

Form 10-K Cell Source, Inc. For: Dec 31
 Tweet
Tweet Share
Share|
☒
☐
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the years ended December 31, 2018
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from _____________ to _____________
|
|
Nevada
|
|
32-0379665
|
|
(State or other jurisdiction of incorporation or organization)
|
|
(I.R.S. Employer Identification No.)
|
|
Large accelerated filer ☐
|
Accelerated filer ☐
|
|
Non-accelerated filer ☒
|
Smaller reporting company ☒
|
|
|
Emerging growth company ☐
|
|
Page
|
||
|
PART I
|
||
|
Item 1.
|
Business.
|
1 |
|
Item 1A.
|
Risk Factors.
|
29 |
|
Item 1B.
|
Unresolved Staff Comments.
|
41 |
|
Item 2.
|
Properties.
|
41 |
|
Item 3.
|
Legal Proceedings.
|
41 |
|
Item 4.
|
Mine Safety Disclosures.
|
41 |
|
PART II
|
||
|
Item 5.
|
Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
|
42 |
|
Item 6.
|
Selected Financial Data.
|
43 |
|
Item 7.
|
Management’s Discussion and Analysis of Financial Condition and Results of Operations.
|
43 |
|
Item 7A.
|
Quantitative and Qualitative Disclosures About Market Risk.
|
46 |
|
Item 8.
|
Financial Statements and Supplementary Data.
|
46 |
|
Item 9.
|
Changes in and Disagreements With Accountants on Accounting and Financial Disclosure.
|
46 |
|
Item 9A.
|
Controls and Procedures.
|
46 |
|
Item 9B.
|
Other Information.
|
47 |
| PART III | |
|
|
Item 10.
|
Directors, Executive Officers and Corporate Governance.
|
48 |
|
Item 11.
|
Executive Compensation.
|
50 |
|
Item 12.
|
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
|
52 |
|
Item 13.
|
Certain Relationships and Related Transactions, and Director Independence.
|
52 |
|
Item 14.
|
Principal Accounting Fees and Services.
|
54 |
| PART IV | |
|
|
Item 15.
|
Exhibits, Financial Statement Schedules.
|
55 |
|
Signatures
|
57 |
|
·
|
Hematological malignancies (leukemias, lymphomas, etc.). One of the most effective treatments for these
conditions is SCT - stem cell transplantation (e.g. bone marrow transplantation). While the challenge finding donors for allogeneic (donor vs. patient derived) SCT can be addressed through haploidentical (partially mismatched donor)
transplants, is a risky and difficult procedure primarily because of potential conflicts between host and donor immune systems and also due to viral infections that often follow even successful SCT while the compromised new immune system
works to reconstitute itself by using the transplanted stem cells.
|
|
·
|
The broader set of cancers, including solid tumors, that can potentially be treated effectively using
genetically modified cells such as CAR-T cells, but also face efficacy and economic constraints due to limited persistence based on immune system issues (i.e., the need to be able to safely and efficiently deliver allogeneic CAR-T
therapy).
|
|
·
|
Organ failure and transplantation. A variety of conditions can be treated by the transplantation of vital
organs. However, transplantation is limited both by the insufficient supply of available donor organs and the need for lifelong, daily anti-reject treatments post-transplant.
|
|
·
|
Non-malignant hematological conditions (such as sickle cell anemia) which could, in many cases, also be
effectively treated by stem cell transplantation if the procedure could be made safer and more accessible by addressing conflicts between host and donor immune systems.
|
Cell Source, Inc. (the "Company") is a Nevada corporation formed on June 6, 2012 under the name Ticket to See, Inc. ("TTSI"). Cell Source Ltd. (“Cell Source Israel”) was founded in 2011 in order to commercialize a suite of inventions that were the result of over ten (10) years of research at the Weizmann Institute of Science in Rehovot, Israel (“Weizmann Institute”). Pursuant to a Research and License Agreement by and between Cell Source Israel and Yeda, dated October 3, 2011, as amended in April, 2014 November, 2016, and, most recently, in March, 2018 (the “Yeda License Agreement”), Yeda, the commercial arm of the Weizmann Institute, granted Cell Source Israel an exclusive license to certain patents, discoveries, inventions, and other intellectual property generated (together with others) by Yair Reisner, Ph.D. (“Dr. Reisner”), former head of the Immunology Department at the Weizmann Institute.
| · |
being permitted to present only two years of audited financial statements and only two years of related Management’s Discussion & Analysis of Financial
Condition and Results of Operations in this report on Form 10-K;
|
| · |
not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, as amended, or the Sarbanes-Oxley
Act; and
|
| · |
reduced disclosure obligations regarding executive compensation in our periodic reports, proxy statements and registration statements.
|
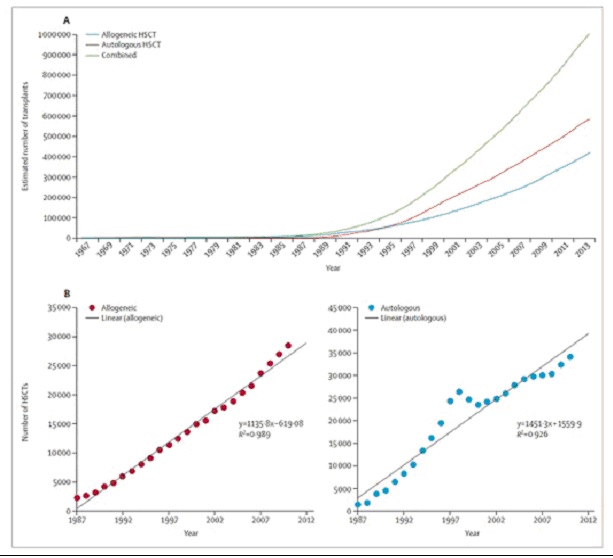
|
a)
|
many blood cancer patients are not candidates for the primary treatment (HSCT) that represents a potential
cure;
|
|
b)
|
there is high mortality among those patients who are candidates for HSCT and do undergo the procedure;
|
|
Initial Malignancy Indications
(note estimates for
North America and EU only)
|
Incidence
(Annual New Cases)
|
Annual Bone Marrow
Transplantations
|
||||||
|
Lymphoma
|
217,491
|
20,291
|
||||||
|
Multiple Myeloma
|
79,067
|
20,884
|
||||||
|
Leukemia
|
155,080
|
14,480
|
||||||
|
Total
|
451,638
|
55,655
|
||||||
|
1)
|
As noted above, increasing incidence of these disorders, largely driven by the aging population.
|
|
2)
|
Improvement and proliferation of HSCT treatments.
|
|
3)
|
A “virtuous circle” of lowered death rate due to better transplantations leading to more aggressive focus on
HSCT.
|
|
4)
|
The growing use of milder conditioning regimens, which makes the procedure more survivable for older patients
(see table below).
|
|
Name: VETO CELLS EFFECTIVE IN PREVENTING GRAFT REJECTION AND DEVOID OF GRAFT VERSUS HOST POTENTIAL
|
||||||||||
|
Country
|
Patent Number
|
Filed
|
Expires
|
Status
|
Assignee
|
|||||
|
USA (Basic)
|
6,544,506
|
05-Jan-2000
|
05-Jan-2020
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
USA (National Phase)
|
7,270,810
|
28-Dec-2000
|
5-Dec-2021
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Israel
|
150440
|
28-Dec-2000
|
28-Dec-2020
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Name: ANTI THIRD PARTY CENTRAL MEMORY T CELLS, METHODS OF PRODUCING SAME AND USE OF SAME IN TRANSPLANTATION
AND DISEASE TREATMENT
|
||||||||||
|
Country
|
Patent Number
|
Filed
|
Expires
|
Status
|
Assignee
|
|||||
|
USA
|
2011-0212071-A1
|
29-Oct-2009
|
29-Oct-2029
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Europe
|
2365823
|
29-Oct-2009
|
29-Oct-2029
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Israel
|
212587
|
29-Oct-2009
|
29-Oct-2029
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
India
|
905/MUMNP/2011
|
29-Oct-2009
|
29-Oct-2029
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
China
|
ZL200980153053.4
|
29-Oct-2009
|
29-Oct-2029
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Russian Federation
|
2506311
|
29-Oct-2009
|
29-Oct-2029
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
| Name: USE OF ANTI THIRD PARTY CENTRAL MEMORY T CELLS FOR ANTI-LEUKEMIA/LYMPHOMA TREATMENT | ||||||||||
|
Country
|
Patent Number
|
Filed
|
Expires
|
Status
|
Assignee
|
|||||
|
USA
|
9,421,228
|
08-Sep-2011
|
08-Sep-2031
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
USA (Continuation)
|
2016-0354410-A1
|
08-Sep-2011
|
08-Sep-2031
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Japan
|
2013-527738
|
08-Sep-2011
|
08-Sep-2031
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Canada
|
2,810,632
|
08-Sep-2011
|
08-Sep-2031
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
China
|
CN 103282047 A 9
|
08-Sep-2011
|
08-Sep-2031
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
China (Divisional)
|
CN 105907713 A
|
08-Sep-2011
|
08-Sep-2031
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
| Israel | 225102 | 08-Sep-2011 | 08-Sep-2031 | Granted | Yeda Research and Development Co. Ltd. | |||||
|
Republic of Korea
|
2013-7008892
|
08-Sep-2011
|
08-Sep-2031
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Brazil
|
BR 11 2013 0057564
|
08-Sep-2011
|
08-Sep-2031
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Mexico
|
357746
|
08-Sep-2011
|
08-Sep-2031
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Singapore
|
188473
|
08-Sep-2011
|
08-Sep-2031
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Europe
|
2613801
|
08-Sep-2011
|
08-Sep-2031
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Hong Kong
|
14100513.2
|
08-Sep-2011
|
08-Sep-2031
|
Granted
|
Yeda Research and Development Co. Ltd.
|
| Name: ANTI THIRD PARTY CENTRAL MEMORY T CELLS, METHODS OF PRODUCING SAME AND USE OF SAME IN TRANSPLANTATION AND DISEASE TREATMENT | ||||||||||
|
Country
|
Patent Number
|
Filed
|
Expires
|
Status
|
Assignee
|
|||||
|
USA
|
15/825,275
|
06-Sep-2012
|
06-Sep-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Europe
|
2753351
|
06-Sep-2012
|
06-Sep-2032
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Hong Kong
|
1200099A
|
06-Sep-2012
|
06-Sep-2032
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Japan
|
2014-529143
|
06-Sep-2012
|
06-Sep-2032
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Canada
|
2,848,121
|
06-Sep-2012
|
06-Sep-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
China
|
CN 103930130 A
|
06-Sep-2012
|
06-Sep-2032
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Australia
|
2012305931
|
06-Sep-2012
|
06-Sep-2032
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Republic of Korea
|
10-2014-7009267
|
06-Sep-2012
|
06-Sep-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
New Zealand
|
622749
|
06-Sep-2012
|
06-Sep-2032
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
South Africa
|
2014/01993
|
06-Sep-2012
|
06-Sep-2032
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
India
|
577/MUMNP/2014
|
06-Sep-2012
|
06-Sep-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Israel
|
231397
|
06-Sep-2012
|
06-Sep-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Russian Federation
|
2014110897
|
06-Sep-2012
|
06-Sep-2032
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Brazil
|
BR 11 2014 005355 3
|
06-Sep-2012
|
06-Sep-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Mexico
|
351226
|
06-Sep-2012
|
06-Sep-2032
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Singapore
|
11201400513P
|
06-Sep-2012
|
06-Sep-2032
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|
Name: GENETICALLY MODIFIED ANTI-THIRD PARTY CENTRAL MEMORY T CELLS AND USE OF SAME IN IMMUNOTHERAPY
|
||||||||||
|
Country
|
Patent Number
|
Filed
|
Expires
|
Status
|
Assignee
|
|||||
|
USA
|
2018-0207272-A1
|
14-July-2016
|
16-Jul-2036
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Europe
|
3322425
|
14-July-2016
|
16-Jul-2036
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
China
|
201680053580.8
|
14-July-2016
|
16-Jul-2036
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Japan
|
2018-501339
|
14-July-2016
|
16-Jul-2036
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Hong Kong
|
18114191.8
|
14-July-2016
|
16-Jul-2036
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Israel
|
256916
|
14-July-2016
|
16-Jul-2036
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Canada
|
2,991,690
|
14-July-2016
|
16-Jul-2036
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Australia
|
2016291825
|
14-July-2016
|
16-Jul-2036
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Name: USE OF ANTI THIRD PARTY CENTRAL MEMORY T CELLS
|
||||||||||
|
Country
|
Patent Number
|
Filed
|
Expires
|
Status
|
Assignee
|
|||||
|
China
|
CN 108025026 A
|
14-Jul-2016
|
16-Jul-2036
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Europe
|
3322424
|
14-Jul-2016
|
16-Jul-2036
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Name: METHODS OF TRANSPLANTATION AND DISEASE TREATMENT
|
||||||||||
|
Country
|
Patent Number
|
Filed
|
Expires
|
Status
|
Assignee
|
|||||
|
USA
|
2018-0207247-A1
|
14-Jul-2016
|
14-Jul-2036
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Name: VETO CELLS GENERATED FROM MEMORY CELLS
|
||||||||||
|
Country
|
Patent Number
|
Filed
|
Expires
|
Status
|
Assignee
|
|||||
|
USA
|
16/313,486
|
27-Jun-2017
|
27-Jun-2037
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Japan
|
2018-567129
|
27-Jun-2017
|
27-Jun-2037
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Canada
|
3,029,001
|
27-Jun-2017
|
27-Jun-2037
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Australia
|
2017289879
|
27-Jun-2017
|
27-Jun-2037
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
India
|
201927002672
|
27-Jun-2017
|
27-Jun-2037
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Israel
|
263924
|
27-Jun-2017
|
27-Jun-2037
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Russian Federation
|
2019101826
|
27-Jun-2017
|
27-Jun-2037
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Mexico
|
MX/a/2019/000022
|
27-Jun-2017
|
27-Jun-2037
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Republic of Korea
|
10-2019-7002824
|
27-Jun-2017
|
27-Jun-2037
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Singapore
|
11201811563R
|
27-Jun-2017
|
27-Jun-2037
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Europe
|
62/354,950
|
27-Jun-2017
|
27-Jun-2037
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
China
|
62/354,950
|
27-Jun-2017
|
27-Jun-2037
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Name: METHODS OF TRANSPLANTATION AND DISEASE TREATMENT USING ANTI-THIRD-PARTY CTL VETO CELLS
|
||||||||||
|
Country
|
Patent Number
|
Filed
|
Expires
|
Status
|
Assignee
|
|||||
|
USA
|
2018-0200300-A1
|
18-Jan-2018
|
18-Jan-2038
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Name: GENETICALLY MODIFIED VETO CELLS AND USE OF SAME IN IMMUNOTHERAPY
|
||||||||||
|
Country
|
Patent Number
|
Filed
|
Expires
|
Status
|
Assignee
|
|||||
|
PCT
|
WO2018/134824
|
18-Jan-2018
|
18-Jan-2038
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Name: A COMBINATION THERAPY FOR A STABLE AND LONG-TERM ENGRAFTMENT
|
||||||||||
|
Country
|
Patent Number
|
Filed
|
Expires
|
Status
|
Assignee
|
|||||
|
Singapore
|
10201801905W
|
20-Dec-2012
|
20-Dec-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Mexico
|
MX/a/2014/007647
|
20-Dec-2012
|
20-Dec-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Russian Federation
|
2014128479
|
20-Dec-2012
|
20-Dec-2032
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Israel
|
233303
|
20-Dec-2012
|
20-Dec-2032
|
Allowed
|
Yeda Research and Development Co. Ltd.
|
|||||
|
India
|
1468/MUMNP/2014
|
20-Dec-2012
|
20-Dec-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
South Africa
|
2014/05071
|
20-Dec-2012
|
20-Dec-2032
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
New Zealand
|
627272
|
20-Dec-2012
|
20-Dec-2032
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Republic of Korea
|
10-2014-7020449
|
20-Dec-2012
|
20-Dec-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Australia
|
2012355990
|
20-Dec-2012
|
20-Dec-2032
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Canada
|
2,859,953
|
20-Dec-2012
|
20-Dec-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Europe
|
2793914
|
20-Dec-2012
|
20-Dec-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
USA
|
2014-0363437-A1
|
20-Dec-2012
|
20-Dec-2032
|
Allowed |
Yeda Research and Development Co. Ltd.
|
|||||
|
Hong Kong
|
15103467.1
|
20-Dec-2012
|
20-Dec-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Name: A COMBINATION THERAPY FOR A STABLE AND LONG TERM ENGRAFTMENT USING SPECIFIC PROTOCOLS FOR T/B
CELL DEPLETION
|
||||||||||
|
Country
|
Patent Number
|
Filed
|
Expires
|
Status
|
Assignee
|
|||||
|
Singapore
|
11201403456U
|
20-Dec-2012
|
20-Dec-2032
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Mexico
|
MX/a/2014/007648
|
20-Dec-2012
|
20-Dec-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Brazil
|
BR 11 2014 015959 9
|
20-Dec-2012
|
20-Dec-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Russian Federation
|
2014129632
|
20-Dec-2012
|
20-Dec-2032
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Israel
|
233302
|
20-Dec-2012
|
20-Dec-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
India
|
1467/MUMNP/2014
|
20-Dec-2012
|
20-Dec-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
South Africa
|
2014/05298
|
20-Dec-2012
|
20-Dec-2032
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
New Zealand
|
627549
|
20-Dec-2012
|
20-Dec-2032
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Australia
|
2012355989
|
20-Dec-2012
|
20-Dec-2032
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Australia (Divisional)
|
2016259415
|
20-Dec-2012
|
20-Dec-2032
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
China
|
CN 104093314 A 4
|
20-Dec-2012
|
20-Dec-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Canada
|
2,859,952
|
20-Dec-2012
|
20-Dec-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Japan
|
6,313,219
|
20-Dec-2012
|
20-Dec-2032
|
Granted
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Europe
|
EP2797421
|
20-Dec-2012
|
20-Dec-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
USA
|
2014-0369974-A1
|
20-Dec-2012
|
20-Dec-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
Hong Kong
|
15103468.0
|
20-Dec-2012
|
20-Dec-2032
|
Pending
|
Yeda Research and Development Co. Ltd.
|
|||||
|
1)
|
Significantly improve outcomes of transplantations by reducing the host (transplant recipient) rejection rate
of T-cell depleted stem cells (e.g. from bone marrow) – thus supporting successful engraftment of the transplanted cells, which is the treatment for the blood cancer itself. In order to improve the safety of this cancer treatment, Veto
Cell technology has shown in preclinical studies that it can markedly reduce both the risk of GVHD and the need for using aggressive amounts of immunosuppression medications, as well as preventing viral infections that typically threaten
patients post transplantation. This safer means of deliver stem cell transplants would significantly reduce the HSCT mortality rate and therefore lead to broader use of this treatment.
|
|
2)
|
Substantively increase the number of transplantations by enabling successful engraftment under lower levels of
immune suppression and therefore making the therapy accessible to older and sicker patients (who today may not survive ablation).
|
|
3)
|
Further increase the number of transplantations by making transplantation appropriate for other indications
(for which today transplantation would be considered an inappropriately risky treatment).
|
| – |
Gene modified cell therapy is considered to be one of the most promising cancer treatment approaches in decades, with companies like Kite Pharma and JUNO
Therapeutics having recently been acquired at multi-billion dollar valuations after having successfully treated relatively small numbers of patients in clinical trials.
|
| – |
While gene modified treatments such as CAR-T have shown remarkable results in cancer treatment trials, their published successes to date have been mostly
limited to “autologous” blood cell cancer treatments using the patient’s own cells. There are concerns that this type of “personalized” treatment may not have favorable economics on a large-scale basis.
|
| – |
The ideal, more lucrative commercial path for CAR-T and similar genetically engineered cell therapies is to become “allogeneic” or off-the-shelf product
with drug-like distribution economics and to treat a broad spectrum of cancers including solid tumors.
|
| – |
Cell Source licenses Yeda’s patent applications for combining Veto Cells with genetically modified T cells and is currently exploring active collaboration
with CAR-T cell providers to move Veto and CAR-T combined cell therapy towards the clinic.
|
| – |
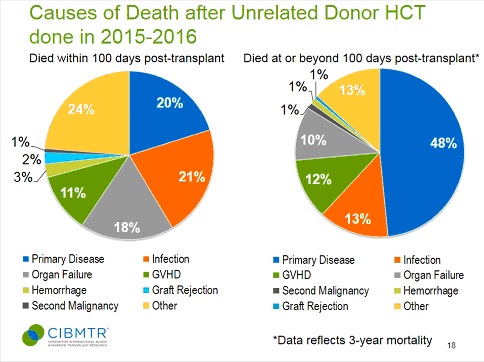
Other than primary disease (typically blood cell cancer) the leading causes of death in unrelated donor bone marrow transplants are rejection, GVHD (Graft
vs. Host Disease), where the donor bone marrow rejects the host or recipient), and infections, which collectively are responsible for 30% of deaths after unrelated donor transplants within the first 100 days post-transplant.
|
| – |
It is well established that GVHD can be prevented by T cell depletion of the bone marrow transplant. However, this procedure is also is associated with an
increased rate of graft rejection. Preclinical studies clearly suggest that this problem can be overcome by adding Veto Cells to the bone marrow transplant. However, viruses such as CMV and EBV remain a major threat to patients
post-transplant.
|
| – |
Cell Source has developed a next generation Veto Cell that not only facilitates mismatched transplants but also protects the transplant recipient against
these common viruses. During the initial period after a stem cell transplantation the patient’s body undergoes an immune system reconstitution period. While the “new” immune system is building up, the patient is particularly
vulnerable to viral infections such as CMV, an infection that is typically development in about half of bone marrow transplant recipients during the first 100 days post transplantation. Veto cells can fend off CMV until such time as
the patient’s own immune system reconstitutes to the point that it can fight off the infection on its own.
|
| – |
Combining GVHD prevention by using T cell depleted transplants with anti-rejection action as well as virus prevention, Veto Cell could potentially
significantly increase survival rates post-transplant.
|
| – |
Based on preclinical data, veto cells can also be used to facilitate organ transplants (e.g. kidney transplant combined with a bone marrow transplant) with
partially mismatched donors and either reduce or eliminate the need for lifelong daily anti-rejection treatment currently given to even fully matched donor organ recipients.
|
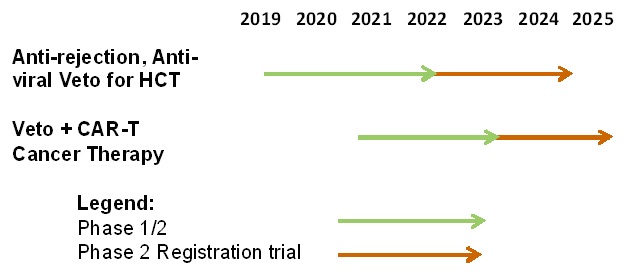
| – |
Cell Source is currently in the process of attaining regulatory validation for the production of its Anti-Viral Veto Cells in Europe and plans to commence
human clinical trials in the US in 2019.
|
|
1)
|
It has an outer surface coating that triggers attack by specific host T-cells (and only those specific
T-cells).
|
|
2)
|
It can annihilate an attacking T-cell without itself being damaged (specifically, it exposes or releases a
death-signaling molecule when an attacking T-cell binds to it).
|
|
3)
|
It has been oriented to attack cells of a simulated third party (i.e., neither host nor donor) and thus
exhibits markedly reduced risk of GVHD or graft rejection.
|
|
4)
|
It is long-lived and endures in the body for extended periods.
|
|
5)
|
It migrates to the thymus and lymph nodes.
|
|
1)
|
It destroys the host T-cells so they will not attack (reject) the donor bone marrow cells.
|
|
2)
|
It makes space in the host bone marrow for the new donor cells.
|
|
3)
|
It destroys diseased host blood cells so that they do not proliferate and cause relapse following the
procedure.
|
|
·
|
Host rejection - the myeloablative conditioning does not destroy all of the host T-cells. Those that remain
may aggressively attack the donor bone marrow cells before they can engraft.
|
|
·
|
“Graft versus Host Disease” (GVHD) - the transplanted cells include donor T-cells which recognize the host's
body as foreign and attack it.
|
|
·
|
Viral infections are a common complication from HSCT and result in 20% of early patients deaths in
unrelated-donor transplants in the US
|
|
1.
|
Inducing chimerism: |
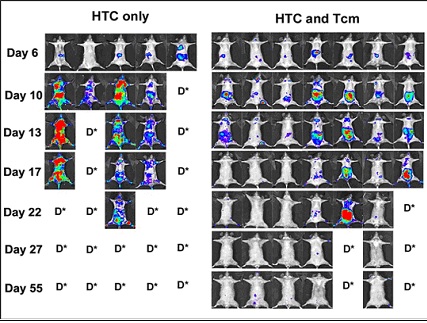
|
Offering
|
Objective
|
Major Activities
|
Estimated Start Date
|
|||
|
Anti-Rejection,
Anti-Viral Veto Cell
|
Validate and introduce new commercial treatment to increase engraftment of allogeneic bone marrow transplantations
|
1. Regulatory approval and treatment protocols
2. Conduct human clinical trials
3. Develop plan for commercial exploitation
|
· Initiate a human clinical trial in the US by 2019
· Commence human trials in Europe in 2020
|
|||
|
Veto – CAR-T Cell Therapy
|
Validate the possibility of combining Veto Cell treatment with CAR-T cell treatment for both blood cell cancer and solid tumor
cancer treatment
|
4. Collaboration with Zelig Eshhar, inventor of Car-T cells
5. Validate combined treatment model in clinical trials
|
· Proof of concept in completion in 2019
· Commence human trials in 2021 or 2022
|
|
1.
|
“Anti-rejection,
Anti-Viral” Veto Cell tolerance therapy for donor mismatched allogeneic bone marrow transplantations.
|
|
2.
|
“Anti-cancer”
Veto + CAR-T cell therapy for blood cell and, eventually, solid tumor cancers.
|
|
3.
|
“Anti-rejection”
Veto Cell tolerance therapy for donor mismatched organ transplantation.
|
| 4. |
Veto Cell tolerance therapy for non-malignant disorders.
|
|
1)
|
Complete development of human treatment protocol (2-5 years)
|
|
2)
|
Apply for and receive approval to commence human trials (9-18 months)
|
|
3)
|
Recruit patients (1-6 months)
|
|
4)
|
Conduct Phase I trials showing safety of product (1-2 years)
|
|
5)
|
Apply for and receive approval to conduct trials showing product efficacy (6-12 months)
|
|
6)
|
Data collecting and analysis (6-12 months)
|
|
7)
|
Conduct Phase II efficacy trials (2-3 years)
|
|
8)
|
Data collecting and analysis (6-12 months)
|
|
9)
|
Apply for and receive approval to conduct trials showing efficacy in larger numbers of patients (6-12 months)
|
|
10)
|
Conduct Phase III efficacy trials with larger numbers of patients (2-4 years)
|
|
11)
|
Data collecting and analysis (6-12 months)
|
|
12)
|
Apply for and receive approval for production scale manufacturing facilities (6-12 months)
|
|
13)
|
Contract third party or establish own production facilities (6-30 months)
|
|
14)
|
Contract third party or establish own distribution platform (6-18 months)
|
|
15)
|
Commence manufacturing and distribution (6-12 months)
|
|
·
|
successfully complete adequate and well-controlled clinical trials that demonstrate statistically significant
safety and efficacy and to obtain all requisite regulatory approvals in a timely and cost-effective manner;
|
|
·
|
effectively use patents and possibly exclusive partnership agreements with important third-party treatment
providers and collaboration partners to maintain a stable competitive stance for our Technology;
|
|
·
|
attract and retain appropriate clinical and commercial personnel and service providers; and
|
|
·
|
establish adequate distribution relationships for our products.
|
|
Strategy
Element
|
Introductory period
(years 1 -3 post FDA approval)
|
Years 4+
|
||
|
Market Segments
|
· Lymphoma and Leukemia
· Multiple Myeloma
|
· Same as before plus broader set of solid tumor cancer targets, kidney
and liver failure, sickle cell anemia beta
thalassemia and other
non-malignant hematological disorders;
|
||
|
Product Rollout
|
· Veto Cell therapy for B-cell malignancies
· Veto+CAR-T Veto Cell therapy for both blood cell cancers
cancers
|
· Veto Cell tolerizing treatment for HSCT and organ transplantation
· Veto Cell therapy for both liquid and solid tumor cancers as well
as non-malignant disorders;
|
|
Customer/ Geographic Focus
|
· North America
· Western Europe
· China
|
· North America, Western & Eastern Europe, Australia/New Zealand,
Russia, Brazil, selected Asian markets
|
||
|
Channels/Go to Market
|
· Direct relationships with leading transplantation centers
· International production and distribution through partners
|
· Partnership with global market leaders
|
||
|
Pricing
|
· Consistent with other cell therapy offerings currently associated with
transplantations and immuno-oncology
|
· Potentially higher volume, lower cost for “off the shelf” offerings
|
||
|
Operations
|
· Three production centers:
- US
- Western Europe
- Far East
· Initial capacity leased from or situated adjacent to major transplantation
center.
|
· Regional production centers owned or JV with partners
|
|
1)
|
Severity of unmet medical need: degree of severity of the indication and the effectiveness of existing
treatments. These criteria help determine the proper regulatory pathway.
|
|
2)
|
Technology relevance: relative value of the ability to manage immune response to the treatment of a given
indication.
|

|
1)
|
Treating patients after the end of Phase 2 (based on US Fast Track approvals and/or European Marketing
Authorization Approvals) with either partial or full insurance reimbursement available); and
|
|
2)
|
Potential upfront and milestone driven licensing revenues from collaborations with third parties.
|
|
a.
|
by January 1, 2022, to have commenced Phase II clinical trials in a respect of a Product;
|
|
b.
|
by January 1, 2025, to have either commenced Phase III clinical trials or to have received FDA or EMA
marketing approval in a respect of a Product (“Marketing Approval”);
|
|
c.
|
within 12 (twelve) months from the date of Marketing Approval, to have made a First Commercial Sale of a
Product; or
|
|
d.
|
in case commercial sale of any Product having commenced, there shall be a period of 12 (twelve) months or more
during which no sales of any Product shall take place by the Company or its Sublicensees (except as a result of force majeure or other factors beyond the control of the Company)."
|
|
·
|
Title.
All right, title and interest in and to the Licensed Information and the Patents (as those terms are defined in the Yeda License Agreement) and all right, title and interest in and to any drawings, plans, diagrams, specifications, other
documents, models, or any other physical matter in any way containing, representing or embodying any of the foregoing, vest and shall vest in Yeda and subject to the license granted in the Yeda License Agreement.
|
|
·
|
Patents.
Both Yeda and the Company shall consult with one another on the filing of patent applications for any portion of Licensed Information and/or corresponding to patent application existing at the time the Yeda License Agreement was executed.
Yeda shall retain outside patent counsel that will be approved by Cell Source, to prepare, file and prosecute patent applications. All applications will be filed in Yeda’s name.
|
|
·
|
Patents;
Patent Infringements. Where the Company determines that a third party is infringing one or more of the Patents or is sued, in prosecuting or defending such litigation, the Company must pay any expenses or costs or other
liabilities incurred in connection with such litigation (including attorney’s fees, costs and other sums awarded to the counterparty in such action). The Company agreed to indemnify Yeda against any such expenses or costs or other
liabilities.
|
|
·
|
License.
With regard to the expiration of Patents, a Product is deemed to be covered by a Patent so long as such Product is protected by “Orphan Drug” status (or the like). The Company has an exclusive worldwide license under the Licensed
Information and the Patents for the development, manufacture and sales of the Products. License remains in force in each country with respect to each Product until the later of (i) the expiration of the last Patent in such country
covering such Product or (ii) the expiration of a 15-year period commencing the day FDA New Drug Approval is received for a Product in such country.
|
|
i.
|
the proposed Sublicense is for monetary consideration only;
|
|
ii.
|
the proposed Sublicense is to be granted in a bona fide arm’s length commercial transaction;
|
|
iii.
|
a copy of the agreement granting the Sublicense and all amendments thereof shall be made available to Yeda, 14
days before their execution and Cell Source shall submit to Yeda copies of all such Sublicenses and all amendments thereof promptly upon execution thereof; and
|
|
iv.
|
the proposed Sublicense is made by written agreement, the provisions of which are consistent with the terms of
the License and contain, inter alia, the following terms and conditions, including: the Sublicense shall expire automatically on the termination of the License for any reason.
|
|
·
|
Termination.
The Yeda License Agreement terminates on the later of: (i) the expiration of the last of the Patents or (ii) the expiry of a continuous period of 20 years during which there shall not have been a First commercial sale of any product in
any country. Yeda may terminate by written notice, effective immediately, if the Company challenges the validity of any of the Patents. If a challenge is unsuccessful, then in addition to Yeda’s right to termination, the Company shall pay
to Yeda liquidated damages in the amount of $8,000,000. Either the Company or Yeda may terminate the Yeda License Agreement and the License by serving a written notice upon (i) occurrence of a material breach or (ii) the granting of a
winding-up order. Additionally, Yeda may terminate for failure to reimburse Yeda for patent application and/or prosecution expenses.
|
Our success may depend, in part, on the extent to which reimbursement for the costs of therapeutic products and related treatments will be available from third-party payers such as government health administration authorities, private health insurers, managed care programs, and other organizations. Over the past decade, the cost of health care has risen significantly, and there have been numerous proposals by legislators, regulators, and third-party health care payers to curb these costs. Some of these proposals have involved limitations on the amount of reimbursement for certain products. Similar federal or state health care legislation may be adopted in the future and any products that we or our collaborators seek to commercialize may not be considered cost-effective. Adequate third-party insurance coverage may not be available for us or our collaborative partners to establish and maintain price levels that are sufficient for realization of an appropriate return on investment in product development.
|
·
|
the degree and range of protection any patents will afford us against competitors, including whether third
parties will find ways to invalidate or otherwise circumvent the patents that we license;
|
|
·
|
whether or not others will obtain patents claiming aspects similar to those covered by the patents
that we license; or
|
|
·
|
whether we will need to initiate litigation or administrative proceedings, which may be costly whether we win
or lose.
|
|
·
|
the duration of the clinical trial;
|
|
·
|
the number of sites included in the trials;
|
|
·
|
the countries in which the trial is conducted;
|
|
·
|
the length of time required and ability to enroll eligible patients;
|
|
·
|
the number of patients that participate in the trials;
|
|
·
|
the number of doses that patients receive;
|
|
·
|
the drop-out or discontinuation rates of patients;
|
|
·
|
per patient trial costs;
|
|
·
|
third party contractors failing to comply with regulatory requirements or meet their contractual obligations
to us in a timely manner;
|
|
·
|
our final product candidates having different properties in humans than in laboratory testing;
|
|
·
|
the need to suspend or terminate our clinical trials;
|
|
·
|
insufficient or inadequate supply of quality of necessary materials to conduct our trials;
|
|
·
|
potential additional safety monitoring, or other conditions required by FDA or comparable foreign regulatory
authorities regarding the scope or design of our clinical trials, or other studies requested by regulatory agencies;
|
|
·
|
problems engaging institutional review boards (“IRB”) to oversee trials or in obtaining and maintaining IRB
approval of studies;
|
|
·
|
the duration of patient follow-up;
|
|
·
|
the efficacy and safety profile of a product candidate;
|
|
·
|
the costs and timing of obtaining regulatory approvals; and
|
|
·
|
the costs involved in enforcing or defending patent claims or other intellectual property rights.
|
|
·
|
delays in the development of manufacturing capabilities for our product candidates to enable their consistent
production at clinical trial scale;
|
|
·
|
delays in the commencement of clinical trials as a result of clinical trial holds or the need to obtain
additional information to complete an Investigational New Drug Application (IND);
|
|
·
|
delays in obtaining regulatory approval to commence new trials;
|
|
·
|
adverse safety events experienced during our clinical trials;
|
|
·
|
insufficient efficacy during trials leading to withdrawal of product candidate;
|
|
·
|
delays in obtaining clinical materials;
|
|
·
|
slower than expected patient recruitment for participation in clinical trials; and
|
|
·
|
delays in reaching agreement on acceptable clinical trial agreement terms with prospective sites or obtaining
institutional review board approval.
|
|
·
|
our clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require
us, to conduct additional clinical and/or preclinical testing or to abandon programs;
|
|
·
|
the results obtained in earlier stage clinical testing may not be indicative of results in future clinical
trials;
|
|
·
|
clinical trial results may not meet the level of statistical significance required by the FDA or other
regulatory agencies;
|
|
·
|
enrollment in our clinical trials for our product candidates may be slower than we anticipate, resulting in
significant delays and additional expense;
|
|
·
|
we, or regulators, may suspend or terminate our clinical trials if the participating patients are being
exposed to unacceptable health risks; and
|
|
·
|
the effects of our product candidates on patients may not be the desired effects or may include undesirable
side effects or other characteristics that may delay or preclude regulatory approval or limit their commercial use, if approved.
|
|
·
|
the therapeutic endpoints chosen for evaluation;
|
|
·
|
the eligibility criteria defined in the protocol;
|
|
·
|
the perceived benefit of the investigational drug under study;
|
|
·
|
the size of the patient population required for analysis of the clinical trial’s therapeutic endpoints;
|
|
·
|
our ability to recruit clinical trial investigators and sites with the appropriate competencies and
experience;
|
|
·
|
our ability to obtain and maintain patient consents; and
|
|
·
|
competition for patients by clinical trial programs for other treatments.
|
|
·
|
variations in our quarterly operating results;
|
|
·
|
announcements that our revenue or income are below analysts’ expectations;
|
|
·
|
general economic slowdowns;
|
|
·
|
sales of large blocks of the Company’s Common Stock; and
|
|
·
|
announcements by us or our competitors of significant contracts, acquisitions, strategic partnerships, joint
ventures or capital commitments.
|
|
·
|
Eleven notes payable with
principal amounts totaling $1,613,000; and
|
|
·
|
Seventeen convertible notes
payable with principal amounts totaling $1,060,000.
|
|
·
|
Only two years of audited financial statements in addition to any required unaudited interim financial
statements with correspondingly reduced "Management's Discussion and Analysis of Financial Condition and Results of Operations" disclosure;
|
|
·
|
Reduced disclosure about our executive compensation arrangements;
|
|
·
|
Exemption from the auditor attestation requirement in the assessment of our internal control over financial
reporting.
|
Management’s efforts to remediate the material weakness included raising funds and seeking new resources to alleviate this material weakness and filing all necessary regulatory reports on a timely basis. Although management concluded that our internal controls over financial reporting were effective as of December 31, 2018, there can be no assurance that we will have the necessary resources to maintain effective controls in future periods.
|
2018 Fiscal Year
|
||||||||
|
High
|
Low
|
|||||||
|
First Quarter ended March 31, 2018
|
$
|
0.43
|
$
|
0.25
|
||||
|
Second Quarter ended June 30, 2018
|
$
|
0.85
|
$
|
0.42
|
||||
|
Third Quarter ended September 30, 2018
|
$
|
1.09
|
$
|
0.55
|
||||
|
Fourth Quarter ended December 31, 2018
|
$
|
0.75
|
$
|
0.45
|
||||
|
2017 Fiscal Year
|
||||||||
|
High
|
Low
|
|||||||
|
First Quarter ended March 31, 2017
|
$
|
0.53
|
$
|
0.35
|
||||
|
Second Quarter ended June 30, 2017
|
$
|
0.45
|
$
|
0.25
|
||||
|
Third Quarter ended September 30, 2017
|
$
|
0.40
|
$
|
0.30
|
||||
|
Fourth Quarter ended December 31, 2017
|
$
|
0.45
|
$
|
0.20
|
||||
|
·
|
Hematological malignancies (leukemias, lymphomas, etc.). One of the most effective treatments for these
conditions is SCT - stem cell transplantation (e.g. bone marrow transplantation). While the challenge finding donors for allogeneic (donor vs. patient derived) SCT can be addressed through haploidentical (partially mismatched donor)
transplants, is a risky and difficult procedure primarily because of potential conflicts between host and donor immune systems and also due to viral infections that often follow even successful SCT while the compromised new immune system
works to reconstitute itself by using the transplanted stem cells.
|
|
·
|
The broader set of cancers, including solid tumors, that can potentially be treated effectively using
genetically modified cells such as CAR-T cells, but also face efficacy and economic constraints due to limited persistence based on immune system issues (i.e., the need to be able to safely and efficiently deliver allogeneic CAR-T
therapy).
|
|
·
|
Organ failure and transplantation. A variety of conditions can be treated by the transplantation of vital
organs. However, transplantation is limited both by the insufficient supply of available donor organs and the need for lifelong, daily anti-reject treatments post-transplant.
|
|
·
|
Non-malignant hematological conditions (such as sickle cell anemia) which could, in many cases, also be
effectively treated by stem cell transplantation if the procedure could be made safer and more accessible by addressing conflicts between host and donor immune systems.
|
On February 7, 2019, MD Anderson submitted an IND application with respect to the Anti-Rejection, Anti-Viral Veto Cell to the FDA.
On February 19, 2019, we entered into an agreement with MD Andersen for the latter to perform cell production and conduct Phase I/II human clinical trials. In connection with that agreement, we committed to fund such work in the amount of approximately $2,000,000 over a two-year period beginning that same date.
Loss on Exchange of Warrants for Common Stock
During the year ended December 31, 2017, we recognized $38,393 of loss on exchange of warrants for common stock related to outstanding warrants exchanged in connection with a note extension during the period.|
December 31,
|
||||||||
|
2018
|
2017
|
|||||||
|
Cash
|
$
|
18,934
|
$
|
371,048
|
||||
|
Working capital deficiency
|
$
|
(4,920,171
|
)
|
$
|
(4,557,374
|
)
|
||
|
(1)
|
Due to a lack of financial resources, we were unable to file our annual reports on Form 10-K for the years
ended December 31, 2017 and December 31, 2016 and the quarterly reports on Form 10-Q for the periods ended March 31, 2017, June 30, 2017, September 30, 2017, March 31, 2018 and June 30, 2018 on a timely basis. Management evaluated the
lack of financial resources in its assessment of our reporting controls and procedures and has concluded that the control deficiency represented a material weakness.
|
|
Name
|
Age
|
Title(s)
|
||
|
Dennis Brown
|
69
|
Director (Chairman)
|
||
|
Itamar Shimrat
|
59
|
Chief Executive Officer, Chief Financial Officer and Director
|
||
|
Yoram Drucker
|
53
|
Director
|
||
|
David Zolty
|
69
|
Director
|
||
|
Ben Friedman
|
60
|
Director
|
| 1. |
any bankruptcy petition filed by or against such person or any business of which such person was a general partner or executive officer either at the time
of the bankruptcy or within two years prior to that time;
|
| 2. |
any conviction in a criminal proceeding or being subject to a pending criminal proceeding (excluding traffic violations and other minor offenses);
|
| 3. |
being subject to any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction, permanently or
temporarily enjoining him from or otherwise limiting his involvement in any type of business, securities or banking activities or to be associated with any person practicing in banking or securities activities;
|
| 4. |
being found by a court of competent jurisdiction in a civil action, the SEC or the Commodity Futures Trading Commission to have violated a Federal or state
securities or commodities law, and the judgment has not been reversed, suspended, or vacated;
|
| 5. |
being subject of, or a party to, any Federal or state judicial or administrative order, judgment decree, or finding, not subsequently reversed, suspended or
vacated, relating to an alleged violation of any Federal or state securities or commodities law or regulation, any law or regulation respecting financial institutions or insurance companies, or any law or regulation prohibiting mail
or wire fraud or fraud in connection with any business entity; or
|
| 6. |
being subject of or party to any sanction or order, not subsequently reversed, suspended, or vacated, of any self-regulatory organization, any registered
entity or any equivalent exchange, association, entity or organization that has disciplinary authority over its members or persons associated with a member.
|
|
Name and
|
Stock
|
Option
|
All Other
|
||||||||||||||||||||||
|
Principal Position
|
Year
|
Salary
|
Bonus
|
Awards
|
Awards
|
Compensation
|
Total
|
||||||||||||||||||
|
Itamar Shimrat
|
2018
|
$
|
165,625
|
$
|
-
|
$
|
-
|
$
|
-
|
$
|
-
|
$
|
165,625
|
||||||||||||
|
Chief Executive Officer
|
2017
|
$
|
179,064
|
$
|
-
|
$
|
-
|
$
|
-
|
$
|
-
|
$
|
179,064
|
||||||||||||
|
Option Awards
|
Stock Awards
|
||||||||||||||||||||||||||||||||
|
Equity
|
|||||||||||||||||||||||||||||||||
|
incentive
|
|||||||||||||||||||||||||||||||||
|
plan
|
|||||||||||||||||||||||||||||||||
|
Equity
|
awards:
|
||||||||||||||||||||||||||||||||
|
incentive
|
Market or
|
||||||||||||||||||||||||||||||||
|
Equity
|
plan
|
payout
|
|||||||||||||||||||||||||||||||
|
incentive
|
awards:
|
value of
|
|||||||||||||||||||||||||||||||
|
plan awards:
|
Number of
|
unearned
|
|||||||||||||||||||||||||||||||
|
Number of
|
Number of
|
Number of
|
Number
|
Market
|
unearned
|
shares,
|
|||||||||||||||||||||||||||
|
securities
|
securities
|
securities
|
of shares
|
value of
|
shares,
|
units or
|
|||||||||||||||||||||||||||
|
underlying
|
underlying
|
underlying
|
or units of
|
shares of
|
units or
|
other
|
|||||||||||||||||||||||||||
|
unexercised
|
unexercised
|
unexercised
|
Option
|
Option
|
stock that
|
units
|
other rights
|
rights
|
|||||||||||||||||||||||||
|
options
|
options
|
unearned
|
exercise
|
expiration
|
have not
|
that have
|
that have
|
that have
|
|||||||||||||||||||||||||
|
Name
|
exercisable
|
unexercisable
|
options
|
price
|
date
|
vested
|
not vested
|
not vested
|
not vested
|
||||||||||||||||||||||||
|
Itamar Shimrat
|
750,000
|
-
|
-
|
$
|
0.75
|
11/10/2019
|
-
|
$
|
-
|
-
|
$
|
-
|
|||||||||||||||||||||
|
Change in
|
|||||||||||||||||||||||||
|
Present Value
|
|||||||||||||||||||||||||
|
and
|
|||||||||||||||||||||||||
|
Fees
|
Nonqualified
|
||||||||||||||||||||||||
|
Earned or
|
Deferred
|
||||||||||||||||||||||||
|
Paid In
|
Stock
|
Option
|
Compensation
|
All Other
|
|||||||||||||||||||||
|
Year
|
Salary
|
Awards
|
Awards
|
Earnings
|
Compensation
|
Total
|
|||||||||||||||||||
|
Dennis Brown
|
2018
|
$ | - | $ | - | $ | - | $ | - | $ | - | $ | - | ||||||||||||
|
Yoram Drucker
|
2018
|
$ | - | $ | - | $ | - | $ | - | $ | - | $ | - | ||||||||||||
|
David Zolty
|
2018
|
$ | - | $ | - | $ | - | $ | - | $ | - | $ | - | ||||||||||||
|
Ben Friedman
|
2018
|
$ | - | $ | - | $ | - | $ | - | $ | - | $ | - | ||||||||||||
|
As of March 27, 2019
|
|||||||||
|
Name and Address of Beneficial Owner (10)
|
Amount and Nature
of
Beneficial Ownership (1)
|
Percentage of
Class (2)
|
|||||||
|
Directors and Officers:
|
|||||||||
|
Yoram Drucker, Director
|
1,125,004
|
(3)
|
4.22
|
%
|
|||||
|
Itamar Shimrat, Chief Executive Officer, Chief Financial Officer and Director
|
1,337,311
|
(4)
|
4.99
|
%
|
|||||
|
David Zolty, Director
|
1,168,318
|
(5)
|
4.47
|
%
|
|||||
|
Ben Friedman, Director (6)
|
4,383,344
|
(7)
|
16.80
|
%
|
|||||
|
Dennis Brown, Director (Executive Chairman)
|
200,000
|
(8)
|
*
|
||||||
|
All directors and executive officers as a group (5 persons)
|
8,213,977
|
29.80
|
%
|
||||||
|
Yeda Research & Development Co. Ltd.
P.O. Box 95
Rehovot, 76100, Israel
|
1,308,684
|
(9)
|
4.99
|
%
|
|||||
| * |
less than 1%
|
| (1) |
Beneficial ownership is determined in accordance with the rules of the SEC and generally includes voting or
investment power with respect to securities. Shares of common stock subject to options or warrants currently exercisable or convertible, or exercisable or convertible within 60 days of March27, 2019 are deemed outstanding for computing
the percentage of the person holding such option or warrant but are not deemed outstanding for computing the percentage of any other person.
|
| (2) |
Based on 26,077,611 shares issued and outstanding as of March 27, 2019.
|
| (3) |
Includes a five-year warrant to purchase 550,000 shares of common stock with an exercise price of $0.75 per share.
|
| (4) |
Includes 762,307 shares of a five-year warrant to purchase 750,000 shares of
common stock with an exercise price of $0.75 per share, which warrant is subject to
a 4.99% conversion limitation.
|
| (5) |
Includes warrants to purchase a total of 72,500 shares of common stock with an exercise price of $0.75 per share.
|
| (6) |
Mr. Friedman’s beneficial ownership includes shares beneficially owned by his wife, Phyllis Friedman.
|
| (7) |
Excludes a five-year warrant to purchase 50,000 shares of common stock with an exercise price of $0.75 per share, which warrant is subject to a 4.99%
conversion limitation.
|
| (8) |
Includes a five-year warrant to purchase 100,000 shares of common stock with an exercise price of $0.75 per share.
|
| (9) |
Includes 148,722 shares of a five-year warrant to purchase 1,995,376 shares of common stock with an exercise price
of $0.001 per share, which warrant is subject to a 4.99% conversion limitation.
|
| (10) |
Except as otherwise indicated, the address of each beneficial owner is c/o Cell Source, Inc., 57 West 57th Street, Suite 400, New York, New York 10019.
|
|
2018
|
2017
|
|||||||
|
Audit Fees
|
$
|
185,900
|
$
|
73,000
|
||||
|
Tax fees
|
--
|
--
|
||||||
|
All other fees
|
--
|
--
|
||||||
|
$
|
185,900
|
$
|
73,000
|
|||||
|
Exhibit
Number
|
Description
|
|
|
2.1 (1)
|
Share Exchange Agreement, dated June 30, 2014, by and between Cell Source, Ltd., and Ticket to See, Inc.
|
|
|
3.1 (1)
|
Articles of Association of Cell Source Limited, dated August 14, 2011, as amended on November 11, 2013
|
|
|
3.2 (2)
|
Articles of Incorporation of Ticket to See, Inc., dated June 6, 2012
|
|
|
3.3 (3)
|
Certificate of Amendment to Articles of Incorporation of Ticket to See, Inc., dated June 23, 2014
|
|
|
3.3 (4)
|
Certificate of Amendment to Articles of Incorporation of Ticket to See, Inc., dated May 20, 2014
|
|
|
3.4 (2)
|
Bylaws of Cell Source, Inc., dated June 6, 2012
|
|
|
3.5 (18)
|
Certificate of Designation with respect to Series A Preferred Stock dated November 14, 2016
|
|
|
10.1 (1)
|
Form of Subscription Agreement
|
|
|
10.2 (1)
|
Form of Registration Rights Agreement
|
|
|
10.3 (1)
|
Form of Investor Warrant
|
|
|
10.4 (1)
|
Form of Consultant Warrant(8)
|
|
|
10.5 (1)
|
Form of Researcher Company Warrant
|
|
|
10.6 (1)
|
Form of Company Warrant
|
|
|
10.7 (1)
|
Form of Lockup Agreement (included in Exhibit 2.1)
|
|
|
10.8 (1)
|
Research and License Agreement by and between Yeda Research and Development Company Limited and Cell Source
Limited, dated October 3, 2011
|
|
|
10.9 (1)
|
Amendment to Research and License Agreement
|
|
|
10.10 (1)
|
Evaluation and Exclusive Option Agreement by and between Yeda Research and Development Company Limited and Cell
Source Limited, dated Oct. 3, 2011 (included in Exhibit 10.7)
|
|
|
10.11 (1)
|
Amendment dated April 1, 2014 to Evaluation and Exclusive Option Agreement by and between Yeda Research and
Development Company Limited and Cell Source Limited
|
|
|
10.12 (1)
|
Second Amendment dated June 22, 2014 to Evaluation and Exclusive Option Agreement by and between Yeda Research and
Development Company Limited and Cell Source Limited
|
|
|
10.13 (1)
|
Consulting Agreement by and between Cell Source Limited and Professor Yair Reisner
|
|
|
10.14 (6)
|
Form of Amendment No. 1 to Registration Rights Agreement
|
|
|
10.15 (7)
|
Bridge Funding Agreement
|
|
|
10.16 (5)
|
Third Amendment dated June 22, 2014 to Evaluation and Exclusive Option Agreement by and between Yeda Research and
Development Company Limited and Cell Source Limited
|
|
|
10.17 (8)
|
Form of Consulting Agreement pursuant to which the Company issued warrants to purchase an aggregate of 2,000,000
shares of the Company’s common stock
|
|
|
10.18 (9)
|
Form of Promissory Note issued to the Company’s Chief Executive Officer
|
|
|
10.19(10)
|
Form of March 2015 Promissory Note
|
|
|
10.20(10)
|
Form of March 2015 Warrant
|
|
|
10.21(11)
|
Form of Note Amendment Letter Agreement
|
|
|
10.22(11)
|
Form of May 2015 Note
|
|
|
10.23(11)
|
Form of May 2015 Warrant
|
|
|
10.24(12)
|
Form of Advisory/Consulting Agreement
|
|
|
10.25(13)
|
Zolty Promissory Note
|
|
|
10.26(13)
|
Zolty Warrant
|
|
|
10.27(13)
|
Form of July 2015 Convertible Promissory Note
|
|
|
10.28(13)
|
Form of July 2015 Warrant
|
|
10.29(15)
|
Form of Bridge Note Subscription Agreement
|
|
|
10.30(15)
|
Form of Convertible Note
|
|
|
10.31(15)
|
Form of March 2016 Note
|
|
|
10.32(15)
|
Form of March 2016 Warrant
|
|
|
10.33(18)
|
Form of July 2016 Warrants
|
|
|
10.34(18)
|
Second Amendment to Research and License Agreement dated as of November 28, 2016 between the Company and Yeda
Research and Development Company Limited
|
|
|
10.35(18)
|
Third Amendment to Research and License Agreement dated as of March 29, 2018 between the Company and Yeda Research
and Development Company Limited
|
|
|
10.36(18)
|
Fourth Amendment to Research and License Agreement dated as of March 30, 2018 between the Company and Yeda Research
and Development Company Limited
|
|
|
10.37(16)
|
Convertible Note due July 27, 2016
|
|
|
10.38(17)
|
Promissory Note dated May 10, 2016
|
|
|
10.39(19) *
|
Sponsored Research Agreement dated November 28, 2018 between The University of Texas M.D. Anderson Cancer Center and Cell Source
Limited
|
|
| 10.40(19) * |
Agreement for Veto Cell Production and Clinical Trial Program dated February 19, 2019 between The University of Texas
M.D. Anderson Cancer Center and Cell Source Limited
|
|
|
16.1(1)
|
Letter from Paritz & Company, P.A.
|
|
|
21(14)
|
Subsidiaries
|
|
|
31.1 *
|
Certification of principal executive and principal financial officer pursuant to Section 302 of the
Sarbanes-Oxley Act of 2002
|
|
|
32.1 *
|
Certification of principal executive and principal financial officer pursuant to Section 906 of the Sarbanes-Oxley
Act of 2002
|
|
|
101.INS *
|
XBRL Instance Document
|
|
|
101.SCH *
|
XBRL Taxonomy Extension Schema Document
|
|
|
101.CAL *
|
XBRL Taxonomy Extension Calculation Linkbase Document
|
|
|
101.DEF *
|
XBRL Taxonomy Extension Definition Linkbase Document
|
|
|
101.LAB *
|
XBRL Taxonomy Extension Label Linkbase Document
|
|
|
101.PRE *
|
XBRL Taxonomy Extension Presentation Linkbase Document
|
| (1) |
Incorporated by reference to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on July 1, 2014.
|
| (2) |
Incorporated by reference to the Company’s Registration Statement on Form S-1 filed with the Securities and Exchange Commission on June 6, 2012.
|
| (3) |
Incorporated by reference to the Company’s Form 8-K filed with the Securities and Exchange Commission on June 26, 2014.
|
| (4) |
Incorporated by reference to the Company’s Form 8-K filed with the Securities and Exchange Commission on June 6, 2014.
|
| (5) |
Incorporated by reference to the Company’s Form 10-Q filed with the Securities and Exchange Commission on August 19, 2014.
|
| (6) |
Incorporated by reference to the Company’s Registration Statement on Form S-1 filed with the Securities and Exchange Commission on August 8, 2014.
|
| (7) |
Incorporated by reference to the Company’s Registration Statement Form S-1/A filed with the Securities and Exchange Commission on September 23, 2014.
|
| (8) |
Incorporated by reference to the Company’s Form 8-K filed with the Securities and Exchange Commission on June 30, 2014.
|
| (9) |
Incorporated by reference to the Company’s Form 8-K filed with the Securities and Exchange Commission on December 2, 2014.
|
| (10) |
Incorporated by reference to the Company’s Form 8-K filed with the Securities and Exchange Commission on April 1, 2015.
|
| (11) |
Incorporated by reference to the Company’s Form 8-K filed with the Securities and Exchange Commission on June 3, 2015.
|
| (12) |
Incorporated by reference to the Company’s Form 8-K filed with the Securities and Exchange Commission on June 10, 2015.
|
| (13) |
Incorporated by reference to the Company’s Form 8-K filed with the Securities and Exchange Commission on July 28, 2015.
|
| (14) |
Incorporated by reference to the Company’s Form 10-K filed with the Securities and Exchange Commission on March 13, 2015.
|
| (15) |
Incorporated by reference to the Company’s Form 10-K filed with the Securities and Exchange Commission on April 14, 2016.
|
| (16) |
Incorporated by reference to the Company’s Form 10-Q filed with the Securities and Exchange Commission on May 13, 2016.
|
| (17) |
Incorporated by reference to the Company’s Form 10-Q filed with the Securities and Exchange Commission on August 15, 2016.
|
|
(18)
(19)
|
Incorporated by reference to the Company’s Form 10-K filed with the Securities and Exchange Commission on July 25, 2018.
Certain portions of this exhibit have been omitted based upon a request for confidential treatment. The non-public information has been filed separately with
the Secrities and Exchange Commission.
|
| * |
Filed Herewith
|
|
CELL SOURCE, INC.
|
|||
|
Dated: March 29, 2019
|
By:
|
/s/ Itamar Shimrat
|
|
|
Name:
|
Itamar Shimrat
|
||
|
Title:
|
Chief Executive Officer and
|
||
|
Chief Financial Officer
|
|||
|
(Principal Executive, Financial and Accounting Officer)
|
|||
|
SIGNATURE
|
TITLE
|
DATE
|
|||
|
By:
|
/s/ Dennis Brown |
Chairman
|
March 29,
2019
|
||
|
Dennis Brown
|
|||||
|
By:
|
/s/ Itamar Shimrat |
Chief Executive Officer, Chief Financial Officer and Director
|
March 29,
2019
|
||
|
Itamar Shimrat
|
(Principal Executive, Financial and Accounting Officer)
|
||||
|
By:
|
/s/ Ben Friedman |
Director
|
March 29,
2019
|
||
|
Ben Friedman
|
|||||
|
By:
|
/s/ Yoram Drucker |
Director
|
March 29,
2019
|
||
|
Yoram Drucker
|
|||||
|
By:
|
/s/ David Zolty |
Director
|
March 29,
2019
|
||
|
David Zolty
|
|||||
|
Page
|
|
|
Report of Independent Registered Public Accounting Firm
|
F-1
|
|
Consolidated Balance Sheets as of December 31, 2017, 2018 and 2017
|
F-2
|
|
Consolidated Statements of Operations for the Years Ended December 31, 2018 and 2017
|
F-3
|
|
Consolidated Statements of Stockholders' Deficiency for the Years Ended December 31, 2018
and 2017
|
F-4 |
|
Consolidated Statements of Cash Flows for the Years Ended December 31, 2018 and 2017
|
F-5
|
|
Notes to Consolidated Financial Statements
|
F-6
|
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Cell Source, Inc. (the “Company”) as of December 31, 2018 and 2017, the related consolidated statements of operations, stockholders’ deficiency and cash flows for each of the two years in the period ended December 31, 2018, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2018 and 2017, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2018, in conformity with accounting principles generally accepted in the United States of America.
Explanatory Paragraph – Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As more fully described in Note 2, the Company has a significant working capital deficiency, has incurred significant losses and needs to raise additional funds to meet its obligations and sustain its operations. These conditions raise substantial doubt about the Company's ability to continue as a going concern. Management's plans in regard to these matters are also described in Note 2. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) ("PCAOB") and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
|
/s/ Marcum llp
|
|
December 31,
|
||||||||
|
2018
|
2017
|
|||||||
|
Assets
|
||||||||
|
Current Assets:
|
||||||||
|
Cash
|
$
|
18,934
|
$
|
371,048
|
||||
|
Prepaid expenses
|
38,926
|
124,693
|
||||||
|
Other current assets
|
7,932
|
35,936
|
||||||
|
Total Assets
|
$
|
65,792
|
$
|
531,677
|
||||
|
Liabilities and Stockholders' Deficiency
|
||||||||
|
Current Liabilities:
|
||||||||
|
Accounts payable
|
$
|
277,786
|
$
|
201,824
|
||||
|
Accrued expenses
|
532,790
|
749,244
|
||||||
|
Accrued expenses - related party
|
72,000
|
72,000
|
||||||
|
Accrued interest
|
345,948
|
248,746
|
||||||
|
Accrued interest - related parties
|
27,559
|
13,310
|
||||||
|
Accrued compensation
|
587,734
|
626,758
|
||||||
|
Accrued compensation - related party
|
55,083
|
19,262
|
||||||
|
Advances payable
|
100,000
|
100,000
|
||||||
|
Advances payable - related party
|
100,000
|
-
|
||||||
|
Notes
payable, net of debt discount of $0 and $89,326, as of December 31, 2018 and 2017, respectively
|
1,463,000
|
1,173,674
|
||||||
|
Notes payable - related parties
|
150,000
|
150,000
|
||||||
|
Convertible notes payable, net of debt discount of $0 and $34,173 as of December 31, 2018 and 2017, respectively
|
835,000
|
800,827
|
||||||
|
Convertible notes payable - related parties, net of debt discount of $0 and $28,356 as of December 31, 2018
and
2017, respectively
|
225,000
|
196,644
|
||||||
|
Derivative liabilities
|
200,500
|
628,200
|
||||||
|
Accrued dividend payable
|
13,563
|
108,562
|
||||||
|
Total Liabilities
|
4,985,963
|
5,089,051
|
||||||
|
Commitments and contingencies (Note 11)
|
-
|
-
|
||||||
|
Stockholders' Deficiency:
|
||||||||
|
Convertible Preferred Stock, $0.001 par value, 10,000,000 shares authorized; Series A Convertible
Preferred Stock, 1,335,000 shares
designated, 860,291 and 643,790 shares issued and outstanding as of December 31, 2018 and 2017,
respectively; liquidation
preference of $6,465,745 and $4,936,987 as of December 31, 2018 and 2017, respectively
|
860
|
644
|
||||||
|
Common Stock, $0.001 par value, 200,000,000 shares authorized, 26,077,611 and 25,349,236 shares issued
and outstanding as of
December 31, 2018 and 2017, respectively
|
26,078
|
25,349
|
||||||
|
Additional paid-in capital
|
11,723,224
|
9,969,520
|
||||||
|
Accumulated deficit
|
(16,670,333
|
)
|
(14,552,887
|
)
|
||||
|
Total Stockholders' Deficiency
|
(4,920,171
|
)
|
(4,557,374
|
)
|
||||
|
Total Liabilities and Stockholders' Deficiency
|
$
|
65,792
|
$
|
531,677
|
||||
|
For the Years Ended December 31,
|
||||||||
|
2018
|
2017
|
|||||||
|
Operating Expenses:
|
||||||||
|
Research and development
|
$
|
274,937
|
$
|
637,318
|
||||
|
Research and development - related party
|
450,151
|
839,538
|
||||||
|
General and administrative
|
1,281,055
|
815,947
|
||||||
|
Total Operating Expenses
|
2,006,143
|
2,292,803
|
||||||
|
Loss From Operations
|
(2,006,143
|
)
|
(2,292,803
|
)
|
||||
|
Other (Expense) Income:
|
||||||||
|
Interest expense
|
(219,122
|
)
|
(174,970
|
)
|
||||
|
Interest expense - related parties
|
(49,875
|
)
|
(3,000
|
)
|
||||
|
Amortization of debt discount
|
(181,299
|
)
|
(389,218
|
)
|
||||
|
Amortization of debt discount - related parties
|
(28,356
|
)
|
(48,944
|
)
|
||||
|
Change in fair value of derivative liabilities
|
485,500
|
590,173
|
||||||
|
Gain on exchange of accrued liabilities for warrants
|
73,100
|
-
|
||||||
|
Loss on exchange of notes payable for Series A Convertible Preferred Stock
|
(191,251
|
)
|
(725,355
|
)
|
||||
|
Loss on exchange of warrants for common stock
|
-
|
(38,393
|
)
|
|||||
|
Total Other Expense
|
(111,303
|
)
|
(789,707
|
)
|
||||
|
Net Loss
|
(2,117,446
|
)
|
(3,082,510
|
)
|
||||
|
Dividend attributable to Series A Convertible Preferred Stockholders
|
(451,283
|
)
|
(240,559
|
)
|
||||
|
Net Loss Applicable to Common Stockholders
|
$
|
(2,568,729
|
)
|
$
|
(3,323,069
|
)
|
||
|
Net Loss Per Common Share - Basic and Diluted
|
$
|
(0.09
|
)
|
$
|
(0.12
|
)
|
||
|
Weighted Average Common Shares Outstanding -
|
||||||||
|
Basic and Diluted
|
27,549,867
|
26,774,860
|
||||||
The accompanying notes are an integral part of these consolidated financial statements.
| Series A Convertible | Total | |||||||||||||||||||||||||||
|
Preferred Stock
|
Common Stock
|
Additional
|
Accumulated
|
Stockholders'
|
||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Paid-In Capital
|
Deficit
|
Deficiency
|
||||||||||||||||||||||
|
Balance, December 31, 2016
|
-
|
$
|
-
|
24,679,256
|
$
|
24,679
|
$
|
5,202,749
|
$
|
(11,470,377
|
)
|
$
|
(6,242,949
|
)
|
||||||||||||||
|
Issuance of Series A Convertible
Preferred
Stock for cash, net of
offering expenses of $54,543
|
306,759
|
307
|
-
|
-
|
2,240,277
|
-
|
2,240,584
|
|||||||||||||||||||||
|
Issuance of Series A Convertible
Preferred
Stock in exchange for
advances payable
|
23,834
|
24
|
-
|
-
|
178,722
|
-
|
178,746
|
|||||||||||||||||||||
|
Issuance of Series A Convertible
Preferred
Stock in exchange for
notes payable
|
281,697
|
282
|
-
|
-
|
2,112,452
|
-
|
2,112,734
|
|||||||||||||||||||||
|
Issuance of Series
A Convertible
Preferred
Stock as debt discount
inconnection
with the issuance
of
notes payable
|
24,000
|
24
|
-
|
-
|
119,976
|
-
|
120,000
|
|||||||||||||||||||||
|
Issuance of
Series A Convertible
Preferred and Common Stock in
connection with the extension
of notes payable
|
7,500
|
7
|
243,750
|
244
|
116,937
|
-
|
117,188
|
|||||||||||||||||||||
|
Series A Convertible Preferred Stock dividends:
|
||||||||||||||||||||||||||||
|
Accrual of earned dividends
|
-
|
-
|
-
|
-
|
(240,559
|
)
|
-
|
(240,559
|
)
|
|||||||||||||||||||
|
Payment of dividends in-kind
|
-
|
-
|
176,230
|
176
|
131,997
|
-
|
132,173
|
|||||||||||||||||||||
|
Issuance of Common
Stock in
exchange forsurrender of
warrants
|
-
|
-
|
250,000
|
250
|
62,250
|
-
|
62,500
|
|||||||||||||||||||||
|
Stock-based compensation:
|
||||||||||||||||||||||||||||
|
Warrants
|
-
|
-
|
-
|
-
|
44,719
|
-
|
44,719
|
|||||||||||||||||||||
|
Net loss
|
-
|
-
|
-
|
-
|
-
|
(3,082,510
|
)
|
(3,082,510
|
)
|
|||||||||||||||||||
|
Balance, December 31, 2017
|
643,790
|
$
|
644
|
25,349,236
|
$
|
25,349
|
$
|
9,969,520
|
$
|
(14,552,887
|
)
|
$
|
(4,557,374
|
)
|
||||||||||||||
|
Issuance of Series
A Convertible
Preferred Stock for cash
|
140,001
|
140
|
-
|
-
|
1,049,860
|
-
|
1,050,000
|
|||||||||||||||||||||
|
Issuance of Series A Convertible
Preferred
Stock in exchange
for
notes payable
|
76,500
|
76
|
-
|
-
|
573,675
|
-
|
573,751
|
|||||||||||||||||||||
| Series A Convertible Preferred Stock
dividends: |
||||||||||||||||||||||||||||
|
Accrual of earned dividends
|
-
|
-
|
-
|
-
|
(451,283
|
)
|
-
|
(451,283
|
)
|
|||||||||||||||||||
|
Payment of dividends in-kind
|
-
|
-
|
728,375
|
729
|
545,552
|
-
|
546,281
|
|||||||||||||||||||||
|
Issuance of
warrants in satisfaction of
accrued liabilities
|
-
|
-
|
-
|
-
|
35,900
|
-
|
35,900
|
|||||||||||||||||||||
|
Net loss
|
-
|
-
|
-
|
-
|
-
|
(2,117,446
|
)
|
(2,117,446
|
)
|
|||||||||||||||||||
|
Balance, December 31, 2018
|
860,291
|
$
|
860
|
26,077,611
|
$
|
26,078
|
$
|
11,723,224
|
$
|
(16,670,333
|
)
|
$
|
(4,920,171
|
)
|
||||||||||||||
|
For The Years Ended December 31,
|
||||||||
|
2018
|
2017
|
|||||||
|
Cash Flows From Operating Activities:
|
||||||||
|
Net loss
|
$
|
(2,117,446
|
)
|
$
|
(3,082,510
|
)
|
||
|
Adjustments
to reconcile net loss to net cash used in operating activities:
|
||||||||
|
Change in fair value of derivative liabilities
|
(485,500
|
)
|
(590,173
|
)
|
||||
|
Amortization of debt discount
|
209,655
|
438,162
|
||||||
|
Loss on exchange of notes payable for Series A Convertible Preferred Stock
|
191,251
|
725,355
|
||||||
|
Loss on exchange of warrants for common shares
|
-
|
38,393
|
||||||
|
Gain on exchange of accrued liabilities for warrants
|
(73,100
|
)
|
-
|
|||||
|
Depreciation
|
-
|
407
|
||||||
|
Stock-based compensation:
|
||||||||
|
Common stock
|
(22,500
|
)
|
-
|
|||||
|
Warrants
|
3,385
|
44,719
|
||||||
|
Changes in operating assets and liabilities:
|
||||||||
|
Prepaid expenses
|
85,767
|
(23,105
|
)
|
|||||
|
Other current assets
|
28,004
|
34,394
|
||||||
|
Accounts payable
|
75,962
|
(17,270
|
)
|
|||||
|
Accrued expenses
|
(24,955
|
)
|
(134,671
|
)
|
||||
|
Accrued interest
|
97,202
|
155,804
|
||||||
|
Accrued interest - related parties
|
14,249
|
3,000
|
||||||
|
Accrued compensation
|
15,912
|
119,681
|
||||||
|
Net Cash Used In Operating Activities
|
(2,002,114
|
)
|
(2,287,814
|
)
|
||||
|
Cash Flows From Financing Activities:
|
||||||||
|
Proceeds from issuance of notes payable
|
500,000
|
135,000
|
||||||
|
Proceeds from issuance of notes payable - related party
|
-
|
225,000
|
||||||
|
Proceeds from issuance of Series A Convertible Preferred Stock
|
1,050,000
|
2,295,127
|
||||||
|
Proceeds from cash advance - related party
|
100,000
|
-
|
||||||
|
Net Cash Provided By Financing Activities
|
1,650,000
|
2,655,127
|
||||||
|
Net (Decrease) Increase In Cash
|
(352,114
|
)
|
367,313
|
|||||
|
Cash - Beginning of Period
|
371,048
|
3,735
|
||||||
|
Cash - End of Period
|
$
|
18,934
|
$
|
371,048
|
||||
|
Supplemental Disclosures of Cash Flow Information:
|
||||||||
|
Cash paid for:
|
||||||||
|
Interest
|
$
|
18,030
|
$
|
-
|
||||
|
Non-cash investing and financing activities:
|
||||||||
|
Preferred
stock issued in exchange for notes and advances payable
|
$
|
573,751
|
$
|
2,291,480
|
||||
|
Reduction
of additional paid-in capital for public offering issuance costs that were previously paid
|
$
|
-
|
$
|
54,543
|
||||
|
Accrual of earned preferred stock dividends
|
$
|
451,283
|
$
|
240,559
|
||||
|
Common stock issued in connection with exchange of warrants
|
$
|
-
|
$
|
62,500
|
||||
|
Common
stock issued in connection with payment of Series A Convertible Preferred Stock dividends in-kind
|
$
|
546,281
|
$
|
132,173
|
||||
|
Stock
issued in connection with issuances and extensions of notes payable
|
$
|
-
|
$
|
237,188
|
||||
|
Warrants
issued in satisfaction of accrued liabilities
|
$
|
35,900
|
$
|
-
|
||||
|
Warrants
and conversion options issued in connection with issuance and extension of notes payable
|
$
|
57,800
|
$
|
67,080
|
||||
The accompanying notes are an integral part of these consolidated financial statements.
|
December 31,
|
||||||||
|
2018
|
2017
|
|||||||
|
Warrants
|
11,414,818
|
11,615,481
|
||||||
|
Convertible notes
|
1,564,110
|
1,530,433
|
||||||
|
Convertible preferred stock
|
8,602,910
|
6,437,900
|
||||||
|
Total
|
21,581,838
|
19,583,814
|
||||||
|
|
Quoted Prices
|
|||||||||||||||
|
In Active
|
Significant
|
|||||||||||||||
|
Markets for
|
Other
|
Significant
|
||||||||||||||
|
Identical
|
Observable
|
Unobservable
|
||||||||||||||
|
Liabilities
|
Inputs
|
Inputs
|
||||||||||||||
|
Total
|
(Level 1)
|
(Level 2)
|
(Level 3)
|
|||||||||||||
|
Accrued compensation - common stock
|
$
|
37,500
|
$
|
-
|
$
|
-
|
$
|
37,500
|
||||||||
|
Accrued compensation - warrants
|
24,741
|
-
|
-
|
24,741
|
||||||||||||
|
Accrued compensation - warrants - related party
|
55,083
|
-
|
-
|
55,083
|
||||||||||||
|
Derivative liabilities
|
200,500
|
-
|
-
|
200,500
|
||||||||||||
|
Balance - December 31, 2018
|
$
|
317,824
|
$
|
-
|
$
|
-
|
$
|
317,824
|
||||||||
|
Accrued compensation - common stock
|
$
|
60,000
|
$
|
-
|
$
|
-
|
$
|
60,000
|
||||||||
|
Accrued compensation - warrants
|
19,262
|
-
|
-
|
19,262
|
||||||||||||
|
Derivative liabilities
|
628,200
|
-
|
-
|
628,200
|
||||||||||||
|
Balance - December 31, 2017
|
$
|
707,462
|
$
|
-
|
$
|
-
|
$
|
707,462
|
||||||||
|
For the Years Ended
December 31,
|
||||||||
|
2018
|
2017
|
|||||||
|
Risk-free interest rate
|
1.73% - 2.91
|
%
|
1.04% - 2.09
|
%
|
||||
|
Expected term (years)
|
0.08 - 5.00
|
0.25 - 4.00
|
||||||
|
Expected volatility
|
110
|
%
|
110
|
%
|
||||
|
Expected dividends
|
0.00
|
%
|
0.00
|
%
|
||||
|
Accrued
|
Derivative
|
|||||||||||
|
Compensation
|
Liability
|
Total
|
||||||||||
|
Balance - December 31, 2016
|
$
|
79,178
|
$
|
1,175,400
|
$
|
1,254,578
|
||||||
|
Issuance of warrants and conversion options
|
-
|
67,080
|
67,080
|
|||||||||
|
Exchange of warrant for common stock
|
-
|
(24,107
|
)
|
(24,107
|
)
|
|||||||
|
Change in fair value
|
85
|
(590,173
|
)
|
(590,088
|
)
|
|||||||
|
Balance - December 31, 2017
|
79,262
|
628,200
|
707,462
|
|||||||||
|
Issuance of warrants
|
-
|
57,800
|
57,800
|
|||||||||
|
Accrued compensation - warrants
|
24,300
|
-
|
24,300
|
|||||||||
|
Accrued compensation - warrants - related party
|
36,000
|
-
|
36,000
|
|||||||||
|
Change in fair value
|
(22,238
|
)
|
(485,500
|
)
|
(507,738
|
)
|
||||||
|
Balance - December 31, 2018
|
$
|
117,324
|
$
|
200,500
|
$
|
317,824
|
||||||
As of December 31, 2018 and 2017, the Company had an obligation to issue 150,000 shares of common stock to a service provider. The shares had a fair value of $37,500 and $60,000, respectively, which was a component of accrued compensation on the consolidated balance sheets and the reduction in fair value of $22,500 was included within stock-based compensation expense during the year ended December 31, 2018.
|
December 31,
|
||||||||
|
2018
|
2017
|
|||||||
|
Accrued research and development
|
$
|
302,433
|
$
|
430,504
|
||||
|
Accrued legal fees
|
122,351
|
119,216
|
||||||
|
Accrued other professional fees
|
70,399
|
50,731
|
||||||
|
Accrued director compensation
|
12,000
|
12,000
|
||||||
|
Accrued Scientific Advisory Board compensation
|
-
|
109,000
|
||||||
|
Other accrued expenses
|
25,607
|
27,793
|
||||||
|
Total accrued expenses
|
$
|
532,790
|
$
|
749,244
|
||||
|
December 31,
|
||||||||
|
2018
|
2017
|
|||||||
|
Witholding tax
|
$
|
116,401
|
$
|
147,082
|
||||
|
Social security
|
34,825
|
54,218
|
||||||
|
Stock-based compensation - common stock
|
37,500
|
60,000
|
||||||
|
Stock-based compensation - warrants
|
24,741
|
-
|
||||||
|
Pension insurance
|
117,897
|
97,221
|
||||||
|
Accrued payroll
|
98,505
|
132,366
|
||||||
|
Vacation
|
46,808
|
38,059
|
||||||
|
Severance
|
111,057
|
97,812
|
||||||
|
$
|
587,734
|
$
|
626,758
|
|||||
|
December 31,
|
|||||||||||
|
2018
|
2017
|
||||||||||
|
i)
|
|
Notes issued on March 26, 2015
|
$
|
500,000
|
$
|
500,000
|
|||||
| ii) |
Note issued on May 15, 2015, net
of debt discount of $0 and $66,532 as of
December 31, 2018 and 2017, respectively
|
250,000
|
183,468
|
||||||||
|
iii)
|
Notes issued on March 8, 2016
|
-
|
300,000
|
||||||||
|
iv)
|
Note issued on May 10, 2016
|
53,000
|
53,000
|
||||||||
| v) |
Notes issued on various dates from July 20, 2016 to October 13, 2016, net of
debt discounts
of $0 and $22,794 as of December 31, 2018 and 2017, respectively
|
160,000
|
137,206
|
||||||||
|
vi)
|
Note issued on December 5, 2017
|
-
|
-
|
||||||||
|
vii)
|
Notes issued on February 21, 2018
|
400,000
|
-
|
||||||||
|
viii)
|
Note issued on February 26, 2018
|
100,000
|
-
|
||||||||
|
$
|
1,463,000
|
$
|
1,173,674
|
||||||||
| i) |
As of December 31, 2018 and
through the date of this filing, the Company is not in compliance with the terms of these notes due to non-payment of principal. On December 21, 2018, the holder of one of those notes issued a notice of default and demanded
repayment. See Note 11, Commitments and Contingencies - Litigation, for
additional disclosure regarding this matter. The holder of the other note has not issued a notice of default.
|
| ii) |
On December 28, 2017, the maturity date of the note was extended from its original maturity date of
September 15, 2015 to March 31, 2018. In connection with that extension, the Company issued 125,000 shares of common stock. Those shares were valued at a total of $31,250 on the date of issuance and was recorded as a debt
discount. Such discount was amortized to expense over the termof the extension period.
|
| iii) |
On March 8, 2017, the holder of one of these notes with a principal amount of $300,000 and accrued interest of
$30,000 exchanged that note for 66,000 shares of the Company's Series A Convertible Preferred Stock. The value of the shares issued exceeded the carrying value of the debt and accrued interest and, as more fully discussed in Note 10, Stockholders’ Deficiency - Series A Convertible Preferred Stock, the difference of $165,000 was recorded in the
consolidated statements of operations as a loss on exchange of notes payable for Series A Convertible Preferred Stock.
|
|
On December 12, 2018, the holder of the remaining note with a principal amount of $300,000 and accrued interest of
$82,500 exchanged that note for 76,500 shares of the Company's Series A Convertible Preferred Stock. The value of the shares issued exceeded the carrying value of the debt and accrued interest and, as more fully discussed in Note
10, Stockholders’ Deficiency - Series A Convertible Preferred Stock, this difference of $191,251 was recorded in the
consolidated statements of operations as a loss on exchange of notes payable for Series A Convertible Preferred Stock.
|
| v) |
On March 8, 2017, the holders of three of these notes with total principal amount of $300,000 and accrued interest of
$19,167 exchanged those notes for 63,833 shares of the Company's Series A Convertible Preferred Stock. The value of the shares issued exceeded the carrying value of the debt and accrued interest and, as more fully discussed in Note
10, Stockholders’ Deficiency - Series A Convertible Preferred Stock, this difference of $159,585 was recorded in the
consolidated statements of operations as a loss on exchange of notes payable for Series A Convertible Preferred Stock.
|
| vi) |
On December 5, 2017, the Company issued a note payable in the aggregate principal amount of $100,000 and a warrant
for the purchase of a total of 50,000 shares of common stock at $0.75 per share for a period of five years. The note did not accrue interest, matured on December 15, 2017 and was repaid on that date. The warrant was 100% vested upon
issuance, valued at $8,200 on the date of issuance, and recorded as a debt discount. The discount was amortized to expense over the term of the note.
|
| vii) |
On February 21, 2018, the Company issued two notes payable in the aggregate principal amount of $400,000 and warrants
for the purchase of a total of 240,000 shares of common stock at $0.75 per share for a period of five years. These notes did not accrue interest, matured on May 21, 2018, and had an effective interest rate of 40% per annum. The
warrants were 100% vested upon issuance, valued at $39,700 on the date of issuance, and recorded as a debt discount. The discount was amortized to expense over the term of those notes.
|
| viii) |
On February 26, 2018, the Company issued a note payable in the aggregate principal amount of $100,000 and a warrant
for the purchase of a total of 60,000 shares of common stock at $0.75 per share for a period of five years. The note did not accrue interest, matured on May 26, 2018, and had an effective interest rate of 40% per annum. The warrant
was 100% vested upon issuance, valued at $9,900 on the date of issuance, and recorded as a debt discount. The discount was amortized to expense over the term of the note.
|
|
December 31,
|
|||||||||||
|
2018
|
2017
|
||||||||||
|
i)
|
|
Note issued on November 26, 2014
|
$
|
50,000
|
$
|
50,000
|
|||||
|
ii)
|
Note issued on July 20, 2015
|
100,000
|
100,000
|
||||||||
|
$
|
150,000
|
$
|
150,000
|
||||||||
|
December 31,
|
|||||||||||
|
2018
|
2017
|
||||||||||
| i) |
Convertible
notes issued on July 24, 2015, net of debt discounts of $0 and $17,159 as of December 31, 2018 and 2017, respectively
|
$
|
145,000
|
$
|
127,841
|
||||||
|
ii)
|
Convertible notes issued on October 7, 2015
|
265,000
|
265,000
|
||||||||
|
iii)
|
Convertible notes issued on November 9 and 17, 2015
|
-
|
-
|
||||||||
| iv) |
Convertible notes issued on
various dates from January 6, 2016 to March 15, 2016
|
290,000
|
290,000
|
||||||||
|
v)
|
|
Convertible note issued on January 14, 2016
|
-
|
-
|
|||||||
| vi) |
Convertible
notes issued on May 18, 2017, net of debt discounts of $0 and $17,014 as of December 31, 2018 and 2017, respectively
|
135,000
|
117,986
|
||||||||
|
$
|
835,000
|
$
|
800,827
|
||||||||
| i) |
On August 15, 2017, the maturity dates of two of these notes were extended to February 15, 2018. In connection with
that extension, the Company issued 93,750 common shares. The shares were valued at $23,438 on the date of issuance and recorded as a debt discount. These discounts were amortized to expense over the remaining term of the notes.
|
| iii) |
On May 18 and 24, 2017, the holders exchanged these notes with total principal and interest of $332,500 and $49,875,
respectively, for 76,475 shares of Series A Convertible Preferred Stock. The value of the shares issued exceeded the carrying value of the debt and accrued interest and, as more fully discussed in Note 10 - Stockholders’ Deficiency - Series A Convertible Preferred Stock, this difference of $191,190 was recorded in the consolidated statements of
operations as a loss on exchange of notes payable for Series A Convertible Preferred Stock.
|
| iv) |
On October 3, 2017, the holder of one of these notes with the principal amount of $100,000 and accrued interest of
$15,417 exchanged that note for 15,389 shares of the Company's Series A Convertible Preferred Stock. There was no gain or loss in connection with this exchange.
|
| v) |
On January 19, 2017, the holder exchanged the note with total principal and interest of $250,000 and $50,000,
respectively, for 60,000 shares of Series A Convertible Preferred Stock. The value of the shares issued exceeded the carrying value of the debt and accrued interest and, as more fully discussed in Note 10, Stockholders’ Deficiency - Series A Convertible Preferred Stock, this difference of $150,000 was recorded in the consolidated statements of operations as a loss on exchange of notes payable for Series A
Convertible Preferred Stock.
|
| vi) |
On May 18, 2017, the Company issued three convertible notes payable in the total principal amount of $135,000 and a
total of 9,000 shares of Series A Convertible Preferred Stock. These notes did not accrue interest and matured on May 18, 2018. Proceeds from the issuance of such notes were allocated proportionately to the value of the notes and
the shares. Consequently, those shares were allocated $45,000 on the date of issuance and recorded as debt discounts. Such discounts were amortized to expense over the term of those notes.
|
|
December 31,
|
||||||||
|
2018
|
2017
|
|||||||
|
Convertible notes issued on May 18, 2017, net of debt discounts
|
||||||||
|
of $0 and $28,356 as of December 31, 2018 and 2017, respectively
|
$
|
225,000
|
$
|
196,644
|
||||
|
For the Years Ended
December 31,
|
||||||||
|
2018
|
2017
|
|||||||
|
Israel
|
$
|
(983,695
|
) |
|
$
|
(1,793,483
|
)
|
|
|
United States
|
(1,133,751
|
) |
(1,289,027
|
) | ||||
|
Income before income taxes
|
$
|
(2,117,446
|
) |
$
|
(3,082,510
|
)
|
||
|
For the Years Ended
December 31,
|
||||||||
|
2018
|
2017
|
|||||||
| Net operating loss carryforwards | $ |
4,536,000 |
$ |
3,712,000 | ||||
| Foreign deferred research and development costs | 224,000 |
331,000 | ||||||
| Stocked-based compensation expense | 26,000 |
26,000 | ||||||
|
Deferred tax assets
|
4,786,000 |
4,069,000
|
||||||
|
Valuation allowance
|
( 4,786,000 | ) |
(4,069,000
|
)
|
||||
|
Deferred tax assets, net
|
$
|
-
|
$
|
-
|
||||
|
For the Years Ended
December 31,
|
||||||||
|
2018
|
2017
|
|||||||
|
Current
|
||||||||
|
Foreign
|
$
|
-
|
$
|
-
|
||||
|
Federal
|
-
|
-
|
||||||
|
U.S State and local
|
-
|
-
|
||||||
|
Deferred
|
||||||||
|
Foreign
|
(226,000 |
) |
(357,000
|
)
|
||||
|
Federal
|
(403,587 |
) |
110,000
|
|||||
|
U.S State and local
|
(87,413 |
) |
(161,000
|
)
|
||||
| (717,000 |
) |
(408,000
|
)
|
|||||
|
Change in valaution allowance
|
717,000 |
408,000
|
||||||
|
Income tax provision (benefit)
|
$
|
-
|
$
|
-
|
||||
|
For The Years Ended
|
||||||||
|
December 31,
|
||||||||
|
2018
|
2017
|
|||||||
|
Expected federal statutory rate
|
(21.0
|
%)
|
(34.0
|
%)
|
||||
|
State and local taxes, net of federal tax benefit
|
(11.3
|
%)
|
(9.4
|
%)
|
||||
|
Statutory rate differential - domestic vs. foreign
|
4.3
|
%
|
11.3
|
%
|
||||
|
Permanent differences - change in fair value of derivatives
|
(7.4
|
%)
|
(8.3
|
%)
|
||||
|
Permanent differences - loss on exchange of notes for preferred stock
|
2.9
|
%
|
10.2
|
%
|
||||
|
Permanent differences - tax rate changes
|
-
|
17.4
|
%
|
|||||
|
Permanent differences - other
|
(0.2
|
%)
|
1.2
|
%
|
||||
|
Other
|
(1.2
|
%)
|
(1.6
|
%)
|
||||
|
Change in valuation allowance
|
33.9
|
%
|
13.2
|
%
|
||||
|
Income tax provision (benefit)
|
0.0
|
%
|
0.0
|
%
|
||||
The Company assesses the likelihood that deferred tax assets will be realized. ASC 740, “Income Taxes” requires that a valuation allowance be established when it is “more likely than not” that all, or a portion of, deferred tax assets will not be realized. A review of all available positive and negative evidence needs to be considered, including the scheduled reversal of deferred tax liabilities, projected future taxable income, and tax planning strategies. After consideration of all the information available, management believes that uncertainty exists with respect to future realization of its deferred tax assets and has, therefore, established a full valuation allowance as of December 31, 2018 and 2017. For the years ended December 31, 2018 and 2017, the increase in the valuation allowance was approximately $717,000 and $408,000, respectively.
|
·
|
93,750 shares of common stock valued at $0.25 per share in connection with the extension of
convertible notes payable, as more fully described in Note 8(c)(i), Notes Payable;
|
|
·
|
25,000 shares of common stock valued at $0.25 per share in connection with the extension of a note
payable, as more fully described in Note 8(a)(v), Notes Payable; and
|
|
·
|
125,000 shares of common stock valued at $0.25 per share in connection with the extension of a
notes payable, as more fully described in Note 8(a)(ii), Notes Payable.
|
|
Weighted
|
||||||||||||||||
|
Weighted
|
Average
|
|||||||||||||||
|
Average
|
Remaining
|
|||||||||||||||
|
Number of
|
Exercise
|
Life
|
Intrinsic
|
|||||||||||||
|
Warrants
|
Price
|
In Years
|
Value
|
|||||||||||||
|
Outstanding, December 31, 2016
|
13,909,316
|
$
|
0.64
|
|||||||||||||
|
Granted
|
50,000
|
0.75 |
||||||||||||||
|
Exercised
|
-
|
-
|
||||||||||||||
|
Exchanged
|
(250,000
|
)
|
0.75
|
|||||||||||||
|
Forfeited
|
-
|
-
|
||||||||||||||
|
Outstanding, December 31, 2017
|
13,709,316
|
$
|
0.64
|
|||||||||||||
|
Granted
|
518,000
|
0.75
|
||||||||||||||
|
Exercised
|
-
|
-
|
||||||||||||||
|
Exchanged
|
-
|
-
|
||||||||||||||
|
Expired
|
(768,663
|
)
|
0.75
|
|||||||||||||
|
Outstanding, December 31, 2018
|
13,458,653
|
$
|
0.64
|
0.9
|
$
|
508,915
|
||||||||||
|
Exercisable, December 31, 2018
|
13,458,653
|
$
|
0.64
|
0.9
|
$
|
508,915
|
||||||||||
|
Warrants Outstanding
|
Warrants Exercisable
|
|||||||||||||
|
Weighted
|
||||||||||||||
|
Outstanding
|
Average
|
Exercisable
|
||||||||||||
|
Exercise
|
Number of
|
Remaining Life
|
Number of
|
|||||||||||
|
Price
|
Warrants
|
In Years
|
Warrants
|
|||||||||||
|
$
|
0.001
|
2,043,835
|
1.9
|
2,043,835
|
||||||||||
|
$
|
0.750
|
11,414,818
|
0.7
|
11,414,818
|
||||||||||
|
13,458,653
|
0.9
|
13,458,653
|
||||||||||||
|
Three Months Ending:
|
Total
|
|||
|
|
||||
|
March 31, 2018
|
$
|
200,000
|
||
|
June 30, 2018
|
150,000
|
|||
|
September 30, 2018
|
50,000
|
|||
|
December 31, 2018
|
50,000
|
|||
|
March 31, 2019
|
25,000
|
|||
|
June 30, 2019
|
25,000
|
|||
|
|
$
|
500,000
|
||
|
-
|
hereinafter called Cell Source –
|
| (2) |
As outlined in the initial budget , MDA undertakes to perform 4 production validation runs and a Phase I/II clincial trial using veto cells in * patients.
Based on the assessment of results in the first * patients further continuation of the study in up to * patients will be considered.
|
| (3) |
MD Anderson, under the supervision of its Principal Investigator Richard Champlin, M.D., shall perform the Program in close co-operation with Cell Source
and shall keep Cell Source informed of the Program’s progress and all major developments concerning the Program.
|
| (4) |
Upon completion of each quarter MDA shall submit a written report to Cell Source describing the results of the Program.
|
| (5) |
Cell Source represents and warrants that it exclusively licenses proprietary scientific knowledge developed at the Weizmann Institute of Science by a team headed by Prof. Yair Reisner, including without limitation, the Anti-viral central memory CD8 veto cells, from Yeda Research and Development Limited
(“Yeda”). The Program will be conducted in cooperation with Prof. Riesner.
|
| (6) |
The rights to any outcomes of the treatments performed on behalf of Cell Source,
to the extent that they may be protected by patent laws, will belong exclusively to Yeda and fall under the existing exclusive license that Cell Source has from the Weizmann Institute through its agreement with Yeda, provided,
however, MD Anderson shall have the right to use such outcomes for MD Anderson’s internal academic, research and patient care purposes.
|
| (7) |
MD Anderson and Cell Source will promptly notify each other upon identifying any aspect of the protocol for the Program or the Program results that may
adversely affect the safety, well-being, or medical care of Program subjects, or that may affect the willingness of subjects to continue participation of the Program, influence the conduct of the Program, or may alter the IRB’s approval
to continue the Program; when possible, such findings shall be submitted to MD Anderson electronically. MD Anderson shall promptly notify the IRB of any such events. When Program subject safety or medical care could be directly
affected by Program results, then notwithstanding any other provision of this Agreement, MD Anderson will send Program subjects a written communication about the results.
|
|
§ 2 – Payment
|
|
§ 3 – Confidentiality
|
|
(1)
|
Any Confidential Information that will be exchanged between the Parties, whether orally, in writing or by any
other medium, during the course of the Program under this agreement, shall be treated confidential by the receiving Party. The receiving Party shall not use such Confidential Information for any purpose other than performance of this
agreement, and shall not make available such Confidential Information to any third party.
|
|
(2)
|
For the purposes of this agreement, “Confidential Information” shall mean any information that is expressly
marked as confidential by appropriate means, or clearly identifiable as being confidential by its nature. However, “Confidential Information” shall not include any information that
|
|
-
|
was known to the receiving Party prior to its disclosure, or
|
|
-
|
was known to the public or was generally available prior to its disclosure, or
|
|
-
|
became known to the public or became generally available after disclosure through no
wrongful act or omission of the receiving Party, or
|
|
-
|
essentially corresponds to information that was disclosed or made available to the receiving
Party at any time by a third party, who, to the knowledge of the receiving Party, had the legal right to disclose the information to the receiving Party (it is agreed that Prof. Reisner, his team or anyone from the Weizmann Institute
deriving its information from the technology developed by Prof. Reisner while at the Weizmann Institute are not considered third party to this matter),
|
|
-
|
was developed independently by the receiving Party without knowledge of the Confidential Information,
|
|
-
|
is required to be disclosed in order to obtain the informed consent from subjects who may wish to enroll
in the Program, provided, however, that the Confidential Information will be disclosed only to the extent necessary and will not be provided in answer to unsolicited inquiries by telephone or to individuals who are not eligible Program
candidates,
|
|
-
|
is disclosed to a Program subject for the safety or well-being of the Program subject.
|
|
(3)
|
Notwithstanding the foregoing, each receiving Party will be permitted to disclose Confidential Information as
required by law or regulation, provided to the extent practicable, prior to such disclosure, the receiving Party will provide reasonable advance notice to the disclosing Party to allow the disclosing Party an opportunity to obtain a
protective order.
|
|
(4)
|
Each Party shall ensure that its respective employees will be bound by these confidentiality obligations.
|
|
§ 4 – Publications
|
|
(1)
|
Notwithstanding anything to the contrary herein, Cell Source ackowledges that MDA has the first right to
publishe the results of Program. Any publication in whatever form on the data and results of the Program by MD Anderson, shall be submitted to Cell Source in advance. Cell Source will review the manuscript and provide comments within 30
days. If Cell Source does not react within such 30 days, Cell Source shall be deemed to not have any comments on the publication. Cell Source will also have the right to publish press releases regarding the Program, provided, however, all
press releases including the name of MD Anderson requires prior written approval from MD Anderson’s Department of External Communications. All such publication and press releases shall be in accordance with generally acceptable scientific
research and/or academic standards.
|
|
(2)
|
Cell Source may request a delay of the publication for a period not to exceed sixty (60) days, or changes to
its content, only insofar as necessary to avoid undue disclosure of Cell Source’s Confidential Information, provided, in no case shall MD Anderson be required to remove any data or results of the Program. Should Cell Source or Yeda elect
to file a patent regarding a new invention that results from this Program, Cell Source may request a delay of the publication for a period not to exceed sixty (60) days if the publication discloses the content of such patent application.
|
|
§ 5 – Liability
|
|
(1)
|
MDA shall perform the Program hereunder with its usual care and on the basis of the current state of the art
in science and technology research and in accordance with all relevant laws and regulations required in the MDA’s domicile. With respect to the Results, no warranty of any kind is given, neither express nor implied, as to the Results’
fitness for any particular purpose or to the non-infringement of any third party’s rights by the Results.
|
|
(2)
|
Any liability among the contracting parties shall be limited to gross negligence and willful misconduct.
Except in cases of willful misconduct, neither party shall be liable for any remote or incidental damage or loss (such as loss of profit or loss of contract).
|
|
(3)
|
This Agreement does not obligate any of the contracting parties to provide medical treatment, except to the
extent required by applicable law, nor does this Agreement obligate either party to provide reimbursement for medical treatment if a Program subject requires medical treatment for physical illness or injury sustained as a direct result of
the treatment of such Program subject in accordance with this Agreement and the protocol for the Program.
|
|
§ 6 – Term
|
|
(1)
|
This agreement shall enter into force upon signature by both parties.
|
|
(2)
|
This agreement may not be terminated prematurely by either Party, except in case of “good cause for
termination” as per the pertinent regulations in MDA’s domicile.
|
|
§ 7 – Miscellaneous
|
|
(1)
|
This agreement may only be amended by written agreement of the Parties.
|
|
EXHIBIT B- BUDGET
Funding Agency:
|
Cell Source Limited
|
||||
|
Principal Investigator:
|
Champlin, Richard
|
||||
|
Title:
|
Role of Veto Cells in Haploidentical Transplantation for Myeloma
|
||||
|
Project Dates:
|
TBD
|
||||
|
Protocol(s)
|
2018-0221
|
||||
|
Total Patients
|
*
|
||||
|
*
|
|||||
| Year 1 | Year 2 |
Grand Total | ||||||||||
|
Total Costs
|
$
|
1,162,760.38
|
$
|
889,624.93
|
$
|
2,052,385.31
|
| Payment (USD) | ||||
| * |
$29,375.00
|
|||
| * |
$177,668.45
|
|||
| * |
$245,766.09
|
|||
| * |
$245,766.09
|
|||
| * |
$245,766.10
|
|||
|
Total
|
$944,341.73
|
|||
|
SPONSOR
|
THE UNIVERSITY OF TEXAS
|
|
M. D. ANDERSON CANCER CENTER
|
|
|
By
|
By
|
|
Name: Itamar Shimrat
|
Name: Jaime Farias
|
|
Title: Chief Executive Officer
|
Title: Assistant Director of Sponsored Programs
|
|
READ AND UNDERSTOOD BY:
|
|
|
Dr. Yair Reisner
|
|
|
Principal Investigator
|
|
Total Costs
|
Year 1
|
Year 2
|
Year 3
|
Grand Total
|
||||||||||||
|
$
|
499,673.60
|
$
|
499,703.81
|
$
|
507,974.92
|
$
|
1,507,352.33
|
|||||||||
|
YEAR 01
|
YEAR 02
|
YEAR 03
|
||||||||||
|
Quarterly payments*
|
$
|
124,918.40
|
$
|
124,925.95
|
$
|
126,993.73
|
||||||
|
Total
|
$
|
499,673.60
|
$
|
499,703.81
|
$
|
507,974.95
|
||||||
|
|
a)
|
designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed
under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is
being prepared;
|
|
|
b)
|
designed such internal control over financial reporting, or caused such internal control over financial reporting
to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting
principles;
|
|
|
c)
|
evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our
conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
|
|
|
d)
|
disclosed in this report any change in the registrant’s internal control over financial reporting that occurred
during the registrant’s most recent fiscal quarter (the registrant's fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over
financial reporting; and
|
|
|
a)
|
all significant deficiencies and material weaknesses in the design or operation of internal control over financial
reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and
|
|
|
b)
|
any fraud, whether or not material, that involves management or other employees who have a significant role in the
registrant’s internal control over financial reporting.
|
|
Dated: March 29, 2019
|
By:
|
/s/ Itamar Shimrat
|
|
Itamar Shimrat
|
||
|
Chief Executive Officer and Chief Financial Officer
|
||
|
(Principal Executive, Financial and Accounting Officer)
|
| (1) |
The Report fully complies with the requirements of section 13(a) or 15(d) of the Securities Exchange Act of 1934; and
|
| (2) |
The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
|
Serious News for Serious Traders! Try StreetInsider.com Premium Free!
You May Also Be Interested In
- Irish Residential Properties Sub-Fund 1: Form 8.3 - Irish Residential Properties REIT PLC
- Nykode Therapeutics Initiates Phase 2 Trial of VB10.16 in Second Line HPV16-Positive Cervical Cancer
- Proposed Increase in Size of Offer for Subscription and Re-Opening of Offer for Subscription to Further Applications
Create E-mail Alert Related Categories
SEC FilingsSign up for StreetInsider Free!
Receive full access to all new and archived articles, unlimited portfolio tracking, e-mail alerts, custom newswires and RSS feeds - and more!
