaborations, distribution or other arrangements and the financial terms of such agreements; and |
| ● | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing possible patent claims, including litigation costs and the outcome of any such litigation. |
Additional funds may not be available when we need them on terms that are acceptable to us, or at all or we may not have sufficient authorized shares to raise additional capital. If adequate funds are not available to us on a timely basis, we may be required to delay, limit, reduce or terminate one or more of our product development programs or future commercialization efforts.
We are substantially dependent on the success of Mytesi, our current lead prescription drug product, and Canalevia-CA1, our conditionally approved prescription drug product for CID in dogs, and Canalevia-CA2, our candidate for EID in dogs. We cannot be certain that necessary approvals will be received for planned Mytesi and Canalevia-CA1 follow-on indications or that these product candidates will be successfully commercialized, either by us or any of our partners.
Other than Mytesi and Canalevia-CA1 (for which we have conditional approval), we currently do not have regulatory approval for any of our prescription drug product candidates. Our current efforts are primarily focused on the ongoing commercialization of Mytesi and Canalevia-CA1, and development efforts related to Mytesi and Canalevia-CA1. With regard to Mytesi, we are focused on marketing the product in the United States as well as on development efforts related to a follow-on indication for Mytesi in CTD, an important supportive care indication for patients undergoing primary or adjuvant chemotherapy for cancer treatment, and symptomatic relief of COVID-related diarrhea. Mytesi is also in development for other possible follow-on indications, including orphan drug indications for symptomatic relief of diarrhea in infants and children with CDD and for adult and pediatric patients with SBS; and for supportive care for diarrhea relief in IBD; IBS-D; and for idiopathic/functional diarrhea. With regard to Canalevia-CA1, we are focused on the launch of the product in the United States for chemotherapy-induced diarrhea in dogs. In addition, a second-generation proprietary anti-secretory agent is in development for cholera. Mytesi previously received orphan-drug designation for SBS. Accordingly, our near term prospects, including our ability to generate material product revenue, obtain any new financing if needed to fund our business and operations or enter into potential strategic transactions, will depend heavily on the success of Mytesi and Canalevia-CA1.
Substantial time and capital resources have been previously devoted by third parties in the development of crofelemer, the active pharmaceutical ingredient, or API, in Mytesi and Canalevia, and the development of the botanical extract used in Equilevia and Neonorm. Both crofelemer and the botanical extract used in Equilevia and Neonorm were originally developed at Shaman Pharmaceuticals, Inc. (“Shaman”), by certain members of our management team, including Lisa A. Conte, our chief executive officer and president, and Steven R. King, Ph.D., our executive vice president of sustainable supply, ethnobotanical research and intellectual property and secretary. Shaman spent significant development resources before voluntarily filing for bankruptcy in 2001 pursuant to Chapter 11 of the U.S. Bankruptcy Code. The rights to crofelemer and the botanical extract used in Equilevia and Neonorm, as well as other intellectual property rights, were subsequently acquired by Napo from Shaman in 2001 pursuant to a court approved sale of assets. Ms. Conte founded Napo in 2001 and was the current interim chief executive officer of Napo and a member of Napo’s board of directors prior to the Merger. While at Napo, certain members of our management team, including Ms. Conte and Dr. King, continued the development of crofelemer. Following the merger of Jaguar and Napo in July 2017, Napo became Jaguar’s wholly-owned subsidiary. If we are not successful in the development and commercialization of Mytesi, our business and our prospects will be harmed.
43
The successful development and commercialization of Mytesi and Canalevia-CA1 will depend on a number of factors, including the following:
| ● | our ability to demonstrate to the satisfaction of the FDA and any other regulatory bodies, the safety and efficacy of Canalevia; |
| ● | our ability and that of our contract manufacturers to manufacture supplies of Mytesi and Canalevia-CA1 and to develop, validate and maintain viable commercial manufacturing processes that are compliant with current good manufacturing practices, or cGMPs, if required; |
| ● | our ability to successfully market Mytesi and Canalevia-CA1, whether alone or in collaboration with others; |
| ● | the availability, perceived advantages, relative cost, relative safety and relative efficacy of our prescription drug product candidates compared to alternative and competing treatments; |
| ● | the acceptance of our prescription drug product candidates and non-prescription products as safe and effective by physicians, veterinarians, patients, animal owners and the human and animal health community, as applicable; |
| ● | our ability to achieve and maintain compliance with all regulatory requirements applicable to our business; and |
| ● | our ability to obtain and enforce our intellectual property rights and obtain marketing exclusivity for our prescription drug product candidates and non-prescription products, and avoid or prevail in any third-party patent interference, patent infringement claims or administrative patent proceedings initiated by third parties or the U.S. Patent and Trademark Office (“USPTO”). |
Many of these factors are beyond our control. Accordingly, we may not be successful in developing or commercializing Mytesi, Neonorm, Equilevia, Canalevia or any of our other potential products. If we are unsuccessful or are significantly delayed in commercializing Mytesi, our business and prospects will be harmed and you may lose all or a portion of the value of your investment in our common stock.
If we are not successful in identifying, licensing, developing and commercializing additional product candidates and products, our ability to expand our business and achieve our strategic objectives could be impaired.
Although a substantial amount of our efforts is focused on the commercial performance of Mytesi and Canalevia-CA1, a key element of our strategy is to identify, develop and commercialize a portfolio of products to serve the gastrointestinal health market. Most of our potential products are based on our knowledge of medicinal plants. Our current focus is primarily on product candidates whose active pharmaceutical ingredient or botanical extract has been successfully commercialized or demonstrated to be safe and effective in human or animal trials. In some instances, we may be unable to further develop these potential products because of perceived regulatory and commercial risks. Even if we successfully identify potential products, we may still fail to yield products for development and commercialization for many reasons, including the following:
| ● | competitors may develop alternatives that render our potential products obsolete; |
| ● | an outside party may develop a cure for any disease state that is the target indication for any of our planned or approved drug products; |
| ● | potential products we seek to develop may be covered by third-party patents or other exclusive rights; |
| ● | a potential product may on further study be shown to have harmful side effects or other characteristics |
44
| that indicate it is unlikely to be effective or otherwise does not meet applicable regulatory criteria; |
| ● | a potential product may not be capable of being produced in commercial quantities at an acceptable cost, or at all; and |
| ● | a potential product may not be accepted as safe and effective by physicians, veterinarians, patients, animal owners, key opinion leaders and other decision-makers in the gastrointestinal health market, as applicable. |
While we are developing specific formulations, including flavors, methods of administration, new patents and other strategies with respect to our current potential products, we may be unable to prevent competitors from developing substantially similar products and bringing those products to market earlier than we can. If such competing products achieve regulatory approval and commercialization prior to our potential products, our competitive position may be impaired. If we fail to develop and successfully commercialize other potential products, our business and future prospects may be harmed and we will be more vulnerable to any problems that we encounter in developing and commercializing our current potential products.
Mytesi faces significant competition from other pharmaceutical companies, both for its currently approved indication and for planned follow-on indications, and our operating results will suffer if we fail to compete effectively.
The development and commercialization of products for human gastrointestinal health is highly competitive and our success depends on our ability to compete effectively with other products in the market. During the ongoing commercialization of Mytesi for its currently approved indication, and during the future commercialization of Mytesi for any planned follow-on indications, if such follow-on indications receive regulatory approval, we expect to compete with major pharmaceutical and biotechnology companies that operate in the gastrointestinal space, such as Takeda Pharmaceuticals, Allergan, Inc., Ironwood Pharmaceuticals, Inc., Synergy Pharmaceuticals Inc., Sebela Pharmaceuticals, Inc. and Salix Pharmaceuticals.
Many of our competitors and potential competitors in the human gastrointestinal space have substantially more financial, technical and human resources and greater ability to lower costs of manufacturing and sales and marketing than we do. Many also have more experience in the development, manufacture, regulation and worldwide commercialization of human gastrointestinal health products.
For these reasons, we cannot be certain that we and Mytesi can compete effectively.
We may be unable to obtain, or obtain on a timely basis, regulatory approval for our existing or future human or animal prescription drug product candidates under applicable regulatory requirements, which would harm our operating results.
The research, testing, manufacturing, labeling, approval, sale, marketing and distribution of human and animal health products are subject to extensive regulation. We are typically not permitted to market our prescription drug product candidates in the United States until we receive approval of the product from the FDA through the filing of an NDA or NADA, as applicable. To gain approval to market a prescription drug, we must provide the FDA with safety and efficacy data from pivotal trials that adequately demonstrate that our prescription drug product candidates are safe and effective for the intended indications. Likewise, to gain approval to market an animal prescription drug for a particular species, we must provide the FDA with safety and efficacy data from pivotal trials that adequately demonstrate that our prescription drug product candidates are safe and effective in the target species (e.g., dogs, cats or horses) for the intended indications. In addition, we must provide manufacturing data evidencing that we can produce our product candidates in accordance with cGMPs. For the FDA, we must also provide data from toxicology studies, also called target animal safety studies, and in some cases environmental impact data. In addition to our internal activities, we will partially rely on contract research organizations (“CROs”), and other third parties to conduct our toxicology studies and for certain other product development activities. The results of toxicology studies, other initial development activities, and/or any previous studies in humans or animals conducted by us or third parties
45
may not be predictive of future results of pivotal trials or other future studies, and failure can occur at any time during the conduct of pivotal trials and other development activities by us or our CROs. Our pivotal trials may fail to show the desired safety or efficacy of our prescription drug product candidates despite promising initial data or the results in previous human or animal studies conducted by others. Success of a prescription drug product candidate in prior animal studies, or in the treatment of humans, does not ensure success in subsequent studies. Clinical trials in humans and pivotal trials in animals sometimes fail to show a benefit even for drugs that are effective because of statistical limitations in the design of the trials or other statistical anomalies. Therefore, even if our studies and other development activities are completed as planned, the results may not be sufficient to obtain a required regulatory approval for a product candidate.
Regulatory authorities can delay, limit or deny approval of any of our prescription drug product candidates for many reasons, including:
| ● | if they disagree with our interpretation of data from our pivotal studies or other development efforts; |
| ● | if we are unable to demonstrate to their satisfaction that our product candidate is safe and effective for the target indication and, if applicable, in the target species; |
| ● | if they require additional studies or change their approval policies or regulations; |
| ● | if they do not approve of the formulation, labeling or the specifications of our current and future product candidates; and |
| ● | if they fail to approve the manufacturing processes of our third-party contract manufacturers. |
Further, even if we receive a required approval, such approval maybe for a more limited indication than we originally requested, and the regulatory authority may not approve the labeling that we believe is necessary or desirable for successful commercialization.
Any delay or failure in obtaining any necessary regulatory approval for the intended indications of our human or animal product candidates would delay or prevent commercialization of such product candidates and would harm our business and our operating results.
The results of our earlier studies of Mytesi may not be predictive of the results in any future clinical trials and species-specific formulation studies, respectively, and we may not be successful in our efforts to develop or commercialize line extensions of Mytesi.
Our human and animal product pipeline includes a number of potential indications of Mytesi, our lead prescription product. The results of our studies and other development activities and of any previous studies in humans or animals conducted by us or third parties may not be predictive of future results of these clinical studies and formulation studies, respectively. Failure can occur at any time during the conduct of these trials and other development activities. Even if our formulation/clinical studies and other development activities are completed as planned, the results may not be sufficient to pursue a particular line extension for Mytesi. Further, even if we obtain promising results from our clinical trials or species-specific formulation studies, as applicable, we may not successfully commercialize any line extension. Because line extensions are developed for a particular market, we may not be able to leverage our experience from the commercial launch of Mytesi in new markets. If we are not successful in developing and successfully commercializing these line extension products, we may not be able to grow our revenue and our business may be harmed.
Development of prescription drug products is inherently expensive, time-consuming and uncertain, and any delay or discontinuance of our current or future pivotal trials would harm our business and prospects.
Development of prescription drug products for human and animal gastrointestinal health remains an inherently lengthy, expensive and uncertain process, and our development activities may not be successful. We do not
46
know whether our current or planned pivotal trials for any of our product candidates will begin or conclude on time, and they may be delayed or discontinued for a variety of reasons, including if we are unable to:
| ● | address any safety concerns that arise during the course of the studies; |
| ● | complete the studies due to deviations from the study protocols or the occurrence of adverse events; |
| ● | add new study sites; |
| ● | address any conflicts with new or existing laws or regulations; or |
| ● | reach agreement on acceptable terms with study sites, which can be subject to extensive negotiation and may vary significantly among different sites. |
Further, we may not be successful in developing new indications for Mytesi and Canalevia-CA1, and Neonorm may be subject to the same regulatory regime as prescription drug products in jurisdictions outside the United States. Any delays in completing our development efforts will increase our costs, delay our development efforts and approval process and jeopardize our ability to commence product sales and generate revenue. Any of these occurrences may harm our business, financial condition and prospects. In addition, factors that may cause a delay in the commencement or completion of our development efforts may also ultimately lead to the denial of regulatory approval of our product candidates, which, as described above, would harm our business and prospects.
We will partially rely on third parties to conduct our development activities. If these third parties do not successfully carry out their contractual duties, we may be unable to obtain regulatory approvals or commercialize our current or future human or animal product candidates on a timely basis, or at all.
We will partially rely upon CROs to conduct our toxicology studies and for other development activities. We intend to rely on CROs to conduct one or more of our planned pivotal trials. These CROs are not our employees, and except for contractual duties and obligations, we have limited ability to control the amount or timing of resources that they devote to our programs or manage the risks associated with their activities on our behalf. We are responsible for ensuring that each of our studies is conducted in accordance with the development plans and trial protocols presented to regulatory authorities. Any deviations by our CROs may adversely affect our ability to obtain regulatory approvals, subject us to penalties or harm our credibility with regulators. The FDA and foreign regulatory authorities also require us and our CROs to comply with regulations and standards, GCPs or GLPs, for conducting, monitoring, recording and reporting the results of our studies to ensure that the data and results are scientifically valid and accurate.
Agreements with CROs generally allow the CROs to terminate in certain circumstances with little or no advance notice. These agreements generally will require our CROs to reasonably cooperate with us at our expense for an orderly winding down of the CROs’ services under the agreements. If the CROs conducting our studies do not comply with their contractual duties or obligations, or if they experience work stoppages, do not meet expected deadlines, or if the quality or accuracy of the data they obtain is compromised, we may need to secure new arrangements with alternative CROs, which could be difficult and costly. In such event, our studies also may need to be extended, delayed or terminated as a result, or may need to be repeated. If any of the foregoing were to occur, regulatory approval, if required, and commercialization of our product candidates may be delayed and we may be required to expend substantial additional resources.
Even if we obtain regulatory approval for planned follow-on indications of crofelemer, Canalevia or our other product candidates, they may never achieve market acceptance. Further, even if we are successful in the ongoing commercialization of Mytesi and Canalevia, we may not achieve commercial success.
If we obtain necessary regulatory approvals for planned follow-on indications of crofelemer or our other product candidates, such products may still not achieve market acceptance and may not be commercially successful. Market acceptance of Mytesi, Canalevia, and any of our other products depends on a number of factors, including:
47
| ● | the safety of our products as demonstrated in our target animal studies; |
| ● | the indications for which our products are approved or marketed; |
| ● | the potential and perceived advantages over alternative treatments or products, including generic medicines and competing products currently prescribed by physicians or veterinarians, as applicable, and, in the case of animal products, products approved for use in humans that are used extra-label in animals; |
| ● | the acceptance by physicians, veterinarians, companion animal owners, as applicable, of our products as safe and effective; |
| ● | the cost in relation to alternative treatments and willingness on the part of physicians, veterinarians, patients and animal owners, as applicable, to pay for our products; |
| ● | the prevalence and severity of any adverse side effects of our products; |
| ● | the relative convenience and ease of administration of our products; and |
| ● | the effectiveness of our sales, marketing and distribution efforts. |
Any failure by Mytesi or Canalevia to achieve market acceptance or commercial success would harm our financial condition and results of operations.
Human and animal gastrointestinal health products are subject to unanticipated post-approval safety or efficacy concerns, which may harm our business and reputation.
The success of our commercialization efforts will depend upon the perceived safety and effectiveness of human and animal gastrointestinal health products, in general, and of our products, in particular. Unanticipated safety or efficacy concerns can subsequently arise with respect to approved prescription drug products, such as Mytesi, or non-prescription products, such as Neonorm, which may result in product recalls or withdrawals or suspension of sales, as well as product liability and other claims. Any safety or efficacy concerns, or recalls, withdrawals or suspensions of sales of our products could harm our reputation and business, regardless of whether such concerns or actions are justified.
Future federal and state legislation may result in increased exposure to product liability claims, which could result in substantial losses.
Under current federal and state laws, companion and production animals are generally considered to be the personal property of their owners and, as such, the owners’ recovery for product liability claims involving their companion and production animals may be limited to the replacement value of the animal. Companion animal owners and their advocates, however, have filed lawsuits from time to time seeking non-economic damages such as pain and suffering and emotional distress for harm to their companion animals based on theories applicable to personal injuries to humans. If new legislation is passed to allow recovery for such non-economic damages, or if precedents are set allowing for such recovery, we could be exposed to increased product liability claims that could result in substantial losses to us if successful. In addition, some horses can be worth millions of dollars or more, and product liability for horses may be very high. While we currently have product liability insurance, such insurance may not be sufficient to cover any future product liability claims against us.
48
If we fail to retain current members of our senior management, or to identify, attract, integrate and retain additional key personnel, our business will be harmed.
Our success depends on our continued ability to attract, retain and motivate highly qualified management and scientific personnel. We are highly dependent upon our senior management, particularly Lisa A. Conte, our president and Chief Executive Officer. The loss of services of any of our key personnel would cause a disruption in our ability to develop our current or future product pipeline and commercialize our products and product candidates. Although we have offer letters with these key members of senior management, such agreements do not prohibit them from resigning at any time. For example, the resignation of our former Chief Financial Officer and Treasurer, Karen Wright, in August 2019, caused us to incur additional expenses and expend resources to ensure a smooth transition with her successor, which diverted management attention away from executing our operational plan during this period. To help attract, retain, and motivate qualified management and other personnel, we use share-based incentive awards such as employee stock options and restricted stock units. However, given the volatility in our stock price, it may be more difficult and expensive to recruit and retain employees, particularly senior management, through grants of stock or stock options. If our share-based compensation ceases to be viewed as a valuable benefit, our ability to attract, retain, and motivate qualified management and other personnel could be weakened, which could harm our results of operations and adversely affect the timing or outcomes of our current and planned studies, as well as the prospects for commercializing our products.
In addition, competition for qualified personnel in the human gastrointestinal health field is intense because there are a limited number of individuals who are trained or experienced in the field. We will need to hire additional personnel as we expand our product development and commercialization activities. Even if we are successful in hiring qualified individuals, as we are a growing organization, we do not have a track record for integrating and retaining individuals. If we are not successful in identifying, attracting, integrating or retaining qualified personnel on acceptable terms, or at all, our business will be harmed.
We are dependent on two suppliers for the raw material used to produce the active pharmaceutical ingredient in Mytesi and Canalevia. The termination of either of these contracts would result in a disruption to product development and our business will be harmed.
The raw material used to manufacture Mytesi and Canalevia-CA1 is CPL derived from the Croton lechleri tree, which is found in countries in South America, principally Peru. The ability of our contract suppliers to harvest CPL is governed by the terms of their respective agreements with local government authorities. Although CPL is available from multiple suppliers, we only have contracts with two suppliers to obtain CPL and arrange the shipment to our contract manufacturer. Accordingly, if our contract suppliers do not or are unable to comply with the terms of our respective agreements, and we are not able to negotiate new agreements with alternate suppliers on terms that we deem commercially reasonable, it may harm our business and prospects. The countries from which we obtain CPL could change their laws and regulations regarding the export of the natural products or impose or increase taxes or duties payable by exporters of such products. Restrictions could be imposed on the harvesting of the natural products or additional requirements could be implemented for the replanting and regeneration of the raw material. Such events could have a significant impact on our cost and ability to produce Mytesi, Canalevia-CA1 and anticipated line extensions.
We are dependent upon third-party contract manufacturers, both for the supply of the active pharmaceutical ingredient in Mytesi and Canalevia-CA1, as well as for the supply of finished products for commercialization.
We are in negotiations with Indena for the purification of the CPL received from our suppliers into the API used to manufacture Canalevia-CA1 and Mytesi , as well as the botanical extract in Neonorm. Indena has never manufactured either such ingredient to commercial scale. Glenmark is the current manufacturer of crofelemer, the active API in Canalevia-CA1and Mytesi. As announced in October of 2015, we have entered into an agreement with Patheon, a provider of drug development and delivery solutions, under which Patheon provides enteric-coated tablets to us for use in humans and animals. We also may contract with additional third parties for the formulation and supply of finished products, which we will use in our planned studies and commercialization efforts.
49
We are dependent upon our contract manufacturers for the supply of the API in Mytesi and Canalevia-CA1. We currently have sufficient quantities of the API used in Mytesi and Canalevia to support our projected sales efforts. We are working with our contract manufacturers to increase API manufacturing capacity of the API to support the sales forecast for 2022 and beyond. If our contract manufacturer cannot manufacture sufficient quantities of the API in a timely manner, we could suffer losses due to lost sales opportunities. We currently have sufficient quantities of the botanical extract used in Neonorm and Equilevia to support planned commercialization efforts for Neonorm and Equilevia. If we are not successful in reaching agreements with third parties on terms that we consider commercially reasonable for manufacturing and formulation of Mytesi and Canalevia-CA1, or if our contract manufacturer and formulator are not able to produce sufficient quantities or quality of the Mytesi and Canalevia-CA1 API or finished product under their agreements, it could delay our plans and harm our business prospects. For example, as a result of the outbreak in 2020 of SARS-CoV-2, the virus that causes COVID-19, that originated in Wuhan, China and then spread globally, our suppliers and contract manufacturer could be disrupted by worker absenteeism, quarantines, or other travel or health-related restrictions or could incur increased costs associated with ensuring the safety and health of their personnel. If our suppliers or contract manufacturer is so affected, our supply chain could be disrupted, our product shipments could be delayed, our costs could be increased and our business could be adversely affected.
The facilities used by our third-party contractors are subject to inspections, including by the FDA, and other regulators, as applicable. We also depend on our third-party contractors to comply with cGMPs. If our third-party contractors do not maintain compliance with these strict regulatory requirements, they and we will not be able to secure or maintain regulatory approval for their facilities, which would have an adverse effect on our operations. In addition, in some cases, we also are dependent on our third-party contractors to produce supplies in conformity to our specifications and maintain quality control and quality assurance practices and not to employ disqualified personnel. If the FDA or a comparable foreign regulatory authority does not approve the facilities of our third-party contractors if so required, or if it withdraws any such approval in the future, we may need to find alternative manufacturing or formulation facilities, which could result in delays in our ability to develop or commercialize our products, if at all. We and our third-party contractors also may be subject to penalties and sanctions from the FDA and other regulatory authorities for any violations of applicable regulatory requirements. The European Medicines Agency (the “EMA”), employ different regulatory standards than the FDA, so we may require multiple manufacturing processes and facilities for the same product candidate or any approved product. We are also exposed to risk if our third-party contractors do not comply with the negotiated terms of our agreements, or if they suffer damage or destruction to their facilities or equipment.
If we are unable to establish sales capabilities on our own or through third parties, we may not be able to market and sell our current or future human products and product candidates, if approved, and generate product or other revenue.
We currently have limited sales, marketing or distribution capabilities, and prior to Napo’s launch of Mytesi for the symptomatic relief of noninfectious diarrhea in adults with HIV/AIDS on antiretroviral therapy, and our launch of Neonorm for pre-weaned dairy calves and Canalevia for chemotherapy-induced diarrhea in dogs, we had no experience in the sale, marketing and distribution of human or animal health products. There are significant risks involved in building and managing a sales organization, including our potential inability to attract, hire, retain and motivate qualified individuals, generate sufficient sales leads, provide adequate training to sales and marketing personnel and effectively oversee a geographically dispersed sales and marketing team. Any failure or delay in the development of our internal sales, marketing and distribution capabilities and entry into adequate arrangements with distributors or other partners would adversely impact the commercialization of Mytesi, and Canalevia-CA1. If we are not successful in commercializing Mytesi and/or Canalevia-CA1, for their respective currently approved or conditionally approved indications or for any potential follow-on indications, either on our own or through one or more distributors, or in generating upfront licensing or other fees, including through the previously announced licensing arrangement between Napo Pharmaceuticals, Inc. and Napo Therapeutics S.p.A., we may never generate significant revenue and may continue to incur significant losses, which would harm our financial condition and results of operations.
50
We will need to increase the size of our organization and may not successfully manage such growth.
As of December 31, 2021, we had 52 employees. Our ability to manage our growth effectively will require us to hire, train, retain, manage and motivate additional employees and to implement and improve our operational, financial and management systems. These demands also may require the hiring of additional senior management personnel or the development of additional expertise by our senior management personnel. If we fail to expand and enhance our operational, financial and management systems in conjunction with our potential future growth, it could harm our business and operating results.
Canalevia-CA1 and, our animal health prescription drug product candidates, if approved, may be marketed in the United States only in the target animals and for the indications for which they are approved, and if we want to expand the approved animals or indications, it will need to obtain additional approvals, which may not be granted.
We may market or advertise Canalevia-CA1 and our animal health prescription drug product candidates are approved by regulatory authorities, only in the specific species and for treatment of the specific indications for which they were approved, which could limit use of the products by veterinarians and animal owners. We intend to develop, promote and commercialize approved products for new animal treatment indications in the future, but we cannot be certain whether or at what additional time and expense we will be able to do so. If we do not obtain marketing approvals for new indications, our ability to expand our animal health business may be harmed.
Under the Animal Medicinal Drug Use Clarification Act of 1994, veterinarians are permitted to prescribe extra-label uses of fully approved animal drugs and approved human drugs for animals under certain conditions. While veterinarians may in the future prescribe and use human-approved products or use our products for extra-label uses, we may not promote our animal health products for extra-label uses. We note that extra-label uses are uses for which the product has not received approval. If the FDA determines that any of our marketing activities constitute promotion of an extra-label use, we could be subject to regulatory enforcement, including seizure of any misbranded or mislabeled drugs, and civil or criminal penalties, any of which could have an adverse impact on our reputation and expose us to potential liability. We will continue to spend resources ensuring that our promotional claims for our animal health products and product candidates remain compliant with applicable FDA laws and regulations, including materials we post or link to on our website. For example, in 2012, our Chief Executive Officer received an “untitled letter” from the FDA while at Napo regarding preapproval promotion statements constituting misbranding of crofelemer, which was then an investigational drug. These statements were included in archived press releases included on Napo’s website. Napo was required to expend time and resources to revise its website to remove the links in order to address the concerns raised in the FDA’s letter.
The misuse or extra-label use of Mytesi, Canalevia and our human or animal prescription drug product candidates approved by regulatory authorities, may harm our reputation or result in financial or other damages.
If our human or animal prescription drug product candidates are approved by regulatory authorities, there may be increased risk of product liability if physicians, veterinarians, patients, animal owners or others, as applicable, attempt to use such products extra-label, including the use of our products for indications or in species for which they have not been approved. Furthermore, the use of an approved human or animal drug such as Mytesi and Canalevia for indications other than those indications for which such products have been approved may not be effective, which could harm our reputation and lead to an increased risk of litigation. If we are deemed by a governmental or regulatory agency to have engaged in the promotion of any approved human or animal product for extra-label use, such agency could request that we modify our training or promotional materials and practices and we could be subject to significant fines and penalties, and the imposition of these sanctions could also affect our reputation and position within the gastrointestinal health industry. Any of these events could harm our reputation and our operating results.
51
We may be unable to obtain, or obtain on a timely basis, a renewal of conditional approval for Canalevia-CA1, or to eventually obtain full regulatory approval of Canalevia-CA1, which would harm our operating results.
On December 21, 2021, the FDA conditionally approved Canalevia-CA1 (crofelemer delayed-release tablets) for the treatment of CID in dogs under application number 141-552. FDA’s conditional approval allows the Company to legally sell Canalevia-CA1 before proving it meets the “substantial evidence” standard of effectiveness for full approval. The Company may request renewal of the conditional approval annually for up to four more years, for a total of five years of conditional approval. To receive a renewal from FDA, the Company must show active progress toward proving “substantial evidence of effectiveness” for full approval.
If FDA grants all four annual renewals, the Company has up to four-and-a-half years to develop and submit the necessary data to complete the effectiveness requirement. If the Company does not submit all necessary information to support full approval of Canalevia-CA1 by this four-and-a-half-year deadline, the conditional approval terminates immediately. The Company would then be required to stop marketing the drug because it would be considered to be unapproved.
If the Company submits the necessary information before the four-and-a-half-year deadline, the conditional approval period runs another six months, for a total of five years, while FDA reviews the application for full approval. The conditional approval automatically terminates five years after the date of the initial conditional approval. If FDA does not fully approve the drug before the five-year termination date, the Company would then have to stop marketing the drug because it would be considered to be unapproved.
We may not maintain the benefits associated with MUMS designation, including market exclusivity.
Although we have received MUMS designation for Canalevia-CA1 for the treatment of CID in dogs, we may not maintain the benefits associated with MUMS designation. MUMS designation is a status similar to “orphan drug” status for human drugs. When we were granted MUMS designation for Canalevia-CA1 for the indication of CID in dogs, we became eligible for incentives to support the approval or conditional approval of the designated use. This designation does not allow us to commercialize a product until such time as we obtain approval or conditional approval of the product.
Because Canalevia-CA1 has received MUMS designation for the identified particular intended use, we are eligible to obtain seven years of exclusive marketing rights upon approval (or conditional approval) of Canalevia-CA1 for that intended use and become eligible for grants to defray the cost of our clinical work. Each designation that is granted must be unique, i.e., only one designation can be granted for a particular API in a particular dosage form for a particular intended use. The intended use includes both the target species and the disease or condition to be treated.
At some point, we could lose MUMS designation. The basis for a lost designation can include but is not limited to, our failure to engage with due diligence in moving forward with a non-conditional approval. In addition, MUMS designation may be withdrawn for a variety of reasons such as where the FDA determines that the request for designation was materially defective, or if the manufacturer is unable to assure sufficient quantity of the prescription drug product to meet the needs of animals with the rare disease or condition. If this designation is lost, it could have a negative impact on the product and us, which includes but is not limited to, market exclusivity related to MUMS designation, or eligibility for grants as a result of MUMS designation.
The market for our human and animal products, and the gastrointestinal health market as a whole, is uncertain and may be smaller than we anticipate, which could lead to lower revenue and harm our operating results.
It is very difficult to estimate the commercial potential of any of our human or animal products because the gastrointestinal health market continues to evolve and it is difficult to predict the market potential for our products. The market will depend on important factors such as safety and efficacy compared to other available treatments, changing standards of care, preferences of physicians, as applicable, the willingness of patients, as applicable, to pay for such products, and the availability of competitive alternatives that may emerge either during the product development process or after commercial introduction. If the market potential for our human or animal products is less
52
than we anticipate due to one or more of these factors, it could negatively impact our business, financial condition and results of operations. Further, the willingness of patients to pay for our products may be less than we anticipate, and may be negatively affected by overall economic conditions.
Insurance coverage for Mytesi for its current approved indication could decrease or end, or Mytesi might not receive insurance coverage for any approved follow-on indications, which could lead to lower revenue and harm our operating results.
For its current approved indication, Mytesi is currently reimbursed by almost all of commercial and Medicare insurance plans. Mytesi is currently covered on Medicaid in all 50 states. However, the nature or extent of coverage for Mytesi by any of these plans or programs could change or be terminated, or Mytesi might not receive insurance coverage for any approved follow-on indications. Either outcome could lead to significantly lower revenue and significantly harm our operating results.
We may engage in future acquisitions that increase our capital requirements, dilute our stockholders, cause us to incur debt or assume contingent liabilities and subject us to other risks.
We may evaluate various strategic transactions, including licensing or acquiring complementary products, technologies or businesses. Any potential acquisitions may entail numerous risks, including increased operating expenses and cash requirements, assimilation of operations and products, retention of key employees, diversion of our management’s attention and uncertainties in our ability to maintain key business relationships of the acquired entities. In addition, if we undertake acquisitions, we may issue dilutive securities, assume or incur debt obligations, incur large one-time expenses and acquire intangible assets that could result in significant future amortization expense. Moreover, we may not be able to locate suitable acquisition opportunities and this inability could impair our ability to grow or obtain access to technology or products that may be important to the development of our business.
Certain of the countries in which we plan to commercialize our products in the future are developing countries, some of which have potentially unstable political and economic climates.
We may commercialize our products in jurisdictions that are developing and emerging countries. This may expose us to the impact of political or economic upheaval, and we could be subject to unforeseen administrative or fiscal burdens. At present, we are not insured against the political and economic risks of operating in these countries. Any significant changes to the political or economic climate in any of the developing countries in which we operate or plan to sell products either now or in the future may have a substantial adverse effect on our business, financial condition, trading performance and prospects.
Fluctuations in the exchange rate of foreign currencies could result in currency transactions losses.
As we expand our operations, we expect to be exposed to risks associated with foreign currency exchange rates. We anticipate that we may commercialize Mytesi and Canalevia-CA1 and its line extensions in jurisdictions outside the United States. As a result, we may also be further affected by fluctuations in exchange rates in the future to the extent that sales are denominated in currencies other than U.S. dollars. We do not currently employ any hedging or other strategies to minimize this risk, although we may seek to do so in the future.
Laws and regulations governing global trade compliance could adversely impact our business.
The U.S. Department of the Treasury’s Office of Foreign Assets Control (“OFAC”), and the Bureau of Industry and Security (“BIS”) at the U.S. Department of Commerce, administer certain laws and regulations that restrict U.S. persons and, in some instances, non-U.S. persons, in conducting activities, transacting business with or making investments in certain countries, governments, entities and individuals subject to U.S. economic sanctions. In addition, engaging in sales activities to foreign governments introduces additional compliance risks, including risks specific to anti-bribery regulations, including the U.S. Foreign Corrupt Practices Act of 1977, as amended, or the FCPA, the U.K. Bribery Act 2010 and other similar statutory requirements prohibiting bribery and corruption in the jurisdictions in which we operate. The FCPA prohibits U.S. corporations and their representatives from offering,
53
promising, authorizing or making payments to any foreign government official, government staff member, political party or political candidate in an attempt to obtain or retain business abroad. The scope of the FCPA includes interactions with certain healthcare professionals in many countries. Other countries have enacted similar anti-corruption laws and/or regulations.
Our international operations subject us to these laws and regulations, which are complex, restrict our business dealings with certain countries, governments, entities, and individuals, and are constantly changing. Further restrictions may be enacted, amended, enforced or interpreted in a manner that materially impacts our operations.
Violations of these regulations are punishable by civil penalties, including fines, denial of export privileges, injunctions, asset seizures, debarment from government contracts and revocations or restrictions of licenses, as well as criminal fines and imprisonment. We have established policies and procedures designed to assist with our compliance with such laws and regulations. However, there can be no assurance that our policies and procedures will prevent us from violating these regulations in every transaction in which we may engage, or that any businesses that we may acquire have complied with such regulations, and such a violation could adversely affect our reputation, business, financial condition, results of operations and cash flows.
There are other gastrointestinal-focused human pharmaceutical companies, and we face competition in the marketplaces in which we operate or plan to operate.
Our commercial success in the human drug arena remains dependent on maintaining or establishing a competitive position in the market for the current, approved specialty indication of Mytesi as well as for planned Mytesi follow-on indications. In the IBS-D market in particular, several competitors have commercially available products approved for our planned IBS-D indication. The availability of our competitors’ products could limit the demand, and the price we are able to charge, for any drug candidate we develop. The inability to compete with existing or subsequently introduced drug candidates would have a material adverse impact on our business, financial condition and prospects.
Our obligations to Streeterville are secured by a security interest in all of Napo’s lechlemer assets, so if we default on those obligations, Streeterville could foreclose on our assets.
Our obligations under the secured promissory note issued to Streeterville Capital, LLC (“Streeterville”) are secured by a first priority security interest in all existing and future lechlemer technology held by Napo, including intellectual property, as provided in the Security Agreement, dated January 19, 2021 between Napo and Streeterville. As a result, if we default on our obligations under these agreements, Streeterville could foreclose on its security interests and liquidate some or all of these assets, which would harm our plans to develop and commercialize lechlemer, financial condition and results of operations and could require us to reduce or cease operations with respect to lechlemer.
Our royalty interests require us to make minimum royalty payments, even if we do not sell a sufficient amount of products to cover such payments, which may strain our cash resources.
Since March 2020, we have sold royalty interests to certain lenders that entitle such lenders to receive future royalties on sales of our products. These royalty interests require us to make minimum royalty payments beginning 2021, even if we do not sell a sufficient amount of product to cover such payments, which may strain our cash resources. The total minimum royalty payments will be $6.0 million in 2022, $18.0 million in 2023, $13.9 million in 2024, $7.10 million 2025, and $3.9 million in 2026.
Failure in our information technology systems, including by cyber-attacks or other data security incidents, could significantly disrupt our operations.
Our operations depend, in part, on the continued performance of our information technology systems. Our information technology systems are potentially vulnerable to physical or electronic break-ins, computer viruses, phishing attacks and other types of disruptions. We have and continue to experience cyber-attacks of varying degrees.
54
Our security measures may also be breached due to employee error, malfeasance, system errors or other vulnerabilities. Such breach or unauthorized access or attempts by outside parties to fraudulently induce employees or users to disclose sensitive information in order to gain access to our data could result in significant legal and financial exposure, and damage to our reputation that could potentially have an adverse effect on our business. Because the techniques used to obtain unauthorized access, or sabotage systems change frequently, become more sophisticated, and often are not recognized until launched against a target, we may be unable to anticipate these techniques or to implement adequate preventative measures. Additionally, cyber-attacks could also compromise trade secrets and other sensitive information and result in such information being disclosed to others and becoming less valuable, which could negatively affect our business. Although we have information technology security systems, a successful cybersecurity attack or other data security incident could result in the misappropriation and/or loss of confidential or personal information, create system interruptions, deploy malicious software that attacks our systems, or result in financial losses. It is possible that a cybersecurity attack might not be noticed for some period of time. The occurrence of a cyber security attack or incident could result in business interruptions from the disruption of our information technology systems, or negative publicity resulting in reputational damage with our shareholders and other stakeholders and/or increased costs to prevent, respond to or mitigate cybersecurity events. In addition, the unauthorized dissemination of sensitive personal information or proprietary or confidential information could expose us or other third-parties to regulatory fines or penalties, litigation and potential liability, or otherwise harm our business.
The novel coronavirus global pandemic could adversely impact our business, including our supply chain, clinical trials and commercialization of Mytesi and Canalevia.
As a result of the outbreak of SARS-CoV-2, the virus that causes COVID-19, we may experience disruptions that could severely impact our supply chain, ongoing and future clinical trials and commercialization of Mytesi and Canalevia. For example, COVID-19 has resulted in increased travel restrictions and the shutdown or delay of business activities in various regions, including certain activities of our contract manufacturers in India and in Italy. To the extent our suppliers and contract manufacturer are unable to comply with their obligations under our agreements with them or they are otherwise unable to deliver or are delayed in delivering raw materials, Mytesi and Canalevia API or finished products to us due to COVID-19, our ability to continue meeting commercial demand for Mytesi and Canalevia in the United States or advancing development of our product candidates may become impaired. Travel restrictions and shutdowns in business operations as a result of the outbreak may also limit our ability to pursue business development activities, including limiting onsite diligence of manufacturing facilities owned or operated by the Company and our contractors.
Such travel restrictions and shutdowns in business operations may also adversely impact our commercialization of Mytesi and Canalevia, including limiting the ability of our marketing and sales force to engage with healthcare providers and patient groups, and could result in patients postponing visits to healthcare provider facilities, healthcare providers temporarily closing their offices or restricting patient visits, pharmacies being closed or suffering supply chain disruptions, healthcare provider and/or pharmacy employees being unavailable and general disruptions in the operations of payors, distributors, logistics providers and other third parties that are necessary for Mytesi to be prescribed and reimbursed.
COVID-19 continues to rapidly evolve. The extent to which COVID-19, and mutated variants of SARS-CoV-2 – the virus that causes COVID-19, may impact our business, including our supply chain, clinical trials, commercialization of Mytesi and Canalevia and distribution channels, will depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the ultimate geographic spread of the pandemic, the duration of the pandemic, travel restrictions and social distancing in the United States and other countries, business closures or business disruptions and the effectiveness of actions taken in the United States and other countries to contain and treat the pandemic.
55
Long-term remote work arrangements may adversely affect our business.
Many of our employees have been working remotely the past year and will continue to do so this year. An extended period of remote work arrangements could strain our business continuity plans, introduce operational risk, including but not limited to cyber-security risks, impair the effectiveness of our internal controls over financial reporting and impact our ability to manage our business.
Substantially all of our revenue for recent periods has been received from three customers.
Substantially all of our revenue has been derived from three customers. Except for the shelter-in-place mandate, we have not been made aware by our customers if they have experienced other issues arising due to COVID-19 that may materially impact our financial condition, liquidity or results of operations. We will continue to have dialogues with our customers.
We are subject to state laws in California that require gender and diversity quotas for boards of directors of public companies headquartered in California.
In September 2018, California enacted SB 826, requiring public companies headquartered in California to maintain minimum female representation on their boards of directors as follows: by December 31, 2019, public company boards must have a minimum of one female director; by December 31, 2021, public company boards with five members will be required to have at least two female directors, and public company boards with six or more members will be required to have at least three female directors.
Additionally, on September 30, 2020, California enacted AB 979, requiring public companies with principal executive offices in California to each have at least one director from an underrepresented community based on ethnicity and sexual orientation by December 31, 2021. By December 31, 2022, each of these companies will be required to have at least two directors from such underrepresented communities if such company has more than four but fewer than nine directors, or at least three directors from underrepresented communities if the company has nine or more directors.
Our board of directors currently includes one female director. As a result, we are currently not in compliance with either law. An initial violation of either law can result in a fine from the California Secretary of State in the amount of $100,000, with each subsequent violation resulting in a fine of $300,000. We cannot assure that we can recruit, attract and/or retain qualified members of the board and continue to meet gender and diversity quotas as required by California law, which may cause certain investors to divert their holdings in our securities and expose us to financial penalties and/or reputational harm.
Risks Related to Intellectual Property
We cannot be certain that our patent strategy will be effective to protect against competition.
Our commercial success depends in large part on obtaining and maintaining patent, trademark and trade secret protection of our human or animal products, both prescription and non-prescription, our current human or animal product candidates and any future human or animal product candidates, and their respective components, formulations, methods used to manufacture them and methods of treatment, as well as successfully defending our patents and other intellectual property rights against third-party challenges. Our ability to stop unauthorized third parties from making, using, selling, offering to sell or importing our products or our product candidates is dependent upon the extent to which we have rights under valid and enforceable patents, trade secrets and other similar intellectual property that cover these activities. The patent prosecution process is expensive and time-consuming, and we may not be able to prepare, file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of inventions made in the course of development and commercialization activities in time to obtain patent protection on them.
56
We have a portfolio of United States and foreign issued patents and pending applications related to our products and product candidates. We have three issued United States patents listed in the FDA’s Orange Book for Mytesi. We plan to rely on certain of these issued patents as protection for Canalevia. The strength of patents in the field of pharmaceuticals and animal health involves complex legal and scientific questions and can be uncertain. We cannot be certain that pending applications will issue as patents. For those patents that are already issued and even if other patents do successfully issue, third parties may challenge their validity, enforceability or scope, which may result in such patents being narrowed, invalidated or held unenforceable. Furthermore, even if they are unchallenged, our patents may not adequately protect our intellectual property or prevent others from designing around their claims. If the patents we have are not maintained or their scope is significantly narrowed or if we are not able to obtain issued patents from pending applications, our business and prospects would be harmed.
The Leahy-Smith America Invents Act, patent reform legislation enacted in 2011, could increase the uncertainties and costs surrounding the prosecution of any patent applications and the enforcement or defense of any patents that issue. The Leahy-Smith Act introduced significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted, redefine prior art, may affect patent litigation, and switch the U.S. patent system from a “first-to-invent” system to a “first-to-file” system. Under a “first-to-file” system, assuming the other requirements for patentability are met, the first inventor to file a patent application generally is entitled to the patent on an invention regardless of whether another inventor had made the invention earlier. The USPTO developed regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first-to-file provisions, became effective on March 16, 2013. Among some of the other changes to the patent laws are changes that limit where a patentee may file a patent infringement suit and that provide opportunities for third parties to challenge any issued patent in the USPTO. The Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our patents and any other patents that issue, all of which could harm our business and financial condition.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance and annuity fees on any issued patent and, in certain jurisdictions, pending applications, are due to be paid to the USPTO and foreign patent agencies in several stages over the lifetime of the patent. The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we fail to maintain the patents and patent applications covering our prescription drug products, prescription drug product candidates and non-prescription products, our competitors might be able to enter the market, which would harm our business.
Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, which would be costly, time-consuming and, if successfully asserted against us, delay or prevent the development and commercialization of our current or future products and product candidates.
Our research, development and commercialization activities may infringe or otherwise violate or be claimed to infringe or otherwise violate patents owned or controlled by other parties. There may be patents already issued of which we are unaware that might be infringed by a product or one of our current or future prescription drug product candidates or non-prescription products. Moreover, it is also possible that patents may exist that we are aware of, but that we do not believe are relevant to our current or future prescription drug product candidates or non-prescription products, which could nevertheless be found to block our freedom to market these products. Because patent applications can take many years to issue and may be confidential for 18 months or more after filing, there may be
57
applications now pending of which we are unaware and which may later result in issued patents that may be infringed by our current or future prescription drug product candidates or non-prescription products. We cannot be certain that our products, current or future prescription drug product candidates or non-prescription products will not infringe these or other existing or future third-party patents. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents.
To the extent we become subject to future third-party claims against us or our collaborators, we could incur substantial expenses and, if any such claims are successful, we could be liable to pay substantial damages, including treble damages and attorney’s fees if we or our collaborators are found to be willfully infringing a third party’s patents. If a patent infringement suit were brought against us or our collaborators, we or they could be forced to stop or delay research, development, manufacturing or sales of the human or animal prescription drug or non-prescription product that is the subject of the suit. Even if we are successful in defending such claims, infringement and other intellectual property claims can be expensive and time-consuming to litigate and divert management’s attention from our business and operations. As a result of or in order to avoid potential patent infringement claims, we or our collaborators may be compelled to seek a license from a third party for which we would be required to pay license fees or royalties, or both. Moreover, these licenses may not be available on acceptable terms, or at all. Even if we or our collaborators were able to obtain such a license, the rights may be nonexclusive, which could allow our competitors access to the same intellectual property. Any of these events could harm our business and prospects.
Our proprietary position depends upon the botanical guidance of our drug approval and patents that are formulation or method-of-use patents, which do not prevent a competitor from using the same human or animal drug for another use.
Composition-of-matter patents on the API in prescription drug products are generally considered to be the strongest form of intellectual property protection because such patents provide protection without regard to any particular method of use or manufacture or formulation of the API used. The composition-of-matter patents for crofelemer, the API in Mytesi and Canalevia-CA1, have expired, and the issued patents and applications relevant to our products and product candidates cover methods of use for crofelemer and the botanical extract in Neonorm and Equilevia.
Method-of-use patents protect the use of a product for the specified method and formulation patents cover formulations of the API or botanical extract. These types of patents do not prevent a competitor from developing or marketing an identical product for an indication that is outside the scope of the patented method or from developing a different formulation that is outside the scope of the patented formulation. Moreover, with respect to method-of-use patents, even if competitors do not actively promote their product for our targeted indications or uses for which we may obtain patents, physicians may recommend that patients use our products extra-label, and veterinarians may recommend that animal owners use these products extra-label, or animal owners may do so themselves. Although extra-label use may infringe or contribute to the infringement of method-of-use patents, the practice is common and such infringement is difficult to prevent or prosecute.
We may be involved in lawsuits to protect or enforce our patents, which could be expensive, time-consuming and unsuccessful, and third parties may challenge the validity or enforceability of our patents and they may be successful.
We intend to rely upon a combination of regulatory exclusivity periods, patents, trade secret protection, and confidentiality agreements to protect the intellectual property related to Mytesi, our current prescription drug product candidates, non-prescription products and our development programs.
If the breadth or strength of protection provided by any patents, patent applications or future patents we may own, license, or pursue with respect to any of our current or future product candidates or products is threatened, it could threaten our ability to commercialize any of our current or future human or animal product candidates or products. Further, if we encounter delays in our development efforts, the period of time during which we could market any of our current or future product candidates or products under any patent protection we obtain would be reduced.
58
Given the amount of time required for the development, testing and regulatory review of new product candidates or products, patents protecting such candidates might expire before or shortly after such product candidates or products are commercialized. The United States Patent and Trademark Office has issued a patent term extension certificate extending the term of US 7,341,744 by 1075 days under 35 USC 156. With respect to requests for patent term extensions, the applicable authorities, including the USPTO and the FDA, and any equivalent regulatory authority in other countries, may not agree with our assessment of whether such extensions are available, and may refuse to grant extensions to patents, or may grant more limited extensions than requested. If this occurs, our competitors may take advantage of our investment in development and trials by referencing our clinical and preclinical data and launch their product earlier than might otherwise be the case.
Even where laws provide protection or we are able to obtain patents, costly and time-consuming litigation may be necessary to enforce and determine the scope of our proprietary rights, and the outcome of such litigation would be uncertain. Moreover, any actions we may bring to enforce our intellectual property against our competitors could provoke them to bring counterclaims against us, and some of our competitors have substantially greater intellectual property portfolios than we have. To counter infringement or unauthorized use of any patents we may obtain, we may be required to file infringement claims, which can be expensive and time-consuming to litigate. In addition, if we or one of our future collaborators were to initiate legal proceedings against a third party to enforce a patent covering one of our products, current product candidates, or one of our future products, the defendant could counterclaim that the patent is invalid or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity or unenforceability are commonplace and challenges to validity of patents in certain foreign jurisdictions is common as well. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness, non-enablement or lack of statutory subject matter. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant material information from the USPTO, or made a materially misleading statement, during prosecution. Under the Hatch-Waxman Act, a competitor seeking to market a generic form of Mytesi before the expiration of any of the patents listed in the FDA’s Orange Book for Mytesi could file an ANDA with a certification under 21 U.S.C. § 3559(j)(2)(A)(iv) that each of these patents (except for those which the ANDA filer states it will market only after its expiration) is either invalid, unenforceable or not infringed. We may assert the patents in Hatch-Waxman litigation against the party filing the ANDA to keep the competing product off of the market until the patents expire but there is a risk that we will not succeed. The party filing the ANDA may also counterclaim in the litigation that our patents are not valid or unenforceable, and the court may find one or more claims of our patents invalid or unenforceable. If this occurs, a competing generic product could be marketed prior to expiration of our patents listed in the Orange Book, which would harm our business.
Third parties may also raise similar validity claims before the USPTO in post-grant proceedings such as ex parte reexaminations, inter partes review, or post-grant review, or oppositions or similar proceedings outside the United States, in parallel with litigation or even outside the context of litigation. The outcome following legal assertions of invalidity and unenforceability is unpredictable. If a defendant were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of any future patent protection on one or more of our products or our current or future product candidates. Such a loss of patent protection could harm our business. We cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution or other basis for a finding of invalidity. Litigation proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be unsuccessful, it could have an adverse effect on the price of our common stock. Finally, we may not be able to prevent, misappropriation of our trade secrets or confidential information, particularly in countries where the laws may not protect those rights as fully as in the United States.
59
If we are unable to prevent disclosure of our trade secrets or other confidential information to third parties, our competitive position may be impaired.
We also rely on trade secret protection and confidentiality agreements to protect proprietary know-how that is not patentable or for which we have not filed patent applications, processes for which patents are difficult to enforce and other elements of our product development processes that involve proprietary know-how, information or technology that is not covered by patents. Although we require all of our employees to assign their inventions to us, and endeavor to execute confidentiality agreements with all of our employees, consultants, advisors and any third parties who have access to our proprietary know-how, information or technology, we cannot be certain that we have executed such agreements with all parties who may have helped to develop our intellectual property or had access to our proprietary information, or that our agreements will not be breached. We cannot guarantee that our trade secrets and other confidential proprietary information will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. If we are unable to prevent disclosure of our intellectual property to third parties, we may not be able to maintain a competitive advantage in our market, which would harm our business.
Any disclosure to or misappropriation by third parties of our confidential proprietary information could enable competitors to quickly duplicate or surpass our technological achievements, and erode our competitive position in our market.
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our products.
As is the case with other human or animal pharmaceutical product companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the human and animal health industries involves both technological and legal complexity. Therefore, obtaining and enforcing patents is costly, time-consuming and inherently uncertain. In addition, the United States has recently enacted and implemented wide-ranging patent reform legislation. The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the federal courts, and the USPTO, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce patents that we have or that we might obtain in the future.
We may not be able to protect our intellectual property rights throughout the world, which could impair our business.
Filing, prosecuting and defending patents on human and animal drug products, product candidates and non-prescription products throughout the world would be prohibitively expensive. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we may obtain patent protection, but where patent enforcement is not as strong as that in the United States. These products may compete with our products in jurisdictions where we do not have any issued or licensed patents and any future patent claims or other intellectual property rights may not be effective or sufficient to prevent them from so competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to animal health products, which could make it difficult for us to stop the infringement of our future patents, if any, or patents we have in licensed, or marketing of competing products in violation of our proprietary rights generally. Further, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the United States. As a result, we may encounter significant problems in protecting and defending our intellectual property both in the United States and abroad. Proceedings to enforce our future patent rights, if any, in foreign
60
jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
Our business could be harmed if we fail to obtain certain registered trademarks in the United States or in other countries.
Our registered and pending U.S. trademarks include MYTESI®, JAGUAR HEALTH®, the Jaguar Health Logo®, NAPO®, Napo Logo®, Napo Therapeutics, CANALEVIA, CANALEVIA-CA1, CANALEVIA-CA2, EQUILEVIA, NEONORM®, JAGUAR ANIMAL HEALTH®, and the Jaguar Animal Health Logo®. We also own registered and pending applications for the CANALEVIA mark in a number of foreign countries. During trademark registration proceedings, we may receive rejections of our trademark applications. If so, we will have an opportunity to respond, but we may be unable to overcome such rejections. In addition, the USPTO and comparable agencies in many foreign jurisdictions may permit third parties to oppose pending trademark applications and to seek to cancel registered trademarks. If opposition or cancellation proceedings are filed against any of our trademark applications or any registered trademarks, our trademarks may not survive such proceedings. Moreover, any name we propose to use with our prescription drug product candidates in the United States, including CANALEVIA and CANALEVIA-CA1, must be approved by the FDA, regardless of whether we have registered or applied to register as a trademark. The FDA typically conducts a review of proposed prescription drug product names, including an evaluation of potential for confusion with other product names. If the FDA objects to any of our proposed proprietary product names, we may be required to expend significant additional resources in an effort to identify a suitable substitute name that would qualify under applicable trademark laws, not infringe the existing rights of third parties and be acceptable to the FDA.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties.
We have received confidential and proprietary information from third parties. In addition, we employ individuals who were previously employed at other biotechnology, pharmaceutical or animal health companies. We may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise improperly used or disclosed confidential information of these third parties or our employees’ former employers. Litigation may be necessary to defend against any such claims. Even if we were successful in defending against any such claims, such litigation could result in substantial cost and be a distraction to our management and employees.
Even if we receive any of the required regulatory approvals for our current or future prescription drug product candidates and non-prescription products, we will be subject to ongoing obligations and continued regulatory review, which may result in significant additional expense and delays.
If the FDA or any other regulatory body approves any of our current or future prescription drug product candidates, or if necessary, our non-prescription products, the manufacturing processes, clinical development, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for the product may be subject to extensive and ongoing regulatory requirements. These requirements could include, but are not limited to, submissions of efficacy and safety and other post-marketing information and reports, establishment registration, and product listing, compliance with new rules promulgated under the FSMA, as well as continued compliance with cGMPs, GLPs and GCPs for any studies that we conduct post-approval. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with our contract manufacturers or manufacturing processes, or failure to comply with regulatory requirements, are reportable events to the FDA and may result in, among other things:
| ● | restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, revised labeling, or voluntary or involuntary product recalls; |
| ● | additional clinical studies, fines, warning letters or holds on target animal studies; |
| ● | refusal by the FDA, or other regulators to approve pending applications or supplements to approved applications filed by us or our strategic collaborators related to the unknown problems, or suspension or |
61
| revocation of the problematic product’s license approvals; |
| ● | product seizure or detention, or refusal to permit the import or export of products; and |
| ● | injunctions and/or the imposition of civil or criminal penalties. |
The FDA or other regulatory agency’s policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates or require certain changes to the labeling or additional clinical work concerning safety and efficacy of the product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability, which would harm our business. In addition, failure to comply with these regulatory requirements could result in significant penalties and delays.
In addition, from time to time, we may enter into consulting and other financial arrangements with physicians or veterinarians, who prescribe or recommend our products, once approved. As a result, we may be subject to state, federal and foreign healthcare and/or veterinary medicine laws. If our financial relationships with veterinarians are found to be in violation of such laws that apply to us, we may be subject to penalties.
Further, our commercial supply is regulated by the FDA, which requires regular filings, annual reports, and may include modifications by the Company to our approvals. Failure to gain agreement from the FDA on a timely basis could adversely affect our commercial supply of product.
Lastly, if we obtain conditional approval for our current or future drug product candidates, this conditional approval is renewable annually for five years and may be withdrawn or terminated under certain circumstances either during or at the end of the five-year period. For example, even though we have obtained conditional approval for Canalevia-CA1, if we do not undertake substantial efforts to do additional clinical research each year for the next five years, the FDA could terminate such conditional approval by refusing to renew the conditional approval.
Any of our current or future prescription drug product candidates or non-prescription products may cause or contribute to adverse medical events that we would be required to report to regulatory authorities and, if we fail to do so, we could be subject to sanctions that would harm our business.
If we are successful in commercializing any of our current or future prescription drug product candidates or non-prescription products, certain regulatory authorities will require that we report certain information about adverse medical events if those products may have caused or contributed to those adverse events. The timing of our obligation to report would be triggered by the date we become aware of the adverse event as well as the nature of the event. We may fail to report adverse events we become aware of within the prescribed timeframe. We may also fail to appreciate that we have become aware of a reportable adverse event, especially if such event is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of our products. If we fail to comply with our reporting obligations, the regulatory authorities could take action including, but not limited to, criminal prosecution, seizure of our products, facility inspections, removal of our products from the market, recalls of certain lots or batches, or cause a delay in approval or clearance of future products.
62
Legislative or regulatory reforms with respect to animal health may make it more difficult and costly for us to obtain regulatory clearance or approval of any of our current or future product candidates and to produce, market, and distribute our products after clearance or approval is obtained.
From time to time, legislation is drafted and introduced in the U.S. Congress or other jurisdictions in which we intend to operate that could significantly change the statutory provisions governing the testing, regulatory clearance or approval, manufacture, and marketing of regulated products. In addition, the FDA’s regulations and guidance are often revised or reinterpreted by the FDA and such other regulators in ways that may significantly affect our business and our products and product candidates. Similar changes in laws or regulations can occur in other countries. Any new regulations or revisions or reinterpretations of existing regulations in the United States or in other countries may impose additional costs or lengthen review times of any of our current or future products and product candidates. We cannot determine what effect changes in regulations, statutes, legal interpretation or policies, when and if promulgated, enacted or adopted may have on our business in the future. Such changes could, among other things, require:
| ● | changes to manufacturing methods; |
| ● | additional clinical trials or testing; |
| ● | new requirements related to approval to enter the market; |
| ● | recall, replacement, or discontinuance of certain products; and |
| ● | additional record keeping or the development of certain regulatory required hazard identification plans. |
Each of these would likely entail substantial time and cost and could harm our financial results. In addition, delays in receipt of or failure to receive regulatory clearances or approvals for any future products would harm our business, financial condition, and results of operations.
We believe that our non-prescription products are not subject to regulation by regulatory agencies in the United States, but there is a risk that regulatory bodies may disagree with our interpretation, or may redefine the scope of their regulatory reach in the future, which would result in additional expense and could delay or prevent the commercialization of these products.
The FDA retains jurisdiction over all animal prescription drug products however, in many instances, the Federal Trade Commission will exercise primary or concurrent jurisdiction with FDA on non-prescription products as to post marketing claims made regarding the product. On April 22, 1996, the FDA published a statement in the Federal Register, 61 FR 17706, that it believes that the Dietary Supplement and Health Education Act (“DSHEA”), does not apply to animal health supplement products, such as our non-prescription products. Accordingly, the FDA’s Center for Veterinary Medicine only regulates those animal supplements that fall within the FDA’s definition of an animal drug, animal food or animal feed additive. The Federal Food Drug and Cosmetic Act defines food as “articles used for food or drink for man or other animals and articles used as components of any such article.” Animal foods are not subject to pre-market approval and are designed to provide a nutritive purpose to the animals that receive them. Feed additives are defined as those articles that are added to an animal’s feed or water as illustrated by the guidance documents. Our non-prescription products are not added to food, are not ingredients in food nor are they added to any animal’s drinking water. Therefore, our non-prescription products do not fall within the definition of a food or feed additive. In light of the pronouncement by the FDA that the DSHEA was not intended to apply to animals, the FDA seeks to regulate such supplements as food or food additives depending on the intended use of the product. The intended use is demonstrated by how the article is included in a food, or added to the animals’ intake (i.e., through its drinking water). If the intended use of the product does not fall within the proscribed use making the product a food, it cannot be regulated as a food. There is no intent to make our non-prescription products a component of an animal food, either directly or indirectly. A feed additive is a product that is added to a feed for any reason including the top dressing of an already prepared feed. Some additives, such as certain forage, are deemed to be Generally Recognized as Safe, or GRAS, and therefore, not subject to a feed Additive Petition approval prior to use. However, the
63
substances deemed GRAS are generally those that are recognized as providing nutrients as a food does. We do not believe that our non-prescription products fit within this framework either. Finally, a new animal drug refers to drugs intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in animals. Our non-prescription Neonorm Foal and Neonorm Calf products are not intended to diagnose, cure, mitigate, treat or prevent disease and therefore, do not fit within the definition of an animal drug. Additionally, because a previously marketed human formulation of the botanical extract in our non-prescription products was regulated as a human dietary supplement subject to the DSHEA (and not regulated as a drug by the FDA), we do not believe that the FDA would regulate the animal formulation used in our non-prescription products in a different manner. We do not believe that our non-prescription products fit the definition of an animal drug, food or food additive and therefore are not regulated by the FDA at this time.
However, despite many such unregulated animal supplements currently on the market, the FDA may choose in the future to exercise jurisdiction over animal supplement products in which case, we may be subject to unknown regulations thereby inhibiting our ability to launch or to continue marketing our non-prescription products. In the past, the FDA has redefined or attempted to redefine some non-prescription non-feed products as falling within the definition of drug, feed or feed additive and therefore subjected those products to the relevant regulations. We have not discussed with the FDA its belief that the FDA currently does not exercise jurisdiction over our non-prescription products. Should the FDA assert regulatory authority over our non-prescription products, we would take commercially reasonable steps to address the FDA’s concerns, potentially including but not limited to, seeking registration for such products, reformulating such products to further distance such products from regulatory control, or ceasing sale of such products. Further, the Animal and Plant Health Inspection Service, an agency of the USDA, may at some point choose to exercise jurisdiction over certain non-prescription products that are not intended for production animals. We do not believe we are currently subject to such regulation, but could be in the future. If the FDA or other regulatory agencies, such as the USDA, try to regulate our non-prescription products, we could be required to seek regulatory approval for our non-prescription products, which would result in additional expense and could delay or prevent the commercialization of these products.
Even if we receive the required regulatory approvals for our current or future prescription drug product candidates and non-prescription products, we will be subject to ongoing obligations and continued regulatory review, which may result in significant additional expense.
If the FDA or any other regulatory body approves any of our current or future prescription drug product candidates, or if necessary, our non-prescription products, the manufacturing processes, clinical development, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for the product is subject to extensive and ongoing regulatory requirements. These requirements could include, but are not limited to, submissions of efficacy and safety and other post-marketing information and reports, establishment registration, and product listing, compliance with new rules promulgated under the FSMA, as well as continued compliance with cGMPs, GLPs and GCPs for any studies that Napo conducts post-approval. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with our contract manufacturers or manufacturing processes, or failure to comply with regulatory requirements, are reportable events to the FDA and may result in, among other things:
| ● | restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, revised labeling, or voluntary or involuntary product recalls; |
| ● | additional clinical studies fines, warning letters or holds on studies; |
| ● | refusal by the FDA, or other regulators to approve pending applications or supplements to approved applications filed by Napo or Napo’s strategic collaborators related to the unknown problems, or suspension or revocation of the problematic product’s license approvals; |
| ● | product seizure or detention, or refusal to permit the import or export of products; and |
| ● | injunctions or the imposition of civil or criminal penalties. |
64
The FDA or other regulatory agency’s policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates or require certain changes to the labeling or require additional clinical work concerning safety and efficacy of the product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability, which would harm our business. In addition, failure to comply with these regulatory requirements could result in significant penalties.
In addition, from time to time, we may enter into consulting and other financial arrangements with physicians, who prescribe or recommend our products, once approved. As a result, we may be subject to state, federal and foreign healthcare laws, including but not limited to anti-kickback laws. If our financial relationships with physicians or veterinarians are found to be in violation of such laws that apply to us, we may be subject to penalties.
Risks Related to Our Common Stock
Our failure to meet the continued listing requirements of The Nasdaq Capital Market could result in a delisting of our common stock.
If we fail to satisfy the continued listing requirements of The Nasdaq Capital Market, such as the minimum closing bid price requirement, Nasdaq may take steps to delist our common stock. The closing bid price for our common stock must remain at or above $1.00 per share to comply with Nasdaq’s minimum bid requirement for continued listing. If the closing bid price for our common stock is less than $1.00 per share for 30 consecutive business days, Nasdaq may send us a notice stating that we will be provided a period of 180 days to regain compliance with the minimum bid requirement or else Nasdaq may make a determination to delist our common stock. Our common stock traded for less than $1.00 for 30 consecutive trading days, and we received notice of this from the Listing Qualifications Staff of The Nasdaq Stock Market LLC on February 17, 2022. Under Nasdaq Listing Rule 5810(c)(3)(A), the Company has been granted a 180 calendar day grace period, or until August 16, 2022, to regain compliance with the minimum bid price requirement. The minimum bid price requirement will be met if our common stock has a minimum closing bid price of at least $1.00 per share for a minimum of 10 consecutive business days during the 180 calendar day grace period. We are diligently working to evidence compliance with the minimum bid requirement for continued listing on Nasdaq; however, there can be no assurance that we will be able to regain compliance or that Nasdaq will grant us a further extension of time to regain compliance, if necessary.
The Company may be eligible for additional time to comply if it does not achieve compliance with the minimum bid price requirement by August 16, 2022. In order to be eligible for consideration for such additional time, the Company will be required to meet the continued listing requirement for market value of publicly held shares and all other initial listing standards for The Nasdaq Capital Market, with the exception of the minimum bid price requirement, and must notify Nasdaq in writing of its intention to cure the deficiency during the second compliance period.
The delisting of our common stock from Nasdaq may make it more difficult for us to raise capital on favorable terms in the future. Such a delisting would likely have a negative effect on the price of our common stock and would impair your ability to sell or purchase our common stock when you wish to do so. Further, if we were to be delisted from The Nasdaq Capital Market, our common stock would cease to be recognized as covered securities and we would be subject to regulation in each state in which we offer our securities. Moreover, there is no assurance that any actions that we take to restore our compliance with the Nasdaq minimum bid requirement would stabilize the market price or improve the liquidity of our common stock, prevent our common stock from falling below the Nasdaq minimum bid price required for continued listing again or prevent future non-compliance with Nasdaq’s listing requirements.
65
If we issue all shares available for issuance pursuant to the ATM Agreement, we will have no shares of common stock available for new securities issuances, which may restrict us from accessing additional capital through the sale of new securities.
Our Third Amended and Restated Certificate of Incorporation, as amended, authorizes us to issue up to 150,000,000 shares of voting common stock, 77,053,990 of which are issued and outstanding and 6,634,077 of which are reserved for issuance upon exercise of options, warrants, vesting of RSUs, and shares to consultants as of March 10, 2022. Accordingly, assuming the sale by us under our ATM Agreement at a price of $0.72 per share, which was the closing price of our Common Stock on The Nasdaq Capital Market on January 28, 2022, the maximum amount of shares that we could issue in this offering pursuant to the ATM Agreement without exceeding our total authorized but unissued shares is approximately $68.1 million. If we were to issue the maximum amount of shares in this offering pursuant to the ATM Agreement, we will have no shares of voting common stock available for additional issuances. Our failure to increase our authorized shares may restrict our ability to access additional capital through the sale of new securities, which may harm our financial position and business prospects.
If our shares become subject to the penny stock rules, it would become more difficult to trade our shares.
The SEC has adopted rules that regulate broker-dealer practices in connection with transactions in penny stocks. Penny stocks are generally equity securities with a price of less than $5.00, other than securities registered on certain national securities exchanges or authorized for quotation on certain automated quotation systems, provided that current price and volume information with respect to transactions in such securities is provided by the exchange or system. If we do not retain a listing on The Nasdaq Capital Market and if the price of our common stock is less than $5.00, our common stock will be deemed a penny stock. The penny stock rules require a broker-dealer, before a transaction in a penny stock not otherwise exempt from those rules, to deliver a standardized risk disclosure document containing specified information. In addition, the penny stock rules require that before effecting any transaction in a penny stock not otherwise exempt from those rules, a broker-dealer must make a special written determination that the penny stock is a suitable investment for the purchaser and receive (i) the purchaser’s written acknowledgment of the receipt of a risk disclosure statement; (ii) a written agreement to transactions involving penny stocks and (iii) a signed and dated copy of a written suitability statement. These disclosure requirements may have the effect of reducing the trading activity in the secondary market for our common stock, and therefore stockholders may have difficulty selling their shares.
The price of our common stock could be subject to volatility related or unrelated to our operations, and purchasers of our common stock could incur substantial losses.
We have experienced and may continue to experience significant volatility in the price of our common stock. From January 29, 2021 through January 28, 2022, the share price of our common stock ranged from a high of $10.74 to a low of $0.70. The reason for the volatility in our stock is not well understood and may continue. Factors that may have contributed to such volatility include, but are not limited to, those discussed previously in this “Risk Factors” section of this report and others, such as:
| ● | delays in the commercialization of Mytesi, Neonorm, Canalevia-CA1, Equilevia or our other current or future prescription drug product candidates and non-prescription products; |
| ● | any delays in, or suspension or failure of, our current and future studies; |
| ● | announcements of regulatory approval or disapproval of any of our current or future product candidates or of regulatory actions affecting our company or our industry; |
| ● | manufacturing and supply issues that affect product candidate or product supply for our studies or commercialization efforts; |
| ● | quarterly variations in our results of operations or those of our competitors; |
66
| ● | changes in our earnings estimates or recommendations by securities analysts; |
| ● | the payment of licensing fees or royalties in shares of our common stock; |
| ● | announcements by us or our competitors of new prescription drug products or product candidates or non-prescription products, significant contracts, commercial relationships, acquisitions or capital commitments; |
| ● | announcements relating to future development or license agreements including termination of such agreements; |
| ● | adverse developments with respect to our intellectual property rights or those of our principal collaborators; |
| ● | commencement of litigation involving us or our competitors; |
| ● | any major changes in our board of directors or management; |
| ● | new legislation in the United States relating to the prescription, sale, distribution or pricing of gastrointestinal health products; |
| ● | product liability claims, other litigation or public concern about the safety of our prescription drug product or product candidates and non-prescription products or any such future products; |
| ● | market conditions in the human or animal industry, in general, or in the gastrointestinal health sector, in particular, including performance of our competitors; |
| ● | future issuances of shares of common stock or other securities; |
| ● | uncertainties related to COVID-19; |
| ● | general economic conditions in the United States and abroad; and |
| ● | market speculation regarding |
In addition, the stock market, in general, or the market for stocks in our industry, in particular, may experience broad market fluctuations, which may adversely affect the market price or liquidity of our common stock. Any sudden decline in the market price of our common stock could trigger securities class-action lawsuits against us. If any of our stockholders were to bring such a lawsuit against us, we could incur substantial costs defending the lawsuit and the time and attention of our management would be diverted from our business and operations. We also could be subject to damages claims if we were found to be at fault in connection with a decline in our stock price.
A possible “short squeeze” due to a sudden increase in demand of our common stock that largely exceeds supply may lead to further price volatility in our common stock.
Investors may purchase shares of our common stock to hedge existing exposure in our common stock or to speculate on the price of our common stock. Speculation on the price of our common stock may involve long and short exposures. To the extent aggregate short exposure exceeds the number of shares of our common stock available for purchase in the open market, investors with short exposure may have to pay a premium to repurchase our common stock for delivery to lenders of our common stock. Those repurchases may in turn, dramatically increase the price of our common stock until investors with short exposure are able to purchase additional shares of common stock to cover their short position. This is often referred to as a “short squeeze.” A short squeeze could lead to volatile price movements in shares of our common stock that are not directly correlated to the performance or prospects of our
67
company and once investors purchase the shares necessary to cover their short position the price of our common stock may decline.
You may not be able to resell our common stock when you wish to sell them or at a price that you consider attractive or satisfactory.
The listing of our common stock on The Nasdaq Capital Market does not assure that a meaningful, consistent and liquid trading market exists. Although our common stock is listed on The Nasdaq Capital Market, trading volume in our common stock has been limited and an active trading market for our shares may never develop or be sustained. If an active market for our common stock does not develop, you may be unable to sell your shares when you wish to sell them or at a price that you consider attractive or satisfactory. The lack of an active market may also adversely affect our ability to raise capital by selling securities in the future, or impair our ability to license or acquire other product candidates, businesses or technologies using our shares as consideration.
If securities or industry analysts do not publish research or reports about our company, or if they issue adverse or misleading opinions regarding us or our stock, our stock price and trading volume could decline.
The trading market for our common stock depends in part on the research and reports that industry or financial analysts publish about us or our business. We do not influence or control the reporting of these analysts. If one or more of the analysts who do cover us downgrade or provide a negative outlook on our company or our industry, or the stock of any of our competitors, the price of our common stock could decline. If one or more of these analysts ceases coverage of our company, we could lose visibility in the market, which in turn could cause the price of our common stock to decline.
You may be diluted by conversions of outstanding shares of non-voting common stock, exercises of outstanding options and warrants and issuances of securities pursuant to our ATM Agreement.
As of March 10, 2022, we had (i) outstanding options to purchase an aggregate of 2,327,368 shares of our common stock at a weighted average exercise price of $9.83 per share, (ii) outstanding options to purchase an aggregate of 127,949 shares of our common stock issuable upon exercise of outstanding inducement options, with a weighted-average exercise price of $4.42 per share, (iii) 563,108 shares of our common stock issuable upon exercise of warrants outstanding, with weighted-average exercise price of $7.17, (iv) 465,194 shares of our common stock issuable upon vesting of outstanding RSUs, (v) 38,333 shares of our common stock issuable to third parties upon exercise of those shares, and (vi) 673 shares of our non-voting common stock issuable at an equivalent share of voting common stock. The exercise of such options, warrants, vesting of RSUs, and conversion of the non-voting common stock will result in further dilution of your investment.
In addition, you may experience further dilution if we issue common stock in the future, including common stock issued pursuant to our existing At The Market Offering Agreement (the “ATM Agreement”). Pursuant to the ATM Agreement with Ladenburg Thalmann & Co. Inc. (“Ladenburg”), we may offer and sell up to $75.0 million of our common stock from time to time through Ladenburg as our sales agent. During the year ended December 31, 2021, we sold 2,261,596 shares of common stock pursuant to the ATM Agreement for gross proceeds of $3.5 million.
As a result of this dilution, you may receive significantly less in net tangible book value than the full purchase price you paid for the shares in the event of liquidation.
68
Provisions in our charter documents and under Delaware law could discourage a takeover that stockholders may consider favorable and may lead to entrenchment of management.
Our third amended and restated certificate of incorporation and amended and restated bylaws contain provisions that could delay or prevent changes in control or changes in our management without the consent of our board of directors. These provisions to include the following:
| ● | a classified board of directors with three-year staggered terms, which may delay the ability of stockholders to change the membership of a majority of our board of directors; |
| ● | no cumulative voting in the election of directors, which limits the ability of minority stockholders to elect director candidates; |
| ● | the exclusive right of our board of directors to elect a director to fill a vacancy created by the expansion of the board of directors or the resignation, death or removal of a director, which prevents stockholders from being able to fill vacancies on our board of directors; |
| ● | the ability of our board of directors to authorize the issuance of shares of preferred stock and to determine the terms of those shares, including preferences and voting rights, without stockholder approval, which could adversely affect the rights of our common stockholders or be used to deter a possible acquisition of our company; |
| ● | the ability of our board of directors to alter our bylaws without obtaining stockholder approval; |
| ● | the required approval of the holders of at least 75% of the shares entitled to vote at an election of directors to adopt, amend or repeal our bylaws or repeal the provisions of our third amended and restated certificate of incorporation regarding the election and removal of directors; |
| ● | a prohibition on stockholder action by written consent, which forces stockholder action to be taken at an annual or special meeting of our stockholders; |
| ● | the requirement that a special meeting of stockholders may be called only by the chairman of the board of directors, the chief executive officer, the president or the board of directors, which may delay the ability of our stockholders to force consideration of a proposal or to take action, including the removal of directors; and |
| ● | advance notice procedures that stockholders must comply with in order to nominate candidates to our board of directors or to propose matters to be acted upon at a stockholders’ meeting, which may discourage or deter a potential acquirer from conducting a solicitation of proxies to elect the acquirer’s own slate of directors or otherwise attempting to obtain control of us. |
These provisions could inhibit or prevent possible transactions that some stockholders may consider attractive.
We are also subject to the anti-takeover provisions contained in Section 203 of the Delaware General Corporation Law. Under Section 203, a corporation generally may not engage in a business combination with any holder of 15% or more of its capital stock unless the holder has held the stock for three years or, among other exceptions, the board of directors has approved the transaction.
69
Our amended and restated bylaws designate the Court of Chancery of the State of Delaware as the sole and exclusive forum for certain actions and proceedings that may be initiated by our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or other employees.
Our amended and restated bylaws provide that, unless we consent in writing to an alternative forum, the Court of Chancery of the State of Delaware will be the sole and exclusive forum for (i) any derivative action or proceeding brought on our behalf, (ii) any action asserting a claim of breach of a fiduciary duty owed by any director, officer or other employees to us or our stockholders, (iii) any action asserting a claim arising pursuant to any provision of the Delaware General Corporation Law, (iv) any action asserting a claim that is governed by the internal affairs doctrine or (v) any action to interpret, apply, enforce or determine the validity of our certificate of incorporation or bylaws. Any person purchasing or otherwise acquiring any interest in any shares of our capital stock shall be deemed to have notice of and to have consented to this provision of our amended and restated bylaws. This choice-of-forum provision may limit our stockholders’ ability to bring a claim in a judicial forum that they find favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits. Alternatively, if a court were to find this provision of our amended and restated bylaws inapplicable or unenforceable with respect to one or more of the specified types of actions or proceedings, we may incur additional costs associated with resolving such matters in other jurisdictions, which could harm our business and financial condition.
We do not intend to pay dividends on our common stock, and your ability to achieve a return on your investment will depend on appreciation in the market price of our common stock.
We currently intend to invest our future earnings, if any, to fund our growth and not to pay any cash dividends on our common stock. Because we do not intend to pay dividends, your ability to receive a return on your investment will depend on any future appreciation in the market price of our common stock. We cannot be certain that our common stock will appreciate in price.
The requirements of being a public company, including compliance with the reporting requirements of the Exchange Act and the requirements of the Sarbanes-Oxley Act, may strain our resources, increase our costs and distract management, and we may be unable to comply with these requirements in a timely or cost-effective manner.
Our initial public offering had a significant, transformative effect on us. Prior to our initial public offering, our business operated as a privately-held company, and we were not required to comply with public reporting, corporate governance and financial accounting practices and policies required of a publicly-traded company. As a publicly-traded company, we incur significant additional legal, accounting and other expenses compared to historical levels. In addition, new and changing laws, regulations and standards relating to corporate governance and public disclosure, including the Dodd-Frank Wall Street Reform and Consumer Protection Act and the rules and regulations thereunder, as well as under the Sarbanes-Oxley Act, the JOBS Act and the rules and regulations of the SEC and The Nasdaq Capital Market, may result in an increase in our costs and the time that our board of directors and management must devote to our compliance with these rules and regulations. These rules and regulations have substantially increased our legal and financial compliance costs and diverted management time and attention from our product development and other business activities.
The Sarbanes-Oxley Act requires, among other things, that we assess the effectiveness of our internal control over financial reporting annually and the effectiveness of our disclosure controls and procedures quarterly. In particular, Section 404 of the Sarbanes-Oxley Act, or Section 404, requires us to perform system and process evaluation and testing of our internal control over financial reporting to allow management to report on, and our independent registered public accounting firm potentially to attest to, the effectiveness of our internal control over financial reporting. We have needed to expend time and resources on documenting our internal control over financial reporting so that we are in a position to perform such evaluation when required. As a smaller reporting company (“SRC”), we expect to avail ourselves of the exemption from the requirement that our independent registered public accounting firm attest to the effectiveness of our internal control over financial reporting under Section 404. However, we may no longer avail ourselves of this exemption when we cease to be an SRC. When our independent registered
70
public accounting firm is required to undertake an assessment of our internal control over financial reporting, the cost of our compliance with Section 404 will correspondingly increase. Our compliance with applicable provisions of Section 404 requires that we incur substantial accounting expense and expend significant management time on compliance-related issues as we implement additional corporate governance practices and comply with reporting requirements. Moreover, if we are not able to comply with the requirements of Section 404 applicable to us in a timely manner, or if we or our independent registered public accounting firm identifies deficiencies in our internal control over financial reporting that are deemed to be material weaknesses, the market price of our stock could decline and we could be subject to sanctions or investigations by the SEC or other regulatory authorities, which would require additional financial and management resources.
We are a smaller reporting company and the reduced reporting requirements applicable to smaller reporting companies may make our common stock less attractive to investors.
We are a smaller reporting company (“SRC”) and a non-accelerated filer, which allows us to take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not SRCs or non-accelerated filers, including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, as amended, reduced disclosure obligations regarding executive compensation in our Annual Report and our periodic reports and proxy statements and providing only two years of audited financial statements in our Annual Report and our periodic reports. We will remain an SRC so long as (a) the aggregate market value of our outstanding common stock held by non-affiliates as of the last business day our most recently completed second fiscal quarter is less than $250 million or (b) (1) we have less than $100 million in annual revenues and (2) the aggregate market value of our outstanding common stock held by non-affiliates as of the last business day our most recently completed second fiscal quarter is less than $700 million. We cannot predict whether investors will find our common stock less attractive if we rely on certain or all of these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile and may decline.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 2. PROPERTIES
Our corporate headquarters are located at 200 Pine Street, Suite 400, San Francisco, California.
ITEM 3. LEGAL PROCEEDINGS
May 2020 Letter from the Committee on Oversight and Reform of the U.S. House of Representatives
On May 4, 2020, Jaguar Health, Inc. received a letter from the Committee on Oversight and Reform of the U.S. House of Representatives (the “Committee”) regarding the list price adjustment of Mytesi. Among other things, the Committee expressed an interest in understanding whether the price adjustment was connected to the Company’s expectation that it could market crofelemer to treat coronavirus patients given the Company’s submission of a request to the U.S. Food and Drug Administration for Emergency Use Authorization (“EUA”) for crofelemer for the symptomatic relief of diarrhea and other gastrointestinal symptoms in patients with COVID-19 and for patients with COVID-19 who have diarrhea associated with certain antiviral treatments, which submission was denied by the FDA on April 7, 2020 as previously disclosed.
The Company has cooperated with the Committee’s inquiry and has prepared a public statement regarding the price adjustment, which is available on the Company’s website at https://jaguarhealth.gcs-web.com/company-statement. In its statement, the Company explains that the decision to adjust the price for crofelemer was made in December 2019 as part of expanding the Company’s comprehensive patient access program, and had the Company received EUA, it would have deferred the price adjustment until after the emergency use period ended.
71
July 2017 Complaint Relating to the Merger
On July 20, 2017, a putative class action complaint was filed in the United States District Court, Northern District of California, Civil Action No. 3:17 cv 04102, by Tony Plant (the “Plaintiff”) on behalf of shareholders of the Company who held shares on April 12, 2017 and were entitled to vote at the 2017 Special Shareholders Meeting, against the Company and certain individuals who were directors as of the date of the vote (collectively, the “Defendants”), in a matter captioned Tony Plant v. Jaguar Animal Health, Inc., et al. (Jaguar Health, Inc. was formerly known as Jaguar Animal Health, Inc.), making claims arising under Section 14(a) and Section 20(a) of the Exchange Act and Rule 14a 9, 17 C.F.R. § 240.14a 9, promulgated thereunder by the SEC. The claims alleged false and misleading information provided to investors in the Joint Proxy Statement/Prospectus on Form S 4 (File No. 333 217364) declared effective by the Commission on July 6, 2017 related to the solicitation of votes from shareholders to approve the merger and certain transactions related thereto.
Following the completion of document discovery, the parties engaged in a mediation that resulted in an agreement in principle to settle the litigation on a class-wide basis for $2.6 million, subject to court approval.
On May 27, 2021, the court gave the final approval to the proposed settlement and the entire settlement consideration was provided by the Company’s director and officer liability insurance carrier. Under the loss recovery model in ASC 450 and in reference to ASC 410, the ultimate net income effect of the recognized loss and the insurance proceeds directly related to the recognized loss is zero.
As of December 31, 2021 and 2020, the Company concluded not to record any loss contingency and insurance recovery.
Other than as described above, there are currently no claims or actions pending against us, the ultimate disposition of which could have a material adverse effect on our results of operations, financial condition or cash flows.
ITEM 4. MINE SAFETY DISCLOSURE
Not applicable.
72
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock trades on The Nasdaq Capital Market under the symbol “JAGX.”
Holders
As of March 10, 2022 there were approximately 28 stockholders of record of our common stock. These figures do not reflect the beneficial ownership or shares held in nominee name, nor do they include holders of any RSUs.
Dividend Policy
We have never paid any cash dividends on our common stock to date. We currently anticipate that we will retain all future earnings, if any, to fund the development and growth of our business and do not anticipate paying any cash dividends for at least the next five years, if ever.
Recent Sales of Unregistered Securities
Other than as provided on our quarterly reports on Form 10-Q filed with the SEC on May 17, 2021, August 13, 2021 and November 17, 2021 and our current reports on Form 8 K filed with the SEC on January 8, 2021, January 22, 2021, March 11, 2021, April 8, 2021, September 17, 2021, there were no unregistered sales of equity securities during the period.
The offers, sales, and issuances of the securities described above were deemed to be exempt from registration under the Securities Act in reliance on Section 3(a)(9) of the Securities Act, Section 4(a)(2) of the Securities Act, or Regulation D or Regulation S promulgated thereunder as transactions by an issuer not involving a public offering. The recipients of securities in each of these transactions acquired the securities for investment only and not with a view to or for sale in connection with any distribution thereof and appropriate legends were affixed to the securities issued in these transactions. Each of the recipients of securities in these transactions was an accredited or sophisticated person and had adequate access, through employment, business or other relationships, to information about us.
ITEM 6. SELECTED FINANCIAL DATA
Not Applicable.
73
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis should be read together with our financial statements and the related notes appearing elsewhere in this report.
Overview
Jaguar Health, Inc. (“Jaguar” or the “Company”) is a commercial stage pharmaceuticals company focused on developing novel, plant-based, non-opioid, and sustainably derived prescription medicines for people and animals with GI distress, including chronic, debilitating diarrhea. Jaguar Animal Health is a tradename of Jaguar Health. Our wholly owned subsidiary, Napo Pharmaceuticals, Inc. (“Napo”), focuses on developing and commercializing proprietary plant-based human pharmaceuticals for the global marketplace from plants or plant products used traditionally in rainforest areas. Napo’s marketed drug Mytesi (crofelemer 125 mg delayed-release tablets) is a first-in-class oral botanical drug product approved by the U.S. Food and Drug Administration (“FDA”) for the symptomatic relief of noninfectious diarrhea in adults with HIV/AIDS on antiretroviral therapy. To date, this is the only oral plant-based botanical prescription medicine approved under the FDA’s Botanical Guidance. Jaguar Animal Health’s Canalevia-CA1 (crofelemer delayed-release tablets) drug is the first and only oral plant-based prescription product that is FDA conditionally approved to treat chemotherapy-induced diarrhea (CID) in dogs. Canalevia-CA1 is a canine-specific formulation of crofelemer. Napo Therapeutics S.p.A., Napo’s majority owned Italian subsidiary, focuses on expanding crofelemer access in Europe.
Jaguar, formerly known as Jaguar Animal Health, Inc., was founded in San Francisco, California as a Delaware corporation on June 6, 2013 (inception). The Company was a majority-owned subsidiary of Napo until the close of the Company's initial public offering on May 18, 2015. The Company was formed to develop and commercialize first-in-class prescription and non-prescription products for companion and production animals and horses. The Company's first non-prescription commercial products, Neonorm Calf and Neonorm Foal, were launched in 2014 and 2016, respectively.
On July 31, 2017, Jaguar completed a merger with Napo pursuant to the Agreement and Plan of Merger dated March 31, 2017, by and among Jaguar, Napo, Napo Acquisition Corporation (“Merger Sub”), and Napo's representative (the “Merger Agreement”). In accordance with the terms of the Merger Agreement, upon the completion of the merger, Merger Sub merged with and into Napo, with Napo surviving as the wholly-owned subsidiary (the “Merger” or “Napo Merger”). Immediately following the Merger, Jaguar changed its name from “Jaguar Animal Health, Inc.” to “Jaguar Health, Inc.” Napo now operates as a wholly-owned subsidiary of Jaguar focused on human health including the ongoing development of crofelemer and commercialization of Mytesi.
On March 15, 2021, Jaguar established Napo EU S.p.A (which changed its name in December 2021 to “Napo Therapeutics”) based in Milan, Italy as a subsidiary of Napo. Napo Therapeutics’ mission is to provide access to crofelemer in Europe to address significant rare/orphan disease indications, including, initially, two key orphan target indications: Short bowel syndrome with intestinal failure (SBS-IF), and congenital diarrheal disorders (CDD). On November 3, 2021, Napo Therapeutics merged with Dragon SPAC S.p.A. (“Dragon SPAC”).
Most of the activities of the Company are focused on the commercialization of Mytesi and Canalevia-CA1 and the ongoing clinical development of crofelemer for the prophylaxis of diarrhea in adult patients receiving targeted cancer therapy. In the field of animal health, we are continuing limited activities related to developing and commercializing first in class gastrointestinal products for dogs, dairy calves, foals, and high value horses.
We believe Jaguar is poised to realize a number of synergistic, value adding benefits—an expanded pipeline of potential blockbuster human follow on indications of crofelemer, and a second generation anti secretory agent—upon which to build global partnerships. Jaguar, through Napo, holds global unencumbered rights for crofelemer, Mytesi, and Canalevia-CA1. Additionally, several of the drug product opportunities in Jaguar’s crofelemer pipeline are backed Phase 2 and proof of concept evidence from human clinical trials.
74
Crofelemer is a novel, first in class anti secretory agent which has a normalizing effect on electrolyte and fluid balance while acting locally in the gut, and this mechanism of action has the potential to benefit multiple disorders that cause gastrointestinal distress, including diarrhea and abdominal discomfort. Crofelemer is also in development for possible follow on indications, including prophylaxis for cancer therapy related diarrhea (“CTD”); for rare disease indications for symptomatic treatment of infants and children with congenital diarrheal disorders (“CDD”) and for adult and pediatric patients with short bowel syndrome with intestinal failure (“SBS-IF”). Crofelemer has received orphan drug designation (ODD) for short bowel syndrome (SBS) in the US and in EU. Furthermore, the drug is being evaluated for management of diarrhea and abdominal discomfort in inflammatory bowel disease (“IBD”); diarrhea-predominant irritable bowel syndrome (“IBS-D”); and for idiopathic/functional diarrhea. A second generation proprietary anti secretory agent, NP-300 (lechlemer), is undergoing preclinical development for symptomatic relief and treatment of diarrhea in patients with acute infection from cholera.
Financial Operations Overview
On a consolidated basis, we have not yet generated enough revenue to date to achieve break even or positive cash flow, and we expect to continue to incur significant research and development and other expenses. Our net loss was $52.6 million and $33.8 million for the years ended December 31, 2021 and 2020, respectively. As of December 31, 2021, we had total stockholders' equity of $11.9 million, an accumulated deficit of $219.5 million, and cash of $17.1 million. We expect to continue to incur losses and experience increased expenditures for the foreseeable future as we expand our product development activities, seek necessary approvals for our product candidates, conduct species-specific formulation studies for our non-prescription products, establish API manufacturing capabilities and begin additional commercialization activities.
Revenue
Our product and collaboration revenue consists of the following:
| ● | Revenues from the sale of our human drug Mytesi, which is sold through distributors and wholesalers. |
| ● | Revenues from the sale of our animal products branded as Neonorm Calf and Neonorm Foal. Our Neonorm and Botanical extract products are primarily sold to distributors, who then sell the products to the end customers. |
| ● | Our policy typically permits returns if the product is damaged, defective, or otherwise cannot be used when received by the customer if the product has expired. Returns are accepted for product that will expire within six months or that have expired up to one year after their expiration dates. Estimates for expected returns of expired products are based primarily on an ongoing analysis of our historical return patterns. |
See “Results of Operations” below for more detailed discussion on revenues
Cost of Revenue
Cost of revenue consists of direct drug substance and drug product materials expense, direct labor, distribution fees, royalties and other related expenses associated with the sale of our products.
75
Research and Development Expense
Research and development expenses consist primarily of clinical and contract manufacturing expense, personnel and related benefit expense, stock-based compensation expense, employee travel expense and reforestation expenses. Clinical and contract manufacturing expense consists primarily of costs to conduct stability, safety and efficacy studies, and manufacturing startup expenses at an outsourced API provider in Italy. It also includes expenses with a third-party provider for the transfer of the Mytesi manufacturing process, and the related feasibility and validation activities.
We typically use our employee and infrastructure resources across multiple development programs. We track outsourced development costs by prescription drug product candidate and non-prescription product but do not allocate personnel or other internal costs related to development to specific programs or development compounds.
The timing and amount of our research and development expenses will depend largely upon the outcomes of current and future trials for our prescription drug product candidates as well as the related regulatory requirements, the outcomes of current and future species-specific formulation studies for our non-prescription products, manufacturing costs and any costs associated with the advancement of our line extension programs. We cannot determine with certainty the duration and completion costs of the current or future development activities.
The duration, costs and timing of trials, formulation studies and development of our prescription drug and non-prescription products will depend on a variety of factors, including:
| ● | the scope, rate of progress, and expense of our ongoing, as well as any additional clinical trials, formulation studies and other research and development activities; |
| ● | future clinical trial and formulation study results; |
| ● | potential changes in government regulations; and |
| ● | the timing and receipt of any regulatory approvals. |
A change in the outcome of any of these variables with respect to the development of a prescription drug product candidate or non-prescription product could mean a significant change in the costs and timing associated with our development activities.
We expect research and development expense to increase due to the start-up costs associated with our clinical trials for other indications.
Sales and Marketing Expense
Sales and marketing expenses consist of personnel and related benefit expense, stock-based compensation expense, direct sales and marketing expense, employee travel expense, and management consulting expense. We currently incur sales and marketing expenses to promote Mytesi. We do not currently have any marketing or promotional expenses related to Neonorm Calf or Neonorm Foal for the years ended December 31, 2021 and 2020.
We expect sales and marketing expense to increase going forward as we focus on expanding our market access activities and commercial partnerships for the development of follow-on indications of Mytesi and crofelemer.
General and Administrative Expense
General and administrative expenses consist of personnel and related benefit expense, stock-based compensation expense, employee travel expense, legal and accounting fees, rent and facilities expense, and management consulting expense.
76
In the near term, we expect general and administrative expense to remain flat as we focus on our pipeline development and market access expansion. This will include efforts to grow the business.
Interest Expense
Interest expense consists primarily of non-cash and cash interest costs related to our borrowings.
Critical Accounting Policies and Significant Judgments and Estimates
The preparation of financial statements in conformity with U.S. generally accepted accounting principles (“U.S. GAAP”) requires the use of estimates and assumptions that affect the reported amounts of assets and liabilities, revenues and expenses, and related disclosures in the financial statements. Critical accounting policies are those accounting policies that may be material due to the levels of subjectivity and judgment necessary to account for highly uncertain matters or the susceptibility of such matters to change, and that have a material impact on financial condition or operating performance. While we base our estimates and judgments on our experience and on various other factors that we believe to be reasonable under the circumstances, actual results may differ from these estimates under different assumptions or conditions. We believe the following critical accounting policies used in the preparation of our financial statements require significant judgments and estimates. For additional information relating to these and other accounting policies, see Note 2 to the consolidated financial statements, appearing elsewhere in this report.
77
Results of Operations
Comparison of the Years Ended December 31, 2021 and 2020
The following table summarizes the Company’s results of operations with respect to the items set forth in such table for the years ended December 31, 2021 and 2020 together with the change in such items in dollars and as a percentage.
Year Ended |
| |||||||||||
December 31, | ||||||||||||
(in thousands) |
| 2021 |
| 2020 |
| Variance |
| Variance % | ||||
Product revenue | $ | 4,335 | $ | 9,385 | $ | (5,050) |
| (53.8) | % | |||
Total revenue |
| 4,335 |
| 9,385 |
| (5,050) |
| (53.8) | % | |||
Operating Expenses |
|
|
|
|
|
|
| |||||
Cost of product revenue |
| 2,333 |
| 3,280 |
| (947) |
| (28.9) | % | |||
Research and development |
| 15,079 |
| 6,413 |
| 8,666 |
| 135.1 | % | |||
Sales and marketing |
| 8,894 |
| 6,609 |
| 2,285 |
| 34.6 | % | |||
General and administrative |
| 17,103 |
| 14,387 |
| 2,716 |
| 18.9 | % | |||
Series 3 warrants inducement expense | 1,462 | 3,696 | (2,234) | (60.4) | % | |||||||
ELOC warrants inducement expense | 172 | — | 172 | 100.0 | % | |||||||
Series B convertible preferred stock inducement expense |
| — |
| 1,647 |
| (1,647) |
| (100.0) | % | |||
Total operating expenses |
| 45,043 |
| 36,032 |
| 9,011 |
| 25.0 | % | |||
Loss from operations |
| (40,708) |
| (26,647) |
| (14,061) |
| 52.8 | % | |||
Interest expense |
| (8,421) |
| (2,792) |
| (5,629) |
| 201.6 | % | |||
Loss on extinguishment of debt and exchange of Series D perpetual preferred stock |
| (753) |
| (1,864) |
| 1,111 |
| (59.6) | % | |||
Change in fair value of financial instruments and hybrid instrument designated at Fair Value Option |
| (1,953) |
| (2,696) |
| 743 |
| (27.6) | % | |||
Other expense, net |
| (765) |
| 190 |
| (955) |
| (502.6) | % | |||
Loss before income tax |
| (52,600) |
| (33,809) |
| (18,791) |
| 55.6 | % | |||
Income tax expense | — | — | — | 100.0 | % | |||||||
Net loss and comprehensive loss | (52,600) | (33,809) | (18,791) | 55.6 | % | |||||||
Deemed dividend attributable to Series C perpetual preferred stock | — | (2,521) | 2,521 | (100) | % | |||||||
Deemed dividend attributable to accretion of Series A redeemable convertible preferred stock | — | (1,332) | 1,332 | (100) | % | |||||||
Deemed dividend attributable to Series 1, Series 2 and Bridge warrant holders | — | (856) | 856 | (100) | % | |||||||
Stock dividend attributable to Series C perpetual preferred stock | — | (130) | 130 | (100) | % | |||||||
Adjusted net loss and comprehensive loss | $ | (52,600) | $ | (38,648) | $ | (13,952) | 36.1 | % | ||||
Net loss attributable to noncontrolling interest | $ | (5) | $ | — | $ | (5) | 100 | % | ||||
Net loss attributable to common shareholders | $ | (52,595) | $ | (38,648) | $ | (13,947) | 36.1 | % | ||||
Revenue
Product revenue
We transitioned from selling to the wholesalers that resell the product to retail pharmacies to the closed Specialty Pharmacy distribution networks throughout the year 2021 and we fully transitioned in the fourth quarter of the same year. The transition caused a one-time inventory draw-down of approximately 1,300 bottles of Mytesi across
78
our third-party logistics warehouse, wholesalers, distributors, and retail stores. This significantly contributed to the decrease of $4.8 million of Mytesi gross revenue for the year 2021 compared to 2020.
Medicaid and AIDS Drug Assistance Program (“ADAP”) rebates accounted for $3.5 million and $1.7 million for the year ended December 31, 2021 and 2020, respectively, an increase of $1.7 million primarily due to the WAC increase implemented by the Company in April 2020 which resulted in higher government rebates from Medicaid, ADAP, public health services programs, and includes approximately $800,000 in chargebacks from the State of California. Sales discounts were $6.2 million and $7.0 million for the year ended December 31, 2021 and 2020, respectively, a decrease of $778,000. We expect the wholesaler fees to decline in line with our switch to the closed Specialty Pharmacy distribution network.
Due to the Company’s arrangements, including elements of variable consideration, gross product sales are reduced in order to reflect the expected consideration to arrive at net product sales. Deductions to reduce gross product sales to net product sales for the years ended December 31, 2021 and 2020 are as follows:
Year Ended |
| |||||||||||
December 31, |
| |||||||||||
(in thousands) |
| 2021 |
| 2020 |
| Variance |
| Variance % | ||||
Gross product sales |
| |||||||||||
Mytesi | $ | 15,657 | $ | 20,434 | $ | (4,777) |
| (23.4) | % | |||
Neonorm | 62 | 77 |
| (15) |
| (19.5) | % | |||||
Total gross product sales | 15,719 | 20,511 |
| (4,792) |
| (23.4) | % | |||||
Medicaid rebates | (3,484) | (1,738) |
| (1,746) |
| 100.5 | % | |||||
Sales discounts | (6,268) | (7,046) | 778 | (11.0) | % | |||||||
Sales returns | (104) | (273) | 169 | (61.9) | % | |||||||
Wholesaler fee | (1,528) | (2,069) | 541 | (26.1) | % | |||||||
Net product sales | $ | 4,335 | $ | 9,385 | $ | (5,050) |
| (53.8) | % | |||
Our gross product revenues were $15.7 million and $20.5 million for the year ended December 31, 2021 and 2020, respectively. These periods reflect revenue from the sale of our human drug Mytesi and our animal products branded as Neonorm Calf and Neonorm Foal. This contributed to the decrease of $4.8 million of Mytesi gross revenue for the year 2021 compared to 2020.
Our Neonorm product revenues were $62,000 and $77,000 for the year ended December 31, 2021 and 2020, respectively. Sales and marketing expenses for Neonorm products are not significant during 2021 and none during the same period in 2020.
Cost of Product Revenue
Year Ended |
| |||||||||||
December 31, |
| |||||||||||
(in thousands) |
| 2021 |
| 2020 |
| Variance |
| Variance % | ||||
Cost of Product Revenue |
| |||||||||||
Material cost |
| $ | 998 |
| $ | 1,800 |
| $ | (802) |
| (44.6) | % |
Direct labor |
| 996 |
| 724 |
| 272 |
| 37.6 | % | |||
Distribution fees |
| 199 |
| 430 |
| (231) |
| (53.7) | % | |||
Other |
| 140 |
| 326 |
| (186) |
| (57.1) | % | |||
Total |
| $ | 2,333 |
| $ | 3,280 |
| $ | (947) |
| (28.9) | % |
The change in cost of product revenue of $947,000 for the year ended December 31, 2021 compared to 2020 was primarily due to:
| ● | Material costs decreased $802,000 from $1.8 million for the year ended December 31, 2020 to $998,000 in 2021, largely attributable to lower sales resulting in less cost of materials for bottles sold and to non- |
79
| recurring write-off of non-conforming inventory in the year ended December 31, 2020. There was no write-off in 2021. |
| ● | Distribution fees decreased $231,000 from $430,000 for the year ended December 31, 2020 to $199,000 in 2021 in line with our transition from Title model to Specialty Pharmacy distribution network. |
| ● | Other costs decreased $186,000 from $326,000 for the year ended December 31, 2020 to $140,000 in 2021 mainly consisting of $118,000 less in write-offs of non-conforming inventory, and a decrease in equipment maintenance of $9,000 in 2020. |
| ● | Direct labor increased $272,000 from $724,000 for the year ended December 31, 2020 to $996,000 in 2021, due to increased resources in manufacturing. |
Research and Development Expense
The following table presents the components of research and development (“R&D”) expense for the years ended December 31, 2021 and 2020:
Year Ended |
| |||||||||||
December 31, |
| |||||||||||
(in thousands) |
| 2021 |
| 2020 |
| Variance |
| Variance % | ||||
Research and Development: |
|
|
|
|
|
|
|
| ||||
Clinical and contract manufacturing | $ | 6,257 | $ | 1,674 | $ | 4,583 |
| 273.8 | % | |||
Personnel and related benefits | 3,954 | 1,771 | 2,183 |
| 123.3 | % | ||||||
Stock-based compensation |
| 1,319 |
| 749 | 570 |
| 76.1 | % | ||||
Materials expense and tree planting |
| 361 |
| 94 | 267 |
| 284.0 | % | ||||
Travel, other expenses |
| 40 |
| 45 | (5) |
| (11.1) | % | ||||
Other |
| 3,148 |
| 2,080 | 1,068 |
| 51.3 | % | ||||
Total | $ | 15,079 | $ | 6,413 | $ | 8,666 |
| 135.1 | % | |||
The change in R&D expense of $8.7 million for the year ended December 31, 2021 compared to 2020 was primarily due to:
| ● | Clinical and contract manufacturing expenses increased $4.6 million from $1.7 million for the year ended December 31, 2020 to $6.3 million in 2021 largely due to increased clinical trial activities related to the start-up of CTD and other indications, additional CMC manufacturing, consulting and contractors’ expenses, and cholera/lechlemer research expenses. |
| ● | Personnel and related benefits increased $2.2 million from $1.8 million for the year ended December 31, 2020 to $4.0 million in 2021 due to increased resources, salary increases in April 2021 and an increase in bonus. |
| ● | Other expenses increased $1.1 million from $2.1 million for the year ended December 31, 2020 to $3.1 million in 2021 mainly consisting of consulting, formulation and regulatory fees. Consulting expenses increased due to increase in clinical trial consultants while direct R&D testing costs also increased due to an increase in R&D work. |
| ● | Stock-based compensation increased $570,000 from $749,000 for the year ended December 31, 2020 to $1.3 million in 2021 primarily due to new options granted in April 2021. |
| ● | Material expense and tree planting increased $267,000 from $94,000 for the year ended December 31, 2020 to $361,000 in 2021 due to increased clinical trials. |
80
Sales and Marketing Expense
The following table presents the components of sales and marketing (“S&M”) expense for the years ended December 31, 2021 and 2020:
Year Ended |
| |||||||||||
December 31, |
| |||||||||||
(in thousands) |
| 2021 |
| 2020 |
| Variance |
| Variance % | ||||
Sales and Marketing: |
|
|
|
|
|
|
|
| ||||
Personnel and related benefits | $ | 3,916 | $ | 3,323 | $ | 593 |
| 17.8 | % | |||
Direct marketing fees and expense | 3,415 | 2,187 | 1,228 |
| 56.1 | % | ||||||
Stock-based compensation |
| 319 |
| 220 |
| 99 |
| 45.0 | % | |||
Other |
| 1,244 |
| 879 |
| 365 |
| 41.5 | % | |||
Total | $ | 8,894 | $ | 6,609 | $ | 2,285 |
| 34.6 | % | |||
The change in S&M expense of $2.3 million for the year ended December 31, 2021 compared to 2020 was primarily due to:
| ● | Direct marketing fees and expense increased $1.2 million from $2.2 million for the year ended December 31, 2020 to $3.4 million in 2021 due to an increase in marketing programs for Mytesi related to the expanding market access through specialty pharmacy channels. |
| ● | Personnel and related benefits increased $593,000 from $3.3 million for the year ended December 31, 2020 to $3.9 million in 2021 due to the addition of four new personnel within Commercial Operations, increase in bonus, and salary increases in April 2021. |
| ● | Other expenses increased $365,000 from $879,000 for the year ended December 31, 2020 to $1.2 million in 2021 largely due to additional marketing consulting costs. |
General and Administrative Expense
The following table presents the components of general and administrative (“G&A”) expense for the years ended December 31, 2021 and 2020:
Year Ended |
| |||||||||||
December 31, |
| |||||||||||
(in thousands) |
| 2021 |
| 2020 |
| Variance |
| Variance % | ||||
General and Administrative: |
|
|
|
|
|
|
|
| ||||
Personnel and related benefits | $ | 3,390 | $ | 1,845 | $ | 1,545 |
| 83.7 | % | |||
Stock-based compensation |
| 2,336 |
| 1,855 |
| 481 |
| 25.9 | % | |||
Legal services |
| 2,303 |
| 2,449 |
| (146) |
| (6.0) | % | |||
Public company expense |
| 2,270 |
| 1,179 |
| 1,091 |
| 92.5 | % | |||
Audit, tax and accounting services |
| 982 |
| 706 |
| 276 |
| 39.1 | % | |||
Third-party consulting services |
| 859 |
| 845 |
| 14 |
| 1.7 | % | |||
Rent and lease expense |
| 282 |
| 690 |
| (408) |
| (59.1) | % | |||
Travel, other expenses |
| 197 |
| 38 |
| 159 |
| 418.4 | % | |||
Other |
| 4,484 |
| 4,780 |
| (296) |
| (6.2) | % | |||
Total | $ | 17,103 | $ | 14,387 | $ | 2,716 |
| 18.9 | % | |||
The change in G&A expenses of $2.7 million for the year ended December 31, 2021 compared to 2020 was due primarily to:
| ● | Personnel and related benefits increased $1.5 million from $1.8 million for the year ended December 31, 2020 to $3.4 million in 2021 due to new hires and increase in bonus. |
81
| ● | Public company expenses increased $1.1 million from $1.2 million for the year ended December 31, 2020 to $2.3 million in 2021 largely attributable to the investor relations and communications consulting expenses, and expenses for the annual shareholder meeting. |
| ● | Stock-based compensation expense increased $481,000 from $1.9 million for the year ended December 31, 2020 to $2.3 million in 2021 primarily due to higher expense incurred for options granted with immediate vesting to existing employees. |
| ● | Audit, tax and accounting services fees increased $276,000 from $706,000 for the year ended December 31, 2020 to $982,000 in 2021, mostly due to the increased audit fees related to complex debt and equity transactions. |
| ● | Travel, other expenses increased $159,000 from $38,000 for the year ended December 31, 2020 to $197,000 in 2021 due to other corporate and investor relations activities. |
| ● | Rent and lease expense decreased $408,000 from $690,000 for the year ended December 31, 2020 to $282,000 in 2021 as a result of the transfer to a lower-cost facility and the occupancy of less space. |
| ● | Other general and administrative expenses decreased $296,000 from $4.8 million for the year ended December 31, 2020 to $4.5 million in 2021 largely due to decreased consulting expenses in 2021. There was a non-recurring charge of $1.0 million for the Atlas trial delay penalty incurred in the year ended December 31, 2020. |
| ● | Legal services decreased $146,000 from $2.4 million for the year ended December 31, 2020 to $2.3 million in 2021 primarily due to a decrease in fees related to legal proceedings and other regulatory filings. |
Series 3 Warrants Inducement Expense
The decrease in the Series 3 Warrants inducement expense of $2.2 million is due to the following:
| ● | In January 2021, the Company issued 135,416 Series 3 Warrants to a certain investor for the exercise of 135,416 Bridge Note Warrants in accordance with the May 2020 Modification of the 2019 Bridge Note Warrants and Inducement Offer. These Series 3 Warrants were valued at $1.5 million using the Black-Scholes-Merton option pricing model on the issuance date. |
| ● | In May 2020, concurrent with the May 2020 modification of the exercise price of the Series 1, Series 2 and Bridge Warrants and inducement offer, the Company issued unregistered Series 3 warrants to purchase 2,890,284 shares of common stock. These Series 3 warrants were valued at $3.7 million using the Black-Scholes-Merton option pricing model on the issuance date. |
ELOC Warrants Inducement Expense
In April 2021, in consideration for Oasis Capital’s entry into the amendment to the March 2020 Equity Line of Credit, the Company issued Oasis Capital a common stock purchase warrant exercisable for 33,333 shares of common stock with an exercise price per share equal to $5.61 on the date of the amendment. These warrants were valued at $172,000 on the issuance date.
Series B Convertible Preferred Stock Inducement Expense
On March 24, 2020, the Company entered into a Warrant Exercise and Preferred Stock Amendment Agreement with a holder of its Series 2 warrants previously issued in the Company’s registered public offering on July 23, 2019, pursuant to which the holder agreed to exercise in cash its warrants to purchase an aggregate of 416,666
82
shares of common stock, at a reduced exercise price of $1.57 per share for gross proceeds to the Company of approximately $653,000. As a further inducement to enter into the Amendment Agreement, the Company agreed to reduce the conversion price of the Company’s Series B Convertible Preferred Stock from $6.00 to $1.34. The modification of the conversion price of the Series B Convertible Preferred shares was qualitatively considered an extinguishment and the Company followed the guidance in ASC 260-10-S99-2 and recorded an expense of $1.6 million and derecognizing the Series B Convertible Preferred shares.
Interest Expense, net
Interest expense increased $5.6 million from $2.8 million for the year ended December 31, 2020 to $8.4 million in 2021 primarily due to additional interest expense incurred on royalty interest agreements.
Loss on Extinguishment of Debt and Exchange of Series D Perpetual Preferred Stock
The loss on extinguishment of debt and conversion of Series D Perpetual Preferred Stock decreased $1.1 million from $1.9 million for the year ended December 31, 2020 to $753,000 in 2021 is due to the following:
| ● | During 2020, the Company recorded a $560,000 extinguishment loss from exchanges of the outstanding Exchange Note 1 for shares of the Company’s common stock; and |
| ● | In December 2020, the Company recorded a $1.3 million loss from exchanges of Series D Perpetual Preferred Stock for shares of the Company’s common stock. |
| ● | In January 2021, the Company recorded a $753,000 loss from the exchange of the outstanding balance of Exchange Note 2 for the Company’s shares common stock. |
Change in Fair Value of Financial Instruments and Hybrid Instrument Designated at FVO
Change in fair value of financial instruments decreased $743,000 from a loss of $2.7 million for the year ended December 31, 2020 to a loss of $2.0 million in 2021 primarily due to fair value adjustments in liability classified warrants and notes payable designated at FVO.
Deemed Dividend Attributable to Series C Perpetual Preferred Stock
The Company recorded a deemed dividend of $2.5 million for the year ended December 31, 2020 that resulted from the series of exchanges of Series C Perpetual Preferred Stock in October and December 2020.
Deemed Dividend Attributable to Accretion of Series A Redeemable Convertible Preferred Stock
The Company recorded a deemed dividend charge of $1.3 million for the year ended December 31, 2020 for the accretion of the redemption amount and carrying value of the Series A Convertible Preferred Stock.
Deemed Dividend Attributable to Series 1, Series 2 and Bridge Warrant Holders
The Company recorded a deemed dividend of $856,000 for the year ended December 31, 2020 that resulted from the modification of the Series 1, Series 2 and Bridge Warrants in May 2020.
Stock Dividend Attributable to Series C Perpetual Preferred Stock
The Company recorded a $130,000 stock dividend attributable to the Series C Perpetual Preferred Stock for the year ended December 31, 2020. The Series C Perpetual Preferred shares were entitled to receive 10% cumulative stock dividends, to be payable in arrears on a monthly basis for 24 consecutive months. Dividends payable on the
83
Series C Perpetual Preferred shares shall be payable through the Company’s issuance of Series C Perpetual Preferred share by delivering to each record holder the calculated number of payment-in-kind dividend shares.
Liquidity and Capital Resources
Sources of Liquidity
We have incurred net losses since our inception. For the years ended December 31, 2021 and 2020, we had net losses of $52.6 million and $33.8 million, respectively, and we expect to incur additional losses in the near-term future. At December 31, 2021, we had an accumulated deficit of $219.5 million. To date, we have generated only limited revenue, and we may never achieve revenue sufficient to offset our expenses. The Company expects to incur substantial losses and negative cash flows in future periods. Further, the Company’s future operations, which include the satisfaction of current obligations, are dependent on the success of the Company’s ongoing development and commercialization efforts, as well as securing of additional financing and generating positive cash flows from operations. There is no assurance that the Company will have adequate cash balances to maintain its operations.
We had cash of $17.1 million as of December 31, 2021 to fund our operating plan through one year from the issuance of these consolidated financial statements.
Although the Company plans to finance its operations and cash flow needs through equity and/or debt financing, collaboration arrangements with other entities, license royalty agreements, as well as revenue from future product sales, the Company does not believe its current cash balances are sufficient to funds its operating plan through one year from the issuance of these consolidated financial statements. The Company has an immediate need to raise cash. There can be no assurance that additional funding will be available to the Company on acceptable terms, or on a timely basis, if at all, or that the Company will generate sufficient cash from operations to adequately fund operating needs. If the Company is unable to obtain an adequate level of financing needed for the long-term development and commercialization of our products, the Company will need to curtail planned activities and reduce costs. Doing so will likely have an adverse effect on our ability to execute our business plan; accordingly, there is substantial doubt about the ability of the Company to continue in existence as a going concern. The accompanying consolidated financial statements do not include any adjustments that might result from the outcome of these uncertainties.
We have funded our operations primarily through the issuance of equity and debt financing, in addition to sales of commercial products. Cash provided by financing activities in 2021 are as follows:
| ● | During January 2021, an aggregate of 416,664 shares of common stock was issued upon the exercise of the December 2019 PIPE Financing Warrants for total proceeds of $975,000. |
| ● | On January 13, 2021, the Company entered into a securities purchase agreement, pursuant to which the Company agreed to issue and sell, in a registered public offering an aggregate of 1,479,290 shares of common stock, at an offering price of $10.14 per share for net proceeds of approximately $13.5 million. |
| ● | On January 19, 2021, the Company entered into a note purchase agreement with Streeterville Capital, LLC (“Streeterville”), pursuant to which the Company issued a secured promissory note in the aggregate principal amount of $6.2 million for an aggregate purchase price of $6.0 million. |
| ● | During January and February 2021, the Company issued an aggregate of 669,850 shares under the ATM Agreement for total net proceeds of $5.4 million. |
| ● | On March 8, 2021, the Company entered into a Royalty Purchase Agreement with Streeterville, pursuant to which the Company sold a royalty interest entitling Streeterville to $10.0 million and any interest, fees, and charges as royalty repayment amount for an aggregate purchase price of $5.0 million. Interest will accrue on the royalty repayment amount at a rate of 5% per annum, compounding quarterly, and will increase to 10% per annum, compounding quarterly on the 12-month anniversary of the closing date. |
84
| ● | Between January to March 2021, an aggregate of 1,383,524 shares of common stock were issued upon the exercise of Series 1, Series 2 and Bridge Note Warrants for total proceeds of $2.0 million. |
| ● | On April 29, 2021, the Company entered into a securities purchase agreement, pursuant to which the Company agreed to issue and sell, in a registered public offering an aggregate of 2,549,000 shares of common stock at an offering price of $4.23 per share for gross proceeds of approximately $10.8 million before deducting placement agent fees and related offering expenses of $948,000. |
| ● | On September 13, 2021, the Company entered into a securities purchase agreement (the “September 2021 PIPE Financing”) with certain investors, pursuant to which the Company agreed to issue and sell to the investors in a private placement an aggregate of 309,242 unregistered shares of the Company’s common stock for an aggregate purchase price of approximately $776,000 or $2.51 per share. |
| ● | On December 10, 2021, the Company entered into another ATM Agreement (“December 2021 ATM Agreement”) with Ladenburg, pursuant to which the Company may offer and sell, from time to time through Ladenburg, shares of common stock having an aggregate offering price of up to $15.0 million. As of December 31, 2021, the Company has issued 2,261,596 shares under the December 2021 ATM Agreement for a total net proceeds of $3.2 million. |
We expect our expenditures will continue to increase as we continue our efforts to develop our products and continue development of our pipeline in the near term. We may seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans. We may also not be successful in entering into partnerships that include payment of upfront licensing fees for our products and product candidates for markets outside the United States, where appropriate. If we do not generate upfront fees from any anticipated arrangements, it would have a negative effect on our operating plan. We still plan to finance our operations and capital funding needs through equity and/or debt financing as well as revenue from future product sales. However, there can be no assurance that additional funding will be available to us on acceptable terms on a timely basis, if at all, or that we will generate sufficient cash from operations to adequately fund operating needs or ultimately achieve profitability. If we are unable to obtain an adequate level of financing needed for the long-term development and commercialization of our products, we will need to curtail planned activities and reduce costs. Doing so will likely have an adverse effect on our ability to execute on our business plan.
Cash Flows for Year Ended December 31, 2021 compared to the Year Ended December 31, 2020
The following table shows a summary of cash flows for the years ended December 31, 2021 and 2020:
Year Ended December 31, | |||||||
(in thousands) |
| 2021 |
| 2020 |
| ||
Total cash used in operating activities | $ | (34,970) | $ | (15,278) | |||
Total cash used in investing activities |
| (6) |
| (7) | |||
Total cash provided by financing activities |
| 43,937 |
| 19,492 | |||
Net increase in cash | $ | 8,961 | $ | 4,207 | |||
Cash Used in Operating Activities
During the year ended December 31, 2021, net cash used in operating activities of $35.0 million resulted from our net loss of $52.6 million adjusted by amortization of debt issuance costs, debt discount, and non-cash interest expense of $5.2 million, stock-based compensation of $4.0 million, change in fair value of financial instruments and hybrid instruments designated at FVO of $2.0 million, depreciation and amortization expenses of $1.7 million, Series 3 and ELOC warrants inducement expenses of $1.6 million, loss on extinguishment of debt of $753,000, amortization of operating lease right-of-use assets of $94,000, derecognition of debt discount on settlement of receivables secured borrowing of $49,000, shares issued in exchange for services of $16,000, and net changes in operating assets and liabilities of $2.3 million.
85
During the year ended December 31, 2020, net cash used in operating activities of $15.3 million resulted from our net loss of $33.8 million adjusted by depreciation and amortization expenses of $1.7 million, interest paid on the conversion of debt to equity of $0.6 million, a loss on extinguishment of debt and conversion of Series D Perpetual Preferred Stock of $1.9 million, a loss on recourse obligation on secured borrowing of $15,000, amortization of operating lease right-of-use assets of $553,000, expense on modifications of warrants of $86,000, inducement charge of $1.6 million on the modification of Series B Convertible Preferred Stock, stock-based compensation of $2.8 million, other stock and warrant payments of $1.1 million, amortization of debt discounts, debt issuance costs, and non-cash interest expense of $2.7 million, $2.5 million in shares issued to Atlas Sciences for settlement of the Trial Delay Fee, an increase in fair value of financial instruments of $2.7 million, and $3.7 million charge for Series 3 Warrants issued as an inducement to exercise equity-classified Series 1, Series 2 and Bridge warrants, offset by changes in operating assets and liabilities of $3.5 million.
Cash Used in Investing Activities
During the year ended December 31, 2021, cash used in investing activities was $6,000 which consisted of cash used to purchase property and equipment.
During the year ended December 31, 2020, cash used in investing activities was $7,000 which consisted of cash used to purchase property and equipment.
Cash Provided by Financing Activities
During the year ended December 31, 2021, net cash provided by financing activities of $43.9 million consisted of $23.2 million in net proceeds received from shares issued in registered public offering, $11.0 million in net proceeds received from issuance of notes payable, $8.6 million in net proceeds from shares issued in an At the Market offering, $2.0 million in net proceeds received from shares issued on conversion of Series 1, Series 2, and 2019 Bridge Note Warrants, $1.8 million in net proceeds received from shares issued in PIPE financing, $247 million noncontrolling interest, and $3,000 in net proceeds from exercise of stock options, offset by $1.8 million repayment of receivables secured borrowing, $943,000 repayment of insurance financing, $100,000 in principal payments of the notes payable and $35,000 payment of ELOC warrants offering costs.
During the year ended December 31, 2020, net cash provided by financing activities of $19.5 million consisted of $12.3 million in net proceeds received from issuances of a notes payable, $7.1 million received from borrowings secured by the Company’s trade receivables, $668,000 in net proceeds received from shares of common stock issued via a PIPE financing, $5.8 million in net proceeds received from shares of common stock issued on exercise of Series 1, Series 2, and 2019 Bridge Note warrants, and $1.3 million in net proceeds received from issuance of other shares of common stock, offset by $7.3 million in principal payments of the notes payable, secured borrowings and insurance premium borrowings, $185,000 million in issuance costs from shares issued as part of the underwriter settlement agreement, and $142,000 other payments of issuance costs.
Off-Balance Sheet Arrangements
Since inception, we have not engaged in the use of any off-balance sheet arrangements, such as structured finance entities, special purpose entities or variable interest entities.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK.
Not applicable.
86
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Jaguar Health, Inc.
Index to Financial Statements
87
Report of Independent Registered Public Accounting Firm
To the Board of Directors and
Shareholders of Jaguar Health, Inc.:
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheet of Jaguar Health, Inc., (the “Company”) as of December 31, 2021, and the related consolidated statements of operations, changes in stockholders’ equity, and cash flows for the year ended December 31, 2021, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2021, and the results of its operations and its cash flows for the year ended December 31, 2021, in conformity with accounting principles generally accepted in the United States of America.
The Company’s Ability to Continue as a Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the Company has an accumulated deficit, recurring losses, and expects continuing future losses. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. Management’s evaluation of the events and conditions and management’s plans regarding these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit, we are required to obtain an understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audit provides a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the consolidated financial statements that was communicated or required to be communicated to the audit committee and that: (1)
88
relates to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of a critical audit matter does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Impairment of Intangible Assets, Net—Refer to Note 2 and Note 7 to the financial statements
Critical Audit Matter Description
As of December 31, 2021, the Company had intangible assets, net, of $22.7 million. Intangible assets are evaluated based on the asset group based on product as well as being evaluated between definite-lived and indefinite-lived intangible assets for the purpose of the impairment assessment. The Company assesses potential impairments whenever events or circumstances indicate that the asset may be impaired. For finite-lived intangibles assets the impairment is based on recoverability. Recoverability of an asset group is measured by a comparison of the carrying amount of an asset group to its forecasted cash flows expected to be generated by the asset group. If the carrying amount of the asset group exceeds its estimated forecasted cash flows, an impairment charge is recognized as the amount by which the carrying amount of the asset group exceeds the fair value of the asset group. An indefinite-lived intangible asset is considered impaired if the carrying amount exceeds the fair value of the asset group. The fair value of the asset group was determined using the income approach. The Company did not recognize an impairment loss in the financial statements for the year ended December 31, 2021.
We identified the evaluation of intangible asset impairment as a critical audit matter because the determination of the forecasted individual asset group’s cash flows, including revenue, expenses, and other items, requires a high degree of auditor judgment and increased extent of effort.
How the Critical Audit Matter Was Addressed in the Audit
The principal considerations for our determination that performing procedures relating to the valuation of intangible assets as a critical audit matter are (1) there was a high degree of auditor judgment and subjectivity in applying procedures relating to the fair value of intangible assets acquired due to the significant judgment by management when developing the estimates and (2) significant audit effort was required in evaluating the significant assumptions relating to the estimates, including the income projections and discount rates. In addition, the audit effort involved the use of professionals with specialized skill and knowledge to assist in performing these procedures and evaluating the audit evidence obtained.
Addressing the matter involved performing procedures and evaluating audit evidence in connection with forming our overall opinion on the consolidated financial statements. These procedures included the following:
| ● | Inquiry of management regarding the development of the assumptions used in the valuation of the intangible assets. |
| ● | Testing management’s process included evaluating the appropriateness of the valuation models, testing the completeness, accuracy, and relevance of underlying data used in the models, and testing the reasonableness of significant assumptions, including the income projections and discount rates. |
| ● | Professionals with specialized skill and knowledge were used to assist in evaluating the reasonableness of significant assumptions. |
| ● | Evaluated the experience, qualifications and objectivity of the Company’s specialist, a third-party valuation firm. |
| ● | Obtained an understanding of the nature of the work the Company’s specialist performed, including the objectives and scope of the specialist’s work; the methods or assumptions used; and a comparison of the methods or assumptions used with those used in the preceding period. Identified and evaluated assumptions developed by the specialist considering assumptions generally used in the specialist’s field; supporting evidence provided by the specialist; existing market data; historical or recent experience and changes in conditions and events affecting the Company. |
89
| ● | Tested the accuracy and completeness of company-produced data used by the specialist, and evaluated the relevance and reliability of externally obtained data. For assumptions provided to the specialist by the company, evaluated whether there is a reasonable basis for using each assumption considering whether other reasonably likely outcomes could materially affect the relevant financial statement assertions. Identified and evaluated significant assumptions used by the specialist for reasonableness. |
| ● | Evaluated the Company’s estimates of future revenue projections by completing a retrospective comparison to historical revenue projections. We tested the significant assumptions discussed above, as well as the completeness and accuracy of the underlying data used in the projected cash flows and valuations. |
| ● | To reflect the uncertainty inherent in the projections, we performed our own sensitivity analyses by increasing or decreasing the significant assumptions and evaluated the potential impact on the fair value. In addition, we tested the reconciliation of the fair value of the asset group developed by management to the market capitalization of the Company as of the valuation date. |
/s/
We have served as the Company's auditor since 2021.
March 11, 2022
PCAOB ID Number
90
Report of Independent Registered Public Accounting Firm
To the Board of Directors and
Stockholders of Jaguar Health, Inc.:
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheet of Jaguar Health, Inc. (“Company”) as of December 31, 2020, and the related consolidated statements of operations, changes in stockholders’ equity, and cash flows for the year ended December 31, 2020, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2020, and the results of its operations and its cash flows for the year ended December 31, 2020, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audit provides a reasonable basis for our opinion.
/s/
We served as the Company's auditor since 2019, which ended in 2021.
March 31, 2021 (except for the effects of the reverse stock split described in Note 1, as to which the date is March 11, 2022)
PCAOB ID Number
91
JAGUAR HEALTH, INC.
CONSOLIDATED BALANCE SHEETS
December 31, | December 31, | |||||
(In thousands, except share and per share data) |
| 2021 |
| 2020 | ||
Assets | ||||||
Current assets: | ||||||
Cash | $ | $ | ||||
Accounts receivable | ||||||
Accounts receivable - pledged |
| — |
| |||
Other receivable | ||||||
Inventory | ||||||
Prepaid expenses and other current assets | ||||||
Total current assets |
|
| ||||
Property and equipment, net | ||||||
Operating lease - right-of-use asset | — | |||||
Intangible assets, net | ||||||
Other assets |
|
| ||||
Total assets | $ | $ | ||||
Liabilities and stockholders' equity | ||||||
Current liabilities: | ||||||
Accounts payable | $ | $ | ||||
Accrued liabilities | ||||||
Warrant liability | ||||||
Operating lease liability, current | — | |||||
Notes payable, current |
|
| ||||
Series D perpetual preferred stock: $ | — | — | ||||
Total current liabilities |
|
| ||||
Operating lease liability, net of current portion | — | |||||
Notes payable, net of discount, net of current portion (includes hybrid instrument designated at Fair Value Option amounting to $ | ||||||
Total liabilities |
|
| ||||
Commitments and contingencies (See Note 6) | ||||||
Stockholders' equity | ||||||
Series B-2 convertible preferred stock: $ | — | — | ||||
Series C perpetual preferred stock: | — | — | ||||
Common stock - voting: $ |
|
| ||||
Common stock - non-voting: $ | — | — | ||||
Additional paid-in capital |
|
| ||||
Noncontrolling interest | — | |||||
Accumulated deficit |
| ( |
| ( | ||
Total stockholders' equity |
|
| ||||
Total liabilities and stockholders' equity | $ | $ | ||||
The accompanying notes are an integral part of these consolidated financial statements.
92
JAGUAR HEALTH, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
Year Ended | ||||||
December 31, | ||||||
(In thousands, except share and per share data) |
| 2021 |
| 2020 | ||
Product revenue | $ | $ | ||||
Operating expenses | ||||||
Cost of product revenue | ||||||
Research and development |
|
| ||||
Sales and marketing | ||||||
General and administrative | ||||||
Series 3 warrants inducement expense | ||||||
ELOC warrants inducement expense | — | |||||
Series B convertible preferred stock inducement expense | — | |||||
Total operating expenses |
|
| ||||
Loss from operations |
| ( |
| ( | ||
Interest expense |
| ( |
| ( | ||
Loss on extinguishment of debt and exchange of Series D perpetual preferred stock | ( | ( | ||||
Change in fair value of financial instruments and hybrid instrument designated at Fair Value Option | ( | ( | ||||
Other income (expense), net | ( | |||||
Loss before income tax | ( | ( | ||||
Income tax expense | ||||||
Net loss and comprehensive loss | ( | ( | ||||
Deemed dividend attributable to Series C perpetual preferred stock | — | ( | ||||
Deemed dividend attributable to accretion of Series A redeemable convertible preferred stock | — | ( | ||||
Deemed dividend attributable to Series 1, Series 2 and Bridge warrant holders | — | ( | ||||
Stock dividend attributable to Series C perpetual preferred stock | — | ( | ||||
Adjusted net loss and comprehensive loss | $ | ( | $ | ( | ||
Net loss attributable to noncontrolling interest | $ | ( | $ | — | ||
Net loss attributable to common shareholders | $ | ( | $ | ( | ||
Net loss per share, basic and diluted | ( | ( | ||||
Weighted-average common shares outstanding, basic and diluted |
| |
| | ||
The accompanying notes are an integral part of these consolidated financial statements.
93
JAGUAR HEALTH, INC.
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY
| Series A | Series B | Series B-2 | Series C | |||||||||||||||||||||||||||||||||||||||
| Convertible | Convertible | Convertible | Perpetual | Common | Common | Additional | Noncontrolling | Accumulated | Total | |||||||||||||||||||||||||||||||||
(In thousands, except share data) |
| Shares |
| Amount |
|
| Shares | Amount | Shares |
| Amount | Shares |
| Amount | Shares | Amount | Shares | Amount | paid-in capital | interest | deficit | Equity | |||||||||||||||||||||
Balances as of January 1, 2020 | | $ | | $ | | $ | — | $ | — | | $ | | $ | $ | $ | — | $ | ( | $ | ||||||||||||||||||||||||
Shares issued on exercise of Series 1, Series 2, and 2019 Bridge Note warrants | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Shares issued on exercise of Series 2 warrants and inducement offer conversion of Series B-1 convertible preferred stock | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Shares issued on exercise of Series 1, Series 2, and 2019 Bridge Note warrants, net of issuance costs of $ | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Shares issued on conversion of Series 1, Series 2, and 2019 Bridge Note warrants; June 2020 | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Modification of Series 1, Series 2 and Bridge warrants | — | — | — | — | — | — | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||||
Deemed dividend attributable to Series 1, Series 2 and Bridge warrant holders | — | — | — | — | — | — | — | — | — | — | — | — | ( | — | — | ( | |||||||||||||||||||||||||||
Shares issued on exercise of Series 3 warrants | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||||
Shares issued on exercise of Series 1 and Series 2 warrants | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Shares issued to Atlas Sciences for settlement of Trial Delay Fee | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Warrants issued and converted to common shares to Atlas Sciences for settlement of Trial Delay Fee | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Issuance of common stock in PIPE financing, net of issuance costs of $ | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Shares issued in Underwriter settlement agreement | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Warrants issued in Underwriter settlement agreement | — | — | — | — | — | — | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||||
Underwriter settlement offering cost | — | — | — | — | — | — | — | — | — | — | — | — | ( | — | — | ( | |||||||||||||||||||||||||||
Conversion of Series B-2 convertible preferred stock into common stock | — | — | — | — | ( | ( | — | — | | — | — | — | — | — | — | ||||||||||||||||||||||||||||
Conversion of Series B convertible preferred stock into common stock | — | — | ( | ( | — | — | — | — | | — | — | — | — | — | — | ||||||||||||||||||||||||||||
Shares issued to Oasis as consideration under the March 2020 equity purchase agreement | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Shares issued to Oasis under the March 2020 equity purchase agreement, put option exercise, net of issuance costs of $ | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Series A convertible preferred stock redeemed and Series C perpetual preferred issued under the exchange transaction | ( | ( | — | — | — | — | | — | — | — | — | — | — | ||||||||||||||||||||||||||||||
Stock dividend attributable to Series C perpetual preferred stock of $ | — | — | — | — | — | — | | — | — | — | — | ( | — | — | — | ||||||||||||||||||||||||||||
Extinguishment of Series C perpetual preferred stock | — | — | — | — | — | — | — | ( | — | — | — | — | — | — | — | ||||||||||||||||||||||||||||
Deemed dividend attributable to Series C perpetual preferred stock | — | — | — | — | — | — | — | — | — | — | — | ( | — | — | — | ||||||||||||||||||||||||||||
Shares issued to third party for services | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Shares issued to Sagard Capital in exchange of services | — | — | — | — | — | — | — | — | | — | — | — | — | ||||||||||||||||||||||||||||||
Shares issued in exchange of CVP Exchange Notes | — | — | — | — | — | — | — | — | | — | — | — | — | ||||||||||||||||||||||||||||||
Warrants issued and converted to common shares to Pacific Capital Management in exchange of promissory note | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Shares issued to PoC Capital in Payment of Contracted Research Fees | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Shares issued to Iliad in exchange of Royalty Interest | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Shares issued on conversion of Series C perpetual preferred stock | — | — | — | — | — | — | ( | ( | | — | — | — | — | — | — | ||||||||||||||||||||||||||||
Warrants issued and converted to common stock in exchange for Series C Preferred Stock | — | — | — | — | — | — | — | — | | — | — | ( | — | — | — | ||||||||||||||||||||||||||||
Shares issued on conversion of Series D convertible preferred stock | — | — | — | — | — | — | — | — | | — | — | — | — | ||||||||||||||||||||||||||||||
Conversion of non-voting common stock to voting common stock | — | — | — | — | — | — | — | — | | — | ( | ( | — | — | — | ||||||||||||||||||||||||||||
Shares issued in Registered Direct Offering Priced Above-the-Market, net of $ | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Accretion to redemption value of redeemable preferred stock | — | — | — | — | — | — | — | — | — | — | — | ( | — | — | ( | ||||||||||||||||||||||||||||
Fractional shares | — | — | — | — | — | — | — | — | | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
Shares issued upon exercise of stock options | — | — | — | — | — | — | — | — | | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
Stock-based compensation | — | — | — | — | — | — | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||||
Net loss | — | — | — | — | — | — | — | — | — | — | — | — | — | — | ( | ( | |||||||||||||||||||||||||||
Balances as of December 31, 2020 | — | $ | — | — | $ | — | — | $ | — | — | $ | — | | $ | | $ | — | $ | $ | — | $ | ( | $ | ||||||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
94
| Series A | Series B | Series B-2 | Series C | |||||||||||||||||||||||||||||||||||||||
| Convertible | Convertible | Convertible | Perpetual | Common | Common | Additional | Noncontrolling | Accumulated | Total | |||||||||||||||||||||||||||||||||
(In thousands, except share data) |
| Shares | Amount |
|
| Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | paid-in capital | interest | deficit | Equity | ||||||||||||||||||||||||
Balances as of January 1, 2021 | — | $ | — | — | $ | — | — | $ | — | — | $ | — | | $ | | $ | — | $ | $ | — | $ | ( | $ | ||||||||||||||||||||
Shares issued on exercise of Series 1, Series 2, and 2019 Bridge Note Warrants | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Shares issued in PIPE financing | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Shares issued in At the Market offering, net of issuance and offering costs of $ | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Shares issued in registered public offering, net of issuance and offering costs of $ | — | — | — | — | — | — | — | — | | — | — | — | — | ||||||||||||||||||||||||||||||
Shares issued in extinguishment of Exchange Note 2 | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Shares issued on exercise of Series 3 warrants | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Shares issued to Iliad in exchange of notes payable and accrued interest | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Warrants issued to Oasis for ELOC amendment, net of offering costs of $ | — | — | — | — | — | — | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||||
Shares issued on conversion of Napo merger common shares | — | — | — | — | — | — | — | — | | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
Shares issued upon exercise of stock options | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Shares issued to third party for services | — | — | — | — | — | — | — | — | | — | — | — | — | — | |||||||||||||||||||||||||||||
Fractional shares | — | — | — | — | — | — | — | — | | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
Merger of subsidiary | — | — | — | — | — | — | — | — | — | — | — | — | — | ||||||||||||||||||||||||||||||
Stock-based compensation | — | — | — | — | — | — | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||||
Net loss | — | — | — | — | — | — | — | — | — | — | — | — | — | ( | ( | ( | |||||||||||||||||||||||||||
Balances as of December 31, 2021 | — | $ | — | — | $ | — | — | $ | — | — | $ | — | | $ | | $ | — | $ | $ | $ | ( | $ | |||||||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements
95
JAGUAR HEALTH, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
Year Ended | ||||||
December 31, | December 31, | |||||
(in thousands) |
| 2021 |
| 2020 | ||
Cash flows from operating activities | ||||||
Net loss | $ | ( | $ | ( | ||
Adjustments to reconcile net loss to net cash used in operating activities: | ||||||
Amortization of debt issuance costs, debt discount, and non-cash interest expense | ||||||
Stock-based compensation |
|
| ||||
Change in fair value of financial instruments and hybrid instrument designated at Fair Value Option | ||||||
Depreciation and amortization expense |
|
| ||||
Series 3 warrants inducement expense | ||||||
Loss on extinguishment of debt and conversion of Series D perpetual preferred stock | ||||||
ELOC warrants inducement expense | — | |||||
Amortization of operating lease - right-of-use-asset | ||||||
Derecognition of debt discount on settlement of receivables secured borrowing | — | |||||
Shares issued in exchange for services | ||||||
Series B convertible preferred stock inducement expense | — | |||||
Expense on modification of warrants | — | |||||
Shares and warrants issued in Underwriter settlement agreement | — | |||||
Shares issued as consideration paid under the Oasis Capital Equity Purchase Agreement | — | |||||
Loss on recourse obligation on secured borrowing | — | |||||
Interest paid on the conversion of debt to equity | — | |||||
Shares issued to Atlas for settlement of Trial Delay Fee | — | |||||
Shares issued on conversion of warrants of Atlas for settlement of Trial Delay Fee | — | |||||
Changes in assets and liabilities |
|
| ||||
Accounts receivable | ( | |||||
Other receivable | ( | ( | ||||
Inventory |
| ( |
| ( | ||
Prepaid expenses and other current assets |
| ( |
| ( | ||
Other assets | ( | |||||
Accounts payable | ( | |||||
Accrued liabilities |
|
| ||||
Operating lease liability | ( | ( | ||||
Total cash used in operating activities |
| ( |
| ( | ||
Cash flows from investing activities | ||||||
Purchase of equipment | ( | ( | ||||
Total cash used in investing activity |
| ( |
| ( | ||
Cash flows from financing activities |
|
| ||||
Proceeds from issuance of shares in registered public offering, net of issuance and offering costs of $ | — | |||||
Proceeds from issuance of notes payable, net of issuance costs of $ | ||||||
Proceeds from issuance of shares in At the Market offering, net of issuance and offering costs of $ | ||||||
Proceeds from issuance of shares on conversion of Series 1, Series 2, and 2019 Bridge Note warrants, net of issuance and offering costs of $ | ||||||
Repayment of receivables secured borrowing | ( | ( | ||||
Proceeds from issuance of shares in PIPE financing | ||||||
Repayment of insurance financing | ( | ( | ||||
Noncontrolling interest | — | |||||
Repayment of notes payable | ( | ( | ||||
Payment of ELOC warrants offering costs | ( | — | ||||
Proceeds from exercise of stock options | — | |||||
Proceeds from sale of receivables, net of debt discount and issuance costs of $ | — | |||||
Issuance costs from shares issued on Underwriter settlement agreement | — | ( | ||||
Payments of deferred offering costs | — | ( | ||||
Proceeds from issuance of common stock on conversion of Oasis Capital an Equity Purchase Agreement put options, net of issuance costs of $ | — | |||||
Total cash provided by financing activities | ||||||
Net increase in cash |
|
| ||||
Cash at beginning of period | ||||||
Cash at end of period | $ | $ | ||||
96
JAGUAR HEALTH, INC.
STATEMENTS OF CASH FLOWS (continued)
Year Ended | ||||||
December 31, | December 31, | |||||
2021 | 2020 | |||||
Supplemental schedule of cash flow information | ||||||
Cash paid for interest | $ | $ | ||||
Supplemental schedule of non-cash financing and investing activities | ||||||
Shares issued in exchange of partial settlement of royalty interest | $ | $ | — | |||
Shares issued on exercise of Series 3 warrants | $ | $ | ||||
Insurance financing | $ | $ | ||||
Recognition of operating lease - right-of-use asset and operating lease liability | $ | $ | — | |||
Lease modification | $ | $ | — | |||
Offering costs included in accounts payable and accrued liabilities | $ | $ | — | |||
Extinguishment of Series A redeemable convertible preferred stock | $ | — | $ | |||
Common stock issued as redemption of Series D perpetual preferred stock | $ | — | $ | |||
Issuance of Series D perpetual preferred stock | $ | — | $ | |||
Common stock issued as redemption of notes payable and related interest | $ | — | $ | |||
Issuance of Series C perpetual preferred stock | $ | — | $ | |||
Deemed dividend attributable to Series C perpetual preferred stock | $ | — | $ | |||
Accretion to redemption value of Series A contingently redeemable convertible preferred stock | $ | — | $ | |||
Conversion of Series B-2 convertible preferred stock into common stock | $ | — | $ | |||
Deemed dividend attributable to Series 1, Series 2 and Bridge warrant holders | $ | — | $ | |||
Shares issued on exercise of Series B convertible preferred shares | $ | — | $ | |||
Shares issued to PoC Capital in payment of contracted research fees | $ | — | $ | |||
Stock dividend attributable to Series C perpetual preferred stock | $ | — | $ | |||
The accompanying notes are an integral part of these consolidated financial statements.
97
Jaguar Health, Inc.
Notes to Financial Statements
1. Organization and Business
Jaguar Health, Inc. (“Jaguar” or the “Company”), formerly known as Jaguar Animal Health, Inc., was incorporated on June 6, 2013 (inception) in Delaware. The Company was a majority-owned subsidiary of Napo until the close of the Company's initial public offering on May 18, 2015. The Company was formed to develop and commercialize first-in-class gastrointestinal products for companion and production animals and horses. The Company's first commercial product, Neonorm Calf, was launched in 2014 and Neonorm Foal was launched in the first quarter of 2016. The Company's activities are subject to significant risks and uncertainties, including failing to secure additional funding in order to timely complete the development and commercialization of products.
On July 31, 2017, Jaguar completed a merger with Napo pursuant to the Agreement and Plan of Merger dated March 31, 2017 by and among Jaguar, Napo, Napo Acquisition Corporation (“Merger Sub”), and Napo's representative (the “Merger Agreement”). In accordance with the terms of the Merger Agreement, upon the completion of the merger, Merger Sub merged with and into Napo, with Napo surviving as the wholly-owned subsidiary (the “Merger” or “Napo Merger”). Immediately following the Merger, Jaguar changed its name from “Jaguar Animal Health, Inc.” to “Jaguar Health, Inc.” Napo now operates as a wholly-owned subsidiary of Jaguar focused on human health and the ongoing commercialization of Mytesi, a Napo drug product approved by the U.S. FDA for the symptomatic relief of noninfectious diarrhea in adults with HIV/AIDS on antiretroviral therapy.
On March 15, 2021, the Company established Napo EU in Italy as a subsidiary of Napo. Napo EU’s mission is to develop and commercialize novel, plant-based, sustainably derived prescription medicines in Europe (excluding Russia) for people with gastrointestinal distress to provide relief and treatment from various gut disorders, their symptoms, and interventions. The initial focus of Napo EU is on pursuing the accelerated conditional marketing authorization pathway from the European Medicines Agency for crofelemer for an important orphan-designated disease: Intestinal failure with short bowel syndrome (“SBS-IF”).
On November 3, 2021, Napo EU and Dragon SPAC S.p.A (“Dragon SPAC”) merged. Dragon SPAC is private company limited by shares with registered office in Italy. Upon close, Dragon SPAC became a controlled subsidiary of the Company and will be a consolidated entity. Further, Napo EU was incorporated into Dragon SPAC with Dragon SPAC as the surviving entity which took over by operation of law all the assets, rights, reasons and actions as well as liabilities, obligations and commitments of NAPO EU. The merged entity was named as Napo Therapeutics S.p.A (“Napo Therapeutics”).
The Company manages its operations through
Nasdaq Communication and Compliance
Minimum Stockholders’ Equity Requirement
On February 17, 2022, the Company received a letter from the Staff of Nasdaq indicating that the bid price of the Company’s common stock for the last
Liquidity and Going Concern
The Company, since its inception, has incurred recurring operating losses and negative cash flows from operations and has an accumulated deficit of $
98
commercialization efforts, as well as securing of additional financing and generating positive cash flows from operations. There is no assurance that the Company will have adequate cash balances to maintain its operations.
Although the Company plans to finance its operations and cash flow needs through equity and/or debt financing, collaboration arrangements with other entities, license royalty agreements, as well as revenue from future product sales, the Company does not believe its current cash balances are sufficient to funds its operating plan through one year from the issuance of these consolidated financial statements. The Company has an immediate need to raise cash. There can be no assurance that additional funding will be available to the Company on acceptable terms, or on a timely basis, if at all, or that the Company will generate sufficient cash from operations to adequately fund operating needs. If the Company is unable to obtain an adequate level of financing needed for the long-term development and commercialization of our products, the Company will need to curtail planned activities and reduce costs. Doing so will likely have an adverse effect on our ability to execute our business plan; accordingly, there is substantial doubt about the ability of the Company to continue in existence as a going concern. The accompanying consolidated financial statements do not include any adjustments that might result from the outcome of these uncertainties.
Reverse Stock Split
On September 3, 2021, the Company filed the Certificate of Fifth Amendment to its Third Amended and Restated Certificate of Incorporation with the Secretary of State of Delaware to effect a -for-3 reverse stock split of the Company’s issued and outstanding shares of voting common stock, effective September 8, 2021. The reverse split has been retrospectively reflected in all voting common stock, warrants, and common stock option shares disclosed in these consolidated financial statements. The non-voting common stock and the convertible preferred stock were excluded from the reverse split.
2. Summary of Significant Accounting Policies
Basis of Presentation
The consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America ("U.S. GAAP").
Principles of Consolidation
The consolidated financial statements have been prepared in accordance with U.S. GAAP and applicable rules and regulations of the Securities and Exchange Commission (“SEC”) and include the accounts of the Company and its subsidiaries. All inter-company transactions and balances have been eliminated in consolidation. The reporting currency of the Company is the U.S. dollar.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires the Company’s management to make judgments, assumptions and estimates that affect the amounts reported in its financial statements and the accompanying notes. The accounting policies that reflect the Company’s more significant estimates and judgments and that the Company believes are the most critical to aid in fully understanding and evaluating its reported financial results are the valuation of stock options, valuation of Series C Perpetual Preferred Stock and Series D Perpetual Stock, valuation of hybrid instruments designated at fair value option (“FVO”), valuation of warrant liability, acquired in-process research and development (“IPR&D”), useful lives assigned to long-lived assets, impairment assessment of intangible assets, valuation adjustments for excess and obsolete inventory, allowance for doubtful accounts, deferred taxes and valuation allowances on deferred tax assets, evaluation and measurement of contingencies, and recognition of revenue, including estimates for product returns. Those estimates could change, and as a result, actual results could differ materially from those estimates.
99
In March 2020, the World Health Organization declared the COVID-19 outbreak to be a pandemic. During the year ended December 31, 2021, the Company’s financial results were not significantly affected by the COVID-19 outbreak. The Company has considered all information available as of the date of issuance of these financial statements and the Company is not aware of any specific events or circumstances that would require an update to its estimates or judgments, or a revision to the carrying value of its assets or liabilities. These estimates may change as new events occur and additional information becomes available. The extent to which the COVID-19 outbreak affects the Company’s future financial results and operations will depend on future developments which are highly uncertain and cannot be predicted, including new information which may emerge concerning the severity of the outbreak, and current or future domestic and international actions to contain and treat it. For a discussion of risks of COVID-19 relating to the Company’s business, see “Item 1A. - Risk Factors- Risks Related to Our Business- The novel coronavirus global pandemic could adversely impact our business, including our supply chain, clinical trials and commercialization of Mytesi and Canalevia.”
Cash
The Company’s cash on deposit may exceed United States federally insured limits at certain times during the year. The Company maintains cash accounts with certain major financial institutions in the United States. The Company does not have cash equivalents as of December 31, 2021 and 2020.
Accounts Receivable
Accounts receivable is recorded net of allowances for discounts for prompt payment and credit losses. The Company estimates an allowance for credit losses by considering factors such as historical experience, credit quality, the age of the accounts receivable balances, and current economic conditions that may affect a customer’s ability to pay. The corresponding expense for the credit loss allowance is reflected in general and administrative expenses. The credit loss allowance was immaterial as of December 31, 2021 and 2020.
Concentrations
Cash is the financial instrument that potentially subjects the Company to a concentration of credit risk as cash is deposited with banks and cash balances are generally in excess of Federal Deposit Insurance Corporation (“FDIC”) insurance limits.
For the years ended December 31, 2021 and 2020, substantially all of the Company’s revenue was derived from the sale of Mytesi. In looking at sales by the Company to distributors whose net revenue percentage of total net revenue was equal to or greater than
Year Ended |
| |||||
December 31, |
| |||||
| 2021 |
| 2020 |
| ||
Customer 1 | | % | | % | ||
Customer 2 | | % | — | % | ||
Customer 3 | | % | — | % | ||
100
The Company is subject to credit risk from its accounts receivable related to its sales. The Company generally does not perform evaluations of customers' financial condition and generally does not require collateral. Accounts receivable balance of the significant customers as a percentage of total accounts receivable is as follows:
December 31, | |||||
| 2021 | 2020 |
| ||
Customer 1 | | % | | % | |
Customer 2 | | % | — | % | |
Customer 3 | | % | — | % | |
The Company is subject to concentration risk from its suppliers. The Company sources raw material used to produce the active pharmaceutical ingredient (“API”) in Mytesi from
Other Risks and Uncertainties
The Company’s future results of operations involve a number of risks and uncertainties. Factors that could affect the Company’s future operating results and cause actual results to vary materially from expectations include, but are not limited to, rapid technological change, obtaining second source suppliers, regulatory approval from the FDA or other regulatory authorities, the results of clinical trials and the achievement of milestones, market acceptance of the Company’s product candidates, competition from other products and larger companies, protection of proprietary technology, strategic relationships and dependence on key individuals.
Fair Value
The Company’s financial instruments include accounts receivable, accounts payable, accrued liabilities, warrant liability, operating lease liability, equity-linked financial instruments, and debt. The recorded carrying amount of accounts receivable, accounts payable and accrued liabilities reflect their fair value due to their short-term nature. Other financial liabilities are initially recorded at fair value, and subsequently measured at either fair value or amortized cost using the effective interest method. See Note 4 for the fair value measurements.
Fair Value Option
ASC 825-10, Financial Instruments, provides FVO election that allows companies an irrevocable election to use fair value as the initial and subsequent accounting measurement attribute for certain financial assets and liabilities. ASC 825-10 permits entities to elect to measure eligible financial assets and liabilities at fair value on an ongoing basis. Unrealized gains and losses on items for which the FVO has been elected are reported in earnings. The decision to elect the FVO is determined on an instrument-by-instrument basis, must be applied to an entire instrument and is irrevocable once elected. Assets and liabilities measured at fair value pursuant to ASC 825-10 are required to be reported separately from those instruments measured using another accounting method. In accordance with the options presented in ASC 825-10, the Company elected to present the aggregate of fair value and non-fair-value amounts in the same line item in the consolidated balance sheets and parenthetically disclose the amount measured at fair value in the aggregate amount.
Inventory
Inventory is stated at the lower of cost or net realizable value. Cost is determined using the first-in, first-out method. Cost is initially recorded at the invoiced amount of raw materials or API, including the sum of qualified expenditures and charges in bringing the inventory to its existing condition and location. The Company calculates inventory valuation adjustments when conditions indicate that net realizable value is less than cost due to physical deterioration, usage, obsolescence, reductions in estimated future demand or reduction in selling price. Inventory write-downs are measured as the difference between the cost of inventory and net realizable value.
101
Property and Equipment
Land is stated at cost, reflecting fair value of the property at July 31, 2017, the date of the Napo merger. Equipment is stated at cost, net of accumulated depreciation. Equipment begins to be depreciated when it is placed into service. Depreciation is calculated using the straight-line method over estimated useful lives ranging between to
Expenditures for repairs and maintenance of assets are charged to expense as incurred. Costs of major additions and betterments are capitalized and depreciated on a straight-line basis over their estimated useful lives. Upon retirement or sale, the cost and related accumulated depreciation of assets disposed of are removed from the accounts and any resulting gain or loss is included in the consolidated statements of operations.
Long-Lived Assets
The Company regularly reviews the carrying value and estimated lives of all of its long-lived assets, including property and equipment and definite-lived intangible assets, to determine whether indicators of impairment exist that warrant adjustments to carrying values or estimated useful lives. The determinants used for this evaluation include management’s estimate of the asset’s ability to generate positive income from operations and positive cash flow in future periods as well as the strategic significance of the assets to the Company’s business objectives. If the Company determines that an impairment trigger has been met, the Company evaluates the realizability of its long-lived assets (asset group) based on a comparison of projected undiscounted cash flows from use and eventual disposition with the carrying value of the related asset. Any write-downs (which are measured based on the difference between the fair value and the carrying value of the asset) are treated as permanent reductions in the carrying amount of the assets (asset group). Based on this evaluation, the Company believes that, as of each of the balance sheet dates presented, none of the Company’s long-lived assets were impaired. The Company’s had
Indefinite-lived Intangible Assets
Acquired IPR&D are intangible assets acquired in the July 2017 Napo merger. Under ASC 805, IPR&D are initially recognized at fair value and classified as indefinite-lived assets until the successful completion or abandonment of the associated research and development efforts. During the development period, these assets will not be amortized as charges to earnings; instead, these assets will be tested for impairment on an annual basis or more frequently if impairment indicators are identified. An impairment loss is measured based on the excess of the carrying amount over the asset’s fair value. The Company recorded an impairment of
Leases
The Company accounts for its leases in accordance with ASC 842, Leases.
At the inception of an arrangement, the Company determines whether the arrangement is or contains a lease based on the unique facts and circumstances present. Operating lease liabilities and their corresponding right-of-use assets are recorded based on the present value of lease payments over the expected lease term. Because the interest rate implicit in lease contracts is typically not readily determinable, the Company utilizes its incremental borrowing rate, which is the rate incurred to borrow on a collateralized basis over a similar term, an amount equal to the lease payments in a similar economic environment. Certain adjustments to the right-of-use asset may be required for items such as initial direct costs paid or incentives received.
Operating Lease
The Company had a non-cancelable operating lease with CA-Mission Street Limited Partnership for its offices in San Francisco, California, through September 30, 2020. The lease agreement called for monthly base rents
102
between $
The Company entered into a sublease agreement with Peacock Construction, Inc., a California corporation, for office space located in San Francisco, California. The term of the sublease began on August 31, 2020 and expired on May 31, 2021. The rent under the sublease was $
In April 2021, the Company entered into an office lease agreement with M & E, LLC, a California Limited Liability Company, to lease approximately
In December 2021, the Company entered into the first amendment to the lease with M & E, LLC whereby the commencement date of one of the leased premises was modified to March 1, 2022. Accordingly, the expiration of the lease was extended to February 28, 2025. The base rent under the original agreement remained the same but will be due starting March 1, 2022. In addition, the rent for one of the leased premises being occupied by the Company will continue to be $
Research and Development Expense
Research and development expense consists of expenses incurred in performing research and development activities including related salaries, clinical trial and related drug and non-drug product costs, contract services and other outside service expenses. Research and development expense is charged to operating expense in the period incurred.
Clinical Trial Accruals
Clinical trial costs are a component of research and development expenses. The Company accrues and expenses clinical trial activities performed by third parties based upon actual work completed in accordance with agreements established with clinical research organizations and clinical sites. The Company determines the costs to be recorded based upon validation with the external service providers as to the progress or stage of completion of trials or services and the agreed-upon fee to be paid for such services.
Revenue Recognition
The Company recognizes revenue in accordance with ASC Topic 606, Revenue from Contracts with Customers (“ASC 606”).
The Company’s policy typically permits returns if the product is damaged, defective, or otherwise cannot be used when received by the customer if the product has expired. Returns are accepted for product that will expire within six months or that have expired up to one year after their expiration dates. Estimates for expected returns of expired products are based primarily on an ongoing analysis of our historical return patterns.
The Company recognizes revenue in accordance with the core principle of ASC 606 or when there is a transfer of control of promised goods or services to customers in an amount that reflects the consideration that the Company expects to be entitled to in exchange for those goods or services.
103
The Company recognizes the incremental costs of obtaining a contract as an expense when incurred if the amortization period of the asset that the Company otherwise would have recognized is one year or less.
The Company does not adjust the amount of consideration for the effects of a significant financing component if, at contract inception, the expected period between the transfer of promised goods or services and customer payment is
The Company has elected to treat shipping and handling activities as fulfillment costs.
Additionally, the Company elected to record revenue net of sales and other similar taxes.
Contracts – Cardinal Health
Effective January 16, 2019, Napo engaged Cardinal Health SPS as its exclusive third-party logistics distribution agent for commercial sales for the Company’s Mytesi product and to perform certain other services which include, without limitation, storage, distribution, returns, customer support, financial support, Electronic Data Interchange and system access support (the “Exclusive Distribution Agreement”).
On September 3, 2021, the Company ended its engagement with Cardinal Health as its exclusive title model customer for commercial sales and fully implemented its limited distribution Specialty Pharmacy model. Cardinal Health continues to provide third-party logistics services for Mytesi.
The Company's Neonorm and botanical extract products are primarily sold to distributors, who then sell the products to the end customers. Since 2014, the Company has entered into several distribution agreements with established distributors such as Animart, Vedco, VPI, RJ Matthews, Covetrus, and Stockmen Supply to distribute the Company's products in the United States, Japan, and China. The distribution agreements and the related purchase order together meet the contract existence criteria under ASC 606-10-25-1. The Company sells directly to its customers without the use of an agent.
Performance obligations
For animal products sold by the Company, the single performance obligation identified is the Company’s promise to transfer the Company’s animal products to distributors based on specified payment and shipping terms in the arrangement. Product warranties are assurance-type warranties that do not represent a performance obligation. For the Company’s human product, Mytesi, the single performance obligation identified above is the Company’s promise to transfer Mytesi to Cardinal Health, based on specified payment and shipping terms as outlined in the Exclusive Distribution Agreement.
Transaction price
For contracts with Cardinal Health, the transaction price is the amount of consideration to which the Company expects to collect in exchange for transferring promised goods or services. The transaction price of Mytesi and Neonorm is the Wholesaler Acquisition Cost (“WAC”), net of discounts, returns, and price adjustments.
Allocate transaction price
For contracts with Cardinal Health, the entire transaction price is allocated to the single performance obligation contained in each contract.
Revenue recognition
For contracts with Cardinal Health, for the Company, a single performance obligation is satisfied at a point in time, upon the FOB terms of each contract when control, including title and all risks, has transferred to the customer.
104
Disaggregation of Product Revenue
Human
Sales of Mytesi are recognized as revenue at a point in time when the products are delivered to the wholesaler. Net revenues from the sale of Mytesi were $
Animal
The Company recognized Neonorm revenues of $
Contracts – Atlas Sciences, LLC
Effective April 15, 2020, the Company entered into a patent purchase agreement with Atlas Sciences, LLC (“Atlas”), pursuant to which Atlas agreed to purchase certain patents and patent applications relating to Napo’s NP-500 drug product candidate (the “Patent Rights”) for an upfront cash payment of $
Concurrent with the Patent Rights sale, the Company entered into a license agreement with Atlas (the “License Agreement”), pursuant to which Atlas granted the Company an exclusive
Included in the arrangement with Atlas, the Company was obligated to initiate a proof of concept Phase 2 study of NP-500 under an investigational new drug (“IND”) application with the U.S. Food and Drug Administration or an IND-equivalent dossier under appropriate regulatory authorities (the “Phase 2 study”) within
In September 2020, the Company made the decision not to initiate the Phase 2 study and negotiated the payment of the trial delay fee of $
The Company derecognized $
The Company evaluated the nature of the consideration payable to the customer and the rights and obligations in the related contract and concluded that the excess payment or loss should be presented as part of the general and administrative expenses due to the following factors:
| ● |
| ● | The Company settled the trial delay fee in full in October 2020, which constitutes termination of the customer relationship considering that Atlas cannot compel the Company or has no recourse to force the Company to initiate the Phase 2 Study. The Company does not anticipate future revenue contract with Atlas. |
105
| ● | The trial delay fee is a penalty in its economic term, subject to accounting for contingencies and provisions under relevant authoritative guidance. |
In October 2020, the Company entered into a fee settlement agreement with Atlas pursuant to which the Company agreed to issue
Contracts – Specialty Pharmacies
Effective October 1, 2020, the Company engaged a private company as its third-party logistics distribution agent for commercial sales of the Company’s Mytesi product. Under the Specialty Product Distribution Agreement, the Company shall supply the products to the private company’s specialty pharmacies, through a designated wholesaler, in such amounts as may be ordered. There is no minimum purchase or inventory requirement. The specialty pharmacies were authorized distributors of record for all National Drug Codes (“NDCs”) of Mytesi.
Effective April 20, 2021, the Company engaged another private company as an authorized specialty pharmacy provider of Mytesi. Under the Specialty Pharmacy Distribution and Services Agreement, the private company shall sell and dispense the Mytesi directly ordered from the Company at the agreed price to patients within the territories identified in the agreement.
The
Performance obligations
The single performance obligation is the Company’s promise to transfer Mytesi to specialty pharmacies, based on specified payment and shipping terms outlined in the agreements.
Transaction price
The transaction price is the amount of consideration to which the Company expects to collect in exchange for transferring the promised goods or services. The transaction price of Mytesi is the WAC, net of estimated discounts, returns, and price adjustments.
Allocate transaction price
The entire transaction price is allocated to the single performance obligation contained in each contract.
Revenue recognition
The single performance obligation is satisfied at a point in time, upon the free on board (“FOB”) terms of each contract, when control, including title and all risks, has transferred to the customer.
Product Revenue
Sales of Mytesi are recognized as revenue at a point in time when the products are delivered to the specialty pharmacies. Net revenues from the sale of Mytesi to the specialty pharmacies were $
106
Collaboration Revenue
Revenue recognition for collaboration agreements requires significant judgment. The Company’s assessments and estimates are based on contractual terms, historical experience and general industry practice. Revisions in these values or estimations have the effect of increasing or decreasing collaboration revenue in the period of revision.
On September 24, 2018, the Company entered into a Distribution, License and Supply Agreement (“License Agreement”) with Knight Therapeutics ("Knight"). The License Agreement has a term of
Modifications to Liability-classified Instruments
In accounting for debt modifications and exchange transactions, it is the Company’s policy to first determine whether it qualifies as a Troubled Debt Restructuring (“TDR”) pursuant to the guidance provided in ASC 470-60. A debt modification or exchange transaction that is not within the scope of the ASC 470-60 is accounted for under ASC 470-50 to determine if the transaction is a mere modification or an extinguishment.
The Company amended the terms of its October 2020 Purchase Agreement and Exchange Note 2 in the year 2021 (see Note 8). The Company also amended the terms of its Exchange Note 1, Exchange Note 2, March 2020 Purchase Agreement, and Series D Perpetual Preferred Stock in the year 2020 (see Note 8).
Modifications to Equity-classified Instruments
In accounting for modifications of equity-classified warrants, it is the Company’s policy to determine the impact by analogy to the share-based compensation guidance of ASC 718, Compensation - Stock Compensation (“ASC 718”). The model for a modified share-based payment award that is classified as equity and remains classified in equity after the modification is addressed in ASC 718-20-35-3. Pursuant to that guidance, the incremental fair value from the modification is recognized as an expense in the statements of operations to the extent the modified instrument has a higher fair value; however, in certain circumstances, such as when an entire class of warrants are modified, the measured increase in fair value may be more appropriately recorded as a deemed dividend, depending upon the nature of the warrant modification.
The Company modified certain equity-classified warrants in the year 2020 (see Note 9). The Company did not modify any equity-classified warrants in the year 2021.
In accounting for amendments to equity-classified preferred stock, it is the Company’s policy to measure the impact by analogy to ASC 470-50 in determining if such an amendment is an extinguishment or a modification. If the amendment results in an extinguishment, the Company follows the SEC staff guidance in ASC 260-10-S99-2 and ASC 470-20. If the amendment results in a modification, the Company follows the model in either ASC 718 or ASC 470-50, depending on the nature of the amendment.
The Company modified the terms of its Series B Convertible Preferred Stock and Series C Perpetual Preferred Stock in the year 2020 (see Note 10). The Company did not modify any equity-classified preferred stock in the year 2021.
107
Stock-Based Compensation
The Company's Stock Incentive Plan (see Note 12) provides for the grant of stock options, restricted stock and restricted stock unit awards. The Company measures stock awards granted to employees, non-employees and directors at estimated fair value on the date of grant and recognizes the corresponding compensation expense of the awards, net of estimated forfeitures, over the requisite service periods, which correspond to the vesting periods of the awards. Forfeitures are estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. The Company issues stock awards with only service-based vesting conditions, and records compensation expense for these awards using the straight-line method.
The Company uses the grant date fair market value of its common stock to determine the grant date fair value of options granted to employees, non-employees and directors. The Company measures and recognizes compensation expense for all stock options and restricted stock units (“RSUs”) granted to its employees and directors based on the estimated fair value of the award on the grant date. The Company uses the Black-Scholes valuation model to estimate the fair value of stock option awards. The fair value is recognized as expense, net of estimated forfeitures, over the requisite service period, which is generally the vesting period of the respective award, on a straight-line basis. The Company believes that the fair value of stock options granted to non-employees is more reliably measured than the fair value of the services received. The determination of the grant date fair value of options using an option pricing model is affected by the Company’s estimated Common Stock fair value and requires management to make a number of assumptions including the expected life of the option, the volatility of the underlying stock, the risk-free interest rate and expected dividends.
The Company estimates the fair value of stock options using the Black-Scholes option valuation model. The fair value of employee stock options is being amortized on a straight-line basis over the requisite service period of the awards. The fair market value of common stock is based on the closing price of the Company’s common stock as reported on the date of the grant.
Income Taxes
The Company uses the asset and liability method of accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on the differences between the financial reporting and the tax bases of assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. A valuation allowance is provided when it is more likely than not that some portion or all of a deferred tax asset will not be realized.
The Company has adopted the provisions of ASC 740, Income Taxes Related to Uncertain Tax Positions. Under these principals, tax positions are evaluated in a two-step process. The Company first determines whether it is more-likely-than-not that a tax position will be sustained upon examination. If a tax position meets the more-likely-than-not recognition threshold, it is then measured to determine the amount of benefit to be recognized in the financial statements. The tax position is measured as the largest amount of benefit that has a greater than 50 percent likelihood of being realized upon ultimate settlement.
Foreign Currency Remeasurement and Translation
The functional currency of Napo Therapeutics is Euro. The Company follows ASC 830, Foreign Currency Matters (“ASC 830”). ASC 830 requires the assets, liabilities, and results of operations of a foreign operation to be measured using the functional currency of that foreign operation. Exchange gains or losses from remeasuring transactions and monetary accounts in a currency other than the functional currency are included in current earnings.
For certain subsidiaries, translation adjustments result from the process of translating the functional currency of subsidiary financial statements into the U.S. Dollar reporting currency. These translation adjustments are reported separately and accumulated in the consolidated balance sheets as a component of accumulated other comprehensive loss.
108
Comprehensive Loss
The Company follows ASC 220, Comprehensive Income, which establishes standards for reporting and displaying comprehensive income and its components (revenue, expenses, gains and losses) in a full set of general-purpose financial statements.
For the year ended December 31, 2020, the comprehensive loss was equal to the net loss; therefore, a separate statement of comprehensive loss was not included in the accompanying consolidated financial statements.
For the year ended December 31, 2021, the amount of other comprehensive loss was only de minimis; hence, a separate statement of comprehensive loss was not also included in the accompanying consolidated financial statements.
Basic and Diluted Net Loss Per Common Share
Basic net loss per common share is computed by dividing net loss attributable to common stockholders for the year by the weighted-average number of common shares outstanding during the year. Diluted net loss per share is computed by dividing the net loss attributable to common stockholders for the year by the weighted-average number of common shares, including potential dilutive shares of common stock assuming the dilutive effect of potential dilutive securities. For years in which the Company reports a net loss, diluted net loss per common share is the same as basic net loss per common share, because their impact would be anti-dilutive to the calculation of net loss per common share. Diluted net loss per common share is the same as basic net loss per common share for the years ended December 31, 2021 and 2020.
Recent Accounting Pronouncements
Recently Adopted Accounting Pronouncements
In December 2019, the FASB issued ASU 2019-12, Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes, which is intended to simplify various aspects related to accounting for income taxes. The standard also removes certain exceptions to the general principles in Topic 740 and clarifies and amends existing guidance to improve consistent application. The Company adopted the standard on January 1, 2021. The adoption of this standard did not have a material effect on the Company’s consolidated financial statements and related disclosures.
Recently Issued Accounting Pronouncements Not Yet Adopted
In June 2016, the FASB issued ASU 2016-13, Financial Instruments – Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments. The main objective of the standard is to provide financial statement users with more decision-useful information about the expected credit losses on financial instruments and other commitments to extend credit held by a reporting entity at each reporting date. To achieve this objective, the amendments in this standard replace the incurred loss impairment methodology in current GAAP with a methodology that reflects expected credit losses and requires consideration of a broader range of reasonable and supportable information to inform credit loss estimates. The update is effective for the Company beginning January 1, 2023 with early adoption permitted. The Company is still evaluating the impact of the adoption of this standard.
In August 2020, the FASB issued ASU 2020-06, Debt – Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging – Contracts in Entity’s Own Equity (Subtopic 815-40): Accounting for Convertible Instruments and Contracts in an Entity’s Own Equity, which simplifies the accounting for certain financial instruments with characteristics of liabilities and equity, including convertible instruments and contracts on an entity’s own equity. The pronouncement is effective for the Company beginning January 1, 2022 with early adoption permitted. The Company is still evaluating the impact of the adoption of this standard.
109
In May 2021, the FASB issued ASU 2021-04, Issuer’s Accounting for Certain Modification or Exchanges of Freestanding Equity-Classified Written Call Options – a consensus of the FASB Emerging Issues Task Force. The ASU provides a principles-based framework to determine whether an issue should recognize the modification or exchange as an adjustment to equity or an expense. The amendments in the update are effective for all entities for fiscal years beginning January 1, 2022, including interim periods within those fiscal years with early adoption permitted. The Company is still evaluating the impact of the adoption of this standard.
Reclassification of Prior Year Presentation
Certain prior period amounts of cash flows from financing activities in the consolidated statements of cash flows have been reclassified within the same category of cash flow activity to be consistent with the current period presentation. There were no reclassifications to other categories of cash flow activity and the reclassification did not impact the profit or loss during the prior period.
3. Napo Therapeutics Subsidiary
As discussed in Note 1 – Organization and Business, Napo EU completed a merger with Dragon SPAC on November 3, 2021, with Dragon SPAC as the surviving entity. Dragon SPAC took over by operation of law all the assets, rights, reasons, and actions as well as liabilities, obligations, and commitments of Napo EU. The merged entity was named Napo Therapeutics. Napo Therapeutics now operates as a subsidiary of Napo, with Napo owning
4. Fair Value Measurements
ASC 820 "Fair Value Measurements," defines fair value, establishes a framework for measuring fair value under U.S. GAAP and enhances disclosures about fair value measurements. Fair value is defined under ASC 820 as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value under ASC 820 must maximize the use of observable inputs and minimize the use of unobservable inputs. The standard describes a fair value hierarchy based on three levels of inputs, of which the first two are considered observable and the last unobservable, that may be used to measure fair value which are the following:
| ● | Level 1 – Observable inputs such as quoted prices (unadjusted) for identical instruments in active markets. |
| ● | Level 2 – Observable inputs such as quoted prices for similar instruments in active markets, quoted prices for identical or similar instruments in markets that are not active, or model‑derived valuations whose significant inputs are observable. |
| ● | Level 3 – Unobservable inputs that reflect the reporting entity’s own assumptions. |
The following tables set forth the fair value of the Company’s consolidated financial instrument that was measured at fair value on a recurring basis as of December 31, 2021 and 2020:
110
December 31, 2021 | ||||||||||||
(in thousands) |
| Level 1 |
| Level 2 |
| Level 3 |
| Total | ||||
Warrant liability | $ | — | $ | — | $ | $ | ||||||
Streeterville note | — | — | ||||||||||
Total fair value | $ | — | $ | — | $ | $ | ||||||
December 31, 2020 | ||||||||||||
(in thousands) |
| Level 1 |
| Level 2 |
| Level 3 |
| Total | ||||
Warrant liability | $ | — | $ | — | $ | $ | ||||||
Total fair value | $ | — | $ | — | $ | $ | ||||||
The change in the estimated fair value of the Level 3 liability is summarized below:
Year Ended | ||||||
December 31, 2021 | ||||||
(in thousands) | Warrant liability |
| Streeterville note | |||
Beginning fair value of Level 3 liability |
| $ |
| $ | — | |
Additions | ||||||
Exercises | ( | — | ||||
Change in fair value |
|
| ||||
Ending fair value of Level 3 liability |
| $ |
| $ | ||
Warrant Liability
The warrants associated with the Level 3 warrant liability were the November 2016 Series A Warrants and the October 2018 Underwriter Warrants, which, at December 31, 2021, were valued at
The November 2016 Series A Warrants
The Series A warrant valuation of
The October 2018 Underwriter Warrants
The October 2018 Underwriter Warrants valuation of $
111
The May 2020 Series 3 Warrants
There were
Streeterville Note
The fair value of the Streeterville Note at January 13, 2021, date of issuance and as of December 31, 2021 amounting to $
The Company determined and performed the valuations of the Streeterville Note with the assistance of an independent valuation service provider. On a quarterly basis, the Company considers the main Level 3 inputs used as follows:
| ● | Discount rate for the Streeterville note was determined using a comparison of various effective yields on bonds as of the valuation date. |
| ● | Market indications for vouchers, which affect the Return Bonus from the sale of Tropical Disease Priority Review Voucher (“TDPRV”) |
| ● | Weighted probability of cash outflows was estimated based on the entity's knowledge of the business and how the current economic environment is likely to impact the timing of the cash outflows, attributed to the different repayment features of the note. |
The following table summarizes the quantitative information about the significant unobservable inputs used in Level 3 fair value measurement:
Range of Inputs | ||||||
(probability-weighted average) | Relationship of unobservable inputs | |||||
Unobservable Inputs | 2021 | 2020 | to fair value | |||
Risk Adjusted Discount Rate |
| N/A | If discount rate is adjusted to total of additional | |||
Sales Proceeds: Amount of comparable TDPRV | $ | N/A | If expected cash flows by management considered the lowest amount of market indications for vouchers, FV would have decreased by $ | |||
112
If expected cash flows by management considered the highest amount of market indications for vouchers, FV would have increased by $ | ||||||
Range of Probability for Timing of Cash Flows: | N/A | If expected cash flows by management considered the scenario with the least amount of indicated value, FV would have decreased by $ |
Fair Value Option
Beginning January 1, 2021, the Company elected to apply the FVO accounting to selected financial instruments to align the measurement attributes of those instruments under U.S. GAAP and to simplify the accounting model applied to those financial instruments. The Company elected to apply FVO accounting to the entire class of hybrid instruments, including structured notes, of which there are assessed embedded derivatives that would be eligible for bifurcation. Changes in the fair value of FVO assets and liabilities as well as the mark-to-market adjustment on the entire class of hybrid instruments, including derivatives and the net realized gains or losses on these instruments are reported in the change in fair value of financial instruments and hybrid instrument designated at FVO in the consolidated statements of operations.
For the year ended December 31, 2021, the Company did not note any fair value movement on FVO liabilities attributable to any instrument-specific credit risk, which should be recorded in other comprehensive income (loss).
Hybrid Instruments
The Company elected to apply FVO accounting to all of the hybrid instruments issued, including structured notes. The valuation of the hybrid instruments is predominantly driven by the derivative features embedded within the instruments. The Company determined and performed the valuations of the hybrid instruments with the assistance of an independent valuation service provider. The valuation methodology utilized is consistent with the income approach for estimating the fair value of the interest-bearing portion of the instrument and the related derivatives. Cash flows of the hybrid instruments in their entirety, including the embedded derivatives, are discounted at an appropriate rate for the applicable duration of the instrument. Interest on the interest-bearing portion of the instrument that is held to maturity is aggregated as gain (loss) on instruments designated at fair value and related derivatives in the change in fair value of financial instruments and hybrid instruments designated at FVO in the consolidated statements of operations.
The following table summarizes the fair value and unpaid principal balance for items the Company accounts under FVO:
(in thousands) | Fair value | Unpaid Principal Balance | Fair Value Over (Under) Unpaid Principal Balance | ||||||
At December 31, 2021 | |||||||||
Hybrid Instrument: | |||||||||
Streeterville note | $ | $ | $ | ||||||
113
5. Related Party Transactions
Management Services Agreement
In March 2018, concurrent with the issuance of the Company’s Series A Convertible Preferred Stock to Sagard Capital Partners, L.P. (“Sagard Capital”), the Company entered into a Management Services Agreement with Sagard Capital. Under the agreement, Sagard Capital was to provide consulting and management advisory service to the Company from March 2018 through March 2021. These services include assistance with strategic planning regarding the Company’s commercial strategy, research and due diligence regarding human resource activities, and strategic advice in financial matters. In consideration for such services, the Company paid Sagard Capital an annual fee of $
Letter of Credit
On March 24, 2020, the Company entered into a letter of credit agreement with Dr. Charles Conte, the brother of Lisa Conte, the Company’s President, CEO and member of the Company’s board of directors (“BOD”), pursuant to which the Company replaced then existing letter of credit in the amount of $
BOD Cash Compensation
Effective May 2021, the Company will pay the BOD cash compensation on a quarterly basis based on the Director Compensation Program for 2021. For the year ended December 31, 2021, the Company paid approximately $
6. Commitments and Contingencies
Commitments
Leases
On August 28, 2018, the Company entered into an office lease extension agreement for approximately
114
On August 31, 2020, the Company entered into an office sublease of approximately
On April 6, 2021, the Company entered into an office lease agreement of approximately
The base rent under the lease were $
On December 24, 2021, the Company entered into the first amendment of the lease of office space in San Francisco. The expiration of the lease was extended to February 28, 2025 due to the change in the commencement date of one of the leased premises to March 1, 2022. The base rent under the lease amendment remained the same but will only be due starting March 1, 2022. The rent in one of the leased premises currently being occupied by the Company was and will still be $
The table below provided additional details of the office space lease presented in the consolidated balance sheet:
December 31, | |||||
(in thousands) | 2021 | 2020 | |||
Operating lease - right-of-use asset | $ | $ | — | ||
Operating lease liability, current | — | ||||
Operating lease liability, net of current portion | — | ||||
Total | $ | $ | — | ||
Weighted-average remaining life (years) | — | ||||
Weighted-average discount rate | — | ||||
Lease cost included in general and administrative expenses in the consolidated statements of operations for the year ended December 31, 2021 was approximately $
For the year ended December 31, 2021 and 2020, cash paid for operating lease liabilities recognized under operating cash flows amounted to $
115
liabilities amounting to $
The following table summarizes the undiscounted cash payment obligations for the operating lease liability:
December 31, | |||||
(in thousands) | 2021 | 2020 | |||
2021 | $ | — | $ | — | |
2022 | — | ||||
2023 | — | ||||
2024 | — | ||||
2025 | — | ||||
Total undiscounted operating lease payments | — | ||||
Imputed interest expenses | ( | — | |||
Total operating lease liability | — | ||||
Less: Operating lease liability, current | — | ||||
Operating lease liability, net of current portion | $ | $ | — | ||
On October 10, 2021, the Company also entered into a short-term office lease in Milan, Italy. The term of the lease began on November 1, 2021 and will expire on April 30, 2022, subject to automatic renewal equal to the present term until terminated by mutual agreement. The Company recognizes rent expense on a straight-line basis over the non-cancellable lease period. Rent expense, included in general and administrative expenses in the consolidated statements of operations, was $
Purchase Commitment
On September 3, 2020, the Company entered into a manufacturing and supply agreement (the “Agreement”) with Glenmark Life Sciences Limited (“Glenmark”), pursuant to which Glenmark will continue to serve as the Company’s manufacturer of crofelemer for use in Mytesi, the Company’s human prescription drug product approved by the U.S. Food and Drug Administration, and for other crofelemer-based products manufactured by the Company or its affiliates for human or animal use. The term of the Agreement is approximately
Master Services Agreement (“MSA”)
On June 24, 2019, the Company entered into an MSA for clinical research organization services (the “2019 MSA”) and a service order under such 2019 MSA with Integrium, LLC (“Integrium”). The service order supports the Company’s study to evaluate the effect of Mytesi on gastrointestinal microbiome in people living with HIV. The 2019 MSA will terminate upon the satisfactory performance of all services to be provided thereunder unless earlier terminated by the parties.
On October 5, 2020, the Company entered into another MSA for clinical research organization services (the “2020 MSA”) and a service order under such 2020 MSA with Integrium. The service order covers the Company’s planned upcoming pivotal Phase 3 clinical trial for cancer-therapy related diarrhea. As consideration for its services,
116
the Company will pay Integrium a total amount of up to approximately $
Asset Transfer and Transition Commitment
On September 25, 2017, Napo entered into the Termination, Asset Transfer and Transition Agreement dated September 22, 2017 with Glenmark. As a result of the agreement, Napo now controls commercial rights for Mytesi for all indications, territories and patient populations globally, and also holds commercial rights to the existing regulatory approvals for crofelemer in Brazil, Ecuador, Zimbabwe and Botswana. In exchange, Napo agrees to pay Glenmark
Revenue Sharing Commitment Update
On December 14, 2017, the Company announced its entry into a collaboration agreement with Seed Mena Businessmen Services LLC (“SEED”) for Equilevia™, the Company's non-prescription, personalized, premium product for total gut health in equine athletes. According to the terms of the Agreement, the Company will pay SEED
Legal Proceedings
On July 20, 2017, a putative class action complaint was filed in the United States District Court, Northern District of California, Civil Action No. 3:17-cv-04102, by Tony Plant (the “Plaintiff”).
The Company answered the complaint on August 2, 2019; the answer denied the material allegations of the second amended complaint. Following the completion of document discovery, the parties engaged in a mediation that resulted in an agreement in principle to settle the litigation on a class-wide basis for $
On May 27, 2021, the court gave the final approval to the proposed settlement and the entire settlement consideration will be provided by the Company’s director and officer liability insurance carrier. Under the loss recovery model in ASC 450 and in reference to ASC 410, the ultimate net income effect of the recognized loss and the insurance proceeds directly related to the recognized loss is
As of December 31, 2021 and 2020, the Company concluded not to record any loss contingency and insurance recovery.
Settlement of Underwriter Fee
In August 2018, the Company entered into an agreement with an underwriter pursuant to which the underwriter would aid the Company in identifying certain financing transactions, in exchange for a percentage fee of any such financing and warrants. In the first quarter of 2020, the Company and the underwriter agreed on a final settlement for the underwriter services comprised of a cash payment, warrants and common stock. The cash payment amount totalled $
117
and, in 2020, issuing an additional
Severance Agreements
In June 2020, the Company entered into certain agreements relating to the payment of severance and other benefits to executive officers of the Company, the severance agreements provide for compensation and benefits if the executive officer is subject to (a) a termination of employment by the Company without cause or (b) a good reason termination, within three months following a change in control.
Contingencies
From time to time, the Company maybe a party to various legal actions, both inside and outside the U.S., arising in the ordinary course of its business or otherwise. The Company accrues amounts, to the extent they can be reasonably estimated, that the Company believes will result in a probable loss (including, among other things, probable settlement value), to adequately address any liabilities related to legal proceedings and other loss contingencies. A loss or a range of loss is disclosed when it is reasonably possible that a material loss will incur and can be estimated, or when it is reasonably possible that the amount of a loss, when material, will exceed the recorded provision. The Company did not have any material accruals for any currently active legal action in its consolidated balance sheets as of December 31, 2021, as the Company could not predict the ultimate outcome of these matters, or reasonably estimate the potential exposure.
7. Balance Sheet Components
Inventory
Inventory at December 31, 2021 and 2020 consisted of the following:
December 31, | ||||||
(in thousands) |
| 2021 |
| 2020 | ||
Raw Material | $ | $ | ||||
Work in Process | ||||||
Finished Goods | ||||||
Inventory | $ | $ | ||||
Property and Equipment, net
Property and equipment at December 31, 2021 and 2020 consisted of the following:
December 31, | ||||||
(in thousands) |
| 2021 |
| 2020 | ||
Land | $ | $ | ||||
Lab equipment | ||||||
Clinical equipment |
|
| ||||
Software | ||||||
Total property and equipment at cost |
|
| ||||
Accumulated depreciation |
| ( |
| ( | ||
Property and equipment, net | $ | $ | ||||
118
Depreciation and amortization expense was $
Intangible assets, net
Intangible assets, net of amortization, at December 31, 2021 and 2020 consisted of the following:
December 31, | ||||||
(in thousands) |
| 2021 |
| 2020 | ||
Developed technology | $ | $ | ||||
Accumulated developed technology amortization |
| ( |
| ( | ||
Developed technology, net |
|
| ||||
In-process research and development | ||||||
In process research and development, net |
|
| ||||
Trademarks |
|
| ||||
Accumulated trademark amortization |
| ( |
| ( | ||
Trademarks, net |
|
| ||||
Total intangible assets, net | $ | $ | ||||
Amortization expense of finite-lived intangible assets was $
The following table summarizes the Company’s estimated future amortization expense of intangible assets with finite lives as of December 31, 2021:
(in thousands) |
| Amounts | |
2022 | $ | ||
2023 | |||
2024 | |||
2025 | |||
2026 | |||
Thereafter | |||
$ | |||
Accrued Liabilities
Accrued liabilities at December 31, 2021 and 2020 consisted of the following:
| December 31, | |||||
(in thousands) |
| 2021 | 2020 | |||
Accrued interest | $ | $ | ||||
Accrued legal costs | ||||||
Accrued chargebacks and discounts | ||||||
Accrued local tax | — | |||||
Accrued vacation | ||||||
Accrued distributor services fees | ||||||
Accrued audit and tax services | ||||||
Accrued payroll and commission |
|
| ||||
Accrued payroll tax | ||||||
Accrued consulting | ||||||
Accrued other | ||||||
Total | $ | $ | ||||
119
Other accrued liabilities as of December 31, 2021 largely consist of other accrued interests, contract fees and scientific advisory board fees while other accrued liabilities as of December 31, 2020 significantly comprise of contract fees and scientific advisory board fees.
8. Debt
Notes payable at December 31, 2021 and 2020 consisted of the following:
December 31, | ||||||
(in thousands) | 2021 | 2020 | ||||
Royalty Interest | $ | $ | ||||
Streeterville Note | — | |||||
Insurance Financing | ||||||
Tempesta Note | ||||||
Oasis Secured Borrowing | — | |||||
Exchange Note 2 | — | |||||
|
|
| ||||
Less: unamortized discount and debt issuance costs |
| ( |
|
| ( | |
Note payable, net of discount | $ |
| $ | |||
Notes payable - non-current, net | $ | $ | ||||
Notes payable - current, net | $ | $ | ||||
Future maturities of the notes payable as of December 31, 2021 are as follows:
(in thousands) | Amounts | ||
Years ended December 31, | |||
2022 | $ | ||
2023 | |||
2024 | |||
2025 | |||
2026 | |||
Less: unamortized discount and debt issuance costs | ( | ||
Total | $ | ||
Future maturities are based on contractual minimum payments. Timing of maturities may fluctuate based on future revenue.
Sale of Future Royalty Interest
March 2020 Purchase Agreement
In March 2020, the Company entered into a royalty interest purchase agreement (the “March 2020 Purchase Agreement”) with Iliad, pursuant to which the Company sold to Iliad a royalty interest entitling Iliad to receive $
Until such time as the Royalty Repayment Amount has been paid in full, the Company will pay Iliad ten percent (
120
the Purchase Price Date, the monthly Royalty Payment shall be the greater of (a) $
The Royalty Interest amount of $
On July 10, 2020, the Company and Iliad entered into an amendment to the March 2020 Purchase Agreement to which the parties agreed that
In November 2020, the Company and Iliad entered into an exchange agreement pursuant to which the Company issued
As of December 31, 2020, the carrying amount of the debt was
October 2020 Purchase Agreement
On October 8, 2020, the Company entered into another royalty interest purchase agreement (the “October 2020 Purchase Agreement”) with Iliad, pursuant to which the Company sold to Iliad a royalty interest entitling Iliad to receive $
Until such time as the Royalty Repayment Amount has been paid in full, the Company will pay Iliad
121
The Royalty Interest amount of $
Pursuant to the October 2020 Purchase Agreement, if the weekly volume weighted average price (“VWAP”) of the Company’s common stock is not equal or greater than the minimum VWAP of $
On April 13, 2021, the Company entered into an exchange agreement with Iliad, pursuant to which the parties agreed to partition $
Interest expense for the years ended December 31, 2021 and 2020 was $
December 2020 Purchase Agreement
On December 22, 2020, the Company entered into a royalty interest purchase agreement (the “December 2020 Purchase Agreement”) with Irving Park Capital, LLC (“Irving”), pursuant to which the Company sold to Irving a royalty interest entitling Irving to receive $
Until such time as the Royalty Repayment Amount has been paid in full, the Company will pay Irving
The Royalty Interest amount of $
122
Interest expense for the years ended December 31, 2021 and 2020 was $
March 2021 Purchase Agreement
On March 8, 2021, the Company entered into a purchase agreement (the “March 2021 Purchase Agreement”) with Streeterville Capital, LLC (“Streeterville”), a company affiliated with CVP, pursuant to which the Company sold a royalty interest entitling Streeterville to $
The Company will be obligated to make minimum royalty payments on a monthly basis beginning at the earlier of (a) 36 months following the closing date or (b)
The Royalty Interest amount of $
Interest expense for the year ended December 31, 2021 was $
Streeterville Note
On January 13, 2021, the Company issued a secured promissory note to Streeterville in the original principal amount of $
123
At any time following the occurrence of a trial failure which refers to any of the following: (i) the Company abandons the clinical trial with lechlemer for an indication for the symptomatic relief of infectious diarrhea for cholera; (ii) the Company fails to start the Phase 1 clinical trial of lechlemer for the symptomatic relief of infectious diarrhea for cholera by July 1, 2022; or (iii) the Company fails to meet all primary endpoints in the pivotal trials of Lechlemer for the symptomatic relief if infectious diarrhea for cholera with statistical significance, Streeterville may elect to increase the outstanding balance as of the date of the trial failure by
Streeterville is entitled to a maximum of
Beginning on the earlier of (a)
After Streeterville becomes aware of the occurrence of any default, Streeterville may accelerate the note, with the outstanding balance becoming immediately due and payable in cash at the Mandatory Default Amount (i.e., the outstanding balance following the application of the Default Effect). Streeterville reserves the right to declare the outstanding balance immediately due and payable at any time following the default. Default Effect means multiplying the outstanding balance as of the date of default by
In connection with the note issuance, the Company has entered into a security agreement with Streeterville, pursuant to which Streeterville will receive a first priority security interest in all existing and future lechlemer technology, and any TDPRV and the sale proceeds therefrom that may be granted to the Company by the FDA in connection with the development of lechlemer for the cholera indication. The Company also agreed, with certain exceptions, not to grant any lien on any of the collateral securing the note and not to grant any license under any of the intellectual property relating to such collateral. The grant of security interest has become effective upon the receipt of the Salix Waiver on April 6, 2021 in observance to the requirement of the settlement agreement previously entered by the Company with Salix Pharmaceuticals, Inc.
The Company irrevocably elected to initially and subsequently apply the FVO accounting to the entire note. The fair value at transaction date was equal to the cash proceeds received of $
124
Insurance Financing
Insurance Premium Financing
In May 2020, the Company entered into a financing agreement for $
March 2021 First Insurance Financing
In March 2021, the Company entered into a premium finance agreement for $
May 2021 First Insurance Financing
In May 2021, the Company entered into another premium finance agreement for $
2019 Tempesta Note
In October 2019, the Company entered into a License Termination and Settlement Agreement with Dr. Michael Tempesta, pursuant to which certain royalty payment disputes between Napo and Tempesta were settled. Per the terms of the Agreement, Tempesta received $
Oasis Secured Borrowing
The Purchase Agreement
In May 2020, the Company, entered into a
In the event Oasis does not receive at least the Threshold Price for a purchased account on or before such account becomes due and payable, the Company will, at Oasis’s election, be obligated to either (i) pay the difference
125
between the Threshold Price and the amount received by Oasis for such account (the “Shortfall”) within
The initial term of the Purchase Agreement is
Per the Purchase Agreement, the Company will service and administer the purchased accounts receivable for Oasis. Oasis appointed the Company to be its agent and servicer for monitoring and collecting the accounts receivable subject to the terms of the Purchase Agreement. The Company will perform its duties in a commercially reasonable manner and agrees that Company will not commence any legal action with respect to such servicing and collection efforts and shall not terminate, discharge, discount or write off any accounts receivable without Oasis's prior written consent.
The Company, having determined that it did not meet the criteria per ASC 860-10-40-5 to account for the transactions under the Purchase Agreement as sales, accounts for such transactions as secured borrowings in accordance with ASC 860-30, “Transfers – Secured Borrowings and Collateral.”
During 2020, the Company made the required payments to Oasis for the first five purchases with total payments equalling to $
In December 2020, for its sixth purchase under the terms of the Purchase Agreement, the Company received cash proceeds of $
The Company recorded the sale to Oasis as a short-term secured borrowing with a principal amount of $
In February 2021, the Company made its final required payment to Oasis under Tranche #6 Secured Note, with total payments equalling the $
Exchange Note 2
In May 2019, CVP and the Company agreed to exchange
126
the Company’s common stock to CVP. The series of exchanges was accounted for as an extinguishment which resulted in a loss of $
In September 2020, the Company and CVP also entered into a global amendment agreement, pursuant to which the maturity date of Exchange Note 2 is extended to December 31, 2021. In consideration of CVP’s grant of extension, together with the related fees and other accommodation set forth, principal debt was increased by
Pursuant to the global amendment agreement, the Company issued
In December 2020, the Company and CVP entered into a note exchange agreement to which the Company made a prepayment of principal amounting to $
In January 2021, the Company and CVP entered into another note exchange agreement to which the Company made a prepayment of the remaining outstanding balance of Exchange Note 2 amounting to $
As of December 31, 2021 and 2020, the carrying value of Exchange Note 2, net of discount, was
127
9. Warrants
The following table summarizes information about warrants outstanding and exercisable into shares of the Company’s common stock for the years ended December 31, 2021 and 2020:
December 31, | ||||
2021 | 2020 | |||
Warrants outstanding, beginning balance | | | ||
Issuances | | | ||
Exercises | ( | ( | ||
Expirations and cancelations | — | ( | ||
Warrants outstanding, ending balance | | | ||
May 2020 Series 3 Warrants
In May 2020, concurrent with the May 2020 modification of the exercise price of the Series 1, Series 2 and Bridge Note Warrants and inducement offer, the Company issued unregistered Series 3 Warrants to purchase
The Series 3 Warrants were initially valued at $
A Special Meeting of Stockholders was held on July 21, 2020, whereupon a proposal to approve the “Alternate Cashless Exercise” settlement method for the Series 3 Warrants was approved. In 2020, certain holders of the Series 3 Warrants agreed to exercise a total of
On January 8, 2021, in accordance with the May 2020 Modification of the 2019 Bridge Note Warrants and Inducement Offer, an investor received
During the year ended December 31, 2021, certain holders of the Series 3 Warrants agreed to exercise a total of
A total of
October 2018 Underwriter Warrants
In October 2018, in consideration of services provided leading up to the Company’s October 2018 public offering, the Company issued warrants to various service providers to purchase an aggregate of
128
of common stock at an exercise price of $
April 2020 Underwriter Warrants
In April 2020, in consideration of the settlement of a dispute regarding underwriting fees (see Note 6), the Company issued warrants to purchase
March 2019 Ladenburg Warrants
In March 2019, in consideration of services provided in the Company’s March 2019 public offering of
March 2019 LOC Warrant
In March 2019, in consideration of a letter of credit cancellation related to the Company’s office lease, the Company issued a warrant to purchase warrant shares equal to a fixed principal amount divided by a variable exercise price. The warrants were initially classified as liabilities pursuant to ASC 480-10 due to their debt-like nature. On July 23, 2019, upon the exercise price of the warrants becoming fixed, the warrants became exercisable into
2019 Bridge Note Warrants
Between March 18, 2019 and June 26, 2019, concurrent to the Company entering into Promissory Notes of $
February 2020 Modification of Certain 2019 Bridge Note Warrants
In February 2020, the Company entered into a warrant exercise agreement with a holder of its Bridge warrants, pursuant to which the holder agreed to exercise
129
May 2020 Modification of the 2019 Bridge Note Warrants and Inducement Offer
In May 2020, the Company reduced the exercise price of all outstanding 2019 Bridge Note Warrants from $
In May 2020, concurrent with the reduction of the exercise price of the Bridge Note Warrants, the Company entered into a warrant exercise inducement offer with certain holders of the Bridge Note Warrants, pursuant to which such holders agreed to exercise for cash Bridge Note Warrants to purchase
During the year ended December 31, 2021, an aggregate of
A total of
July 2019 Series 1 Warrants
In July 2019, the Company entered into an underwriting agreement, relating to a public offering, which was comprised of (1)
The Series 1 Warrants had an exercise price of $
In the offering, the Company sold (i)
September 2019 Modification of the July 2019 Series 1 Warrants
In September 2019, the Company reduced the exercise price of all
February 2020 Modification of the July 2019 Series 1 Warrants
In February 2020, the Company entered into a warrant exercise agreement with a holder of its Series 1
130
Warrants, pursuant to which the holder agreed to exercise
May 2020 Modification of the July 2019 Series 1 Warrants and Inducement Offer
In May 2020, the Company reduced the exercise price of all outstanding Series 1 Warrants from $
In May 2020, concurrent with the reduction of the exercise price of the Series 1 Warrants, the Company entered into a warrant exercise inducement offer with certain holders of the Series 1 Warrants, pursuant to which such holders agreed to exercise for cash Series 1 Warrants to purchase
During the year ended December 31, 2021, an aggregate of
A total of
July 2019 Series 2 Warrants
The Series 2 Warrants had an exercise price of $
In the July 2019 offering, the Company sold (i)
March 5, 2020 Modification of the July 2019 Series 2 Warrants
On March 5, 2020, the Company entered into a warrant exercise agreement with a holder of its Series 2 Warrants, pursuant to which the holder agreed to exercise
131
the impact of modifications to equity-classified warrants by analogy to share-based compensation guidance per ASC 718, Compensation – Stock Compensation. Pursuant to that guidance, and due to the modification being applicable only to a single holder of the Series 2 Warrants, the incremental increase of $
March 23, 2020 Modification of the July 2019 Series 2 Warrants
On March 23, 2020, the Company entered into a Warrant Exercise and Preferred Stock Amendment Agreement (see Note 10) with Ionic Ventures of its Series 2 Warrants, pursuant to which the holder agreed to exercise in cash its Series 2 Warrants to purchase an aggregate of
May 2020 Modification of the July 2019 Series 2 Warrants and Inducement Offer
In May 2020, the Company reduced the exercise price of all outstanding Series 2 Warrants from $
In May 2020, concurrent with the reduction of the exercise price of the Series 2 Warrants, the Company entered into a warrant exercise inducement offer with certain holders of the Series 2 Warrants, pursuant to which such holders agreed to exercise for cash Series 2 Warrants to purchase
During the year ended December 31, 2021, an aggregate of
A total of
December 2019 PIPE Financing Warrants
In December 2019, the Company entered into a securities purchase agreement with certain investors pursuant to which the Company, in a Private Placement, sold (i) an aggregate of
The warrants were valued at $
132
and warrants being allocated $
During January 2021, an aggregate of
April 2021 ELOC Warrants
On April 7, 2021, in consideration for Oasis Capital’s entry into the March 2020 ELOC amendment, the Company issued Oasis Capital a common stock purchase warrant (“ELOC Warrants”) exercisable for
10. Preferred Stock
At December 31, 2021 and 2020, preferred stock consisted of the following:
Liquidation | ||||||||||||
(in thousands, except share and per share data) | Shares |
| Issued and |
| Carrying | Preference | ||||||
Series | Authorized | Outstanding | Value | per Share | ||||||||
B-2 | | — | $ | — | $ | — | ||||||
C | | — | — | | ||||||||
Total | | — | $ | — | ||||||||
Series A Convertible Preferred Stock
In March 2018, the Company entered into a stock purchase agreement with Sagard Capital pursuant to which the Company, in a private placement, agreed to issue and sell to Sagard Capital
Holders of the Series A Convertible Preferred shares were entitled to participate equally and ratably with the holders of shares of common stock in all dividends paid and distributions made to the holders of the common stock as if, immediately prior to each record date of the common stock, the shares of Series A Convertible Preferred Stock then outstanding were converted into shares of common stock.
In the event of any voluntary or involuntary liquidation, dissolution or winding up of the Company or deemed liquidation event, the holders of Series A Convertible Preferred shares then outstanding were entitled to be paid in cash out of the assets of the Company before any payment shall be made to the holders of common stock or shares of any series or class of preferred or other capital stock then outstanding that by its terms is junior to the Series A Convertible Preferred Stock in respect of the preferences as to distributions and payments upon such liquidation event by reason of their ownership, an amount per share of Series A Convertible Preferred Stock equal to one times the Series A Convertible Preferred Stock original issue price.
133
The Series A Convertible Preferred shares were redeemable by Sagard Capital upon a Redemption Event that is not solely within the control of the Company. Were a Redemption Event to occur as of the Measurement Date (the later of April 30, 2021 and the date on which the Company files its Form 10-Q for the three months ending March 31, 2021, but in no event later than September 30, 2021), the holders of at least a majority of the shares of Series A Convertible Preferred Stock then outstanding may require the Company to redeem all Series A shares for cash at a per share purchase price equal to $
The preferred stock was classified outside of stockholders' equity in accordance with authoritative guidance for the classification and measurement of potentially redeemable securities.
In September 2020, the Company and Sagard Capital entered into an exchange agreement, by which the remaining Series A Convertible Preferred shares were exchanged for (i)
During the year ended December 31, 2020, the Company determined that a Redemption Event was probable. The Company recorded a deemed dividend charge of $
In September 2020, the Company filed a certificate with the Secretary of State of Delaware effecting the retirement and cancellation of the Series A Convertible Preferred Stock. As of December 31, 2020, there were
Series B Convertible Preferred Stock
In July 2019, the Company entered into an underwriting agreement relating to the public offering comprised of (1)
The Company sold
Holders of the Series B shares are entitled to participate equally and ratably with the holders of shares of common stock in all dividends paid and distributions made to the holders of the common stock as if, immediately prior to each record date of the common stock, the shares of Series B then outstanding were converted into shares of common stock. With certain exceptions, the shares of Series B Convertible Preferred Stock have
134
However, as long as any shares of Series B Convertible Preferred Stock remain outstanding, the Company shall not, without the affirmative vote of holders of a majority of the then outstanding shares of Series B Convertible Preferred Stock, (a) alter or change adversely the powers, preferences or rights given to the Series B Convertible Preferred Stock or alter or amend the Series B Certificate of Designation or (b) enter into any agreement with respect to any of the foregoing. Each share of Series B Convertible Preferred Stock is convertible at any time at the holder’s option into
In March 2020, the Company entered into a Warrant Exercise and Preferred Stock Amendment Agreement (“Amendment Agreement”) with a Ionic Ventures of its Series 2 Warrants, pursuant to which the holder agreed to exercise in cash its Series 2 Warrants to purchase an aggregate of
In September 2020, the Company filed a certificate with the Secretary of State of Delaware effecting the retirement and cancellation of the Series B Convertible Preferred Stock. As of December 31, 2020, there were
Series B-2 Convertible Preferred Stock
In December 2019, the Company entered into an exchange agreement with Oasis Capital, pursuant to which Oasis Capital gave up (i) its remaining unexercised Prepaid Forward contracts exercisable for
Holders of the Series B-2 Convertible Preferred Stock are entitled to receive dividends on shares of Series B-2 Convertible Preferred Stock equal (on an as-if-converted-to-common-stock basis) to and in the same form as dividends actually paid on shares of the common stock when, as and if such dividends are paid on shares of the common stock.
The shares of Series B-2 Convertible Preferred Stock have
Upon any liquidation, dissolution or winding-up of the Company, whether voluntary or involuntary, the Holders of the Series B-2 Convertible Preferred Stock are entitled to receive out of the assets, whether capital or surplus, of the Company the same amount that a holder of common stock would receive if the Series B-2 Convertible Preferred Stock were fully converted to common stock which amounts shall be paid pari passu with all holders of common stock.
Each share of Series B-2 Convertible Preferred Stock is convertible at any time at the holder’s option into
135
transactions as specified in the Series B-2 Certificate of Designation. The Series B-2 Convertible Preferred Stock was classified in stockholders' equity in accordance with authoritative guidance.
In January 2020, a holder of the Series B-2 Convertible Preferred Stock converted
As of December 31, 2020, there were
Series C Perpetual Preferred Stock
In September 2020, the Company entered into an exchange agreement with Iliad to issue
Holders of the Series C Perpetual Preferred Stock were not entitled to voting rights. However, as long as any Series C Perpetual Preferred share is outstanding, the Company is restricted to alter, change, or enter into an agreement to alter or change adversely the powers, preferences, or rights given to the shareholders.
In the event of any voluntary or involuntary liquidation, dissolution or winding up of the Company or deemed liquidation event, the holders of Series C Perpetual Preferred shares then outstanding would be entitled to be paid in cash out of the assets of the Company before any payment shall be made to the holders of common stock or shares of any series or class of preferred or other capital stock then outstanding that by its terms is junior to the Series C Perpetual Preferred shares in respect of the preferences as to distributions and payments upon such liquidation event by reason of their ownership, an amount per share of Series C equal to one times the Series C original issue price.
The Series C Perpetual Preferred shares were redeemable upon the option or discretion of the Company.
The Series C Perpetual Preferred shares were entitled to receive
The Series C Perpetual Preferred shares were initially measured at fair value using the income approach, which considered the weighted probability of discounted cash flows at various scenarios of redemption by the Company or liquidation event and perpetual holding of the shares. As of the date of exchange, total fair value of the Series C Perpetual Preferred shares amounted to $
The preferred stock has been classified as permanent stockholders' equity in accordance with authoritative guidance for the classification and measurement of perpetual shares without mandatory redemption period because the redemption option was ultimately in the control of the Company.
In October 2020, the Company entered into an exchange agreement with Iliad pursuant to which the Company agreed to issue a total of
136
exercised in full. The nominal exercise price of each pre-funded warrant was $
As of December 31, 2020, there were
11. Stockholders’ Equity
As of December 31, 2021 and 2020, the Company had reserved shares of common stock, on an as-if converted basis, for issuance as follows:
|
| |||
December 31, | ||||
2021 | 2020 | |||
Options issued and outstanding |
| |
| |
Inducement options issued and outstanding | | | ||
Options available for grant under stock option plans |
| |
| |
Restricted stock unit awards issued and outstanding |
| |
| |
Warrants issued and outstanding |
| |
| |
Total |
| |
| |
Common Stock
The holders of common stock are entitled to
The holders of non-voting common stock are not entitled to vote, except on an as converted basis with respect to any change of control of the Company that is submitted to the stockholders of the Company for approval. Shares of the Company's non-voting common stock have the same rights to dividends and other distributions and are convertible into shares of the Company's common stock on a
The Company is authorized to issue a total number of
Reverse Stock-Split
On December 22, 2020, the Company obtained approval through a special shareholders meeting held on December 9, 2020 to effect a reverse split of the Company’s issued and outstanding voting common stock at a ratio not less than -for-2 and not greater than -for-20.
On September 3, 2021, the reverse stock split became effective. Upon effectivity, every shares of the Company’s issued and outstanding common stock immediately prior to the effective time shall automatically be reclassified into one share of common stock without any change in the par value. The reverse stock split reduces the number of shares of common stock issuable upon the conversion of the Company’s outstanding non-voting common stock and the exercise or vesting of its outstanding stock options and warrants in proportion to the ratio of the reverse stock split and causes a proportionate increase in the conversion and exercise prices of such non-voting common
137
stock, stock options and warrants. In addition, the number of shares reserved for issuance under the Company’s equity compensation plans immediately prior to the effective time will be reduced proportionately. The reverse stock split did not change the total number of authorized shares of common stock or preferred stock.
March 2020 ELOC (Equity Line of Credit)
In March 2020, the Company entered into an equity purchase agreement (the “March 2020 ELOC”) with Oasis Capital, which provides that Oasis Capital is committed to purchase up to an aggregate of $
Pursuant to the terms and conditions of the March 2020 ELOC, on any trading day selected by the Company (such date the “Put Date”), after the SEC has declared effective the registration statement registering the sale of the shares of common stock that may be issued to Oasis Capital under the March 2020 ELOC, the Company has the right, in its sole discretion, to present to Oasis Capital with a purchase notice (each a “Put Notice”), directing Oasis Capital to purchase up to the lesser of (i)
In addition, on any date on which Oasis Capital receives shares of common stock in connection with a Put Notice (the “Clearing Date”), the Company also has the right, in its sole discretion, to present to Oasis Capital with a Put Notice (each an “Option 2 Put”) directing Oasis Capital to purchase an amount of common stock equal to the lesser of (i) such amount that equals
On April 15, 2020, the SEC declared effective the registration statement registering the sale of the shares of common stock issued to Oasis Capital under the March 2020 ELOC. The Company will control the timing and amount of sales of common stock to Oasis Capital. Oasis Capital has no right to require any sales by the Company but is obligated to make purchases from the Company as directed by the Company in accordance with the March 2020 ELOC.
In connection with the equity line, the Company agreed to pay Oasis Capital a commitment fee and in April 2020, in settlement of the commitment fee, the Company issued to Oasis Capital
Per the terms of the equity purchase agreement, the Option Put 1 and Option Put 2 may be exercised only at a price that is always above the trading price of the underlying common stock at the exercise date, thereby rendering any exercise by the Company being out-of-the-money. At inception of the equity line on March 24, 2020, the Put Options were classified as derivative assets with a fair value of
In April 2020, the Company sold
On April 7, 2021, the Company entered into an amendment to the March 2020 ELOC with Oasis Capital, pursuant to which the parties agreed to increase (i) the purchase price from $
138
from $
March 2020 PIPE Financing
In March 2020, Company entered into a securities purchase agreement (the “PIPE Purchase Agreement”) with certain investors, pursuant to which the Company agreed to issue and sell to the Investors in a private placement an aggregate of
At The Market Offering (“ATM”)
October 2020 ATM Agreement
On October 5, 2020, the Company entered into an ATM Agreement (“October 2020 ATM Agreement”) with Ladenburg, pursuant to which the Company may offer and sell, from time to time through Ladenburg, shares of common stock, subject to the terms and conditions of the October 2020 ATM Agreement. The October 2020 ATM Agreement will terminate upon the earlier of (i) October 5, 2022 and (ii) termination of the October 2020 ATM Agreement as permitted therein. In 2020, the Company sold
During January and February 2021, the Company issued an aggregate of
December 2021 ATM Agreement
On December 10, 2021, the Company entered into another ATM Agreement (“December 2021 ATM Agreement”) with Ladenburg, pursuant to which the Company may offer and sell, from time to time through Ladenburg, shares of common stock having an aggregate offering price of up to $
As of December 31, 2021, the Company has issued
PoC Capital Registered Direct Offering
On October 6, 2020, the Company entered into a Stock Plan Agreement for payment of contracted research fees (the “SPA”) with PoC Capital, LLC (“PoC”), pursuant to which the Company issued to PoC an aggregate of
Securities Purchase Agreement
On January 13, 2021, the Company entered into a securities purchase agreement, pursuant to which the Company agreed to issue and sell, in a registered public offering an aggregate of
139
On April 29, 2021, the Company entered into another securities purchase agreement, pursuant to which the Company agreed to issue and sell, in a registered public offering through Ladenburg as the placement agent, an aggregate of
Subscription Agreement
On June 1, 2021, the Company entered into a subscription agreement with the SPAC and its sponsor, pursuant to which Dragon SPAC agreed to issue and sell, in a private placement by Dragon SPAC directly to the Company, units of Dragon SPAC, with each unit consisting of
On November 3, 2021, Dragon SPAC issued
September 2021 PIPE Financing
On September 13, 2021, the Company entered into a securities purchase agreement (the “September 2021 PIPE Financing”) with certain investors, pursuant to which the Company agreed to issue and sell to the investors in a private placement an aggregate of
Noncontrolling Interest
As a result of the merger last November 3, 2021 between Napo EU and Dragon SPAC, the Company assumed a non-controlling interest amounting to $
12. Stock-Based Compensation
2013 Equity Incentive Plan
In November 2013, the Company's board of directors and sole stockholder adopted the Jaguar Health, Inc. 2013 Equity Incentive Plan (the “2013 Plan”). The 2013 Plan allows the Company's board of directors to grant stock options, restricted stock awards and restricted stock unit awards to employees, officers, directors and consultants of the Company. Following the effective date of the IPO and after effectiveness of any grants under the 2013 Plan that were contingent on the IPO,
140
2014 Stock Incentive Plan
Effective May 12, 2015, the Company adopted the Jaguar Health, Inc. 2014 Stock Incentive Plan (“2014 Plan”). The 2014 Plan provides for the grant of options, restricted stock and restricted stock units to eligible employees, directors and consultants to purchase the Company's common stock. The term of an incentive stock option may not exceed , except that with respect to any participant who owns more than 10% of the voting power of all classes or our outstanding stock, the term must not exceed . The 2014 Plan provides for automatic share increases on the first day of each fiscal year in the amount of
As of December 31, 2021, there were
2020 New Employee Inducement Award Plan
Effective June 16, 2020, the Company adopted the Jaguar Health, Inc. New Employee Inducement Award Plan (“2020 Inducement Award Plan”) and, subject to the adjustment provisions of the Inducement Award Plan, reserved
As of December 31, 2021, there were
Stock Options and Restricted Stock Units (“RSUs”)
The following table summarized the incentive plan activity for the year ended December 31, 2021:
Weighted | Weighted Average | |||||||||||||
Shares | Stock | Average | Remaining | Aggregate | ||||||||||
Available | Options | RSUs | Stock Option | Contractual Life | Intrinsic | |||||||||
(in thousands, except share and per share data) |
| for Grant |
| Outstanding |
| Outstanding |
| Exercise Price |
| (Years) |
| Value* | ||
Outstanding at December 31, 2020 | | | | $ | | $ | ||||||||
Additional shares authorized | | — | — | — | — | |||||||||
Options granted | ( | | — | | — | |||||||||
Options exercised | — | ( | — | | — | |||||||||
Options canceled | | ( | — | | — | |||||||||
RSUs granted | ( | — | | — | — | |||||||||
Outstanding at December 31, 2021 | | | | $ | | $ | ||||||||
Exercisable at December 31, 2021 |
| | $ | |
| $ | ||||||||
Vested and expected to vest at |
| | $ | |
| $ | ||||||||
*Fair market value of Jaguar stock on December 31, 2021 was $
141
The intrinsic value is calculated as the difference between the exercise price of the underlying options and the fair market value of the Company's common stock for options that were in-the-money.
The number of options exercised during the year ended December 31, 2021 and 2020 were
The weighted average grant date fair value of stock options granted was $
The number of options that vested in the years ended December 31, 2021 and 2020 was
December 31, 2021 and 2020 was $
Stock-Based Compensation
The following table summarizes stock-based compensation expense related to stock options, inducement stock options and RSUs for the years ended December 31, 2021 and 2020, and are included in the consolidated statements of operations as follows:
Year Ended | |||||||
December 31, | |||||||
(in thousands) |
| 2021 |
| 2020 | |||
Research and development expense | $ | $ | |||||
Sales and marketing expense |
|
| |||||
General and administrative expense |
|
| |||||
Total | $ | $ | |||||
As of December 31, 2021, the Company had $
The fair value of options granted during the years ended December 31, 2021 and 2020, respectively, were calculated using the range of assumptions set forth below:
Year Ended | ||||
December 31, | ||||
| 2021 |
| 2020 | |
Volatility | ||||
Expected term (years) |
| |||
Risk-free interest rate |
| |||
Expected dividend yield | — | — | ||
401(k) Plan
The Company sponsors a 401(k) defined contribution plan covering all employees. There were
142
13. Net Loss Per Share Attributable to Common Stockholders
The following table presents the calculation of basic and diluted net loss per common share for the years ended December 31, 2021 and 2020:
Year Ended December 31, | ||||||
(In thousands, except share and per share data) |
| 2021 |
| 2020 | ||
Net loss attributable to common shareholders (basic and diluted) | ( | ( | ||||
Shares used to compute net loss per common share, basic and diluted | | | ||||
Net loss per share attributable to common shareholders, basic and diluted | ( | ( | ||||
Basic net loss per share is calculated by dividing net loss by the weighted-average number of common shares outstanding during the period. Diluted net loss per share is computed by dividing net loss by the weighted-average number of common shares and common share equivalents outstanding for the period. Common stock equivalents are only included when their effect is dilutive. The Company's potentially dilutive securities which include stock options, convertible preferred stock, RSUs and common stock warrants have been excluded from the computation of diluted net loss per share as they would be anti-dilutive. For all periods presented, there is no difference in the number of shares used to compute basic and diluted shares outstanding due to the Company's net loss position.
The following outstanding common stock equivalents have been excluded from diluted net loss per common share for the years ended December 31, 2021 and 2020 because their inclusion would be anti-dilutive:
December 31, | ||||
| 2021 | 2020 | ||
Options issued and outstanding | | | ||
Inducement options issued and outstanding | | | ||
Restricted stock units issued and outstanding | | | ||
Warrants issued and outstanding | | | ||
Total | | | ||
As of March 10, 2022 there were
14. Income Taxes
The Company's loss before provision for income taxes during the years ended December 31, 2021 and 2020, was a domestic loss of $
The effective tax rate for 2021 and 2020 was
143
The Company’s effective tax during the years ended December 31, 2021 and 2020, differed from the federal statutory rate as follows:
December 31, |
| ||||
| 2021 |
| 2020 |
| |
Statutory rate | ( | % | ( | % | |
Intercompany transactions | | % | — | % | |
Valuation allowance | | % | | % | |
Nondeductible warrant expense | | % | — | % | |
Book loss on debt extinguishment | | % | | % | |
Foreign rate differential | ( | % | — | % | |
Other | | % | | % | |
Effective tax rate | ( | % | — | % | |
Net deferred tax assets as of December 31, 2021 and 2020 consisted of the following:
| December 31, | |||||
(In thousands) | 2021 | 2020 | ||||
Non-current deferred tax assets: | ||||||
Net operating losses | $ | $ | ||||
Tax credits | ||||||
Stock compensation | ||||||
Other | ||||||
| ||||||
Valuation allowance | ( | ( | ||||
Net non-current deferred tax assets | ||||||
Non-current deferred tax liabilities: | ||||||
Other | ( | — | ||||
Property and equipment | ( | ( | ||||
Net non-current deferred tax liability | ( | ( | ||||
Net non-current deferred tax asset (liability) | $ | — | $ | — | ||
A valuation allowance is provided when it is more likely than not that the deferred tax assets will not be realized. The Company has established a valuation allowance to offset net deferred tax assets as of December 31, 2021 and 2020, due to the uncertainty of realizing future tax benefits from its NOL carryforwards and other deferred tax assets.
The valuation allowance increased by $
As of December 31, 2021, the Company had federal and California NOL carryovers of approximately $
As of December 31, 2021, the Company had California research credit carryovers of approximately $
Utilization of the domestic NOL and tax credit forwards may be subject to a substantial annual limitation due to ownership change limitations that may have occurred or that could occur in the future, as required by the Internal Revenue Code Section 382, as well as similar state provisions. In general, an "ownership change," as defined by the code, results from a transaction or series of transactions over a three-year period resulting in an ownership change of more than 50 percentage points of the outstanding stock of a company by certain stockholders or public groups. Any
144
limitation may result in expiration of all or a portion of the NOL or tax credit carryforwards before utilization. The Company has previously reduced its federal and California R&D credit carryforwards by $
Enacted on March 27, 2020, the Coronavirus Aid, Relief, and Economic Security Act (“the CARES Act”) authorizes more than $2.0 trillion to battle COVID-19 and its economic effects, including immediate cash relief for individual citizens, loan programs for small business, support for hospitals and other medical providers, and various types of economic relief for impacted businesses and industries. The CARES Act does not have a material impact on the Company’s financial results for the year ended December 31, 2021.
The Consolidated Appropriations Act, 2021 (the "Act") was enacted in the United States on December 27, 2020. The Act enhances and expands certain provisions of the CARES Act. The Act does not have a material impact on the Company’s financial results for the year ended December 31, 2021.
Uncertain Tax Positions
The Company has adopted the provisions of ASC 740, “Income Taxes Related to Uncertain Tax Positions.” Under these principals, tax positions are evaluated in a two-step process. The Company first determines whether it is more-likely-than-not that a tax position will be sustained upon examination. If a tax position meets the more-likely-than-not recognition threshold it is then measured to determine the amount of benefit to be recognized in the financial statements. The tax position is measured as the largest amount of benefit that has a greater than 50 percent likelihood of being realized upon ultimate settlement.
As of December 31, 2021, all unrecognized tax benefits were offset against deferred tax assets which are subject to a full valuation allowance, and if recognized, will not affect the Company's tax rate.
The Company does not anticipate that the total amounts of unrecognized tax benefits will significantly increase or decrease in the next 12 months.
The Company's policy is to include interest and penalties related to unrecognized tax benefits within its provision for income taxes. Due to the Company's net operating loss position, the Company has not recorded an accrual for interest or penalties related to uncertain tax positions for the years ended December 31, 2021 or 2020.
The following is a reconciliation of the beginning and ending amount of the Company’s total gross unrecognized tax benefit liabilities:
December 31, | ||||||
(In thousands) |
| 2021 |
| 2020 | ||
Gross Unrecognized Tax Benefit--Beginning Balance | $ | $ | ||||
Increases Related to Tax Positions from Prior Years | ||||||
Increases Related to Tax Positions Taken During the Current Year | — | — | ||||
Gross Unrecognized Tax Benefit--Ending Balance | $ | $ | ||||
15. Segment Data
The Company has
145
The Company’s reportable segments sales and net income consisted of:
Year Ended | ||||||
December 31, | ||||||
(in thousands) |
| 2021 |
| 2020 | ||
Revenue from external customers |
|
|
|
| ||
Human Health | $ | $ | ||||
Animal Health |
|
| ||||
Consolidated Totals | $ | $ | ||||
Segment net loss |
|
|
|
| ||
Human Health | $ | ( | $ | ( | ||
Animal Health |
| ( |
| ( | ||
Consolidated Totals | $ | ( | $ | ( | ||
The Company’s reportable segments assets consisted of the following:
December 31, | ||||||
(in thousands) |
| 2021 |
| 2020 | ||
Segment assets |
|
|
| |||
Human Health | $ | $ | ||||
Animal Health |
|
| ||||
Total | $ | $ | ||||
The reconciliation of segments assets to the consolidated assets is as follows:
December 31, | ||||||
(in thousands) |
| 2021 |
| 2020 | ||
Total assets for reportable segments | $ | $ | ||||
Less: Investment in subsidiary |
| ( |
| ( | ||
Less: Intercompany loan |
| ( |
| ( | ||
Consolidated Totals | $ | $ | ||||
16. Subsequent Events
Issuances under the December 2021 ATM Agreement
During January to March 2022, the Company issued an aggregate of
Amendment to December 2021 ATM Agreement
On February 2, 2022, the Company entered into an amendment to the December 2021 ATM Agreement, pursuant to which, the aggregate offering amount of the shares of the Company’s common stock which the Company may sell and issue through Ladenburg, as the sales agent, was increased from $
3a9 Exchange Agreements
On February 11, 2022, the Company entered into a privately-negotiated exchange agreement with Iliad, pursuant to which, the Company issued
146
Royalty in exchange for the Exchange Shares, which will be free of any restrictive securities legend. Other than the surrender of the Partitioned Royalty,
On March 2, 2022, the Company entered into another exchange agreement with Iliad, pursuant to which, the Company issued
On March 4, 2022, the Company entered into another exchange agreement with Iliad, pursuant to which, the Company issued
On March 9, 2022, the Company entered into another exchange agreement with Iliad, pursuant to which, the Company issued
Notice of Delisting
On February 17, 2022, the Company received a letter from the Staff of Nasdaq indicating that the bid price of the Company’s common stock for the last
Under Nasdaq Listing Rule 5810(c)(3)(A), the Company has been granted a
The Company is diligently working to evidence compliance with the minimum bid price requirement for continued listing on Nasdaq; however, there can be no assurance that the Company will be able to regain compliance or that Nasdaq will grant the Company a further extension of time to regain compliance, if necessary. If the Company fails to regain compliance with the Nasdaq continued listing standards, its common stock will be subject to delisting from Nasdaq.
147
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Disclosure Controls and Procedures
Our management, Chief Executive Officer and Principal Financial and Accounting Officer, evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2021. The term "disclosure controls and procedures," as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC's rules and forms.
Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is accumulated and communicated to our management, including our Chief Executive Officer and Principal Financial and Accounting Officer, as appropriate, to allow timely decisions regarding required disclosure. Based on this evaluation, our Chief Executive Officer and Principal Financial and Accounting Officer concluded that our disclosure controls and procedures were effective at the reasonable assurance level as of December 31, 2021.
Material Weaknesses
A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim consolidated financial statements will not be prevented or detected and corrected on a timely basis. As previously reported in our annual report on Form 10-K for the year ended December 31, 2020, management concluded that, as of such date, our disclosure controls and procedures were not effective due to the existence of a material weakness in the design and operating effectiveness of internal controls related to staff turnover in our accounting department and inadequate internal technical staffing levels.
In connection with our preparation of our annual financial statements for the year ended December 31, 2019, we identified a material weakness in our internal control over financial reporting related to staff turnover in our accounting department. We did not maintain a sufficient complement of internal personnel with appropriate knowledge, experience and/or training commensurate with our financial reporting requirements. We relied on outside consulting technical experts and did not maintain adequate internal qualified personnel to properly supervise and review the information provided by the outside consulting technical experts to ensure certain significant complex transactions and technical matters were properly accounted for. In addition, we identified inadequate internal technical staffing levels and expertise to properly supervise and review the information of the outside consulting technical experts to properly apply ASC 815-40 for liability classification of certain warrants and ASC 470-50 and ASC 470-60 to properly reflect the accounting impact to multiple modifications of the Company’s debt instruments. In connection with the preparation of our annual financial statements for the year ended December 31, 2020, we did not have adequate policies and procedures in place to ensure the timely, effective review of assumptions used in measuring the fair value of certain financial instruments. In addition, we did not have adequate policies and procedures in place to ensure the timely, effective review of compliance with contractual covenants in certain financial instruments. As discussed below in Remediation Efforts to Address Material Weaknesses, we believe these material weaknesses have been remediated as of December 31, 2021.
148
Remediation Efforts to Address Material Weaknesses
To remediate the material weaknesses described above, management added controls to further enhance and revise the design of the existing controls including:
| ● | Established policies and procedures to ensure timely review, by qualified personnel, of assumptions used in measuring fair value of certain financial instruments. |
| ● | Reassessed the design and operation of internal controls over financial reporting and review procedures over the preparation of our financial statements. |
| ● | Hired permanent accounting personnel and used consultants to provide support during our quarterly and annual preparation, review, and reporting of our financial statements. |
| ● | Maintained adequate internal qualified personnel to properly supervise and review the information provided by the outside consulting technical experts to ensure certain significant complex transactions and technical matters were properly accounted for. |
Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rule 13a-15(f) and 15d-15(c) under the Exchange Act. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of the effectiveness of internal control to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with policies or procedures may deteriorate. Under the supervision and with the participation of our management, including our Chief Executive Officer and Principal Financial and Accounting Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2021 using the criteria established in Internal Control-Integrated Framework (“2013 Framework”) issued by the Committee of Sponsoring Organization of the Treadway Commission (“COSO”). Based on our evaluation using those criteria, our management has concluded that, as of December 31, 2021, our internal control over financial reporting was effective to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles for the reasons discussed above.
This Annual Report on Form 10-K does not include an attestation report of our registered public accounting firm on our internal control over financial reporting because we are an SRC and are not subject to auditor attestation requirements under applicable SEC rules.
Changes in Internal Control over Financial Reporting
Other than the changes disclosed above regarding the remediation efforts to address the material weaknesses, there were no changes in our internal control over financial reporting that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting during the year ended December 31, 2021.
ITEM 9B. OTHER INFORMATION
None.
149
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this item is incorporated by reference from the Proxy Statement for the 2022 Annual Meeting of Stockholders to be filed with the SEC within 120 days of the fiscal year ended December 31, 2021.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this item is incorporated by reference from the information under the captions “Compensation of Directors and Executive Officers” contained in the Proxy Statement for the 2022 Annual Meeting of Stockholders to be filed with the SEC within 120 days of the fiscal year ended December 31, 2021.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this item is incorporated by reference from the information under the captions “Security Ownership of Certain Beneficial Owners and Management” and “Compensation of Directors and Executive Officers—Equity Compensation “contained in the Proxy Statement for the 2022 Annual Meeting of Stockholders to be filed with the SEC within 120 days of the fiscal year ended December 31, 2021.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this item is incorporated by reference from the information under the caption “Proposal 1—Election of Directors—Director Independence” and “Certain Relationships and Related Transactions” contained in the Proxy Statement for the 2022 Annual Meeting of Stockholders to be filed with the SEC within 120 days of the fiscal year ended December 31, 2021.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information required by this item is incorporated by reference from the information under the caption “Proposal 2—Ratification of the Appointment of Independent Registered Public Accounting Firm—Principal Accountant Fees and Services” contained in the Proxy Statement for the 2022 Annual Meeting of Stockholders to be filed with the SEC within 120 days of the fiscal year ended December 31, 2021.
150
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
Exhibit No. |
| Description | ||
|---|---|---|---|---|
2.1 | ||||
3.1 | ||||
3.2 | ||||
3.3 | ||||
3.4 | ||||
3.5 | ||||
3.6 | ||||
3.7 | ||||
3.8 | ||||
3.9 | ||||
3.10 | Corrected Certificate of Amendment of the Third Amended and Restated Certificate of Incorporation (incorporated by reference to Exhibit 3.1 to the Form 8-K of Jaguar Health, Inc. filed December 10, 2020, File No. 001-36714). | |||
3.11 | ||||
4.1 | ||||
4.2 | ||||
4.3 | ||||
4.4 | ||||
151
Exhibit No. |
| Description | ||
|---|---|---|---|---|
4.5 | ||||
4.6 | ||||
4.7 | ||||
4.8 | ||||
4.9 | ||||
4.10 | ||||
4.11 | ||||
4.12 | ||||
4.13 | ||||
4.14 | ||||
4.15 | ||||
4.16 | ||||
4.17 | ||||
4.18 | ||||
4.19 | ||||
4.20 | ||||
4.21 | ||||
4.22 | ||||
4.23 | ||||
10.1‡ | ||||
152
Exhibit No. |
| Description | ||
|---|---|---|---|---|
10.2‡ | ||||
10.3‡ | ||||
10.4‡ | ||||
10.5‡ | ||||
10.6‡ | ||||
10.7† | ||||
10.8 | ||||
10.9 | ||||
10.10 | ||||
10.11† | ||||
10.12† | ||||
10.13† | ||||
10.14† | ||||
10.15 | ||||
10.16 | ||||
153
Exhibit No. |
| Description | ||
|---|---|---|---|---|
10.17 | ||||
10.18 | ||||
10.19 | ||||
10.20 | ||||
10.21 | ||||
10.22# | ||||
10.23 | ||||
10.24 | ||||
10.25 | ||||
10.26# | ||||
10.27 | ||||
10.28 | ||||
10.29 | ||||
10.30 | ||||
10.31 | ||||
10.32‡ | ||||
154
Exhibit No. |
| Description | ||
|---|---|---|---|---|
10.33 | ||||
10.34 | ||||
10.35 | ||||
10.36 | ||||
10.37‡ | ||||
10.38‡ | ||||
10.39‡ | ||||
10.40 | ||||
10.41 | ||||
10.42‡ | ||||
10.43 | ||||
10.44 | ||||
10.45 | ||||
10.46 | ||||
10.47 | ||||
10.48 | ||||
10.49 | ||||
155
Exhibit No. |
| Description | ||
|---|---|---|---|---|
10.50 | ||||
10.51# | ||||
10.52 | ||||
10.53# | ||||
10.54 | ||||
10.55 | ||||
10.56 | ||||
10.57# | ||||
10.58 | ||||
10.59# | ||||
10.60 | ||||
10.61 | ||||
10.62 | ||||
10.63# | ||||
10.64# | ||||
10.65 | ||||
10.66 | ||||
10.67 | ||||
156
Exhibit No. |
| Description | ||
|---|---|---|---|---|
16.1 | ||||
21.1* | ||||
23.1* | Consent of RBSM LLP, Independent Registered Public Accounting Firm. | |||
23.2* | Consent of Mayer Hoffman McCann P.C., Independent Registered Public Accounting Firm. | |||
31.1* | ||||
31.2* | ||||
32.1** | Certification Pursuant to 18 U.S.C. § 1350 (Section 906 of Sarbanes-Oxley Act of 2002). | |||
32.2** | Certification Pursuant to 18 U.S.C. § 1350 (Section 906 of Sarbanes-Oxley Act of 2002). | |||
101.INS | Inline XBRL Instance Document | |||
101.SCH | Inline XBRL Taxonomy Extension Schema | |||
101.CAL | Inline XBRL Taxonomy Extension Calculation Linkbase | |||
101.DEF | Inline XBRL Taxonomy Extension Definition Linkbase | |||
101.LAB | Inline XBRL Taxonomy Extension Label Linkbase | |||
101.PRE | Inline XBRL Taxonomy Extension Presentation Linkbase | |||
104 | Cover Page Interactive Data File (formatted as inline XBRL and contained in Exhibit 101) | |||
* Filed herewith.
** In accordance with Item 601(b)(32)(ii) of Regulation S-K and SEC Release No. 34-47986, the certifications furnished in Exhibits 32.1 and 32.2 hereto are deemed to accompany this Form 10-K and will not be deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934 (the “Exchange Act”) or deemed to be incorporated by reference into any filing under the Exchange Act or the Securities Act of 1933 except to the extent that the registrant specifically incorporates it by reference.
† Confidential treatment granted as to portions of the exhibit. Confidential materials omitted and filed separately with the Securities and Exchange Commission.
‡ Management contract or compensatory plan or arrangement.
# | Portions of this exhibit have been omitted pursuant to Item 601 of Regulation S-K promulgated under the Securities Act because the information (i) is not material and (ii) would be competitively harmful if publicly disclosed. |
157
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
JAGUAR HEALTH, INC. | ||
By: | /s/ LISA A. CONTE | |
Lisa A. Conte | ||
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Lisa A. Conte and Carol Lizak, jointly and severally, his or her attorneys-in-fact, each with the power of substitution, for him or her in any and all capacities, to sign any amendments to this Annual Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that each of said attorneys-in-fact, or his or her substitute or substitutes, may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Act of 1933, this Registration Statement has been signed by the following persons in the capacities and on the date indicated.
Signature | Title | Date | ||||||
/s/ LISA A. CONTE Lisa A. Conte | Chief Executive Officer, President and Director (Principal Executive Officer) | March 11, 2022 | ||||||
/s/ CAROL LIZAK Carol Lizak | Chief Financial Officer and Treasurer (Principal Financial and Accounting Officer) | March 11, 2022 | ||||||
/s/ JAMES J. BOCHNOWSKI James J. Bochnowski | Chairman of the Board | March 11, 2022 | ||||||
/s/ JOHN MICEK III John Micek III | Director | March 11, 2022 | ||||||
/s/ JONATHAN B. SIEGEL Jonathan B. Siegel | Director | March 11, 2022 | ||||||
/s/ GREG J. DIVIS Greg J. Divis | Director | March 11, 2022 |
158
Serious News for Serious Traders! Try StreetInsider.com Premium Free!
You May Also Be Interested In
- Jaguar Health Satisfies Conditions for Receipt of Non-Dilutive $2 Million Holdback Payment from Future Pak for Commercial License to Mytesi and Canalevia-CA1
- BRX Coin to Launch on PancakeSwap August 9 With $500,000 Liquidity and 100% Locked LP
- Tyros Biopharma Accelerates Expansion of Women’s Health Portfolio With Four New Product Introductions in 2026
Create E-mail Alert Related Categories
SEC FilingsSign up for StreetInsider Free!
Receive full access to all new and archived articles, unlimited portfolio tracking, e-mail alerts, custom newswires and RSS feeds - and more!