

Form 6-K Ascendis Pharma A/S For: Mar 31
 Tweet
Tweet Share
ShareExhibit 99.2
ASCENDIS PHARMA A/S
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND
RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations in conjunction with our unaudited condensed consolidated interim financial statements, including the notes thereto, included with this report and the section contained in our Annual Report on Form 20-F for the year ended December 31, 2021 – “Item 5. Operating and Financial Review and Prospects”. The following discussion is based on our financial information prepared in accordance with International Accounting Standard 34, “Interim Financial Reporting.” Certain information and disclosures normally included in the consolidated financial statements prepared in accordance with International Financial Reporting Standards (“IFRS”) have been condensed or omitted. IFRS as issued by the International Accounting Standards Board, and as adopted by the European Union, might differ in material respects from generally accepted accounting principles in other jurisdictions.
Special Note Regarding Forward-Looking Statements
This report contains forward-looking statements concerning our business, operations and financial performance and condition, as well as our plans, objectives and expectations for our business operations and financial performance and condition. Any statements contained herein that are not statements of historical facts may be deemed to be forward-looking statements. In some cases, you can identify forward-looking statements by terminology such as “aim,” “anticipate,” “assume,” “believe,” “contemplate,” “continue,” “could,” “due,” “estimate,” “expect,” “goal,” “intend,” “may,” “objective,” “plan,” “predict,” “potential,” “positioned,” “seek,” “should,” “target,” “will,” “would,” and other similar expressions that are predictions or indicate future events and future trends, or the negative of these terms or other comparable terminology. These forward-looking statements include, but are not limited to, statements about:
| • | the timing or likelihood of regulatory filings and approvals for our product candidates; |
| • | our expectations regarding the commercial availability of TransCon Growth Hormone, or TransCon hGH, and related patient support services; |
| • | the commercialization of TransCon hGH and our other product candidates, if approved; |
| • | our commercialization, marketing and manufacturing capabilities of TransCon hGH and our other product candidates and associated devices; |
| • | the scope, progress, results and costs of developing our product candidates or any other future product candidates, and conducting preclinical studies and clinical trials; |
| • | our pursuit of oncology as our second of three independent therapeutic areas of focus, and our development of a pipeline of product candidates related to oncology; |
| • | our expectations regarding the potential market size and the size of the patient populations for TransCon hGH and our other product candidates, if approved for commercial use; |
| • | our expectations regarding the potential advantages of TransCon hGH and our other product candidates over existing therapies; |
| • | our ability to enter into new collaborations; |
| • | our expectations with regard to the ability to develop additional product candidates using our TransCon technologies and file Investigational New Drug Applications, or INDs, or similar for such product candidates; |
| • | our expectations with regard to the ability to seek expedited regulatory approval pathways for our product candidates, including the potential ability to rely on the parent drug’s clinical and safety data with regard to our product candidates; |
1
| • | our expectations with regard to our current and future collaboration partners to pursue the development of our product candidates and file INDs or similar for such product candidates; |
| • | our development plans with respect to TransCon hGH and our other product candidates; |
| • | our ability to develop, acquire and advance product candidates into, and successfully complete, clinical trials; |
| • | the implementation of our business model and strategic plans for our business, TransCon hGH and our other product candidates and technologies, including global commercialization strategies; |
| • | the scope of protection we are able to establish and maintain for intellectual property rights covering TransCon hGH and our other product candidates; |
| • | estimates of our expenses, future revenue, capital requirements, our needs for additional financing and our ability to obtain additional capital; |
| • | our financial performance; |
| • | developments and projections relating to our market conditions, competitors and industry; and |
| • | the potential effects on our business of the worldwide COVID-19 pandemic. |
These forward-looking statements are based on senior management’s current expectations, estimates, forecasts and projections about our business and the industry in which we operate and involve known and unknown risks, uncertainties and other factors that are in some cases beyond our control. As a result, any or all of our forward-looking statements in this report may turn out to be inaccurate. Factors that may cause actual results to differ materially from current expectations include, among other things, those listed under the section in our Annual Report on Form 20-F for the year ended December 31, 2021 — “Item 3.D. Risk Factors”. You are urged to consider these factors carefully in evaluating the forward-looking statements. These forward-looking statements speak only as of the date of this report. Except as required by law, we assume no obligation to update or revise these forward-looking statements for any reason, even if new information becomes available in the future. Given these risks and uncertainties, you are cautioned not to rely on such forward-looking statements as predictions of future events.
You should read this report and the documents that we reference in this report and have filed as exhibits to this report completely and with the understanding that our actual future results may be materially different from what we expect. You should also review the factors and risks we describe in the reports we will file or submit from time to time with the Securities and Exchange Commission after the date of this report. We qualify all of our forward-looking statements by these cautionary statements.
Overview
We are applying our innovative TransCon technologies to build a leading, fully integrated, global biopharmaceutical company and develop a pipeline of product candidates with potential best-in-class profiles to address unmet medical needs.
Our product candidates combine our TransCon technologies with clinically-validated parent drugs and pathways, with the goal of optimizing therapeutic effect and improving tolerability and convenience.
We have applied these technologies in combination with a clinically-validated parent drug or pathway using our algorithm for product innovation with the goal of creating product candidates with the potential to be best-in-class in endocrinology rare diseases and oncology. In addition, we plan to apply this algorithm for product innovation and selection in new therapeutic areas. We believe our approach to product innovation may reduce the risks associated with traditional drug development, and that our TransCon technologies have been validated by non-clinical and clinical programs completed to date.
We had a net loss of €125.5 million for the three months ended March 31, 2022, and a net loss of €383.6 million for the year ended December 31, 2021. Our total equity was €672.8 million as of March 31, 2022 compared to €883.6 million as of December 31, 2021.
2
Ascendis Algorithm for Product Innovation
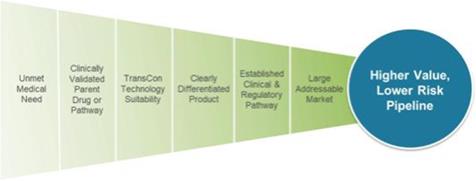
When we apply our TransCon technologies to already approved drug compounds, we may benefit from established clinical safety and efficacy data, which we believe increases the probability of success compared to traditional drug development. As presented above, our algorithm for product innovation focuses on identifying indications that have an unmet medical need, have a clinically-validated parent drug or pathway, are suitable to our TransCon technologies, have potential for creating a clearly differentiated product, have an established development pathway and have a large potentially addressable market.
We currently have one marketed product and a diversified portfolio of five product candidates in clinical development in the areas of endocrinology rare diseases and oncology. We are also evaluating additional therapeutic areas and indications.
| • | First Marketed Product – Our first marketed product is SKYTROFA® (lonapegsomatropin-tcgd), developed as TransCon Growth Hormone (“TransCon hGH”), which has received regulatory approval in the United States for the treatment of pediatric patients one year and older who weigh at least 11.5 kg and have growth failure due to inadequate secretion of endogenous growth hormone, also known as growth hormone deficiency (“GHD”) and which is now commercially available for prescription in the United States. TransCon hGH is also approved in the European Union (“EU”) under the name Lonapegsomatropin Ascendis Pharma for the treatment of children and adolescents aged from 3 years up to 18 years with growth failure due to insufficient endogenous growth hormone secretion. |
| • | Endocrinology Rare Disease Pipeline – We are developing three product candidates in our endocrinology rare disease portfolio spanning seven different clinical programs. These include TransCon hGH for pediatric GHD in Japan; TransCon hGH for adults with GHD; TransCon PTH for adult hypoparathyroidism; the last classical hormone deficiency for which complete hormone replacement has been elusive; and TransCon CNP for pediatric achondroplasia, the most common form of dwarfism. VISEN Pharmaceuticals (“VISEN”) is also developing TransCon hGH, TransCon PTH and TransCon CNP in China. In addition, we are planning new trials in other endocrinology rare disease indications, including TransCon hGH for Turner Syndrome; TransCon PTH for pediatric hypoparathyroidism; and TransCon CNP for infants (age 0-2 years) with achondroplasia. |
| • | Oncology Pipeline – In oncology, we are leveraging our TransCon technologies in effort to enhance anti-tumor effects of clinically-validated parent drugs and pathways and to provide sustained modulation of tumor microenvironments and activate cytotoxic immune cells. We have initiated clinical development of two product candidates: TransCon TLR7/8 Agonist, an investigational, long-acting prodrug of resiquimod, a small molecule agonist of Toll like receptors (“TLR”) 7 and 8 for intratumoral delivery and TransCon IL-2 ß/g for systemic delivery, which is designed for prolonged exposure to an IL-2 variant that selectively activates the IL-2Rß/g, with minimal binding to IL-2Ra. Our clinical development program for these product candidates also includes evaluation of them as a potential combination therapy. |
3
TransCon Product and Product Candidate Pipeline
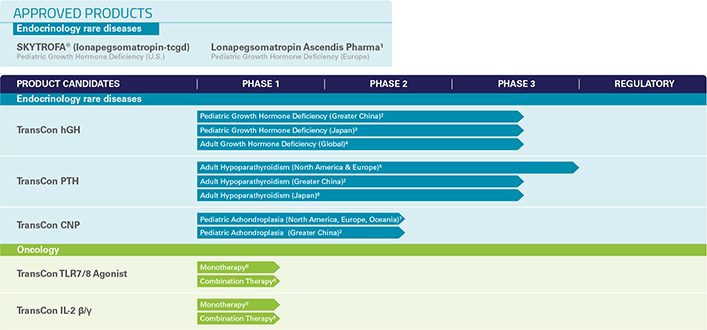
| 1 | Developed under the name TransCon hGH |
| 2 | In development in Greater China through strategic investment in VISEN Pharmaceuticals. |
| 3 | Japanese riGHt Trial. |
| 4 | Global foresiGHt Trial. |
| 5 | Top-line results from the North American and European PaTHway Trial were reported on March 13, 2022. |
| 6 | Japanese PaTHway Japan Trial. |
| 7 | North America, Europe, and Oceania ACcomplisH Trial. |
| 8 | transcendIT-101 Trial. |
| 9 | IL-ßeliege Trial. |
TransCon Technologies
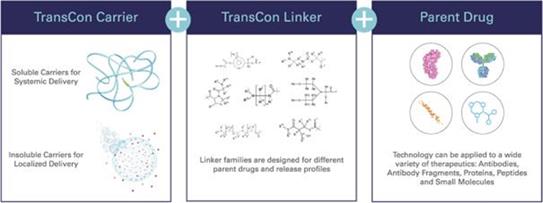
Our TransCon technologies are designed to combine the benefits of conventional prodrug and sustained release technologies to solve the fundamental limitations seen in other approaches to extending duration of a drug’s action in the body with the goal of developing highly differentiated product candidates based on potential safety and efficacy. In addition to retaining the original mode of action of the parent drug and potentially supporting dosing frequency from daily up to six months or more, we believe that predictable release over time can improve treatment efficacy, increase the likelihood of clinical development success, and provide intellectual property benefits.
TransCon molecules have three components: a parent drug, an inert carrier that protects it, and a linker that temporarily binds the two. When bound, the carrier inactivates and shields the parent drug from clearance. When injected into the body, the unmodified parent drug is released in a predictable manner. Depending upon the type of TransCon carrier we employ, we can design our TransCon prodrugs for sustained localized or systemic delivery.
4
TransCon Growth Hormone
TransCon hGH is a long-acting prodrug of somatropin (“hGH”) composed of an unmodified somatropin that is transiently bound to a carrier and proprietary linker. TransCon hGH is designed to maintain the same mode of action as daily therapies by releasing the same recombinant growth hormone molecule, somatropin, as used in extensively proven daily hGH therapy which is the current standard of care for growth hormone deficiency.
On August 25, 2021, the FDA approved TransCon hGH, known by its brand name SKYTROFA (lonapegsomatropin-tcgd), for the treatment of pediatric patients one year and older who weigh at least 11.5 kg and have growth failure due to inadequate secretion of endogenous growth hormone, also known as GHD. Once-weekly SKYTROFA (lonapegsomatropin-tcgd) is the first FDA approved product that delivers somatropin, or growth hormone, by sustained release over one week.
The FDA approval of SKYTROFA (lonapegsomatropin-tcgd) was based on results from the Phase 3 heiGHt Trial, a 52-week, global, randomized, open-label, active-controlled, parallel-group trial that compared once-weekly TransCon hGH to daily somatropin (Genotropin®) in 161 treatment-naïve children with GHD. The primary endpoint was annualized height velocity (“AHV”) at 52 weeks for weekly SKYTROFA (lonapegsomatropin-tcgd) and the daily hGH treatment groups. Other endpoints included adverse events, injection-site reactions, incidence of anti-hGH antibodies, annualized height velocity, change in height standard deviation score (“SDS”), proportion of subjects with IGF-1 SDS (0.0 to +2.0), PK/PD in subjects > 3 years, and preference for and satisfaction with SKYTROFA (lonapegsomatropin-tcgd).
We believe once-weekly SKYTROFA (lonapegsomatropin-tcgd) offers patients benefits compared to daily growth hormone:
| • | A national study has shown 66%, or 2/3 of patients miss more than one injection per week. We believe reducing injection frequency is associated with better adherence and thus may improve height velocity. |
| • | In a Phase 3 clinical study, TransCon hGH demonstrated higher AHV compared to daily somatropin with similar safety profile in treatment-naïve children with GHD. |
| • | With a weekly injection, patients switching from daily injections can experience up to 86% fewer injection days per year. |
| • | After first removed from a refrigerator, SKYTROFA (lonapegsomatropin-tcgd) can be stored at room temperature for up to six months. |
On January 12, 2022, the European Commission granted marketing authorization for Lonapegsomatropin Ascendis Pharma (developed under the name TransCon hGH) as a once-weekly subcutaneous injection for the treatment of children and adolescents ages 3 to 18 years with growth failure due to insufficient secretion of endogenous growth hormone.
In September 2020, we filed a Clinical Trial Notification, or CTN, with the Pharmaceuticals and Medical Devices Agency (“PMDA”), in Japan, to initiate our Phase 3 riGHt Trial of lonapegsomatropin for the treatment of pediatric GHD. The primary objective of the riGHt Trial is to evaluate and compare the AHV of 40 Japanese prepubertal treatment naïve children with GHD treated with weekly lonapegsomatropin to that of a commercially available daily somatropin formulation at 52 weeks.
In October 2019, we received Orphan Designation (“OD”) from the European Commission for TransCon hGH for GHD. OD is granted to therapies aimed at the treatment, prevention or diagnosis of a disease that is life-threatening or chronically debilitating, affects no more than five in 10,000 persons in the EU, or the product, without the benefits derived from orphan status, would not
5
generate sufficient return in the EU to justify investment and for which no satisfactory method of diagnosis, prevention, or treatment has been authorized (or if such a method exists, the product would provide significant additional benefit over existing therapies). We received Orphan Drug Designation (“ODD”) from the FDA for TransCon hGH as a treatment for GHD in April 2020.
Additionally, we continue to enroll patients in the foresiGHt Trial, a global Phase 3 study with the aim to demonstrate the metabolic benefits of lonapegsomatropin in adults. The ongoing conflict in the region surrounding Ukraine and Russia has impacted our ability to continue clinical trial activities in those countries. As a result, we are now targeting completion of enrollment in the foresiGHt Trial in the fourth quarter of 2022. In Greater China, VISEN completed the patient enrollment of 154 treatment-naïve, prepubertal children for the ongoing Phase 3 pivotal trial of lonapegsomatropin in patients with pediatric GHD. We plan to submit a protocol to FDA in the second quarter of 2022 to evaluate TransCon hGH for Turner Syndrome. In addition, we are also considering other potential indications for TransCon hGH where a long-acting hGH therapy may offer a best-in-class option for patients with rare growth disorders.
U.S. Commercialization Strategy
We have developed a multi-faceted commercial organization and strategy to optimize U.S. market adoption of SKYTROFA (lonapegsomatropin-tcgd).
| • | Sales Force: In the United States, the sales team covers approximately 1,400 prescribers who represent approximately 80% of the prescription volume. |
| • | Medical Affairs: Our Medical Affairs organization is educating stakeholders and broadening SKYTROFA (lonapegsomatropin-tcgd) awareness. |
| • | Market Access: Payor coverage and reimbursement are important factors which can influence market adoption. Recognizing this importance, our Market Access organization has been engaging national and regional payors in an effort to garner reimbursement for SKYTROFA (lonapegsomatropin-tcgd). |
As of December 31, 2021, 369 SKYTROFA prescriptions had been written by 139 prescribers. In addition, 42% of the prescribers had written prescriptions for more than one patient. As of April 29, 2022, 1,231 SKYTROFA prescriptions had been written by 404 prescribers. In addition, approximately 50% of the prescribers had written prescriptions for more than one patient.
TransCon Parathyroid Hormone
TransCon Parathyroid Hormone (“PTH”) is an investigational long-acting prodrug of parathyroid hormone that is designed as a novel replacement therapy for PTH dosed once-daily to achieve and maintain a steady concentration of PTH in the bloodstream within the normal range, at levels similar to those observed in healthy individuals. TransCon PTH is designed to restore physiologic levels of PTH 24 hours per day, thereby more fully addressing all aspects of the disease including normalizing serum and urinary calcium and serum phosphate levels. Pharmacokinetic data from our Phase 1 trial of TransCon PTH in healthy subjects demonstrated a half-life of approximately 60 hours, supporting a continuous infusion-like profile with daily administration. With once-daily dosing, we believe this substantial half-life extension of PTH could more closely reflect the physiological levels of PTH observed in healthy individuals thereby maintaining blood calcium levels and normalizing urinary calcium excretion. Pharmacokinetic data from multiple ascending dose cohorts in our Phase 1 trial of TransCon PTH in healthy subjects demonstrated a continuous infusion-like profile of free PTH. By providing steady levels of PTH in the physiological range, we believe TransCon PTH can address the fundamental limitations of short-acting PTH molecules and become a highly differentiated therapy for hypoparathyroidism (“HP”).
Current conventional therapy for HP patients primarily consists of active vitamin D and oral calcium supplementation. However, since PTH is not present at the kidney to facilitate calcium reabsorption from the urine, the goal of conventional therapy is to maintain serum calcium (“sCa”) levels just below or within the lower part of the normal range and thereby limit as much as possible the damage from excess urinary calcium excretion. Nonetheless, conventional therapy frequently leads to significant sCa fluctuations accompanied by symptomatic hyper- or hypocalcemia. In addition, conventional therapy with active vitamin D and calcium have been shown to contribute to the risk of renal disease.
HP also poses a high burden on the healthcare system despite current conventional therapy. For example, one survey of 374 patients showed that 72% experienced more than ten symptoms in the preceding twelve months, with symptoms experienced for a mean of 13 ± 9 hours a day. Other studies showed that 79% of HP cases require hospitalizations and that patients with the disease results have a four-fold increase in the risk of renal disease compared to healthy controls. Patients often experience decreased quality of life. We conducted a survey of 42 patients which found that 100% of subjects reported negative psychological impacts, interference with daily life and impact on physical functioning from HP, and that 76% were either no longer able to work or experienced interference with work productivity.
6
On March 13, 2022, the Company announced that top-line data from the randomized, double-blind, placebo-controlled portion of its Phase 3 PaTHway Trial of TransCon PTH in adults with hypoparathyroidism demonstrated statistically significant improvement with TransCon PTH compared to control on the primary composite endpoint and all key secondary endpoints. The primary endpoint – defined as serum calcium levels in the normal range (8.3– 10.6 mg/dL) and independence from conventional therapy (active vitamin D and >600 mg/day of calcium supplements) with no increase in prescribed study drug within the 4 weeks prior to the Week 26 visit – was achieved by 78.7% of TransCon PTH-treated patients (48 of 61), compared to 4.8% for patients (1 of 21) in control group (p-value <0.0001).
Highlights of the Phase 3 PaTHway Trial Top-Line Data
The PaTHway Trial is a Phase 3 double-blind, placebo-controlled trial of 82 dosed adults with chronic hypoparathyroidism randomized 3:1 (TransCon PTH:placebo).
Primary Composite Endpoint:
| • | 78.7% of TransCon PTH-treated patients (48 of 61) achieved serum calcium levels in the normal range (8.3–10.6 mg/dL) and independence from therapeutic levels of conventional therapy, compared to 4.8% for patients (1 of 21) in control group (p-value <0.0001). |
Key Pre-Specified Secondary Endpoints:
| • | Statistically significant decrease in patient-reported, disease-specific physical and cognitive symptoms compared to patients in control group, as shown on Hypoparathyroidism Patient Experience Scales (“HPES”) Symptom-Physical domain scores (p-value = 0.0038) and HPES Symptom-Cognitive domain scores (p-value = 0.0055). |
| • | Statistically significant reduction in patient-reported disease impact compared to patients in control group, as shown on HPES Impact-Physical Functioning domain scores (p-value = 0.0046) and HPES Impact-Daily Life domain scores (p-value = 0.0061). |
| • | Statistically significant improvements in patient-reported physical functioning compared to patients in control group, as shown on the SF-36v2® survey Physical Functioning subscale (p-value = 0.0347). |
Selected other Analyses:
| • | At Week 26, 95% of TransCon PTH-treated patients were able to discontinue conventional treatments with therapeutic levels of calcium supplements and active vitamin D. |
| • | PaTHway patients had low levels of bone turnover at baseline. TransCon PTH-treated patients demonstrated increased levels of bone turnover markers at Week 26. |
Safety Summary:
| • | TransCon PTH was generally well tolerated, with no discontinuations related to study drug. Three patients discontinued during the treatment period – 2 from the placebo arm and 1 from the TransCon PTH arm. |
| • | 82% of TransCon PTH patients and 100% of patients in control group reported treatment-emergent adverse events (“TEAEs”), the majority of which were Grade 1, 2 in severity. |
| • | One serious related TEAE in the TransCon PTH arm was reported due to a dosing error. |
| • | One death in the TransCon PTH arm was assessed as unrelated to study drug. |
7
| • | TransCon PTH-treated patients showed a mean decrease in 24-hour urine calcium excretion into the normal range, from 390 mg/24 hours down to 220 mg/24 hours. Despite a higher mean serum calcium at Week 26, there was a significantly greater decrease in mean 24-hour urine calcium for TransCon PTH-treated patients compared to patients in control group. |
Following an initial blinded study period of 26 weeks, for which top-line data are reported here, all 79 patients completing the blinded period opted to receive treatment with TransCon PTH in the ongoing open-label extension portion of the study for up to 3 years (156 weeks). As of May 1, 2022, all 79 patients continued in the open label extension portion of the PaTHway Trial.
In November 2021, we announced week 84 top-line data from the Phase 2 PaTH Forward Trial. Week 84 results from the PaTH Forward OLE demonstrated:
| • | Mean serum calcium levels remained stable and in the normal range. |
| • | All study subjects discontinued active vitamin D supplements in the earliest weeks of the trial and have remained off it since then. In addition, 93% of subjects were taking calcium supplements <600 mg per day. |
| • | Mean urinary calcium excretion remained stable and in the normal range. |
| • | TransCon PTH was well-tolerated at all doses administered. No treatment-related serious or severe adverse events occurred, and no treatment-emergent adverse events (“TEAEs”) led to discontinuation of study drug. |
| • | Injections were well-tolerated using pen injector planned for commercial presentation. |
At week-58, quality-of-life and bone mineral density data were collected. The data demonstrated:
| • | All mean summary and subdomain SF-36 Health Survey scores continued normalization between week 26 and week 58 despite all mean scores starting below norms at baseline. |
| • | Bone mineral density Z-scores trended towards normalization and stabilization over 58 weeks in PaTH Forward. |
As of May 1, 2022, 57 out of the 59 patients continued in the open-label extension portion of the trial, where they receive an individualized maintenance dose of TransCon PTH (6 to 30 µg per day). In addition, all 57 subjects have exceeded two years of follow-up in the PaTH Forward Trial. Two patients withdrew from the trial for reasons unrelated to safety or efficacy of the study drug.
In the second quarter of 2021, we submitted a Clinical Trial Notification (“CTN”) to the MHLW for PaTHway Japan Trial, a Phase 3 trial to evaluate the safety, tolerability, and efficacy of TransCon PTH. In July 2021, the Japanese Pharmaceuticals and Medical Devices Agency accepted the CTN for the PaTHway Japan Trial, a single-arm, Phase 3 trial of TransCon PTH in a minimum of 12 Japanese subjects with HP. Subjects will start with an 18 µg dose of TransCon PTH and be followed over a 26-week period during which they will be titrated to an optimal dose. The minimum enrollment target of 12 patients was achieved in April 2022.
In October 2020, the EC granted orphan designation to TransCon PTH for the treatment of HP.
In June 2018, we were granted orphan drug designation by the FDA for TransCon PTH for the treatment of hypoparathyroidism.
TransCon C-Type Natriuretic Peptide
TransCon C-Type Natriuretic Peptide (“CNP”) is an investigational long-acting prodrug of C-type natriuretic peptide designed to provide continuous CNP exposure at therapeutic levels with a well-tolerated and convenient once-weekly dose. It is being developed for the treatment of children with achondroplasia. TransCon CNP is designed to provide effective shielding of CNP from neutral endopeptidase degradation in subcutaneous tissue and the blood compartment, minimize binding of CNP to the NPR-C receptor to decrease clearance, reduce binding of CNP to the NPR-B receptor in the cardiovascular system to avoid hypotension, and release unmodified CNP, which is small enough in size to allow effective penetration into growth plates. We believe TransCon CNP offers advantages over shorter-acting CNP and CNP analogs in development that result in high Cmax levels which may cause adverse cardiovascular events. In addition, we expect a more constant CNP exposure at lower Cmax to correlate with better therapeutic outcomes.
In July 2019, we initiated the Phase 2 ACcomplisH Trial, a randomized, double-blind, placebo-controlled, sequential rising dose trial to evaluate the safety and efficacy of TransCon CNP in approximately 60 children with achondroplasia (ages two to ten years). Subjects are randomized to receive either TransCon CNP or placebo in a 3:1 ratio. The primary efficacy endpoint is annualized height
8
velocity at twelve months. Key secondary and additional endpoints include body proportionality and change in BMI, both evaluated after twelve months of weekly TransCon CNP treatment, and patient reported outcome measures. In December 2021, we announced that enrollment in ACcomplisH Trial was completed.
We are assessing additional trials evaluating the safety and efficacy of TransCon CNP in ACH patients.
In collaboration with VISEN, we are sponsoring the ACcomplisH China Trial, a randomized, double-blind, placebo-controlled, Phase 2 dose expansion trial to evaluate the safety and efficacy of TransCon CNP in subjects with achondroplasia. The primary endpoint is to evaluate the safety of treatment and its effect on 12-month annualized height velocity. In January 2021, China Center for Drug Evaluation of National Medical Products Administration approved VISEN’s IND application to conduct the ACcomplisH China Trial.
In July 2020, we received orphan designation from the EC for TransCon CNP for treatment of achondroplasia.
In February 2019, we were granted orphan drug designation by the FDA for TransCon CNP for the treatment of achondroplasia.
TransCon Products Candidates – Oncology
In January 2019, we established oncology as our second independent therapeutic area of focus for our TransCon technologies. Our goal is to improve treatment efficacy while limiting or reducing toxicity by applying TransCon technologies to clinically validated drugs, using our unique algorithm for product innovation.
We are currently investigating two clinical-stage product candidates designed to activate the patients’ own immune system to eradicate malignant cells. We believe our approach, if successfully developed, has the potential to optimize the efficacy of systemically administered, clinically validated therapies while limiting adverse effects.
Our TransCon product candidates in oncology are designed to provide sustained systemic or intratumoral administration, which we believe could provide potent and durable anti-tumor efficacy. Our nonclinical studies have showed sustained activation of cytotoxic immune cells that resulted in robust anti-tumor responses by TransCon product candidates using infrequent administration.
TransCon TLR 7/8 Agonist
TransCon TLR7/8 Agonist is an investigational long-acting prodrug, designed for sustained release of resiquimod, a small molecule agonist of Toll-like receptors (“TLR”) 7 and 8. It is designed to provide sustained and potent activation of the innate immune system in the tumor and tumor draining lymph node and to have a low risk of systemic toxicity for weeks or months following a single intratumoral injection. Enrollment continues in the Phase 1/2 transcendIT-101 Trial for which we submitted an IND in 2020.
TransCon IL-2 ß/g
TransCon IL-2 ß/g is an investigational long-acting prodrug designed to improve cancer immunotherapy through sustained release of an IL-2 variant that selectively activates the IL-2Rß/g, with minimal binding to IL-2Rα. The Phase 1/2 IL- ßeliege Trial evaluating TransCon IL-2 ß/g monotherapy in patients with advanced cancer is enrolling.
We are evaluating additional TransCon product candidates in nonclinical research studies with potential to enhance anti-tumor immune responses for the treatment of multiple tumor types. We are exploring product candidates using both systemic and intratumoral administration as monotherapies and as components of combination regimens. We believe these programs have the potential to make a positive impact to the lives of many patients with cancer.
9
Results of Operations
Impact from COVID-19 Pandemic
The COVID-19 pandemic has affected countries where we are operating, where we have planned or have ongoing clinical trials, and where we rely on third-parties to manufacture preclinical, clinical and commercial supply.
Since COVID-19 started to spread around the world, we have closely monitored the development, and implemented several measures to accommodate impacts on our business, and to ensure the safety of our employees, including:
| • | Encouraging employees to work remotely, reduce travel activity and minimize face-to-face meetings; |
| • | Establishing home offices, and ensuring proper and secure IT infrastructure to improve the safety and efficiency of the remote work environment; |
| • | Implementing remote visits for patients enrolled in our clinical trials, including ensuring safe delivery of clinical drugs; and |
| • | Addressing COVID-19 in relation to logistics and manufacturing at Joint Steering Committees with manufacturing partners. |
While COVID-19 had an impact on how we work and conduct our activities, we have managed to avoid significant disruptions to our clinical and manufacturing operations. As a result of governmental restrictions, field-based sales personnel primarily have worked under a remote engagement model with healthcare professionals and patient care organizations, and similarly, patients have not been able to see their physicians. As restrictions ceases, field-based sales personnel have begun in person engagements when interacting with healthcare professionals and patient care organizations, as well as patients having easier access to their physicians. The impact on the commercial product revenue is uncertain and difficult to quantify.
We monitor the risks from this pandemic closely, and work with relevant stakeholders to avoid and limit disruptions, and to develop and establish working measures. However, while COVID-19 continues to impact global societies, the uncertainty related to the duration and direction of the pandemic makes the future impact from COVID-19, including the magnitude of any impact on our operational results, highly uncertain and unpredictable.
For additional description of COVID-19 related risks, please refer to “Item 3D. Risk Factors”, set forth in our 2021 Annual Report on Form 20-F.
10
Comparison of the Three Months Ended March 31, 2022 and 2021 (unaudited):
Summary
Following the launch of SKYTROFA (lonapegsomatropin-tcgd) in the U.S. in the fourth quarter of 2021, we continued to increase sale of commercial products and we closely monitor the uptake of our products in the market. As of April 29, 2022, over 1,200 patients have been prescribed SKYTROFA since launch.
We continue to see progress in our clinical trials within all our product candidates, and on March 13, 2022 we reported top-line data from our Phase 3 PaTHway Trial of TransCon PTH in adults for hypoparathyroidism, which demonstrated statistically significantly improvement with TransCon PTH compared to control on the primary composite endpoint and all key secondary endpoints.
At the end of March 2022, we completed a U.S. $575 million convertible bond offering strengthening our balance sheet and support the runway to create a sustainable leading global biopharma company.
We realized a net loss of €125.5 million for the three months ended March 31, 2022 compared to €62.8 million for the same period last year.
| Three Months Ended March 31, |
||||||||
| 2022 | 2021 | |||||||
| (EUR’000) | ||||||||
| Revenue |
6,828 | 746 | ||||||
| Cost of sales |
4,246 | — | ||||||
|
|
|
|
|
|||||
| Gross profit / (loss) |
2,582 | 746 | ||||||
| Research and development costs |
83,193 | 88,149 | ||||||
| Selling, general and administrative expenses |
47,418 | 37,247 | ||||||
|
|
|
|
|
|||||
| Operating profit / (loss) |
(128,029 | ) | (124,650 | ) | ||||
| Share of profit / (loss) of associate |
(4,873 | ) | 28,106 | |||||
| Finance income |
13,044 | 34,430 | ||||||
| Finance expenses |
5,399 | 869 | ||||||
|
|
|
|
|
|||||
| Profit / (loss) before tax |
(125,257 | ) | (62,983 | ) | ||||
|
|
|
|
|
|||||
| Tax on profit / (loss) for the period |
(241 | ) | 191 | |||||
|
|
|
|
|
|||||
| Net profit / (loss) for the period |
(125,498 | ) | (62,792 | ) | ||||
|
|
|
|
|
|||||
Revenue
The following table summarizes our revenue for the three months ended March 31, 2022 and 2021.
| Three Months Ended March 31, |
||||||||
| 2022 | 2021 | |||||||
| (EUR’000) | ||||||||
| Commercial sale of products |
1,888 | — | ||||||
| Rendering of services |
372 | 169 | ||||||
| Sale of clinical supply |
3,936 | — | ||||||
| Licenses |
632 | 577 | ||||||
|
|
|
|
|
|||||
| Total revenue |
6,828 | 746 | ||||||
|
|
|
|
|
|||||
Revenue for the three months ended March 31, 2022 was €6.8 million, an increase of €6.1 million, compared to €0.7 million for the three months ended March 31, 2021, and comprised commercial sale of SKYTROFA (lonapegsomatropin-tcgd), and sale of clinical supply, rendering of services, and recognition of internal profit deferred from November 2018 when we entered into license agreements with VISEN. The increase in revenue was primarily attributable to the €1.9 million commercial sale of products, and €3.9 million higher sales of clinical supply to VISEN compared to the same period last year. As we did not have any commercial sales of SKYTROFA (lonapegsomatropin-tcgd) in the three months ended March 31, 2021, no revenue from commercial sale of products were reported for that period.
11
Cost of Sales
Cost of sales was €4.2 million for the three months ended March 31, 2022, and comprised cost of commercial products sold and cost of clinical supply delivered to VISEN. As we did not have any commercial sales of SKYTROFA (lonapegsomatropin-tcgd) in the three months ended March 31, 2021, no similar costs were reported for that period.
Research and Development Costs
The following table specifies our external development costs on our principal development pipeline and other research and development costs.
| Three Months Ended March 31, |
||||||||
| 2022 | 2021 | |||||||
| (EUR’000) | ||||||||
| External project costs |
||||||||
| TransCon hGH |
19,939 | 26,578 | ||||||
| TransCon PTH |
11,686 | 8,393 | ||||||
| TransCon CNP |
9,555 | 8,310 | ||||||
| TransCon IL-2 ß/g |
1,446 | 4,191 | ||||||
| TransCon TLR7/8 |
2,137 | 3,572 | ||||||
| Other project costs |
602 | 300 | ||||||
|
|
|
|
|
|||||
| Total external project costs |
45,365 | 51,344 | ||||||
| Other research and development costs |
||||||||
| Employee costs |
30,417 | 31,846 | ||||||
| Other external costs |
4,691 | 2,809 | ||||||
| Depreciation |
2,720 | 2,150 | ||||||
|
|
|
|
|
|||||
| Total other research and development costs |
37,828 | 36,805 | ||||||
|
|
|
|
|
|||||
| Total research and development costs |
83,193 | 88,149 | ||||||
|
|
|
|
|
|||||
Research and development costs were €83.2 million for the three months ended March 31, 2022, a decrease of €4.9 million, or 6%, compared to €88.1 million for the three months ended March 31, 2021.
External project costs related to TransCon hGH decreased by €6.6 million compared to the same period last year; primarily reflecting lower costs for manufacturing of commercial product supply. Following the U.S. FDA approval of SKYTROFA (lonapegsomatropin-tcgd) on August 25, 2021, manufacturing of commercial product supply is recognized as inventory, whereas such costs were recognized as research and development costs prior to the U.S. FDA approval, and as such included in the external development costs for TransCon hGH for the three months ended March 31, 2021.
External project costs related to TransCon PTH increased by €3.3 million, primarily reflecting increased costs for commercial manufacturing, partly offset by lower costs of clinical trials and clinical supplies, compared to the same period last year.
External project costs related to TransCon CNP increased by €1.2 million, primarily reflecting an increase in manufacturing costs, partly offset by lower costs for clinical trials and clinical supplies.
External project costs related to our TransCon TLR7/8 Agonist decreased by €1.5 million, reflecting lower manufacturing costs partly offset by higher clinical trial costs.
External project costs related to our TransCon IL-2 ß/g decreased by €2.8 million, primarily reflecting lower manufacturing costs partly offset by higher clinical trial costs.
Other research and development costs increased by €1.0 million, including an increase in personnel costs of €1.2 million, a decrease in non-cash share-based payment of €2.6 million, and increases in travel costs of €0.9 million, depreciation of €0.6 million and other costs of €0.9 million.
Research and development costs included non-cash share-based payment of €11.6 million for the three months ended March 31, 2022, compared to €14.2 million for the three months ended March 31, 2021.
12
Selling, General and Administrative Expenses
Selling, general and administrative expenses were €47.4 million for the three months ended March 31, 2022, an increase of €10.2 million, or 27%, compared to €37.2 million for the three months ended March 31, 2021. The higher expenses were primarily due to an increase in personnel costs of €5.9 million for additional commercial and administrative personnel, an increase in commercial costs of €3.4 million, and a net increase in other costs allocated to selling, general and administrative expenses of €0.9 million, including an increase in travel costs of €0.8 million.
Selling, general and administrative expenses included non-cash share-based payment of €8.3 million for the three months ended March 31, 2022, compared to €8.9 million for the three months ended March 31, 2021.
Net Profit / (Loss) of Associate
Net loss of associate was €4.9 million for the three months ended March 31, 2022, compared to a net profit of €28.1 million for the three months ended March 31, 2021. The net loss represents our share of net result from VISEN. For the three months ended March 31, 2021, the net profit of associate included a non-cash gain of €42.3 million as a result of the Series B financing in VISEN on January 8, 2021, and the Company’s share of loss of €14.2 million.
Finance Income and Finance Expenses
Finance income was €13,0 million for the three months ended March 31, 2022, compared to €34.4 million for the three months ended March 31, 2021. Finance expenses were €5.4 million for the three months ended March 31, 2022, compared to €0.9 million for the same period in 2021. As we hold positions of marketable securities and cash and cash equivalents in U.S. Dollars, we are affected by exchange rate fluctuations when reporting our financial results in Euro. For the three months ended March 31, 2022, as well as for the three months ended March 31, 2021, we recognized net exchange rate gains when reporting our U.S. Dollar positions in Euro, reflecting positive exchange rate fluctuations.
Finance expenses for the three months ended March 31, 2022, included €4.2 million transaction costs related to the Convertible Senior Notes (“convertible notes”) financing completed on March 29, 2022, representing the part of the total transaction costs that is attributable to the derivative component of the convertible note financing.
Tax for the Period
Tax for the three months ended March 31, 2022 was a net tax expense of €0.2 million compared to a net tax credit of €0.2 million for the three months ended March 31, 2021. Taxes for the three months ended March 31, 2022 comprised estimated tax expenses of €0.3 million in one of our U.S. subsidiaries and €0.1 million in our German subsidiary, partly offset by an estimated tax credit of €0.2 million in the group of Danish companies.
Liquidity and Capital Resources
Our liquidity and capital resources comprise cash, cash equivalents and marketable securities.
As of March 31, 2022, these amounted to €1,065 million, and are specified as follows:
| Carrying amount |
Fair value |
|||||||
| (EUR’000) | ||||||||
| March 31, 2022 |
||||||||
| Liquidity and capital resources |
||||||||
| Marketable securities |
309,542 | 306,728 | ||||||
| Cash and cash equivalents |
755,643 | 755,643 | ||||||
|
|
|
|
|
|||||
| Total liquidity and capital resources |
1,065,185 | 1,062,371 | ||||||
|
|
|
|
|
|||||
| Classification in consolidated statement of financial position |
||||||||
| Non-current assets |
86,487 | 84,909 | ||||||
| Current assets |
978,698 | 977,462 | ||||||
|
|
|
|
|
|||||
| Total liquidity and capital resources |
1,065,185 | 1,062,371 | ||||||
|
|
|
|
|
|||||
13
Marketable securities have a weighted average duration of 5.7 and 15.5 months, for current (i.e., those maturing within twelve months after the reporting date) and non-current positions, respectively. The entire portfolio of marketable securities (current and non-current) has a weighted average duration of 8.4 months.
We have historically funded our operations primarily through issuance of preference shares, ordinary shares, including our initial public offering, follow-on offerings and exercise of warrants, convertible debt securities, and payments to us made under collaboration agreements.
In February 2015, we announced the closing of our initial public offering, with net proceeds of $111.5 million (or €101.4 million). In addition, we have completed follow-on public offerings of American Depositary Shares (“ADSs”) as specified below:
| • | In 2016, with net proceeds of $127.1 million (or €116.6 million); |
| • | In 2017, with net proceeds of $145.2 million (or €123.1 million); |
| • | In 2018, with net proceeds of $242.5 million (or €198.6 million); |
| • | In 2019, with net proceeds of $539.4 million (or €480.3 million); |
| • | In 2020, with net proceeds of $654.6 million (or €580.5 million); and |
| • | In 2021, with net proceeds of $436.5 million (or €367.9 million). |
On March 29, 2022, we issued an aggregate principal amount of $575.0 million of fixed rate 2.25% Convertible Senior Notes (the “convertible notes”). The net proceeds from the offering of the convertible notes were $557.9 million (or €503.3 million), after deducting the initial purchasers’ discounts and commissions and expected offering expenses. The convertible notes rank equally in right of payment with all of our existing and future senior unsecured indebtedness and are redeemable by us no earlier than on or after April 7, 2025. Unless earlier converted or redeemed, the convertible notes will mature on April 1, 2028. For further description of the convertible notes, and a maturity analysis (on an un-discounted basis) for non-derivative financial liabilities, recognized in the unaudited condensed consolidated statements of financial position on March 31, 2022, please refer to Note 10, “Financial Assets and Financial Liabilities”.
We used $116.7 million (€105.2 million) of the net proceeds from the offering to repurchase 1,000,000 ADSs representing the Company’s ordinary shares. Total holding of treasury shares is disclosed in Note 9, “Treasury Shares”.
Our operational expenditures are primarily related to research and development activities and general and administrative activities to support our therapeutic areas within endocrinology and oncology. In addition, expenditures relate to building our sales and marketing capabilities, and inventories, to support the launch of SKYTROFA (lonapegsomatropin-tcgd), as well as preparation for future product launches.
We manage our liquidity risk by maintaining adequate cash reserves and banking facilities, and by matching the maturity profiles of financial assets including marketable securities, with cash-forecasts including payment profiles on liabilities. We monitor the risk of a shortage of funds using a liquidity planning tool, to ensure sufficient funds are available to settle liabilities as they become due.
Based on our current operating plan, we believe that our existing cash, cash equivalents and marketable securities as of March 31, 2022, will be sufficient to meet our projected cash requirements for at least twelve months from the date of this report. However, our operating plan may change as a result of many factors currently unknown to us, and we may need to seek additional funds sooner than planned.
For additional description of our cash requirements, expense structure and commitments, please refer to “Item 5B. Liquidity and Capital Resources”, set forth in our 2021 Annual Report on Form 20-F.
Our future funding requirements will depend on many factors, including, but not limited to:
| • | the manufacturing, selling and marketing costs associated with TransCon hGH and with our other product candidates, if approved, including the cost and timing of building our sales and marketing capabilities; |
| • | the timing, receipt, and amount of sales of, or royalties on, TransCon hGH and any future products; |
14
| • | the sales price and the availability of adequate third-party coverage and reimbursement for TransCon hGH and for our other product candidates, if approved; |
| • | our ability to establish and maintain strategic partnerships, licensing or other arrangements and the financial terms of such agreements; |
| • | our ability to collect payments which are due to us from collaboration partners (if any), which in turn is impacted by the financial standing of any such collaboration partners; |
| • | the progress, timing, scope, results and costs of our preclinical studies and clinical trials and manufacturing activities for our product candidates that have not been licensed, including the ability to enroll patients in a timely manner for clinical trials; |
| • | the time and cost necessary to obtain regulatory approvals for our product candidates and the costs of post-marketing studies that could be required by regulatory authorities; |
| • | the cash requirements of any future acquisitions or discovery of product candidates; |
| • | the number and scope of preclinical and discovery programs that we decide to pursue or initiate; |
| • | the potential acquisition and in-licensing of other technologies, products or assets; |
| • | the time and cost necessary to respond to technological and market developments, including further development of our TransCon technologies; |
| • | the achievement of development, regulatory and commercial milestones resulting in the payment to us from collaboration partners of contractual milestone payments and the timing of receipt of such payments, if any; |
| • | our progress in the successful commercialization and co-promotion of TransCon hGH and of our other product candidates, if approved, and our efforts to develop and commercialize our other existing product candidates; and |
| • | the costs of filing, prosecuting, maintaining, defending and enforcing any patent claims and other intellectual property rights, including litigation costs and the outcome of such litigation, including costs of defending any claims of infringement brought by others in connection with the development, manufacture or commercialization of our product candidates. |
Additional funds may not be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available to us on a timely basis, we may be required to delay, limit, scale back or cease our research and development activities, preparing for potential commercialization, preclinical studies and clinical trials for our product candidates for which we retain such responsibility and our establishment and maintenance of sales and marketing capabilities or other activities that may be necessary to commercialize our product candidates. The following table summarizes our cash flows for each of the unaudited three month periods ended March 31, 2022 and 2021:
| Three Months Ended March 31, |
||||||||
| 2022 | 2021 | |||||||
| (EUR’000) | ||||||||
| Cash flow from / (used in) |
||||||||
| Operating activities |
(130,788 | ) | (81,619 | ) | ||||
| Investing activities |
38,542 | (30,117 | ) | |||||
| Financing activities |
397,735 | 20 | ||||||
|
|
|
|
|
|||||
| Net increase / (decrease) in cash and cash equivalents |
305,489 | (111,716 | ) | |||||
|
|
|
|
|
|||||
Cash Flow from / (Used in) Operating Activities
Cash flow used in operating activities represents all cash flow from other than those classified as either investing or financing activities. Cash flow used in operating activities includes net loss for the period adjusted for non-cash net financial income and taxes, changes to provisions, other non-cash items and net change in working capital items.
Net cash used in operating activities for the three months ended March 31, 2022, was €130.8 million. The net loss for the three months ended March 31, 2022, of €125.5 million included non-cash charges of €29.3 million, comprising share-based payment, share of loss in associate, depreciation and amortization, and non-cash net income, including non-cash revenue, net financial income, increase in provisions and taxes, of €5.8 million. The net change in working capital contributed negatively to cash flows by €28.8 million, due to net increases in inventories of €17.0 million, trade payables, accrued expenses and other payables of €4.3 million, contract liabilities of €2.3 million and receivables and prepayments of €5.1 million.
Net cash used in operating activities for the three months ended March 31, 2021, was €81.6 million. The net loss for the three months ended March 31, 2021, of €62.8 million included non-cash charges of €26.8 million, comprising share-based payment, depreciation and amortization, and non-cash net income, including non-cash revenue, net financial income and taxes, of €61.9 million. The net change in working capital contributed positively to cash flows by €16.3 million, primarily due to a net increase in trade payables, accrued expenses and other payables of €17.6 million, an increase in prepayments and receivables of €1.2 million, and a decrease in deferred income of €0.1 million.
The €49.2 million increase in cash flow used in operating activities for the three months ended March 31, 2022 compared to the same period last year, was attributable to increase in net loss for the period adjusted for net financial income, taxes, and non-cash items of €4.1 million, an increase in working capital items of €45.1 million, primarily attributable to inventories of €17.0 million due to the launch of SKYTROFA (lonapegsomatropin-tcgd) and trade payables and accrued expenses of €21.9 million primarily due to increased activity and changes in payment patterns.
15
Cash Flow from / (Used in) Investing Activities
Cash flow from investing activities for the three months ended March 31, 2022, includes capital expenditures for property, plant and equipment of €3.8 million, offset by reimbursement of such acquisition of €3.8 million, and purchase and settlement of marketable securities with a maturity of three months or less after the date of acquisition (trade-date) contributing positively to the cash position by €38.6 million.
Cash flow used in investing activities for the three months ended March 31, 2021, of €30.1 million were related to the acquisition of marketable securities of €39.4 million and the settlement of marketable securities of €24.1 million, to Series B investment in VISEN of €10.2 million, to the acquisition of property, plant and equipment of €4.0 million, primarily related to leasehold improvements and equipment for use in the United States, and to the acquisition of software of €0.5 million.
The €68.6 million increase in cash flow from investing activities for the three months ended March 31, 2022 compared to the same period last year, was primarily attributable to:
| • | additional net settlements of marketable securities of €53.9 million in line with our liquidity management strategy; |
| • | reimbursement from property, plant and equipment of €4.0 million primarily related to leasehold improvements for our U.S. facilities; and |
| • | the Series B investment in VISEN of €10.2 million made in January 2021. |
Cash Flow from / (Used in) Financing Activities
Cash flow from financing activities for the three months ended March 31, 2022 of €397.7 million includes €1.9 million payment of principal portion of lease liabilities, €504.5 million in net proceeds from issuance of convertible notes, €0.4 million in proceeds from warrants exercises and €105.2 million acquisition of treasury shares.
Cash flows from financing activities for the three months ended March 31, 2021 of €20 thousand were comprised of €2.0 million in net proceeds from warrant exercises in March 2021, offset by payments on lease liabilities of €2.0 million.
The €397.8 million increase in cash flow from financing activities for the three months ended March 31, 2022 compared to the same period last year was primarily attributable to net proceeds from issuance of convertible notes, net of paid transaction costs, of €504.5 million, partly offset by acquisition of treasury shares of €105.2 million.
Off-balance Sheet Arrangements
We have not entered into any off-balance sheet arrangements or any holdings in variable interest entities.
Qualitative Disclosures about Market Risk
Our activities expose us to the financial risks of changes in foreign currency exchange rates and interest rates. We do not enter into derivative financial instruments to manage our exposure to such risks. Further, we are exposed to credit risk and liquidity risk. For a description of our exposure to liquidity risks and processes for managing these risks, please refer to “Liquidity and Capital Resources”, set forth above.
Foreign Currency Risk
We are exposed to foreign exchange risk arising from various currency exposures, primarily with respect to the U.S. Dollar, the British Pound and the Danish Krone. We have received payments in U.S. Dollars under our collaborations, and the proceeds from our Series D financing in November 2014, our initial public offering in February 2015, and our follow-on offerings were in U.S. Dollars. In addition, our outstanding convertible notes, due April 2028, are in U.S. Dollars. We seek to minimize our exchange rate risk by maintaining cash positions in the currencies in which we expect to incur the majority of our future expenses and we make payments from those positions.
16
Interest Rate Risk
Outstanding convertible notes comprise a 2.25% coupon fixed rate structure. In addition, interest rate on lease liabilities is fixed at the lease commencement date. Future indebtedness including those related to lease arrangements, if any, may be subject to higher interest rates. In addition, future interest income from interest-bearing bank deposits and marketable securities may fall short of expectations due to changes in interest rates.
Derivative liabilities are measured at fair value through profit or loss. Accordingly, since the fair value is exposed from the development in interest rates, the profit or loss is exposed to volatility from such development.
Credit Risk
We have adopted an investment policy with the primary purpose of preserving capital, fulfilling our liquidity needs and diversifying the risks associated with cash, cash equivalents and marketable securities. Our investment policy establishes minimum ratings for institutions with which we hold cash, cash equivalents and marketable securities, as well as rating and concentration limits for marketable securities held.
All material counterparties are considered creditworthy. While the concentration of credit risk may be significant, the credit risk for each individual counterpart is considered to be low. Our exposure to credit risk primarily relates to cash, cash equivalents, and marketable securities. The credit risk on our bank deposits is limited because the counterparties, holding significant deposits, are banks with high credit-ratings (minimum A3/A-) assigned by international credit-rating agencies. The banks are reviewed on a regular basis and deposits may be transferred during the year to mitigate credit risk. In order to mitigate the concentration of credit risks on bank deposits and to preserve capital, a portion of the bank deposits have been placed into primarily U.S. government bonds, corporate bonds and agency bonds. Our investment policy, approved by the Board of Directors, only allows investment in marketable securities having investment grade credit-ratings, assigned by international credit-rating agencies. Accordingly, the risk from probability of default is low. On each reporting date, we consider the risk of expected credit loss on bank deposits and marketable securities, including the hypothetical impact arising from the probability of default, which is considered in conjunction with the expected loss caused by default by banks or securities with similar credit-ratings and attributes. In line with previous periods, this assessment did not reveal a material impairment loss, and accordingly no provision for expected credit loss has been recognized.
Equity Risk
We are exposed from the development in the Company’s own share price, when remeasuring derivative liabilities at their fair value. Derivative liabilities relate to foreign currency conversion options embedded in the convertible notes, and are measured at fair value through profit or loss. Fair value cannot be measured based on quoted prices in active markets, or other observable input, and accordingly, derivative liabilities are measured by use of valuation techniques in form of the Black-Scholes Option Pricing model, where the pricing is exposed from changes in the Company’s own share price. Sensitivity analysis over derivative liabilities is disclosed in Note 10, “Financial Assets and Financial Liabilities.”
17
Serious News for Serious Traders! Try StreetInsider.com Premium Free!
You May Also Be Interested In
- Ascendis Pharma (ASND) Announces UK Approval of YORVIPATH
- Log10 Secures $7.2 Million in Seed Funding Led by TQ Ventures and Quiet Capital
- Iruka Hawaii Dolphin and Pearl Haven Celebrate Successful Marine Educational Event for Youth
Create E-mail Alert Related Categories
SEC FilingsSign up for StreetInsider Free!
Receive full access to all new and archived articles, unlimited portfolio tracking, e-mail alerts, custom newswires and RSS feeds - and more!
