

Form 20-F Centogene N.V. For: Dec 31
 Tweet
Tweet Share
Share
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 20-F
| (Mark One) | ||
o |
REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
| OR | ||
ý |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
For the fiscal year ended December 31, 2019 |
||
| OR | ||
o |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
| OR | ||
o |
SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
Date of event requiring this shell company report: |
||
For the transition period from t o |
||
Commission file number: 001-39124
Centogene N.V.
(Exact name of Registrant as specified in its charter)
The Netherlands
(Jurisdiction of incorporation or organization)
Am Strande 7
18055 Rostock, Germany
(+49) 381 80113 500
(Address of principal executive offices)
Arndt Rolfs,
Chief Executive Officer
Tel: (+49) 381 80113 500
Email: [email protected]
Am Strande 7
18055 Rostock, Germany
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered | ||
|---|---|---|---|---|
| Common shares, par value €0.12 per share | CNTG | The Nasdaq Stock Market LLC |
Securities registered or to be registered pursuant to Section 12(g) of the Act.
None
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act.
None
Indicate the number of outstanding shares of each of the issuer's classes of capital or common stock as of the close of the period covered by the annual report.
The number of outstanding common shares as of December 31, 2019 was:
| Title of each class | Number of Shares Outstanding at December 31, 2019 | |
|---|---|---|
| Common shares, par value of €0.12 per share | 19,861,340 |
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes o No ý
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934.
Yes o No ý
Note—Checking the box above will not relieve any registrant required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 from their obligations under those Sections.
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes ý No o
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Yes o No o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or an emerging growth company. See definition of "large accelerated filer," "accelerated filer," and "emerging growth company" in Rule 12b-2 of the Exchange Act.
Large Accelerated Filer o Accelerated Filer o Non-accelerated Filer ý Emerging growth company ý
- †
- The term "new or revised financial accounting standard" refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012.
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards† provided pursuant to Section 13(a) of the Exchange Act.
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
- o
- U.S. GAAP
- ý
- International
Financial Reporting Standards as issued by the International Accounting Standards Board
- o
- Other
If "Other" has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow.
o Item 17 o Item 18
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
Yes o No ý
TABLE OF CONTENTS
i
Except otherwise indicated and except where the context otherwise requires, references in this Annual Report on Form 20-F to:
| "Centogene", the "Company", "we", "our", "ours", "us" or similar terms | are to Centogene N.V. or Centogene N.V. together with its subsidiaries, as the context may require; | |
"Exchange Act" |
are to the United States Securities Exchange Act of 1934, as amended; |
|
"FDA" |
are to the United States Food and Drug Administration; |
|
"IASB" |
are to International Accounting Standards Board; |
|
"IFRS" |
are to International Financial Reporting Standards; |
|
"SEC" |
are to the United States Securities and Exchange Commission; |
|
"Securities Act" |
are to the Securities Act of 1933, as amended; |
|
"€", "EUR" and "euro" |
are to the European currency euro; and |
|
"$," "USD," "US$" and "U.S. dollar" |
are to the United States dollar. |
1
PRESENTATION OF FINANCIAL INFORMATION
We report under International Financial Reporting Standards ("IFRS"), as issued by the International Accounting Standards Board (the "IASB"). We present our consolidated financial statements in accordance with IFRS. We have made rounding adjustments to some of the figures included in this Annual Report. Accordingly, numerical figures shown as totals in some tables may not be an arithmetic aggregation of the figures that preceded them.
Our financial statements included in this Annual Report are presented in euro and, unless otherwise specified, all monetary amounts are in euro. All references in this Annual Report to "$", "U.S. dollars" and "dollars" means U.S. dollars and all references to "€", "EUR" and "euro" mean euro, unless otherwise noted.
In this Annual Report, unless otherwise indicated, some euro amounts related to our initial public offering, which closed on November 8, 2019, have been translated into U.S. dollars at the rate of $1.1129 to €1.00, the official exchange rate quoted as of October 17, 2019 by the U.S. Federal Reserve Bank. For information on the exchange rate used for the Group's consolidated financial statements as of December 31, 2019 and 2019 and for the three years ended December 31, 2017, 2018 and 2019, included in this Annual Report, please see "Note 5(a)—Foreign currency and currency translations" to such financial statements.
We have historically conducted our business through Centogene AG, and therefore our historical financial statements present the results of operations and financial condition of Centogene AG and its controlled subsidiaries. In connection with our initial public offering, Centogene N.V. became the holding company of Centogene AG, and the historical consolidated financial statements of Centogene AG became the historical consolidated financial statements of Centogene N.V. Centogene AG was acquired by Centogene B.V., which subsequently converted into Centogene N.V., on November 7, 2019, as part of our corporate reorganization in connection with our initial public offering, and the historical consolidated financial statements of Centogene AG included in this Annual Report became the historical consolidated financial statements of Centogene N.V. On March 5, 2020, the Company resolved that Centogene AG shall be converted into a German limited liability company and renamed Centogene GmbH. Such conversion will become effective upon the registration in the German commercial register which is expected to be completed in the financial year ending December 31, 2020.
USE OF TRADEMARKS, TRADE NAMES AND SERVICE MARKS
CENTOGENE™ is our main trademark. The trademarks, trade names and service marks appearing in this Annual Report are property of their respective owners. Solely for convenience, the trademarks and trade names in this Annual Report are referred to without the symbols ® and ™, but such references should not be construed as any indication that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto.
This Annual Report contains statements that constitute forward-looking statements. All statements other than present and historical facts and conditions contained in this Annual Report, including statements regarding our future results of operations and financial position, business strategy, plans and our objectives for future operations, are forward-looking statements. When used in this Annual Report, the words "anticipate," "believe," "can," "could," "estimate," "expect," "intend," "is designed to," "may," "might," "plan," "potential," "predict," "objective," "should," or the negative of these and similar expressions identify forward-looking statements.
Forward-looking statements are based on our management's beliefs and assumptions and on information currently available to our management. Such statements are subject to risks and
2
uncertainties, and actual results may differ materially from those expressed or implied in the forward-looking statements due to various factors, including, but not limited to, those identified under "Item 3. Key Information—D. Risk Factors" in this Annual Report. These risks and uncertainties include factors relating to:
- •
- our ability to effectively manage our future growth and to execute our business strategy;
- •
- our ability to generate sufficient revenue from our relationships with our pharmaceutical partners and clients, and to otherwise maintain our
current relationships, or enter into new relationships, with pharmaceutical partners and clients;
- •
- the effects of the COVID-19 pandemic on our business and results of operations;
- •
- economic, political or social conditions and the effects of these conditions on our pharmaceutical partners' and diagnostics clients'
businesses and levels of business activity;
- •
- our expectations for our products and solutions achieving commercial market acceptance, and our ability to keep pace with the rapidly evolving
industry in which we operate;
- •
- our assumptions regarding market size in the rare disease industry and our growth potential;
- •
- our pharmaceutical partners' and clients' need for rare disease information products and solutions and any perceived advantage of our products
over those of our competitors;
- •
- our ability to manage our international expansion, including our exposure to new and complex business, regulatory, political, operational,
financial, and economic risks, and numerous and conflicting legal and regulatory requirements;
- •
- our continued reliance on our senior management team, in particular our CEO, and other qualified personnel and our ability to retain such
personnel;
- •
- our ability to obtain, maintain, protect and enforce sufficient patent and other intellectual property protection for any products or solutions
we develop and for our technology;
- •
- the ongoing protection of our trade secrets, know-how, and other confidential and proprietary information;
- •
- our ability to remediate our material weakness on internal control over financial reporting;
- •
- general economic, political, demographic and business conditions in North America, the Middle East, Europe and other regions in which we
operate;
- •
- changes in government and industry regulation and tax matters;
- •
- other factors that may affect our financial condition, liquidity and results of operations; and
- •
- other risk factors discussed under "Item 3. Key Information—D. Risk Factors."
You should refer to the section of this Annual Report titled "Item 3. Key Information—D. Risk Factors" for a discussion of important factors that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. As a result of these factors, we cannot assure you that the forward-looking statements in this Annual Report will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame or at all. We undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law.
3
We are incorporated under the laws of the Netherlands, and our headquarters are located in Germany. Substantially all of our assets are located outside the United States. The majority of our management board and supervisory board reside outside the United States. As a result, it may not be possible for investors to effect service of process within the United States upon such persons or to enforce against them or us in U.S. courts, including judgments predicated upon the civil liability provisions of the federal securities laws of the United States.
There is currently no treaty between the United States and the Netherlands for the mutual recognition and enforcement of judgments (other than arbitration awards) in civil and commercial matters. Therefore, a final judgment for the payment of money rendered by any federal or state court in the United States based on civil liability, whether or not predicated solely upon the U.S. federal securities laws, would not be enforceable in the Netherlands unless the underlying claim is relitigated before a Dutch court of competent jurisdiction. Under current practice, however, a Dutch court will generally, subject to compliance with certain procedural requirements, grant the same judgment without a review of the merits of the underlying claim if such judgment (i) is a final judgment and has been rendered by a court, which has established its jurisdiction vis-à-vis the relevant Dutch companies or Dutch company, as the case may be, on the basis of internationally accepted grounds of jurisdiction, (ii) has not been rendered in violation of principles of proper procedure (behoorlijke rechtspleging), (iii) is not contrary to the public policy of the Netherlands, and (iv) is not incompatible with (a) a prior judgment of a Netherlands court rendered in a dispute between the same parties, or (b) a prior judgment of a foreign court rendered in a dispute between the same parties, concerning the same subject matter and based on the same cause of action, provided that such prior judgment is capable of being recognized in the Netherlands and except to the extent that the foreign judgement contravenes Dutch public policy (openbare orde). Dutch courts may deny the recognition and enforcement of punitive damages or other awards. Moreover, a Dutch court may reduce the amount of damages granted by a U.S. court and recognize damages only to the extent that they are necessary to compensate actual losses or damages. Enforcement and recognition of judgments of U.S. courts in the Netherlands are solely governed by the provisions of the Dutch Code of Civil Procedure. Based on the foregoing, there can be no assurance that U.S. investors will be able to enforce any judgments obtained in U.S. courts in civil and commercial matters, including judgments under the U.S. federal securities.
The United States and Germany currently do not have a treaty providing for the reciprocal recognition and enforcement of judgments, in civil and commercial matters. Consequently, a final judgment for payment or declaratory judgments given by a court in the United States, whether or not predicated solely upon U.S. securities laws, would not automatically be recognized or enforceable in Germany. German courts may deny the recognition and enforcement of a judgment rendered by a U.S. court if they consider the U.S. court not to be competent or the decision to be in violation of German public policy principles. For example, judgments awarding punitive damages are generally not enforceable in Germany. A German court may reduce the amount of damages granted by a U.S. court and recognize damages only to the extent that they are necessary to compensate actual losses or damages.
In addition, actions brought in a German court against us, our management board and supervisory board and the experts named herein to enforce liabilities based on U.S. federal securities laws may be subject to certain restrictions. In particular, German courts generally do not award punitive damages. Litigation in Germany is also subject to rules of procedure that differ from the U.S. rules, including with respect to the taking and admissibility of evidence, the conduct of the proceedings and the allocation of costs. German procedural law does not provide for pre-trial discovery of documents, nor does Germany support pre-trial discovery of documents under the 1970 Hague Evidence Convention. Proceedings in Germany would have to be conducted in the German language and all documents submitted to the court would, in principle, have to be translated into German. For these reasons, it may be difficult for a U.S. investor to bring an original action in a German court predicated upon the civil liability provisions of the U.S. federal securities laws against us, our management board and supervisory board and the experts named in this Annual Report.
4
Item 1. Identity of Directors, Senior Management and Advisers
Not applicable
Item 2. Offer Statistics and Expected Timetable
Not applicable
A. Selected Financial Data
The following selected consolidated statements of financial position as of December 31, 2018 and 2019, and the consolidated statements of comprehensive loss for the years ended December 31, 2017, 2018 and 2019 are derived from the consolidated financial statements appearing elsewhere in this Annual Report, which have been audited by Ernst & Young GmbH Wirtschaftsprüfungsgesellschaft ("Ernst & Young"). The following selected consolidated statement of financial position data as of December 2017, and consolidated statement of comprehensive loss data for the year ended December 31, 2016 are derived from our audited consolidated financial statements included in our registration statement on Form F-1 (File No. 333-234177).
Financial information presented in the consolidated financial statements of Centogene N.V. for periods prior to the completion of our corporate reorganization is that of Centogene AG, our wholly-owned subsidiary. The consolidated financial statements of Centogene N.V. are a continuation of the historical consolidated financial statements of Centogene AG.
The information presented below is qualified by the more detailed historical consolidated financial statements set forth in this Annual Report, and should be read in conjunction with those consolidated financial statements, the notes thereto and the discussion under "Item 5. Operating and Financial Review and Prospects" included elsewhere in this Annual Report.
We maintain our books and records in euros, and we prepare our financial statements under IFRS as issued by the IASB.
We have not included selected consolidated financial data as of December 31, 2015 and 2016 and for the year ended December 31, 2015 in the tables below as we qualify as an emerging growth
5
company under the Jumpstart Our Business Startups Act of 2012, or the JOBS Act, and as defined in Section 2(a)(19) of the Securities Act, and we make use of an accommodation for reduced reporting.
| |
For the Years Ended December 31, | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2016 | 2017 | 2018 | 2019 | |||||||||
| |
(€ in thousands) |
||||||||||||
Consolidated statement of comprehensive loss: |
|||||||||||||
Revenue |
27,669 | 31,689 | 40,478 | 48,780 | |||||||||
Cost of sales |
12,856 | 14,939 | 19,941 | 26,005 | |||||||||
| | | | | | | | | | | | | | |
Gross profit |
14,813 | 16,750 | 20,537 | 22,775 | |||||||||
| | | | | | | | | | | | | | |
Research and development expenses |
5,885 | 6,396 | 6,300 | 9,590 | |||||||||
General administrative expenses |
8,888 | 9,498 | 18,610 | 23,160 | |||||||||
Selling expenses |
5,364 | 5,897 | 7,474 | 9,254 | |||||||||
Other operating income |
1,295 | 1,043 | 2,306 | 3,781 | |||||||||
Other operating expenses |
908 | 457 | 1,065 | 2,036 | |||||||||
Real estate transfer tax expenses |
— | — | — | 1,200 | |||||||||
| | | | | | | | | | | | | | |
Operating loss |
(4,937 | ) | (4,455 | ) | (10,606 | ) | (18,684 | ) | |||||
| | | | | | | | | | | | | | |
Interest and similar income |
26 | 14 | 33 | 16 | |||||||||
Interest and similar expense |
856 | 1,021 | 1,075 | 2,029 | |||||||||
| | | | | | | | | | | | | | |
Finance costs, net |
(830 | ) | (1,007 | ) | (1,042 | ) | (2,013 | ) | |||||
| | | | | | | | | | | | | | |
Loss before taxes |
(5,767 | ) | (5,462 | ) | (11,648 | ) | (20,697 | ) | |||||
Income tax (benefits)/expenses |
(408 | ) | 14 | (310 | ) | 158 | |||||||
| | | | | | | | | | | | | | |
Loss for the year |
(5,359 | ) | (5,476 | ) | (11,338 | ) | (20,855 | ) | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Other comprehensive income/(loss) |
9 | 10 | (8 | ) | 16 | ||||||||
| | | | | | | | | | | | | | |
Total comprehensive loss for the period |
(5,350 | ) | (5,466 | ) | (11,346 | ) | (20,839 | ) | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Loss per share—Basic and diluted(1) |
(0.5 | ) | (0.4 | ) | (0.8 | ) | (1.3 | ) | |||||
Weighted average number of outstanding shares |
10,542,749 | 12,065,714 | 14,112,841 | 16,409,285 | |||||||||
- (1)
- Basic and diluted loss per share is calculated by dividing loss for the year attributable to our equity holders by the weighted average number of shares outstanding during the same period, adjusted for the effect of the corporate reorganization and is represented in euros per share.
| |
As of December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2017 | 2018 | 2019 | |||||||
| |
(€ in thousands) |
|||||||||
Consolidated statement of financial position: |
||||||||||
Cash and cash equivalents |
3,157 | 9,222 | 41,095 | |||||||
Total assets |
55,486 | 76,674 | 117,510 | |||||||
Total current liabilities |
23,808 | 24,283 | 29,000 | |||||||
Total non-current liabilities |
15,324 | 25,867 | 29,588 | |||||||
Total equity |
16,354 | 26,524 | 58,922 | |||||||
B. Capitalization and Indebtedness
Not applicable
C. Reason for the Offer and Use of Proceeds
Not applicable
6
D. Risk Factors
You should carefully consider the following risks and uncertainties and all of the other information in this Annual Report before making any investment decision. Our business, financial condition or results of operations could be materially and adversely affected if any of these risks occurs, and as a result, the market price of our common shares could decline and you could lose all or part of your investment. The risks described below are those that we currently believe may materially affect us. We may face additional risks and uncertainties not currently known to us or that we currently deem to be immaterial.
Certain Factors Relating to Our Business and Strategy
We may fail to generate sufficient revenue from our relationships with our clients or pharmaceutical partners to achieve and maintain profitability.
We believe our commercial success is dependent upon our ability to successfully market and sell our products and solutions to clients and pharmaceutical partners, to continue to sell our suite of diagnostic tests, to continue to expand our current relationships and to develop new relationships with pharmaceutical partners. The demand for our existing services may decrease or may not continue at historical rates for a number of reasons, including, among others, the development by competitors of new products or solutions that we are not able to commercialize, and increased competition from companies that offer similar products and solutions. In addition to reducing our revenue, if our pharmaceutical partners or clients decide to decrease or discontinue their partnerships or relationships with us, and their use of our knowledge and interpretation-based solutions, this may reduce our access to research and patient data that facilitates the incorporation of newly developed information about rare diseases into our data repository. Our business model and strategy depend on the continued input of new data into our repository, and any such reduction in access to research and patient data could affect our ability to offer the same quality and scope of solutions to our pharmaceutical partners and other clients, which could adversely affect or business, prospects, financial condition and results of operations.
We are currently not profitable. Even if we succeed in increasing adoption of our existing solutions by pharmaceutical partners or tests by our clients or pharmaceutical partners, we may fail to generate sufficient revenue to achieve and maintain profitability.
The COVID-19 pandemic could adversely impact our business and results of operations.
In December 2019, the COVID-19 virus, commonly known as "coronavirus", surfaced in Wuhan, China. In March 2020, the World Health Organization declared the COVID-19 outbreak a pandemic. The COVID-19 disease has spread from China to many other countries including the U.S., the UK and the EU, with the number of reported cases and related deaths increasing daily and, in many countries, at a very rapid pace.
Many governments, including in the United States and Germany, have imposed increasingly stringent restrictions to seek to mitigate, or slow down, the spread of COVID-19, including restrictions on international and local travel, public gatherings and participation in business meetings, as well as closures of workplaces, schools, and other sites, and are requesting "social distancing." In addition to those government measures, we have also taken a series of actions aimed at safeguarding the Company's employees and business associates, including implementing a work-from-home policy for employees except for those related to our laboratory operations. The duration of such measures is highly uncertain, but could be prolonged, and even stricter measures may be put in place.
In March and April 2020, the COVID-19 pandemic, coupled with a sharp material decline in the oil price, caused financial markets globally to experience material declines and very elevated volatility, signaling a likely economic downturn and adverse impact on GDP and broader economic conditions,
7
including in Germany and the United States. There is no assurance that the responses from central banks (which include reductions in interest rates and liquidity support) and financial support and fiscal spending by certain governments will be sufficient to support the U.S. or other economies or that financial markets will return to normal.
There is significant uncertainty relating to the potential effect of the COVID-19 virus on our business. Any of the factors above could result in significant volatility in, and have a material adverse effect on, our business, financial condition, rating and results of operations. In particular, they could result in increased costs of execution with regards to operational plans. In addition, COVID-19 may disrupt our supply chain, particularly as it relates to the United States (from where a significant proportion of our sequencing products are sourced) as well other countries in which we operate and from where we receive tests, and otherwise adversely affect international trade and business activities. The magnitude of the impact on us will depend on future developments, which are highly uncertain and cannot be predicted with confidence, including, among other things, the duration of the outbreak and counter-measures. The effects of COVID-19 on us, and on the environment in which we operate, has resulted, and may continue to result, in significant volatility of the trading price of our common shares.
To the extent the COVID-19 pandemic adversely affects our business and financial results, it may also have the effect of heightening many of the other risks described under "—We may face restrictions or delays in the receipt of patient samples to our laboratories for genetic testing" and "—We may be adversely affected by volatile, negative or uncertain economic, political or social conditions and the effects of these conditions on our pharmaceutical partners' and diagnostics clients' businesses and levels of business activity."
We may fail to maintain our current relationships with pharmaceutical companies, or enter into new relationships on a similar scale.
Our success in the future depends in part on our ability to maintain relationships and to enter into new relationships with pharmaceutical partners. Partnerships are complex and time-consuming to negotiate and document. Whether we reach a definitive agreement for a partnership will depend on a number of factors, including, among other things, upon our partners' assessment of our industry knowledge, data repository, logistical resources and expertise, the terms and conditions of the proposed partnership, and our partners' evaluation of the potential value added from our rare disease knowledge and insights. If we are unable to do so, we may have to curtail our research on a particular rare disease or increase our expenditures and undertake research and development activities at our own expense. Further, there have been a significant number of recent business combinations among large pharmaceutical companies that have resulted in a reduced number of potential future partners.
Our ability to maintain our current relationships with our pharmaceutical partners, or enter into new relationships, can be difficult due to several factors, including that:
- •
- our products and solutions are focused towards facilitating the development of rare disease treatments which limits our market to
pharmaceutical partners active in the rare disease space;
- •
- orphan drug development is complex, expensive and time-consuming due to limited identified patient populations and limited industry knowledge
of rare diseases;
- •
- our pharmaceutical partners may decide to decrease or discontinue their use of our rare disease information platform due to circumstances outside of our control, including changes in their research and development plans, whether they can obtain positive data or regulatory approval in clinical trials or successfully commercialize a treatment, changes in the regulatory environment, or utilization of internal testing resources or genetic tests performed by other parties, among others;
8
- •
- internal and external constraints may be placed on potential pharmaceutical partners that can limit the number and type of relationships with
companies like us they can consider and consummate; and
- •
- our pharmaceutical partners may be dissatisfied with our products or solutions or that we may fail to deliver expected benefits from our products or solutions.
Additionally, some of our pharmaceutical partners have contracted with us to provide testing for large numbers of samples or to focus our research on a particular rare disease, which could restrict our ability to perform tests for other clients or pharmaceutical partners or limit our ability to expand our data repository outside of a specified patient population or rare disease. If we fail to maintain our current relationships with our pharmaceutical partners, or enter into new partnerships, our business could suffer.
Because the identified patient populations for rare diseases are relatively small, it may be difficult to successfully identify patients for our pharmaceutical partners.
Our inability to identify a sufficient number of patients for our partners' clinical trials could result in significant delays and could require our partners to abandon one or more clinical trials altogether. Enrollment delays in our partners' clinical trials may result in increased development costs for our partners' drug candidates, which would cause the value of the solutions which we offer to our pharmaceutical partners to decline. If we are unable to identify patients with a specified driver of disease or applicable genomic alteration, this could compromise our ability to add value to our partners' clinical trials by accelerating clinical development and regulatory timelines. In addition, our projections of both the number of people who have these diseases, as well as the subset of people with these diseases who have the potential to benefit from treatment with our partners' existing treatments or drug candidates, are based on our internal estimates derived from data in our repository. These estimates may prove to be incorrect, and new studies may reduce the estimated incidence or prevalence of these diseases. The number of patients in the United States, European Union and elsewhere may turn out to be lower than expected, may not be otherwise amenable to treatment with our partners' drug candidates or patients may be difficult to identify and access, all of which would adversely affect our business, prospects and ability to achieve or sustain profitability.
We may fail to generate sufficient volumes of data from our diagnostic tests for inclusion in our data repository.
Our business model assumes that we will be able to continue to generate significant diagnostic test volume in order to maintain the generation of data that feeds into our data repository, which is necessary for the development of new products and solutions for our pharmaceutical partners and clients. We may not succeed in continuing to drive clinical adoption of our tests to achieve sufficient volumes. Inasmuch as detailed genetic data from our tests have only recently become available at relatively affordable prices, the pace and degree of clinical acceptance of the utility of such testing is uncertain. Specifically, it is uncertain how much genetic data will be accepted as necessary or useful, as well as how detailed that data should be, particularly since medical practitioners may have become accustomed to genetic testing that is specific to one or a few genes. To generate demand for our tests, we will need to continue to make our diagnostics clients, as well as physicians and key opinion leaders, aware of the benefits of our tests, including the price, the breadth of our testing options, and the benefits of having additional genetic data available from which to make treatment decisions. In addition, physicians in other areas of medicine may not adopt genetic testing for certain rare diseases as readily as it has been adopted for some more well-known rare diseases and our efforts to sell our tests to physicians outside of a set number of rare diseases may not be successful. A lack of or delay in increased clinical acceptance of our diagnostic tests would negatively impact sales and market acceptance of our tests and limit our ability to expand on the scope and quality of knowledge and
9
interpretation-based solutions offered to our pharmaceutical partners, which could in turn impact our revenue growth and potential profitability.
In addition, genetic testing is still relatively expensive and many potential pharmaceutical partners and clients may be sensitive to pricing concerns. Potential pharmaceutical partners or clients may not adopt our tests if adequate reimbursement is unavailable, or if we are not able to maintain low prices in the future relative to our competitors. If we are not able to generate demand for our tests at sufficient volume, or if it takes significantly more time to generate this demand than we anticipate, our business, prospects, financial condition and results of operations could be materially harmed.
We derive a large proportion of our revenues from agreements with a limited number of pharmaceutical partners and clients.
We have historically earned a large proportion of our revenue from a limited number of pharmaceutical partners and diagnostic testing clients. In the years ended December 31, 2019 and 2018, our top five pharmaceutical partners, in the aggregate, accounted for 39.3% and 39.0% of our revenues, respectively. The loss of, or material reduction in, revenues from any one of our major pharmaceutical partners or clients could materially reduce our total revenues, harm our reputation in the industry and/or reduce our ability to accurately predict our revenue, net income and cash flow. The loss of, or material reduction, in revenue from any one of our major pharmaceutical partners or clients could also adversely affect our gross profit and utilization as we seek to redeploy resources previously dedicated to that partner. We cannot assure you that revenue from our major pharmaceutical partners or clients will not be significantly reduced in the future. We also may not be able to maintain our relationships with our major pharmaceutical partners or clients on existing or on continued favorable terms and our major pharmaceutical partners or clients may not renew their agreements with us, in which case our business, financial condition and results of operations would be adversely affected.
In particular, during the year ended December 31, 2019, our collaboration with Shire International GmbH ("Shire"), now a subsidiary of Takeda Pharmaceutical Company Limited, represented 24.3% of our total revenues. We expect that our collaboration with Shire will continue to account for a material portion of our revenue in 2020. The revenue attributable to Shire may fluctuate in the future, which could have an adverse effect on our financial condition and results of operations. In addition, changes in the terms of our agreements with Shire, or a modification or termination of our relationship with Shire, could result in delays in the receipt of revenue by us, or a temporary or permanent loss of revenue to us. In addition, certain pharmaceutical companies, including those with which we currently have agreements, may choose not to do business with us or may seek out other partners for genetic rare disease information due to our strategic collaboration with Shire, particularly if they are actual or potential competitors with Shire. If we are unable to continue to grow our business with other pharmaceutical companies, our business and results of operations would be adversely affected.
Our client concentration may also subject us to perceived or actual leverage that our pharmaceutical partners or clients may have, given their relative size and importance to us. If our pharmaceutical partners or clients seek to negotiate their agreements on terms less favorable to us and we accept such unfavorable terms, this may have a material adverse effect on our business, financial condition and results of operations. Accordingly, unless and until we diversify and expand our client base, our future success will significantly depend upon the timing and volume of business from our largest pharmaceutical partners and clients and the financial and operational success of these pharmaceutical partners and clients.
10
We may be adversely affected by volatile, negative or uncertain economic, political or social conditions and the effects of these conditions on our pharmaceutical partners' and diagnostics clients' businesses and levels of business activity.
Global economic conditions affect our pharmaceutical partners' and diagnostic clients' businesses and the markets they serve, and volatile, negative or uncertain economic conditions may have an adverse effect on our revenue growth and profitability. Volatile, negative or uncertain economic conditions in our significant markets, in particular in our North America, Middle East or European regions, where we generated 47.7%, 28.9% and 15.3%, respectively, of our total revenues for the year ended December 31, 2019, could undermine business confidence, both in those markets and other markets, and cause our pharmaceutical partners or clients to reduce or defer their spending on new technologies or initiatives or terminate existing contracts, which would negatively affect our business. Growth in the markets we serve could be at a slow rate, or could stagnate, for an extended period of time. Differing economic conditions and patterns of economic growth and contraction in the geographical regions in which we operate and the industries we serve may affect demand for our products and solutions. Weakening in these markets as a result of high government deficits, credit downgrades or otherwise could have a material adverse effect on our results of operations. Ongoing economic volatility and uncertainty affects our business in a number of other ways, including making it more difficult to accurately forecast partner demand beyond the short term and effectively build our revenue and resource plans, particularly given the iterative nature of the negotiation of new contracts with our pharmaceutical partners. This could result, for example, in us not having the level of appropriate personnel where they are needed, and could have a significant negative impact on our results of operations.
Moreover, acts of terrorist violence, political unrest, armed regional and international hostilities and international responses to these hostilities, natural disasters, global health risks or pandemics or the threat of or perceived potential for these events could have a negative impact on us. These events could adversely affect our pharmaceutical partners' levels of business activity and precipitate sudden significant changes in regional and global economic conditions and cycles. These events also pose significant risks to our people and to physical facilities and operations around the world, whether the facilities are ours or those of our distributors, pharmaceutical partners or physicians that utilize our diagnostic testing services. By disrupting communications and travel and increasing the difficulty of obtaining and retaining highly skilled and qualified personnel, these events could make it difficult or impossible for us to deliver products and solutions to our clients and pharmaceutical partners. Extended disruptions of electricity, other public utilities or network services at our facilities, as well as system failures at, or security breaches in, our facilities or systems, could also adversely affect our ability to serve our clients and pharmaceutical partners. We might be unable to protect our people, facilities and systems against all such occurrences. We generally do not have insurance for losses and interruptions caused by terrorist attacks, conflicts and wars. If these disruptions prevent us from effectively serving our clients and pharmaceutical partners, our results of operations could be adversely affected.
We may face restrictions or delays in the receipt of patient samples to our laboratories for genetic testing.
Our business depends on our ability to quickly and reliably receive samples from physicians. Our CentoCard product is typically sent from locations worldwide to our laboratory in Rostock, Germany as well as our Cambridge, Massachusetts facility. Disruptions in delivery, whether due to factors beyond our control such as natural disasters, terrorist threats, political instability, governmental policies, failures by physicians to properly label or package the samples, failure by postage services, labor disruptions, bad weather or other factors could adversely affect the receipt by us of samples or specimen integrity and could impact our ability to process samples in a timely manner and to provide our services to our clients and pharmaceutical partners. In particular, there is a general trend in certain countries, for
11
example in China and certain countries in South America, where policies have been introduced that restrict the processing of genetic testing outside the country in which the patient is located. This could disrupt the transportation of samples to our testing facilities in Germany and the United States from such countries, and could adversely impact our current business operations or prevent us from expanding into certain new regions.
In addition, the majority of our samples are delivered to us via regular postal services worldwide. If such services are disrupted, or if we are unable to continue to obtain expedited delivery services or specialized delivery services for certain products, such as our prenatal algorithmic test, on commercially reasonable terms, our operating results may be adversely affected.
We may become subject to substantial product liability or professional liability claims that could exceed our resources.
The marketing, sale and use of our products and solutions could lead to the filing of product liability claims if someone were to allege that our products and solutions identified inaccurate or incomplete information regarding the genomic alterations of the rare disease indication analyzed, reported inaccurate or incomplete information concerning the available treatments for a certain type of rare disease or otherwise failed to perform as designed. For example, we have been subject to a claim from a client that our prenatal diagnostic test conducted at their request failed to identify a specific mutation present in a patient. See "Item 4. Information On The Company—B. Business Overview—Legal Proceedings" and "Item 8. Financial Information—A. Consolidated Statements and Other Financial Information—Legal Proceedings." We may also be subject to liability for errors in, a misunderstanding of, or inappropriate reliance upon, the information we provide in the ordinary course of our business activities. A product liability or professional liability claim could result in substantial damages and be costly and time-consuming for us to defend.
Our service and professional liability insurance may not fully protect us from the financial impact of defending against product liability or professional liability claims. Any product liability or professional liability claim brought against us, with or without merit, could increase our insurance rates or prevent us from securing insurance coverage in the future. Additionally, any product liability lawsuit could damage our reputation or cause current clients or pharmaceutical partners to terminate existing agreements and potential clients or pharmaceutical partners to seek other partners, any of which could impact our results of operations.
If the validity of a consent from a patient was challenged, we could be forced to stop using certain of our data resources, which would impede our rare disease information development efforts.
We provide diagnostic testing services to patients of our pharmaceutical partners and diagnostics clients worldwide. We also provide products and solutions, including biomarker development and testing, to our pharmaceutical partners. Such products and solutions involve the aggregation of data obtained from patients in our existing data repository and data obtained from new tests conducted both on patients whose samples remain in our biobank or new patients from whom we collect samples.
To a large extent, we also rely upon our pharmaceutical partners, our clients and, in some cases, third-party laboratories to collect the subject's informed consent and comply with applicable local laws and international regulations. Although we maintain policies and procedures designed to monitor the collection of consents by both ourselves and such third parties, we or third parties may not obtain the required consents in a timely manner, or at all. In addition, consents that we have obtained or will obtain may not meet the existing or future standards required by relevant governmental authorities.
The collection of data and samples in many different countries results in complex legal questions regarding the adequacy of consent and the status of genetic material under a large number of different legal systems. In some jurisdictions, tissue samples that contain a person's DNA might irrevocably
12
qualify as personal data, as in theory such samples can never be completely anonymized. Legitimate interests of the donor might cause a "revival" of his or her personal rights in the future and limit our rights of utilization. The subject's consent obtained in any particular country could be withdrawn or challenged in the future, and those consents could prove invalid, unlawful, or otherwise inadequate for our purposes. Furthermore, we may face disputes with patients should their data be used in a manner which they did not expect or if the consent was recorded incorrectly or obtained fraudulently. Any findings against us, or our pharmaceutical partners, clients or distributors, could deny us access to or force us to stop using certain of our clinical data or samples, which would impede our genetic information solution development efforts. We could become involved in legal challenges, which could consume our management and financial resources.
If access to our highly specialized laboratory facilities, storage facilities or equipment is interrupted or damaged, our business could be negatively impacted.
Our diagnostic testing products and pharmaceutical solutions are rendered at our laboratory facilities. We currently run the majority of our diagnostic testing at our laboratory in Rostock, Germany, and we also commenced operations at our laboratory in Cambridge, Massachusetts in August 2018. If one or more of our laboratories, and particularly our facility in Rostock, become inoperable or some or all of our key equipment ceases to function even for a short period of time, we may be unable to perform our genetic tests or develop solutions in a timely manner or at all, which may result in the loss of clients and pharmaceutical partners or harm to our reputation, and we may be unable to regain those clients and pharmaceutical partners or repair our reputation in the future. Our facilities and equipment could be harmed or rendered inoperable by natural or man-made disasters, including war, fire, earthquake, flood, power loss, communications or internet failure or interruption, or terrorism, which may render it difficult or impossible for us to operate our genetic rare disease information platform for some period of time.
In particular, the biomaterials that are stored in our biobank are located in our Rostock facility. Should the biomaterials that we store there be damaged or destroyed, we would lose part or all of our existing biomaterials and as a result we would not be able to retest this material for future research and development uses.
Furthermore, our facilities and the equipment we use to perform our research and development work could be unavailable or costly and time-consuming to repair or replace. It would be difficult, time-consuming, and expensive to rebuild any of our facilities or license or transfer our proprietary technology to a third party, particularly in light of the licensure and accreditation requirements and specific equipment needed for laboratories like ours. Even in the unlikely event we are able to find a third party with such qualifications to enable us to perform our genetic tests or develop our solutions, we may be unable to negotiate commercially reasonable terms with such third parties. Any interruption of our laboratory operations could harm relationships with our clients and pharmaceutical partners or regulatory authorities, which could adversely affect our ability to generate revenue or maintain compliance with regulatory standards.
While we carry insurance for damage to our property and laboratory and the disruption of our business, such insurance may not cover all of the risks associated with damage to our property or laboratory or disruption to our business, may not provide coverage in amounts sufficient to cover our potential losses, may be challenged by insurers underwriting the coverage, and may not continue to be available to us on acceptable terms, if at all.
13
We depend upon our information technology systems, and any failure of these systems could harm our business.
We depend on information technology and telecommunications systems for significant elements of our operations, including our repository, our CentoMD database, our CentoPortal client-facing platform, our laboratory information management system, our third-party datacenter solutions, our broadband connections and our client relationship management system. We have installed a number of enterprise software systems that affect a broad range of business processes and functional areas, including, for example, systems handling human resources, financial controls and reporting, contract management and other infrastructure operations. These information technology systems support a variety of functions, including laboratory operations, test validation, sample tracking, quality control, customer service support, billing and reimbursement, research and development activities, scientific and medical curation, and general administrative activities. In addition, our system is backed up by two offsite data centers that offer a disaster recovery system for our database in separate locations near Frankfurt. Any technical problems that may arise in connection with third-party data center hosting facilities could result in interruptions in our service.
Our information technology systems are vulnerable to damage from a variety of sources, including network failures, malicious human acts, and natural disasters. Our business will also be harmed if our laboratory partners and potential laboratory partners believe our service is unreliable. Moreover, despite network security and back-up measures, some of our servers are potentially vulnerable to physical or electronic break-ins, malicious computer software (malware), and similar disruptive problems. Failures or significant downtime of our information technology systems, or those used by our third-party service providers, could prevent us from conducting our comprehensive genomic analyses, preparing and providing reports and data to partners and physicians, billing payors, processing reimbursement appeals, handling patient or physician inquiries, conducting research and development activities, and managing the administrative aspects of our business. Additionally, to the extent that any disruption or security breach results in a loss or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we may incur significant liability. Any disruption or loss of information technology or telecommunications systems on which critical aspects of our operations depend could have an adverse effect on our business.
We rely on a limited number of suppliers, or, in some cases, a sole supplier, for some of our laboratory equipment and may not be able to find replacements or immediately transition to alternative suppliers.
We believe that there are only a few equipment manufacturers that are currently capable of supplying and servicing the sequencing equipment necessary for our laboratory operations. For example, we rely on a key supplier, Illumina, for certain sequencing equipment used for our processes. We may not be able to obtain acceptable substitute equipment from another supplier on the same basis or at all. Even if we are able to obtain acceptable substitutes from replacement suppliers, their use could require us to significantly alter our laboratory operations. An interruption in our laboratory operations could occur if we encounter delays or difficulties in securing or maintaining the proper function of this laboratory equipment. Any such interruption could negatively impact research and development and launches of new products or solutions, and significantly affect our business, financial condition, results of operations, and reputation.
The loss or transition of any member of our senior management team, in particular our CEO, or our inability to attract and retain new talent, could adversely affect our business.
Our success depends on the skills, experience, and performance of key members of our senior management team, and in particular our CEO, Prof. Arndt Rolfs. The individual and collective efforts of these employees will be important as we continue to develop our rare disease genetic information platform and additional products and solutions, and as we expand our commercial activities. The loss
14
or incapacity of existing members of our senior management team could adversely affect our operations if we experience difficulties in hiring qualified successors.
The complexity inherent in integrating a new key member of the senior management team with existing senior management may limit the effectiveness of any such successor or otherwise adversely affect our business. Leadership transitions can be inherently difficult to manage and may cause uncertainty or a disruption to our business or may increase the likelihood of turnover of other key officers and employees. Specifically, a leadership transition in the commercial team may cause uncertainty about or a disruption to our commercial organization, which may impact our ability to achieve sales and revenue targets.
Our research and development programs and laboratory operations depend on our ability to attract and retain highly skilled scientists and technicians. We may not be able to attract or retain qualified scientists and technicians in the future due to the intense competition for qualified personnel among life science businesses globally. We also face competition from universities and public and private research institutions in recruiting and retaining highly qualified scientific personnel. We may have difficulties locating, recruiting, or retaining qualified sales people. Recruitment and retention difficulties can limit our ability to support our research and development and sales programs.
International expansion of our business exposes us to new and complex business, regulatory, political, operational, financial, and economic risks.
Our business strategy incorporates plans for significant expansion in the countries in which we currently operate and internationally. Doing business internationally involves a number of risks, including:
- •
- multiple, conflicting, and changing laws and regulations such as data protection laws, privacy regulations, tax laws, export and import
restrictions, employment laws, regulatory requirements (including requirements related to patient consent, testing of genetic material and reporting the results of such testing) and other governmental
approvals, permits, and licenses, or government delays in issuing such approvals, permits, and licenses;
- •
- failure to obtain regulatory approvals for the manufacture and sale of our products and use of our products and solutions in various countries;
- •
- transition and management of our former distribution relationships in various countries;
- •
- potentially relevant third-party intellectual property rights;
- •
- difficulties in staffing and managing foreign operations;
- •
- complexities and difficulties in obtaining, maintaining, protecting and enforcing our intellectual property rights;
- •
- logistics and regulations associated with preparing, shipping, importing and exporting tissue and blood samples, including infrastructure
conditions, transportation delays, and customs;
- •
- limits in our ability to penetrate new geographical regions due to competition;
- •
- logistical issues or increases in costs of transporting tests and samples since our diagnostic tests are conducted primarily in Germany;
- •
- financial risks, such as the impact of local and regional financial crises on demand and payment for our products and solutions, and exposure
to foreign currency exchange rate fluctuations;
- •
- risks associated with operations in countries which have experienced, or are currently experiencing, high rates of inflation which increase our costs, inhibit economic growth and could lead to reduced demand for our products and solutions;
15
- •
- natural disasters, political, and economic instability, including wars, terrorism, and political unrest, outbreak of disease, boycotts,
curtailment of trade, and other business restrictions; and
- •
- regulatory and compliance risks that relate to maintaining accurate information and control over sales and distribution activities that may fall within the purview of the United States Foreign Corrupt Practices Act (the "FCPA") or comparable foreign regulations, including its books and records provisions, or its anti-bribery provisions.
Any of these factors could significantly harm our future international expansion and operations and, consequently, our revenue and results of operations. The difference in regulations under the laws of the countries in which we may expand and the laws of the countries in which we currently operate may be significant and, in order to comply with such new laws, we may have to implement global changes to our products and solutions or business practices. Such changes may result in additional expense to us and either reduce or delay development of our products and solutions, commercialization of our biomarkers and other solutions or expansion of our data repository and biobank. In addition, any failure to comply with applicable legal and regulatory obligations could affect us in a variety of ways that include, but are not limited to, significant criminal, civil and administrative penalties and restrictions on certain business activities. Also, the failure to comply with applicable legal and regulatory obligations could result in the disruption of our activities in these countries.
Failure to manage these and other risks may have a material adverse effect on our operations in any particular country and on our business as a whole.
Implementation of partnership agreements with our pharmaceutical partners may result in material unanticipated problems, expenses, liabilities, competitive responses, loss of client relationships and diversion of management's attention.
The negotiation of our existing partnership agreements, as well as any new partnership agreements that we enter into, take up significant management time and resources. Moreover, in part due to the complex nature of our partnership agreements, which typically provide for research and development collaboration as well as utilization of our genetic patient screening processes, we may need to expend capital and dedicate manpower to meeting the requirements of our pharmaceutical partners. Any partnership agreements that we enter into in the future may contain restrictions on our ability to enter into potential collaborations with other third parties, or to otherwise provide products and solutions in connection with a particular rare disease indication. As a result of these and other factors, our partnership agreements may result in material unanticipated problems, expenses, liabilities, competitive responses, loss of client relationships and diversion of management's attention.
Many of these factors will be outside of our control, and any one of them could result in increased costs, decreases in the amount of expected revenues and diversion of management's time and energy, which could materially impact our business, financial condition and results of operations. As a result, we cannot assure you that our relationship with any pharmaceutical partner will result in the realization of the anticipated benefits.
If our products and solutions do not perform as expected, we may fail to achieve or maintain sales of our products and solutions.
Our success depends on the market's confidence that we can provide accurate diagnostic testing products and reliable, high-quality rare disease information solutions. Our partnerships with our pharmaceutical partners and clients are typically designed to provide results in respect of a particular rare disease, and our preliminary assessments or knowledge about such disease may necessarily be limited by the amount of information currently available. As a result, the work we undertake on behalf of our pharmaceutical partners and clients may not yield the results that our pharmaceutical partners and clients expect or anticipate. We believe that our pharmaceutical partners and clients are likely to
16
be particularly sensitive to solution and testing service defects and errors, including if our products or services fail to detect genomic alterations with high accuracy from clinical specimens or if we fail to accurately develop a biomarker.
Moreover, we may fail to maintain the accuracy and reproducibility we have demonstrated to date with our genetic testing services, particularly for clinical samples, as our test volume increases. The sequencing process yields that we achieve depend on the design and operation of our sequencing process, which uses a number of complex and sophisticated biochemical, informatics, optical, and mechanical processes, many of which are highly sensitive to external factors. An operational or technological failure in one of these complex processes or fluctuations in external variables may result in sequencing processing yields that are lower than we anticipate or that vary between sequencing runs. In addition, we are regularly evaluating and refining our sequencing process. These refinements may initially result in unanticipated issues that further reduce our sequencing process yields or increase the variability of our sequencing process yields. Errors, including if our products or solutions fail to detect genomic variants with high accuracy, or mistakes, including if we fail to or incompletely or incorrectly identify the significance of gene variants, could have a significant adverse impact on our business.
Hundreds of genes can be implicated in some disorders, and overlapping networks of genes and symptoms can be implicated in multiple conditions. As a result, a substantial amount of judgment is required in order to interpret testing results for an individual patient and to develop an appropriate patient report. As a result, we may make errors in our interpretation of testing results, which could impair the results of our tests and (as such results are typically stored in our CentoMD database) adversely impact the quality of our overall knowledge base. The failure of our products or solutions to perform as expected would significantly impair our operating results and our reputation. We may also be subject to legal claims arising from, or loss of business as a result of, any defects or errors in our products and solutions.
We may fail to manage our future growth effectively, which could make it difficult to execute our business strategy.
We anticipate growth in our business operations. This future growth could create strain on our organizational, administrative and operational infrastructure, including laboratory operations, quality control, customer service, and sales force management. We may fail to maintain the quality or expected turnaround times of our products and services, or satisfy customer demand as it grows. Our ability to manage our growth properly will require us to continue to improve our operational, financial and management controls, as well as our reporting systems and procedures.
We also plan to expand our laboratory and technical operations as our business grows. In August 2018, we opened a new facility in Cambridge, Massachusetts, in the United States and recently expanded our clinical studies team to support our U.S. operations, and in April 2020, in connection with the COVID-19 pandemic, we acquired the laboratory facilities and equipment of a former cancer immunotherapy company and leased their former laboratory space in Hamburg, Germany. These or other future expansion strategies and any future growth could create strain on our organizational, administrative and operational infrastructure, including laboratory operations, quality control, customer service and sales force management. We may not be able to maintain the quality or expected turnaround times of our testing services or satisfy client demand as our business grows. Our ability to manage our growth properly will require us to continue to improve our operational, financial, and managerial controls, as well as our reporting systems and procedures, and to obtain appropriate regulatory approvals and meet regulatory standards applicable for the operation of our business.
17
The development of new products and solutions is a complex process, and we may be unable to successfully commercialize new products or solutions on a timely basis or at all.
New diagnostic test products and our interpretation-based solutions, including our biomarkers, take time to develop and commercialize. We may fail to develop and commercialize new diagnostic tests or solutions on a timely basis. Moreover, there can be no assurance that our products or solutions will be capable of meeting the needs of our clients and pharmaceutical partners, or that we will be able to commercialize them at all. Before we can commercialize any new products or solutions, we need to expend significant funds in order to:
- •
- conduct substantial research and development, including epidemiology and validation studies and potentially patient scope analyses;
- •
- further develop our laboratory processes or equipment;
- •
- allocate laboratory space for new solutions or further scale our infrastructure to accommodate research and development or new equipment;
- •
- in the case of products or solutions for which we are seeking regulatory or marketing approval, such as biomarkers, pursue such regulatory approval.
The development of new products and solutions involves risk, and development efforts may fail for many reasons, including the failure of any product or solution to perform as expected, a lack of validation or reference data, failure to demonstrate utility of a test or solution, or, in the case of solutions for which we are seeking or have received the Food and Drug Administration ("FDA"), European Commission and European Medicines Agency ("EMA"), German Federal Institute for Medicinal Products and Medical Devices (Bundesinstitut für Arzneimittel und Medizinprodukte), or comparable authorities' or agencies' approval, the inability to obtain such approval or the loss of such approval. In particular, our biomarker development and patent processes are subject to review by regulatory agencies and governing bodies. We cannot predict whether or when we will successfully complete development of each biomarker and if we will receive patent protection on any biomarkers that we develop.
As we develop new products and solutions, we will have to make significant investments in development, marketing, and selling resources. Any failure to develop or deliver adequate products or solutions to our clients and pharmaceutical partners on a timely basis or at all could significantly affect our business, financial condition, results of operations, and reputation.
We have limited experience in marketing and selling our products and solutions and we may fail to expand our direct sales and marketing force to adequately address our pharmaceutical partners' and clients' needs.
We have limited experience in marketing and selling our products and solutions to pharmaceutical partners, and currently rely on our CEO and our Chief Business Officer ("CBO") along with a small sales force to sell our products and solutions. We may not be able to market, sell, or distribute our existing products and solutions or other services we may develop effectively enough to support our planned growth.
Our future sales and further business growth will depend in large part on our ability to develop, and expand, our sales force and to increase the scope of our marketing efforts, particularly in the United States. Our target market of pharmaceutical partners and clients is a diverse market with particular, individualized needs. As a result, we believe it is necessary to develop a sales force that includes sales representatives with specific rare disease technical backgrounds. We will also need to attract and develop marketing personnel with industry expertise. Competition for such employees is intense. We may not be able to attract and retain personnel or be able to build an efficient and effective sales and marketing force, which could negatively impact sales and market acceptance of our products or solutions and limit our revenue growth and potential profitability. Our expected future growth will impose significant added responsibilities on members of management, including the need to identify, recruit, maintain, and integrate additional employees. Our future financial performance will depend in part on our ability to manage this potential future growth effectively, without compromising quality.
18
If we believe a significant market opportunity for our products or solutions exists in a particular jurisdiction in which we do not have direct access through one of our existing offices, from time to time we may enlist distribution partners and local laboratories to assist with sales, distribution, and client support. We may not be successful in finding, attracting, and retaining distribution partners or laboratories, or we may not be able to enter into such arrangements on favorable terms. Sales practices utilized by our distribution partners that are locally acceptable may not comply with sales practices standards required under German, Dutch, United States or other laws that apply to us, which could create additional compliance risk. If these additional sales and marketing efforts are not successful, we may not achieve significant market acceptance for our solutions in these markets, which could harm our business.
The knowledge and interpretation-based solutions we provide to our pharmaceutical partners may not achieve significant commercial market acceptance.
Our knowledge and interpretation-based solutions may not gain significant acceptance in the orphan drug development market and, therefore, may not generate substantial revenue or profits for us. Our ability to achieve increased commercial market acceptance for our existing knowledge and interpretation-based solutions will depend on several factors, including:
- •
- our ability to convince the medical and pharmaceutical community of the clinical utility of our solutions and their potential advantages over
existing and new solutions;
- •
- the willingness of our pharmaceutical partners, as well as their physicians and patients, to utilize our solutions; and
- •
- the agreement by commercial third-party payors and government payors to reimburse any treatments provided by our pharmaceutical partners, the scope and amount of which will affect a partners' willingness or ability to pay for our solutions and will influence physicians' decisions to recommend our solutions.
We believe that the successful completion of clinical trials by partners that use our solutions, publication of scientific and medical results based on the information gained from our repository in peer-reviewed journals, and presentations at leading conferences are critical to the broad adoption of our solutions. Publication in leading medical journals is subject to a peer-review process, and peer reviewers may not consider the results of studies involving our solutions sufficiently novel or worthy of publication.
The failure to be listed in physician guidelines or the failure of our solutions to produce favorable results for our partners or to be published in peer-reviewed journals could limit the adoption of our solutions. Failure to achieve widespread market acceptance of our solutions would materially harm our business, financial condition, and results of operations.
Failure to keep pace with the rapidly evolving industry in which we operate could make us obsolete.
Our business relies on commercial activities in the rare disease genetic testing and diagnosis field. In recent years, there have been numerous advances in methods used to analyze very large amounts of genomic information and the role of genetics and gene variants in rare diseases and treatments, including through the development of biomarkers. Our industry has and will continue to be characterized by rapid technological change, increasingly larger amounts of data, frequent new testing service introductions and evolving industry standards. Our future success will also depend on our ability to keep pace with the evolving needs of our clients and pharmaceutical partners on a timely and cost-effective basis and to pursue new market opportunities that develop as a result of technological and scientific advances. Our current products and solutions could become obsolete unless we continually update our offerings to reflect new scientific knowledge about genes and genetic variations and their role in rare diseases and treatments. If we fail to anticipate or respond adequately to
19
technological developments, demand for our products and solutions will not grow and may decline, and our business, revenue, financial condition and operating results could suffer materially.
Moreover, many companies in this market are offering, or may soon offer, products and solutions that compete with our products and solutions, in some cases at a lower cost than ours. We cannot assure you that research and discoveries by other companies will not render our existing or potential products and solutions uneconomical or result in tests superior to our existing tests and those we may develop. We also cannot assure you that any of our existing products and solutions, or those that we develop in the future, will be preferred by our clients, pharmaceutical partners, physicians or other payors to any existing or newly developed technologies or tests. If we fail to maintain competitive test products, our business, prospects, financial condition and results of operations could be adversely affected.
We may fail to successfully respond to increasing demand for our products and solutions.
As our sales volume grows, we will need to continue to increase our infrastructure for sample intake, customer service, billing and general process improvements, expand our internal quality assurance program, and extend our platform to support comprehensive genomic analyses at a larger scale within expected turnaround times. We will need additional certified laboratory scientists and other scientific and technical personnel to process higher volumes of our products and solutions. Portions of our process cannot be fully automated and will require additional personnel to scale. We will also need to purchase additional equipment, some of which can take a long time to procure, set up, and validate, and increase our software and computing capacity to meet increased demand.
We may fail to successfully implement any of these increases in scale, expansion of personnel, equipment, software and computing capacities, or process enhancements and we may have inadequate space in our laboratory facilities to accommodate such required expansion.
As additional products and solutions are commercialized, we will need to incorporate new equipment, implement new technology systems and laboratory processes, and hire new personnel with different qualifications. Failure to manage this growth or transition could result in turnaround time delays, higher product costs, declining product quality, deteriorating customer service, and slower responses to competitive challenges. A failure in any one of these areas could make it difficult or impossible for us to meet market expectations for our products and solutions, and could damage our reputation and the prospects for our business.
We may fail to obtain favorable pricing for our products and solutions and to meet our profitability expectations.
If we are not able to obtain favorable pricing for our products and solutions to enable us to meet our profitability expectations, our revenues and profitability could materially suffer. The rates we are able to charge for our products and solutions are affected by a number of factors, including:
- •
- general economic and political conditions in the countries in which we operate;
- •
- the competitive environment in our industry, as described below;
- •
- our clients' and pharmaceutical partners' cost sensitivities;
- •
- our ability to accurately estimate, attain and sustain revenues and royalties, margins and cash flows over the full partnership period for our
solutions, which includes our ability to estimate the impact of inflation and foreign exchange on our margins over long-term contracts; and
- •
- procurement practices of our pharmaceutical partners and clients and their use of third-party advisors.
20
The competitive environment in our industry affects our ability to obtain favorable pricing in a number of ways, all of which could have a material negative impact on our results of operations. The less we are able to clearly convey the value of our products and solutions or differentiate our products and solutions, the more risk we have that they will be seen as commodities, with price being the driving factor in selecting us as a partner. Competitors may be willing, at times, to price contracts or products lower than we do in an effort to enter the market or increase market share. Further, if competitors develop and implement methodologies that yield greater efficiency or efficacy, they may be able to offer products and solutions similar to ours at lower prices.
Ethical, legal and social concerns related to the use of genomic information could reduce demand for our genetic rare disease knowledge and interpretation-based products and solutions.
Genomic testing, like that conducted for our pharmaceutical partners and clients using our genetic rare disease information platform, has raised ethical, legal and social issues regarding privacy and the appropriate uses of the resulting information. Governmental authorities could, for social or other purposes, limit or regulate the use of genomic information or genomic testing or prohibit testing for genetic predisposition to certain conditions, particularly for those that have no known cure. Similarly, these concerns may lead patients to refuse to use genomic tests even if permissible.
Ethical and social concerns may also influence United States and foreign patent offices and courts with regard to patent protection for technology relevant to our business. These and other ethical, legal and social concerns may limit market acceptance of our products and solutions or reduce the potential markets for products and solutions enabled by our genetic rare disease information platform, either of which could have an adverse effect on our business, financial condition, or results of operations.
We have limited resources to be expended on research programs and biomarker development. Our resource allocation decisions may lead us to focus on research programs and biomarkers which are not commercially viable, and as a result we may be unable to recover the costs incurred under these efforts.
Because we have limited financial and managerial resources, we focus on research programs and biomarker development that we identify for rare diseases in collaboration with our pharmaceutical partners, or based on our assessment of the market needs. As a result, we may forego or delay pursuit of opportunities with other orphan drug candidates or for other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial drugs or profitable market opportunities. Our spending on current and future research and development programs and biomarker development for specific diseases may not yield any relevant results that are helpful to our existing programs or assist in the creation of any commercially viable drugs. If we do not accurately evaluate the commercial potential or target market for a particular drug candidate, we may relinquish valuable rights to that drug candidate through collaboration, licensing or other royalty arrangements.
If we fail to compete successfully with our competitors, including new entrants in the market, we may be unable to increase or sustain our revenue or achieve and sustain profitability.
While personalized genomic diagnostics is a relatively new area of science, we face competition from companies that offer tests or have conducted research to profile genes and gene expression in various rare diseases. Our principal competition comes from diagnostic companies that offer diagnostic tests that capture genetic, phenotypic and epidemiological data, as well as laboratories and academic research centers. Many hospitals and academic medical centers may also seek to perform the type of genetic testing and knowledge and interpretation-based solutions we offer at their own facilities or using their own research capabilities.
Some of our present and potential competitors may have substantially greater financial, marketing, technical or manufacturing resources than we do. Our competitors may also be able to respond more
21
quickly to new technologies or processes and changes in client demands. They may also be able to devote greater resources towards the development, promotion and sale of their products or solutions for pharmaceutical partners than we can. As competition in our market increases, we may also be subject to increased litigation risk, including in connection with patents as well as our marketing practices and other promotional activities. In addition, our current and potential competitors may make strategic acquisitions or establish cooperative relationships among themselves or with third parties that increase their ability to address the needs of our physicians or partners. If we fail to compete successfully against current or future competitors, our business will be harmed.
Because our genetic testing and knowledge and interpretation-based solutions and products, in particular our CentoMD database, have limited patent protection, new and existing companies worldwide could seek to develop genetic tests or similar products and solutions that compete with ours. These competitors could have technological, financial, and market access advantages that are not currently available to us and they could develop and commercialize competing products and solutions faster than we are able to do so. Increased competition, including price competition, could have a material adverse impact on our net revenues and profitability.
If our pharmaceutical partners experience any of a number of possible unforeseen events in connection with their clinical trials, our ability to commercialize future solutions or improvements to existing solutions could be delayed or prevented.
Our pharmaceutical partners may experience numerous unforeseen events during, or as a result of, clinical trials that could delay or prevent their ability to continue or conduct further clinical trials or obtain regulatory approval of or commercialize future orphan drugs. Unforeseen events that could delay or prevent our pharmaceutical partners' ability to conduct or support clinical trials, obtain regulatory approval of or commercialize future orphan drugs include:
- •
- regulatory authorities or ethical review boards, including IRBs, may not authorize the commencement of a clinical trial or may not accept
clinical trial protocols;
- •
- clinical trials may produce negative or inconclusive results, and our pharmaceutical partners may decide, or regulatory authorities may require
them to, to abandon development programs;
- •
- the number of patients, or amount of data, required for clinical trials may be larger than we or our pharmaceutical partners anticipate, or
patient enrollment in clinical trials may be slower than we or our pharmaceutical partners anticipate or patients may drop out of these clinical trials at a higher rate than we or our pharmaceutical
partners anticipate;
- •
- failure to conduct our clinical trials in accordance with applicable regulatory requirements of the FDA and of the regulatory authorities
responsible for authorization or oversight of the conduct of clinical trials in other countries;
- •
- inability to develop companion diagnostic tests for a particular rare disease or to add companion diagnostic claims to existing tests, and/or
obtain regulatory approval to market any such test on a timely basis or at all;
- •
- clinical trials of our pharmaceutical partners for which we are developing companion diagnostic tests may suggest or demonstrate that our
partners' treatments are not as efficacious and/or as safe as other similar treatments or that our companion diagnostic test is not essential to determine which patients would benefit from these
treatments; and
- •
- our pharmaceutical partners may decide, or regulatory authorities or institutional review boards may require them, to suspend or terminate clinical research for various reasons, including cost, adequate end market size, available data or noncompliance with regulatory requirements.
22
If our pharmaceutical partners choose not to conduct clinical trials for treatments in the rare disease space due to the above factors or otherwise, they may have less need of our products and solutions and may therefore choose not to partner with us. Our ability to continually expand our existing data repository depends on our ability to maintain partnerships with our pharmaceutical clients. Should our partners delay or cancel their ongoing existing trials or choose not to begin new trials for treatments in the rare disease industry, our ability to commercialize future solutions or improvements to existing solutions could be delayed or prevented.
Our employees, principal investigators, consultants, and commercial partners may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements and insider trading.
We are exposed to the risk of fraud or other misconduct by our employees, principal investigators, consultants, and commercial partners, including our distributors in our diagnostics business and pharmaceutical partners in our pharmaceutical business. Misconduct by these parties could include intentional failures to comply with the regulations of applicable regulatory authorities (including the FDA and the European Commission and EMA), comply with healthcare fraud and abuse laws and regulations, report financial information or data accurately, or disclose unauthorized activities to us. In particular, sales, marketing, and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, bribery, kickbacks, self-dealing, and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, client incentive programs, and other business arrangements. Such misconduct could also involve the improper use of information obtained in the course of clinical studies, which could result in regulatory sanctions and cause serious harm to our reputation. We currently have an insider trading policy as well as a code of conduct applicable to all of our employees and conduct a background check before entering into any new contracts with third party distributors, but it is not always possible to identify and deter employee or third party misconduct, and our insider trading policy and code of conduct, due diligence and the other precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses, or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could result in the imposition of significant fines or other sanctions, which could have a significant impact on our business. Whether or not we are successful in defending against such actions or investigations, we could incur substantial costs, including legal fees, and divert the attention of management in defending ourselves against any of these actions or investigations.
We may lose the support of key thought leaders and fail to establish our products and solutions as a standard of care for patients with rare diseases, which may limit our revenue growth and ability to achieve future profitability.
We have established relationships with leading rare disease thought leaders at premier institutions and rare disease networks. If we suffer harm to our reputation, whether due to actions outside of our control or otherwise, our relationships with these persons may suffer which could adversely impact our business, including our key pharmaceutical partnerships and diagnostic client relationships. Moreover, if these key thought leaders determine that our platform (including CentoMD), our existing products or solutions or other new products or solutions that we develop are not useful to our partners' development of treatments for rare diseases, that alternative technologies are more effective, or if they elect to use internally developed products or solutions, we could encounter significant difficulty validating our testing platform, driving adoption, or establishing our genetic knowledge and interpretation-based solutions and tests as a standard of care, which would limit our revenue growth and our ability to achieve profitability.
23
Security breaches, loss of data, and other disruptions could compromise sensitive information related to our business or prevent us from accessing critical information and expose us to liability, which could adversely affect our business and our reputation.
In the ordinary course of our business, we collect and store sensitive data, including legally protected health information, personally identifiable information, intellectual property, and proprietary business information owned or controlled by us or physicians, pharmaceutical partners and other clients. We manage and maintain our applications and data utilizing a combination of on-site systems, managed data center systems, and cloud-based data center systems. We also communicate, and facilitate the exchange of, sensitive patient data to and between ourselves and physicians of the patients for whom we conduct diagnostic tests through an online client-facing portal, CentoPortal. These applications and related data encompass a wide variety of business-critical information including legally protected health information, personally identifiable information, research and development information, commercial information, and business and financial information. We face a number of key risks related to the protection of this information, including: unauthorized access risk; inappropriate or unauthorized disclosure risk; inappropriate modification risk; and the risk of our being unable to adequately monitor our controls.
The secure processing, storage, maintenance, and transmission of this critical information is vital to our operations and business strategy. Our information technology and infrastructure, and that of our third-party disaster recovery back-up providers, may be vulnerable to attacks by hackers or malicious software or breached due to personnel error, unauthorized access, malfeasance, or other disruptions. Any such breach or interruption could compromise the security or integrity of our networks, and the information stored there could be accessed by unauthorized parties, publicly or incorrectly disclosed, corrupted, lost, or stolen. Any such access, disclosure, corruption, other loss, or theft of information could result in governmental investigations, class action legal claims or proceedings, liability under laws that protect the privacy of personal information, such as but not limited to the Health Insurance Portability and Accountability Act ("HIPAA"), the General Data Protection Regulation (EU 2016/679) ("GDPR") and regulatory penalties. Although we have implemented security measures and a formal, dedicated enterprise security program to prevent unauthorized access to patient data, applications such as our online client-facing portals are currently accessible through public web portals and may, in the future, be accessible through dedicated mobile applications, and there is no guarantee we can absolutely protect our online portals or our mobile applications from breach. Unauthorized access to, or loss or dissemination of, the data embedded in or transferred via these applications could also disrupt our operations, including our ability to conduct our analyses, provide test results, bill our pharmaceutical or other partners, provide client assistance solutions, conduct research and development activities, collect, process, and prepare company financial information, provide information about our products and solutions and other pharmaceutical partner and physician education and outreach efforts through our website, manage the administrative aspects of our business, and damage our reputation, any of which could adversely affect our business.
We are a "covered entity" as defined under HIPAA, and the United States Office of Civil Rights may impose penalties on a covered entity for a failure to comply with a requirement of HIPAA. Penalties will vary significantly depending on factors such as the date of the violation, whether the covered entity knew or should have known of the failure to comply, or whether the covered entity's failure to comply was due to willful neglect. A person who knowingly obtains or discloses individually identifiable health information in violation of HIPAA may face a criminal penalty of up to $50,000 and imprisonment up to one year. The criminal penalties increase to $100,000 and up to five years' imprisonment if the wrongful conduct involves false pretenses, and to $250,000 and up to 10 years' imprisonment if the wrongful conduct involves the intent to sell, transfer, or use identifiable health information for commercial advantage, personal gain, or malicious harm. The United States Department of Justice (the "DOJ") is responsible for criminal prosecutions under HIPAA. Furthermore, in the event of a breach as defined by HIPAA, the covered entity has specific reporting
24
requirements under HIPAA regulations. In the event of a significant breach, the reporting requirements could include notification to the general public.
In addition, the interpretation and application of consumer, health-related, and data protection laws in the United States, Europe, and elsewhere are often uncertain, contradictory, and in flux. It is possible that these laws may be interpreted and applied in a manner that is inconsistent with our practices. If so, this could result in government-imposed fines or orders requiring that we change our practices, which could adversely affect our business. In addition, these privacy regulations may differ from country to country, and may vary based on whether testing is performed in the United States or in the local country. Our operations or business practices may not comply with these regulations in each country, and complying with these various laws could cause us to incur substantial costs or require us to change our business practices and compliance procedures in a manner adverse to our business.
We are subject to significant foreign currency exchange controls in certain countries in which we operate.
We are in some countries, and could become elsewhere, subject to strict restrictions on the movement of cash and the exchange of foreign currencies, which limits our ability to use this cash across our global operations. We also face risks related to the collection of payments due to us from our major pharmaceutical partners or clients that are located in certain geographical regions with foreign currency or international monetary controls. This risk could increase as we continue our geographic expansion. In particular, for the years ended December 31, 2018 and 2019, we derived 30.6% and 28.9%, respectively, of our total revenues from our Middle East region. Certain Middle East economies have adopted or been subject to international restrictions on the ability to transfer funds out of the country and convert local currencies into euros. This may increase our costs and limit our ability to convert local currency into euros and transfer funds out of certain countries. Any shortages or restrictions may impede our ability to convert these currencies into euros and to transfer funds, including for the payment of dividends or interest or principal on our outstanding debt.
We may acquire assets or other businesses that could negatively affect our operating results, dilute our shareholders' ownership or increase our debt.
In addition to organic growth, we may pursue growth through the acquisition of assets or other businesses that may enable us to enhance our technologies and capabilities, expand our geographic market, add experienced management personnel or add new or improve our existing products and solutions. We also may pursue strategic alliances and joint ventures that leverage our technical platform and industry knowledge to expand our products and solutions. Negotiating these transactions and the formation of strategic alliances or joint ventures can be time-consuming and expensive, and may be subject to third-party approvals as well as approvals from governmental authorities, which are beyond our control. In addition, some third parties may choose not to enter into partnership or collaboration agreements with us because of our existing relationships with other pharmaceutical partners. Consequently, we may not be able to complete any contemplated transactions on favorable terms or at all, and we can make no assurance that such transactions, once undertaken and announced, will close.
An acquisition or investment may result in unforeseen operating difficulties and expenditures, including in integrating businesses, products and solutions, personnel, operations, and financial, accounting and other controls and systems, and retaining key employees, with the assumption of unknown liabilities or known liabilities that prove greater than anticipated, and in retaining the clients of any acquired business. Any such difficulties could disrupt our ongoing operations or require management resources that we would otherwise focus on developing our existing business. Future acquisitions could result in the use of our available cash and marketable securities, potentially dilutive issuances of equity securities, the incurrence of debt, contingent liabilities, or impairment expenses related to goodwill, and impairment or amortization expenses related to other intangible assets, which could harm our financial condition. As a result, we may not realize the anticipated benefits of any
25
acquisition, technology license, strategic alliance, or joint venture. These challenges related to acquisitions or investments could adversely affect our business, results of operations, and financial condition.
Certain Factors Relating to Our Industry
Regulatory Risks
Our global operations expose us to numerous and sometimes conflicting legal and regulatory requirements, and violation of these requirements could harm our business.
We are subject to numerous, and sometimes conflicting, legal regimes in the countries in which we operate, including on matters as diverse as health and safety standards, marketing and promotional activities, anticorruption, import/export controls, content requirements, trade restrictions, tariffs, taxation, sanctions, immigration, internal and disclosure control obligations, securities regulation, anti-competition, data privacy and labor relations. This includes in emerging markets where legal systems may be less developed or familiar to us. We strive to abide by and maintain compliance with these laws and regulations. Compliance with diverse legal requirements is costly, time-consuming and requires significant resources. Violations of one or more of these regulations in the conduct of our business could result in significant fines, criminal sanctions against us or our supervisory board or officers, prohibitions on doing business and damage to our reputation. Violations of these regulations in connection with the performance of our obligations to our clients or pharmaceutical partners also could result in liability for significant monetary damages, fines and/or criminal prosecution, unfavorable publicity and other reputational damage, restrictions on our ability to process information and allegations by our clients or pharmaceutical partners that we have not performed our contractual obligations. Due to the varying degrees of development of the legal systems of the countries in which we operate, local laws might be insufficient to protect our rights.
Our international operations could be affected by changes in laws, trade regulations, labor and employment regulations, and procedures and actions affecting approval, products and solutions, pricing, reimbursement and marketing of our products and solutions, as well as by inter-governmental disputes. Any of these changes could adversely affect our business. The imposition of new laws or regulations, including potential trade barriers, may increase our operating costs, impose restrictions on our operations or require us to spend additional funds to gain compliance with the new rules, if possible, which could have an adverse impact on our financial condition.
Current and future legislation, in particular legislation related to orphan drugs, may impact overall investment and activity in the rare disease space or our ability to obtain regulatory approvals.
In the United States, the European Union, its member states and some other foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system. These changes could affect our ability to sell profitably any products for which we require approvals. Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access to healthcare.
Specifically, regulatory authorities in some jurisdictions, including the United States and the European Union, may designate drugs for relatively small patient populations as orphan drugs. Under the Orphan Drug Act, the FDA may designate a drug as an orphan drug if it is a drug intended to treat a rare disease or condition, which is generally defined as a patient population of fewer than 200,000 individuals annually in the United States, or a patient population of greater than 200,000 in the United States where there is no reasonable expectation that the cost of developing the drug will be recovered from sales in the United States. In the United States, orphan drug designation entitles a
26
party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages and user-fee waivers.
Similarly, in the European Union, the European Commission grants orphan drug designation after receiving the opinion of the EMA's Committee for Orphan Medicinal Products on an orphan drug designation application. Orphan drug designation is intended to promote the development of drugs that are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than five in 10,000 persons in the European Union and for which no satisfactory method of diagnosis, prevention, or treatment has been authorized (or the product would be a significant benefit to those affected). In addition, designation is granted for drugs intended for the diagnosis, prevention, or treatment of a life-threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the drug in the European Union would be sufficient to justify the necessary investment in developing the drug. In the European Union, orphan drug designation entitles a party to financial incentives, such as reduction of fees or fee waivers, and a ten-year market exclusivity once the drug is on the market.
These legislative initiatives have led to an increase in investment and activity in the rare disease drug development space. If these and other legislative initiatives were to change to become less favorable to orphan drug developers and researchers, it could harm our business, results of operations and financial condition.
We may fail to comply with the complex federal, state, local and foreign laws and regulations that apply to our business and become subject to severe financial and other consequences.
Our laboratory in the United States is subject to the Clinical Laboratory Improvement Amendments of 1998 ("CLIA"), a United States federal law that regulates all clinical diagnostic laboratories that perform testing on specimens derived from humans for the purpose of providing information for the diagnosis, prevention, or treatment of disease. CLIA regulations mandate specific standards in the areas of personnel qualifications, administration, participation in proficiency testing, patient test management, quality control, quality assurance, and inspections. Our laboratory facilities located in Rostock, Germany and Cambridge, Massachusetts, United States each have a current certificate of accreditation under CLIA to conduct all genetic and biochemical analyses offered through our accreditation by the College of American Pathologists ("CAP"). To renew the CLIA certificates, we are subject to survey and inspection every two years. Moreover, CLIA inspectors may make unannounced inspections of our clinical laboratories at any time.
Any sanction imposed under CLIA, its implementing regulations, or state or foreign laws or regulations governing licensure, or our failure to renew a CLIA certificate, a state or foreign license, or accreditation, could have a material adverse effect on our business. Most CLIA deficiencies are not classified as "condition-level" deficiencies, and there are no adverse effects upon the laboratory operations as long as the deficiencies are corrected. Remediation of these deficiencies are routine matters, with corrections occurring within several hours or weeks. More serious CLIA deficiencies could rise to the level of "condition-level" deficiencies, and CMS has the authority to impose a wide range of sanctions, including revocation of the CLIA certification along with a bar on the ownership or operation of a CLIA certified laboratory by any owners or operators of the deficient laboratory. There is an administrative hearing procedure that can be pursued by the laboratory in the event of imposition of such sanctions, during which the sanctions are stayed, but the process can take a number of years to complete. If we were to lose our CLIA certification or CAP accreditation, we would not be able to operate our clinical laboratories and perform our genetic tests, which would result in material harm to our business and results of operations.
We are also required to maintain a license for our Cambridge laboratory facility to perform testing in Massachusetts. Massachusetts laws establish standards for day-to-day operation of our clinical laboratory, including the training and skills required of personnel and quality control over and above
27
that required by CLIA. We are also licensed to perform testing in our Cambridge laboratory facility by the states of California, Pennsylvania and Maryland. We are in the process of obtaining a New York State license to perform testing and deliver the related test report for specimens originating from New York.
For samples tested in the U.S., we are also subject to HIPAA, under which the Department of Health and Human Services established comprehensive federal standards with respect to the privacy and security of protected health information and requirements for the use of certain standardized electronic transactions; certain of our services, including our online client-facing portals for reporting and research, are subject to these standards and requirements. Amendments to HIPAA under the Health Information Technology for Economic and Clinical Health Act (the "HITECH Act"), and related regulatory amendments, which strengthen and expand HIPAA privacy and security standards, increase penalties for violators, extend enforcement authority to state attorneys general, and impose requirements for breach notification.
We furnish to pharmaceutical partners genomic information that has been de-identified in accordance with HIPAA or pseudonymized in accordance with GDPR and relevant international health information privacy regulations. The laws of certain states and countries may require specific consent from the individual either to retain or utilize certain genetic information for research or other purposes even if such information has been de-identified, or may require that we obtain a waiver of such consent from an ethical or privacy review board. Even where we furnish to pharmaceutical partners and academic researchers genomic information that has been de-identified or pseudonymized in accordance with applicable laws and regulations, pharmaceutical partners or academic researchers may use technology or other methods to link that de-identified or pseudonymized genomic information to the patient from whom it was obtained in contravention of one or more applicable laws and regulations. Similarly, as we expand our decision support applications and offerings, we may encounter greater regulatory risk, such as compliance with HIPAA, GDPR and other regulations governing the use of protected health information and the promotion of FDA approved drugs. A finding that we have failed to comply with any such laws and any remedial activities required to ensure compliance with such laws could cause us to incur substantial costs, to be subject to unfavorable publicity or public opinion, to change our business practices, or to limit the retention or use of genetic information in a manner that, individually or collectively, could be adverse to our business.
In the European Union, various regulations apply to genetic testing and the use of genomic information. In Germany, the Genetic Diagnosis Act (Gendiagnostikgesetz) (the "GenDG") and guidelines and written opinions on novel genetic screenings developed by the Commission on Genetic Testing, an interdisciplinary independent commission established in 2009 in accordance with the GenDG, apply to such testing. The GenDG prohibits us from communicating results of genetic tests directly to a patient located within Germany. Instead, the results may only be provided to a physician who is a qualified genetic counsellor under applicable rules.
In addition to CLIA, GDPR, HIPAA and the GenDG, our operations are subject to other extensive federal, state, local, and foreign laws and regulations, all of which are subject to change. Our failure to comply with any such laws and regulations could lead to civil or criminal penalties, exclusion from participation in government healthcare programs, or prohibitions or restrictions on our ability to conduct commercial activities. We believe that we are in material compliance with all statutory and regulatory requirements, but there is a risk that one or more government agencies could take a contrary position. These laws and regulations are complex and are subject to interpretation by the courts and by government agencies. If one or more such agencies allege that we may be in violation of any of these requirements, regardless of the outcome, it could damage our reputation and adversely affect important business relationships with third parties.
28
We may fail to comply with evolving European and other privacy laws.
On May 25, 2018, Regulation (EU) 2016/679 of the European Parliament and of the Council of April 27, 2016 on the protection of natural persons with regard to the processing of personal data and on the free movement of such data (the "GDPR") went into effect. The GDPR imposes a broad range of strict requirements on companies subject to the GDPR, such as us, including requirements relating to having legal bases for processing personal information relating to identifiable individuals and transferring such information outside the European Economic Area (the "EEA"), including to the United States, providing details to those individuals regarding the processing of their personal information, keeping personal information secure, having data processing agreements with third parties who process personal information, responding to individuals' requests to exercise their rights in respect of their personal information, reporting security breaches involving personal data to the competent national data protection authority and affected individuals, appointing data protection officers, conducting data protection impact assessments, and record-keeping. The GDPR increases substantially the penalties to which we could be subject in the event of any non-compliance, including fines of up to 10,000,000 Euros or up to 2% of our total worldwide annual turnover for certain comparatively minor offenses, or up to 20,000,000 Euros or up to 4% of our total worldwide annual turnover for more serious offenses. Given the new law, we face uncertainty as to the exact interpretation of the new requirements and we may be unsuccessful in implementing all measures required by data protection authorities or courts in interpretation of the new law.
In particular, national laws of member states of the European Union are still in the process of being adapted to the requirements under the GDPR, thereby implementing national laws which may partially deviate from the GDPR and impose different obligations from country to country, so that we do not expect to operate in a uniform legal landscape in the European Union. Also, in the field of handling genetic and health data, the GDPR specifically allows national laws to impose additional and more specific requirements or restrictions, and European laws have historically differed quite substantially in this field, leading to additional uncertainty.
Further complicating compliance efforts, on January 31, 2020, the United Kingdom left the European Union, commonly referred to as "Brexit." Until December 31, 2020, the European Union and the United Kingdom are in a transition period during which the European Union and the United Kingdom will negotiate new arrangements for their future relationship. The negotiations are currently in a very preliminary stage and it is not clear yet the extent to which legal regulations based on European Union law will continue to apply to the United Kingdom in the same or similar form as previously. In particular, since the regulatory framework for pharmaceutical products in the United Kingdom covering quality, safety and efficacy of pharmaceutical products, clinical trials, marketing authorization, commercial sales and distribution of pharmaceutical products is derived from European Union directives and regulations, Brexit could materially impact the future regulatory regime which applies to products and the approval of product candidates in the United Kingdom. It remains to be seen how, if at all, Brexit will impact regulatory requirements for product candidates and products in the United Kingdom.
We must also ensure that we maintain adequate safeguards to enable the transfer of personal data outside of the EEA, in particular to the United States, in compliance with European data protection laws. We expect that we will continue to face uncertainty as to whether our efforts to comply with our obligations under European privacy laws will be sufficient. If we are investigated by a European data protection authority, we may face fines and other penalties. Any such investigation or charges by European data protection authorities could have a negative effect on our existing business and on our ability to attract and retain new clients or pharmaceutical partners. We may also experience hesitancy, reluctance, or refusal by European or multinational clients or pharmaceutical partners to continue to use our products and solutions due to the potential risk exposure as a result of the current (and, in particular, future) data protection obligations imposed on them by certain data protection authorities in
29
interpretation of current law, including the GDPR. Such clients or pharmaceutical partners may also view any alternative approaches to compliance as being too costly, too burdensome, too legally uncertain, or otherwise objectionable and therefore decide not to do business with us. Any of the foregoing could materially harm our business, prospects, financial condition and results of operations.
We could be adversely affected by violations of worldwide anti-bribery laws, including the U.S. Foreign Corrupt Practices Act.
We are subject to a variety of anti-bribery and anti-corruption laws in the jurisdictions in which we operate. In particular, we are subject to Germany's Anti-Bribery Act of 2015 (Gesetz zur Bekämpfung der Korruption im Gesundheitswesen), which implements EU anti-corruption laws and the European legislation and the Criminal Law Convention on Corruption of the Council of Europe into German law, and the FCPA, which prohibits companies and their intermediaries from making payments in violation of law to non-United States government officials for the purpose of obtaining or retaining business or securing any other improper advantage. We are also subject to similar anti-bribery laws in the jurisdictions in which we operate, including the United Kingdom's Bribery Act of 2010, which prohibits commercial bribery and makes it a crime for companies to fail to prevent bribery.
We use third-party collaborators, strategic partners, law firms and other representatives for patent registration and other purposes in a variety of countries, including those that are known to present a high corruption risk. We also use third-party distributors worldwide as part of our diagnostics business. Our reliance on third parties to sell our products and solutions internationally demands a high degree of vigilance because we can be held liable for the corrupt or other illegal activities of these third-party collaborators, or their or our employees, representatives, contractors, partners, and agents, even if we do not explicitly authorize such activities. In addition, although we have implemented policies and procedures to ensure compliance with anti-corruption and related laws and maintain a code of conduct, there can be no assurance that all of our employees, representatives, contractors, partners, or agents will comply with these laws at all times. Other United States companies in the medical device and pharmaceutical fields have faced criminal penalties under the FCPA for allowing their agents to deviate from appropriate practices in doing business with these individuals.
These laws are complex and far-reaching in nature, and, as a result, we cannot assure you that we would not be required in the future to alter one or more of our practices to be in compliance with these laws, any changes in these laws, or the interpretation thereof. Non-compliance with these and other relevant laws could subject us to whistleblower complaints, investigations, sanctions, settlements, prosecution, other enforcement actions, disgorgement of profits, significant fines, damages, other civil and criminal penalties or injunctions, suspension and debarment from contracting with certain governments or other persons, the loss of export privileges, reputational harm, adverse media coverage, and other collateral consequences. If any subpoenas or investigations are launched, or governmental or other sanctions are imposed, or if we do not prevail in any possible civil or criminal litigation, our business, results of operations, and financial condition could be materially harmed. In addition, responding to any action will likely result in a materially significant diversion of management's attention and resources and significant defense costs and other professional fees. Enforcement actions and sanctions could further harm our business, results of operations, and financial condition.
Transactions involving Iran or other countries or parties that are targets of U.S. or other economic sanctions could expose us to certain risks and may lead some potential customers and investors to avoid doing business with us or investing in our securities.
U.S. law generally prohibits U.S. persons, and in some cases non-U.S. entities owned or controlled by U.S. persons, from doing business with countries, territories, individuals and entities that are the target of sanctions administered by the U.S. Department of the Treasury's Office of Foreign Assets Control, including Iran. Other countries also maintain certain economic sanctions targeting certain
30
counties, territories and parties. The United States has also implemented certain sanctions targeting non-U.S. persons for activities conducted outside the United States "secondary sanctions" that involve specific sanctions targets or certain activities, including, among other things, certain transactions related to Iran. Further, certain countries maintain and enforce export controls regulating trade in items that originate in, incorporate content from, or are produced on the basis of technology developed in such country "export controls."
Centogene AG, which is not a U.S. person and is not owned or controlled by U.S. persons, has contracts with several laboratories and one distributor in Iran through which it provides diagnostic tests to patients in Iran, primarily non-invasive prenatal testing ("NIPT") for pregnant women. To our knowledge, neither we nor our distributor have entered into any arrangements with or sold any products to persons included on the Specially Designated Nationals and Blocked Persons List maintained by the U.S. Department of the Treasury's Office of Foreign Asset Control. During the years ended December 31, 2017, 2018 and 2019, revenues from Iran amounted to €300 thousand, €2,950 thousand and €1,277 thousand, respectively. Our assets receivable from or attributable to our contacts in Iran as of December 31, 2017, 2018 and 2019 amounted to €77 thousand, € 1,351 thousand and €1,471 thousand, respectively. We had no liabilities due from or attributable to our contacts in Iran for these periods. Centogene believes that its business with Iranian parties is conducted in compliance with all applicable sanctions and export controls and that such activities, which involve providing genetic testing services to patients, are not sanctionable under U.S. secondary sanctions targeting Iran. However, U.S. sanctions are subject to change and if we were then determined to have engaged in activities targeted by certain U.S. sanctions, we could be exposed to the possible imposition of sanctions on us. We may also face reputational damage due to our sales to Iran. The above circumstances could have an adverse effect on our business or results of operations.
We may fail to adhere to regulations of promotional claims and activities regarding our products and solutions.
Once a patient has been identified and diagnosed through our diagnostics testing, we provide each patient's physician with a diagnostic report. If a positive diagnosis is confirmed, we provide the physician with information on relevant treatment options, although the physician is responsible for ultimately making clinically relevant decisions for the treatment of his or her patient.
In the United States, the FDA and other regulatory agencies strictly regulate the promotional claims that may be made about prescription drugs and devices. In particular, a device may not be promoted for uses or indications beyond those contained in the device's approved labeling, or "off-label" uses. Similar laws and regulations exist in other jurisdictions where we promote our products. If the FDA determines that we have promoted our products for off-label use, it could request that we modify those promotional materials or take regulatory or enforcement actions, including the issuance of an untitled letter, warning letter, injunction, seizure, civil fine and criminal penalties. It is also possible that other federal, state or foreign enforcement authorities may take action if they consider our promotional or training materials to constitute promotion of an unapproved use. If not successfully defended, enforcement actions related to off-label promotion could result in significant fines or penalties. The U.S. government has levied large civil and criminal fines against companies for alleged improper promotion and has entered into corporate integrity agreements and deferred prosecution agreements with companies that engaged in off-label promotion. The FDA has also requested that such companies enter into consent decrees and has taken other enforcement action. If the DOJ or FDA determines that we have engaged in off-label promotion in our test reports, we may be subject to civil or criminal fines. Although our policy is to refrain from statements that could be considered off-label promotion of third parties, the regulatory standards regarding off-label promotion are ambiguous, and the FDA or another regulatory agency could conclude that we have engaged in off-label promotion.
31
In addition to promoting our devices in a manner consistent with their approved indications, we must have adequate substantiation for the claims we make for our products or solutions. If any of our claims are determined to be false, misleading or deceptive, our products or solutions could be considered to be misbranded under the Federal Food, Drug, and Cosmetic Act (the "FDC Act") or to violate the Federal Trade Commission Act. We could also face lawsuits from our competitors under the Lanham Act, alleging that our marketing materials are false or misleading. Such lawsuits, whether with or without merit, are typically time-consuming, costly to defend, and could harm our reputation.
Federal and state legislation regulate interactions between medical device manufacturers and healthcare professionals. We are subject to federal and state laws targeting fraud and abuse in healthcare, including anti-kickback laws, false claims laws, and other laws constraining or otherwise related to financial arrangements manufacturers may enter into with healthcare professionals. For example, the Physician Payments Sunshine Act requires device manufacturers to report and disclose payments or other transfers of value made to physicians and teaching hospitals. Violations of these laws can result in criminal or civil sanctions, including fines, imprisonment, and exclusion from government reimbursement programs, all of which could materially harm our business.
In addition, incentives exist under applicable laws that encourage competitors, employees, and physicians to report violations of law governing promotional activities for pharmaceutical products and solutions. These incentives could lead to so-called whistleblower lawsuits as part of which such persons seek to collect a portion of monies allegedly overbilled to government agencies due to, for example, promotion of pharmaceutical products and solutions beyond labeled claims. These incentives could also lead to lawsuits that claim we have mischaracterized a competitor's service in the marketplace and, as a result, we could be sued for alleged damages to our competitors. Such lawsuits, whether with or without merit, are typically time-consuming and costly to defend. Such lawsuits may also result in related shareholder lawsuits, which may also be costly to defend.
Changes in the way that the FDA and the European Union regulate laboratory developed tests, manufactured, validated, and performed by laboratories like ours could result in additional expense in offering our current and any future products and solutions or even possibly delay or suspend development, manufacture, or commercialization of such products and solutions.
The FDA does not currently regulate most laboratory developed tests ("LDTs"). We believe that the tests we currently offer meet the definition of LDTs, as they have been designed, developed and validated for use in a single CLIA-certified laboratory. If our tests are qualified as LDTs, they are currently not subject to FDA regulation as medical devices. Since the early 1990s, the FDA has taken the position that, although LDTs are medical devices, it would exercise enforcement discretion by not requiring compliance with the FDC Act, or its regulations for LDTs. That remains the guidance of the FDA today. However, the FDA has taken certain actions in the past that, if renewed by the FDA, could result in a new regulatory approach for LDTs. In October 2014, the FDA published two draft guidance documents that, if finalized, would implement a regulatory approach for most LDTs. The draft guidance documents proposed to impose a risk-based, phased-in approach for LDTs similar to the existing framework for in vitro diagnostic devices. In January 2017, the FDA released a discussion paper synthesizing public comments on the 2014 draft guidance documents and outlining an updated possible approach to regulation of LDTs. Although the discussion paper has no legal status and does not represent a final version of the LDT draft guidance documents, it proposes a risk-based framework that would require most LDTs to comply with most of the FDA's regulatory requirements for medical devices. In March 2017, a discussion draft of the Diagnostic Accuracy and Innovation Act ("DAIA") was circulated, which, if enacted, would implement a regulatory scheme for all diagnostic tests, including both in vitro diagnostic devices and LDTs. Under DAIA, CMS would have jurisdiction over laboratory operations under an amended CLIA, and the FDA would regulate the design, development and validation of diagnostic tests under an amended FDC Act. We cannot predict whether this bill or any other any other legislative proposal will be enacted into law or the impact such new legal
32
requirements would have on our business. We also cannot predict whether the FDA will take action to regulate LDTs or what approach the FDA will seek to take.
In addition, in November 2013, the FDA finalized guidance regarding the sale and use of products labeled for research or investigational use only. Among other things, the guidance states that the FDA continues to be concerned about distribution of research- or investigational-use only products intended for clinical diagnostic use. The guidance states that the FDA will assess whether a manufacturer of such research- or investigational-use only products intends that its products be used for clinical diagnostic purposes by examining the totality of circumstances, including advertising, instructions for clinical interpretation, presentations that describe clinical use, and specialized technical support such as assistance performing clinical validation, surrounding the distribution of the product in question. The FDA has advised that if evidence demonstrates that a product is inappropriately labeled for research-or investigational-use only, the device could be deemed misbranded and adulterated within the meaning of the FDC Act. If the FDA were to undertake enforcement actions, some of our suppliers may cease selling research-use only ("RUO") products to us, and any failure to obtain an acceptable substitute could significantly and adversely affect our business, financial condition and results of operations.
In the European Union LDTs are similarly exempt from the regulations that govern medical devices and in-vitro diagnostics ("IVDs") under certain conditions. The European Union and German legislation on in-vitro diagnostic medical devices ("IVD-MDD") applies. According to the recitals of the Council Directive 98/79/EC on IVD-MDD, reagents which are produced within "health-institution laboratories" for use in that environment and which are not subject to commercial transactions are not covered by the Directive. However, the legal framework for applying the exemption clauses for LDTs is not entirely clear as the IVD-MDD lacks an explicit definition and there is no related case law. On May 26, 2022, when the new Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in-vitro diagnostic medical devices ("IVD-MDR") becomes applicable, the general safety and performance requirements set out in Annex I of the IVD-MDR are applicable also to devices manufactured and used only within health institutions. Overall the exemptions for LDTs will be narrowed, as even in relation to LDTs, health institutions—among others—have to provide information upon request on the use of such devices to their competent authority and each health institution will have to draw up a declaration which it will make publicly available. If these conditions are not met and/or diagnostic tests are manufactured and used only within health institutions but "on an industrial scale", such tests will qualify as IVDs with the full applicability of the IVD-MDR. If we were not able to qualify for an exemption, we would be subject to regulation in the European Union. We also cannot predict whether the EU will amend or implement new laws which may impact our current operations.
For tests that are subject to FDA or EU regulation, we may not be able to obtain timely approvals for our tests or for modifications to our tests, which could delay or prevent us from commercializing our tests and harm our business.
The diagnostic tests we currently offer might meet the definition of LDTs, as they have been designed, developed and validated for use in a single CLIA-certified laboratory. If our tests are LDTs, they are currently not subject to FDA or EU regulation as an in-vitro-diagnostic. In May 2022 when the new IVD Regulation 2017/746/EU comes into force in the European Union, a qualification of our diagnostic tests as IVD-MDs becomes more likely as the manufacture of diagnostic tests "on an industrial scale" will not qualify as LDTs. If the FDA or EU takes action to finalize and implement a regulatory system for LDTs, or if legislation is enacted that subjects LDTs to FDA regulation, we would need to comply with the FDA regulatory requirements for our LDTs. If the FDA takes action to regulate LDTs as devices, we believe that our LDTs would likely be regulated as Class II devices.
In the EU, genetic tests on humans and prenatal tests for genetically caused disorders are regulated as Class C devices under the IVD Regulation. If our LDTs are subject to the IVD
33
Regulation, our tests that qualify as Class C devices will be subject to conformity assessments performed by a notified body.
If services that are currently marketed as LDTs become subject to FDA requirements for in-vitro-diagnostics or are qualified as being subject to the European Union regulations on in vitro diagnostic medical devices, including requirements for premarket clearance or approval, we may not be able to obtain such clearance or approvals on a timely basis, or at all. Our business could be negatively impacted if we are required to stop selling genetic rare disease knowledge and interpretation-based products and solutions pending their clearance or approval, or the launch of any new products and solutions that we develop could be delayed. Likewise, for tests that are regulated as medical devices, we may not be able to obtain clearance or approval of new devices or modifications to marketed devices on a timely basis, or at all, which could delay or prevent us from commercializing our tests and harm our business.
Class II medical devices must obtain FDA clearance of a premarket notification, or 510(k), prior to marketing, unless the FDA has exempted the device from this requirement. Under the 510(k) process, we must demonstrate that our test is substantially equivalent in technological characteristics and intended use to a legally marketed predicate device. The FDA's review and clearance of a 510(k) usually takes from four to twelve months, but it can take longer. Any modifications to an FDA-cleared device that could significantly affect its safety or effectiveness or that would constitute a major change in its intended use would require a new 510(k) clearance or, if the modified device is not substantially equivalent, possibly a de novo classification request or a premarket approval application ("PMA").
If we are unable to identify an appropriate predicate that is substantially equivalent to our device, we would be required to submit a PMA application or a de novo reclassification request, because devices that have not been classified are automatically categorized as Class III. Under the de novo process, we may request that the FDA classify a new low or moderate risk device that lacks an appropriate predicate as a Class I or Class II device. The de novo process typically requires the development of clinical data and usually takes between six to twelve months from the time of submission of the de novo application, but it can take longer.
For tests that are subject to FDA or EU regulation, if we do not comply with FDA or EMA regulatory requirements, we may be subject to enforcement action, with severe consequences for our business.
After approval, devices subject to FDA or EMA regulation are required to comply with post-market requirements. Among the requirements, we and our suppliers must comply with the FDA's Quality System Regulations ("QSRs"), which set forth requirements for the design and manufacture of devices, including the methods and documentation for the design, control testing, quality assurance, labeling, packaging, storage, and shipping of our devices. Our limited experience in complying with these requirements may lead to operational challenges as we increase the scale of our QSR-compliant operations in the United States and develop and refine our policies and procedures for evaluating and mitigating issues we encounter with our processes. Further, if there are any modifications made to the manufacturing of our PMA-approved marketed solutions, a PMA supplement may be required to be submitted to, and approved by, the FDA before the modified device may be marketed.
Other post-market requirements include the reporting of adverse events and malfunctions of which we become aware within the prescribed time frame to the FDA, post-approval studies, establishment registration and device listing, and restrictions on advertising and promotion. We may fail to meet these requirements, which could subject our business to further regulatory risks and costs.
The FDA enforces the post-market requirements of the FDC Act through announced and unannounced inspections. Failure to comply with applicable regulatory requirements could require us to expend time and resources to respond to the FDA's observations and to implement corrective and preventive actions, as appropriate. If we cannot resolve such issues to the satisfaction of the FDA, we
34
may be subject to enforcement actions, including untitled or warning letters, fines, injunctions, or civil or criminal penalties. In addition, we could be subject to a recall or seizure of current or future solutions, operating restrictions, a partial suspension, or a total shutdown of service. Any such enforcement action would have a material adverse effect on our business, financial condition, and results of operations.
In the future, we may fail to achieve coverage or adequate reimbursement for our products and solutions by commercial third-party payors or government payors.
As we expand our operations globally, and in particular to the United States, sales of our existing and any future products and solutions we develop, in particular our diagnostic testing services, in the future may depend upon the availability of adequate reimbursement from third-party payors. These third-party payors include government healthcare programs and/or statutory health insurance schemes in various markets, such as Medicare and Medicaid in the United States and statutory health funds in Germany (the "GKV"), managed care providers, accountable care organizations, private health insurers, and other organizations. We believe that obtaining a positive Medicare Local Coverage Determination, or National Coverage Determination and a favorable Medicare reimbursement rate, and obtaining the agreement of established commercial third-party payors to provide coverage and adequate payment, for each of our existing diagnostic testing services, and any future products and solutions we develop, will be an important element in achieving material commercial success in the United States. Physicians may not order our products and solutions unless commercial third-party payors and government payors authorize coverage and pay for all, or a substantial portion, of the rates established for our products and solutions.
Commercial third-party payors and government payors internationally increasingly attempt to contain healthcare costs by lowering reimbursement rates, limiting coverage of diagnostic test services, and creating conditions of reimbursement, such as requiring participation in clinical evidence development involving research studies and the collection of physician decision impact and patient outcomes data. As a result of these cost-containment trends, commercial third-party payors and government payors that currently provide, or in the future may provide, reimbursement for one or more of our services may propose and/or actually reduce, suspend, revoke, or discontinue payments or coverage at any time. Payors may also create conditions for coverage or may contract with third-party vendors to manage laboratory benefits, in both cases creating administrative hurdles for ordering physicians and patients that may make our products and solutions more difficult to sell. The percentage of submitted claims that are ultimately paid, the length of time to receive payment on claims, and the average reimbursement of those paid claims is likely to vary from period to period.
There is significant uncertainty surrounding whether the use of diagnostic tests that incorporate new technology will be eligible for coverage by commercial third-party payors and government payors or, if eligible for coverage, what the reimbursement rates will be for these services. In Germany, the majority of patients are insured via the GKV. The benefit catalogue defining which services in medical care are reimbursed by the GKV is specified by the directives of the Federal Joint Committee as the highest decision-making body of the joint self-government of physicians, dentists, hospitals and health insurance funds in Germany. The fact that a diagnostic test has been approved for reimbursement in the past, has received approval from the FDA or has been certified by a notified body, or has obtained coverage for any particular rare disease indication or in any particular jurisdiction, does not guarantee that such diagnostic service will remain covered and/or reimbursed or that similar or additional diagnostic tests and/or related rare disease types will be covered and/or reimbursed in the future.
35
As a result, if adequate third-party coverage and reimbursement are unavailable, we may not be able to maintain volume and price levels sufficient to realize an appropriate return on investment in our diagnostic testing services or to advance our research and development solutions for our pharmaceutical partners.
We cannot predict what future healthcare initiatives will be introduced or implemented in the jurisdictions in which we operate, or how any future legislation or regulation may affect us. Any taxes imposed by legislation, as well as changes to the reimbursement amounts paid by payors for our existing and future products and solutions, could have a material adverse effect on our business, financial condition and results of operations.
Intellectual Property Risks Related to Our Business
If we are unable to obtain and maintain patent and other intellectual property protection for any products or solutions we develop and for our technology, or if the scope of intellectual property protection obtained is not sufficient, our competitors could develop and commercialize products and solutions similar or identical to ours, and our ability to successfully commercialize any products or solutions we may develop may be adversely affected.
Our success depends in large part on our ability to obtain and maintain patent and other intellectual property protection in the United States and other countries for our biomarkers and other products and solutions. Patent law relating to the scope of claims in the fields in which we operate is complex and uncertain, so we cannot make any assurances that we will be able to obtain or maintain patent or other intellectual property rights, or that the patent and other intellectual property rights we may obtain will be valuable, provide an effective barrier to competitors or otherwise provide competitive advantages. In particular, our Lyso-Gb3 biomarker, which we use to support the diagnosis of Fabry disease, is not protected by any patents or included in any pending patent applications, and its successful commercialization by one of our competitors or by other third parties could materially harm our business or results of operations. Moreover, patent applications that we have made in the past have been subject to comment and revision by the relevant patent offices, which have resulted in our withdrawal of certain patent applications. If we are unable to obtain or maintain patent or other intellectual property protection with respect to our proprietary products and solutions, our business, financial condition, results of operations, and prospects could be materially harmed.
The scope of patent protection outside of the United States is uncertain. Changes in either the patent laws or their interpretation in the United States and other countries may diminish our ability to protect our inventions, obtain, maintain, and enforce our intellectual property rights and, more generally, could affect the value of our intellectual property or narrow the scope of our patents. We cannot predict whether the patent applications we are currently pursuing will issue as patents in any particular jurisdiction or whether the claims of any issued patents will provide sufficient protection from competitors.
The patent prosecution process is expensive, time-consuming, and complex, and we may not be able to file, prosecute, maintain, enforce, or license all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of our research and development output in time to obtain patent protection. Parties who have access to confidential or patentable aspects of our research and development output, such as our employees, advisors, and other third parties, and who are party to non-disclosure and confidentiality agreements with us, may breach such agreements and disclose such output before a patent application is filed, thereby jeopardizing our ability to seek patent protection. In addition, publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing, or in some cases not at all. Therefore, we cannot be certain that we were the first to make the inventions claimed in our
36
patents or pending patent applications, or that we were the first to file for patent protection of such inventions.
The patent position of companies in our industry generally is unsettled, involves complex legal and factual questions, and has been the subject of much litigation in recent years. As a result, the issuance, scope, validity, enforceability, and commercial value of our patent rights are highly uncertain. Our pending and future patent applications may not result in patents being issued that protect our products or solutions or which effectively prevent others from commercializing competitive products and solutions.
Moreover, the coverage claimed in a patent application can be significantly reduced before the patent is issued, and its scope can be reinterpreted after issuance. Even if patent applications issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors or other third parties from competing with us, or otherwise provide us with any competitive advantage. Any patents that we hold may be challenged, narrowed, circumvented, or invalidated by third parties. In particular, for more information regarding U.S. patent law decisions that negatively impact the patentability of biomarkers, diagnostic products and diagnostic methods, and the validity of granted U.S. patents covering such subject matter, see "—Developments in patent law could have a negative impact on our business" below. Consequently, we do not know whether any of our biomarkers or other products and solutions will be protectable or remain protected by valid and enforceable patents. Our competitors or other third parties may be able to circumvent our patents by developing similar or alternative products and solutions in a non-infringing manner. Any of the foregoing could have a material adverse effect on our business, financial condition, results of operations, and prospects.
If we are unable to protect the confidentiality of our trade secrets, know-how, and other confidential and proprietary information, our business and competitive position would be harmed.
In addition to seeking patent protection for our products and solutions, we also rely upon trade secret protection and non-disclosure agreements and invention assignment agreements with our employees, consultants and other third parties to protect our unpatented know-how, technology, and other confidential or proprietary information. For example, significant elements of our proprietary platform and some of our tests, including aspects of sample preparation, computational-biological algorithms, and related processes and software, are based on unpatented trade secrets and know-how that to our knowledge are not publicly disclosed. In addition to contractual measures, we try to protect the confidential nature of our proprietary information using physical and technological security measures. Such measures may not provide adequate protection for our proprietary information; for example, in the case of misappropriation of intellectual rights by an employee, consultant, or other third party with authorized access.
Trade secrets and know-how can be difficult to protect. We cannot guarantee that we have entered into applicable non-disclosure agreements and invention assignment agreements with our employees, consultants and other third parties who have had access to our trade secrets or other proprietary information. Our security and contractual measures may not prevent an employee, consultant, or other third party from misappropriating our trade secrets and providing them to a competitor, and any recourse we take against such misconduct, including litigation, may not provide an adequate remedy to protect our interests fully. Enforcing a claim that a party illegally disclosed or misappropriated intellectual property can be difficult, expensive, and time-consuming, and the outcome is unpredictable. Due to variation in the degree of protection afforded to intellectual property of this nature under the laws and regulations applicable to different international markets where our services are sold, our ability to pursue and obtain an adequate remedy may depend significantly on the jurisdiction in which the misconduct takes place and our ability to enforce a favorable judgment against the offending party in a jurisdiction in which such party has substantial assets. In addition, trade secrets may be
37
independently developed by others in a manner that could prevent legal recourse by us. If any of our confidential or proprietary information, such as our trade secrets, were to be disclosed or misappropriated, or if any such information were independently developed by a competitor, our competitive position could be harmed.
Patents covering our products or solutions could be found invalid or unenforceable if challenged.
The issuance of a patent is not conclusive as to its inventorship, scope, validity, or enforceability, and our patents may be challenged in the courts or patent offices in the United States and abroad. Others have filed, and in the future are likely to file, patent applications that are similar or identical to ours. To determine the priority of inventions, demonstrate that we did not derive our invention from another individual or entity, or defend third-party challenges to the validity or enforceability of our patent rights, we may have to participate in opposition, derivation, revocation, reexamination, post-grant and inter partes review, or interference proceedings at the U.S. Patent and Trademark Office (the "USPTO") or similar offices or respective courts in Europe or other jurisdictions. For example, we are aware of an opposition proceeding filed in the European Patent Office ("EPO") by Sanofi against EP Patent No. 2 718 725 B1 (the "'725 Patent"), a European patent that we own relating to our biomarker for Gaucher disease. The EPO opposition proceeding challenges the patentability of the '725 Patent in its entirety. Although the EPO has rejected the opposition in the first instance in the hearing held on February 4, 2020, Sanofi may appeal the opposition decision to the Board of Appeal at the EPO and the '725 Patent may still be revoked or maintained in amended form, in whole or in part, if the Boards of Appeal do not uphold the opposition decision. Revoking the '725 Patent may limit our ability to stop others from using or commercializing similar or identical products and solutions to ours, or limit the duration of the patent protection of our products and solutions. See "Item 4. Information On The Company—B. Business Overview—Legal Proceedings" and "Item 8. Financial Information—A. Consolidated Statements and Other Financial Information—Legal Proceedings." Sanofi or other third parties may file future oppositions or other challenges, in Europe or other jurisdictions, against other patents that we own and may also challenge or attack the validity of the national parts of the '725 Patent before national patent courts in parallel or after the proceedings before the EPO. An adverse determination in any such proceeding or litigation could reduce the scope of, or invalidate, our patent rights, allow third parties to commercialize our products or solutions and compete directly with us, without payment to us, or result in our inability to commercialize our products or solutions without infringing third-party patent rights. Moreover, we may have to participate in interference proceedings declared by the USPTO to determine priority of invention or in post-grant challenge proceedings, such as oppositions in a foreign patent office or nullity or entitlement proceedings, that challenge priority of invention or other features of patentability. Such challenges may result in loss of patent rights, loss of exclusivity, or in patents being cancelled, narrowed, amended, invalidated, revoked or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical products and solutions, or limit the duration of the patent protection of our products and solutions. Such proceedings could also result in substantial costs in legal fees and require significant time from our management and employees, even if the eventual outcome is favorable to us. In the event of entitlement proceedings, purported co-inventors may bring claims for compensation or damages. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments, and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common shares. Any of the foregoing could have a material adverse effect on our business, financial condition, results of operations, and prospects.
In addition, if we initiate legal proceedings against a third party to enforce a patent covering our products or solutions, the defendant could counterclaim that such patent is invalid or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several
38
statutory requirements, including lack of novelty, obviousness, or non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement, during prosecution. In other jurisdictions, defendants have and/or may have comparable grounds for defending against such claims, especially with regard to claims that a patent is invalid. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art of which we and the patent examiner were unaware during prosecution. Such challenges could result in the revocation of, cancellation of, or amendment to our patents in such a way that they no longer sufficiently cover our products and solutions. If a third party were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our products or solutions. Such a loss of patent protection would materially harm our business, prospects, financial condition and results of operations.
Litigation or other proceedings or third-party claims of intellectual property infringement could require us to spend significant time and money and could prevent us from selling our products and solutions or impact our share price.
Our commercial success depends upon our ability to develop and commercialize products and solutions and use our proprietary technologies without infringing, misappropriating or otherwise violating the intellectual property and proprietary rights of third parties. We could become party to, or threatened with, adversarial proceedings or litigation regarding intellectual property rights with respect to our technology and any products or solutions we may develop, including interference proceedings, post-grant review, inter partes review, and derivation proceedings before the USPTO and similar proceedings in foreign jurisdictions, such as oppositions before the EPO or nullity or entitlement proceedings. Third parties may assert infringement and other claims against us based on existing patents or patents that may be granted in the future, regardless of their merit, and we may assert infringement and other claims against third parties. As we continue to commercialize our genetic rare disease information solutions (including our biomarkers), launch new solutions and enter new markets, we expect that competitors will claim that our products or solutions infringe or otherwise violate their intellectual property rights, including as part of business strategies designed to impede our successful commercialization and entry into new markets. Third parties may have obtained, and may in the future obtain, patents under which such third parties may claim that the use of our technologies constitutes patent infringement. Third parties have in the past asserted and may in the future assert that we are employing their proprietary technology without authorization, and we occasionally receive letters from third parties inviting us to take licenses under, or alleging that we infringe, their patents. Depending upon the circumstances, we may elect to remove a particular biomarker from one of our products or solutions.
Even if we believe that third-party intellectual property claims are without merit, there is no assurance that a court would find in our favor on questions of infringement, validity, enforceability, or priority. A court of competent jurisdiction could hold that these third-party patents are valid, enforceable, and infringed, which could materially and adversely affect our ability to commercialize any products or solutions we may develop. In order to successfully challenge the validity of any such U.S. patent in federal court or in courts in other jurisdictions, we would need to overcome a presumption of validity. As this burden is a high one, requiring us to present clear and convincing evidence as to the invalidity of any such U.S. patent claim, there is no assurance that a court of competent jurisdiction would invalidate the claims of any such U.S. patent. The same applies to other jurisdictions. If we are found to infringe a third party's intellectual property rights, and we are unsuccessful in demonstrating that such patents are invalid or unenforceable, we could be required to obtain a license from such third party to continue commercializing our products or solutions. However, we may not be able to obtain any required license on commercially reasonable terms, or at all and therefore may be unable to
39
develop, sell or otherwise commercialize our products or solutions. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our competitors and other third parties access to the same technologies licensed to us, and it could require us to make substantial licensing, royalty and other payments. Furthermore, parties making claims against us may be able to obtain injunctive or other relief, which could block our ability to develop, commercialize, and sell our products and solutions, and could result in the award of substantial damages against us. In the event of a successful claim of infringement, misappropriation or other intellectual property violation against us, we may be required to render account for and pay damages and attorneys' fees, recall or destroy stocks and obtain one or more licenses from third parties, or be prohibited from developing, commercializing and selling certain products or solutions. In addition, we could be found liable for significant monetary damages, including treble damages and attorneys' fees, if we are found to have willfully infringed a patent or other intellectual property right.
Even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims may cause us to incur significant expenses and could distract our personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions, or other interim proceedings or developments, and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our shares. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing, or distribution activities. We may not have sufficient financial or other resources to conduct such litigation or proceedings adequately. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources and more mature and developed intellectual property portfolios. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. We also could incur substantial costs and divert the attention of our management and other employees in participating in litigation or proceedings of this nature, and an adverse ruling or perception of an adverse ruling in could have a material adverse impact on our cash position and share price. Any of the foregoing could materially harm our business, prospects, financial condition and results of operations.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment, and other requirements imposed by government patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Obtaining and maintaining a patent portfolio entails significant expense and resources. Part of the expense includes periodic maintenance fees, renewal fees, annuity fees and various other governmental fees associated with patents and patent applications due in several stages over the lifetime of patents and patent applications. The USPTO and various non-U.S. government agencies require compliance with several procedural, documentary, fee payment, and other similar provisions during the patent application process. We may or may not choose to pursue or maintain protection for particular inventions. In addition, there are situations in which failure to make certain payments or noncompliance with certain requirements in the patent process can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. If we choose to forego patent protection or allow a patent application or patent to lapse purposefully or inadvertently, our competitive position could suffer. In such an event, potential competitors might be able to enter the market with similar or identical products and solutions. If we fail to obtain, maintain, protect or enforce our intellectual property rights successfully, our competitive position could suffer. Any of the foregoing could materially harm our business, prospects, financial condition and results of operations.
40
Our rights to develop and commercialize our technology, products and solutions may in the future be subject, in part, to the terms and conditions of licenses granted to us by others.
In connection with the development of new products and solutions we may license intellectual property from third parties in the future, or may deem it necessary to do so in order to commercialize our products or solutions. We may be unable to obtain these licenses at a reasonable cost, or at all. We could, therefore, incur substantial costs related to royalty payments or other payments for licenses obtained from third parties. We may also be unable to obtain exclusive rights to use such intellectual property or technology in all relevant fields of use and in all territories in which we may wish to develop or commercialize our products and solutions in the future and, as a result, we may not be able to prevent competitors from developing and commercializing competitive products or solutions. Moreover, we could encounter delays in introducing new products or solutions while we attempt to develop alternative products and solutions, and the defense of any lawsuit or failure to obtain any of these licenses on favorable terms could prevent us from commercializing our products and solutions, which would materially affect our ability to grow.
Our licensors might conclude that we have materially breached our license agreements and might therefore terminate the license agreements, thereby removing our ability to develop and commercialize products and solutions covered by such agreements. If these in-licenses are terminated, or if the underlying patents fail to provide the intended exclusivity, competitors might have the freedom to market competing products and solutions identical or similar to ours. Disputes may arise regarding intellectual property subject to a licensing agreement, including:
- •
- the scope of rights granted under the license agreement and other interpretation-related issues;
- •
- the extent to which our products and solutions infringe on intellectual property of the licensor that is not subject to the licensing
agreement;
- •
- the sublicensing of patent and other rights under our collaborative development relationships;
- •
- our diligence obligations under the license agreement and what activities satisfy those diligence obligations;
- •
- the inventorship and ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors
and us and our partners; and
- •
- the priority of invention of patented technology.
In addition, agreements under which we license intellectual property or technology from third parties could be complex, and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property or technology, or increase what we believe to be our financial or other obligations under the relevant agreement. Moreover, if disputes over intellectual property or technology that we have licensed prevent or impair our ability to maintain other licensing arrangements on commercially acceptable terms, defending our position could materially harm our business, prospects, financial condition and results of operations.
Developments in patent law could have a negative impact on our business.
Changes in either the patent laws or interpretation of patent laws could increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents. From time to time, the United States Supreme Court (the "Supreme Court"), other federal courts, the U.S. Congress, the USPTO, or other foreign patent offices or courts may change the standards of patentability and any such changes could have a negative impact on our business. Assuming that other requirements for patentability are met, prior to March 2013, in the United States, the first to invent the claimed invention was entitled to the patent, while outside the United States, the first to file a patent application was entitled to the patent. After March 2013, under the Leahy-Smith
41
America Invents Act (the "America Invents Act"), enacted in September 2011, the United States transitioned to a first inventor to file system in which, assuming that other requirements for patentability are met, the first inventor to file a patent application will be entitled to the patent on an invention regardless of whether a third party was the first to invent the claimed invention. The America Invents Act also includes a number of significant changes that affect the way patent applications will be prosecuted and also may affect patent litigation. These include allowing third-party submission of prior art to the USPTO during patent prosecution and additional procedures to attack the validity of a patent by USPTO-administered post-grant proceedings, including post-grant review, inter partes review, and derivation proceedings. However, the America Invents Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business, financial condition, results of operations, and prospects.
In addition, the patent positions of companies in our industry are particularly uncertain. Recent U.S. Supreme Court rulings have narrowed the scope of patent protection available in certain circumstances and weakened the rights of patent owners in certain situations. For example, diagnostic method claims and "gene patents" were considered in two landmark Supreme Court cases, Mayo Collaborative v. Prometheus Laboratories ("Prometheus"), and Association for Molecular Pathology v. Myriad Genetics ("Myriad"). In Prometheus, a case involving patent claims over a medical testing method directed to optimizing the amount of drug administered to a specific patient, Prometheus' claims failed to incorporate sufficient inventive content above and beyond merely describing underlying natural correlations to allow the claimed processes to qualify as patent-eligible processes that apply natural laws. In Myriad, a case brought by multiple plaintiffs challenging the validity of patent claims held by Myriad Genetics, Inc. relating to the breast cancer susceptibility genes BRCA1 and BRCA2, the court held that isolated genomic DNA that exists in nature, such as the DNA constituting the BRCA1 and BRCA2 genes, is not patentable subject matter, but that cDNA, which is an artificial construct created from RNA transcripts of genes, may be patent eligible. The Federal Circuit has begun to apply the holdings in Prometheus and Myriad. In 2015, the Federal Circuit, in Ariosa v. Sequenom, applying Prometheus, found claims to a prenatal diagnostic method that relied on a natural product to be patent ineligible, and clarified that the absence of preemption of a natural phenomenon was not sufficient to demonstrate patent eligibility.
In response to the Supreme Court decisions in Prometheus, Myriad, and Alice Corporation Pty. Ltd. v. CLS Bank International ("Alice Corp."), and others, the USPTO has updated the Manual of Patent Examination Procedure to provide guidance to USPTO personnel in determining the eligibility of patent claims reciting judicially recognized exceptions to patentable subject matter, including laws of nature, natural phenomena, or abstract ideas, for patent eligibility. The USPTO guidance indicates that claims reciting a judicial exception to patent-eligible subject matter must amount to significantly more than the judicial exception itself in order to be patent-eligible subject matter. We cannot assure you that our efforts to seek patent protection for our products and solutions will not be negatively impacted by this interim guidance issued by the USPTO, the decisions described above, rulings in other cases, or changes in guidance or procedures issued by the USPTO.
We cannot fully predict what impact the Supreme Court's decisions in Prometheus, Myriad, Alice Corp., and other decisions may have on our ability or the ability of companies or other entities to obtain or enforce patents relating to DNA, genes, or genomic-related discoveries in the future. Despite the USPTO's interim guidance and Federal Circuit cases described above, the contours of when claims reciting laws of nature, natural phenomena, or abstract ideas may meet the patent eligibility requirements are not clear and may take years to develop via interpretation at the USPTO and in the courts. There are many previously issued patents claiming nucleic acids and diagnostic methods based on natural correlations that issued before the recent Supreme Court decisions discussed, and although many of these patents may be invalid under the standards set forth in the Supreme Court's recent decisions, until successfully challenged, these patents are presumed valid and enforceable, and certain
42
third parties could allege that we infringe, or request that we obtain a license to, these patents. Whether based on patents issued prior to or after these Supreme Court decisions, we might have to defend ourselves against claims of patent infringement, or choose to license rights, if available, under patents claiming such methods. In particular, although the Supreme Court has held in Myriad that isolated genomic DNA is not patent-eligible subject matter, certain third parties could allege that activities that we may undertake infringe other classes of gene-related patent claims, and we could have to defend ourselves against these claims by asserting non-infringement and/or invalidity positions, or pay to obtain a license to these claims. In any of the foregoing or in other situations involving third-party intellectual property rights, if we are unsuccessful in defending against claims of patent infringement, we could be forced to pay damages or be subjected to an injunction that would prevent us from utilizing the patented subject matter in question if we are unable to obtain a license on reasonable terms or at all. Such outcomes could materially affect our ability to offer our products and solutions and have a material adverse impact on our business. Even if we are able to obtain a license or successfully defend against claims of patent infringement, the cost and distraction associated with the defense or settlement of these claims could have a material adverse impact on our business. Any of the foregoing could materially harm our business, prospects, financial condition and results of operations.
We may not be able to enforce our intellectual property rights throughout the world.
Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. Accordingly, we may face an increased risk in these jurisdictions that unauthorized parties may attempt to copy or otherwise obtain or use our patented technology, trademarks, formulations or other intellectual property. The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of Germany or the United States. Specifically, the legal systems of some countries, particularly developing countries, do not favor the enforcement of patents and other intellectual property protection, especially those relating to biotechnology. This could make it difficult for us to stop the infringement of our patents or other intellectual property rights and to prevent third parties from selling or importing products made using our inventions in and to the United States, Germany or other jurisdictions. Competitors may use our technologies in jurisdictions where we have not obtained patent or other protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection but enforcement is not as strong as that in Germany or the United States. These products may compete with our products and solutions, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing. Additionally, many foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties or limit the enforceability of patents against third parties, including government agencies or government contractors. In these countries, patents may provide limited or no benefit. Patent protection must ultimately be sought on a country-by-country basis, which is an expensive and time-consuming process with uncertain outcomes. Accordingly, we may choose not to seek patent protection in certain countries, and we will not have the benefit of patent protection in such countries.
Monitoring infringement and misappropriation of intellectual property can be difficult and expensive, and we may not be able to detect every instance of infringement or misappropriation of our proprietary rights. Even if we do detect infringement or misappropriation of our proprietary rights, proceedings to enforce our intellectual property rights in foreign jurisdictions could result in substantial costs, divert the efforts and attention of our employees and management from other aspects of our business, put our patents at risk of being invalidated or construed narrowly or provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property and proprietary rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop. In addition, changes in the law and legal decisions by courts in Germany, the United States and other jurisdictions may affect our ability to obtain adequate protection for our products and solutions and to enforce our intellectual property rights. Any of the foregoing could materially harm our business, prospects, financial condition and results of operations.
43
Third parties may assert ownership or commercial rights to inventions we develop.
Third parties may in the future make claims challenging the inventorship or ownership of our intellectual property. For example, we rely on certain third parties to provide us with biological materials that we use to conduct our genomic analyses. We have written agreements with collaborators that provide for the ownership of intellectual property arising from our collaborations. These agreements provide that we must negotiate certain commercial rights with collaborators with respect to joint inventions or inventions made by our collaborators that arise from the results of the collaboration. In some instances, there may not be adequate written provisions to address clearly the resolution of intellectual property rights that may arise from a collaboration. If we cannot successfully negotiate sufficient ownership and commercial rights to the inventions that result from our use of a third-party collaborator's materials where required, or if disputes otherwise arise with respect to the intellectual property developed with the use of a collaborator's samples, we may be limited in our ability to capitalize on the market potential of these inventions. In addition, we may face claims that our agreements with employees, contractors, or consultants obligating them to assign intellectual property to us are ineffective, or in conflict with prior or competing contractual obligations of assignment, which could result in ownership disputes regarding intellectual property we have developed or will develop and interfere with our ability to capture the commercial value of such inventions. Litigation may be necessary to resolve an ownership dispute, and if we are not successful, we may be precluded from using certain intellectual property, or may lose our exclusive rights in that intellectual property. Any of the foregoing could materially harm our business, prospects, financial condition and results of operations.
Most of our employees and inventions are subject to German law.
Most of our personnel, including our directors, work in Germany and are subject to German employment law. Inventions which may be the subject of a patent or of protection as a utility model and which are or were made by personnel working in Germany (except for legal representatives of our respective legal entities, for example managing directors) are subject to the provisions of the German Act on Employees' Inventions (Gesetz über Arbeitnehmererfindungen) (the "German Inventions Act"), which regulates the ownership of, and compensation for, inventions made by employees. We face the risk that disputes may occur between us and our current or past employees pertaining to the sufficiency of compensation paid by us, allocation of rights to inventions under this act or alleged non-adherence to the provisions of this act, any of which may be costly to resolve and take up our management's time and efforts whether we prevail or fail in such dispute. In addition, under the German Inventions Act, certain employees retain rights to patents they invented or co-invented and disclosed to us prior to October 1, 2009. If we do not manage to have the respective third-party interests transferred to us or are unable to obtain an exclusive license to any such third-party co-owners' or owners' interest in such patents, such co-owners or owners may be able to transfer or license their rights to other third parties, including our competitors. In addition, we may need the cooperation of any such co-owners or owners to enforce any such patents against third parties, and such cooperation may not be provided to us. While we believe that all of our current and past German employee inventors have subsequently assigned to us their interest in inventions or patents they invented or co-invented, there can be no assurance that all such assignments are fully effective, which can lead to unexpected costs or economic disadvantages. Even if we lawfully own all inventions created by our employees who are subject to the German Inventions Act, we are required under German law to reasonably compensate such employees for the use of the inventions and intellectual property rights related thereto. If we are required to pay compensation or face other disputes under the German Inventions Act, our results of operations could be adversely affected. Legal representatives of legal entities, for example managing directors, whose contractual relationships with the respective entity are subject to German law and that are not subject to the German Inventions Act as well as consultants must assign and transfer their interest in inventions and/or patents they invent or co-invent to us in order for us to have any rights to such
44
inventions or patents. While we believe that all assignments have been made, there can be no assurance that all such assignments are fully effective, which may harm our business, prospects, financial condition and results of operations.
If any of our current or past employees, legal representatives of our legal entities or consultants obtain or retain ownership or co-ownership of any inventions or related intellectual property rights that we believe we own, we may lose valuable intellectual property rights and be required to acquire the respective third-party interests or to obtain and maintain licenses from such employees or legal representatives of legal entities or consultants to such inventions or intellectual property rights, which may not be available on commercially reasonable terms or at all, or may be non-exclusive. If we are unable to acquire the respective third-party interests or to obtain and maintain a license to any such employee's, legal representative's of legal entities or consultant's interest in such inventions or intellectual property rights, we may need to cease the development, manufacture, and commercialization of one or more of the products or solutions we may develop or may have developed. In addition, any loss of exclusivity of our intellectual property rights could limit our ability to stop others from using or commercializing similar or identical products and solutions. We may also face compensation and damages claims from our current or past employees, legal representatives of our legal entities or consultants owning or co-owning any inventions or related intellectual property rights that we believe we own. Any of the foregoing events could materially harm our business, prospects, financial condition and results of operations.
Third parties may assert that our employees or consultants have wrongfully used or disclosed confidential information or misappropriated trade secrets.
Many of our employees and consultants are currently or were previously employed at universities or other diagnostic or biopharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees and consultants do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or these individuals have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of a current or former employer or other third parties. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees. Such claims could materially harm our business, prospects, financial condition and result of operations.
In addition, while it is our policy to require our employees and consultants who may be involved in the conception or development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who, in fact, conceives or develops intellectual property that we regard as our own. The assignment of intellectual property rights may not be self-executing, or the assignment agreements may be breached, and we may be forced to bring claims against third parties, or defend claims that they may bring against us, to determine the ownership of what we regard as our intellectual property. Such claims could materially harm our business, prospects, financial condition and results of operations.
45
Intellectual property rights do not necessarily address all potential threats.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations and may not adequately protect our business or permit us to maintain our competitive advantage. For example:
- •
- others may be able to make products or solutions that are similar to any products or solutions we develop or commercialize or utilize similar
technology but that are not covered by the claims of our patents or patents that we might own or license in the future;
- •
- we might not have been the first to make the inventions covered by the issued patents or pending patent applications that we own or may own or
license in the future;
- •
- we might not have been the first to file patent applications covering certain of our inventions;
- •
- others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual
property rights;
- •
- it is possible that our pending patent applications or those that we may own or license in the future will not lead to issued patents;
- •
- our issued patents may be held invalid or unenforceable, including as a result of legal challenges by our competitors;
- •
- our competitors might conduct research and development activities in countries where we do not have patent rights and then use the information
learned from such activities to develop competitive products or solutions for sale in our major commercial markets;
- •
- we may not develop additional proprietary technologies that are patentable;
- •
- the patents of others may harm our business; and
- •
- we may choose not to file a patent in order to maintain certain trade secrets or know-how, and a third party may subsequently file a patent covering such intellectual property.
Should any of these events occur, they could materially harm our business, prospects, financial condition and results of operations.
Risks Relating to Our Financial Condition and Capital Requirements
We have a history of losses and we may incur losses in the future.
We have historically incurred losses, including total comprehensive losses of €20,839 thousand, €11,346 thousand and €5,466 thousand in the years ended December 31, 2019, 2018 and 2017, respectively. We expect our losses to continue as a result of ongoing research and development expenses and increased selling and marketing costs. These losses have had, and will continue to have, an adverse effect on our working capital, total assets, and shareholders' equity. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our inability to achieve and then maintain profitability would negatively affect our business, financial condition, results of operations, and cash flows.
Failure to meet covenants under any loan facility may limit our liquidity and could result in the lenders accelerating amounts we owe to them under the facility.
Our debt agreements contain covenants that restrict our ability to, among other things, use the funds for specified purposes, incur additional indebtedness, pay dividends or engage in certain business activities. We did not satisfy certain covenants under our syndicated loan facility during the years ended
46
December 31, 2017 and 2018, and in order to remedy such breaches, we obtained waivers for the relevant years from the various lenders under this facility. During the year ended December 31, 2019, we repaid a significant portion of outstanding bank loans upon the completion of the sale and leaseback transaction and placed a further cash deposit of €1,500 thousand relating to the remaining outstanding bank loans. Accordingly, all covenants and related restrictions under the syndicated loan facility were removed as of December 31, 2019.
We may enter into new debt facilities in the future. The breach of any covenants under these new facilities, if not remedied or waived in time, could result in a default on our obligations and/or cause the acceleration of our outstanding debt by our lenders, which could have a material adverse impact on our business, financial condition, results of operations, cash flows, and the trading price of our securities. If we are not able to repay the loans, this may lead to the commencement of foreclosure or other enforcement actions against any of our assets securing such debt. Even if the bank would waive a covenant breach, we may be subject to an increase of interest rates or margins, respectively, as well as the payment of a waiver fine. Furthermore, the covenants as well as the breach of the covenants may impose restrictions on the way we can operate and may limit our ability to finance our future operations and capital needs and our ability to pursue business activities that may be in our interests.
We may need to raise additional capital to fund our existing operations, develop our genetic information platform, commercialize new products and solutions and expand our operations.
If our available cash balances and anticipated cash flow from operations are insufficient to satisfy our liquidity requirements, including because of lower demand for our products or solutions as a result of other risks described herein, we may seek to sell common or preferred equity or convertible debt securities, enter into another credit facility or another form of third-party funding, or seek other debt financing.
Our ongoing efforts to expand our business will require substantial cash resources. We may consider raising additional capital in the future to expand our business, to pursue strategic investments, to take advantage of financing opportunities, or for other reasons, including to:
- •
- increase our sales and marketing efforts to drive market adoption of our products and solutions and address competitive developments;
- •
- fund development and marketing efforts of any future products and solutions;
- •
- further expand our laboratory operations;
- •
- expand our technologies into other types of diseases;
- •
- obtain, maintain, protect and enforce existing or new intellectual property rights;
- •
- acquire, license or invest in technologies, including information technologies;
- •
- acquire or invest in complementary businesses or assets; and
- •
- finance capital expenditures and general and administrative expenses.
Our present and future funding requirements will depend on many factors, including:
- •
- our ability to achieve revenue growth;
- •
- the cost of expanding our laboratory operations and offerings, including our sales and marketing efforts;
- •
- our rate of progress in, and cost of the sales and marketing activities associated with, establishing adoption of our products and solutions;
47
- •
- our rate of progress in, and cost of research and development activities associated with, products and solutions in research and early
development;
- •
- the effect of competing technological and market developments;
- •
- costs related to international expansion; and
- •
- the potential cost of and delays in research and development as a result of any regulatory oversight applicable to our products and solutions.
If we raise funds by issuing debt securities, those debt securities would have rights, preferences, and privileges senior to those of holders of our common shares. The terms of debt securities issued or borrowings pursuant to a credit or similar agreement could impose significant restrictions on our operations. Such financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring debt, making capital expenditures or declaring dividends. If additional funds are raised by issuing equity securities, existing shareholders may suffer significant dilution. If we raise funds through collaborations and licensing arrangements, we might be required to relinquish significant rights to our platform technologies or solutions, or grant licenses on terms that are not favorable to us.
Additional equity or debt financing might not be available on reasonable terms or at all. Because of our potential long-term capital requirements, we may access the public or private equity or debt markets whenever conditions are favorable, even if we do not have an immediate need for additional capital at that time. If we cannot secure additional funding when needed, we may have to delay, reduce the scope of, or eliminate one or more research and development programs or sales and marketing initiatives. In addition, we may have to work with a partner on one or more of our development programs, which could lower the economic value of those programs to us. Lastly, if we are unable to obtain the requisite amount of financing needed to fund our planned operations, it could have a material adverse effect on our business and financial position.
We may be required to refund grants and subsidies.
We have received various grants and subsidies to fund our research and development programs from various funding organizations. However, the Company continues to engage in efforts to secure further grants and subsidies for the next development steps of its product candidates. Some of these grants and subsidies provide for certain requirements in respect of the utilization of proceeds generated as a result of the publicly sponsored projects. For example, we received grants from the European Regional Development Fund in order to fund our Rostock facility, which grants are limited in purpose to development and innovation in the state of Mecklenburg-Western Pomerania, Germany. Other grants which we obtain may impose restrictions on our operations, and if we are in noncompliance with the restrictions and conditions of any grant or subsidy program, a partly or complete repayment cannot be excluded. This may also apply to grants and subsidies we may apply for in the future. If we are required to refund grants or subsidies, this could have a material adverse effect on our liquidity and cash flow position and may negatively affect our business, prospects and financial conditions. In the year ended December 31, 2019, we have received a total of €2,181 thousand in grants for our activities, and refunded €358 thousand of grants received for the land on which our Rostock facilities are located following the sale and leaseback transaction.
We incur significant costs as a result of operating as a public company and our management needs to devote substantial time to public company compliance programs.
As a public company, we incur significant legal, accounting, and other expenses due to our compliance with regulations and disclosure obligations applicable to us, including compliance with the Sarbanes-Oxley Act, as well as rules implemented by the SEC, and the Nasdaq Global Market
48
("Nasdaq"). The SEC and other regulatory authorities have continued to adopt new rules and regulations and make additional changes to existing regulations that require our compliance. In July 2010, the Dodd-Frank Wall Street Reform and Consumer Protection Act (the "Dodd-Frank Act") was enacted. There are significant corporate governance- and executive compensation-related provisions in the Dodd-Frank Act that have required the SEC to adopt additional rules and regulations in these areas. Shareholder activism, the current political environment, and the current high level of government intervention and regulatory reform may lead to substantial new regulations and disclosure obligations, which may lead to additional compliance costs and impact, in ways we cannot currently anticipate, and the manner in which we operate our business. Our management and other personnel will need to devote a substantial amount of time to these compliance programs and monitoring of public company reporting obligations, and as a result of the new corporate governance- and executive compensation-related rules, regulations, and guidelines prompted by the Dodd-Frank Act, and further regulations and disclosure obligations expected in the future, we will likely need to devote additional time and costs to comply with such compliance programs and rules. These rules and regulations will cause us to incur significant legal and financial compliance costs and will make some activities more time-consuming and costly.
To comply with the requirements of being a public company, we may need to undertake various actions, including implementing new internal controls and procedures and hiring new accounting or internal audit staff. The Sarbanes-Oxley Act requires that we maintain effective disclosure controls and procedures and internal control over financial reporting. We are continuing to develop and refine our disclosure controls and other procedures that are designed to ensure that information required to be disclosed by us in the reports that we file with the SEC is recorded, processed, summarized, and reported within the time periods specified in SEC rules and forms, and that information required to be disclosed in reports under the Exchange Act is accumulated and communicated to our principal executive and financial officers. Our current controls and any new controls that we develop may become inadequate, and weaknesses in our internal control over financial reporting may be discovered in the future. Any failure to develop or maintain effective controls could adversely affect the results of periodic management evaluations and annual independent registered public accounting firm attestation reports regarding the effectiveness of our internal control over financial reporting, which we may be required to include in the periodic reports we file with the SEC under Section 404 of the Sarbanes-Oxley Act, and could harm our operating results, cause us to fail to meet our reporting obligations, or result in a restatement of our prior period financial statements. In the event that we are not able to demonstrate compliance with the Sarbanes-Oxley Act, that our internal control over financial reporting is perceived as inadequate, or that we are unable to produce timely or accurate financial statements, investors may lose confidence in our operating results, and the price of our common shares could decline.
We are required to comply with certain of the SEC rules that implement Section 404 of the Sarbanes-Oxley Act, which requires management to certify financial and other information in our annual reports and provide an annual management report on the effectiveness of our internal control over financial reporting, commencing with our second annual report. This assessment needs to include the disclosure of any material weaknesses in our internal control over financial reporting identified by our management or our independent registered public accounting firm. During the evaluation and testing process, if we identify one or more material weaknesses in our internal control over financial reporting or if we are unable to complete our evaluation, testing, and any required remediation in a timely fashion, we will be unable to assert that our internal control over financial reporting is effective.
Our independent registered public accounting firm will not be required to formally attest to the effectiveness of our internal control over financial reporting until the first annual report required to be filed with the SEC following the date we are no longer an "emerging growth company" as defined in the JOBS Act. We cannot assure you that there will not be material weaknesses or significant
49
deficiencies in our internal controls in the future. If we are unable to assert that our internal control over financial reporting is effective, or if our independent registered public accounting firm is unable to express an opinion on the effectiveness of our internal control over financial reporting, we could lose investor confidence in the accuracy and completeness of our financial reports, which could have a material adverse effect on the price of our common shares.
If we fail to implement effective internal controls over financial reporting, such failure could result in material misstatements in our financial statements, cause investors to lose confidence in our reported financial and other public information and have a negative effect on the trading price of our common shares.
Effective internal controls over financial reporting are necessary for us to provide reliable financial reports and, together with adequate disclosure controls and procedures, are designed to prevent fraud. Any failure to implement required new or improved controls, or difficulties encountered in their implementation could cause us to fail to meet our reporting obligations. Section 404 of the Sarbanes-Oxley Act of 2002 requires management of public companies to develop and implement internal controls over financial reporting and evaluate the effectiveness thereof. If we fail to design and operate effective internal controls, it could result in material misstatements in our financial statements, impair our ability to raise revenue, result in the loss of investor confidence in the reliability of our financial statements and subject us to regulatory scrutiny and sanctions, which in turn could harm the market value of our common shares.
We will be required to disclose changes made in our internal controls and procedures and our management will be required to assess the effectiveness of these controls annually. However, for as long as we are an "emerging growth company" under the JOBS Act, our independent registered public accounting firm will not be required to attest to the effectiveness of our internal controls over financial reporting pursuant to Section 404. We could be an "emerging growth company" for up to five years after our initial public offering. An independent assessment of the effectiveness of our internal controls could detect problems that our management's assessment might not. Undetected material weaknesses in our internal controls could lead to financial statement restatements and require us to incur the expense of remediation.
We have identified a material weakness in our internal control over financial reporting and may identify additional material weaknesses in the future that may cause us to fail to meet our reporting obligations or result in material misstatements of our financial statements. If we fail to remediate our material weakness or if we fail to establish and maintain an effective system of internal control over financial reporting, we may not be able to report our financial results accurately or to prevent fraud.
Effective internal controls over financial reporting are necessary for us to provide reliable financial reports and, together with adequate disclosure controls and procedures, are designed to prevent fraud. Any failure to implement required new or improved controls, or difficulties encountered in their implementation could cause us to fail to meet our reporting obligations. Section 404 of the Sarbanes-Oxley Act of 2002 requires management of public companies to develop and implement internal controls over financial reporting and evaluate the effectiveness thereof. A material weakness is a deficiency or a combination of deficiencies in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of our financial statements will not be prevented or detected on a timely basis.
In connection with the preparation of our unaudited interim condensed consolidated financial statements as of and for the nine month period ended September 30, 2018, we identified a material weakness in our internal controls as of December 31, 2017, related to the lack of effective review controls over judgmental and complex areas of the financial statement close process and a lack of
50
routine financial statement close process controls. The material weakness was not fully remediated as of December 31, 2019.
In response to such material weakness, management hired additional senior accounting and financial professionals with the experience and knowledge necessary to supervise the financial statement closing process and review the accounting records and necessary adjustments. An additional experienced professional was also engaged to begin establishing and implementing relevant internal control procedures to address the material weakness identified. At the end of 2019, we completed an optimization of our accounting system and we commenced automation of certain control activities and report functionality.
In the year ending December 31, 2020, we intend to further formalize the internal control environment for our key accounting processes, including formal definition and documentation of control activities, monitoring of control testing and integration with our updated IT environment. Further hires of senior staff to support the additional reporting and compliance requirements that listed companies are subject to have also been contemplated. Although we are working to remediate the material weakness as quickly and efficiently as possible, we cannot estimate how long it will take to effectively remediate the material weakness.
If we are unable to successfully remediate our identified material weakness, if we discover additional material weaknesses or if we otherwise are unable to report our financial statements accurately or in a timely manner, we would be required to continue disclosing such material weaknesses in future filings with the SEC, which could adversely affect our business, investor confidence in our company and the market price of our common shares and could subject us to litigation or regulatory enforcement actions. As a result, shareholders could lose confidence in our financial and other public reporting, which would harm our business and the market value of our common shares.
Raising additional capital may cause dilution to our shareholders, restrict our operations or require us to relinquish rights to our technologies or drug candidates.
To the extent that we choose or need to raise additional capital through the sale of common shares or securities convertible or exchangeable into common shares, the ownership interest of our shareholders will be diluted, and the terms of these securities may include liquidation or other preferences that materially adversely affect the rights of our common shareholders. Debt financing, if available, would increase our fixed payment obligations and may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends.
If we raise funds through additional collaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our intellectual property, future revenue streams, research programs or drug candidates or to grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our drug development or future commercialization efforts or grant rights to develop and market drug candidates that we would otherwise prefer to develop and market ourselves.
51
Our results of operations could be materially adversely affected by fluctuations in foreign currency exchange rates.
Although we report our results of operations in euro, not all of our net revenues are denominated in the euro. Unfavorable fluctuations in foreign currency exchange rates could have a material adverse effect on our results of operations.
Because our consolidated financial statements are presented in euro, we must translate revenues, expenses and income, as well as assets and liabilities, into euros at exchange rates in effect during or at the end of each reporting period. Therefore, changes in the value of the euro against other currencies will affect our net revenues, operating income and the value of balance-sheet items originally denominated in other currencies. These changes cause our growth in consolidated earnings stated in euro to be higher or lower than our growth in local currency when compared against other periods.
As we continue to leverage our global delivery model, more of our expenses are incurred in currencies other than those in which we bill for the related services. An increase in the value of certain currencies against the euro could increase costs for delivery of services at off-shore sites by increasing labor and other costs that are denominated in local currency. There can be no assurance that our contractual provisions will offset their impact, or that our currency hedging activities, which are designed to partially offset this impact, will be successful. In addition, our currency hedging activities are themselves subject to risk. These include risks related to counterparty performance under hedging contracts and risks related to currency fluctuations. We also face risks that extreme economic conditions, political instability or hostilities or disasters of the type described below could impact our underlying exposures, perhaps eliminating them. Such an event could lead to losses being recognized on the currency hedges then in place, not offset by anticipated changes in the underlying hedge exposure.
Certain Factors Relating to Our Common Shares
Our share price might fluctuate, as a result of which you could lose a significant part of your investment.
The market price of our common shares may fluctuate significantly in response to numerous factors, many of which are beyond our control, including:
- •
- financial analysts ceasing to cover our common shares or changes in financial estimates by analysts;
- •
- actual or anticipated variations in our operating results;
- •
- changes in financial estimates by financial analysts, or any failure by us to meet or exceed any of these estimates, or changes in the
recommendations of any financial analysts that elect to follow our common shares or the shares of our competitors;
- •
- announcements by us or our competitors of significant contracts or acquisitions;
- •
- future sales of our shares; and
- •
- investor perceptions of us and the industries in which we operate.
In addition, the stock market in general has experienced substantial price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of particular companies affected. These broad market and industry factors may materially harm the market price of our common shares, regardless of our operating performance. In the past, following periods of volatility in the market price of certain companies' securities, securities class action litigation has been instituted against these companies. This litigation, if instituted against us, could adversely affect our financial condition or results of operations.
52
Sales of substantial amounts of our common shares in the public market, or the perception that these sales may occur, could cause the market price of our common shares to decline.
Sales of substantial amounts of our common shares in the public market, or the perception that these sales may occur, could cause the market price of our common shares to decline. This could also impair our ability to raise additional capital through the sale of our equity securities. Under our articles of association, we are authorized to issue up to 79,000,000 common shares, of which 19,861,340 common shares were outstanding as of December 31, 2019. We, our management board members, supervisory board members and certain of our shareholders have agreed with the underwriters, subject to certain exceptions, not to offer, sell, or dispose of any shares of our share capital or securities convertible into or exchangeable or exercisable for any shares of our share capital during the 180-day period following our initial public offering up until May 4, 2020. If, after the end of such lock-up agreements, these shareholders sell substantial amounts of common shares in the public market, or the market perceives that such sales may occur, the market price of our common shares and our ability to raise capital through an issue of equity securities in the future could be adversely affected. We have also entered into a registration rights agreement in connection with our initial public offering pursuant to which we agreed under certain circumstances to file a registration statement to register the resale of the common shares held by certain of our existing shareholders, as well as to cooperate in certain public offerings of such common shares. In addition, we have registered on a Form S-8 registration statement all common shares that we may issue under our new omnibus equity incentive plan under which we have discretion to grant a broad range of equity-based awards to eligible participants. As a result, these shares can be freely sold in the public market upon issuance, subject to volume limitations applicable to affiliates. If a large number of our common shares or securities convertible into our common shares are sold in the public market after they become eligible for sale, the sales could reduce the trading price of our common shares and impede our ability to raise future capital. We cannot predict the size of future issuances of our shares or the effect, if any, that future sales and issuances of shares would have on the market price of our common shares.
We have broad discretion in the use of our cash on hand and may invest or spend it in ways with which you do not agree and in ways that may not yield a return on your investment.
As of December 31, 2019, we had €41.1 million in cash and cash equivalents. Our management will have broad discretion in the use of such cash and could spend it in ways that do not improve our results of operations or enhance the value of our common shares. You will not have the opportunity to influence our decisions on how to use our cash on hand. The failure by our management to apply these funds effectively could result in financial losses that could harm our business, cause the price of our common shares to decline and delay the development of our product candidates. Pending its use, we may invest our cash on hand in a manner that does not produce income or that loses value.
Our recent transformation into a public company is causing an increase of our costs and may disrupt the regular operations of our business.
Our business historically has operated as a privately owned company, and we may incur significant additional legal, accounting, reporting and other expenses as a result of being a publicly traded company. We might also incur costs which we have not incurred previously, including, but not limited to, costs and expenses for managing directors' and supervisory directors' fees, increased directors and officers insurance, investor relations, and various other costs of a public company.
We also expect to incur costs associated with corporate governance requirements, including requirements under the Sarbanes-Oxley Act, as well as rules implemented by the SEC and Nasdaq. We expect these rules and regulations to increase our legal and financial compliance costs and make some management and corporate governance activities more time-consuming and costly, particularly after we are no longer an "emerging growth company." These rules and regulations may make it more difficult
53
and more expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. This could have an adverse impact on our ability to retain, recruit and bring on a qualified management board and a qualified independent supervisory board. We expect that the additional costs we will incur as a public company, including costs associated with corporate governance requirements, will be considerable relative to our costs as a private company.
The additional demands associated with being a public company may disrupt regular operations of our business by diverting the attention of some of our senior management team away from revenue producing activities to management and administrative oversight, adversely affecting our ability to attract and complete business opportunities and increasing the difficulty in both retaining professionals and managing and growing our businesses. Any of these effects could harm our business, financial condition and results of operations.
Furthermore, after the date we are no longer an emerging growth company, our independent registered public accounting firm will only be required to attest to the effectiveness of our internal control over financial reporting depending on our market capitalization. Even if our management concludes that our internal controls over financial reporting are effective, our independent registered public accounting firm may still decline to attest to our management's assessment or may issue a report that is qualified if it is not satisfied with our controls or the level at which our controls are documented, designed, operated or reviewed, or if it interprets the relevant requirements differently from us. In addition, in connection with the implementation of the necessary procedures and practices related to internal control over financial reporting, we may identify deficiencies that we may not be able to remediate in time to meet the deadline imposed by the Sarbanes-Oxley Act for compliance with the requirements of Section 404. Failure to comply with Section 404 could subject us to regulatory scrutiny and sanctions, impair our ability to raise revenue, cause investors to lose confidence in the accuracy and completeness of our financial reports and negatively affect our share price.
We are an "emerging growth company" and we cannot be certain if the reduced disclosure requirements applicable to emerging growth companies will make our common shares less attractive to investors.
We are an "emerging growth company," as defined in the JOBS Act, and we are taking advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not "emerging growth companies." For example, for as long as we are an "emerging growth company" under the recently enacted JOBS Act, our independent registered public accounting firm will not be required to attest to the effectiveness of our internal control over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act. We could be an emerging growth company for up to five years. We cannot predict if investors will find our common shares less attractive because we will rely on these exemptions. If some investors find our common shares less attractive as a result, there may be a less active trading market for our common shares and our share price may be more volatile.
In addition, Section 107 of the JOBS Act provides that an emerging growth company can use the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. Given that we currently report and expect to continue to report under IFRS as issued by the IASB, we have irrevocably elected not to avail ourselves of this extended transition period and, as a result, we will adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required by the IASB. Since IFRS makes no distinction between public and private companies for purposes of compliance with new or revised accounting standards, the requirements for our compliance as a private company and as a public company are the same.
54
We are a foreign private issuer and, as a result, we are not subject to U.S. proxy rules and are subject to Exchange Act reporting obligations that, to some extent, are more lenient and less frequent than those of a U.S. domestic public company.
We report under the Exchange Act as a non-U.S. company with foreign private issuer status. Because we qualify as a foreign private issuer under the Exchange Act, we are exempt from certain provisions of the Exchange Act that are applicable to U.S. domestic public companies, including (i) the sections of the Exchange Act regulating the solicitation of proxies, consents or authorizations in respect of a security registered under the Exchange Act, (ii) the sections of the Exchange Act requiring insiders to file public reports of their share ownership and trading activities and liability for insiders who profit from trades made in a short period of time and (iii) the rules under the Exchange Act requiring the filing with the SEC of quarterly reports on Form 10-Q containing unaudited financial and other specified information, or current reports on Form 8-K, upon the occurrence of specified significant events. In addition, foreign private issuers are not required to file their annual report on Form 20-F until four months after the end of each fiscal year, while U.S. domestic issuers that are accelerated filers are required to file their annual report on Form 10-K within 75 days after the end of each fiscal year. Foreign private issuers are also exempt from the Regulation Fair Disclosure, aimed at preventing issuers from making selective disclosures of material information. As a result of the above, you may not have the same protections afforded to shareholders of companies that are not foreign private issuers.
We may lose our foreign private issuer status, which would then require us to comply with the Exchange Act's domestic reporting regime and cause us to incur significant legal, accounting and other expenses.
We are a foreign private issuer and therefore we are not required to comply with all of the periodic disclosure and current reporting requirements of the Exchange Act applicable to U.S. domestic issuers. If in the future we are not a foreign private issuer as of the last day of the second fiscal quarter in any fiscal year, we would be required to comply with all of the periodic disclosure, current reporting requirements and proxy solicitation rules of the Exchange Act applicable to U.S. domestic issuers. In order to maintain our current status as a foreign private issuer, either (a) a majority of our common shares must be either directly or indirectly owned of record by non-residents of the United States or (b)(i) a majority of our directors and executive officers may not be United States citizens or residents, (ii) more than 50% of our assets cannot be located in the United States and (iii) our business must be administered principally outside the United States. If we were to lose this status, we would be required to comply with the Exchange Act reporting and other requirements applicable to U.S. domestic issuers, which are more detailed and extensive than the requirements for foreign private issuers. We may also be required to make changes in our corporate governance practices in accordance with various SEC and stock exchange rules. The regulatory and compliance costs to us if we are required to comply with the reporting requirements applicable to a U.S. domestic issuer may be significantly higher than the costs we would incur as a foreign private issuer. As a result, we expect that a loss of foreign private issuer status would increase our legal and financial compliance costs and would make some activities highly time consuming and costly. These rules and regulations could also make it more difficult for us to attract and retain qualified directors.
As a foreign private issuer and as permitted by the listing requirements of Nasdaq, we follow certain home country governance practices rather than the corporate governance requirements of the Nasdaq.
We are a foreign private issuer. As a result, in accordance with the listing requirements of Nasdaq, we are relying on home country governance requirements and certain exemptions thereunder rather than on the corporate governance requirements of Nasdaq. In accordance with Dutch law and generally accepted business practices, our Articles of Association do not provide quorum requirements generally applicable to general meetings of shareholders. To this extent, our practice varies from the requirement of Nasdaq Listing Rule 5620(c), which requires an issuer to provide in its bylaws for a generally
55
applicable quorum, and that such quorum may not be less than one-third of the outstanding voting shares. Although we must provide shareholders with an agenda and other relevant documents for the general meeting of shareholders, Dutch law does not have a regulatory regime for the solicitation of proxies and the solicitation of proxies is not a generally accepted business practice in the Netherlands, thus our practice varies from the requirement of Nasdaq Listing Rule 5620(b). As permitted by the listing requirements of Nasdaq, we have also opted out of the requirements of Nasdaq Listing Rule 5605(d), which requires, among other things, an issuer to have a compensation committee that consists entirely of independent directors, Nasdaq Listing Rule 5605(e), which requires independent director oversight of director nominations, and Nasdaq Listing Rule 5605(b)(2), which requires an issuer to have a majority of independent directors on its board. We also rely on the phase-in rules of the SEC and Nasdaq with respect to the independence of our audit committee. These rules require that a majority of our directors must be independent and all members of our audit committee must meet the independence standard for audit committee members by November 6, 2020, the date that is one year from the date of effectiveness of our Form F-1 registration statement in connection with our initial public offering. In addition, we have opted out of shareholder approval requirements, as included in the Nasdaq Listing Rules, for the issuance of securities in connection with certain events such as the acquisition of shares or assets of another company, the establishment of or amendments to equity-based compensation plans for employees, a change of control of the Company and certain private placements. To this extent, our practice varies from the requirements of Nasdaq Rule 5635, which generally requires an issuer to obtain shareholder approval for the issuance of securities in connection with such events. Accordingly, you may not have the same protections afforded to shareholders of companies that are subject to these Nasdaq requirements.
Insiders continue to have substantial control over us and could limit your ability to influence the outcome of key transactions, including a change of control.
Our principal shareholders, directors and executive officers and entities affiliated with them own approximately 79.6% of the outstanding common shares. As a result, these shareholders, if acting together, would be able to influence or control matters requiring approval by our general meeting of shareholders, including the election of managing directors and supervisory directors, changes to our articles of association and the approval of mergers or other extraordinary transactions. They may also have interests that differ from yours and may vote in a way with which you disagree and which may be adverse to your interests. The concentration of ownership may have the effect of delaying, preventing or deterring a change of control of our company, could deprive our shareholders of an opportunity to receive a premium for their common shares as part of a sale of our company and might ultimately affect the market price of our common shares.
We do not anticipate paying any cash dividends in the foreseeable future.
We currently intend to retain our future earnings, if any, for the foreseeable future, to repay indebtedness and to fund the development and growth of our business. We do not intend to pay any dividends to holders of our common shares. As a result, capital appreciation in the price of our common shares, if any, will be your only source of gain on an investment in our common shares.
If we do pay dividends, we may need to withhold tax on such dividends payable to holders of our shares in both Germany and the Netherlands.
As an entity incorporated under Dutch law, any dividends distributed by us are subject to Dutch dividend withholding tax on the basis of Dutch domestic law. On the basis of the 2012 Convention between the Federal Republic of Germany and the Kingdom of the Netherlands for the avoidance of double taxation with respect to taxes on income (the "double tax treaty between Germany and the Netherlands"), however, the Netherlands will be restricted in imposing these taxes if the Company is
56
also a tax resident of Germany and its effective management is located in Germany. See "Item 3. Key Information—D. Risk Factors—We may become taxable in a jurisdiction other than Germany and this may increase the aggregate tax burden on us." Dutch dividend withholding tax is, however, still required to be withheld from dividends if and when paid to Dutch resident holders of our common shares (and non-Dutch resident holders of our common shares that have a permanent establishment in the Netherlands to which their shareholding is attributable). As a result, upon a payment of dividends, we will be required to identify our shareholders in order to assess whether there are Dutch residents (or non-Dutch residents with a permanent establishment in the Netherlands to which the common shares are attributable) in respect of which Dutch dividend tax has to be withheld. Such identification may not always be possible in practice. As the dividend withholding tax liability of a Dutch resident shareholder can generally be credited against such shareholder's (corporate) income tax liability, we may approach the Dutch tax authorities prior to a payment of dividends to apply for a tax ruling confirming that no withholding of any Dutch withholding tax is required. The outcome of such tax ruling request is, however, uncertain. If a favorable tax ruling cannot be obtained and if the identity of our shareholders cannot be determined, withholding of both German and Dutch dividend tax may occur upon a payment of dividends.
Shareholders may not be able to exercise preemptive rights and, as a result, may experience substantial dilution upon future issuances of common shares
In the event of an issuance of common shares, subject to certain exceptions, each shareholder will have a pro rata preemptive right in proportion to the aggregate nominal value of the common shares held by such holder. These preemptive rights may be restricted or excluded by a resolution of the general meeting of shareholders or by another corporate body designated by the general meeting of shareholders. Prior to the closing of our initial public offering, our management board, subject to approval of our supervisory board, was authorized, for a period of five years to issue shares or grant rights to subscribe for shares up to our authorized share capital from time to time and to limit or exclude preemptive rights in connection therewith. This could cause existing shareholders to experience substantial dilution of their interest in us.
If equity and industry research analysts publish negative evaluations of or downgrade our common shares, the price of our common shares could decline.
The trading market for our common shares relies in part on the research and reports that equity and industry research analysts publish about us or our business. We do not control these analysts. If one or more of the analysts covering our business downgrade their evaluations of our common shares, the price of our common shares could decline. If one or more of these analysts cease to cover our common shares, we could lose visibility in the market for our common shares, which in turn could cause our common shares price to decline.
We may become taxable in a jurisdiction other than Germany and this may increase the aggregate tax burden on us.
Since our incorporation we have had, on a continuous basis, our place of "effective management" in Germany. We will therefore qualify as a tax resident of Germany on the basis of German domestic law. As an entity incorporated under Dutch law, however, we also qualify as a tax resident of the Netherlands on the basis of Dutch domestic law. However, based on our current management structure and the current tax laws of the United States, Germany and the Netherlands, as well as applicable income tax treaties, and current interpretations thereof, we should qualify solely as a tax resident of Germany for the purposes of the double tax treaty between Germany and the Netherlands due to the "effective management" tie-breaker. The test of "effective management" is largely a question of fact and degree based on all the circumstances, rather than a question of law. Nevertheless, the relevant
57
case law and OECD guidance suggest that the Company is likely to be regarded as a German tax resident from incorporation and remaining so if, as the Company intends, (i) most meetings of its management board are held in Germany (and none will be held in the Netherlands) with a majority of directors present in Germany for those meetings; (ii) at those meetings there are full discussions of, and decisions are made regarding, the key strategic issues affecting the Company and its subsidiaries; (iii) those meetings are properly minuted; (iv) at least some of the directors of the Company, together with supporting staff, are based in Germany; and (v) the Company has permanent staffed office premises in Germany. We may, however, become subject to limited income tax liability in other countries with regard to the income generated in the respective other country, for example, due to the existence of a permanent establishment or a permanent representative in such other country.
The applicable tax laws or interpretations thereof may change. Furthermore, whether we have our place of effective management in Germany and are as such tax resident in Germany is largely a question of fact and degree based on all the circumstances, rather than a question of law, which facts and degree may also change. Changes to applicable laws or interpretations thereof and changes to applicable facts and circumstances (for example, a change of board members or the place where board meetings take place), may result in us becoming a tax resident of a jurisdiction other than Germany. As a consequence, our overall effective income tax rate and income tax expense could materially increase, which could have a material adverse effect on our business, results of operations, financial condition and prospects, which could cause our share price and trading volume to decline. However, if there is a double tax treaty between Germany and the respective other country, the double taxation of income may be reduced or avoided entirely.
Our ability to use our net operating loss carryforwards and other tax attributes may be limited.
Our ability to utilize our net operating losses ("NOLs") is currently limited, and may be limited further, under Section 8c of the German Corporation Income Tax Act (Körperschaftsteuergesetz, the "KStG") and Section 10a of the German Trade Tax Act (Gewerbesteuergesetz, the "GewStG"). These limitations apply if a qualified ownership change, as defined by Section 8c KStG, occurs and no exemption is applicable.
Generally, a qualified ownership change occurs if more than 50% of the share capital or the voting rights are directly or indirectly transferred to a shareholder or a group of shareholders within a period of five years. A qualified ownership change may also occur in case of a transaction comparable to a transfer of shares or voting rights or in case of an increase in capital leading to a respective change in the shareholding. In the case of such a qualified ownership change tax loss carryforwards expire in full. To the extent that the tax loss carryforwards do not exceed hidden reserves (stille Reserven) taxable in Germany, they may be further utilized despite a qualified ownership change. In case of a qualified ownership change within a group, tax loss carryforwards will be preserved if certain conditions are satisfied. In case of a qualified ownership change, tax loss carryforwards will be preserved (in the form of a "fortführungsgebundener Verlustvortrag") if the business operations have not been changed and will not be changed within the meaning of Section 8d KStG.
According to another appeal filed by the fiscal court of Hamburg dated August 29, 2017, Section 8c, paragraph 1, sentence 1 KStG is not in line with the German constitution. The appeal is still pending. It is unclear when the Federal Constitutional Court will decide this case. According to statements in German legal literature, there are good reasons to believe that the Federal Constitutional Court may come to the conclusion that Section 8, paragraph 1, sentence 1 KStG is not in line with the German constitution.
As of December 31, 2019, we had unrecognized NOL carryforwards for German tax purposes of €41,570 thousand available. Future changes in share ownership may also trigger an ownership change and, consequently, a Section 8c KStG or a Section 10a GewStG limitation. Any limitation may result in
58
the expiration of a portion or the complete tax operating loss carryforwards before they can be utilized. As a result, if we earn net taxable income, our ability to use our pre-change net operating loss carryforwards to reduce German income tax may be subject to limitations, which could potentially result in increased future cash tax liability to us.
Although we do not believe that we were a "passive foreign investment company," or a PFIC, for U.S. federal income tax purposes in 2019, we may be a PFIC in 2020 or one or more future taxable years. If we were a PFIC in any taxable year, U.S. shareholders may be subject to adverse U.S. federal income tax consequences.
Under the Internal Revenue Code of 1986, as amended (the "Code"), we will be a PFIC for any taxable year in which, after the application of certain look-through rules with respect to subsidiaries, either (i) 75% or more of our gross income consists of passive income or (ii) 50% or more of the average quarterly value of our assets consists of assets that produce, or are held for the production of, passive income. Passive income includes, among other things, dividends, interest, certain non-active rents and royalties, and capital gains. Based on our current operations, income, assets and certain estimates and projections, including as to the relative values of our assets, we do not believe that we were a PFIC for our 2019 taxable year. However, there can be no assurance that the Internal Revenue Service (the "IRS") will agree with our conclusion. In addition, whether we will be a PFIC in 2020 or any future taxable year is uncertain because, among other things, (i) we currently own, and expect to continue to own, a substantial amount of passive assets, including cash, (ii) the valuation of our assets that generate non-passive income for PFIC purposes, including our intangible assets, is uncertain and may vary substantially over time and (iii) the composition of our income may vary substantially over time. Accordingly, there can be no assurance that we will not be a PFIC for any taxable year.
If we are a PFIC for any taxable year during which a U.S. investor holds common shares, we would continue to be treated as a PFIC with respect to that U.S. investor for all succeeding years during which the U.S. investor holds common shares, even if we ceased to meet the threshold requirements for PFIC status, unless certain exceptions apply. Such a U.S. investor may be subject to adverse U.S. federal income tax consequences, including (i) the treatment of all or a portion of any gain on disposition as ordinary income, (ii) the application of a deferred interest charge on such gain and the receipt of certain dividends and (iii) compliance with certain reporting requirements. We do not intend to provide the information that would enable investors to make a qualified electing fund election (a "QEF Election") that could mitigate the adverse U.S. federal income tax consequences should we be classified as a PFIC.
As a Dutch public company, the rights of our shareholders may be different from the rights of shareholders in companies governed by the laws of U.S. jurisdictions and may not protect investors in a similar fashion afforded by incorporation in a U.S. jurisdiction.
We are a Dutch public company (naamloze vennootschap) organized under the laws of the Netherlands. Our corporate affairs are governed by our articles of association and by the laws governing companies incorporated in the Netherlands. However, there can be no assurance that Dutch law will not change in the future or that it will serve to protect investors in a similar fashion afforded under corporate law principles in the United States, which could adversely affect the rights of investors.
The rights of shareholders and the responsibilities of managing directors and supervisory directors may be different from the rights and obligations of shareholders and board members in companies governed by the laws of U.S. jurisdictions. In the performance of their duties, our managing directors and supervisory directors are required by Dutch law to consider the interests of our company, its shareholders, its employees and other stakeholders, in all cases with due observation of the principles of reasonableness and fairness. It is possible that some of these parties will have interests that are different from, or in addition to, your interests as a shareholder.
59
Provisions of our articles of association or Dutch corporate law might deter acquisition bids for us that might be considered favorable and prevent, delay or frustrate any attempt to replace or remove the members of our management board or supervisory board.
Under Dutch law, various protective measures are possible and permissible within the boundaries set by Dutch law and Dutch case law. In this respect, certain provisions of our articles of association may make it more difficult for a third party to acquire control of us or effect a change in our management board and supervisory board. These include:
- •
- a provision that our managing directors and supervisory directors are appointed on the basis of a binding nomination prepared by our
supervisory board which can only be overruled by a two-thirds majority of votes cast representing more than 50% of our issued share capital;
- •
- a provision that our managing directors and supervisory directors may only be dismissed by the general meeting of shareholders by a two-thirds
majority of votes cast representing more than 50% of our issued share capital (unless the dismissal is proposed by the supervisory board in which case a simple majority of the votes would be
sufficient); and
- •
- a requirement that certain matters, including an amendment of our articles of association, may only be brought to our shareholders for a vote upon a proposal by our management board with the approval of our supervisory board.
We are not obligated to, and do not, comply with all best practice provisions of the Dutch Corporate Governance Code.
As a Dutch public company, we are subject to the DCGC. The DCGC contains both principles and best practice provisions that regulate relations between the management board, the supervisory board and the shareholders. The DCGC is based on a "comply or explain" principle. Accordingly, companies are required to disclose in their annual reports, filed in the Netherlands, whether they comply with the provisions of the DCGC. If they do not comply with those provisions (for example, because of a conflicting Nasdaq requirement), the company is required to give the reasons for such non-compliance. The DCGC applies to Dutch companies listed on a government-recognized stock exchange, whether in the Netherlands or elsewhere, including Nasdaq. We do not comply with all best practice provisions of the DCGC. This may affect your rights as a shareholder and you may not have the same level of protection as a shareholder in a Dutch company that fully complies with the DCGC.
Claims of U.S. civil liabilities may not be enforceable against us.
We are incorporated under the laws of the Netherlands, and our headquarters is located in Germany. Most of our assets are located outside the United States. The majority of our management board and supervisory board reside outside the United States. As a result, it may not be possible for investors to effect service of process within the United States upon such persons or to enforce against them or us in U.S. courts, including judgments predicated upon the civil liability provisions of the federal securities laws of the United States.
There is currently no treaty between the United States and the Netherlands for the mutual recognition and enforcement of judgments (other than arbitration awards) in civil and commercial matters. Therefore, a final judgment for the payment of money rendered by any federal or state court in the United States based on civil liability, whether or not predicated solely upon the U.S. federal securities laws, would not be enforceable in the Netherlands unless the underlying claim is relitigated before a Dutch court of competent jurisdiction. Under current practice, however, a Dutch court will generally, subject to compliance with certain procedural requirements, grant the same judgment without a review of the merits of the underlying claim if such judgment (i) is a final judgment and has been rendered by a court which has established its jurisdiction vis-à-vis the relevant Dutch companies or
60
Dutch company, as the case may be, on the basis of internationally accepted grounds of jurisdiction, (ii) has not been rendered in violation of principles of proper procedure (behoorlijke rechtspleging), (iii) is not contrary to the public policy of the Netherlands, and (iv) is not incompatible with (a) a prior judgment of a Netherlands court rendered in a dispute between the same parties, or (b) a prior judgment of a foreign court rendered in a dispute between the same parties, concerning the same subject matter and based on the same cause of action, provided that such prior judgment is capable of being recognized in the Netherlands and except to the extent that the foreign judgement contravenes Dutch public policy (openbare orde). Dutch courts may deny the recognition and enforcement of punitive damages or other awards. Moreover, a Dutch court may reduce the amount of damages granted by a U.S. court and recognize damages only to the extent that they are necessary to compensate actual losses or damages. Enforcement and recognition of judgments of U.S. courts in the Netherlands are solely governed by the provisions of the Dutch Code of Civil Procedure.
The United States and Germany currently do not have a treaty providing for the reciprocal recognition and enforcement of judgments, in civil and commercial matters. Consequently, a final judgment for payment or declaratory judgments given by a court in the United States, whether or not predicated solely upon U.S. securities laws, would not automatically be recognized or enforceable in Germany. German courts may deny the recognition and enforcement of a judgment rendered by a U.S. court if they consider the U.S. court not to be competent or the decision to be in violation of German public policy principles. For example, judgments awarding punitive damages are generally not enforceable in Germany. A German court may reduce the amount of damages granted by a U.S. court and recognize damages only to the extent that they are necessary to compensate actual losses or damages.
In addition, actions brought in a German court against us, our directors, our senior management and the experts named herein to enforce liabilities based on U.S. federal securities laws may be subject to certain restrictions. In particular, German courts generally do not award punitive damages. Litigation in Germany is also subject to rules of procedure that differ from the U.S. rules, including with respect to the taking and admissibility of evidence, the conduct of the proceedings and the allocation of costs. German procedural law does not provide for pre-trial discovery of documents, nor does Germany support pre-trial discovery of documents under the 1970 Hague Evidence Convention. Proceedings in Germany would have to be conducted in the German language and all documents submitted to the court would, in principle, have to be translated into German. For these reasons, it may be difficult for a U.S. investor to bring an original action in a German court predicated upon the civil liability provisions of the U.S. federal securities laws against us, our directors, our senior management and the experts named in this Annual Report.
Based on the foregoing, there can be no assurance that U.S. investors will be able to enforce against us or directors, executive officers or certain experts named herein who are residents of or possessing assets in the Netherlands, Germany, or other countries other than the United States any judgments obtained in U.S. courts in civil and commercial matters, including judgments under the U.S. federal securities.
61
Item 4. Information on the Company
A. History and Development of the Company
Centogene AG was founded by our CEO, Prof. Arndt Rolfs, in 2006 in Rostock, Germany. In connection with our initial public offering, which closed on November 12, 2019, we executed a corporate reorganization whereby Centogene B.V., which was incorporated on October 11, 2018, was converted into Centogene N.V. and Centogene N.V. became the holding company for Centogene AG, which remains our principal operating subsidiary. Centogene N.V. is a Dutch public company (naamloze vennootschap) organized under the laws of the Netherlands and our legal and commercial name is Centogene N.V.
Our principal executive offices are located at Am Strande 7, 18055 Rostock, Germany and our additional offices are in Berlin (Germany), Cambridge (Massachusetts, United States), Vienna (Austria), Dubai (United Arab Emirates) and Delhi (India). Since November 7, 2019, our common shares have traded on Nasdaq under the symbol "CNTG." Our agent for service of process in the United States is Cogency Global, located at 10 East 40th Street, 10th Floor, New York, NY 10016.
We are an emerging growth company and as such, we are eligible to, and intend to, take advantage, for up to five years, of certain exemptions from various reporting requirements applicable to other public companies that are not Emerging Growth Companies, such as not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002.
We will remain an emerging growth company until the earliest of: (i) the last day of our fiscal year during which we have total annual gross revenues of at least $1.07 billion; (ii) the last day of our fiscal year following the fifth anniversary of the closing of our initial public offering; (iii) the date on which we have, during the previous three-year period, issued more than $1.0 billion in non-convertible debt; (iv) the date on which we are deemed to be a Large Accelerated Filer under the Exchange Act, with at least $700 million of equity securities held by non-affiliates.
Our capital expenditures for 2019, 2018 and 2017 amounted to €7,576 thousand, €11,769 thousand and €18,035 thousand, respectively. These expenditures were primarily for property, plant and equipment, intangible assets and right-of-use assets.
The SEC maintains an Internet website that contains reports and other information about issuers, like us, that file electronically with the SEC. The address of that website is www.sec.gov. Our website can be found at www.centogene.com. The information on our website is not incorporated by reference into this Annual Report, and you should not consider information contained on our website or any websites mentioned in this Annual Report to be part of this Annual Report.
B. Business Overview
We are a commercial-stage company focused on rare diseases that transforms real-world clinical and genetic data into actionable information for patients, physicians and pharmaceutical companies. Our goal is to bring rationality to treatment decisions and to accelerate the development of new orphan drugs by using our knowledge of the global rare disease market, including epidemiological and clinical data and innovative biomarkers. We have developed a global proprietary rare disease platform based on our real-world data repository with over 2.5 billion weighted data points from nearly 500,000 patients representing over 120 different countries as of December 31, 2019, or an average of over 590 data points per patient. Our platform includes epidemiologic, phenotypic and genetic data that reflects a global population, and also a biobank of these patients' blood samples. We believe this represents the only platform that comprehensively analyzes multi-level data to improve the understanding of rare hereditary diseases, which can aid in the identification of patients and improve our pharmaceutical partners' ability to bring orphan drugs to the market. As of December 31, 2019, we collaborated with 39 pharmaceutical partners for over 45 different rare diseases.
62
A rare disease, by definition in the United States, is a disease that affects 200,000 or fewer people. However, with over 7,000 currently identified rare diseases, they in aggregate affect more than 350 million people globally. Rare diseases can be severe and often take years to diagnosis - on average it takes six to eight years for a patient with a rare disease to be diagnosed. This underscores the significant unmet need for high-quality genetic information in the rare disease space for the early identification and effective treatment of patients. Despite legislative initiatives and continued investment in rare disease drug development, significant unmet need still exists. Of the 7,000 identified rare diseases, it is estimated that 80%, or 5,600, have a genetic origin and, of these rare hereditary diseases, only approximately 250 rare hereditary diseases, or less than 5%, have an FDA approved treatment. The introduction of new treatments and development of cost-effective drugs are constrained by a number of factors including: a lack of high-quality information regarding the clinical heterogeneity of medical symptoms, lack of comprehensive and curated medical data, difficulties in the early identification of patients, lack of biomarkers and difficulties in understanding market size and epidemiology.
Our business is comprised of complementary solutions for both physicians and their patients, as well as pharmaceutical companies. Our diagnostics solution typically starts with specialist physicians requesting diagnostic information to identify or confirm a rare disease by sending us their patients' blood samples on our proprietary dried blood spot collection kit that bears the CE Mark - the CentoCard. With highly advanced technology, our proprietary database and our team of medical experts, we then deliver reports back to the physicians that contain what we believe is critical information containing genetic, proteomic, metabolomic information, or some combination, depending on what is most salient for each case. We also input this data to our CentoMD platform, which enriches our understanding of rare diseases broadly.
For our pharmaceutical partners, we are able to provide various valuable information using our platform. For instance, with the access to the data in our repository and biomaterials in our biobank, we have successfully developed biomarkers by applying highly sophisticated tools, including mass spectrometry technologies, together with artificial intelligence capabilities in an efficient and cost effective manner. Biomarkers are important in orphan drug development as well as post commercialization monitoring, by demonstrating the efficacy of the drugs, performing longitudinal monitoring and informing necessary titration for individual rare disease patients. As of December 31, 2019, we had 58 biomarker programs, of which 14 biomarkers covering eight rare diseases, including AADC deficiency, Cystic Fibrosis, Fabry disease, Faber disease, Gaucher disease, Hereditary Angioedema Disease ("HAE"), Niemann-Pick Type A/B and Niemann-Pick Type C diseases, have completed their development.
In December 2018, the FDA issued a statement that supports the use of real-word evidence to accelerate drug development and to monitor the safety of drugs after they have been commercialized. Moreover, in February 2019, the FDA also issued a revised draft guidance for drug discovery in rare diseases, including a discussion of the benefits of using biomarkers as surrogate endpoints (the outcomes of which can be measured against therapy effectiveness in clinical trials). We believe that this new guidance from FDA, acknowledging the benefits of the use of both real-world evidence and biomarkers, further validates the value of our global proprietary rare disease platform and our biomarkers.
We offer solutions to our pharmaceutical parties and clients through two business segments. Our pharmaceutical segment provides a variety of services to our pharmaceutical partners, including target discovery, early patient recruitment and identification, epidemiological and patient population sizing insights, biomarker discovery and patient monitoring and follow-up. Our information platforms, our access to rare diseases patients and their biomaterials and our ability to develop proprietary technologies including biomarkers enable us to provide services to our pharmaceutical partners in all phases of the drug development process as well as post-commercialization. Revenues in our
63
pharmaceutical segment are generated primarily from collaboration agreements with our pharmaceutical partners, which are structured on a fee per sample basis, milestone basis, fixed fee basis, royalty basis or a combination of these. For the year ended December 31, 2019, €21.5 million, or 44.1%, of our total revenues were derived from our pharmaceutical segment. For the year ended December 31, 2018, €17.3 million, or 42.8%, of our total revenues were derived from our pharmaceutical segment.
Our clinical diagnostics segment provides targeted genetic sequencing and diagnostics services to patients through our distribution partners and clients, who are typically physicians, labs or hospitals. As of December 31, 2019, we believe we offer the broadest diagnostic testing portfolio for rare diseases, covering over 7,000 genes using over 10,000 different tests.
Revenues from our diagnostics segment are typically generated by set fees per diagnostic test or per bundle of diagnostic tests under contracts with our clients. For the year ended December 31, 2019, €27.3 million, or 55.9%, of our total revenues were derived from our diagnostics segment. For the year ended December 31, 2018, €23.2 million, or 57.2%, of our total revenues were derived from our diagnostics segment.
We continuously work on expanding our medical and genetic knowledge of rare genetic diseases. We work with renowned international scientific and academic institutions on a variety of groundbreaking research projects involving a significant number of rare genetic disease patients.
The tests that we receive from our customers in our pharmaceutical segment, our diagnostics segment, as well as from research projects yield a rich collection of genetic and biochemical data which are used to map out phenotype-genotype correlations and continuously enrich and improve the quality of our database.
For the year ended December 31, 2019, we received over 133,800 test requests in total for both our pharmaceutical and diagnostics segments, as well as for our internal research projects, bringing the total number of test requests received in the period from January 1, 2016 to December 31, 2019 to approximately 404,000. Compared to the total number of patients in our repository as of December 31, 2019, this shows that approximately 80% of our data and biomaterials came from the last four years, which is an important factor when it comes to recruiting patients for clinical trials and clinical studies, considering the short average life expectancy of rare disease patients.
64
The graphic below shows the cumulative test requests for the diagnostics and pharmaceutical segments, as well as test requests received for our internal research projects during the period from January 1, 2016 to December 31, 2019. The testing expenses relating to requests received for our internal research projects were included in Corporate as they did not generate any revenue and cannot be allocated to either of our two business segments.
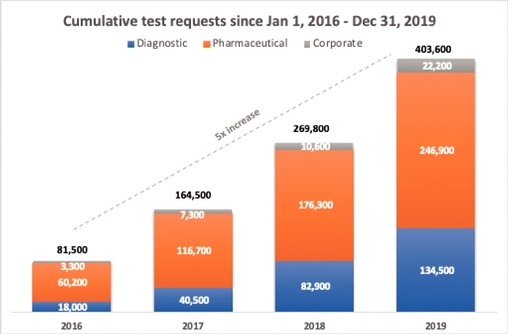
For the majority of these tests, we have express consent from the patients in our CentoMD, which offers the ability to retest their biomaterials in our biobank.
The graphic below shows the cumulative 134,500 test requests received from our diagnostics segment in the period from January 1, 2016 to December 31, 2019, split by different type of analysis.
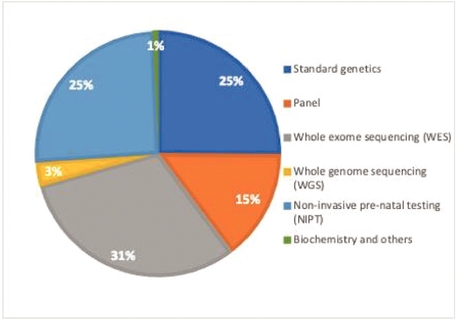
"Standard genetic" testing includes our single gene, CNV and mutation quantification products.
65
From our inception in 2006, Centogene has been focused on changing the way patients with rare diseases are treated. These efforts have been led by our management team, in particular our CEO and founder, Prof. Arndt Rolfs. Our laboratory at our headquarters in Rostock, Germany, as well as our Cambridge, Massachusetts facility, are equipped with the most advanced technologies from thirteen different diagnostic platforms and, as of December 31, 2019, together employ over 440 highly qualified personnel (including consultants) from over 55 nationalities. In addition to our laboratories, we have sales and administrative offices located in Berlin (Germany), Cambridge (Massachusetts, United States), Vienna (Austria), Dubai (United Arab Emirates) and Delhi (India), allowing us to further expand our international footprint.
Strategy
Our objective is to improve the diagnosis and treatment of rare diseases by transforming real-world clinical and genetic data into actionable information for patients, physicians, and pharmaceutical companies, and unlocking critical knowledge that will guide and accelerate orphan drug development. To achieve this objective, our strategy is to:
- •
- Transform the rare disease landscape by applying precision medicine more
comprehensively. Rare diseases affect patients of all ages and ethnicities, across the world. We are focused on creating broader awareness of the
challenges these patients and their families face, including the lack of accurate and up-to-date diagnostic solutions and the lack of effective therapies. We leverage our global network to access
patient populations of varying ethnicities and continue to expand our existing data repository, which we believe is the world's largest repository for rare hereditary diseases. We believe this central
source of knowledge will allow us to apply precision medicine more comprehensively, which will enable more accurate diagnosis as well as support the more efficient discovery and development of
effective new treatment solutions for rare hereditary disease patients.
- •
- Further our leadership position in rare diseases and continue to build upon the largest, most comprehensive repository
for rare disease patient data. Since our Company's founding in 2006, we have been focused on collecting clinical, phenotypic and genomic data for patients with rare hereditary
diseases. As of December 31, 2019, our data repository included nearly 500,000 patient samples from over 120 different countries. We plan to continue growing this repository of information
through the identification of additional patients by expanding our clinical network, which will facilitate more effective new drug development. This synergistic model will allow us to maintain our
competitive advantage of having what we believe is the world's largest curated data repository for rare hereditary diseases.
- •
- Accelerate the discovery and development of orphan drugs for new and existing pharmaceutical
partners. We are focused on leveraging our vast knowledge-base to support drug development for the rare disease industry in various ways. As of December 31, 2019, we
collaborated with 39 pharmaceutical partners. In 2019, we entered into 28 new collaborations, resulting in a total of 76 active/completed collaborations for over 45 different rare diseases. We intend
to continue expanding the scope of these collaborations as well as our network of partners, and in particular commencing our collaboration at an earlier stage of the drug development by our partners.
Our services span the full spectrum of drug development, including in vitro molecular screening (or target discovery), epidemiological studies,
biomarker development as well as patient recruitment and identification. We believe these services support the speed and efficiency of our pharmaceutical partners' drug development efforts and
accelerate bringing new diagnostic and treatment solutions to rare hereditary disease patients.
- •
- Evolve our business to share in more of the value we provide to our pharmaceutical partners. Our database is also valuable beyond drug discovery as the biomarkers can be relevant for
66
patient stratification and monitoring. Our database has multiple additional applications such as patient identification for therapeutic trials and treatment. For example, by identifying patients with a specific rare disease that are eligible for a clinical trial, which can reduce the time of clinical trial patient enrollment for our pharmaceutical partners. Reducing this enrollment time is often critically important in rare disease as the small number of patients of each disease can cause long enrollment periods.
Rare Disease Overview
Overview
The Rare Diseases Act of 2002 defines a rare disease as having a prevalence of fewer than 200,000 affected individuals in the United States. In the European Union, orphan drug designation is intended to promote the development of drugs for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than five in 10,000 persons in the European Union and for which no satisfactory method of diagnosis, prevention, or treatment has been authorized (or the product would be a significant benefit to those affected).
The National Institutes of Health lists more than 7,000 disorders that qualify as rare diseases. A wide range of conditions qualify as rare diseases and include, but are not limited to:
- •
- Lysosomal storage disorders such as Gaucher disease, Fabry disease, Pompe disease, the mucopolysaccharidosis disorders, Farber disease,
Niemann-Pick disease and Metachromatic leukodystrophy;
- •
- Neurologic and neuromuscular disorders such as Huntington's disease, Spinal Muscular Atrophy, Duchenne Muscular Dystrophy and Neuronal
ceroid-lipofuscinosis type 2; and
- •
- Non-malignant hematological disorders such as paroxysmal nocturnal hemoglobinuria, atypical hemolytic uremic syndrome, hemophilia and hemoglobinopathies such as sickle cell disease and b-thalassemia.
According to research published in the European Journal of Human Genetics in September 2019, a conservative, evidence-based estimate for the population prevalence of rare diseases is 3.5-5.9%, which equates to approximately 300 to 500 million people affected globally at any point in time. According to the International Rare Diseases Research Consortium, there were over 800 new rare diseases identified between 2010 and 2019.
Cause of Rare Diseases
While there are many causes of rare diseases, approximately 5,600 are due to genetic mutations which are hereditary and passed from one generation to the next. Genes direct the production of proteins make up body structures like organs and tissue, as well as control chemical reactions and carry signals between cells. If a cell's DNA is mutated, a dysfunctional protein may be produced, which can lead to a disease. Therefore, one way in which rare diseases can be diagnosed is by identifying the specific mutations in a patient's DNA, even without the manifestation of physical symptoms. To date, there are estimated to be approximately 4,500 rare genetic diseases that can be diagnosed by diagnostic sequencing tools. Despite these advancements in science and availability of next-generation sequencing ("NGS") technologies, rare diseases are complex and an underlying genetic cause for approximately 1,100 rare diseases is still unknown.
In addition, new genetic mutations associated with identified rare diseases are discovered every year, and as a result, rare genetic diseases that can be diagnosed need to be continuously updated with the new information, otherwise the diagnosis provided becomes inaccurate over time. From January 1,
67
2010 to December 31, 2019, there were over 4,000 new genetic mutations discovered and linked to rare diseases.
Manifestation and Diagnosis of Rare Diseases
Because of phenotypic heterogeneity, rare disease manifestations vary in onset and severity and many rare diseases exhibit a number of variations or sub-types. Almost 70% of the rare genetic diseases are pediatric onset, which means symptoms may be observed at birth or in childhood, as is the case with Spinal Muscular Atrophy, neurofibromatosis and chondrodysplasia. The remaining rare genetic diseases manifest symptoms during adulthood. Given the delayed onset and large variance in the symptoms that can manifest, the vast majority of these patients are misdiagnosed.
Given the multifaceted genetic and phenotypical nature of rare diseases, diagnosis is complex and requires specialist knowledge. It is often difficult for rare disease patients to find healthcare professionals with adequate experience. If diagnosis, treatment and management are not led by specialists, it may result in an incorrect diagnosis and inappropriate treatment, which can result in poorer patient outcomes. In addition, comprehensive phenotypical information on rare diseases is not always captured, and as a result, symptoms are often misinterpreted and patients are often not properly diagnosed, in particular when patients with such symptoms present to physicians who have never encountered rare diseases before. Even though genetic testing is the current accepted standard for making a diagnosis, there are still knowledge barriers that prevent the full interpretation of data obtained from such tests.
Delay to diagnosis is commonly experienced by patients and is due to poor awareness of rare diseases by health professionals and the small number of patients affected. This delay in diagnosis can be significant for many patients and may lead to irreversible progression of the patient's condition, in particular when children are suffering from rare diseases. For example, in the United Kingdom and the United States, the average time to obtain a correct diagnosis for rare diseases was found to be five to seven years, and in this time there were two to three incorrect diagnoses for a given condition. Pediatric rare disease patients can experience an even more significant delay in diagnosis. Across both pediatric and adult patient populations, approximately 90% of rare disease patients are typically undiagnosed. For example, the National Fabry Disease Foundation estimates that there are approximately 50,000 Fabry disease patients in the United States, whereas only 4,000 to 5,000 are currently diagnosed. As a result of incorrect and delayed diagnosis, unnecessary tests and treatments are often carried out and in some cases treatment windows are missed entirely.
Regulatory Environment and Current Market
The COVID-19 pandemic, which began in December 2019 in China and has spread worldwide, has caused many governments to implement measures to slow the spread of the outbreak through quarantines, travel restrictions, heightened border scrutiny and other measures. We have taken a series of actions aimed at safeguarding our employees and business associates, including implementing a work-from-home policy for employees except for those related to our laboratory operations. These disruptions could result in increased costs of execution of operational plans or may negatively impact our business due to its negative impact on the global economy.
For more information on the impact of the COVID-19 outbreak on our business and financial results, please see "Item 3. Key Information—D. Risk Factors—The COVID-19 pandemic could adversely impact our business and results of operations."
Orphan drug legislation in the United States has made significant improvements in encouraging the development of new drugs to treat rare diseases. Since the passage of the Orphan Drug Act and subsequent amendments to the orphan drug regulations, the FDA granted over 5,000 orphan drug designations to December 31, 2019. Moreover, the FDA's Center for Drug Evaluation and Research
68
("CDER") approved 48 novel drugs in 2019, among which 44% were orphan drugs. During the year ended December 31, 2019, CDER has approved more than twice as many orphan drugs than in the previous 8 years, with a total of 142 novel orphan drugs approved from 2012 to 2019, as compared to 63 approvals for the period from 2004 to 2011.
The success of orphan drug legislation in the United States led to the adoption of similar legislation in other key markets, most notably in the European Union, where the European Commission grants orphan drug designation after receiving the opinion of the EMA committee, with over 2,200 orphan drug designations granted from 2000 to 2019.
In the United States, orphan drug designation allows the drug sponsor to benefit from incentives for the development of these products up to marketing approval. The measures apply to all stages of drug development and include tax credits on clinical research, waiver of certain application fees and marketing exclusivity for seven years. In addition, more than $400 million was provided by the FDA's Orphan Products Clinical Trials Grants Program over the last three decades and led to the approval of more than 60 different drugs for rare diseases. In the European Union, financial incentives including fee reductions or waivers are available and market exclusivity is granted for ten years.
Due to these legislative initiatives, there has been an increase in investment and activity in the rare disease drug development space. In 2017, over $45 billion is estimated to have been spent on discovery and development efforts in the U.S. for the treatment of rare diseases. This represents 10% of overall drug spending in 2017, up from 4% of overall drug spending in 1997. In addition, as of April 30, 2019, there were approximately 610 non-oncology orphan designated products being developed worldwide. These investments are expected to lead to the approval of new rare disease drugs, which, according to market research, are expected to grow at a CAGR of 12.3% from 2019 to 2024 to $242 billion, capturing approximately 20% of worldwide prescription sales.
Key Challenges in Rare Disease Drug Development
Despite the legislative initiatives to encourage orphan drug development and the consequent increase in investment and activity in the rare disease drug development space, significant unmet needs still exist. Of the 5,600 rare hereditary diseases, only approximately 250 rare hereditary diseases have an FDA approved treatment. The limited number of treatments available for rare diseases is the greatest challenge for patient care and is based on the lack of research on rare diseases and barriers in developing and commercializing treatments.
We believe the following summarizes the key challenges clinicians and the pharmaceutical industry are facing today:
- •
- Lack of phenotypic understanding. For many diseases, the symptoms are
non-specific and often do not fit the typical picture of the disease. Due to their phenotypic heterogeneity, rare diseases have highly diverse clinical manifestations and unpredictable progression
rates. These factors make it difficult for physicians to make an accurate diagnosis and determine an optimal treatment strategy.
- •
- Lack of comprehensive and curated information. A full understanding of the causes of a rare disease requires proteomic, metabolomic and genomic information at a genetic level, as well as detailed clinical information. Moreover, thorough medical validation processes must be conducted to ensure the quality of this information. While there are a few, limited rare disease databases available to the market, such as parts of ClinVar and HGMD, they are not specifically set up to service the rare disease industry and, due to their nature, lack medical curation.
Lack of high-quality medical data as a result of:
69
- •
- Lack of ethnically diverse datasets. The majority of existing rare disease datasets only capture individuals in developed regions of the world, where healthcare expenditure is disproportionately higher. This disparity yields population datasets that are specific to such regions and does not capture the full ethnic and hereditary nature that may be present in various rare diseases. For example, as published in Nature, despite the fact that unique genetic mutations are present across many different ethnicities, 87% of all genetic datasets are of European descent.
Consequently, this limits the accuracy and utility of that data for clinical diagnoses and decision-making.
Difficulties in the early identification of patients. Identifying rare disease patients is difficult given the small patient population for each rare disease. In addition, the population for each of the rare diseases is typically also scattered and diverse, which makes it more difficult to gain access to patients and collect sufficient real-world data to perform meaningful analyses to obtain a better understanding of the rare diseases. The lack of sufficient understanding of the clinical manifestations of rare disease makes it even more challenging to derive accurate diagnoses. The ability to access relevant patients with a particular rare disease and to access appropriate expertise, a network and dataset via the biobank, improves the accuracy of disease identification and facilitates the development of new treatments and diagnostic procedures.
Lack of biomarkers. The small patient populations, phenotypic heterogeneity, homogenous datasets and lack of curated information for rare diseases all impede biomarker discovery. Without an identified biomarker, the ability to diagnose and ultimately treat a patient in a timely manner is diminished. Delayed diagnoses and limited knowledge of available treatments can lead to incorrect patient management, further disease progression and/or invasive or detrimental treatments. For example, patients suffering from Gaucher disease and Cystic Fibrosis can have average life expectancies of only eleven years and one year, respectively, if no treatments are available, leaving limited time for effective treatment if not diagnosed early. In addition, the lack of an identified biomarker can create hurdles in obtaining drug approval as biomarkers can be beneficial in clinical development, specifically in monitoring how effectively a patient is treated by a drug.
- •
- Clinical Trial Recruitment. Relevant patient populations are typically
spread across large geographical regions, making adequate patient recruitment for clinical trials particularly difficult, which can delay development.
- •
- Trial Design and Dose Selection. Small patient populations do not allow for
multiple parallel studies in the same indication. This also applies to dosages, where the number of dose levels studied may be limited by the practical considerations of running a trial. As a result
of these limitations, careful thought must be given to study design in order to optimize clinical trial success.
- •
- Patient Management. In an orphan drug trial, clinical management of
individual patients can be difficult. Understanding the burden of disease and managing the patient and family experience within a study is key. Because of the progressive nature of many rare diseases,
it is crucial to enroll patients at a time where treatment has the highest potential to be effective.
- •
- Eligibility Criteria. Eligibility criteria influences the type of patient
eligible to participate in a clinical study. Consequently, this dynamic interferes with the establishment of a database that captures clinical efficacy and safety data which can be extrapolated to a
larger network of patients with the same disorder.
- •
- Understanding the End Market. Obtaining accurate epidemiological data is crucial for pharmaceutical companies to appropriately size the ultimate end market for a given drug in
Difficulties in orphan drug development and commercialization as a result of:
70
- •
- Market Traction. Once a rare disease drug is commercialized, the limited number of identified patients and challenges associated with diagnosis make it difficult for physicians and pharmaceutical companies to find individuals who would benefit from an approved therapy. In order to more successfully market a commercial drug, improved datasets are needed to aid in patient identification.
development. Given the small patient populations, it can be a challenge for pharmaceutical companies to recover the costs of rare disease drug development. As a result, this may impede initial investment in rare disease therapies.
Our Vision
We have an integrated approach with a detailed, global understanding of the genetic basis and the clinical phenotype of rare hereditary diseases, which we believe will unlock the ability to target rare diseases and provide critical knowledge that will guide drug development and monitoring, and ultimately improve patient care. We perform analysis on the patients' data that we receive from our pharmaceutical and diagnostics businesses as well as from our research projects using a multi-omics approach, which combines genomics, proteomics, metabolomics and phenomics. The combination of multi-omics provides deep insights in the pathogenesis of rare hereditary diseases. The value in such a holistic diagnostics process has resulted in a shift from data generation to interpretation-based diagnostics, whereby the development of biomarkers is the central element to bring rationality to treatment decisions for rare disease patients. High-quality, standardized clinical information supporting medical interpretation is a crucial element of the diagnostic process and leads to greater knowledge of the causes and symptoms of rare diseases. We believe a combination of worldwide data and detailed access to phenotype, genotype, proteomics and metabolomics data will aid in the development of new treatments and reduce the costs associated with orphan drug development.
These fundamental principles were the basis of our founding in 2006 by our CEO and founder, Prof. Arndt Rolfs.
Our Platform—An Integrated, Knowledge-Based System
To deliver on this vision, we have developed a global real-world data based proprietary platform that we believe will improve methods for identifying and monitoring rare diseases and provide solutions that accelerate the development of orphan drugs.
At the core of our platform is our data repository, which included, as of December 31, 2019, epidemiologic, phenotypic and genetic data of nearly 500,000 patients representing over 120 countries, and allows us to assemble an extensive knowledge base in rare hereditary diseases. We collect this detailed level of data in our repository through our easy-to-use CentoCard, a CE-Marked dried blood spot collection kit, captures blood samples of potential rare disease patients with a low cost of distribution, accompanied by the patients' medical histories and completed consent forms from the physicians. The data is then validated by professionals using a systematic and scientific approach prior to feeding it into our repository and our central CentoMD database, which we believe is the world's largest curated mutation database for rare diseases. As of December 31, 2019, we had over 2.5 billion weighted data points, or an average of over 590 data points per patient, to draw upon for insights which includes CentoMD data, clinical data, analyses performed, biochemistry data and clinical study data.
This systematic and thorough process results in information-based services that are beneficial for our pharmaceutical partners. This includes the ability to derive diagnostic solutions to accurately identify rare disease patients and the ability to identify new biomarkers, which help streamline and accelerate the path to approval for new drugs. As we facilitate the development of new drugs and the identification of more patients, an increasing number of patients are involved in clinical trials, which
71
leads to even more diagnostic information being added to our repository. This synergistic model allows us to continuously enhance our own expertise and support pharmaceutical knowledge in the rare disease field. A graphical description of our system is shown below:
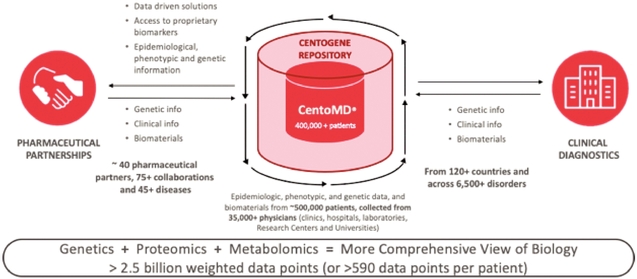
The Strengths of Our Platform
Access to these insights and knowledge through our platform is intended to streamline and accelerate the development of treatments for rare diseases, and aids in the understanding of how to identify new rare disease patients and how to recognize and quantify market opportunities in patient populations. We believe we offer the following solutions for the rare disease industry:
- •
- Extensive repository to identify rare disease patients: Our platform includes epidemiologic, phenotypic and genetic data that reflects a global population from nearly 500,000 patients, and also a biobank of these patients' blood samples. This capability has been facilitated by our development of the CentoCard, a convenient logistical solution. CentoCard is CE-Marked and easily stored, allowing for massive amounts of data aggregation from around the world. Additionally, we have express consent from the majority of patients in our CentoMD, which offers the ability to retest their biomaterials in our biobank. We are able to provide information about available treatment options to the physicians in our medical reports, therefore adding to the physician's decision-making tools in determining treatment for the patients. We believe this solution reflects the largest repository of rare disease patient data, thereby allowing us to assemble a knowledge base from which to derive accurate diagnoses and epidemiological information. We have relationships with a global network of specialists at rare disease "centers of excellence," including over 35,000 physicians worldwide. With these relationships and the logistical advantages of our CentoCard product, we are able to continuously grow our repository from the collection of new patient samples and related patient data.
72
- •
- Ethnically diverse datasets: Our repository has the advantage of holding samples from a broad range of ethnicities. Our repository covers a substantial majority of ethnicities, as we have performed diagnostic tests for patients in over 120 countries. Without the ability to recognize ethnicity-specific patterns, the interpretation of genetic variants in patients is difficult and a patient's physician may fail to find an accurate diagnosis. The mutation frequency distribution within one ethnicity can vary significantly from that of other ethnic groups with the same rare disease population. For example, a mutation in the Caucasian population might have a significant functional impact and cause a disease, but the exact same mutation in the Mongolian population might be without any functional consequence. Also, studies have shown that of the more than 1,893 Cystic Fibrosis mutations identified, patients of different ethnicities were subject to different types of genetic mutations. With access to data from a more diverse patient population, we are able to improve the interpretation of genetic variants, whether benign or causative. As of December 31, 2019, the patients from which we held data in our CentoMD 5.7 were located among different geographical regions as follows:
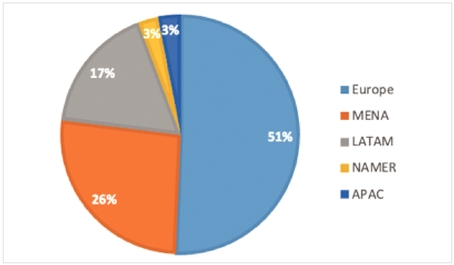
- •
- Curated information in
CentoMD: We focus on achieving the highest level of quality through data curation and process standardization. As of December 31, 2019,
our CentoMD 5.7 database included curated data from over 400,000 patients with over 12.2 million unique variants. Whenever sufficient information is available, another layer of manual curation
will be performed by our professional scientists with strong backgrounds in human genetics for genotype-phenotype association, supported by computer-based tools. Our team of scientists collects,
annotates and reviews the phenotypic, genetic and epidemiologic data of patient samples to ensure the highest medical validity of each sample. We also employ Human Phenotype Ontology ("HPO") coding to
accurately track and standardize sample phenotype and genotype data. Our methodological approach to information curation ensures we provide highly accurate data relevant to clinical diagnoses and
decision-making. CentoMD brings rationality to the interpretation of global genetic data.
- •
- Our detailed genetic, proteomic and metabolic analysis is the key to fueling the knowledge base of rare disease patient populations needed to lead the pharmaceutical industry towards the successful development of additional rare disease treatments. Since all phenotypes have been HPO coded, researchers can access the database and query by keywords and identifiers. For example, with the term "renal insufficiency," our system can directly analyze which genes and which pathogenic variants have been found to be causative for this phenotype. By combining multiple HPO codes such as "headache, diplopia, unsteady gait," a list of relevant genes associated with these clinical symptoms with corresponding real
73
- •
- Discovering biomarkers: The
interpretation of curated data in our repository and the ready access to biomaterials in our biobank are the initial steps in the identification of biomarkers. Our access to a large number of patients
with the same disease enables us to build a homogenous sub-cohort of those patients.
- •
- We can apply our highly sophisticated tools, including mass spectrometry technologies and artificial intelligence capabilities, to
compare this homogenous patient sub-cohort to a matched control cohort of healthy individuals. The combination of these steps allows us to identify biomarkers in a rapid and efficient manner.
- •
- As of December 31, 2019, we have 58 biomarker programs, of which we have commercialized fourteen proprietary biomarkers for eight rare diseases. Biomarkers further support the diagnosis and monitoring of patients in a cost-effective manner, which is important to our pharmaceutical partners during the drug development process, and also can be used to help physicians make informed predictions regarding the progression of a particular disease in order to optimize treatment.
diagnosed patients can be extracted and used for further follow-up analysis on a biomarker, which thereby refines our and our client's understanding of variation in rare diseases. We believe this resource speeds up research projects dealing with the in-depth analysis of rare genotypes and phenotypes, which cannot be found in other databases with this level of convenience and reliability. As of December 31, 2019, CentoMD 5.7 contained over 3,700 associated phenotypes and approximately 175,000 individual HPO associations, covering the 12 therapeutic areas as described below. These 12 therapeutic areas include over 3,700 diseases.
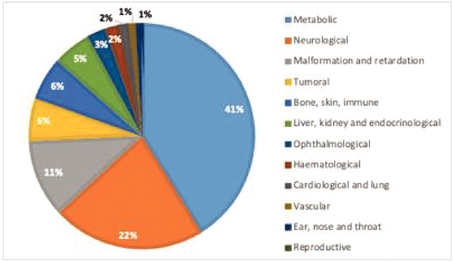
Based on the strengths of our platform, we are well placed to address the needs of the pharmaceutical industry. The following examples capture solutions that we have provided to our pharmaceutical partners covering epidemiological study, biomarker development and pharmaceutical diagnostics.
- •
- Fabry Disease. We published research in 2014 demonstrating that Fabry disease is the most frequent monogenic etiology in stroke patients under 55 years of age. Such insight is highly important for both patients and physicians in order to make an accurate and early diagnosis, and for our pharmaceutical partners trying to appropriately size the ultimate end market for Fabry disease. As of December 31, 2019, we have identified over 6,500 Fabry patients. We also published additional research in January 2020 showing proteostasis regulators represent a promising therapeutic target.
74
- •
- Gaucher Disease. We have been able to demonstrate that a mutation within
the Gaucher gene (glucocerebrosidase gene) increases the likelihood of developing Parkinson's disease. We believe our biomarker, Lyso-Gb1, has the potential to demonstrate the highest sensitivity and
specificity for the diagnosis and monitoring of Gaucher disease, allowing clinicians and our pharmaceutical partners to gain a better understanding of the disease pathophysiology. A paper published in
June 2019 in the International Journal of Molecular Sciences reported a four-year study of 81 children suffering from Gaucher disease of varying
severity. This paper suggested that Lyso-Gb1 has the potential to be used as an accurate biomarker for monitoring children suffering from Gaucher disease. In addition, the data generated from Gaucher
disease has stimulated new research and treatment strategies for Parkinson's disease.
- •
- Niemann-Pick Type C. Through our studies, we have been able to demonstrate that the majority of adult patients suffering from Niemann-Pick Type C also exhibit psychiatric symptoms. In addition, our preliminary data suggests our biomarker, Lyso-SM509, is a feasible biomarker for Niemann-Pick Type C. As we further analyze the sensitivity and specificity of Lyso-SM509, we believe this biomarker has the potential to provide an earlier and more simplified diagnosis of patients with Niemann-Pick Type C. Our recent report in the Journal of Biochemical and Clinical Genetics demonstrated that our Lyso-SM509 is able to facilitate the genetic diagnosis of Niemann-Pick disease by enabling classification of novel SMPD1 variants.
Our Commercialization Strategy
We are committed to improving the lives of rare disease patients by improving methods for identifying and monitoring rare diseases and providing solutions that accelerate the development of orphan drugs.
Our solutions are offered to our clients via two channels:
- •
- Pharmaceutical: Our
pharmaceutical solutions provide a variety of services to our pharmaceutical partners, including target discovery, early patient recruitment and identification, epidemiological insights, biomarker
discovery and patient monitoring. Our information platforms, deep access to rare disease patients and ability to develop proprietary technologies and biomarkers enable us to provide services to our
pharmaceutical partners in all phases of the drug development process as well as post-commercialization. Revenues from our pharmaceutical segment are generated primarily from collaboration agreements
with our pharmaceutical partners, which can be structured on a fee per sample basis, milestone basis, fixed fee basis, royalty basis or a combination of these. For the year ended December 31,
2019, €21.5 million, or 44.1%, of our total revenues were derived from our pharmaceutical segment. For the year ended December 31, 2018,
€17.3 million, or 42.8%, of our total revenues were derived from our pharmaceutical segment.
- •
- Diagnostics: Our clinical diagnostics segment provides targeted genetic sequencing and diagnostics services to patients through our distribution partners or our clients, who are typically physicians, labs or hospitals. As of December 31, 2019, we believe we offer the broadest diagnostic testing portfolio for rare diseases, covering over 7,000 genes using over 10,000 different tests. Revenues from our diagnostics segment are typically generated by set fees per diagnostic test or per bundle of diagnostic tests under contracts with our clients. In turn, the data collected from our diagnostics services allow us to continue to grow our repository and our CentoMD database. For the year ended December 31, 2019, €27.3 million, or 55.9%, of our total revenues were derived from our diagnostics segment. For the year ended December 31, 2018, €23.2 million, or 57.2%, of our total revenues were derived from our diagnostics segment.
75
Pharmaceutical Solutions
We are committed to accelerating the orphan drug development process for our pharmaceutical partners by providing our unique insights into rare diseases. As of December 31, 2019, we collaborated with over 39 pharmaceutical partners. In 2019, we entered into 28 new collaborations, resulting in a total of 76 active/completed collaborations, covering over 45 different rare diseases as of December 31, 2019. We provide information solutions and diagnostic services to our pharmaceutical partners in all phases of orphan drug development and treatment, including discovery, preclinical development and clinical development, as well as post-market care. The below chart demonstrates the scope of our pharmaceutical services to each stage of the drug development process:
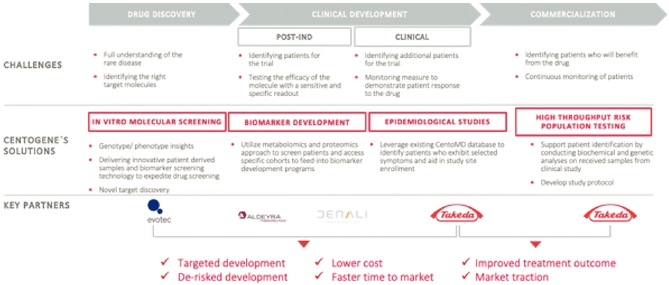
In Vitro Molecular Screening
A full understanding of a given rare disease and the ability to identify and target the right molecules is essential for drug development. With access to our biological samples, we are able to aid in vitro molecular screening efforts which can accelerate drug discovery efforts. Combined with access to our biobank and our data repository, our pharmaceutical partners are able to gain novel insights into the natural history of rare diseases, the broad spectrum of the different clinical symptoms as well as the genotype-phenotype correlation. Moreover, in situations where several genes can cause the same clinical symptoms and therefore, potentially cloud an accurate diagnosis, we believe we are able to identify additional genes that aid in the accurate diagnosis with the knowledge gathered in our database.
Epidemiological Studies
The ability of pharmaceutical companies to identify patients early and to optimize their clinical trials is key to the development of treatments for rare diseases. We offer epidemiological studies that will provide our partners with a more accurate picture and understanding of the scope and size of a particular rare disease population. We can also target these studies to a specific country or region of interest. This detailed epidemiological data can then aid our partners' clinical study enrollment efforts.
After a pharmaceutical partner specifies the rare disease for the clinical trial, we identify the available epidemiological data and enhance the data with genetic and phenotypic information from our repository and curated CentoMD database. From there, our pharmaceutical partner can create a defined list of specific conditions that patients must meet for a clinical study.
76
We then perform a patient selection and identification program. We start by identifying existing patients in our database who fit the defined criteria. If a patient sample is included in our sample repository but not yet tested to the level required for the trial, we run a diagnostics test to confirm if the patient meets the study criteria. If we need to find a larger cohort of patients than is currently included in our database or in our sample repository, we leverage our global network of partners, key opinion leaders, clinical labs and specialist physicians to help identify new patients who are at risk of developing, or have developed, the particular disease, in line with our pharmaceutical partners' defined patient cohort criteria. As a result, we are able to help our pharmaceutical partners optimize their clinical trials by more effectively selecting relevant patient groups and by leveraging our detailed understanding of the epidemiological data of the specific disease.
Biomarker Development
Biomarkers are key in rare disease drug development, as they can be used to support a diagnosis, demonstrate the efficacy of a treatment and monitor the progress of the rare disease patients. Biomarkers can also be used to enhance treatment solutions and guide dose titration. Biomarkers enable more efficient and economical patient diagnosis than genetic testing does and enables mass screening programs of a large patient cohort.
To develop a high-quality biomarker for a given rare disease, a homogeneous cohort of patients with known phenotypic and genotypic aspects is needed to simplify the process and increase efficiency. We believe our CentoMD database is the largest curated mutation database for rare diseases, as well as a vast source of healthy control individuals against whom to identify the biomarker characteristics. Therefore, we believe we are ideally positioned to lead the market in rare disease biomarker development.
We have developed a suite of biomarkers, and we plan to further expand the development programs in 2020 and going forward. As of December 31, 2019, we had 58 biomarker programs, of which 14 biomarkers covering eight rare diseases, including AADC deficiency, Cystic Fibrosis, Fabry disease, Faber disease, Gaucher disease, Hereditary Angioedema Disease ("HAE"), Niemann-Pick Type A/B and Niemann-Pick Type C diseases, have completed their development.
The table below shows a selected list of our biomarkers that had completed development as of December 31, 2019.
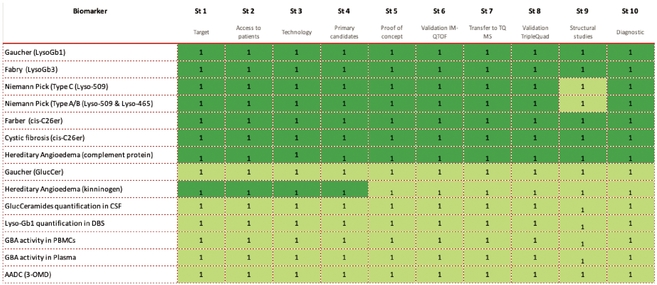
Light green in the table above indicates the developments made in 2019.
77
With proprietary biomarkers, we can also qualitatively measure a patient's response to approved drugs and to drugs in clinical trials, and using this data helps to determine the optimum treatment dosage for each patient. This not only helps to accelerate the development of orphan drugs by demonstrating the efficacy of the drugs in clinical trials, but also allows patients, physicians and reimbursement agencies to better understand the impact of the drugs. The below graphs demonstrate how Lyso-Gb1, our first commercialized biomarker, can be used for patient screening and monitoring in the context of Gaucher disease:
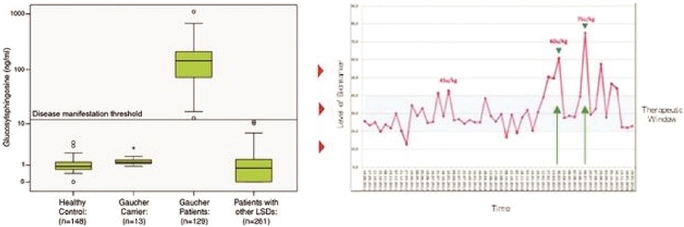
- *
- Based on a combination of our biomarker and a genetic confirmatory test
(Rolfs et. al., 2013.)
The left graph demonstrates the sensitivity and specificity of our Lyso-Gb1 biomarker for Gaucher disease. According to a 2017 study, patients who are not suffering from Gaucher disease present with a Lyso-Gb1 level of less than 12 nanograms per ml, whereas patients with Gaucher disease display elevated levels of Lyso-Gb1. Based on the definition of the cut-off of 12ng/ml Lyso-Gb1, we can demonstrate a 100% sensitivity and close to 100% specificity, which means our Lyso-Gb1 biomarker, when combined with a confirmatory genetic test, can provide 100% accuracy in identifying patients suffering from Gaucher disease, and also those who are not suffering from the disease.
The right graph demonstrates how our Lyso-Gb1 biomarker can also be used to titrate the proper enzyme replacement therapy dosage in each individual patient. An increase of the Lyso-Gb1 level signals that the dosage of the enzyme replacement therapy needs to be adjusted. After adjustment, Lyso-Gb1 levels decreased to an almost normal level. This is valuable for demonstrating drug efficacy to relevant authorities for approval, and also for demonstrating to reimbursement agencies that individualized treatment and dosage may be required for the patient.
High-Throughput Risk Population Testing
Once a treatment is available for a rare disease, early identification of patients is critical so that patients can be treated before they have reached the stage of irreversible progression. We are able to support our pharmaceutical partners in their patient identification efforts by leveraging our knowledge and performing mass-spectrometry screening on a much broader group of patients with the risk profile of a given rare disease. We do this by using our biomarkers, which is economically efficient. If a positive diagnosis is concluded, we provide physicians with information on relevant treatment options, which helps physicians make clinically relevant decisions for the treatment of their patients. For negative diagnoses, no further confirmatory genetic testing is necessary. We provide each patient's physician with a diagnostic report.
78
Research and Development Validation
Based on our extensive expertise in rare diseases and our access to detailed genetic data, our pharmaceutical partners can approach us for guidance during their drug development endeavors. More specifically, our pharmaceutical partners can ask us to review their clinical trial design, evaluate clinical data from an ongoing or recently completed clinical trial and validate related biomarkers. All of these services are aimed at optimizing their clinical development efforts.
Key Partnerships
Shire
In January 2015, we entered into an agreement with Shire, now a subsidiary of Takeda Pharmaceutical Company Limited, to provide certain diagnostic testing capabilities to Shire and its affiliates in order to enhance early diagnosis of patients suffering from lysosomal storage and other rare diseases, including Fabry disease, Gaucher disease and Hunter syndrome. Our unique expertise and repository of data contributes to Shire's mission to shorten the time it takes for rare disease patients to get diagnosed. In connection with this agreement, we receive a fixed annual fee plus additional service-based payments related to regulatory and diagnostic sequencing activities.
In addition, in 2018, we entered into a new research agreement with Shire relating to their ongoing drug development efforts in HAE. As part of this agreement, we are conducting an extensive epidemiological study leveraging our data repository and network of physicians at centers of excellence to gain unique insights into HAE and to support Shire's ongoing clinical development efforts.
In July 2019, we entered into a collaborative research agreement with Shire related to HAE Kininogen assay mass spectrometry testing and screening. Both parties obtained the limited right to use each other's data and intellectual property related to such testing and screening for the sole purpose of performing research under the agreement.
Evotec International GmbH ("Evotec")
In July 2018, we entered into an agreement with Evotec to support and expedite their identification of new small molecule treatments. Evotec identifies active pharmaceutical ingredients based on the induced pluripotent stem cells ("iPSC") that are generated from fibroblasts we obtain from skin biopsies of patients. We believe our collaboration will aid in the acceleration of drug development through the adoption and application of more accurate cellular models of the target disease and specific biomarkers to monitor such diseases. Our collaboration combines Evotec's iPSC platform and drug discovery capabilities with our medical and genetic insights to develop a high throughput platform to test innovative small molecules in rare hereditary metabolic diseases. In connection with this agreement, we received an initial payment in 2018 and milestone payments in 2018 and 2019, as well as further royalty fees on net sales of products developed from this collaboration.
Denali Therapeutics ("Denali")
In September 2018, we entered into a strategic collaboration with Denali for the global identification and recruitment of LRRK2 positive Parkinson's disease patients. We will utilize our CentoCard and extensive network with centers of excellence to conduct a targeted global recruitment campaign focused on the early identification and characterization of LRRK2 positive Parkinson's patients for the recruitment into Denali's clinical trials. Given that we believe Denali's study is the lead clinical investigation of LRRK2 inhibitors for the treatment of Parkinson's disease, there is no large global existing cohort of identified patients with the LRRK2 mutation in the early phase of the disease. We aim to overcome those challenges and accelerate the enrollment of further patients into this clinical study and consequently facilitate Denali's drug development process. In connection with this
79
collaboration, we received an initial payment, and are eligible for success-based and commercial milestones and reimbursement of selected costs.
Pfizer Inc. ("Pfizer")
In July 2019, we entered into a strategic collaboration with Pfizer, pursuant to a global master scientific services agreement ("MSSA"). In addition to the MSSA, we entered into a statement of work ("SOW") in November 2019 to provide sequencing services to Pfizer for patients in the United States or Puerto Rico with transthyretin amyloid cardiomyopathy ("ATTR-CM"), patients suspected of having ATTR-CM, or individuals with a confirmed family history of hereditary ATTR-CM.
In October 2019, we entered into a data access and collaboration agreement ("DACA") with Pfizer, pursuant to which we granted Pfizer access to our data repository, which may be used in the discovery and validation of novel genetic and biochemical targets for the potential development of new therapies for rare diseases.
Our Diagnostic Solutions
Overview and Product Offering
Our diagnostic solutions channel provides diagnostic testing services to patients exclusively through our network of distribution partners and our diagnostics clients, who are typically physicians, labs or hospital facilities. Our patient outreach includes over 120 countries due in part to our CentoCard solution enabling an efficient and simple transfer of the sample from the point of care to the lab. Additionally, our online platform, CentoPortal, allows our clients to quickly and easily place orders and obtain information related to their patients' test results and benefit from advancements in rare disease research, which we update on a regular basis. We provide a high-quality, end-to-end clinical diagnostics solution, which includes pretest clinical counseling performed by our medical experts whenever necessary, sample preparation, sequencing using NGS technology, medical interpretation using our manual and automated bioinformatics pipelines and medical reporting by our specialists.
Of the more than 5,600 identified rare hereditary diseases, in many cases not only is there no treatment available but even the natural course of the disease and the relevant tests to diagnose the disease are unknown or underdeveloped. In order to further improve the understanding of rare hereditary diseases and to provide a better and earlier diagnosis for rare disease patients, in 2018 we instituted a program with the aim of characterizing at least 50 new genes per year.
In addition, we continuously develop new testing products to provide the most effective diagnosis products to our physician clients, leveraging insights from our platform and our deep medical expertise. For example, in 2018 we launched CentoDx, one of the largest panels currently available for rare hereditary disease diagnosis, based on next-generation sequencing technology and covering over 7,000 clinically relevant genes and more than 3,700 rare diseases. In addition to traditional genetic testing, we have continued developing innovative diagnostics tests to provide patients with faster, more reliable and more complete solutions. For example, we have developed CentoMetabolic, a panel product covering almost 200 metabolic disorders, developed specifically for patients with suspected metabolic disorders or presenting complex, overlapping symptoms, a metabolic crisis or neurological conditions of unknown etiology. The panel also includes enzyme-activity or biomarker testing, where applicable, which help to determine the disease status and severity. As of December 31, 2019, we offered a comprehensive testing portfolio of over 10,000 genetic sequencing tests covering more than 7,000 genes, from single gene WGS-based products, using Sanger, MLPA, qPCR, array and NGS technologies. Of this total, we offer over 9,000 standard genetic sequencing products, over 200 panel products, 26 whole exome sequencing ("WES") products and 11 whole genome sequencing ("WGS") products. We also offer differentiated comprehensive testing solutions including 30 biochemistry testing products, over 1,460
80
copy number variation tests ("CNVs") and 5 NIPT products (single and twin). The graphic below outlines the scope of the diagnostics products that we currently offer.
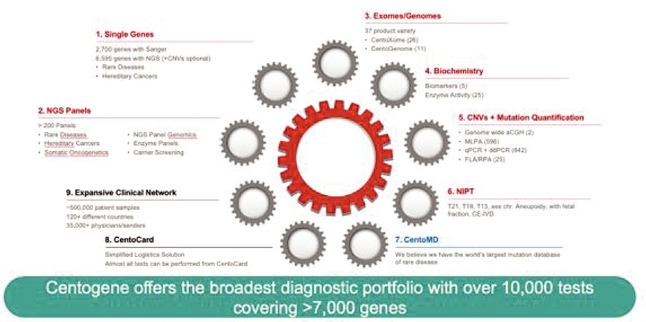
Overview of the Diagnostic Process
Our diagnostics processes are designed with the aim of providing the highest-quality diagnosis within the shortest turnaround time. We currently perform a majority of the diagnostic services for our diagnostics and pharmaceutical businesses in our clinical laboratory located in Rostock, Germany, which is certified under CLIA and accredited by the CAP. Additionally, we perform certain of these services in our Cambridge, Massachusetts facility, which is also certified under CLIA, accredited by the CAP and permitted by the Massachusetts Clinical Laboratory Program.
We strive to provide the best quality of diagnostics testing, not only by following the strictest quality criteria complying with CAP, CLIA and ISO 15189 certifications supported by our multidiscipline quality management system ("QMS"), but also applicable and market standard Good Laboratory Practice ("GLP") and Good Manufacturing Practice Regulations ("GMP") guidelines. Our processes are highly efficient and have been designed to deliver our medical report back to the physician within 30 days from receipt of the sample.
81
Our diagnostics process is defined by our five-step process:
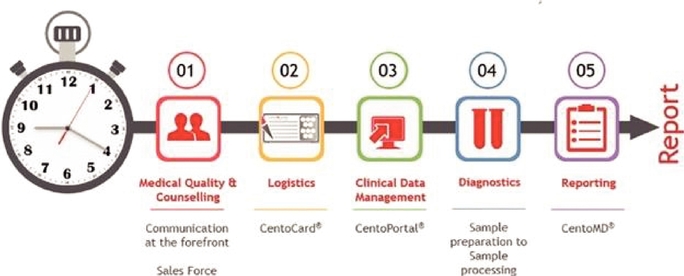
- •
- Medical Quality and
Counseling: Genetic specialists review the patient's clinical records and confirm that the physician has requested the appropriate genetic test
with regard to the patient's individual circumstances and medical history. In all cases, the physician is required to provide us with a completed patient consent form, which our staff review for
adequacy prior to the performance of any diagnostic services.
- •
- Logistics: We use the CentoCard
collection method for obtaining the majority of our samples. This standardized procedure allows us to extract high-quality biological material from dried blood spots on the CentoCard, including DNA
(for molecular diagnostics), protein (for enzymatic and biomarker assays) and metabolites (for biomarker assays).
- •
- Clinical Data
Management: Physicians are able to order our diagnostic tests for a particular patient either online through our CentoPortal platform or by email
or mail.
- •
- Diagnostics: Once a patient
sample is received, we prepare the biological material for testing by taking an extract of the DNA from the relevant sample. Depending on the test requested by the physician, we would then proceed to
run any number of our diagnostic services listed above.
- •
- Once produced, the data is entered into a sophisticated series of our proprietary computational algorithms designed to detect and
identify known pathogenic variants. The sequenced data is analyzed using our fully validated and automated bioinformatics pipeline and annotated with information from our mutation database, CentoMD.
The database is key to the diagnostics process as it is used as the basis of comparison with the patient's sequenced data. This analyzed genetic information together with the patient's medical history
and clinical data is then interpreted by our medical experts, a team of trained human geneticists and doctors. All identified mutations along with their annotations will undergo a manual validation
against the medical history of the patient in order to ensure accuracy.
- •
- Additionally, our bioinformatics pipelines provide a highly automated approach to analysis of variant classification, CNV identification and other genetic data. To augment our bioinformatics pipelines, we have developed a database to store all variant information, which, in addition to CentoMD, is the basis for our evaluation and interpretation of genetic data. We have developed an in-house variant prioritization and classification system, named CentoPrio, to enhance our interpretation capabilities. CentoPrio takes advantage of the vast amount of genotypic and phenotypic data stored in our databases. Through the use of
82
- •
- Reporting: Our test reports
deliver clinically relevant information in a manner that seamlessly integrates into physician practices. A standard report contains a summary of the test result, provides our analysis, recommendations
and detailed description of the patient's relevant genomic alterations and a full data record for consolidation with the patient's medical records. The report also identifies noteworthy absences of
genomic alterations and summaries of, and references to, supporting data from peer-reviewed publications. If requested by the physician, we also provide information on variants in genes not associated
with the patient's disease or symptoms but that nonetheless contain medically actionable information (such as incidental or secondary findings).
- •
- All of our medical reports are written by professional medical experts facilitated by our automated report writing technology and are reviewed and approved by our Chief Medical Officer before distribution. Physicians obtain one report per patient diagnosis while our pharmaceutical partners obtain genomic information that has been provided with express patient consent and de-identified in accordance with HIPAA and other relevant health information privacy procedures. All reports are easily accessible through our online platform, CentoPortal.
proprietary algorithms and machine learning algorithms (artificial intelligence), we combine this data with current medical knowledge to prioritize particular variants that have been identified in previously closed patient cases.
Our Solutions for Providing High-Quality Data
CentoCard
Our sample collection method is a CE-Marked dried blood spot collection kit, the CentoCard (as shown below), which is translated into more than 30 languages and registered in more than 50 countries. The CentoCard is sent to physicians as part of a five-component kit: (1) the CentoCard, (2) a genetic testing informed consent form, (3) an instruction leaflet, (4) a self-addressed return envelope and (5) a plastic sleeve for the used CentoCard to be sealed in once the sample is obtained. In order to obtain the sample, a small amount of blood is drawn from a patient by his or her physician and placed on designated spots on the CentoCard. This sample is then left to dry for approximately two hours, during which time the sample stabilizes. Each CentoCard produced has a unique barcode that allows for the card to be traced at all times. It is delivered to our laboratory in Rostock, Germany, along with a signed consent form, from anywhere in the world via regular post. Samples collected on CentoCard are considered non-biohazardous materials, which allows them to be mailed across many borders without the need for certain customs declarations.
We use the CentoCard collection method to obtain the majority of our samples. This standardized procedure allows us to extract high-quality biological material and perform most of our diagnostic tests from a portion of a single dried blood spot on the CentoCard. Using the CentoCard, we are able to provide a solution where necessary molecular and biochemical tests can be run simultaneously using the same patient sample. Given that the biomaterial stabilizes on the CentoCard, we are able to retest the existing patient samples multiple times for more than 10 years from initial sample collection.
83
CentoCard sample:
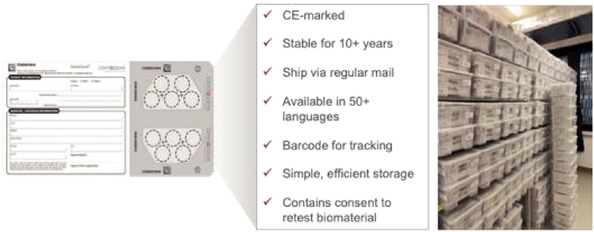
CentoPortal
After a physician creates an online account on CentoPortal by following a few easy steps, the physician can order a test product of his or her choosing, provide and sign a patient consent online, provide an overview of the patient's medical history, track the samples and progress of the diagnostic test and download the final medical report once the process is complete. Access to the CentoPortal requires secured authentication. This helps prevent unauthorized access, unauthorized use or loss of patient data.
CentoMD
We believe our CentoMD database is the world's largest curated mutation database for rare diseases. All approved curated individual data is anonymized and released to CentoMD on a regular basis, offering the most complete and up-to-date information possible. The patient data we have collected in CentoMD 5.7 cover 12 therapeutic areas with over 3,700 diseases. Our CentoMD 5.7 includes curated data from over 400,000 patients with over 12.2 million unique variants and over 3,700 associated phenotypes.
Through CentoMD, we are able to combine variant information with proteomic and metabolomics information, in particular for high-throughput genes where such a functional assay is available. Thus, crucial functional information necessary to support classification decisions, such as variant expression, can be reviewed by users.
CentoPharma
Powered by CentoMD, CentoPharma is an online tool which offers an additional tailored interface where our pharmaceutical partners can generate customized datasets combining phenotype, genotype and biochemistry information. Our pharmaceutical partners can dynamically query the database using any combination of six criteria: HPO, requested tests, genetically confirmed diagnoses, home country or geographical region and certain screened genes.
While CentoPharma and CentoMD are powered by the same database, the dynamic search capability of CentoPharma allows the user to identify cohorts based on clinical symptoms, country, screened gene and/or genetically confirmed diagnosis, and the resulting datasets help identify patient cohorts exhibiting specific combinations of attributes, enabling the discovery of new targets for drug development and the assessment of market opportunities. CentoPharma also supports the design of clinical trials and the feasibility of recruiting patients to studies operated by our pharmaceutical
84
partners. We grant pharmaceutical partners access to CentoPharma through a singular stand-alone license or as an add-on to part of broader contracted collaboration.
CentoLSD
We have launched CentoLSD in January 2019, which we believe is the world's largest knowledge-driven lysosomal storage disease ("LSD") database, for the purpose to facilitate sharing our rare disease knowledge and enhance the diagnostic and treatment opportunities for rare disease patients. CentoLSD is free and accessible via our website and allows for researchers, pharmaceutical partners and clinicians to access a comprehensive database of genetic variants classified through a standardized curation workflow. Every variant reported in CentoLSD is linked to at least one clinically described case tested against Gaucher or Fabry disease through a validated and accredited laboratory workflow. CentoLSD's interface is easy to use—users first select a gene of interest and can further filter based on cDNA change, protein change, gDNA change, location of DNA change, coding effect, clinical significance and other variables.
MyLSD app
In parallel with CentoLSD, we are also developing MyLSDapp, a smartphone application, which is currently in its pilot testing phase. MyLSDapp is designed to drive the management of personalized treatment for Gaucher disease patients and to support Gaucher specialists around the world. For patients, MyLSDapp is designed to provide an overview of their personal treatment plan and their biomarker (Lyso-Gb1) monitoring results, and allows them to share treatment and result information with physicians. The app is designed to help physicians monitor the treatment of their patients and to develop insights into new treatments.
MyLSDapp combines patients' medical and quality-of-life information in one simple system, bringing a comprehensive view of treatment progress to patients and their authorized physicians. With the ability to request blood tests, monitor treatment dosage, and track Lyso-Gb1 levels from within one smartphone app, Gaucher disease patients and their physicians are able to examine the efficacy of therapy and if necessary, explore additional or alternative treatment methods.
Biobank
We have established a high quality biorepository for use in our research and development collaborations with our pharmaceutical partners. Our biobank provides a large diversity of positive test cases in the field of inherited rare diseases and comprises original patient materials characterized through our genetic and/or biochemical diagnostics and with associated clinical information. The biobank operates under robust quality standards and is the first CAP accredited repository outside the United States. It is also compliant with the new ISO20387 standard and comprises materials from patients who have consented to the use of such materials in research. All of our samples have gone through our rigorous process of documentation, analysis and data evaluation by our in-house experts.
Biomarker Development Process
So long as an adequate patient cohort exists for any of the 7,000 identified rare diseases, of which approximately 5,600 have a genetic origin, we believe a biomarker can be developed. We may either develop a biomarker on our own, in which case we choose the rare disease to be mapped by the biomarker, or we may develop a biomarker at the request of a pharmaceutical company, in which case we typically map a biomarker for a specific rare disease identified by the pharmaceutical company. In both cases, we own the rights to the biomarker, but in circumstances where a pharmaceutical company is funding the biomarker development process, we may agree to parameters for use of the biomarker going forward.
85
The first step to the biomarker development process is analyzing the data taken from our repository to perform a biomarker target validation. Patients with a phenotype and/or genotype known to be an indicator of the particular rare disease for which we plan to develop the biomarker are compared with a large cohort of healthy control individuals. We can conduct this process with a disease cohort of as few as five to ten patients, although it is our experience that a higher number of patients (i.e., approximately 40) could result in a more specific biomarker target validation. The samples included in the study (patients and controls) must be of the same type (e.g., blood, plasma, tissue). The extraction is performed in a highly standardized manner as the results of the study depend on the stability of the samples and the uniformity of the extraction process. We then run the samples through an untargeted high resolution hybrid mass spectrometric analysis either of the small molecules (full metabolic profile) or of the peptides (full proteomics profile). The resulting differences found between the patient cohort profiles and the control cohort profiles are identified using statistical and mathematical algorithms.
We then use artificial intelligence to facilitate the biomarker development process. Artificial intelligence helps us to identify correlations between different data in an efficient and more accurate manner, and discover patterns that would not be discovered manually. Artificial intelligence also allows us to perform fully automated pattern recognition on multidimensional data (e.g., retention time, collision cross section, monoisotopic ion mass, fragmentation pattern) obtained from mass spectrometry.
While it may take weeks to months of manual data comparisons, biomarker candidates can be found in just a few days with our in-house artificial intelligence discovery system. Furthermore, this innovative approach enables us to use multi-peak biomarkers to evaluate even the most difficult patterns in the patient's metabolism. The differences (signals) measured from the mass spectrometer are retested for confirmation, then investigated with another mass spectrometric techniques (fragmentation and targeted mass spectrometry). Proof of concept is performed on anonymized samples using targeted mass spectrometry, in which the identified biomarker candidates are quantified. This part of the process (from project selection to proof of concept) could be completed in three-months per biomarker project. We can then use the biomarkers to develop standardized tests for other services such as our patient screening processes.
Validation Tests
As more patients are enrolled to the clinical trials, we are also able to perform further validation tests for the biomarker so that it could be used for longitudinal monitoring. Validation is a three to six month process during which the biomarker and its characteristics are assessed, which helps to determine the range of conditions under which the biomarker will give reproducible and accurate data. Approximately 50 to 100 patients in a disease cohort are needed to complete the validation process and approximately 8,000 different measurements are needed to comply with all CAP/CLIA/ISO requirements.
Research and Development
We are dedicated to scientific research and development in order to continuously improve the industry's understanding of epidemiology and its analysis of clinical heterogeneity as an aid to the diagnosis of rare diseases. We have organized various conferences with experts and patient advocacy groups in the rare disease field from all over the world to exchange and promote knowledge related to rare diseases, such as our "Recent Advances in Rare Diseases (RARD)" conference which we hold annually.
In addition, we also undertake scientific research and clinical studies, both independently and together with our pharmaceutical partners, with the aim of positively contributing to the global
86
understanding of rare diseases, as well as to improve the accuracy of diagnosis and to support the development of effective treatments for rare diseases.
We published over 45 scientific papers in 2019. Our major on-going clinical studies, other than those related to biomarker development, are as follows as of December 31, 2019.
In February 2020, we commenced our Rare Disease Day 2020 in Lahore, Pakistan at the Children's Hospital of Lahore with presentations, talks and panel discussion reflecting the diagnostics and everyday challenges of patients and patient organizations. This was followed by a group discussion in Berlin, Germany involving policy makers, public authorities, researchers, health professionals and community members about innovative approaches to shorten the diagnostic odyssey of rare disease patients. The event ended in Mexico with discussions held by patients, patient organizations, physicians, politicians and community members from all over Latin America relating to a collaborative approach towards, and modern technologies used in, the creation of life-changing solutions for rare disease patients and their families. All of these activities serve to raise public awareness of rare diseases and shorten the diagnostic odyssey of rare disease patients.
Two Year Global Study on Colon- and Pancreas-Carcinoma (PICOP-Global)
In June 2019, we announced the initiation of a 24-month global proof-of-concept study focusing on the identification of tumor-specific neoantigens, which we anticipate will be used by our partners as the basis for developing a personalized, immune-based therapy to trigger patients' own immune responses against tumors.
The study aims to enroll approximately 130 participants with pancreas or colorectal carcinoma in order to analyze the molecular characteristics of tumors and to subsequently identify tumor-specific antigens (neoepitopes). In addition, the tumor and normal samples will be tested genetically to identify tumor associated mutations and MS/MS-based mutations, which will be used to develop neoepitopes and biomarkers. We believe that the collected data can be used in a multi-omics approach to potentially predict neoepitopes suitable for use in vaccines.
The PICOP-GLOBAL Study will be conducted without a treatment arm and in collaboration with the Surgical Oncology Society Pakistan. We are also partnering on this study with the University of Rostock, Germany and University of Greifswald, Germany. After this study is completed, a follow-up Phase I study will be conducted by Miltenyi Biotec.
Induced pluripotent stem cells (iPSC) program
Since early 2019, we have independently conducted an iPSC program (the "iPSC Program") with the aim of supporting orphan drug development in a more cost-effective and efficient manner, in particular for the development of orphan drugs related to rare neurodegenerative, metabolic, and cardiovascular diseases. Human iPSCs, first reported in 2007, are reprogrammed from somatic cells and are self-renewal cells that can produce different types of cells. In the drug discovery process for rare diseases, iPSC technology is particularly important in providing information on the clinical spectrum of such diseases by generating disease-specific cells.
As of December 31, 2019, we had collected approximately 850 skin biopsies from patients with rare diseases from around the world. As part of our iPSC Program, we are currently in the process of reprogramming these biopsies into iPSC for a number of metabolic rare diseases such as Gaucher, Niemann Pick Type A and C, Polycystic kidney disease, Fabry disease, Glycogenosis, Sanfilippo (MPS III), Maroteaux-Lamy Disease (MPS VI), Morquio (MPS IV) Pompe and Wilson diseases. Once completed, the iPSC Program will also further support orthogonal target validation as well as further biomarker discovery that we undertake.
87
Rostock International Parkinson's Disease Study (ROPAD)
In May 2019, we initiated a 24-month global study (the "ROPAD Study") to investigate the genetic factors in Parkinson's disease ("PD"), one of the most common neurodegenerative disorders that affects approximately 1% of individuals globally over the age of 60. The ROPAD Study, which is being conducted in cooperation with the University of Lübeck, Germany, aims to enroll approximately 10,000 participants worldwide in order to provide a study cohort with a broad genetic background that mirrors the global population. The objective of the ROPAD Study is to gain a comprehensive understanding of how many and which genetic mutations in PD-associated genes are linked to the development of the disease. We plan to utilize our CentoCard product to identify participants with a mutation in LRRK2, GBA and other PD-associated genes. As of December 31, 2019, over 850 patients were tested as part of this study.
ROPAD Study participants that display mutations in PD genes will have the option to undergo further clinical assessment in a supplementary study, "LRRK2 International Parkinson's Disease Project (LIPAD)", conducted at the University of Lübeck, where a detailed phenotyping of participants will be performed in order to describe the frequency of all important clinical PD signs and symptoms. Patients enrolled in ROPAD with a LRRK2 mutation may also be offered participation in future clinical studies with one of our major collaboration partners, Denali.
Epidemiological Analysis for Hereditary Angioedema Disease (EHA)
In September 2018, we commenced the EHA study, which is a prospective, multicenter study in Germany, Poland, Turkey and the United Kingdom. The study focuses on patients with HAE, or patients with high-grade suspicion of suffering from HAE. The aim of the study is to estimate the epidemiological prevalence of disease in a population of 5,000 patients with repetitive abdominal pain attacks of unknown origin, as well as other clinical symptoms that could indicate the presence of HAE. HAE is a rare autosomal dominant disease resulting from mutations in the SERPING1 gene, leading to the deficient (type 1) or nonfunctional (type 2) C1 inhibitor protein. Clinical manifestations in all HAE types include acute attacks of non-urticarial edemas affecting the upper airway, face, extremities, genitals, and gastrointestinal system. As of December 31, 2019, over 850 patients were tested under the study.
European Alpha-Mannosidosis Patient Epidemiological Program (EUMAP)
In August 2018, we commenced the EUMAP study, which is an international, multicenter, epidemiological study. The study aims to further explore and analyze the prevalence of Alpha-Mannosidosis disease in a clinical study cohort of 1,000 patients that are potentially suffering from Alpha-Mannosidosis disease, based on the patient's clinical symptoms. Alpha-Mannosidosis is a very rare, hereditary lysosomal storage disorder, closely related to Mucopolysaccharidoses, which is estimated to occur in approximately 1 in 500,000 people worldwide. The disease is caused by mutations in the MAN2B1 gene, which interferes with the activity of the alpha-mannosidase enzyme and results in accumulation mannose-containing oligosaccharides in the lysosomes. The course of the disease is progressive in general, although symptoms can vary significantly between individual patients. It has been categorized into "mild", "moderate" and "severe" depending on the severity of the symptoms and age of onset.
Hereditary Transthyretin-Related Amyloidosis Study (TRAM2)
In April 2018, we commenced TRAM2 study, which is a prospective, multicenter study in Germany, Austria and Switzerland, focusing on patients with polyneuropathy or cardiomyopathy of undetermined etiology. The study is a continuance of TRAM1 and aims to estimate the prevalence of hereditary transthyretin-Related Amyloidosis ("hATTR") in a study cohort of 5,000 patients with
88
polyneuropathy or cardiomyopathy of unknown etiology. As of December 31, 2019, over 3,600 patients were tested under the study. hATTR is an autosomal dominant inherited variable penetrance disease that we believe is often under- or mis-diagnosed, and it is caused by mutations in the receptor gene TTR. The clinical spectrum of hATTR varies greatly from exclusive neurological involvement to predominant cardiac manifestations, and without treatment hATTR can lead to heart failure. This disease typically affects people in their 30s to 50s and may lead to death within 10 years if left untreated.
Screening for the Transthyretin-Related Familial Amyloidotic Small Fiber Polyneuropathy (TRAP)
In December 2016, we commenced the TRAP study, an international, multicentre, epidemiological study. The study aims to determine the prevalence of patients with transthyretin-related familial amyloidotic-polyneuropathy ("TTR-FAP") in a clinical study cohort of 500 patients with a polyneuropathy of undetermined etiology, based on medical history (no anamnesis for carcinoma, no continuous alcohol consumption, no anamnesis for heavy metal exposure, no significant comorbidities), and the normal results of laboratory data. As of December 31, 2019, over 260 patients were tested under the study.
TTR-FAP is an autosomal dominant rare disease, the exact prevalence of which is unknown but is estimated to occur in around 1 in 100,000 people to 1 in 1,000,000 people. The TTR-FAP has a very heterogeneous phenotype which can manifest starting at the age of 18 and may lead to death within 10 years.
Our Operations
Sales and Marketing
As of December 31, 2019, our CBO led a team of six dedicated employees for business development in our pharmaceutical segment. With the importance of the segment, it has been historically closely supported by our CEO due to his network with different pharmaceutical partners, as well as the appreciation of his knowledge of rare diseases by the industry. We anticipate growing our team to support the growing number of partnership opportunities.
As of December 31, 2019, we had a sales force of approximately 25 employees and 26 consultants in our diagnostics business. Our sales employees are all trained in key account management and/or genetic diagnostics and are able to discuss the different diagnostic and workflow needs of doctors, physicians and genetic counselors. To further develop our footprint and to support the rare disease patients in the United States, we established a presence in the United States at the end of 2017 with the hiring of a sales team and the opening of a new laboratory in Cambridge, Massachusetts in October 2018.
In addition, we will continue to expand our sales force and our distribution network in order to further increase the sample volumes in targeted geographic areas, particularly in the North America, Latin America and Asia Pacific regions.
Information Technology Platforms
Our IT infrastructure platform is based on state-of-the-art standardized components. We run our systems according to the following hybrid production model in an effort to optimize cost and service levels:
- •
- Systems that require a short distance-to-lab infrastructure are run in-house in separate, protected server rooms;
89
- •
- Tailored systems with special requirements and heightened security use outsourced infrastructure as a service provided by Datagroup AG, which
is GDPR-compliant. These services are provided by two datacenters in Frankfurt and our lab in Rostock, which are connected by two independent and encrypted 10GB landlines; and
- •
- Highly standardized, high volume requirements use cloud services provided by Amazon Web Services and Microsoft.
All services are based on virtualized server systems with central storage components accompanied by backup and restore services, centrally managed network services, firewall systems, internet, databases and workplace services. System monitoring and events are implemented for all relevant systems with a central monitoring solution and central network scanner controls. Centrally managed user accounts are handled in the directory system.
Information security is highly valued and the principles of confidentiality, integrity and availability of information are a part of our core values. Information is protected by a variety of controls and procedures, including firewalls, password protections, data encryption (in storage and in transit) and malware protection tools. All internet-facing applications are security tested. All personal data processing services are evaluated by our data protection officer and documented in accordance with GDPR. Additionally, our data services are certified across a variety of industry security standards, including ISO 9001 (which aims to ensure we consistently provide services and products that meet customer and regulatory security expectations) and ISO 27001 (which standards ensure the data in our database are secured).
Our workflows and processes are supported by various specialized applications. For example, via our user-friendly online portal "CentoPortal," analyses ranging from individual diagnostic requests to requests for pharmaceutical projects with high throughput testing can be ordered. Physicians can view the status of the samples they submitted and download a complete medical report. Upon receiving samples, we digitalize all information to support a fully digital internal workflow. This starts with a web application for sample entries, where information is transferred automatically by interfaces to our laboratory information system. This information forms the basis of our medical reports, which are made available to doctors for download. Data is shared between CentoPortal and our laboratory information systems through a fully automated interface.
Artificial Intelligence
Since 2018, we have been using artificial intelligence to further automate our processes, obtain new insights about rare diseases from mass data sets and generate new knowledge-driven business models. For example, we use artificial intelligence to enhance our biomarker discovery process. This allows us to shorten data analysis time from weeks to minutes and to identify multiple biomarkers or additional biomarker patterns in our phenotypic, genomic, transcriptomic, proteomic and metabolomic datasets. We also use artificial intelligence to automate our curation process and the identification of genetic and/or metabolic modifiers. We currently have five employees dedicated to this artificial intelligence effort.
We believe that our data repository provides us with a competitive advantage for driving the development of new and effective artificial intelligence tools, as the foundation of any successful artificial intelligence program is high quality data in a volume that can effectively generate results. The higher quality the data and the more data that are available, the better chance we have of building a machine learning model with high predictive power and accuracy.
90
As of December 31, 2019, we deployed the following artificial intelligence programs:
Intelligent Character Recognition
Intelligent character recognition ("ICR") at the sample entry stage enables us to fully digitize all information contained in sample order paperwork. Even on handwritten texts, our ICR technology achieves significant performance. This allows us to obtain accurate patient information at the initial stage of the diagnostics process and reduces the likelihood of human error.
Variant Prioritization
We have deployed a new variant prioritization tool based on our in-house artificial intelligence capability. This tool identifies the most likely disease-causing genes based on our repository, in order to further accelerate our and our partners' diagnostics processes, and is in particular aimed to enhance the diagnostics process for whole exome and clinical exome sequencings.
With our clinical exome panel, which covers over 7,000 genes with known associated clinical phenotypes and covers over 3,700 diseases, the result of the sequencing process usually discovers between 70,000 and 150,000 variants per individual. However, the majority of these variants are benign or unrelated to the observed disease phenotype of the patient. With our huge data repository built up from the last 12 years, and a curated database with standardized HPO terms, our tool is able to rank the variants from most to least relevant. Based on such "ranked" variants, we can then compare the HPO terms of a new patient with the results of prior, anonymized patients included in our repository with variants in the same gene. This allows us provide a diagnosis in a more rapid, comprehensive and accurate manner, especially for patients with very rare or as yet undescribed diseases.
Automated Curation Report
Our curators are responsible for the collection, association, update and review of genetic and phenotypic data of cases analyzed at Centogene to assure the highest level of data quality in CentoMD. The automated curation process supports our curation process with a set of rules encoding the expert knowledge and classifying newly incoming cases as well as reclassifying the old ones if new genomic insights result from research.
Biomarker Discovery
Artificial intelligence enables results that previously would have been practically impossible. The direct analysis of multiple measurements that previously would have been impossible due to diverging experimental conditions, enables the detection of combined biomarkers, and drastically improves speed and reliability.
Healthcare Reimbursement
Reimbursement of genetic testing differs markedly among countries and evolves rapidly based on advancements in technologies and cost. It is a challenge for insurers or public payors to decide when to reimburse for genetic tests that are offered by healthcare providers. One of the reasons this is difficult is that often there are alternative treatments with differing results, which insurers may not be able to easily evaluate.
Depending on the billing arrangement and applicable law, we may be reimbursed for genetic testing services by third-party payors that provide coverage to the patient, such as an insurance company or managed care organization, or by physicians or other authorized parties (such as hospitals or independent laboratories) that order our tests or refer tests to us. We do not receive reimbursement from any United States federal healthcare program, including Medicare or Medicaid. In the years
91
ended December 31, 2019 and 2018, we derived less than 1% of our total revenue from United States third-party payers that includes managed care organizations and other healthcare providers. In the years ended December 31, 2019 and 2018, we derived less than 1% of our total revenue from EU insurance companies and managed care organizations based in the European Union.
We have strategically determined to focus on countries around the globe where the prevalence of rare hereditary diseases is high or the availability of national genetic testing and interpretation is to some extent limited and therefore the complete reimbursement or partial payment by the government for our services is more likely. Therefore, the major markets for our diagnostics business currently include the Middle East and North Africa region, Scandinavia, parts of Central and Eastern Europe, Latin America, North America and parts of Asia. In most of our markets, our diagnostics tests are billable directly to the party submitting the request for a test to us and we have less than 1% bad debts written off since the inception of our business.
Data Management
Data is the basis for all of our diagnostic and research processes. We are generating approximately up to 25TB of new data in the lab every month. The data is stored in our own infrastructure as well as in a certified third party data center and with Amazon Web Services. The software solutions supporting these processes are based on modern database architecture, and all of our critical systems are fully redundant and backed up in real-time to these facilities.
Further, we implement our big data concept based on architecture. Because we store a vast amount of raw data in our repository, we are able to aggregate data to gain new insights. We are currently using this for biomarker research and will stepwise roll it out for the entire company in the next 15 months. Data gathering and variant curation are procedures developed and implemented in a web-based software (developed and maintained by Centogene N.V.) that is compliant with the HUGO Gene Nomenclature Committee (the "HGNC"), the Human Genome Variant Society (the "HGVS") and HPO nomenclatures. The software integrates in-house sample management systems and analysis platforms with external databases, utilizes a combination of computer-based tools and manual review in order to assure the accuracy, efficiency and quality of curation process.
All approved curated individual data is then anonymized and released to CentoMD on a regular basis, offering the most complete and up-to-date information possible.
Quality Management System
We have developed and maintained a QMS that integrates the compliance of our processes with various medical device regulations, clinical trial requirements and clinical laboratory requirements. Our QMS is supported by standard operating procedures, educational and staff training plans, internal and external proficiency and competency programs, internal and external auditing, quality improvement indicators and pre-post analytical quality controls, including equipment maintenance, negative and positive controls, change management, employees and customer health and safety and document control programs. Our QMS integrates the compliance of our processes with the following requirements:
- •
- the GLP regulations, which are intended to ensure compliance with quality and integrity of the safety data filed pursuant to certain sections
of the FDC Act and Public Health Service Act in the United States;
- •
- the GMP and the Good Clinical Practice Regulations, which exist to control the safety and efficacy of manufacturing operations and conduct of clinical trials;
92
- •
- the Code of Federal Regulations Title 21 part 820 and part 821 as amended by ISO 13485:2016, which set forth the
requirements for a comprehensive quality management system for the manufacture and tracking of medical devices;
- •
- CAP and CLIA requirements (see "Intellectual Property—Regulations—United States Regulation—CLIA and State
Regulation");
- •
- Massachusetts Department of Public Health clinical laboratory program standards (Chapter 105, Section 180.00 et. seq. of the Code
of Massachusetts Regulations);
- •
- ISO 15189:2012 requirements, which specify requirements for quality and competence in medical laboratories; and
- •
- over 47 different country-specific medical device registration requirements.
We believe our QMS was built to withstand the rigorous review and auditing of medical device regulations, clinical trial requirements and clinical laboratory requirements to ensure our patients and clients receive the highest quality level of care and service.
Client data protection is of high importance to us, as we provide solutions to our clients in more than 120 different countries with varying requirements. We protect our clients and employees through an informed consent process, which goes through a rigorous legal review with in-country specialists and our internal HIPAA and GDPR compliance policies. We continuously monitor all electronically archived and incoming data through these channels.
Data Acquisition and Curation
Curation is the process of collection, association, updating and review of epidemiologic, phenotypic and genetic data of patients analyzed by us into a structured and standardized format. It uses a combination of computer-based tools and manual review in order to assure the accuracy, efficiency and quality of the curation process.
Data acquisition. Data gathering and variant curation procedures are developed and implemented in a web-based software which is compliant with the HGNC, HGVS and HPO nomenclatures allowing collection of variants detected in nuclear coding, nuclear non-coding and mitochondrial genes. The software integrates in-house sample management systems and analysis platforms with external databases providing the curator with a comprehensive and straightforward overview of the evidences regarding genotype-phenotype correlation available both in-house and external.
The data is gathered by a combination of manual submission and data importation following an individual-oriented model where characteristics belonging to a particular individual (including patient information, clinical data, methodology and detected genetic variants) are stored and associated together.
Our uniform classification of variants is an important step in improving our understanding of disease pathogenicity. There are approximately 3 billion base pairs in an individual genome, which translates to approximately 200 gigabytes of data that can be obtained from a single sequencing process. Based on the variants in CentoMD 5.7 (released in September 2019), a comparison against the list of variants in other industry databases such as ClinVar (version ClinVar 2019, September 2019) and HGMD Pro (version HGMD Pro 2019.1, January 2019) shows that of the shared variants among CentoMD 5.7, ClinVar and HGMD Pro, approximately 40% and approximately 61% of such shared variants in ClinVar and HGMD Pro, respectively, are discordantly classified. The classification of variants which we record in our CentoMD database follow the American College of Medical Genetics and Genomics guidelines for variant classification, differentiated into five categories: pathogenic, likely pathogenic, uncertain clinical significance, neutral or likely neutral. If a diagnostic test is finalized without a pathogenic indication, we still include the data in CentoMD under an "uncurated"
93
classification. This information can then be used as comparative data for future diagnostic tests. This uniform classification of variants is based on a highly qualified and standardized curation process, which allows us to provide our clients with high-quality clinical interpretations of newly identified variants, and also ensures that changes in variant classification will be communicated and reflected in our clinical interpretations in a timely manner.
As industry knowledge on variant frequencies increases, we reevaluate the variant classifications contained in our database on a regular basis to ensure our system incorporates the most up-to-date information. Additionally, given the number of rare diseases that have yet to be fully diagnosed and the speed of advancements in the rare disease industry, we regularly revisit "uncertain" patient data to reassess prior clinical interpretations against this new industry knowledge.
Database curators. Our CentoMD curators are scientists with strong backgrounds in human genetics. They continuously undergo extensive training to ensure curation consistency and standardization. They assure that data is properly associated and interpreted and that there are no inconsistencies or discrepancies against detected in-house observations and from external sources. They close the curation process by manual approval that reviewed and curated data comply with standard in-house procedures.
Curation workflow. To provide high-quality data, our curation process is divided in three phases: variant-wise, individual-wise and warnings-wise procedures.
- •
- Curation by variant. To begin the curation process, the variant-linked
information is reviewed. This includes approval of variant nomenclature, terminology, accuracy, consistency and record completeness.
- •
- Curation by individual. In order to start curation on a patient-by-patient
basis, all variants detected in an individual must be approved. This process aims to assure that the data belonging to an individual follows the guidelines for clinical reporting closely and that all
associated data is in agreement with our established guidelines and applicable industry standards. The following factors are considered critical for the clinical statement: variant clinical
significance, patient genotype, inheritance pattern of the disorder, the sex of the patient and the phenotypic description, when available.
- •
- Curation by warning. The database generates warnings at different levels (variant, individual, gene database levels) to detect errors, invalid terms and nomenclatures and inconsistencies. These warnings are triggered by additional evidence obtained internally, such as medical reports, or detected externally, such as articles, publications and external databases. Each warning is then manually documented and resolved.
All approved curated individual data is then anonymized and released to CentoMD on a regular basis, offering the most complete and up to date information possible to its users. CentoMD is a constantly growing and enriching its database. As of December 31, 2019, CentoMD included curated data from over 400,000 patients. In addition, whenever additional evidence provided by our in house medical professionals or by external peer-reviewed literature becomes available, specific variants are revised and reclassified accordingly.
Intellectual Property
Our success depends in part on our ability to obtain and maintain proprietary protection for our genetic rare disease information platform, proprietary biomarkers, products and solutions and other know-how related to our business, defend and enforce our intellectual property rights, in particular, our patent rights, preserve the confidentiality of our trade secrets, and operate without infringing valid and enforceable intellectual property rights of others. We seek to protect our proprietary position by, among other things, filing EU, U.S. and certain foreign patent applications related to our biomarkers,
94
where patent protection is available. Our policy is to seek patent protection and trademark registration for commercially valuable assets we develop, as appropriate, and maintain as trade secrets other aspects of our genetic rare disease information platform, processes and know-how. We also rely on proprietary technologies, methods and processes, product designs and branding that we have developed.
Notwithstanding these efforts, we cannot be sure that patents will be granted with respect to any patent applications we have filed or may file in the future, and we cannot be sure that any issued patents will not be challenged, invalidated, or circumvented or that such patents will be commercially useful in protecting our technology. Moreover, trade secrets can be difficult to protect. While we have confidence in the measures we take to protect and preserve our trade secrets, such measures can be breached, and we may not have adequate remedies for any such breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. For more information regarding the risks related to our intellectual property, please see "Item 3. Key Information—D. Risk Factors—Intellectual Property Risks Related to Our Business."
Patents
Each patent family in our patent portfolio typically includes one or more priority-forming patent applications on the basis of which an international patent application (an application filed under the Patent Cooperation Treaty ("PCT")) is filed, after which national and regional patent applications are prosecuted in various jurisdictions. As of December 31, 2019, our patent portfolio was as follows:
- •
- With regard to our biomarker for Gaucher disease, we own two pending U.S. non-provisional patent applications, issued patents in Australia,
China, Europe, Israel, Japan and Russia, and seven pending patent applications in the following foreign jurisdictions: Australia, Brazil, Canada, Europe, Israel, India, and Russia. The issued European
patent is currently being validated in one or more contracting states of the European Patent Convention. These issued patents, and any patents granted from such applications, are expected to expire in
2032, without taking potential patent term extensions or adjustments into account.
- •
- With regard to our biomarker for metachromatic leukodystrophy, we own two separate patent families. The first patent family consists of one
pending U.S. non-provisional patent application, issued European, Australian and Israeli patents, and one pending patent application in Brazil. The issued patent, and any patents granted from such
applications, are expected to expire between 2033 and 2039, without taking potential patent term extensions or adjustments into account.
- •
- With regard to our biomarker for Niemann-Pick disease, we own three issued U.S. patents, two pending U.S. non-provisional patent applications,
issued patents in Europe, Japan and Mexico, and pending patent applications in the following foreign jurisdictions: Australia, Brazil, Canada, Europe, Israel, India, Japan, Mexico and Saudi Arabia.
The issued European patent is currently being validated in one or more contracting states of the European Patent Convention. These issued patents, and any patents granted from such applications, are
expected to expire between 2032 and 2035, without taking potential patent term extensions or adjustments into account.
- •
- With regard to our biomarker for Farber's disease, we own one pending U.S. non-provisional patent application and nine pending patent
applications in the following foreign jurisdictions: Australia, Brazil, Canada, China, Europe, Hong Kong, Mexico, Saudi Arabia and the United Arab Emirates. Any patents granted from such applications
are expected to expire in 2036, without taking potential patent term extensions or adjustments into account.
- •
- With regard to our biomarker for Cystic Fibrosis, we own one pending U.S. non-provisional patent application and seven pending patent applications in the following foreign jurisdictions: Australia, Brazil, Canada, Europe, Israel, Hong Kong and India. Any patents granted from such
95
applications are expected to expire in 2037, without taking potential patent term extensions or adjustments into account.
The term of individual patents depends upon the legal term for patents in the countries in which they are granted. In most countries, including the United States, the patent term is 20 years from the earliest claimed filing date of a non-provisional patent application in the applicable country. In the United States, a patent's term may, in certain cases, be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the USPTO in examining and granting a patent, or may be shortened if a patent is terminally disclaimed over a commonly owned patent or a patent naming a common inventor and having an earlier expiration date.
We have entered into agreements with the University of Rostock and a related scientific institute pursuant to which such parties have fully transferred to us interests that they had co-owned with us with respect to patents and patent applications relating to our biomarkers for Gaucher disease, metachromatic leukodystrophy and Niemann-Pick disease or to the treatment of cancer or lysosomal storage disorders. Pursuant to the terms of these agreements, we were required to pay a total of €150,000 in upfront transfer fees and are obligated to pay royalties below 1% on net sales generated by the applicable patents in the future.
Trade Secrets and Trademarks
In addition to patent protection, we also rely on trade secrets, know-how, continuing technological innovation, and confidential information to develop and maintain our proprietary position and protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection, including, our genetic rare disease information platform. We seek to protect our proprietary technology and processes, in part, by entering into confidentiality agreements and invention assignment agreements with our employees, consultants, scientific advisors, contractors and commercial partners. In addition, we take other appropriate precautions, such as physical and technological security measures, to guard against misappropriation of our proprietary technology by third parties.
Our brand is very important to us, as it is a symbol of our reputation and representative of the goodwill we seek to generate with our customers. Consequently, we have invested significant resources in the protection of our trademarks. We seek trademark protection in the United States and in foreign jurisdictions where available and when appropriate. We own registered trademarks for both "Centogene" and "CentoMD" in Europe, the United States and other jurisdictions, including Canada and Japan.
Regulation
Our diagnostics and pharmaceutical businesses are highly regulated due to our operation of clinical laboratories in Rostock, Germany and Cambridge, Massachusetts and because of our provision of diagnostic services and our development of proprietary biomarkers. In addition, we are subject to a variety of regulations and industry standards worldwide governing, among other things, data privacy, distribution of our products and patents and trademark licensing.
The key U.S. and European regulations that are applicable to our business are discussed in more detail below. Whether or not we obtain FDA clearance or approval or a CE Mark for a product, we must obtain the requisite approvals from regulatory authorities in foreign countries prior to the use of a diagnostic or other product in those countries. The requirements and processes governing patient consents, product registration and pricing vary from country to country.
96
United States Regulation
Our business is subject to and impacted by extensive and frequently changing laws and regulations in the United States at both the federal and state levels. These laws and regulations include regulations particular to our business and laws and regulations relating to conducting business generally. We also are subject to inspections and audits by governmental agencies. Set forth below are highlights of the key United States regulatory schemes applicable to our business.
CLIA and State Regulation
Because we operate clinical laboratories, we are required to hold certain United States federal and state licenses and certifications to conduct our business. We are subject to CLIA regulations in the United States, which establish quality standards for all laboratory testing to ensure the accuracy, reliability and timeliness of patient test results regardless of where the test is performed. Our laboratories in Rostock, Germany and Cambridge, Massachusetts are CLIA-certified and accredited by CAP, as well as CAP ISO 15189 accredited. In addition, we are required to meet certain laboratory licensing requirements for states with regulations beyond CLIA. For more information on state licensing requirements, see "—Regulation—United States Regulation—State Laboratory Testing."
Under CLIA, a laboratory is any facility that performs laboratory testing on specimens derived from humans for the purpose of providing information for the diagnosis, prevention or treatment of disease, or the impairment of, or assessment of health. CLIA also requires that we hold a certificate applicable to the type of work we perform and comply with certain standards. CLIA further regulates virtually all clinical laboratories by requiring that they be certified by the federal government and comply with various operational, personnel, facilities administration quality and proficiency requirements intended to ensure that their clinical laboratory testing services are accurate, reliable and timely. Laboratories must register and list their tests with the Centers for Medicare & Medicaid Services, or CMS, the agency that oversees CLIA. CLIA compliance and certification is also a prerequisite to be eligible to bill for services provided to governmental payor program beneficiaries and for many private payors. CLIA is user-fee funded. Therefore, all costs of administering the program must be covered by the regulated facilities, including certification and survey costs.
We are subject to survey and inspection every two years to assess compliance with program standards, and may be subject to additional unannounced inspections. Laboratories performing high-complexity testing are required to meet more stringent requirements than laboratories performing less complex tests. In addition, a laboratory like ours that is certified as "high-complexity" under CLIA may develop, manufacture, validate and use proprietary tests referred to as LDTs. While laboratories that offer LDTs are subject to the FDC Act, in addition to CLIA, the FDA has generally exercised enforcement discretion towards these tests. In compliance with CLIA requirements to establish performance specifications, including accuracy, precision, specificity, sensitivity and a reference range for any LDT used in clinical testing, our LDTs have undergone full analytical validation.
In addition to CLIA requirements, we elect to participate in the accreditation program of CAP. CMS has deemed CAP standards to be equally or more stringent than CLIA regulations and has approved CAP as a recognized accrediting organization. Inspection by CAP is performed in lieu of CMS for accredited laboratories. Because we are accredited by the CAP Laboratory Accreditation Program, we are deemed to also comply with CLIA.
State Laboratory Testing
CLIA also provides that a state may adopt laboratory regulations that are more stringent than those under federal law, and a number of states have implemented their own more stringent laboratory regulatory schemes. State laws may require that laboratory personnel meet certain qualifications, specify certain quality controls, or prescribe record maintenance requirements. Our clinical operations at our
97
Cambridge laboratory are required to meet certain state laboratory licensing and other requirements, which in some areas are more stringent than CLIA requirements. Our Cambridge, Massachusetts lab is also subject to Massachusetts Department of Public Health clinical laboratory permitting requirements. In October 2018, we received our CLIA permit to perform high complexity genetic testing in our Cambridge, Massachusetts lab. Our Massachusetts Department of Public Health clinical laboratory permit application was reviewed and the lab was inspected. It passed accreditation with no deficiencies and was issued a Massachusetts license for high complexity testing in November 2018. We were permitted to begin testing in November 2018. Two states, New York and Washington, are CLIA-exempt, however, and as such have their own regulatory requirements to which we may be subject. CMS deemed both New York and Washington as CLIA-exempt because their licensing and supervisory programs are more stringent than that run by CMS and the CDC. New York requires clinical laboratories that accept specimens from New York residents to have both a CLIA and New York Clinical Laboratory Evaluation Program ("CLEP") permit. CLEP approval can take up to a year, and can be costly and time-consuming. Washington State does not require clinical laboratories to have a CLIA permit, but does require the clinical laboratory to apply for a Washington State lab permit.
Several states in the United States require the licensure of out-of-state laboratories that accept specimens from those states. For example, New York requires a laboratory to hold a permit which is issued after an on-site inspection and approval of testing methodology, and has various requirements over and above CLIA and CAP, including those for personnel qualifications, proficiency testing and physical facility, equipment and quality control standards. Each of our CLIA laboratory locations, including our site in Massachusetts, holds the appropriate licensure for the activities performed at that location. CLEP permit requires LDTs that are offered to New York State patients must be submitted for approval before they can be marketed or offered in New York. The Company is in the process of obtaining the requisite approvals for its LDTs.
From time to time, other states, such as California, Rhode Island, Maryland, New York and Pennsylvania, may require out-of-state laboratories to obtain licensure in order to accept specimens from the state, even though the laboratory is not located in such state. From time to time, other states may require out of state laboratories to obtain licensure in order to accept specimens from the state. If we identify any other state with such requirements, or if we are contacted by any other state advising us of such requirements, we intend to follow instructions from the state regulators as to how we should comply with such requirements. We are currently licensed in Pennsylvania, Maryland and California and are in the process of obtaining a New York State license.
Many states have also implemented genetic testing and privacy laws imposing specific patient consent requirements and protecting test results. In some cases, we are prohibited from conducting certain tests without a certification of patient consent by the physician ordering the test. Requirements of these laws and penalties for violations vary widely. We review our obligations regarding genetic testing and consent periodically. If we identify states with such requirements, or if we are contacted by any other state advising us of such requirements, we intend to follow instructions from the state regulators as to how we should comply with such requirements.
FDA
In the United States, medical devices are subject to extensive regulation by the FDA, under the FDC Act, and its implementing regulations, and other federal and state statutes and regulations. The laws and regulations govern, among other things, medical device development, testing, labeling, storage, premarket clearance or approval, advertising and promotion and product sales and distribution. To be commercially distributed in the United States, medical devices must receive from the FDA prior to marketing, unless subject to an exemption, either approval of a PMA (for most Class III devices), clearance of a 510(k) premarket notification or classification pursuant to a de novo submission.
98
IVDs are types of medical devices that can be used in the diagnosis or detection of diseases, conditions or infections, including, without limitation, the presence of certain chemicals, genetic information or other biomarkers. Predictive, prognostic and screening tests, such as carrier screening tests, can also be IVDs. A subset of IVDs is known as analyte-specific reagents ("ASRs"). ASRs consist of single reagents, and are intended for use in a diagnostic application for the identification and quantification of an individual chemical substance in biological specimens. ASRs are medical devices, but most are exempt from 510(k) review. As medical devices, ASRs have to comply with some QSR provisions and other device requirements, such as establishment registration, device listing and medical device reporting.
The FDC Act classifies medical devices into one of three categories based on the risks associated with the device and the level of control necessary to provide reasonable assurance of safety and effectiveness. Class I devices are deemed to be low risk and are subject to the fewest regulatory controls. Many Class I devices are exempt from FDA premarket review requirements. Class II devices, including some software products to the extent that they qualify as a device, are deemed to be moderate risk, and generally require clearance through the premarket notification, or 510(k) clearance, process in order to be commercially distributed. Class III devices are generally the highest risk devices and are subject to the highest level of regulatory control to provide reasonable assurance of the device's safety and effectiveness. Class III devices typically require approval of a PMA by the FDA before they are marketed. A clinical study is almost always required to support a PMA application and is sometimes required for 510(k) clearance. All clinical studies of investigational devices must be conducted in compliance with any applicable FDA and Institutional Review Board requirements. Devices that are exempt from FDA premarket review requirements must nonetheless comply with general post-market controls as described below, unless the FDA has chosen to exercise enforcement discretion and not regulate them.
510(k) clearance pathway. To obtain 510(k) clearance, a manufacturer must submit a premarket notification demonstrating to the FDA's satisfaction that the proposed device is substantially equivalent to a previously 510(k)-cleared device or a device that was in commercial distribution before May 28, 1976 for which the FDA has not yet called for submission of PMA applications. The previously cleared device is known as a predicate. The FDA's 510(k) clearance pathway usually takes from three to 12 months, but it can take longer, particularly for a novel type of product.
PMA pathway. The PMA pathway requires proof of the safety and effectiveness of the device to the FDA's satisfaction. The PMA pathway is costly, lengthy and uncertain. A PMA application must provide extensive preclinical and clinical trial data as well as information about the device and its components regarding, among other things, device design, manufacturing and labeling. As part of its PMA review process, the FDA will typically inspect the manufacturer's facilities for compliance with QSR requirements, which impose elaborate testing, control, documentation and other quality assurance procedures. The PMA review process typically takes one to three years but can take longer.
De novo pathway. If no predicate device can be identified, the product is automatically classified as Class III, requiring a PMA application. However, the FDA can reclassify, or use "de novo classification," for a device for which there was no predicate device if the device is low or moderate risk. The FDA will identify "special controls" that the manufacturer must implement, which often include labeling and other restrictions. Subsequent applicants can rely on the de novo product as a predicate for a 510(k) clearance. The de novo route is less burdensome than the PMA process. A device company can ask the FDA at the outset if the de novo route is available and submit the application as one requesting de novo classification. The de novo route has been used for many IVD products.
Post-market general controls. After a device, including a device exempt from FDA premarket review, is placed on the market, numerous regulatory requirements apply. These include the QSR,
99
labeling regulations, registration and listing, the Medical Device Reporting regulation (which requires that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur) and the Reports of Corrections and Removals regulation (which requires manufacturers to report recalls and field actions to the FDA if initiated to reduce a risk to health posed by the device or to remedy a violation of the FDC Act).
The FDA enforces these requirements by inspection and market surveillance. If the FDA finds a violation, it can institute a wide variety of enforcement actions, ranging from an untitled or public warning letter to more severe sanctions such as fines, injunctions and civil penalties; recall or seizure of products; operating restrictions and partial suspension or total shutdown of production; refusing requests for 510(k) clearance or PMA approval of new products; withdrawing 510(k) clearance or PMAs already granted; and criminal prosecution.
Research use only. Research use only ("RUO") products belong to a separate regulatory classification under a long-standing FDA regulation. RUO products are not regulated as medical devices and are therefore not subject to the regulatory requirements discussed above. The products must bear the statement: "For Research Use Only. Not for Use in Diagnostic Procedures." RUO products cannot make any claims related to safety, effectiveness or diagnostic utility, and they cannot be intended for human clinical diagnostic use. A product labeled RUO but intended to be used diagnostically may be viewed by the FDA as adulterated and misbranded under the FDC Act and is subject to FDA enforcement activities, including requiring the supplier to seek clearance or approval for the products. Our LDT uses instruments and reagents labeled as RUO in our laboratories.
Laboratory-developed tests. LDTs have generally been considered to be tests that are designed, developed, validated and used within a single laboratory. The FDA takes the position that it has the authority to regulate such tests as medical devices under the FDC Act. The FDA has historically exercised enforcement discretion and has not required clearance or approval of LDTs prior to marketing. In addition, New York CLEP separately approves certain LDTs offered to New York State patients. The Company is in the process of obtaining the requisite approvals for its LDTs in New York.
On October 3, 2014, the FDA issued two draft guidance documents regarding oversight of LDTs. These draft guidance documents proposed more active review of LDTs. The draft guidances have been the subject of considerable controversy, and in November 2016, the FDA announced that it would not be finalizing the 2014 draft guidance documents. On January 13, 2017, the FDA issued a discussion paper which laid out elements of a possible revised future LDT regulatory framework, but did not establish any regulatory requirements.
The FDA's efforts to regulate LDTs have prompted the drafting of legislation governing diagnostic products and services that sought to substantially revamp the regulation of both LDTs and IVDs. Congress may still act to provide further direction to the FDA on the regulation of LDTs.
We believe that the majority of the tests we currently offer meet the definition of LDTs, as they have been designed, developed and validated for use in a single CLIA-certified laboratory. If our tests are LDTs, they are currently not subject to FDA regulation as IVDs.
HIPAA and HITECH
Under the administrative simplification provisions of HIPAA, as amended by the HITECH Act, the United States Department of Health and Human Services issued regulations that establish uniform standards governing the conduct of certain electronic healthcare transactions and protecting the privacy and security of protected health information used or disclosed by healthcare providers and other covered entities. Three principal regulations with which we are required to comply have been issued in final form under HIPAA: privacy regulations, security regulations and standards for electronic
100
transactions, which establish standards for common healthcare transactions. The privacy and security regulations were extensively amended in 2013 to incorporate requirements from the HITECH Act.
The privacy regulations cover the use and disclosure of protected health information by healthcare providers and other covered entities. They also set forth certain rights that an individual has with respect to his or her protected health information maintained by a healthcare provider, including the right to access or amend certain records containing protected health information, or to request restrictions on the use or disclosure of protected health information. The security regulations establish requirements for safeguarding the confidentiality, integrity and availability of protected health information that is electronically transmitted or electronically stored. The HITECH Act, among other things, established certain protected health information security breach notification requirements. A covered entity must notify affected individual(s) and the United States Department of Health and Human Services when there is a breach of unsecured protected health information. The HITECH Act also strengthened the civil and criminal penalties that may be imposed against covered entities, business associates, and individuals, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys' fees and costs associated with pursuing federal civil actions. In addition, other federal and state laws may govern the privacy and security of health and other information in certain circumstances, many of which differ from each other in significant ways and may not be preempted by HIPAA, thus complicating compliance efforts. The HIPAA privacy and security regulations establish a uniform federal "floor" that healthcare providers must meet and do not supersede state laws that are more stringent or provide individuals with greater rights with respect to the privacy or security of, and access to, their records containing protected health information. Massachusetts, for example, has a state law that protects the privacy and security of personal information of Massachusetts residents that is more prescriptive than HIPAA.
These laws contain significant fines and other penalties for wrongful use or disclosure of protected health information. Additionally, to the extent that we submit electronic healthcare claims and payment transactions that do not comply with the electronic data transmission standards established under HIPAA and the HITECH Act, payments to us may be delayed or denied.
United States Federal and State Fraud and Abuse Laws
In the United States, there are various fraud and abuse laws with which we must comply and we are potentially subject to regulation by various federal, state and local authorities, including CMS, other divisions of the U.S. Department of Health and Human Services (e.g., the Office of Inspector General), the DOJ and individual U.S. Attorney offices within the DOJ, and state and local governments. We also may be subject to foreign fraud and abuse laws.
In the United States, the federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for patient referrals for, or purchasing, leasing, ordering or arranging for the purchase, lease or order of, any healthcare item or service reimbursable under a governmental payor program. Courts have stated that a financial arrangement may violate the Anti-Kickback Statute if any one purpose of the arrangement is to encourage patient referrals or other federal healthcare program business, regardless of whether there are other legitimate purposes for the arrangement. Violations may result in imprisonment, criminal fines, civil money penalties and exclusion from participation in federal healthcare programs. Many states also have anti-kickback statutes, some of which may apply to items or services reimbursed by any third-party payor, including commercial insurers.
In addition to the administrative simplification regulations discussed above, HIPAA also created two new federal crimes: healthcare fraud and false statements relating to healthcare matters. The healthcare fraud statute prohibits knowingly and willfully executing a scheme to defraud any healthcare
101
benefit program, including private payors. A violation of this statute is a felony and may result in fines, imprisonment or exclusion from governmental payor programs such as the Medicare and Medicaid programs. The false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact, or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. A violation of this statute is a felony and may result in fines, imprisonment or exclusion from governmental payor programs.
Finally, another development affecting the healthcare industry is the increased enforcement of the federal False Claims Act and, in particular, actions brought pursuant to the False Claims Act's "whistleblower" or qui tam provisions. The False Claims Act imposes liability on any person or entity that, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment by a federal governmental payor program. The qui tam provisions of the False Claims Act allow a private individual to bring actions on behalf of the federal government alleging that the defendant has defrauded the federal government by submitting a false claim to the federal government and permit such individuals to share in any amounts paid by the entity to the government in fines or settlement. When an entity is determined to have violated the False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties ranging from $11,181 to $22,363 for each false claim. These civil penalties are also adjusted for inflation periodically.
In addition, various states have enacted false claim laws analogous to the federal False Claims Act, although many of these state laws apply where a claim is submitted to any third-party payor and not merely a governmental payor program.
Physician Referral Prohibitions
Under a United States federal law directed at "self-referral," commonly known as the "Stark Law," there are prohibitions, with certain exceptions, on referrals for certain designated health services, including laboratory services, that are covered by the Medicare and Medicaid programs by physicians who personally, or through a family member, have an investment or ownership interest in, or a compensation arrangement with, an entity performing the tests. The prohibition also extends to payment for any testing referred in violation of the Stark Law. A person who engages in a scheme to circumvent the Stark Law's referral prohibition may be fined up to $100,000 for each such arrangement or scheme. In addition, any person who presents or causes to be presented a claim to the Medicare or Medicaid programs in violation of the Stark Law is subject to civil monetary penalties of up to $15,000 per bill submission, an assessment of up to three times the amount claimed and possible exclusion from participation in federal governmental payor programs. Bills submitted in violation of the Stark Law may not be paid by Medicare or Medicaid, and any person collecting any amounts with respect to any such prohibited bill is obligated to refund such amounts. Many states have comparable laws that are not limited to Medicare and Medicaid referrals.
Corporate Practice of Medicine
Approximately 30 states in the United States have enacted laws prohibiting business corporations, such as us, from practicing medicine and employing or engaging physicians to practice medicine, generally referred to as the prohibition against the corporate practice of medicine. These laws are designed to prevent interference in the medical decision-making process by anyone who is not a licensed physician. For example, California's Medical Board has indicated that determining what diagnostic tests are appropriate for a particular condition and taking responsibility for the ultimate overall care of the patient, including providing treatment options available to the patient, would constitute the unlicensed practice of medicine if performed by an unlicensed person. Violation of these corporate practice of medicine laws may result in civil or criminal fines, as well as sanctions imposed against us and/or the professional through licensure proceedings.
102
Other United States Regulatory Requirements
Our laboratories are subject to United States federal, state and local regulations relating to the handling and disposal of regulated medical waste, hazardous waste and biohazardous waste, including chemical, biological agents and compounds, blood samples and other human tissue. Typically, we use outside vendors who are contractually obligated to comply with applicable laws and regulations to dispose of such waste. These vendors are licensed or otherwise qualified to handle and dispose of such waste.
The U.S. Occupational Safety and Health Administration has established extensive requirements relating to workplace safety for healthcare employers, including requirements to develop and implement programs to protect workers from exposure to blood-borne pathogens by preventing or minimizing any exposure through needle stick or similar penetrating injuries.
The U.S. federal Physician Payments Sunshine Act requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children's Health Insurance Program, with specific exceptions, to annually report to CMS information related to payments or other transfers of value made to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members.
European Regulation
European sales of medical and diagnostic devices are subject to European regulations. The time required to obtain clearance or approval by a foreign country may be longer or shorter than that required for FDA clearance or approval, and the requirements may be different. Set forth below are highlights of the key European regulatory schemes applicable to our business.
European Conformity Marking ("CE Mark") and Certifications
The primary regulatory body in Europe is the European Commission, which has adopted numerous directives and has promulgated standards regulating the design, manufacture, clinical trials, labeling and adverse event reporting for medical and diagnostic devices. Devices that comply with the requirements of a relevant directive will be entitled to bear the CE Mark indicating that the device conforms to the essential requirements of the applicable directives and, accordingly, can be commercially distributed throughout the member states of the European Union, and other countries that comply with or mirror these directives. The method of assessing conformity varies depending on the type and class of the product, but normally involves a combination of self-assessment by the manufacturer and a third-party assessment by a notified body, an independent and neutral institution appointed by a country to conduct the conformity assessment. This third-party assessment may consist of an audit of the manufacturer's quality system, review of technical documentation and specific testing of the manufacturer's device. Such an assessment may be required in order for a manufacturer to commercially distribute the product throughout these countries. ISO 13485 certification is a voluntary standard. Quality systems that implement relevant harmonized standards establish the presumption of conformity with the essential requirements for a CE Mark. We have the authorization to affix the CE Mark to our test kit products including CentoCard, CentoNPC, CentoFarber, CentoMD, CentoGaucher, CentoFabry, and MyLSDApp and to commercialize our devices in the European Union. We currently are able to use CE labels on our CentoCard, CentoMD, CentoGaucher, CentoFabry and MyLSDApp products. The final form of the EU MDR, which will replace the EU MDD, was adopted on May 25, 2017 and will become applicable in its main part on May 26, 2020. Additionally, a new version of ISO 13485 was recently published, effective March 31, 2019. Diagnostic products which qualify as in vitro diagnostic medical devices would be subject to European Union legislation on medical devices, IVD-MDD and from May 26, 2022 IVD-MDR. According to IVD-MDD and IVD-MDR, marketing of in vitro diagnostic medical devices requires a CE mark.
103
Laboratory-Developed Tests
As currently a majority of our diagnostic testing is run at our laboratory in Rostock, Germany, the European Union and German legislation on in vitro diagnostic medical devices applies. According to the recitals of the IVD-MDD, reagents which are produced within "health-institution laboratories" for use in that environment and which are not subject to commercial transactions are not covered by the Directive. However, the legal framework for applying the exemption clauses for LDTs is not entirely clear as the IVD-MDD lacks an explicit definition and there is no related case law. As of May 2022, when the new IVD-MDR becomes applicable, the general safety and performance requirements set out in Annex I IVD-MDR are applicable also to devices manufactured and used only within health institutions. Overall the exemptions for LDTs will be narrowed, as even health institutions that use LDTs, among other institutions, will have to provide information upon request on the use of such devices to their relevant authorities and the particular health institution will have to draw up a declaration which it is required to make publicly available. If those conditions are not met and/or diagnostic tests are manufactured and used only within health institutions but not "on an industrial scale", such tests will qualify as IVDs with the IVD-MDR applying with full applicability. Additionally, U.S. regulation applies to our laboratory-developed tests (see "Regulation—Regulation States Regulation—Laboratory-developed tests" for more information).
General Data Protection Regulation
In May 2016, the European Union formally adopted the GDPR, which applied to all EU member states as of May 25, 2018 and replaced the EU Data Protection Directive. The GDPR imposes strict requirements on controllers and processors of personal data, including special protections for "sensitive information," which includes health and genetic information of data subjects residing in the European Union. The GDPR grants individuals the opportunity to object to the processing of their personal information, allows them to request deletion of personal information in certain circumstances, and provides an individual with an express right to seek legal remedies in the event the individual believes his or her rights have been violated. Further, the GDPR imposes strict rules on the transfer of personal data out of the European Union to the United States or other regions that have not been deemed to offer "adequate" privacy protections. It has increased our responsibility and liability in relation to personal data that we process and we may be required to put in place additional mechanisms ensuring compliance with the new EU data protection rules.
The GDPR is a complex law and the regulatory guidance is still evolving, including with respect to how the GDPR should be applied in the context of transactions from which we may gain access to personal data. Furthermore, many of the countries within the European Union are still in the process of drafting supplementary data protection legislation in key fields where the GDPR allows for national variation, including the fields of clinical study and other health-related information. There is still significant uncertainty related to the manner in which data protection authorities will seek to enforce compliance with GDPR in the medical and research fields. For example, it is not yet clear if such authorities will conduct random audits of companies subject to the GDPR or will only respond to complaints filed by individuals who claim their rights have been violated. Enforcement actions to date in other industries has resulted in significant fines and other penalties. Failure to comply with the requirements of the GDPR and the related national data protection laws of EU member states, which may deviate slightly from the GDPR, may result in material fines.
European Fraud and Abuse Laws
In Europe, various countries have adopted anti-bribery laws providing for severe consequences, in the form of criminal penalties and/or significant fines, for individuals and/or companies committing a bribery offense. Violations of these anti-bribery laws, or allegations of such violations, could have a negative impact on our business, results of operations and reputation. For instance, in the United
104
Kingdom, under the Bribery Act 2010, which went into effect in July 2011, a bribery occurs when a person offers, gives or promises to give a financial or other advantage to induce or reward another individual to improperly perform certain functions or activities, including any function of a public nature. Bribery of foreign public officials also falls within the scope of the Bribery Act 2010. Under the new regime, an individual found in violation of the Bribery Act 2010 faces imprisonment of up to 10 years. In addition, the individual can be subject to an unlimited fine, as can commercial organizations for failure to prevent bribery.
Competition
We believe we are the company offering the most comprehensive services to both diagnostics and pharmaceutical partners in the rare disease field, with highly curated data combining genetic, epidemiological and phenotypical information and proprietary biomarkers. Our principal competitors are existing mainstream diagnostics companies or companies specializing in certain rare diseases as well as cloud-based bioinformatic companies and entities that offer open source uncurated genetic databases. However, these companies do not offer curated information or as broad of a testing portfolio for rare diseases in as many geographical regions as we do. For example, we have found that the genetic mutation causing the same rare diseases and the phenotypical patterns may vary depending on the ethnicity of the patients, which we have identified based on our global data sets. Such unique insights may not be available to other companies that do not have the same global scope of patient data.
Our principal competitors in our diagnostics segment include mainstream diagnostic testing companies as well as labs or hospital conglomerates which offer the same services. In our pharmaceutical segment, our competitors include diagnostic testing companies and large pharmaceuticals.
With the continuous development in the NGS technology, the cost of genetic sequencing is anticipated to decrease and there may be companies intending to compete with us by performing massive sequencing at lower prices in order to obtain the relevant data to construct a similar database and repository. However, given the current limitations in the rare diseases fields, as well as the required quantity and quality of the data in order to make any relevant analysis, we are not aware of any competitors that will be able to build up to such scale in the near term.
Legal Proceedings
For more information, see "Item 8. Financial Information—A. Consolidated Statements and Other Financial Information—Legal Proceedings."
C. Organizational Structure
Our parent company is Centogene N.V. (the "Company"). Centogene B.V. was incorporated on October 11, 2018. In connection with our initial public offering which closed on November 12, 2019, we executed a corporate reorganization whereby Centogene B.V. was converted into Centogene N.V., and Centogene N.V. became the holding company for Centogene AG.
105
Our major subsidiaries are listed below.
| |
|
Equity interests (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| |
Country in which primary activities are pursued |
||||||||
Name
|
Dec 31, 2018 | Dec 31, 2019 | |||||||
Centogene AG |
Germany | 100 | 100 | ||||||
Centogene IP GmbH |
Germany | 100 | 100 | ||||||
Centogene Shared Service GmbH |
Germany | 100 | 100 | ||||||
Centogene Fzllc, Dubai |
United Arab Emirates | 100 | 100 | ||||||
Ludewig Wasserbau GmbH(1) |
Germany | 100 | — | ||||||
Centogene US LLC, Burlington, USA |
USA | 100 | 100 | ||||||
Centogene GmbH, Vienna |
Austria | 90 | 90 | ||||||
Centogene India Pvt. Ltd |
India | 51 | 51 | ||||||
LPC GmbH |
Germany | 51 | 51 | ||||||
- (1)
- The 100% interest in Ludewig Wasserbau GmbH was sold as part of the sale and leaseback transaction during the year ended December 31, 2019. See "Note 13.1—Sale and Leaseback transaction" to the consolidated financial statements for the year ended December 31, 2019 included elsewhere in this Annual Report.
D. Property, Plants and Equipment
Our headquarters are located in Rostock, Germany, where we occupy approximately 8,500 square meters of office and laboratory space that was originally constructed by us. In July 2019, Centogene AG entered into a sale and leaseback transaction, pursuant to which we sold our Rostock headquarters building to a third party for €24,000 thousand. We then leased the building from the third party for a period of 12 years at a fixed rate per month with the option to extend twice. In addition, a bank guarantee of €3,000 thousand (which we have secured by cash deposit of €1,500 thousand) is required to be maintained during the lease period. In February 2020, we entered into another lease contract for the further expansion of our Rostock headquarters. The new lease contract will cover a total area of approximately 2,850 square meters of offices, staff facilities and storage spaces, and will commence during the first quarter of 2023, when the building is expected to be completed by the lessor. The lease is charged at a fixed rate and covers a fixed period of eight years, with the option to extend twice. The lease cannot be terminated during the fixed eight-year period, but we are permitted to sub-lease to a third party.
In September 2018, we also opened an office and laboratory facility in Cambridge, Massachusetts. We rented the premises with a two-year lease covering approximately 168 square meters. In June 2019, we rented additional premises of approximately 194 square meters. The contract provides for, and we have exercised, an option to extend for a period of two years after the current term ends on June 30, 2020.
Both laboratories in Rostock, Germany and in Cambridge, Massachusetts, are equipped with the most advanced technologies for clinical diagnostics, clinical studies and research and development. We strive to follow the strictest quality criteria at all times and both laboratories are certified by the Centers for Medicare and Medicaid Services and accredited by the College of American Pathologists. To further enhance flexibility in capital management, we may purchase some of the leased laboratory equipment. These leases usually cover a period of two to four years, and our obligations under these leases are secured by the lessor's title to the leased assets.
In March 2020, we announced the commencement of testing for COVID-19. In order to increase the testing capacity, we acquired the laboratory facilities and equipment of a former cancer immunotherapy company and leased their former laboratory space in Hamburg, Germany. The lease is charged at a fixed rate and covers a fixed period of five years, with an option to extend. We do not
106
have the right to terminate the lease during the fixed five-year period, but we are permitted to sub-lease to a third party.
In addition to our laboratories, we have sales and administrative offices located in Berlin (Germany), Cambridge (Massachusetts, United States), Vienna (Austria), Dubai (United Arab Emirates) and Delhi (India), allowing us to further expand our international footprint. Considering the continuous expansion of our business, we relocated our office to Berlin, Germany in October 2019. The new office covers an area of approximately 1,770 square meters and was leased for a period of 12 years without an extension option. A rental deposit of EUR 257,000 is required to be maintained, and has been made by us, for the lease of the office in Berlin, Germany until the lease expires.
As of December 31, 2019, we employ over 440 highly qualified personnel (including consultants) from over 55 nationalities.
Item 4A. Unresolved Staff Comments
None
Item 5. Operating and Financial Review and Prospects
A. Operating Results
The following discussion of our financial condition and results of operations should be read in conjunction with Centogene's audited consolidated financial statements as of December 31, 2018 and 2019 and for the years ended December 31, 2017, 2018 and 2019 and the notes thereto, included elsewhere in this Annual Report as well as the information presented under "Item 3—A. Selected Financial Data." Financial information presented in the consolidated financial statements for periods prior to the completion of our corporate reorganization is that of Centogene AG, our wholly-owned subsidiary. The consolidated financial statements of Centogene N.V. are a continuation of the historical consolidated financial statements of Centogene AG. Centogene AG was acquired by Centogene B.V., which subsequently converted into Centogene N.V., on November 7, 2019 as part of our corporate reorganization. Centogene B.V. had no assets, liabilities or contingent liabilities until the completion of our corporate reorganization. Following the corporate reorganization, Centogene N.V. became the holding company of Centogene AG and the historical consolidated financial statements of Centogene AG included in this Annual Report became the historical consolidated financial statements of Centogene N.V. The following discussion is based on our financial information prepared in accordance with IFRS as issued by the IASB, which may differ in material respects from generally accepted accounting principles in the United States and other jurisdictions. The following discussion includes forward-looking statements that involve risks, uncertainties and assumptions. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of many factors, including but not limited to those described under "Item 3—D. Risk Factors" and elsewhere in this Annual Report.
Overview
We are a commercial-stage company focused on rare diseases that transforms real-world clinical and genetic data into actionable information for patients, physicians and pharmaceutical companies. Our goal is to bring rationality to treatment decisions and to accelerate the development of new orphan drugs by using our knowledge of the global rare disease market, including epidemiological and clinical data and innovative biomarkers. We have developed a global proprietary rare disease platform based on our real-world data repository with over 2.5 billion weighted data points from nearly 500,000 patients representing over 120 different countries as of December 31, 2019, or an average of over 590 data points per patient. Our platform includes epidemiologic, phenotypic and genetic data that reflects a global population, and also a biobank of these patients' blood samples. We believe this represents the only platform that comprehensively analyzes multi-level data to improve the understanding of rare
107
hereditary diseases, which can aid in the identification of patients and improve our pharmaceutical partners' ability to bring orphan drugs to the market.
We have identified two reportable segments:
- •
- Pharmaceutical. Our
pharmaceutical solutions provide a variety of services to our pharmaceutical partners, including target discovery, early patient recruitment and identification, epidemiological insights, biomarker
discovery and patient monitoring. Our information platforms, access to rare disease patients and their biomaterials, and ability to develop proprietary technologies and biomarkers enable us to provide
services to our pharmaceutical partners in all phases of the drug development process as well as post-commercialization. Revenues from our pharmaceutical segment are generated primarily from
collaboration agreements with our pharmaceutical partners. As of December 31, 2019, we collaborated with 39 pharmaceutical partners for over 45 different rare diseases. In addition, as of
December 31, 2019, we had 58 biomarker programs, of which 35 were used in connection with our pharmaceutical collaborations.
- •
- Diagnostics. Our diagnostics segment provides targeted genetic sequencing and diagnostics services to our clients worldwide, who are typically physicians, laboratories or hospitals, either directly or through distributors. As of December 31, 2019, we believe we offer the broadest diagnostic testing portfolio for rare diseases, covering over 7,500 genes using over 10,000 different tests. In turn, the data collected from our diagnostics services, together with the biomaterials, allow us to continue to grow our repository and our CentoMD database.
Our business has continuously seen notable expansion in recent years. In the year ended December 31, 2019, we received over 133,800 test requests, representing an approximate 27.1% increase as compared to approximately 105,300 test requests received during the year ended December 31, 2018, and an approximate 61.2% increase as compared to 83,000 test requests received during the year ended December 31, 2017. The increase in test requests received in the year ended December 31, 2018 compared the year ended December 31, 2017 was 26.9%.
Our revenue for the year ended December 31, 2019 was €48,780 thousand, an increase of €8,302 thousand, or 20.5%, from €40,478 thousand for the year ended December 31, 2018. Our revenue for the year ended December 31, 2018 was €40,478 thousand, an increase of €8,789 thousand, or 27.7%, from €31,689 thousand for the year ended December 31, 2017. Our pharmaceutical and diagnostics segments contributed 44.1% and 55.9%, respectively, of our total revenues for the year ended December 31, 2019, as compared to 42.8% and 57.2%, respectively, of our total revenues for the year ended December 31, 2018 and 44.0% and 56.0%, respectively, of our total revenues for the year ended December 31, 2017. Test requests received by our pharmaceutical and diagnostics segments for the year ended December 31, 2019 were approximately 70,600 and 51,600, respectively, representing increases of approximately 18.5% and 21.7%, respectively, as compared to approximately 59,600 and 42,400 test requests, respectively, received for the year ended December 31, 2018. Test requests received by our pharmaceutical and diagnostics segments for the year ended December 31, 2018 represented increases of approximately 5.5% and 88.4%, respectively, as compared to approximately 56,500 and 22,500 test requests, respectively, received for the year ended December 31, 2017.
Since the inception of our business, our research and development has been substantially devoted to our biomarkers and interpretation-based solutions. For the year ended December 31, 2019, we incurred research and development expenses of €9,590 thousand, an increase of €3,290 thousand, or 52.2%, from €6,300 thousand for the year ended December 31, 2018. Our research and development expenses for the year ended December 31, 2018 decreased by €96 thousand, or 1.5%, from €6,396 thousand for the year ended December 31, 2017. During the years ended December 31, 2019, 2018 and 2017, we received test requests of approximately 11,600, 3,300 and 4,000, respectively, for our internal research and development projects.
108
Our loss before taxes for the year ended December 31, 2019 was €20,697 thousand, an increase of €9,049 thousand, or 77.7%, from €11,648 thousand for the year ended December 31, 2018. Our loss before taxes for the year ended December 31, 2018 increased by €6,186 thousand, or 113.3%, from €5,462 thousand for the year ended December 31, 2017. Our loss before taxes for the year ended December 31, 2019 included real estate transfer taxes of €1,200 thousand related to the sale and leaseback transaction for our Rostock headquarter building and initial public offering expenses of €1,092 thousand. Our loss before taxes for the year ended December 31, 2019 also included share-based compensation expenses of €6,418 thousand, as compared to €5,521 thousand for the year ended December 31, 2018, and €894 thousand for the year ended December 31, 2017.
Financial Operations Overview
Revenue
Our revenue is principally derived from the provision of pharmaceutical solutions and diagnostic tests enabled by our knowledge and interpretation-based platform.
We expect our revenue to increase over time as we continue to expand our commercial efforts internationally with a focus on further growth in our pharmaceutical segment. As a result, we expect revenue from the pharmaceutical segment to increase as a proportion of total revenue over time. We expect revenue from our diagnostics segment to grow in absolute terms but decrease as a percentage of revenue as we focus on growth in our pharmaceutical segment.
Changes in revenue mix between our pharmaceutical and diagnostics segments can impact our results period over period. We typically incur lower costs for the provision of solutions in our pharmaceutical segment and therefore generate higher returns from our pharmaceutical segment contracts than from our diagnostics segment contracts. As a result, we anticipate our gross profit as a percentage of revenues to improve in the future.
Pharmaceutical
We generate revenue in our pharmaceutical segment from the solutions we provide to our pharmaceutical partners to accelerate their development of treatments for rare hereditary diseases. Our data-driven studies are not only able to provide valuable information for drug target discovery, but also allow a better and more targeted design of clinical trials afterwards. Our biomarkers can be used not only in effective identification of rare disease patients, but also used to demonstrate the efficacy of the drugs, perform longitudinal monitoring and titrate the dosage needed of individual rare disease patients. Our partnership agreements are structured on a fee per sample basis, milestone basis, fixed fee basis, royalty basis or a combination of these. We recognize our revenue from the rendering of solutions to our pharmaceutical partners as such service is performed, or upon the achievement of certain milestones if applicable to the partnership agreement.
During the year ended December 31, 2019, we entered into 10 new collaborations with nine new pharmaceutical partners, and 18 new collaborations with existing pharmaceutical partners, resulting in a total of 76 (active or completed) collaborations, covering 46 disorders.
109
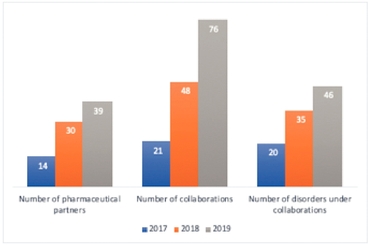
The timing of entry into new contracts with our pharmaceutical partners can be difficult to predict. Accordingly, we can experience different revenue patterns quarter-to-quarter and year-over-year due to the satisfaction of performance obligations involving significant upfront and milestone fees due from our pharmaceutical partners. We recognize revenue for upfront fees at a point in time when the right to use the intellectual property is transferred to the customer, while revenue for milestone payments is recognized over time using an input method based on the work rendered by us, or at a point in time when the applicable provisions for over-time recognition are not present (e.g., the sale of CentoCards).
During the year ended December 31, 2019, we entered into collaboration agreements with certain pharmaceutical partners, of which upfront fees of €1,930 thousand, representing the transaction prices allocated to the one-off transfer of our intellectual property, were recognized as revenues. For the year ended December 31, 2018, we entered into two collaboration agreements with Evotec and Denali. Under the terms of these collaboration agreements, we received upfront payments totaling €4,000 thousand in relation to the licensing by Evotec and Denali of certain of our intellectual property. We expect such fluctuations will increase as we expand our pharmaceutical segment.
Diagnostics
We generate revenue in our diagnostics segment primarily from genetic sequencing and diagnostics services, such as WES and WGS. The test requests received by our diagnostics segment for the years ended December 31, 2019, 2018 and 2017 were split amongst our primary testing products as follows:
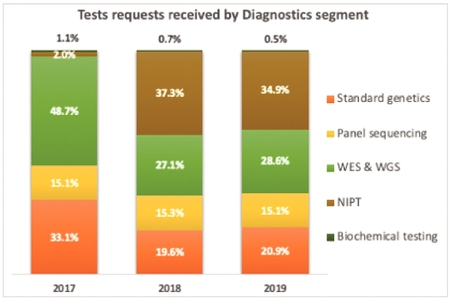
110
We provide these services in over 120 countries either through third party distributors or directly to our diagnostics clients, who are typically physicians, labs or hospital facilities. Revenues are based on a negotiated price per test or on the basis of agreements to provide certain testing volumes over defined periods. Revenue from the rendering of clinical diagnostic services (sequencing, interpretation and reporting) is recognized over time by reference to the percentage of completion of the service on the reporting date, assessed on the basis of the work rendered. We strategically focus on countries around the globe where the prevalence of rare hereditary diseases is high or the availability of national genetic testing and interpretation is to some extent limited and therefore the complete reimbursement or partial payment by the government for our services is more likely. The major markets for our diagnostics business currently include the Middle East and North Africa region, Scandinavia, parts of Central and Eastern Europe, Latin America, North America and parts of Asia. In most of our markets, our diagnostics tests are billable directly to the party submitting the request for a test to us and we have less than 1% bad debts written off since the inception of our business.
Cost of Sales and Operating Expenses
Our cost of sales and our operating expenses support all of the products and services that we provide to our customers and, as a result, are presented in an aggregate total for both business segments. We allocate certain overhead expenses, such as maintenance and depreciation to cost of sales and operating expense categories based on headcount and facility usage. As a result, overhead expense allocation is reflected in cost of sales and each operating expense category.
Cost of Sales
Cost of sales consists of cost of consumables, supplies and other direct costs such as personnel expenses, depreciation of laboratory equipment, amortization of biomarkers, repair and maintenance costs, shipping costs and certain allocated overhead expenses. The share-based compensation expenses included in cost of sales for the years ended December 31, 2019, 2018 and 2017 amounted to €1,153 thousand, €646 thousand and €nil, respectively, mainly related to options granted to an employee.
We expect these costs in absolute terms will increase as we grow our revenue but decrease as a percentage of revenue over time as our pharmaceutical segment revenue increases and as we continue to implement operational efficiencies. During the year ended December 31, 2019, our cost of sales represented 53.3% of our total revenue, as compared to 49.3% for the year ended December 31, 2018, and 47.1% for the year ended December 31, 2017.
Research and Development Expenses
Our research and development ("R&D") expenses consist primarily of costs incurred for the development of new products and solutions, in particular our biomarkers, and the development of our IT driven and interpretation-based solutions, including our CentoMD database. In the three fiscal years ended December 31, 2019, we spent €35,096 thousand on research and development, of which €12,810 thousand was capitalized as intangible assets.
Expenses for research activities are recognized through profit or loss in the period in which they are incurred, unless they reach the development stage and prove to be technically and commercially feasible, upon which the expenses are capitalized. With respect to biomarkers, expenses are capitalized when the target validation process is completed and commercialization is probable. With respect to IT driven solutions, expenses are capitalized upon the completion of our internal validation test. Before such dates, any development costs are recognized in profit or loss.
Research and development which we conduct pursuant to our pharmaceutical partnership agreements is typically limited to a specified rare disease. As a result, our research and development
111
expenses may vary substantially from period to period based on the timing of our research and development activities or our pharmaceutical partners, including due to the entry into, renegotiation of or termination of our partnership agreements. Our research and development expenses may also be impacted by changes in regulatory requirements and healthcare policies globally, particularly in respect of the validation and patent application processes that we conduct for our biomarkers.
During the year ended December 31, 2019, our research and development expenses represented 19.7% of our total revenue, as compared to 15.6% for the year ended December 31, 2018, and 20.2% for the year ended December 31, 2017. We expect that our overall research and development expenses will increase in absolute terms as we continue to innovate our information platform, develop additional products and solutions and expand our data management resources.
General Administrative Expenses
Our general administrative expenses include costs for our personnel, premises, IT operations, accounting and finance, legal and human resources functions. These expenses consist principally of salaries, bonuses, employee benefits, travel, and share-based compensation, as well as professional services fees such as consulting, audit, tax and legal fees and general corporate costs and allocated overhead expenses. We account for all general administrative expenses as incurred.
During the year ended December 31, 2019, our general administrative expenses represented 47.5% of our total revenue, as compared to 46.0% for the year ended December 31, 2018, and 30.0% for the year ended December 31, 2017. The share-based compensation expenses included in general administrative expenses for the years ended December 31, 2019, 2018 and 2017 amounted to €5,265 thousand, €4,875 thousand and €894 thousand, respectively. As a result of our continued international growth, including the expansion of our laboratory in Rostock, Germany and the opening of our new laboratory in Cambridge, Massachusetts in October 2018, we expect our general administrative costs to increase relative to prior periods. We also expect that our general administrative expenses will increase due to the costs of operating as a public company, such as additional legal, accounting, corporate governance and investor relations expenses, and higher directors' and officers' insurance premiums.
Selling Expenses
Our selling expenses consist of costs from our sales organization, which includes our direct sales force and sales management, client services, distributor relations, marketing and business development personnel. These expenses primarily include salaries, commissions, bonuses, employee benefits and travel, as well as marketing and educational activities and allocated overhead expenses. We expense all sales and marketing costs as incurred.
During the year ended December 31, 2019, selling expenses accounted for 19.0% of our total revenue, as compared to 18.5% for the year ended December 31, 2018, and 18.6% for the year ended December 31, 2017. We expect that our selling expenses will continue to grow as we continue to increase our business footprint and expand our business development efforts in our pharmaceutical segment.
Other Operating Income / (Expenses)
Other operating income primarily includes government grants, gain on disposal of property, plant and equipment and exchange rate gains.
Other operating expenses include currency losses, expected credit loss allowances on trade receivables and loss on the sale of property, plant and equipment, among others.
112
Government grants contain performance-based grants to subsidize research, development and innovation in the state of Mecklenburg-Western Pomerania from funds granted by the European Regional Development Fund ("R&D Grants"). Furthermore, government grants contain investment grants related to the construction of our new headquarters in Rostock, Germany in prior years and purchase of equipment for laboratory automization ("Investment Grants"). R&D Grants that compensate for our research and development expenses are recognized directly in profit or loss, while R&D Grants relating to an asset and Investment Grants are initially recognized as deferred income and subsequently released to profit or loss on a systematic basis over the useful life of the related asset. We received different government grants in the state of Mecklenburg-Western Pomerania from funds granted by the European Regional Development Fund to subsidize our research, development and innovation.
During the year ended December 31, 2019, we received R&D Grants and Investment Grants of €1,463 thousand and €792 thousand, respectively, as compared to €378 thousand and €3.0 million, respectively, for the year ended December 31, 2018, as well as €224 thousand and €6.8 million, respectively, for the year ended December 31, 2017. The government grants, which we receive, can fluctuate from period to period.
Results of Operations
Year Ended December 31, 2018 Compared to Year Ended December 31, 2019
| |
For the Years Ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2018 | 2019 | |||||
| |
(€ in thousands) |
||||||
Consolidated statement of comprehensive loss: |
|||||||
Revenue |
40,478 | 48,780 | |||||
Cost of sales |
19,941 | 26,005 | |||||
| | | | | | | | |
Gross profit |
20,537 | 22,775 | |||||
| | | | | | | | |
Research and development expenses |
6,300 | 9,590 | |||||
General administrative expenses |
18,610 | 23,160 | |||||
Selling expenses |
7,474 | 9,254 | |||||
Other operating income |
2,306 | 3,781 | |||||
Other operating expenses |
1,065 | 2,036 | |||||
| | | | | | | | |
Real estate transfer tax expenses |
— | 1,200 | |||||
| | | | | | | | |
Operating loss |
(10,606 | ) | (18,684 | ) | |||
| | | | | | | | |
Interest and similar income |
33 | 16 | |||||
Interest and similar expenses |
1,075 | 2,029 | |||||
| | | | | | | | |
Finance costs, net |
(1,042 | ) | (2,013 | ) | |||
| | | | | | | | |
Loss before taxes |
(11,648 | ) | (20,697 | ) | |||
Income tax expenses/(benefits) |
(310 | ) | 158 | ||||
| | | | | | | | |
Loss for the period |
(11,338 | ) | (20,855 | ) | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Other comprehensive income/(loss) |
(8 | ) | 16 | ||||
| | | | | | | | |
Total comprehensive loss for the period |
(11,346 | ) | (20,839 | ) | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Revenue
Revenue increased by €8,302 thousand, or 20.5%, to €48,780 thousand for the year ended December 31, 2019 from €40,478 thousand for the year ended December 31, 2018, mainly driven by the
113
continuous robust growth in our pharmaceutical segment (an increase in revenues of 24.4% for the year ended December 31, 2019), backed by our knowledge platform and repository which also grew swiftly. The diagnostics segment also demonstrated strong performance with an increase in revenues of 17.6% for the year ended December 31, 2019, which not only contributed to the growth of the revenues in 2019, but also supported the continuous expansion of the repository and helped to enrich our medical and genetic knowledge of rare genetic diseases.
The breakdown of our revenue by segment was as follows:
| |
For the Years Ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2018 | 2019 | |||||
| |
(€ in thousands) |
||||||
Revenue by segment: |
|||||||
Pharmaceutical |
17,307 | 21,522 | |||||
Diagnostics |
23,171 | 27,258 | |||||
| | | | | | | | |
Total Revenue |
40,478 | 48,780 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Revenues from our pharmaceutical segment were €21,522 thousand for the year ended December 31, 2019, an increase of €4,215 thousand, or 24.4%, from €17,307 thousand for the year ended December 31, 2019. This increase was primarily attributable to new pharmaceutical partnerships. As of December 31, 2019, we collaborated with 39 pharmaceutical partners, as compared to over 30 pharmaceutical partners as of December 31, 2018.
During the year ended December 31, 2019, we entered into 28 new collaborations, increasing the total number of active/completed collaborations to 76, from 48 active/completed collaborations as of December 31, 2018. Revenues from our new collaborations totalled €6,995 thousand for the year ended December 31, 2019, of which €1,930 thousand were upfront fees arising from several collaborations with two of our pharmaceutical partners. Such upfront fees were recognized as revenues as they represent the transaction price allocated to the one-off transfer of our intellectual property - epidemiological insights of relevant rare diseases and relevant data. During the year ended December 31, 2018, we received upfront payments totaling €4,000 thousand related to certain of our intellectual property from our collaborations with Evotec and Denali. These upfront fees were recognized as revenues as they represented the transaction price to be allocated to the grant of licenses, which are distinct and allow for use of such intellectual property for an unlimited period or for the time specified in the agreements.
We have been successful in entering into collaborations with pharmaceutical partners in the early stages of drug development, which puts us in a position to provide more support to the development process and increases our potential to secure further collaborations for the same drugs, such as biomarker developments. The graphic below shows our revenues for the year ended December 31, 2018 and 2019 resulting from our collaborations with our pharmaceutical partners split between the six drug development stages. For further details on each of our drug development stages, please see "Item 4. Information on the Company—B. Business Overview—Pharmaceutical Solutions."
114
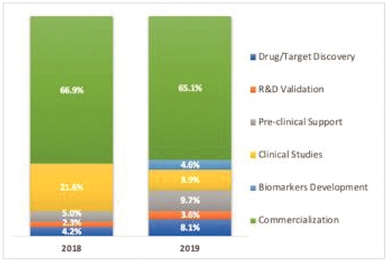
During the year ended December 31, 2019, revenues from one pharmaceutical partner represented 24.3% of our total revenues, as compared to 27.3% for the year ended December 31, 2018.
Diagnostics segment
Revenues from our diagnostics segment were €27,258 thousand for the year ended December 31, 2019, an increase of €4,087 thousand, or 17.6%, from €23,171 thousand for the year ended December 31, 2018. We received approximately 51,600 test requests in our diagnostics segment during the year ended December 31, 2019, representing an increase of approximately 21.7% as compared to approximately 42,400 test requests received for the year ended December 31, 2018.
For the years ended December 31, 2018 and 2019, our total diagnostic segment revenues were split amongst our primary testing products as follows:
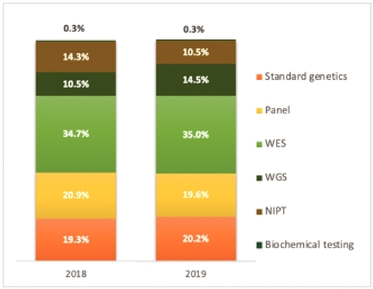
The increase in revenues was primarily driven by our WES, WGS as well as standard genetic testing, which are driven by an increase in test requests during the year ended December 31, 2019. Total revenues from WES, WGS and standard genetic testings for the year ended December 31, 2019 amounted to €18,989 thousand, representing an increase of 27.0% as compared to €14,950 thousand for the year ended December 31, 2018. The total number of WES, WGS and standard genetics test requests received in the diagnostics segment for the year ended December 31, 2019 was approximately
115
25,600, representing an increase of 29.6% as compared to approximately 19,760 test requests received for the year ended December 31, 2018.
The breakdown of our revenue from both of our segments, in the aggregate, by geographical region was as follows:
| |
For the Years Ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2018 | 2019 | |||||
| |
(€ in thousands) |
||||||
Revenue by geographical region: |
|||||||
Europe |
6,850 | 7,447 | |||||
of which: Germany |
1,061 | 508 | |||||
of which: Netherlands |
— | 25 | |||||
Middle East |
12,401 | 14,099 | |||||
of which: Saudi Arabia |
5,475 | 7,417 | |||||
North America |
18,113 | 23,276 | |||||
of which: United States |
17,296 | 22,778 | |||||
Latin America |
2,185 | 2,987 | |||||
Asia Pacific |
929 | 971 | |||||
| | | | | | | | |
Total Revenue |
40,478 | 48,780 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
In cases where our pharmaceutical partners are developing a new rare disease treatment, we generally anticipate that the final approved treatment will be made available globally. As a result, we allocate the revenues of our pharmaceutical segment by geographical region by reference to the location where each pharmaceutical partner mainly operates, which is based on the region from which most of their revenues are generated. The allocation of revenues in our diagnostics segment is based on the location of each customer. Our North America region contributed €23,276 thousand to revenue for the year ended December 31, 2019, an increase of €5,163 thousand, or 28.5%, from €18,113 thousand for the year ended December 31, 2018, primarily driven by the increase in revenues from our pharmaceutical segment, of which over 95% are allocated to the North America region. Revenues from the North America region represented 47.7% of our total revenues for the year ended December 31, 2019 as compared to 44.7% for the year ended December 31, 2018.
Our Middle East region contributed €14,099 thousand to revenue for the year ended December 31, 2019, an increase of €1,698 thousand, or 13.7%, from €12,401 thousand for the year ended December 31, 2018. This revenue growth was primarily attributable to the increase in sales of WGS and standard genetic testing in our diagnostics segment, due to successful account management with major hospitals under the Ministry of Health, in particular in Saudi Arabia and the United Arab Emirates, as well as a further expansion of our footprint by our internal sales force and through our distributors. The increase is partially offset by the decrease in sales of NIPT tests after we cancelled our fixed fee contract in September 2019.
Our Europe region contributed €7,447 thousand to revenue for the year ended December 31, 2019, an increase of €597 thousand, or 8.7%, from €6,850 thousand for the year ended December 31, 2018, primarily driven by an increase in sales of WES in our diagnostics segment.
Cost of Sales
Cost of sales increased by €6,064 thousand, or 30.4%, to €26,005 thousand for the year ended December 31, 2019, from €19,941 thousand for the year ended December 31, 2018. Cost of sales for the year ended December 31, 2019 represented 53.3% of total revenue, an increase of 4.0 percentage points as compared to 49.3% for the year ended December 31, 2019.
116
Cost of sales incurred by our pharmaceutical and diagnostics segments for the year ended December 31, 2019 represented 27.8% and 73.5% of revenues from the respective segments, an increase of 6.3 percentage points and 3.7 percentage points, respectively, as compared to 21.5% and 69.8%, respectively, for the year ended December 31, 2018. The 6.3 percentage point increase for our pharmaceutical segment was mainly due to higher upfront fee revenues recognized for the year ended December 31, 2018, of which no significant additional costs were incurred. If such impact had been excluded, the cost of sales for the pharmaceutical segment would have increased by only 2.5 percentage points, mainly driven by additional direct costs, such as personnel and travelling expenses incurred for the pre-clinical support and clinical study related collaborations. The 3.6 percentage point increase for the diagnostics segment was the result of increased operating costs caused by the expansion of the laboratory operations in both Rostock, Germany, and Cambridge, Massachusetts. Certain share-based compensation amounting to €1,153 thousand for the year ended December 31, 2019 (as compared to €646 thousand for the year ended December 31, 2018) related to options granted to the management member overseeing the diagnostic process which also contributed to the increase.
Gross Profit
As a result of these and other factors, our gross profit increased by €2,238 thousand, or 10.9%, to €22,775 thousand for the year ended December 31, 2019, from €20,537 thousand for the year ended December 31, 2018.
Research and Development Expenses
The table below gives a breakdown of our research and development expenses for the years ended December 31, 2019 and 2018.
| |
For the Years Ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2018 | 2019 | |||||
| |
(€ in thousands) |
||||||
Wages and salaries and social security expenses |
2,559 | 2,806 | |||||
Laboratory supplies and consumable costs |
212 | 2,221 | |||||
IT development costs |
1,933 | 2,963 | |||||
Depreciation and amortization expenses |
736 | 1,266 | |||||
Others |
860 | 334 | |||||
| | | | | | | | |
Total research and development expenses |
6,300 | 9,590 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Research and development expenses increased by €3,290 thousand, or 52.2%, to €9,590 thousand for the year ended December 31, 2019, from €6,300 thousand for the year ended December 31, 2018. This mainly represents personnel costs, consumable costs and IT-related expenses incurred in our research that do not qualify for capitalization. This also includes software and hardware costs, consultation and legal expenses and depreciation of equipment.
117
General Administrative Expenses
The table below gives a breakdown of our general administrative expenses for the years ended December 31, 2019 and 2018.
| |
For the Years Ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2018 | 2019 | |||||
| |
(€ in thousands) |
||||||
Wages and salaries, social security and termination expenses |
6,017 | 6,788 | |||||
Share-based payment expenses |
4,875 | 5,256 | |||||
Legal and consulting expenses |
707 | 1,446 | |||||
Travelling, corporate communication and event expenses |
1,224 | 2,165 | |||||
IT operational costs |
649 | 1,010 | |||||
Insurance premiums |
138 | 963 | |||||
Depreciation and amortization expenses |
1,379 | 1,973 | |||||
Others |
3,621 | 3,559 | |||||
| | | | | | | | |
Total general administrative expenses |
18,610 | 23,160 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
General administrative expenses increased by €4,550 thousand, or 24.4%, to €23,160 thousand for the year ended December 31, 2019, from €18,610 thousand for the year ended December 31, 2018, principally due to an increase in personnel costs and operating expenses as a result of the expansion of the business. The increase is also the result of additional investments in IT infrastructure and data security. The general administrative expenses included share-based compensation expenses of €5,265 thousand for the year ended December 31, 2019, an increase of €390 thousand as compared to €4,875 thousand for the year ended December 31, 2018.
Selling Expenses
Selling expenses increased by €1,780 thousand, or 23.8%, to €9,254 thousand for the year ended December 31, 2019, from €7,474 thousand for the year ended December 31, 2018, principally due to the expansion of our business development team for the pharmaceutical segment, as well as additional marketing expenses.
Other Operating Income / (Expenses)
Other operating income increased by €1,475 thousand, or 64.0%, to €3,781 thousand or the year ended December 31, 2019, from €2,306 thousand for the year ended December 31, 2018, principally due to an increase in recognition of grant income, as well as the gain of €532 thousand from the sale and leaseback transaction related to our Rostock headquarters building.
Other operating expenses increased by €971 thousand, or 91.2%, to €2,036 thousand for the year ended December 31, 2019, from €1,065 thousand for the year ended December 31, 2018, principally due to expenses of €1,092 thousand incurred in connection with our initial public offering which cannot be offset with the proceeds of such offering.
Real estate transfer tax
In June 2019, we sold our Rostock headquarters building, which had a carrying value of €22,778 thousand, to a subsidiary in preparation of the sale and leaseback transaction. As the sale and leaseback transaction with a third party was entered into in July 2019, the intercompany transaction was irreversible and a real estate transfer tax expense of €1,200 thousand related to the intercompany transaction was recognized accordingly.
118
Interest and Similar Income / (Expenses)
Interest and similar income decreased by €17 thousand to €16 thousand for the year ended December 31, 2019, from €33 thousand for the year ended December 31, 2018.
Interest and similar expenses increased by €954 thousand, or 88.7%, to €2,029 thousand for the year ended December 31, 2019, from €1,075 thousand for the year ended December 31, 2018. Interest and similar expenses for the year ended December 31, 2019 included additional interest of €1,159 thousand, resulting from the early repayment of loans related to the construction of the Rostock headquarters with the consideration received from the sale and leaseback transaction.
Loss Before Taxes for the Year
As a result of the factors described above, our loss before taxes for the year ended December 31, 2019 was €20,697 thousand, an increase of €9,049 thousand, or 77.7%, from €11,648 thousand for the year ended December 31, 2018.
Segment Adjusted EBITDA
Our Segment Adjusted EBITDA was as follows:
| |
For the Years Ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2018 | 2019 | |||||
| |
(€ in thousands) |
||||||
Segment Adjusted EBITDA: |
|||||||
Pharmaceutical |
13,641 | 14,956 | |||||
Diagnostics |
2,285 | 2,306 | |||||
| | | | | | | | |
|
15,926 | 17,262 | |||||
| | | | | | | | |
Adjusted EBITDA from our pharmaceutical segment was €14,956 thousand for the year ended December 31, 2019, an increase of €1,315 thousand, or 9.6%, from €13,641 thousand for the year ended December 31, 2018. This increase was primarily attributable to increase in revenues during the year, partially offset by the increase in cost of sales.
Adjusted EBITDA from our diagnostics segment was €2,306 thousand for the year ended December 31, 2019, which remained largely unchanged when compared to €2,285 thousand for the year ended December 31, 2018. The increase in revenues during the year was offset by the increase in cost of sales in 2019, as a result of increase in direct personnel costs and consumable costs.
For the discussion of our results of operations for the year ended December 31, 2017 compared to year ended December 31, 2018, see "Management's Discussion and Analysis of Financial Condition and Results of Operations—Overview—Results of Operations—Year Ended December 31, 2017 Compared to Year Ended 31, 2018" included in our F-1/A registration statement (File No. 333-234177) filed with the SEC on October 28, 2019.
B. Liquidity and Capital Resources
Overview
Our cash requirements are principally for working capital and capital expenditures, including expansions and improvements to our laboratory facilities, technology infrastructure and research and development activities. In fiscal year 2020 and beyond, we anticipate that our capital expenditures will increase from prior periods as we continue to increase our research and development efforts. Historically, our main source of liquidity has been our secured loans, municipal loans and government
119
funding of research programs, proceeds from our initial public offering, our shareholders and the private financings in 2017 and 2018.
Our financial condition and liquidity are and will continue to be influenced by a variety of factors, including our ability to continue to generate cash flows from our operations, our capital expenditure requirements and changes in exchange rates which will impact our generation of cash flows from operations when measured in euros.
Our known material liquidity needs for periods beyond the next twelve months are described below in "—F. Tabular Disclosure of Contractual Obligations." We believe cash generated from our operations, cash equivalents and financial instruments, together with government funding of research programs will be sufficient to fund our operations for at least 12 months.
Comparative Cash Flows
Comparison of the Year Ended December 31, 2018 and 2019
The following table sets forth our cash flows for the periods indicated:
| |
For the Years Ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2018 | 2019 | |||||
| |
(€ in thousands) |
||||||
Consolidated statement of cash flows: |
|||||||
Cash flow used in operating activities |
(4,577 | ) | (7,775 | ) | |||
Cash flow (used in)/from investing activities |
(8,694 | ) | 14,175 | ||||
Cash flow from financing activities |
19,336 | 25,473 | |||||
| | | | | | | | |
Net increase in cash and cash equivalents |
6,065 | 31,873 | |||||
| | | | | | | | |
Cash and cash equivalents at the beginning of the period |
3,157 | 9,222 | |||||
| | | | | | | | |
Cash and cash equivalents at the end of the period |
9,222 | 41,095 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Operating Activities
Our cash flow used in operating activities primarily relates to changes in the components of our working capital, including cash received from our pharmaceutical partners and diagnostics clients, and payments made to our suppliers.
For the year ended December 31, 2019, cash used in operating activities was €7,775 thousand, an increase of €3,198 thousand as compared to €4,577 thousand for the year ended December 31, 2018. This change was principally due to an increase in trade receivables from our diagnostics clients, particularly for those in the Middle East region that typically require a longer period to settle the trade receivables. The change was also the result of an increase in trade receivables from pharmaceutical partners which were not yet due as of December 31, 2019.
Investing Activities
Our cash flow used in/from investing activities for the year ended December 31, 2019 consists of investments in intangible assets, plant, property and equipment and right-of-use assets, grants received for investments in property, plant and equipment and cash received from disposals of property, plant and equipment.
For the year ended December 31, 2019, cash flow from investing activities was €14,175 thousand, as compared to €8,694 thousand used in investing activities for the year ended December 31, 2018. Cash used in investment activities included mainly investment, incurred in the development of new
120
products and solutions, in particular our biomarkers, and the development of our IT driven and interpretation-based solutions, including our CentoMD database. It also includes investment in property, plant and equipment used in the laboratories and other business operations. The cash flow from investing activities for the year ended December 31, 2019 included consideration received from the disposal of our Rostock headquarters building as part of the sale and leaseback transaction of €24,000 thousand, offset by €1,200 thousand for the real estate transfer tax and €1,500 thousand used to secure a bank guarantee for the lease. The investing cash flow for the year ended December 31, 2018 also included investment related to the development of the Rostock headquarters building, which was completed in early 2018.
Financing Activities
Our cash flow provided by financing activities for the year ended December 31, 2019 consists of proceeds of our initial public offering after deducting the underwriting commission and transaction costs, an overdraft facility drawn during the year, net of repayment of secured bank loans related to the construction of our new facility in Rostock, repayment of lease liabilities and interest expenses.
For the year ended December 31, 2019, cash generated from financing activities was €25,473 thousand, an increase of €6,137 thousand as compared to €19,336 thousand for the year ended December 31, 2018.
On November 7, 2019, we offered and sold a total of 4,000,000 of our common shares, €0.12 nominal value per share, at a public offering price of $14.00 (€12.58) per share, raising aggregate net offering proceeds of €41,899 thousand, after deduction of underwriting discounts and commissions as well as transaction costs.
In 2018, we received an investment of €20,000 thousand (€19,974 thousand net of bank charges) from certain of the investors that had participated in our external private financing in 2017 (for further information, see "Item 7. Major Shareholders and Related Party Transactions—Related Party Transactions—Investment and Shareholders Agreement").
For the discussion of our cash flows for the year ended December 31, 2017 compared to year ended December 31, 2018, please see "Management's Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources—Comparative Cash Flows—Comparison of the Year Ended December 31, 2017 and 2018" included in our F-1/A registration statement (File No. 333-234177) filed with the SEC on October 28, 2019.
Indebtedness
Syndicated Loan Facility
On August 4, 2015, we entered into a loan agreement (as amended or supplemented to date, the "Syndicated Loan Facility") with certain German commercial banks. The Syndicated Loan Facility consists of four tranches. As of December 31, 2019, we had €3,930 thousand outstanding under the Syndicated Loan Facility, of which €1,770 thousand was outstanding under Tranche B and €2,160 thousand was outstanding under the Tranche D Loan. As of December 31, 2018, we had €15,757 thousand outstanding under the Syndicated Loan Facility, of which €13,842 thousand was outstanding under Tranche A and Tranche B and €1,915 thousand was outstanding under the Tranche D Loan. As of December 31, 2019 and 2018, there were no balances outstanding under Tranche C.
The Syndicated Loan Facility consists of:
- •
- a Tranche A loan in an aggregate principal amount of €12,000 thousand, which is subdivided into a Tranche A1 loan and a Tranche A2 loan that are scheduled to mature on June 30, 2030 (the "Tranche A Loans"), and bear interest at a fixed rate of 2.5% per annum until June 30,
121
- •
- a Tranche B loan in an aggregate principal amount of €5,410 thousand that is scheduled to mature on
December 30, 2022 (the "Tranche B Loan") and bears interest at a floating rate of EURIBOR plus a margin of 2.95% per annum. Since December 2019, we have pledged
€1,500 thousand in cash in connection with amounts outstanding thereunder;
- •
- a Tranche C loan in an aggregate principal amount of up to €2,500 thousand as overdraft facility that matured and
was fully repaid on June 30, 2018 (the "Tranche C Loan") and bore interest at a floating rate of 6.25% per annum (adjusted in line with the respective Deutsche Bundesbank reference
interest rate). The Company repaid all outstanding amounts under the Tranche C Loan in June 2018; and
- •
- a Tranche D loan with an aggregate principal amount of up to €2,500 thousand as overdraft facility (the "Tranche D Loan") and bears interest at EURIBOR plus a margin of 3.5% per annum. Pursuant to a cash pledge that we entered into in January 2018 with the lenders under the Tranche D loan, as of July 1, 2019, we had pledged €2,500 thousand in cash in connection with amounts outstanding thereunder.
2025, thereafter to be amended in consideration of the development of the capital markets as well as of our financial situation and the value of the collateral. The Company repaid the entire amount outstanding (€10,776 thousand) under the Tranche A Loan in September 2019;
The Tranche A Loans were granted to finance the development of our laboratory in Rostock. This includes financing the acquisition of land, construction of the building and purchase of laboratory equipment. The Tranche B Loan is used to purchase laboratory equipment on a pro rata basis. In addition, it serves to refinance rental purchases for short-term investments in laboratory equipment and IT equipment. The Tranche C Loan is used for advance and interim financing of investment grants. The Tranche D Loan serves us as a working capital line and for the repayment of certain facilities with Commerzbank AG in an aggregate amount of € 2,500 thousand.
As of December 31, 2018, the Syndicated Loan Facility was secured by assignments of certain laboratory equipment, by global assignments of our trade and other receivables and by pledge of a bank account with OstseeSparkasse Rostock. The Syndicated Loan Facility also contained certain financial covenants and other provisions which impose restrictions on the way we operate our business. In addition, our CEO, Prof. Arndt Rolfs, must obtain the consent of the lenders prior to the sale of more than 10% of his shares in our company.
In prior years, the financial covenants of the Syndicated Loan Facility were not met by us and we obtained waivers from the various lenders under this facility for the year ended December 31, 2018. During the year ended December 31, 2019, after Tranche A loan was fully repaid following our sale and leaseback transaction, the terms of the Syndicated Loan Facility were renegotiated with the relevant banks and €1,500 thousand in cash was arranged and pledged for the Tranche B Loan. In exchange, the assignment of assets and receivables was released, and the requirement of compliance with covenants and restrictions were also removed. As of December 31, 2019, there was no requirement to comply with the covenants and no liens on assets, except for cash deposits pledged, remaining under the Syndicated Loan Facility.
Revolving Credit Agreements
We have entered into two further secured bank overdraft agreements totaling €1,500 thousand which we use to finance our day-to-day business operations. €476 thousand was utilized as of December 31, 2019 and none was utilized as of December 31, 2018.
- •
- Our €1,000 thousand revolving credit agreement has an initial floating interest rate of 3.85% (adjusted on EURIBOR) when utilized as an overdraft facility. It is partially secured by separate guarantees provided by a German development bank in an amount up to €210,000 (guarantee
122
- •
- Our €500 thousand revolving credit agreement has an initial floating interest rate of 4.5% per annum, an up-front fee of 0.25% per annum and is secured by two guarantees of up to €250 thousand. Prof. Arndt Rolfs and Christoph Ehlers are guarantors pursuant to the revolving credit agreement. In case there is a change in our shareholder structure, the lender is entitled to terminate the revolving credit agreement if we are unable to agree with the lender on the continuation of the loan under amended terms. Subsequent to the year ended December 31, 2019, we pledged €500 thousand in cash to secure the amounts outstanding under the revolving credit agreement and as a result the guarantees as well as the restriction on the change in our shareholder structure were removed.
fee of 1.25% per annum) and, in an amount of €100,000 each, by our CEO Prof. Arndt Rolfs, and Christoph Ehlers. In case there is a change in our shareholder structure, the lender is entitled to request further collateral from us. Subsequent to the year ended December 31, 2019, the revolving credit facility was reduced to €500 thousand and the guarantees as well as the restriction on the change in our shareholder structure were removed.
Municipal Loans
We entered into four financings, structured as silent participation agreements, with Mittelständische Beteiligungsgesellschaft Mecklenburg-Vorpommern mbH ("MBMV") (the "Municipal Loans"), pursuant to which MBMV participates in the Company as a silent partner on the following material terms:
The silent partnership agreement dated May 18, 2011 (the "Municipal Loan 1") provides for a cash contribution of €500 thousand which matures on December 31, 2021. MBMV is entitled to a fee consisting of an annual non-profit-related remuneration of 8.25% of the contribution per annum and an annual share in our profits of 1.5% of the investment value. If a two year loss is reported, the annual non-profit-related remuneration is increased by 0.75% of the contribution per annum. MBMV is entitled to terminate the Municipal Loan 1 if we do not comply with the contractual obligations under the agreement, including if the contribution is not used in accordance with its designated purposes. If the Municipal Loan 1 is terminated early, we will pay a surcharge fee to MBMV. Arndt Rolfs and Christoph Ehlers (each in the amount of €500 thousand) guarantee our obligations under the Municipal Loan 1 under separate agreements with MBMV. In addition, Bürgschaftsbank Mecklenburg-Vorpommern GmbH provided a guarantee to MBMV for the repayment of up to 80% of its contribution and up to 80% of the fees in accordance with a separate guarantee agreement. We repaid the outstanding amount of the contribution in full in February 2020.
The silent partnership agreement dated March 20, 2013 (the "Municipal Loan 2") provides a cash contribution of €360 thousand which matures on December 30, 2022. MBMV is entitled to a fee consisting of an annual non-profit related remuneration of 8.0% of the contribution per annum and an annual share in our profits of 1.5% of the investment value. If a two year loss is reported, the annual non-profit related remuneration is increased by 0.75% of the contribution per annum. The Municipal Loan 2 contains a covenant to maintain an equity ratio of 20% calculated on a consolidated basis. If this agreed ratio is not achieved, the annual non-profit-related remuneration will be increased by 1.5% per annum. MBMV is entitled to terminate the Municipal Loan 2 if we do not comply with the contractual obligations under the agreement, including if the contribution is not used in accordance with its designated purposes. If the Municipal Loan 2 is terminated early, we will pay a surcharge fee to MBMV. Arndt Rolfs and Christoph Ehlers (each in the amount of €150 thousand) as well as Hans-Bodo Hartmann, Michael Schlenk and Stefan Maeser (each in the amount of €50 thousand) guarantee our obligations under the Municipal Loan 2 under separate agreements with MBMV. In addition, provided a guarantee to MBMV for the repayment of up to 80% of its contribution and up to 80% of the fees in accordance with a separate guarantee agreement. We repaid the outstanding amount of the contribution in full in February 2020.
123
The silent partnership agreement dated August 5, 2015 (the "Municipal Loan 3") between us, certain of our shareholders and MBMV provided for a cash contribution of €140 thousand which would have matured on May 30, 2021. We repaid the outstanding amount of the contribution in full on June 29, 2018.
The silent partnership agreement dated July 8, 2016 (the "Municipal Loan 4") between us, certain shareholders and MBMV provided for a cash contribution of €1,000 thousand which would have matured on December 31, 2023. On April 25, 2018, we and MBMV agreed to terminate the Municipal Loan 4. We repaid the outstanding amount of the contribution in full on June 29, 2018.
C. Research and Development, Patents and Licenses, etc.
For a description of our research and development policies for the last three years see "Item 4. Information on the Company—B. Business Overview—Research and Development." For a description of our intellectual property, see "Item 4. Information on the Company—B. Business Overview—Intellectual Property."
D. Trend Information
For a discussion of trend information, see "Item 5. Operating and Financial Review and Prospects."
E. Off-balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements, as defined in the rules and regulations of the Securities and Exchange Commission.
F. Tabular Disclosure of Contractual Obligations
The table below presents the residual contractual terms of the financial liabilities as of December 31, 2019, including estimated interest payments. The figures are undiscounted gross amounts, including estimated interest payments and interest on undrawn loan funds, but without showing the impact of offsetting.
| |
Payments due by Period | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Carrying amount |
Total contractual cash flows |
Less than 1 year |
Between 1 and 3 years |
Between 3 and 5 years |
More than 5 years |
|||||||||||||
| |
(€ in thousands) |
||||||||||||||||||
Secured bank loans |
1,770 | 1,866 | 860 | 1,006 | — | — | |||||||||||||
Bank overdraft |
2,636 | 2,636 | 2,636 | — | — | — | |||||||||||||
Lease liabilities(1) |
21,704 | 25,934 | 3,789 | 5,700 | 3,874 | 12,571 | |||||||||||||
Municipal loans |
860 | 1,022 | 327 | 695 | — | — | |||||||||||||
Trade payables |
8,554 | 8,554 | 8,554 | — | — | — | |||||||||||||
| | | | | | | | | | | | | | | | | | | | |
Total |
35,524 | 40,012 | 16,166 | 7,401 | 3,874 | 12,571 | |||||||||||||
- (1)
- Lease liabilities include leases related to lease contracts for land and buildings, offices as well as various items motor vehicles and other equipment which are accounted for according to IFRS 16, and measured at the present value of lease payments over the lease term at the commencement date of the leases.
In addition to the contractual obligations disclosed above, we also have various non-cancellable lease contracts of office equipment and storage spaces which have a lease term of less than 12 months or are related to leases of low-value assets. The future lease payments for these non-cancellable lease contracts are €72 thousand within one year and an additional €36 thousand within five years.
124
During 2019, we entered into agreements with suppliers, for goods and services to be provided in 2020 with a total payment obligation of around €802 thousand.
G. Safe Harbor
This Annual Report contains forward-looking statements within the meaning of Section 27A of the Securities Act and Section 21E of the Exchange Act and as defined in the Private Securities Litigation Reform Act of 1995. See the section titled "FORWARD-LOOKING STATEMENTS" of this Annual Report.
H. Critical Accounting Policies and Estimates
Our consolidated financial statements are prepared in accordance with IFRS as issued by the IASB. Some of the accounting methods and policies used in preparing the financial statements under IFRS are based on complex and subjective assessments by our management or on estimates based on past experience and assumptions deemed realistic and reasonable based on the circumstances concerned. The actual value of our assets, liabilities and shareholders' equity and of our earnings could differ from the value derived from these estimates if conditions changed and these changes had an impact on the assumptions adopted.
Our significant accounting policies that we believe to be critical to the judgments and estimates used in the preparation of our financial statements are included in "Note 6—Accounting judgments and estimates" and "Note 20—Share-based payments" to our consolidated financial statements as of and for the year ended December 31, 2019.
Item 6. Directors, Senior Management and Employees
A. Directors and Senior Management
Board Structure
We have a two-tier board structure consisting of a management board (bestuur) and a separate supervisory board (raad van commissarissen).
Management Board
Our management board consists of four members, who we refer to as our managing directors (and who are also our executive officers). Each managing director of Centogene N.V. holds office for the term set by our general meeting of shareholders (as set forth in the table below), except in the case of his earlier death, resignation or removal. Our managing directors do not have a retirement age requirement under our articles of association.
Our managing directors are responsible for the management and representation of our company. We have a strong centralized management team led by Prof. Arndt Rolfs, our CEO, with broad experience in information technology, strategy, operations, finance, sales, communications and training. Our senior management has an average of 18 years of experience in the biopharmaceutical industry. Many of the members of our management team have worked together as a team for many years.
125
The following table lists the current members of our management board all of whom we consider key executive officers, as well as the year of expiration of their terms as management board members of Centogene N.V.:
Name
|
Position | Age | Year of Expiration of Term |
||||||
|---|---|---|---|---|---|---|---|---|---|
Prof. Arndt Rolfs, M.D. |
Chief Executive Officer | 60 | 2022 | ||||||
Dirk H. Ehlers, Ph.D(1). |
Chief Operating Officer | 59 | 2020 | ||||||
Richard Stoffelen |
Chief Financial Officer | 52 | 2021 | ||||||
Volkmar Weckesser, Ph.D. |
Chief Information Officer | 52 | 2022 | ||||||
- (1)
- Mr. Ehlers has decided to leave the Company for personal reasons with effect after the Company's upcoming Annual General Meeting in June 2020.
The following is a brief summary of the business experience of our managing directors. Unless otherwise indicated, the current business addresses for our managing directors is Am Strande 7, 18055 Rostock, Germany.
Prof. Arndt Rolfs, M.D. Prof. Rolfs is one of our co-founders. He has served as Chief Executive Officer since 2014 and Chief Medical Officer since 2006. He previously served as Director of the Albrecht-Kossel-Institute for Neuroregeneration at the University of Rostock from 2008 to 2018. Prior to founding Centogene, Prof. Rolfs was Vice-Director of the Clinic and Outpatients Department for Neurology at the Centre for Neurology, University of Rostock, from 1997 to 2008. He also served as a Senior Consultant at the Department of Neurology and as Head of the Neurobiological Research Laboratory from 1993 to 2008 at the University of Rostock. From 1991 to 1993, he worked at the Psychiatric Clinic at the University Hospital Rudolf-Virchow in Berlin. From 1989 to 1993 he was the Head of the Laboratory for Neurochemistry at the Free University of Berlin. From 1988 to 1989, Prof. Rolfs worked at the Max-Planck-Institute for Molecular Genetics and from 1985 to 1988 at the Department of Neurology at the Free University of Berlin. Prof. Rolfs is the principal investigator of several international epidemiological studies in the area of rare diseases and actively engaged in biomarker research for several metabolic diseases. He has an extensive track record in medical and scientific publications and is the author of or contributor to more than 300 peer-reviewed publications. Prof. Rolfs received his approbation as physician from the University of Mainz in 1985.
Dirk H. Ehlers, Ph.D. Dr. Ehlers has served as chief operating officer and president of Clinical Diagnostics since April 2018. Prior to joining us, he served as senior vice president and president of the Surgical Solutions Division and as a member of the Group Executive Team at Hill-Rom Holdings, Inc. in Chicago, Illinois. From 2015 to 2018, he was a non-executive board member of Protagen AG. From 2010 to 2014, Dr. Ehlers served as president and CEO of Eppendorf AG. Prior to that, he served as head of Professional Diagnostics and as a member of the Diagnostics Executive Committee at Hoffmann La Roche from 2007 to 2010. From 2001 to 2007, he was chief financial officer and an executive board member of Evotec AG. From 2000 to 2001, Dr. Ehlers served as an Executive Board member and president of the Enteral Nutrition Division of Fresenius Kabi AG. From 1995 to 2000, he served as Diagnostic Systems Division head at Olympus Europe. From 1989 to 1994, he was a management consultant and an engagement manager at McKinsey & Co. Inc. Dr. Ehlers holds a M.Sc. in Physics and received his Ph.D. in Physics with distinction, summa cum laude, both from the University RWTH in Aachen.
Richard Stoffelen. Mr. Stoffelen has served as chief financial officer ("CFO") since 2016. He has over 30 years of experience in international roles focused on finance, governance and risk management. Prior to joining us, he was head of Internal Audit at Holcim Group Services from 2013 to 2016. From 2000 to 2013, Mr. Stoffelen worked in various audit and management positions as a partner at KPMG, where he was responsible for audits of clients in a wide variety of industries, including the
126
pharmaceutical industry. Mr. Stoffelen graduated as a Dutch chartered accountant with the NBA and Tilburg University in the Netherlands, with further executive education programs at Harvard Business School, Insead (Executive MBA), the IMD and the IESE business school.
Volkmar Weckesser, Ph.D. Dr. Weckesser has served as chief information officer since 2016. Prior to joining us, he served as chief executive officer and group chief information officer at Gothaer Systems GmbH from 2014 to 2016. He served as a Member of the Executive Council and head of Information Technology at Dekabank Deutsche Girozentrale from 2009 to 2014, as a central division manager of the Information Technology Division of HSH Nordbank AG from 2003 to 2009, division manager of Landesbank Schleswig-Holstein Girozentrale from 1999 to 2003, personal assistant to the CEO at Deutsche Apotheker und Ärtzebank EG from 1998 to 1999, project manager at Mitchell Madison Group from 1996 to 1998, consultant and project manager at Monitor Company from 1993 to 1996 and lecturer at Universitat Karlsruhe 1991 to 1993. He holds a Ph.D. from Universitat Karlsruhe.
The following table lists those of our key executive officers who are not members of our management board:
Name
|
Position | Age | Year of Expiration of Term |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Oved Amitay | Chief Business Officer | 54 | — | ||||||
| Sun Kim | Chief Strategy & Investor Relations Officer | 48 | — | ||||||
| Philip Lambert, Ph.D(1) | Chief Scientific Officer | 52 | — | ||||||
| Prof. Peter Bauer, M.D. | Chief Genomic Officer | 50 | — | ||||||
- (1)
- Mr. Lambert was appointed as Chief Scientific Officer on December 6, 2019.
Oved Amitay. Mr. Amitay has served as our chief business officer since November 2018, prior to which he held the position of president and chief operating officer of Arrett Neuroscience from 2016 to 2018, having led the company's strategy for developing therapies for Rett syndrome. Prior to this role, he served as Vice President, Head of Commercial at Alnylam Pharmaceuticals from 2012 to 2016, with a lead role in the organization's transition from a technology-platform focus to a patient-centric drug development and pre-launch enterprise. Mr. Amitay had a long tenure at Genzyme Corporation (now Sanofi Genzyme) from 1998 to 2011 as Vice President, Strategic Development, responsible for program management, business development, market assessment and business planning for the rare genetic diseases franchise, and as General Manager of the Gaucher and MPS business. He is also a Founding Advisor of Splisense Therapeutics, Israel, where he held this role from 2016 to present. Mr. Amitay holds a M.Sc., Pharmacology, from Northeastern University, Boston, MA and a B.Sc., Pharmacy, from the Hebrew University, Jerusalem, Israel.
Sun Kim. Mr. Kim has served as our chief strategy and investor relations officer since May 2019. Prior to his appointment, Mr. Kim was Head of Corporate Strategy at Shire Plc from August 2018 to April 2019, where he also oversaw Shire Plc's investor relations activities in the US and, from October 2017 to August 2018, he was Vice President, Investor Relations. Prior to joining Shire Plc, he served as Global Head of Business Service Excellence at Novartis from March 2015 to September 2017, as General Manager at Alcon Singapore from July 2013 to March 2015, and as Global Head of Strategy for Alcon from September of 2011 to July 2013. Mr. Kim also worked at Bausch & Lomb from August 2009 to September 2011, heading up its strategy function for the pharmaceutical business unit. Prior to his experience in the pharmaceutical industry, Mr. Kim spent seven years as a Management Consultant at McKinsey & Company and A.T. Kearney. Mr. Kim holds a Ph.D. and M.S. in chemical engineering from Stanford University, and B.S. in chemical engineering from Seoul National University.
127
Philip Lambert, Ph.D Dr. Lambert has served as our chief scientific officer since December 2019. Prior to his appointment, Dr. Lambert was Senior Vice President of Discovery and Labs of Life Biosciences, a global company focused on extending the healthy lifespan of individuals by investigating eight biological pathways believed to increase longevity. He also served as Chief Operating Officer of Selphagy Therapeutics and Spotlight Biosciences, subsidiaries of Life Biosciences. Prior to Life Biosciences, Dr. Lambert served as Scientific Partner and Head of Discovery at Charles River Laboratories, where he was responsible for establishing scientific credibility and delivering the business to client companies and venture capital firms. He also co-founded VivoPath, a preclinical screening company, offering clients discovery and development services for proof-of-concept testing. He holds a PhD in Neuropharmacology from Imperial College, University of London and a BSc in Pharmacology from the University of Liverpool
Prof. Peter Bauer, M.D. Prof. Bauer has served as our chief genomic officer since December 2019, prior to which he served as our chief scientific officer from January 2017 to November 2019 and chief operating officer since joining us in 2016. Prof. Bauer is a professor of human genetics at the University of Tübingen and a board-certified human geneticist with expertise in molecular genetics, diagnostic testing, genetic counselling, functional validation of genetic variants and bioinformatics tools for medical interpretation of clinical sequencing. Prior to joining us, he served as head of the diagnostic and research laboratory at the Institute of Medical Genetics and Applied Genomics, University Hospital Tübingen from 2001 to 2015. Prof. Bauer has been vice president of the German Society of Neurogenetics since 2004. Prof. Bauer received a degree in medicine from the Freie University Berlin and the approbation as physician (German official license to practice medicine) from the Board of Physicians in Berlin in 1998.
Supervisory Board
Our supervisory board consists of eight members. Each supervisory director holds office for the term set by our general meeting of shareholders (as set forth in the table below), except in the case of his earlier death, resignation or removal. Our supervisory directors do not have a retirement age requirement under our articles of association.
The following table presents the names of the current members of our supervisory board, as well as the year of expiration of their terms as supervisory board members of Centogene N.V.:
Name
|
Position | Age | Year of Expiration of Term |
||||||
|---|---|---|---|---|---|---|---|---|---|
Flemming Ornskov |
Chairman of the Supervisory Board | 62 | 2022 | ||||||
Hubert Birner, Ph.D. |
Member of the Supervisory Board | 53 | 2022 | ||||||
Peer M. Schatz(1) |
Member and Vice-Chairman of the Supervisory Board | 54 | NA | ||||||
Christoph Ehlers |
Member of the Supervisory Board | 62 | 2022 | ||||||
Holger Friedrich(2) |
Member of the Supervisory Board | 53 | 2022 | ||||||
Guido Prehn |
Member of the Supervisory Board | 42 | 2022 | ||||||
Eric Souêtre |
Member of the Supervisory Board | 64 | 2022 | ||||||
Berndt Modig |
Member of the Supervisory Board | 61 | 2022 | ||||||
- (1)
- In
March 2020, following the resignation of Mr. Jacob Kaluski from his office as a member of the supervisory board, Mr. Peer M. Schatz joined as an
interim member and as the interim vice-chairman of the supervisory board effective March 16, 2020. Mr. Schatz will be nominated for appointment to the shareholders at the Company's
upcoming Annual General Meeting in June 2020.
- (2)
- On November 21, 2019, Holger Friedrich took a temporary leave of absence from the supervisory board. He resumed his duties in March 2020.
128
The following is a brief summary of the business experience of our supervisory directors. Unless otherwise indicated, the current business addresses for our directors is Am Strande 7, 18055 Rostock, Germany.
Flemming Ornskov, M.D., MPH, MBA. Dr. Ornskov has served as the Chairman of our supervisory board since April 2019. He has served as the Chief Executive Officer of Galderma S.A. since October 2019. He served as Chief Executive Officer and Executive Director of Shire Plc from April 2013 to January 2019, when Shire was acquired by Takeda. Dr. Ornskov has extensive international, strategic and operational experience in the pharmaceutical and biotech sectors, as well as medical expertise as a physician with training in pediatrics. He was appointed Non-Executive Director and Chairman of the Board of Recordati S.p.A. in February 2019. He has been a Non-Executive Director for the Waters Corporation since 2017. Previously, Dr. Ornskov was Non-Executive Chairman of Evotec from 2008 to 2012 and Non-Executive Director of PCI Biotech Holding from 2008 to 2013. From 2010 to 2013, he was Chief Marketing Officer and Global Head, General and Specialty Medicine at Bayer. He also previously held positions as Global President, Pharmaceuticals and Over-the Counter at Bausch & Lomb; Chairman, President and Chief Executive Officer of LifeCycle Pharma A/S, now Veloxis Pharma A/S; President and Chief Executive Officer of Ikaria; and various roles at Merck and Novartis. Dr. Ornskov received his M.D. from the University of Copenhagen, MBA from INSEAD, and Masters of Public Health from Harvard University.
Hubert Birner, Ph.D. Dr. Birner joined the supervisory board of Centogene as Chairman from July 2017 to March 30, 2019, and as Vice-Chairman from April 1, 2019 to March 16, 2020. He currently serves as a managing partner at TVM Capital, and is responsible for its overall investment strategy and fund operations in North American and Europe. Dr. Birner joined TVM Capital in 2000 as an investment manager. He currently also serves as Chairman of the supervisory board of SpePharm Holding B.V., leon-nanodrugs GmbH and AL-S Pharma AG. He is a member of the board of directors of Argos Therapeutics, Inc., Proteon Therapeutics Inc, Noxxon Pharma and Acer Therapeutics Inc. Dr. Birner previously served on the board of directors of Horizon Pharma, Inc., Bioxell SA, Evotec AG, Probiodrug AG and Jerini AG. Prior to his current tenure, he was Head of Business Development Europe and Director of Marketing for Germany at Zeneca Agrochemicals. Dr. Birner joined Zeneca from McKinsey & Company's European Health Care and Pharmaceutical practice and as Assistant Professor for biochemistry at the Ludwig-Maximilian-University ("LMU"). He holds a summa cum laude doctoral degree in biochemistry at LMU. His doctoral thesis was honored with the Hoffmann-La Roche prize for outstanding basic research in metabolic diseases. Dr. Birner also holds an MBA from Harvard Business School.
Peer M. Schatz Mr. Schatz joined the supervisory board of Centogene in March 2020 as interim member and interim vice-chairman of the supervisory board, subject to approval of the shareholders at the Company's upcoming Annual General Meeting in June 2020. He joins Centogene from his recent position as long-time Chief Executive Officer of QIAGEN N.V. (Nasdaq: QGEN; Frankfurt: QIA), a leading provider of molecular sample and assay technologies. From 1993 to 2019, he led QIAGEN's rapid expansion from a start-up company with $2 million in sales into a global leader in molecular testing with over $1.6 billion in revenues. Mr. Schatz also served as a founding member of the German Corporate Governance Commission and as a supervisory board member of Evotec AG (Frankfurt: EVT). Mr. Schatz graduated from the University of St. Gallen, Switzerland with a Masters Degree in Finance and from the University of Chicago Graduate School of Business with an MBA.
Christoph Ehlers, LL.M. Mr. Ehlers is a co-founder of Centogene. After having been in the executive management from 2008 to 2014, he joined our board as a supervisory director in 2014. Mr. Ehlers, by profession a lawyer, founded Equicore Beteiligungsgesellschaft GmbH in 1997 as a specialized consulting and investment vehicle to assist in the development of early stage LifeScience companies. Since 1999, Mr. Ehlers has also served as one of two founding board members on the
129
board of Stiftung Ordnungspolitik, a leading European economic think-tank. As part of the Equicore business, he holds management positions in other early stage portfolio companies. Prior to Equicore he held various positions at Commerzbank AG from 1984 to 1996, including functions in investment banking, the Chairman's office and leading the southwestern branches. He studied law at the University of Constance, was admitted in 1983 to the German bar and holds an LL.M. from the University of San Diego Law School.
Holger Friedrich. Mr. Friedrich joined our board as a supervisory director in 2017. Since 2010, Mr. Friedrich has served as managing director of CORE SE's consulting unit. Prior to this role, he served as chairman of SPM Technologies (acquired by SAP) from 1993 to 2003 and as SAP senior vice president, IT Architecture, from 2003 to 2005. He served as partner at McKinsey from 2005 to 2008 and was responsible for their European Enterprise Architecture practice. He served as board member at Software AG from 2009 to 2010. Mr. Friedrich studied computer science and German studies and he was one of the founding members of the Institute for Theoretical Computer Science at the University of Potsdam, which is known today as the Hasso Plattner Institute.
Jacob Kaluski, M.Sc. Mr. Kaluski joined our board as a supervisory director in 2015. He has served as chairman of Danaka AB since 2005. Prior to joining Danaka AB, he served as co-founder of TKT Europe-5S AB from 2000 to 2004 and in various business and management positions at Pharmacia & Upjohn from 1985 to 1999. He has served on the boards of Belina AB since 2018, Glactone AB since 2014, Pulsetten AB from 2012 to 2016, Bioimics AB from 2010 to 2013, DuoCort AB from 2009 to 2012, Alligator Bioscience from 2007 to 2010, Jederstrom Pharmaceuticals AB from 2006 to 2009, TKT Europe-5S AB from 2000 to 2004 and 5S Pharma AB from 1999 to 2008. He holds an M.Sc. in Pharmaceutical Science from Uppsala University.
Guido Prehn. Mr. Prehn joined our board as supervisory director in 2017. Mr. Prehn has over 15 years of experience in the private equity industry. He currently serves on the boards of Omniamed Holding GmbH, Pharmazell GmbH, Calvias GmbH, Everest TopCo B.V., Auerbach Holding AG, Kohlspitz Holding AG, AWK Group and VTU Group. Mr. Prehn is a managing director of DPE Deutsche Private Equity where he joined in 2008, shortly after its foundation. Between 2002 and 2008, he worked in various positions at Allianz Capital Partners, TPG Capital and Merrill Lynch. Mr. Prehn studied business administration at the European Business School, Oestrich-Winkel, De Paul University Chicago and Universidad Argentina de la Empresa, Buenos Aires.
Eric Souêtre, M.D. Dr. Souêtre joined our board as a supervisory director in 2017. After various research positions at National Institute of Mental Health, Dr. Souêtre founded "BENEFIT" in 1990, a research and consulting company in health economics (subsequently acquired by QUINTILES Inc. (USA) in 1995). He then served as a board member at QUINTILES Inc, where he was responsible for the global consulting function. In 2003, Dr. Souêtre co-founded LABCO - a network of clinical laboratories - and led the company to a European leadership as chairman and CEO until late 2010. He remained as an active board member until LABCO was sold to CINVEN in 2015. Dr. Souêtre has since co-founded a private equity fund, Careventures, focused on pan European healthcare service ventures. He currently serves on the board of OPERA SA. Dr. Souêtre holds a Ph.D. in neurosciences by the Marseille University, an M.D. by the Medical University of Nice and an MBA from HEC school of Paris.
Berndt Modig, MBA. Mr. Modig joined our board as a supervisory director in April 2018. He also serves as chief executive officer of Pharvaris B.V. He served as chief financial officer of Prosensa Holding N.V., a public pharmaceutical company, from March 2010 until its acquisition by BioMarin Pharmaceutical Inc. in January 2015. From October 2003 to November 2008, Mr. Modig was chief financial officer at Jerini AG where he directed private financing rounds, its initial public offering in 2005, and its acquisition by Shire Plc in 2008. Before that, Mr. Modig served as chief financial officer at Surplex AG from 2001 to 2003 and as finance director Europe of U.S.-based Hayward Industrial
130
Products Inc. from 1999 to 2001. In previous positions, Mr. Modig was a partner in the Brussels-based private equity firm, Agra Industria, from 1994 to 1999 and a senior manager in the Financial Services Industry Group of Price Waterhouse LLP in New York from 1991 to 1994. Mr. Modig currently serves as a director and member of the audit committee of Axovant Sciences Ltd, supervisory board director and member of the audit committee of Affimed N.V., and vice-chairman of the supervisory board and chairman of the audit committee of Kiadis Pharma N.V., all of which are publicly held pharmaceutical companies, and he was a director of Mobile Loyalty plc from 2012 to 2013. Mr. Modig received a bachelor's degree in business administration, economics and German language from the University of Lund, Sweden, and an MBA from INSEAD, Fontainebleau, France. He is a certified public accountant (inactive).
B. Compensation
Compensation of Managing Directors, Supervisory Directors and Officers
We are a foreign private issuer. As a result, in accordance with Nasdaq listing requirements, we are complying with home country compensation requirements and certain exemptions thereunder rather than with Nasdaq compensation requirements. Dutch law does not provide for limitations with respect to the aggregate annual compensation paid to members of our management board or supervisory board, provided that such compensation is consistent with our compensation policy. Such compensation policy requires approval by our general meeting of shareholders. The supervisory board determines the remuneration of individual managing directors with due observance of the remuneration policy. A proposal with respect to remuneration schemes in the form of shares or rights to shares in which managing directors may participate is subject to approval by our general meeting of shareholders. Such a proposal must set out at least the maximum number of shares or rights to subscribe for shares to be granted to the management board of directors and the criteria for granting or amendment. The compensation for our supervisory directors is set by the general meeting of shareholders.
The compensation, including benefits in kind, accrued or paid to our managing directors and supervisory directors with respect to the year ended December 31, 2019, for services in all capacities is shown below on an individual basis. Further details for the compensation for our managing directors and supervisory directors are given in note 25 to our consolidated financial statements as of and for the year ended December 31, 2019.
Directors Compensation 2019
Supervisory Directors
| |
F. Ornskov | H. Birner | C. Ehlers | H. Friedrich(1) | J. Kaluski | G. Prehn | E. Souetre | R. Modig | D. Parera(2) | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
(in € thousands) |
|||||||||||||||||||||||||||
Periodically paid compensation |
102 | 35 | 42 | 20 | 26 | 46 | 29 | 75 | 7 | |||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Total cash compensation |
102 | 35 | 42 | 20 | 26 | 46 | 29 | 75 | 7 | |||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
2019 Equity Incentive Plan(3) |
704 | — | — | — | — | — | — | — | — | |||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
- (1)
- On
November 21, 2019, Holger Friedrich took a temporary leave of absence from the supervisory board and resumed duties in March 2020, and during which leave
of absence he did not receive any remuneration.
- (2)
- Mr. Parera
is not a member of the supervisory board since March 2019.
- (3)
- This amount represents the portion of the grant date fair value of the option award recognized as an expense in 2019 under the provisions of IFRS 2.
131
Managing Directors
| |
A. Rolfs | D. Ehlers | R. Stoffelen | V. Weckesser | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
(in € thousands) |
||||||||||||
Periodically paid compensation |
584 | 360 | 514 | 385 | |||||||||
Bonuses |
224 | 90 | 188 | 119 | |||||||||
| | | | | | | | | | | | | | |
Total cash compensation |
808 | 450 | 702 | 504 | |||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
2016 Virtual Share Option Plan(1) |
— | — | 198 | 128 | |||||||||
2017 Virtual Share Option Plan and 2019 Equity Incentive Plan(2) |
493 | 1,169 | 850 | 447 | |||||||||
| | | | | | | | | | | | | | |
Total share-based payment expense |
493 | 1,169 | 1,048 | 575 | |||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
- (1)
- This
amount represents the change in fair value of the option awards issued under 2016 Virtual Share Option Plan between December 31, 2018 and the date of
exercise of the option awards, which is the date of completion of our initial public offering, recognized under the provisions of IFRS 2.
- (2)
- The option awards issued under 2017 Plan were replaced by the awards issued under 2019 Plan at the date of our initial public offering. This amount represents the change in fair value of the option awards issued under 2017 Plan between December 31, 2018 and the date when they were replaced, offset by the difference in the fair value of the option awards issued under 2017 Plan and grant date fair value of options awards issued under 2019 Plan at the date of replacement, recognized under the provisions of IFRS 2.
For the years ended December 31, 2018 and 2019, our performance-based compensation programs are described below.
The amount accrued by us to provide pension, retirement or similar benefits to members of its management board, supervisory board or officers with executive responsibilities amounted to a total of €10 thousand in each of the years ended December 31, 2017, 2018 and 2019.
2016 Virtual Share Option Plan
Under Centogene AG's virtual share option program 2016 (our "2016 Plan"), we have granted virtual share options to our employees, management board members and selected consultants. Immediately prior to the consummation of our initial public offering, the outstanding awards under the 2016 Plan covered an aggregate of 8,223 common shares of Centogene AG derived from 802,283 options. Under this program, holders of vested options are entitled to receive a direct cash payment from Centogene N.V, which payment to the option holders will be reimbursed by the original shareholders of Centogene AG at the same time as the obligation to pay the option holders arises.
Upon the completion of our initial public offering, all options granted under the 2016 Plan were vested immediately in full and the cash payment of each option holder is calculated with reference to the initial public offering price of the shares of Centogene N.V. and the exercise prices of the vested options. As of December 31, 2019, all options under the 2016 Plan were considered vested and exercised. Such entitlements remain outstanding as of the date of this Annual Report. We have not issued and do not intend to issue any additional awards under the 2016 Plan.
132
The table below shows the cash payments our Managing Directors are entitled as a result of the vested 2016 Plan.
Beneficiary
|
Number of Options at the Consummation of the Initial Public Offering |
Strike Price EUR | Cash Entitlement EUR Thousand |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
Richard Stoffelen |
272,683 | 400 | 702 | |||||||
Richard Stoffelen |
46,200 | 100 | 261 | |||||||
Volkmar Weckesser |
138,600 | 200 | 641 | |||||||
| | | | | | | | | | | |
Total |
457,483 | 1,604 | ||||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
2017 Virtual Share Option Plan
In December 2016, Centogene AG established an additional virtual share option program (our "2017 Plan") for virtual share options granted to employees and management board members. Immediately prior to our initial public offering, the outstanding awards under the 2017 Plan covered an aggregate of 16,349 common shares of Centogene AG derived from 16,374 virtual share options which, in connection with our initial public offering, have been converted into awards exercisable for common shares of Centogene N.V. on a 1.00 to 49.2578 basis. Therefore, as of December 31, 2019, there were no options outstanding under 2017 Plan. We have not issued and we do not intend to issue any additional awards under the 2017 Plan. The 2017 Plan was cancelled and replaced with the 2019 Plan as discussed below.
2019 Equity Incentive Plan
In conjunction with the consummation of our initial public offering, we have established a new long-term incentive plan (our "2019 Plan") with the purpose of advancing the interests of our shareholders and other stakeholders by enhancing our ability to attract, retain and motivate individuals who are expected to make important contributions to us. The 2019 Plan governs issuances of equity and equity-based incentive awards from and after the consummation of this offering. The maximum number of common shares underlying awards granted pursuant to the 2019 Plan (other than replacement awards under the 2019 Plan) shall not exceed 13% of the Company's issued share capital immediately following our initial public offering. Such maximum number has been increased on January 1, 2020 and will be increased on January 1 of each calendar year thereafter, with an additional number of common shares equal to 3% of the Company's issued share capital on such date (or any lower number of common shares as determined by the management board or supervisory board, where appropriate on the basis of a recommendation of the compensation committee (as the case may be, as prescribed by the 2019 Plan and, collectively, the "Committee").
Plan Administration. The 2019 Plan is administered by the Committee.
Eligibility. Awards under the 2019 Plan may be granted to our employees, the members of our management board and supervisory boards, consultants or other advisors.
Awards. Awards under the 2019 Plan may be granted in the form of stock options, stock appreciation rights, restricted stock, restricted stock units, other share-based awards or a combination of the foregoing. The Committee may condition the right of an individual to exercise his or her awards upon the achievement or satisfaction of performance criteria.
Vesting. The vesting conditions for awards under the 2019 Plan are determined by the Committee and are set forth in the applicable award documentation.
133
Termination of Service. In the event of a good leaver's (as defined in the 2019 Plan) termination of employment or service, all vested awards will be exercised or settled in accordance with their terms within a period specified by the Committee and all unvested awards shall be cancelled automatically unless decided otherwise by the Committee. In the event of a bad leaver's (as defined in the 2019 Plan) termination of employment or service, all vested and unvested awards will be cancelled automatically without compensation.
Change in Control. In the event of a change in control of the Company (as defined in the 2019 Plan), outstanding awards that will be substituted or exchanged for equivalent replacement awards, will be cancelled. If outstanding rewards are not substituted or exchanged for equivalent replacement awards, the awards shall immediately vest and settle in full, unless otherwise decided by the Committee.
During the year ended December 31, 2019, the following options were granted under 2019 Plan:
Replacement of 2017 Plan
As discussed above, the options granted under 2017 Plan of Centogene AG have been converted into awards exercisable for common shares of Centogene N.V. immediately prior to the completion of our initial public offering. Accordingly, 805,308 new share options were granted pursuant to the 2019 Plan, with each option representing one common share of Centogene N.V., and an exercise price equal to €0.12 per share. The options were considered vested upon the completion of the initial public offering, but are not exercisable in the first 180 days subsequent to the initial public offering.
The following table shows the options granted to our Managing Directors under the converted 2017 Plan:
Beneficiary
|
Grant Date | Number of Options | Strike Price EUR | Expiration Date | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
Prof. Arndt Rolfs |
November 9, 2019 | 81,151 | 0.12 | October 31, 2029 | ||||||
Dirk Ehlers |
November 9, 2019 | 145,382 | 0.12 | October 31, 2029 | ||||||
Richard Stoffelen |
November 9, 2019 | 140,169 | 0.12 | October 31, 2029 | ||||||
Volkmar Weckesser |
November 9, 2019 | 73,773 | 0.12 | October 31, 2029 | ||||||
| | | | | | | | | | | |
Total |
440,475 | |||||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Options to Flemming Ornskov
During the year ended December 31, 2019, an agreement was entered into between the Company and Flemming Ornskov in connection with his appointment to the supervisory board. According to this agreement, Mr. Ornskov was granted a total of 396,522 options for his services as chairman of the supervisory board, with each option representing one common share with an exercise price of €12.58 per option. The vesting period commenced on the day of the grant and ends on March 31, 2022, with one-third of the granted options vesting on March 31 in each year of the vesting period.
C. Board Practices
Date of expiration
For the year of expiration of the current term of the supervisory and management board members of Centogene N.V. see "—A. Directors and Senior Management."
Service Agreements
As of the consummation of our initial public offering, we entered into service agreements with all of the managing and supervisory directors of the Company. All of these agreements provide for notice
134
of termination periods after certain minimum appointment periods and all of them include restrictive covenants.
Committees
Audit Committee
The audit committee, which consists of Berndt Modig, Peer Schatz and Guido Prehn, assists the supervisory board in overseeing our accounting and financial reporting processes and the audits of our financial statements. In addition, the audit committee is responsible for the appointment, compensation, retention and oversight of the work of our independent registered public accounting firm. Our supervisory board has determined that Berndt Modig and Peer Schatz each satisfy the "independence" requirements set forth in Rule 10A-3 under the Exchange Act and Berndt Modig qualifies as an "audit committee financial expert," as such term is defined in the rules of the SEC. Peer Schatz has assumed Jacob Kaluski's role as independent audit committee member following Jacob Kaluski's resignation from the supervisory board effective March 16, 2020. The composition of our audit committee is consistent with the best practice provisions of the DCGC.
The audit committee's responsibilities include:
- •
- monitoring the management board with respect to (i) the relations with, and the compliance with recommendations and follow-up of
comments made by, the Company's internal audit function and the Dutch independent auditor, (ii) the Company's funding, (iii) the application of information and communication technology
by the Company, including risks relating to cybersecurity and (iv) the Company's tax policy;
- •
- issuing recommendations concerning the appointment and the dismissal of the head of the Company's internal audit function;
- •
- reviewing and discussing the performance of the Company's internal audit function;
- •
- the Company's compliance with applicable legal and regulatory requirements;
- •
- the operation of the Company's code of conduct and ethics and its other internal policies;
- •
- reviewing and discussing the Company's audit plan, including with the Dutch independent auditor and the internal audit function;
- •
- reviewing and discussing the essence of the audit results, including (i) flaws in the effectiveness of the internal controls,
(ii) findings and observations with a material impact on the Company's risk profile and (iii) failings in the follow-up of recommendations made previously by the internal audit function;
- •
- receiving from the Dutch independent auditor a formal written statement at least annually delineating all relationships between the Dutch
independent auditor and the Company consistent with applicable requirements of the Public Company Accounting Oversight Board (PCAOB) regarding the communications of the Dutch independent auditor with
the Audit Committee concerning independence;
- •
- reviewing and discussing with the Dutch independent auditor, at least annually (i) the scope and materiality of the Company's audit plan and the principal risks of the Company's annual financial reporting identified in such audit plan, (ii) the findings and outcome of the Dutch independent auditor's audit of the Company's financial statements and the management letter and (iii) significant findings from the audit and any problems or difficulties encountered, including restrictions on the scope of the Dutch independent auditor's activities or on access to requested information, as well as significant disagreements with the Company's management;
135
- •
- determining whether and, if so, how the Dutch independent auditor should be involved in the content and publication of financial reports other
than the Company's financial statements;
- •
- resolving disagreements between management and the Dutch independent auditor regarding the Company's financial reporting;
- •
- reviewing and discussing with the Dutch independent auditor any audit problems or difficulties and the response of the Company's management
thereto, including those matters required to be discussed with the Audit Committee by the Dutch independent auditor pursuant to established auditing standards, such as (i) restrictions on the
scope of the activities of the Dutch independent auditor or on access to requested information, (ii) accounting adjustments that were noted or proposed by the Dutch in-dependent auditor but
were "passed" (as immaterial or otherwise), (iii) communications between the audit team and the audit firm's national office regarding auditing or accounting issues presented by the engagement
and (iv) management or internal control letters issued, or proposed to be issued, by the Dutch independent auditor;
- •
- reviewing and discussing the effectiveness of the design and operation of the internal controls with the management board, the chief executive
officer and the chief financial officer, as appropriate, including identified material failings, deficiencies or material weaknesses in the internal controls and material changes made to, and material
improvements planned for, the internal controls;
- •
- assisting the Company in preparing the disclosure to be included in the Company's applicable filings as required by the Securities Act, the
Exchange Act and their related rules;
- •
- advising the management board regarding the Dutch independent auditor's nomination for (re)appointment or dismissal (including confirmation and
evaluation on the rotation of the audit partners on the audit engagement team as required by applicable laws) and preparing the selection of the Dutch independent auditor for such purpose, as
relevant;
- •
- reviewing and discussing the terms of engagement of the Dutch independent auditor to audit the Company's financial statements, to prepare or
issue an audit report, or to perform other audit, review or attest services, including the scope of the audit, the materiality standard to be applied, and causing the Company, without further action,
to pay the compensation of the Dutch independent auditor as approved by the audit committee;
- •
- engagement of such independent legal, accounting and other advisors as the audit committee deems necessary or appropriate to carry out its
responsibilities, including causing the Company, without further action, to pay the reasonable compensation of such advisors as approved by the audit committee;
- •
- causing the Company to pay, without further action, the ordinary administrative expenses of the audit committee that are necessary or
appropriate in carrying out its duties;
- •
- preparing the audit committee report that the SEC rules require to be included in the Company's annual proxy statement (if and when the Company
would become subject to those rules);
- •
- establishing policies for the Company's hiring of current or former employees of the Dutch independent auditor;
- •
- establishing procedures for (i) the receipt, retention and treatment of complaints received by the Company regarding accounting, internal accounting controls or auditing matters and (ii) the confidential, anonymous submission by employees of the Company of concerns regarding questionable accounting or auditing matters;
136
- •
- reviewing potential conflicts of interest involving managing or supervisory directors, including whether they may take part in the
deliberations in the decision-making process on any issue as to which there may be a conflict; and
- •
- developing and recommending to the supervisory board the Company's related person transaction policy.
We are relying on the phase-in rules of the SEC and Nasdaq with respect to the independence of our audit committee. These rules require that all members of our audit committee must meet the independence standard for audit committee membership by November 6, 2020, the date that is one year from the date of effectiveness of our Form F-1 registration statement in connection with our initial public offering. The audit committee is governed by a charter that complies with applicable Nasdaq rules, which charter is available on our website.
Compensation Committee
The compensation committee consists of Hubert Birner, Guido Prehn and Eric Souêtre. The compensation committee assists the supervisory board in determining compensation for our executive officers and the members of our management board and supervisory board. The composition of our compensation committee is consistent with the best practice provisions of the DCGC.
The compensation committee's responsibilities include:
- •
- reviewing and evaluating the Company's compensation policy and benefits policies generally, including the review and recommendation of
incentive-compensation and equity-based plans of the Company that are subject to approval of the supervisory board, as well as the compensation of the chief executive officer and the Company's other
executive officers;
- •
- submitting proposals to the supervisory board concerning changes to the Company's compensation policy, as relevant;
- •
- submitting proposals to the supervisory board concerning the compensation of individual managing directors and the Company's other executive
officers, at least covering (i) the compensation structure, (ii) the amount of the fixed and variable compensation components, (iii) the applicable performance criteria,
(iv) the scenario analyses that have been carried out, (v) the pay ratios within the Company's group, (vi) the views of the managing director concerned with regard to the amount
and structure of his own compensation and (vii) if considered appropriate by the management board or the compensation committee, the views of the executive officer concerned with regard to the
amount and structure of his own compensation;
- •
- submitting proposals to the supervisory board concerning the compensation of individual supervisory directors;
- •
- the review and assessment of risks arising from the Company's employee compensation policies and practices and whether any such risks are
reasonably likely to have a material adverse effect on the Company;
- •
- the preparation of the Company's compensation report for the supervisory board;
- •
- the preparation of the compensation committee report required by SEC rules or the rules of any other regulatory body; and
- •
- the retention of or obtaining advice from a compensation consultant, legal counsel or other advisor as the compensation committee deems necessary or appropriate to carry out its responsibilities, including the appointment of such consultant, counsel or advisor and the ability to cause the Company, without further approval, to pay with Company funds the reasonable
137
compensation of such consultant, counsel or advisor as approved by the compensation committee, provided, however, that (i) in retaining or obtaining the advice of such consultant, counsel or advisor, other than in-house legal counsel, the compensation committee shall take into consideration the factors affecting independence required by applicable SEC rules and Nasdaq rules and (ii) the compensation committee will be responsible for the oversight of the work of any such consultant, counsel or advisor.
Under SEC and Nasdaq rules, there are heightened independence standards for members of the compensation committee, including a prohibition against the receipt of any compensation from us other than standard director fees. As permitted by the listing requirements of Nasdaq, we have opted out of Nasdaq Listing Rule 5605(d), which requires that a compensation committee consist entirely of independent directors. The compensation committee is governed by a charter that is available on our website.
Nomination and Corporate Governance Committee
The nomination and corporate governance committee consists of Flemming Ornskov, Peer Schatz, Guido Prehn and Hubert Birner. The nomination and corporate governance committee assists our supervisory board in identifying individuals qualified to become members of our management board or supervisory board consistent with criteria established by us and in developing our code of business conduct and ethics. The composition of our nomination and corporate governance committee is consistent with the best practice provisions of the DCGC.
The nomination and corporate governance committee's responsibilities include:
- •
- drawing up selection criteria and appointment procedures for the managing directors and supervisory directors;
- •
- reviewing the size and composition of the management board and the supervisory board and submitting proposals for the composition profile of
the supervisory board;
- •
- making recommendations to the supervisory board as to determinations of supervisory director independence;
- •
- reviewing the functioning of individual managing directors and supervisory directors and reporting on such review to the supervisory board;
- •
- drawing up a plan for the succession of managing directors and supervisory directors;
- •
- submitting proposals for (re)appointment of managing directors and supervisory directors;
- •
- supervising the policy of the management board regarding the selection criteria and appointment procedures for the Company's executive
officers;
- •
- overseeing the self-evaluation of the management board and the supervisory board to determine whether it and its committees are functioning
effectively; and
- •
- developing and recommending to the management board the code of conduct and ethics and overseeing compliance with the code of conduct and ethics, including - at least annually - reviewing and reassessing the adequacy of the code of conduct and ethics and recommending any proposed changes to the management board.
As permitted by the listing requirements of Nasdaq, we have opted out of Nasdaq Listing Rule 5605(e), which requires independent director oversight of director nominations. The nominating and corporate governance committee is governed by a charter that is available on our website.
138
D. Employees
As of December 31, 2019, we employed over 440 highly qualified personnel (including consultants) from over 55 nationalities. All of our employees are engaged in either general and administrative, sale or research and development functions. None of our employees are covered by collective bargaining agreements. The following tables provide breakdowns of our full-time equivalent employees as of December 31, 2019 by function and by region:
| |
As of December 31, 2019 |
|||
|---|---|---|---|---|
Diagnostics Operations |
156 | |||
Pharmaceutical Operations |
42 | |||
Sales and Marketing |
57 | |||
Pharmaceutical Business Development |
7 | |||
Research and Development |
70 | |||
Corporate and other supporting functions |
110 | |||
| | | | | |
Total employees (including consultants and apprentices) |
442 | |||
| | | | | |
| | | | | |
| | | | | |
| |
As of December 31, 2019 |
|||
|---|---|---|---|---|
Germany |
377 | |||
United States |
24 | |||
India |
10 | |||
Consultants (located mainly in Asia, Middle East and Latin America) |
31 | |||
| | | | | |
Total employees (including consultants and apprentices) |
442 | |||
| | | | | |
| | | | | |
| | | | | |
E. Share Ownership
See "Item 7. Major Shareholders and Related Party Transactions—A. Major Shareholders."
Item 7. Major Shareholders and Related Party Transactions
A. Major Shareholders
As of the date of this Annual Report, our authorized share capital was €9,480,000, consisting of 79,000,000 common shares, par value €0.12 per share. Each of our common shares entitles its holder to one vote. The following table presents information relating to the beneficial ownership of our common shares as of the date of this Annual Report by:
- •
- each person, or group of affiliated persons, known by us to own beneficially 5% or more of our outstanding shares;
- •
- each member of our management board and supervisory board; and
- •
- all members of our management board and supervisory board as a group.
139
| |
Number of Common Shares beneficially owned |
||||||
|---|---|---|---|---|---|---|---|
Shareholder
|
Number | Percentage | |||||
>5% Shareholders: |
|||||||
Entities affiliated with DPE Deutsche Private Equity GmbH(1) |
5,124,289 | 25.8 | % | ||||
Careventures Fund II S.C.Sp(3) |
3,249,093 | 16.4 | % | ||||
Entities affiliated with TVM Life Science Innovation I, L.P.(4) |
2,791,248 | 14.1 | % | ||||
Equicore Beteiligungs GmbH(2) |
1,852,161 | 9.3 | % | ||||
Management Board Members and Key Officers: |
|||||||
Arndt Rolfs(5) |
2,401,978 | 12.1 | % | ||||
Dirk Ehlers |
8,305 | * | |||||
Richard Stoffelen |
16,611 | * | |||||
Volkmar Weckesser |
6,644 | * | |||||
Peter Bauer |
3,322 | * | |||||
Oved Amitay |
— | — | |||||
Philip Lambert |
— | — | |||||
Sun Kim |
— | — | |||||
Supervisory Board Members: |
|||||||
Guido Prehn(9) |
5,124,289 | 25.8 | % | ||||
Eric Souêtre(10) |
3,249,093 | 16.4 | % | ||||
Hubert Birner(6) |
2,791,248 | 14.1 | % | ||||
Christoph Ehlers(7) |
1,852,161 | 9.3 | % | ||||
Holger Friedrich(8) |
361,408 | 1.82 | % | ||||
Flemming Ornskov |
— | — | |||||
Peer Schatz(11) |
— | — | |||||
Berndt Modig |
— | — | |||||
- *
- Less
than 1% ownership.
- (1)
- The
5,124,289 common shares held by entities affiliated with DPE Deutsche Private Equity GmbH consist of (a) 3,367,423 common shares held by DPE
Deutschland II A GmbH & Co. KG ("DPE II A") and (b) 1,756,866 common shares held by DPE Deutschland II
B GmbH & Co. KG ("DPE II B"). DPE Deutsche Private Equity GmbH is the managing limited partner of DPE II A and DPE II B and
may be deemed to beneficially own the common shares held by such entities, but disclaims beneficial ownership of such shares. Marc Thiery is the managing director of DPE Deutsche Private
Equity GmbH and has sole power to vote the common shares beneficially owned by DPE Deutsche Private Equity GmbH. Guido Prehn has decision-making power over the common shares beneficially
owned by DPE Deutsche Private Equity GmbH but disclaims beneficial ownership of such shares. The address for DPE Deutsche Private Equity GmbH, DPE II A and
DPE II B is Ludwigstrasse 7, 80539 Munich, Germany.
- (2)
- The
1,852,161 common shares held by Equicore Beteiligungs GmbH consisted of 1,852,161 shares which Christoph Ehlers may be deemed to beneficially own. The
address for Equicore Beteiligungs GmbH is WeiherhofstraBe 5, 79104 Freiburg im Breisgau, Germany.
- (3)
- Careventures
Fund II S.C.Sp ("Careventures II") is managed by Careventures Fund II GP Sarl ("Careventures Fund"), and Eric Souêtre, the
founder of Careventures Fund, may be deemed to have voting and investment power over the shares held by Careventures II. The address for Careventures II is 42-44,
Avenue de la Gare, 1610 Luxembourg, Luxembourg.
- (4)
- The 2,791,248 common shares held by entities affiliated with TVM Life Science Innovation I, L.P. consist of (a) 1,934,105 common shares held by TVM Life Science Innovation I, L.P and (b) 857,143 common shares held by TVM Life Science Innovation II SCSp. The governance, investment strategy and decision-making with respect to investments held by TVM Life Science
140
Innovation I, L.P. is directed by TVM Life Science Innovation I (GP) Limited, whose directors are Reshentha Beeby, Hubert Birner and Gary Leatt, and who have shared power to vote the common shares beneficially owned by TVM Life Science Innovation I, L.P. As a result, each may be deemed to beneficially own the shares beneficially owned by TVM Life Science Innovation I, L.P. The address for TVM Life Science Innovation I, L.P. is 500-1 Place Ville-Marie, Montréal (Québec) H3B 1R1, Canada. The governance, investment strategy and decision-making process with respect to investments held by TVM Life Science Innovation II, SCSp. is directed by TVM Life Science Innovation II (GP) S.à r.l., whose directors are Monica Morsch, Ganash Lokanathen and Jens Hoellermann, and who have shared power to vote the common shares beneficially owned by TVM Life Science Innovation II SCSp. As a result, each may be deemed to beneficially own the shares beneficially owned by TVM Life Science Innovation II SCSp. The address for TVM Life Science Innovation II SCSp is 8 rue Lou Hemmer, L-1748, Senningerberg, Grand Duchy of Luxembourg
- (5)
- Prof.
Arndt Rolfs, our CEO and a member of our management board, also directly owns 10% of the shares in Centogene GmbH, Vienna, Austria ("Centogene Vienna"),
an immaterial subsidiary of ours. The remaining 90% of the shares in Centogene Vienna are directly owned by Centogene AG.
- (6)
- Hubert
Birner beneficially owns common shares through his indirect ownership of interests in TVM Life Science Innovation I, L.P and TVM Life Science
Innovation II (GP) S.à r.l.
- (7)
- Christoph
Ehlers beneficially owns common shares through his direct ownership of interests in Equicore Beteiligungs GmbH.
- (8)
- Common
shares are held by CCG-Commercial Coordination Germany GmbH and are beneficially owned by Holger Friedrich. The address for CCG-Commercial Coordination
Germany GmbH is Mauerstraße 78, 10117 Berlin, Germany.
- (9)
- Guido
Prehn has decision-making power over the common shares beneficially owned by DPE Deutsche Private Equity GmbH but disclaims beneficial ownership of such
shares.
- (10)
- Eric
Souêtre beneficially owns common shares through his indirect ownership of interests in Careventures Fund.
- (11)
- In March 2020, following the resignation of Mr. Jacob Kaluski from his office as a member of the supervisory board, Mr. Peer M. Schatz joined as an interim member and as the interim vice-chairman of the supervisory board effective March 16, 2020. Mr. Schatz will be nominated for appointment to the shareholders at the Company's upcoming Annual General Meeting in June 2020.
Each of our shareholders is entitled to one vote per common share. None of the holders of our shares have different voting rights from other holders of shares. We are not aware of any arrangement that may, at a subsequent date, result in a change of control of our company.
B. Related Party Transactions
The following is a description of related party transactions we have entered into since January 1, 2017 with any of our officers, directors and the holders of more than 5% of our voting securities, or any member of the immediate family of any of the foregoing persons.
Preferred and Common Share Financing
Series A Financing
On June 9, 2017, we entered into investment and shareholders agreements with certain investors, including existing shareholders such as Prof. Arndt Rolfs, our CEO, Dr. Peter Bauer, our chief scientific officer, Richard Stoffelen, our CFO, Dr. Volkmar Weckesser, our chief information officer, all of whom are members of our management board, Holger Friedrich and Christoph Ehlers, members of
141
our supervisory board, Michael Schlenk, TVM Life Science Ventures VII L.P., DPE Deutschland II A GmbH & Co. KG, DPE Deutschland II B GmbH & Co. KG, Careventures S.A., Careventures CG and CM-CIC Investissement SCR, pursuant to which we agreed to issue and sell an aggregate of 31,390 Series A preferred shares of Centogene AG in exchange for a further contribution of € 15.0 million from such investors and increased the authorized amount of Series A preferred shares of Centogene AG by up to 34,010.
On May 22, 2018, pursuant to the Series A Shareholders Agreement we issued an additional 34,010 Series A preferred shares of Centogene AG from the authorized shares to certain investors in exchange for a contribution of €10.0 million from such investors. On November 7, 2018, pursuant to the Series A Extension Agreement (as described below) we issued an additional 26,162 Series A preferred shares of Centogene AG to certain investors in exchange for a contribution of €10.0 million from such investors.
In conjunction with our corporate reorganization, the preferred shares of Centogene AG were converted into common shares of Centogene N.V.
Transactions Involving Members of Our Supervisory or Management Board
As registered in the German commercial register on July 23, 2018, Centogene AG issued 250 common shares to Dirk Ehlers, our Chief Operating Officer and member of our management board. In addition to the nominal value of such shares (€250), Mr. Ehlers made cash contributions into the Company's capital reserves of €99,750.
Investment and Shareholders Agreement
Series A Shareholders Agreement
We and the shareholders who subscribed for Series A preferred shares in the Series A financing entered into a shareholders agreement, dated June 9, 2017 (the "Shareholders Agreement"). The Shareholders Agreement provides for certain restrictions on the shareholders party thereto, including restrictions on transfer of the Series A preferred shares, as well as certain tag-along rights, drag-along rights, demand rights, rights of first offer and rights of first refusal. The Shareholders Agreement was terminated as a result of the corporate reorganization.
Series A Investment Agreement
We and the shareholders who subscribed for Series A preferred shares in the Series A financing entered into an investment agreement, dated June 9, 2017 (the "Investment Agreement"). The Investment Agreement provides for the shareholders' subscription obligations and payment obligations in connection with the Series A financing. According to the agreement, the initial investors were entitled to subscribe for 34,010 additional authorized Series A preferred shares at a price subject to adjustment based on certain thresholds. Such additional shares were issued on May 22, 2018. The Investment Agreement was terminated as a result of the corporate reorganization.
Series A Extension Agreement
We and select shareholders who subscribed for Series A preferred shares in the Series A financing entered into an extension investment agreement, dated October 1, 2018 (the "Series A Extension Agreement"). The Series A Extension Agreement provides for the shareholders' subscription obligations and payment obligations in connection with the Series A extension financing. According to the agreement, the initial investors were entitled to subscribe for 26,162 additional authorized Series A preferred shares at a price subject to adjustment based on certain thresholds. Such additional shares
142
were issued on November 7, 2018. The Series A Extension Agreement was terminated as a result of the corporate reorganization.
Payments for IT and Consulting Services
In the years ended December 31, 2017, 2018 and 2019, we incurred costs of €476 thousand, €nil and €nil, respectively, from CORE SE, an IT provider owned by Holger Friedrich, a member of our supervisory board, for information technology services provided to us. In the years ended December 31, 2017, 2018 and 2019, we incurred costs of €14 thousand, €64 thousand and €nil, respectively, from Equicore Beteiligungsgesellschaft GmbH, a shareholder of ours that is beneficially owned by Christoph Ehlers, a member of our supervisory board, for consultancy services provided to us. In April 2019, we signed a consulting contract with Flemming Ornskov, the chairman of our supervisory board, for corporate strategy services outside the scope of his services as the chairman of our supervisory board to be provided to us. For the year ended December 31, 2019, total costs incurred in respect of this contract were €152 thousand.
Indemnification Agreements
Our articles of association require us to indemnify members of our management board and supervisory board to the fullest extent permitted by law. We intend to enter into indemnification agreements with all members of our supervisory and management board.
C. Interests of Experts and Counsel
Not applicable.
A. Consolidated Statements and Other Financial Information
See "Item 18. Financial Statements", which contains our audited financial statements prepared in accordance with IFRS.
Legal Proceedings
Sanofi has filed an opposition proceeding in the EPO against the '725 Patent, a European patent that we own relating to our biomarker for Gaucher disease. The EPO opposition proceeding challenges the patentability of the '725 Patent in its entirety. The EPO rejected the opposition in the first instance in the oral proceedings on February 4, 2020. However, as the opposition decision may be appealed to the Board of Appeal at the EPO, we cannot predict the outcome of the proceedings. If the opponent appeals the opposition decision and the Board of Appeal does not uphold the opposition decision, the '725 Patent may still be revoked or maintained in amended form, in whole or in part, which could materially harm our business. For more information regarding risks related to intellectual property, including this opposition proceeding, see "Item 3. Key Information—D. Risk Factors—Intellectual Property Risks Related to Our Business."
In May 2016, we were informed in writing by the Universitair Medisch Centrum Utrecht ("UMCU") that a claim had been initiated against UMCU regarding a prenatal diagnostic test that we conducted at UMCU's request which failed to identify a specific mutation present in a patient. On October 1, 2018, the UMCU and Neon Underwriting Limited brought an action at the Regional Court of Rostock (Landgericht Rostock), Germany against us for damages, alleging that our negligence in performing the test resulted in the misdiagnosis of the patient. With the action, UMCU is seeking recovery for compensatory damages as a result of the alleged misdiagnosis. By court order of November 8, 2018, the Regional Court of Rostock set the amount in dispute at €880 thousand and
143
opened the written preliminary proceedings against the Company. On November 12, 2018, we submitted a notice to the Regional Court of Rostock of our intention to defend against the claim. On January 3, 2019 we filed a motion to dismiss in which we denied the merits of the claim. UMCU and Neon Underwriting Limited responded to this motion on March 15, 2019 with a statement of reply, and the parties made several court filings setting out their arguments since. By order dated June 3, 2019, the Regional Court of Rostock provided a first set of questions to be answered by an expert witness. Following a request by the Court, the Director of the Institute of Genetics at the University of Bonn recommended a professor for human genetics from the University of Aachen to be appointed as such expert witness in this case. We agreed to such recommendation. For more information regarding risks related to liability claims, including this proceeding, see "Item 3. Key Information—D. Risk Factors—We may become subject to substantial product liability or professional liability claims that could exceed our resources."
Dividends and Dividend Policy
Under Dutch law, we may only pay dividends to the extent our shareholders' equity (eigen vermogen) exceeds the sum of the paid-up and called-up share capital plus the reserves required to be maintained by Dutch law or by our articles of association. Subject to such restrictions, the amount of any distributions will depend on many factors, such as our results of operations, financial condition, cash requirements, prospects and other factors deemed relevant by our management board and supervisory board. We have not adopted a formal dividend policy with respect to future dividends. We may adopt such a policy in the future, in which case we intend either to place a discussion of such policy on the agenda for our annual general meetings of shareholders, consistent with the DCGC, or to disclose a deviation from the DCGC in this respect in our statutory annual report.
B. Significant Changes
Except as disclosed in this Annual Report, there have been no other significant changes to our business since December 31, 2019.
A. Offer and Listing Details
Our common shares, with a par value of €0.12 per share, have traded on Nasdaq under the symbol "CNTG" since November 7, 2019.
B. Plan of Distribution
Not applicable.
C. Markets
For a description of our publicly traded common shares, see "—A. Offer and Listing Details."
D. Selling Shareholders
Not applicable.
E. Dilution
Not applicable.
144
F. Expenses of the Issue
Not applicable.
Item 10. Additional Information
A. Share Capital
Not applicable.
B. Memorandum and Articles of Association
Our shareholders adopted the Articles of Association included as Exhibit 3.1 to our revised registration statement (the "Amendment No. 1") on Form F-1 (file no. 333-234177), filed with the SEC on October 28, 2019.
We incorporate by reference into this Annual Report our Articles of Association included as Exhibit 3.1 to Amendment No. 1 (File No. 333-234177) filed with the SEC on October 28, 2019. Such description sets forth an English translation of the official Dutch version of our articles of association as currently in effect.
C. Material Contracts
Shire
We have entered into a strategic collaboration with Shire International GmbH ("Shire"), now a subsidiary of Takeda Pharmaceutical Company Limited, pursuant to a global master services agreement originally entered into in January 2015, as subsequently amended and further extended in December 2019, a supply agreement with Shire Pharmaceuticals Ireland Ltd. originally entered into in December 2013, as subsequently amended and further extended in December 2019, and two project services agreements with Shire, respectively, each as described below.
Global Master Services Agreement
Under the global master services agreement with Shire, we provide diagnostic services to Shire and its affiliates. Shire makes an annual flat-fee payment to us for performance of an unlimited number of diagnostic tests for Morbus Fabry, Morbus Gaucher, Morbus Hunter, MPS1, MPS2, MPS3, MPS4, MPS6 and MPS7. Tests for some of these diseases are eligible for incremental payments from Shire upon meeting a minimum quantity threshold. Pursuant to the most recent amendment of December 2019, the annual flat-fee payment for the year 2020 has increased and the minimum quantity threshold for the tests of some diseases has been removed.
Supply Agreement
Under the supply agreement with Shire, we develop, manufacture and supply customized CentoCards and kits for use in approximately 50 countries as requested by Shire. These kits are language-specific and include a filter-card with requested patient/clinician information, self-addressed and labeled envelopes, barcode/tracking stickers, an informed consent form and instructions. Payments are calculated at fixed and variable rates, including fixed rates per newly designed language-specific kits and related storage and quality control fees. Kits are then billed at variable rates based on volumes, with minimum order requirements. We granted Shire and its affiliates a non-exclusive license to use any intellectual property in our existing kits or these custom-developed kits as necessary to distribute and provide the kits pursuant to the agreement. The supply agreement has been extended together with the extension of the Global Master Services Agreement.
145
Project Services Agreement
We have also entered into a project services agreement with Shire dated March 2018 for a collaborative research project on HAE dried-blood-spot-based diagnostic testing screening algorithm. All charges under the agreement are to be paid by Shire. We and Shire have granted each other the limited right to use each other's data and intellectual property related to such testing and screening for the sole purpose of performing research under the agreement. Any subsequent research results and inventions achieved under the agreement will be co-owned by us and Shire, and Shire has the exclusive right, for six months after our delivery of a research report, to negotiate with us to purchase ownership of or a license to our rights in any research results or inventions on commercially reasonable terms.
In July 2019, we entered into a collaborative research agreement with Shire related to HAE Kininogen assay mass spectrometry testing and screening. We and Shire granted each other the limited right to use each other's data and intellectual property related to such testing and screening for the sole purpose of performing research under the agreement. Any subsequent research results and inventions achieved under the agreement will be co-owned by us and Shire, and Shire has the exclusive right, for six months following the delivery of our research report, to negotiate with us to purchase ownership of or a license to our rights in any such research results or inventions on commercially reasonable terms. This project described under the agreement is funded by Shire and is anticipated to be completed before December 2022.
Pfizer
Global Master Scientific Services Agreement
In July 2019, we entered into a strategic collaboration with Pfizer Inc. ("Pfizer"), pursuant to a global master scientific services agreement ("MSSA"). Further to the MSSA, we entered into a statement of work ("SOW") in November 2019 to provide sequencing services to Pfizer, for patients in the United States or Puerto Rico with transthyretin amyloid cardiomyopathy ("ATTR-CM"), patients suspected of having ATTR-CM, or individuals with a confirmed family history of hereditary ATTR-CM. Pfizer makes an annual flat-fee payment to us for performance of a limited number of diagnostic tests. In addition, CentoCards and kits will be delivered to qualified healthcare providers for sample collection. These will be billed at a fixed rate per kit to Pfizer and the fee includes access to CentoPortal. The number of diagnostic tests covered in SOW was further increased under an amended SOW entered into as of December 2019, as well as another SOW for the customization and manufacturing of CentoCards.
Data access and collaboration
In October 2019, we entered into a DACA with Pfizer, pursuant to which we granted Pfizer access to our data repository, which may be used in the discovery and validation of novel genetic and biochemical targets for the potential development of new therapies for rare diseases. See "Item 4. Information on the Company—B. Business Overview—Key Partnerships."
D. Exchange Controls
Under Dutch law, there are no exchange controls applicable to the transfer to persons outside of the Netherlands of dividends or other distributions with respect to, or of the proceeds from the sale of, shares of a Dutch company, subject to applicable restrictions under sanctions and measures, including those concerning export control, pursuant to European Union regulations, the Sanctions Act 1977 (Sanctiewet 1977) or other legislation, applicable anti-boycott regulations and similar rules.
However, we are present in some countries, and could become elsewhere, subject to strict restrictions on the movement of cash and the exchange of foreign currencies, which limits our ability to
146
use this cash across our global operations. We also face risks related to the collection of payments due to us from our major pharmaceutical partners or clients that are located in certain geographical regions with foreign currency or international monetary controls. This risk could increase as we continue our geographic expansion. In particular, for the years ended December 31, 2018 and December 31, 2019, we derived 30.9% and 28.9%, respectively, of our total revenues from our Middle East region. Certain Middle East economies have adopted or been subject to international restrictions on the ability to transfer funds out of the country and convert local currencies into euros. This may increase our costs and limit our ability to convert local currency into euros and transfer funds out of certain countries. Any shortages or restrictions may impede our ability to convert these currencies into euros and to transfer funds, including for the payment of dividends or interest or principal on our outstanding debt.
E. Taxation
The following summary contains a description of certain U.S. federal, Dutch and German income tax consequences of the acquisition, ownership and disposition of common shares, but it does not purport to be a comprehensive description of all the tax considerations that may be relevant to a decision to purchase common shares. The summary is based upon the tax laws of the United States and regulations thereunder, the tax laws of the Netherlands and regulations thereunder and the tax laws of Germany and regulations thereunder as of the date hereof, which are subject to change.
Material U.S. Federal Income Tax Considerations for U.S. Holders
The following is a description of the material U.S. federal income tax consequences to the U.S. Holders, as defined below, of owning and disposing of common shares. It does not set forth all tax considerations that may be relevant to a particular person's decision to invest in our common shares.
This section applies only to a U.S. Holder that holds common shares as capital assets for U.S. federal income tax purposes. In addition, it does not set forth all of the U.S. federal income tax consequences that may be relevant in light of the U.S. Holder's particular circumstances, including alternative minimum tax consequences, the potential application of the provisions of the Code known as the Medicare contribution tax and tax consequences applicable to U.S. Holders subject to special rules, such as:
- •
- certain financial institutions;
- •
- dealers or traders in securities who use a mark-to-market method of tax accounting;
- •
- persons holding common shares as part of a hedging transaction, straddle, wash sale, conversion transaction or other integrated transaction or
persons entering into a constructive sale with respect to the common shares;
- •
- persons whose functional currency for U.S. federal income tax purposes is not the U.S. dollar;
- •
- entities classified as partnerships for U.S. federal income tax purposes;
- •
- tax-exempt entities, including an "individual retirement account" or "Roth IRA";
- •
- persons that own or are deemed to own 10% or more of our shares (by vote or value); or
- •
- persons holding common shares in connection with a trade or business conducted outside of the United States.
If an entity that is classified as a partnership for U.S. federal income tax purposes holds common shares, the U.S. federal income tax treatment of a partner will depend on the status of the partner and the activities of the partnership. Partnerships holding common shares and partners in such partnerships should consult their tax advisers as to the particular U.S. federal income tax consequences of owning and disposing of the common shares.
147
This section is based on the Code, administrative pronouncements, judicial decisions, final, temporary and proposed Treasury regulations, and the income tax treaty between Germany and the United States (the "Treaty") all as of the date hereof, any of which is subject to change or differing interpretations, possibly with retroactive effect.
A "U.S. Holder" is a holder who, for U.S. federal income tax purposes, is a beneficial owner of common shares, who is eligible for the benefits of the Treaty and who is:
- •
- a citizen or individual resident of the United States;
- •
- a corporation, or other entity taxable as a corporation, created or organized in or under the laws of the United States, any state therein or
the District of Columbia; or
- •
- an estate or trust the income of which is subject to U.S. federal income taxation regardless of its source.
U.S. Holders should consult their tax advisers concerning the U.S. federal, state, local and non-U.S. tax consequences of owning and disposing of common shares in their particular circumstances.
Taxation of Distributions
Subject to the passive foreign investment company rules described below, distributions of cash or other property, if any, that are made on our common shares, other than certain pro rata distributions of common shares, will be treated as dividends to the extent paid out of our current or accumulated earnings and profits (as determined under U.S. federal income tax principles). For so long as our common shares are listed on Nasdaq or another established securities market in the United States or we are eligible for benefits under the Treaty, dividends paid to certain non-corporate U.S. Holders will be eligible for taxation as "qualified dividend income," which is taxable at rates not in excess of the long-term capital gain rate applicable to such U.S. Holders. U.S. Holders should consult their tax advisers regarding the availability of the reduced tax rate on dividends in their particular circumstances. The amount of a dividend will include any amounts withheld by us in respect of German income taxes. The amount of the dividend will be treated as foreign-source dividend income to U.S. Holders and will not be eligible for the dividends-received deduction available to U.S. corporations under the Code. Dividends will be included in a U.S. Holder's income on the date of the U.S. Holder's receipt of the dividend. The amount of any dividend income paid in euros will be the U.S. dollar amount calculated by reference to the exchange rate in effect on the date of actual or constructive receipt, regardless of whether the payment is in fact converted into U.S. dollars at that time. A U.S. Holder may have foreign currency gain or loss if the dividend is converted into U.S. dollars after the date of receipt.
German income taxes withheld from dividends on common shares at a rate not exceeding the rate provided by the Treaty will be eligible for credit against the U.S. Holder's U.S. federal income tax liability. German taxes withheld in excess of the rate applicable under the Treaty will not be eligible for credit against a U.S. Holder's federal income tax liability. The rules governing foreign tax credits are complex and U.S. Holders should consult their tax advisers regarding the creditability of foreign taxes in their particular circumstances. In lieu of claiming a foreign tax credit, U.S. Holders may deduct foreign taxes, including any German income tax, in computing their taxable income. An election to deduct foreign taxes instead of claiming foreign tax credits applies to all foreign taxes paid or accrued in the taxable year. If Dutch income taxes are withheld from dividends payable to U.S. Holders, U.S. Holders are urged to consult their tax advisers regarding the creditability of such Dutch income taxes against their U.S. federal income tax liabilities. See "Item 3. Key Information—D. Risk factors—Certain Factors Relating to Our Common Shares—If we do pay dividends, we may need to withhold tax on such dividends payable to holders of our shares in both Germany and the Netherlands."
148
Sale or Other Disposition of Common Shares
Subject to the passive foreign investment company rules described below, gain or loss realized on the sale or other disposition of common shares will be capital gain or loss, and will be long-term capital gain or loss if the U.S. Holder held the common shares for more than one year. The amount of the gain or loss will equal the difference between the U.S. Holder's tax basis in the common shares disposed of and the amount realized on the disposition, in each case as determined in U.S. dollars.
Passive Foreign Investment Company ("PFIC") Rules
Under the Code, we will be a PFIC for any taxable year in which, after the application of certain "look-through" rules with respect to subsidiaries, either (i) 75% or more of our gross income consists of "passive income" or (ii) 50% or more of the average quarterly value of our assets consist of assets that produce, or are held for the production of, "passive income." For purposes of the above calculations, we will be treated as if we hold our proportionate share of the assets of, and receive directly our proportionate share of the income of, any other corporation in which we directly or indirectly own at least 25%, by value, of the shares of such corporation.
Passive income includes, among other things, interest, dividends, rents, certain non-active royalties and capital gains. Based on our current operations, income, assets and certain estimates and projections, including as to the relative values of our assets, we do not believe that we were a PFIC for our 2019 taxable year. However, there can be no assurance that the IRS will agree with our conclusion. In addition, whether we will be a PFIC in 2020 or any future taxable year is uncertain because, among other things, (i) we currently own, and expect to continue to own, a substantial amount of passive assets, including cash, (ii) the valuation of our assets that generate non-passive income for PFIC purposes, including our intangible assets, is uncertain and may vary substantially over time and (iii) the composition of our income may vary substantially over time. Accordingly, there can be no assurance that we will not be a PFIC for any taxable year. If we are a PFIC for any year during which a U.S. Holder holds common shares, we would continue to be treated as a PFIC with respect to that U.S. Holder for all succeeding years during which the U.S. Holder holds common shares, even if we ceased to meet the threshold requirements for PFIC status, unless the U.S. Holder makes a valid deemed sale or deemed dividend election under the applicable Treasury regulations with respect to its common shares.
If we were a PFIC for any taxable year during which a U.S. Holder held common shares (assuming such U.S. Holder has not made a timely mark-to-market election, as described below), gain recognized by a U.S. Holder on a sale or other disposition (including certain pledges) of the common shares would be allocated ratably over the U.S. Holder's holding period for the common shares. The amounts allocated to the taxable year of the sale or other disposition and to any year before we became a PFIC would be taxed as ordinary income. The amount allocated to each other taxable year would be subject to tax at the highest rate in effect for individuals or corporations, as appropriate, for that taxable year, and an interest charge would be imposed on the amount allocated to that taxable year. Further, to the extent that any distribution received by a U.S. Holder on its common shares exceeds 125% of the average of the annual distributions on the common shares received during the preceding three years or the U.S. Holder's holding period, whichever is shorter, that distribution would be subject to taxation in the same manner as gain, described immediately above.
A U.S. Holder can avoid certain of the adverse rules described above by making a mark-to-market election with respect to its common shares, provided that the common shares are "marketable." Common shares will be marketable if they are "regularly traded" on a "qualified exchange" or other market within the meaning of applicable Treasury regulations. If a U.S. Holder makes the mark-to-market election, it will recognize as ordinary income any excess of the fair market value of the common shares at the end of each taxable year over their adjusted tax basis, and will recognize an
149
ordinary loss in respect of any excess of the adjusted tax basis of the common shares over their fair market value at the end of the taxable year (but only to the extent of the net amount of income previously included as a result of the mark-to-market election). If a U.S. Holder makes the election, the U.S. Holder's tax basis in the common shares will be adjusted to reflect the income or loss amounts recognized. Any gain recognized on the sale or other disposition of common shares in a year when we are a PFIC will be treated as ordinary income and any loss will be treated as an ordinary loss (but only to the extent of the net amount of income previously included as a result of the mark-to-market election).
In addition, in order to avoid the application of the foregoing rules, a United States person that owns stock in a PFIC for U.S. federal income tax purposes may make a QEF Election with respect to such PFIC if the PFIC provides the information necessary for such election to be made. If a United States person makes a QEF Election with respect to a PFIC, the United States person will be currently taxable on its pro rata share of the PFIC's ordinary earnings and net capital gain (at ordinary income and capital gain rates, respectively) for each taxable year that the entity is classified as a PFIC and will not be required to include such amounts in income when actually distributed by the PFIC. We do not intend to provide information necessary for U.S. Holders to make QEF Elections.
In addition, if we were a PFIC or, with respect to a particular U.S. Holder, were treated as a PFIC for the taxable year in which we paid a dividend or for the prior taxable year, the preferential dividend rates discussed above with respect to dividends paid to certain non-corporate U.S. Holders would not apply.
If a U.S. Holder owns common shares during any year in which we are a PFIC, the U.S. Holder must file annual reports, containing such information as the U.S. Treasury may require on IRS Form 8621 (or any successor form) with respect to us, with the U.S. Holder's federal income tax return for that year, unless otherwise specified in the instructions with respect to such form.
U.S. Holders should consult their tax advisers concerning our potential PFIC status and the potential application of the PFIC rules.
Information Reporting and Backup Withholding
Payments of dividends and sales proceeds that are made within the United States or through certain U.S.-related financial intermediaries are subject to information reporting, and may be subject to backup withholding, unless (i) the U.S. Holder is a corporation or other exempt recipient or (ii) in the case of backup withholding, the U.S. Holder provides a correct taxpayer identification number and certifies that it is not subject to backup withholding. The amount of any backup withholding from a payment to a U.S. Holder will be allowed as a credit against the holder's U.S. federal income tax liability and may entitle it to a refund, provided that the required information is timely furnished to the IRS.
Reporting With Respect to Foreign Financial Assets
Certain U.S. Holders who are individuals and certain entities may be required to report information relating to an interest in our common shares by filing a Form 8398 with their U.S. federal income tax return, subject to certain exceptions (including an exception for common shares held in accounts maintained by certain U.S. financial institutions). Failure to file a Form 8398 where required can result in monetary penalties and the extension of the relevant statute of limitations with respect to all or a part of the relevant U.S. tax return. U.S. Holders should consult their tax advisers regarding this reporting requirement.
150
Material Dutch Tax Considerations
The following is a summary of certain material Dutch tax consequences of the acquisition, holding and disposal of common shares. This summary does not purport to describe all possible tax considerations or consequences that may be relevant to a holder or prospective holder of common shares and does not purport to deal with the tax consequences applicable to all categories of investors, some of which (such as trusts or similar arrangements) may be subject to special rules. In view of its general nature, this summary should be treated with corresponding caution.
Except as otherwise indicated, this summary is based on the tax laws of the Netherlands, published regulations thereunder and published authoritative case law, all as in effect on the date hereof, and all of which are subject to change, possibly with retroactive effect. Where the summary refers to "the Netherlands" or "Dutch" it refers only to the part of the Kingdom of the Netherlands located in Europe. The applicable tax laws or interpretations thereof may change, or the relevant facts and circumstances may change, and such changes may affect the contents of this section, which will not be up-dated to reflect any such changes.
This discussion is for general information purposes only and is not Dutch tax advice or a complete description of all Dutch tax consequences relating to the acquisition, holding and disposal of the common shares. Holders or prospective holders of common shares should consult their own tax ad-visors regarding the Dutch tax consequences relating to the acquisition, holding and disposal of the common shares in light of their particular circumstances.
Please note that the summary does not describe the Dutch tax consequences for:
(i) holders of common shares if such holders, and in the case of individuals, such holder's partner or certain of its relatives by blood or marriage in the direct line (including foster children), have a substantial interest (aanmerkelijk belang) or deemed substantial interest (fictief aanmerkelijk belang) in us under the Dutch Income Tax Act 2001 (Wet inkomstenbelasting 2001). A holder of securities in a company is considered to hold a substantial interest in such company, if such holder alone or, in the case of individuals, together with such holder's partner (as defined in the Dutch Income Tax Act 2001), directly or indirectly, holds (i) an interest of 5% or more of the total issued and outstanding capital of that company or of 5% or more of the issued and outstanding capital of a certain class of shares of that company; or (ii) rights to acquire, directly or indirectly, such interest; or (iii) certain profit sharing rights in that company that relate to 5% or more of the company's annual profits or to 5% or more of the company's liquidation proceeds. A deemed substantial interest may arise if a substantial interest (or part thereof) in a company has been disposed of, or is deemed to have been dis-posed of, on a non-recognition basis;
(ii) holders of common shares, if the common shares held by such holders qualify or qualified as a participation (deelneming) for purposes of the Dutch Corporate Income Tax Act 1969 (Wet op de vennootschapsbelasting 1969). Generally, a holder's shareholding of 5% or more in a company's nominal paid-up share capital qualifies as a participation. A holder may also have a participation if such holder does not have a shareholding of 5% or more but a related entity (statutorily defined term) has a participation or if the company in which the shares are held is a related entity (statutorily defined term).
(iii) pension funds, investment institutions (fiscale beleggingsinstellingen), exempt investment institutions (vrijgestelde beleggingsinstellingen) (as defined in the Dutch Corporate Income Tax Act 1969) and other entities that are, in whole or in part, not subject to or exempt from Dutch corporate income tax as well as entities that are exempt from corporate income tax in their country of residence, such country of residence being another state of the European Union, Norway, Liechtenstein, Iceland or any other state with which the Netherlands has agreed to exchange information in line with international standards; and
151
(iv) holders of common shares who are individuals for whom the common shares or any benefit derived from the common shares are a remuneration or deemed to be a remuneration for activities performed by such holders or certain individuals related to such holders (as defined in the Dutch Income Tax Act 2001).
Withholding tax
We are required to withhold Dutch dividend withholding tax at a rate of 15% from dividends distributed by us (which withholding tax will not be borne by us, but will be withheld by us from the gross dividends paid on the common shares). However, as long as we continue to have our place of effective management in Germany, and not in the Netherlands, under the Convention between the Federal Republic of Germany and the Netherlands for the avoidance of double taxation with respect to taxes on income of 2012, we will be considered to be exclusively tax resident in Germany and we will not be required to withhold Dutch dividend withholding tax. This exemption from withholding does not apply to dividends distributed by us to a holder who is resident or deemed to be resident in the Netherlands for Dutch income tax purposes or Dutch corporate income tax purposes or to holders of common shares that are neither resident nor deemed to be resident of the Netherlands if the common shares are attributable to a Dutch permanent establishment of such non-resident holder, in which case the following paragraph applies. See "Item 3. Key Information—D. Risk Factors—If we do pay dividends, we may need to withhold tax on such dividends payable to holders of our shares in both Germany and the Netherlands"
Dividends distributed by us to individuals and corporate legal entities who are resident or deemed to be resident in the Netherlands for Dutch (corporate) income tax purposes ("Dutch Resident Individuals" and "Dutch Resident Entities," as the case may be) or to holders of common shares that are neither resident nor deemed to be resident of the Netherlands if the common shares are attributable to a Dutch permanent establishment of such non-resident holder are subject to Dutch dividend withholding tax at a rate of 15%.
The expression "dividends distributed" includes, among other things:
- •
- distributions in cash or in kind, deemed and constructive distributions and repayments of paid-in capital not recognized for Dutch dividend
withholding tax purposes;
- •
- liquidation proceeds, proceeds of redemption of shares, or proceeds of the repurchase of shares by us or one of our subsidiaries or other
affiliated entities to the extent such proceeds exceed the average paid-in capital of those shares as recognized for purposes of Dutch dividend withholding tax;
- •
- an amount equal to the par value of shares issued or an increase of the par value of shares, to the extent that it does not appear that a
contribution, recognized for purposes of Dutch dividend withholding tax, has been made or will be made; and
- •
- partial repayment of the paid-in capital, recognized for purposes of Dutch dividend withholding tax, if and to the extent that we have net profits (zuivere winst), unless (i) the general meeting has resolved in advance to make such repayment and (ii) the par value of the shares concerned has been reduced by an equal amount by way of an amendment to our articles of association.
Dutch Resident Individuals and Dutch Resident Entities can generally credit the Dutch dividend withholding tax against their income tax or corporate income tax liability and to a refund of any residual Dutch dividend withholding tax. The same generally applies to holders of common shares that are neither resident nor deemed to be resident of the Netherlands if the common shares are attributable to a Dutch permanent establishment of such non-resident holder.
152
Pursuant to legislation to counteract "dividend stripping", a reduction, exemption, credit or refund of Dutch dividend withholding tax is denied if the recipient of the dividend is not the beneficial owner as described in the Dutch Dividend Withholding Tax Act 1965 (Wet op de dividendbelasting 1965). This legislation generally targets situations in which a shareholder retains its economic interest in shares but reduces the withholding tax costs on dividends by a transaction with another party. It is not required for these rules to apply that the recipient of the dividends is aware that a dividend strip-ping transaction took place. The Dutch State Secretary of Finance takes the position that the definition of beneficial ownership introduced by this legislation will also be applied in the context of a double taxation convention.
Taxes on income and capital gains
Dutch Resident Entities
Generally speaking, if the holder of common shares is an entity that is a resident or deemed to be resident of the Netherlands for Dutch corporate income tax purposes, any payment under the common shares or any gain or loss realized on the disposal or deemed disposal of the common shares is subject to Dutch corporate income tax at a rate of 16.5% with respect to taxable profits up to €200,000 and 25% with respect to taxable profits in excess of that amount (rates and brackets for 2020).
Dutch Resident Individuals
If the holder of common shares is an individual resident or deemed to be resident of the Netherlands for Dutch income tax purposes, any payment on the common shares or any gain or loss realized on the disposal or deemed disposal of the common shares is taxable at the progressive Dutch income tax rates (with a maximum of 49.50% in 2020), if:
(i) the common shares are attributable to an enterprise from which the holder of common shares derives a share of the profit, whether as an entrepreneur (ondernemer) or as a person who has a co-entitlement to the net worth (medegerechtigd tot het vermogen) of such enterprise without being a shareholder (as defined in the Dutch Income Tax Act 2001); or
(ii) the holder of shares is considered to perform activities with respect to the common shares that go beyond ordinary asset management (normaal, actief vermogensbeheer) or derives benefits from the common shares that are taxable as benefits from other activities (resultaat uit overige werkzaamheden).
If the above-mentioned conditions (i) and (ii) do not apply to the individual holder of common shares, such holder will be taxed annually on a deemed return (with a maximum of 5.33% in 2020) on the individual's net investment assets (rendementsgrondslag) for the year, insofar the individual's net investment assets for the year exceed a statutory threshold (heffingvrij vermogen). The deemed return on the individual's net investment assets for the year is taxed at a rate of 30%. Actual income, gains or losses in respect of the common shares are as such not subject to Dutch income tax.
The net investment assets for the year are the fair market value of the investment assets less the allowable liabilities on January 1 of the relevant calendar year. The common shares are included as investment assets. For the net investment assets on January 1, 2020, the deemed return ranges from 1.7991% up to 5.33% (depending on the aggregate amount of the net investment assets of the individual on January 1, 2020). The deemed return will be adjusted annually on the basis of historic market yields.
Non-residents of the Netherlands
A holder of common shares that is neither a Dutch Resident Entity nor a Dutch Resident Individual will not be subject to Dutch taxes on income or capital gains in respect of any payment
153
under the common shares or in respect of any gain or loss realized on the disposal or deemed disposal of the common shares, provided that:
(i) such holder does not have an interest in an enterprise or deemed enterprise (as defined in the Dutch Income Tax Act 2001 and the Dutch Corporate Income Tax Act 1969) which, in whole or in part, is either effectively managed in the Netherlands or carried on through a permanent establishment, a deemed permanent establishment or a permanent representative in the Netherlands and to which enterprise or part of an enterprise the common shares are attributable; and
(ii) in the event the holder is an individual, such holder does not carry out any activities in the Netherlands with respect to the common shares that go beyond ordinary asset management and does not derive benefits from the shares that are taxable as benefits from other activities in the Netherlands.
Gift and inheritance taxes
Residents of the Netherlands
Gift or inheritance taxes will arise in the Netherlands with respect to a transfer of common shares by way of a gift by, or on the death of, a holder of such common shares who is resident or deemed resident of the Netherlands at the time of the gift or the holder's death.
Non-residents of the Netherlands
No gift or inheritance taxes will arise in the Netherlands with respect to a transfer of the shares by way of gift by, or on the death of, a holder of common shares who is neither resident nor deemed to be resident of the Netherlands, unless:
(i) in the case of a gift of common shares by an individual who at the date of the gift was neither resident nor deemed to be resident of the Netherlands, such individual dies within 180 days after the date of the gift, while being resident or deemed to be resident of the Netherlands; or
(ii) the transfer is otherwise construed as a gift or inheritance made by, or on behalf of, a person who, at the time of the gift or death, is or is deemed to be resident of the Netherlands.
For purposes of Dutch gift and inheritance taxes, amongst others, a person that holds the Dutch nationality will be deemed to be resident of the Netherlands if such person has been resident in the Netherlands at any time during the ten years preceding the date of the gift or such person's death. Additionally, for purposes of Dutch gift tax, amongst others, a person not holding the Dutch nationality will be deemed to be resident of the Netherlands if such person has been resident in the Netherlands at any time during the twelve months preceding the date of the gift. Applicable tax treaties may override deemed residency.
Value added tax (VAT)
No Dutch VAT will be payable by a holder of common shares in respect of any payment in consideration for the holding or disposal of the common shares.
Other taxes and duties
No Dutch registration tax, stamp duty or any other similar documentary tax or duty will be payable by a holder of common shares in respect of any payment in consideration for the holding or disposal of the common shares.
154
Material German Tax Considerations
The following section is the opinion of Taylor Wessing Partnerschaftsgesellschaft mbB ("German Tax Counsel") of the material German tax considerations that become relevant when purchasing, holding or transferring the Company's shares. The Company expects and intends to have its sole place of management in Germany and, therefore, qualifies as a corporation subject to German unlimited income taxation; however, because a company's tax residency depends on future facts regarding the location in which the company is managed and controlled, German Tax Counsel cannot opine as to whether the Company qualifies as a corporation subject to German unlimited income taxation. This section does not set forth all German tax aspects that may be relevant for shareholders. The section is based on the German tax law applicable as of the date of this Prospectus. It should be noted that the law may change following the issuance of this Prospectus and that such changes may have retroactive effect.
The material German tax principles of purchasing, owning and transferring of shares are set forth in the following. This section does not purport to be a comprehensive or complete analysis or listing of all potential tax effects of the purchase, ownership or disposition of shares and does not set forth all tax considerations that may be relevant to a particular person's decision to acquire common shares. All of the following is subject to change. Such changes could apply retroactively and could affect the consequences set forth below. This section does not refer to any U.S. Foreign Account Tax Compliance Act aspects.
Shareholders are advised to consult their own tax advisers with regard to the application of German tax law to their particular situations, in particular with respect to the procedure to be complied with to obtain a relief of withholding tax on dividends and on capital gains (Kapitalertragsteuer) and with respect to the influence of double tax treaty provisions, as well as any tax consequences arising under the laws of any state, local or other foreign jurisdiction. For German tax purposes, a shareholder may include an individual who or an entity that does not have the legal title to the shares, but to whom nevertheless the shares are attributed, based either on such individual or entity owning a beneficial interest in the shares or based on specific statutory provisions.
This section does not constitute a particular tax advice. Potential purchasers of the Company's shares are urged to consult their own tax advisers regarding the tax consequences of the purchase, ownership and disposition of shares in light of their particular circumstances.
Taxation of Dividends
Withholding Tax on Dividends
Dividends distributed from a company to its shareholders are subject to withholding tax, subject to certain exemptions (for example, repayments of capital from the tax equity account (steuerliches Einlagekonto)), as described in the following. The withholding tax rate is 25% plus 5.5% solidarity surcharge (Solidaritätszuschlag) thereon (in total 26.375%) of the gross dividend approved by the ordinary shareholders' meeting. Withholding tax is to be withheld and passed on for the account of the shareholders by a domestic branch of a domestic or foreign credit or financial services institution (Kredit- und Finanzdienstleistungsinstitut), by the domestic securities trading company (inländisches Wertpapierhandelsunternehmen) or a domestic securities trading bank (inländische Wertpapierhandelsbank) which keeps and administers the shares and disburses or credits the dividends or disburses the dividends to a foreign agent, or by the securities custodian bank (Wertpapiersammelbank) to which the shares were entrusted for collective custody if the dividends are distributed to a foreign agent by such securities custodian bank (which is referred to as the "Dividend Paying Agent"). In case the shares are not held in collective deposit with a Dividend Paying Agent, the Company is responsible for withholding and remitting the tax to the competent tax office.
155
Such withholding tax is levied and withheld irrespective of whether and to what extent the dividend distribution is taxable at the level of the shareholder and whether the shareholder is a person residing in Germany or in a foreign country.
In the case of dividends distributed to a company within the meaning of Art. 2 of the amended EU Directive 2011/96/EU of the Council of November 30, 2011 (the "EU Parent Subsidiary Directive") domiciled in another Member State of the European Union, an exemption from withholding tax will be granted upon request if further prerequisites are satisfied (Freistellung im Steuerabzugsverfahren). This also applies to dividends distributed to a permanent establishment located in another Member State of the European Union of such a parent company or of a parent company tax resident in Germany if the participation in the Company is effectively connected with this permanent establishment. The key prerequisite for the application of the EU Parent Subsidiary Directive is that the shareholder has held a direct participation in the share capital of the Company of at least 10% for at least one year.
The withholding tax on distributions to other foreign resident shareholders is reduced in accordance with a double taxation treaty if Germany has concluded such double taxation treaty with the country of residence of the shareholder and if the shareholder does not hold his shares either as part of the assets of a permanent establishment or a fixed place of business in Germany or as business assets for which a permanent representative has been appointed in Germany. The reduction of the withholding tax is procedurally granted in such a manner that the difference between the total amount withheld, including the solidarity surcharge, and the tax liability determined on the basis of the tax rate set forth in the applicable double taxation treaty (15% unless further qualifications are met) is refunded by the German tax administration upon request (Federal Central Office for Taxes (Bundeszentralamt für Steuern), main office in Bonn-Beuel, An der Küppe 1, 53225 Bonn, Germany).
In the case of dividends received by corporations whose statutory seat and effective place of management are not located in Germany and who are therefore not tax resident in Germany, two-fifths of the withholding tax deducted and remitted are refunded without the need to fulfill all prerequisites required for such refund under the EU Parent Subsidiary Directive or under a double taxation treaty or if no double taxation treaty has been concluded between the state of residence of the shareholder.
In order to receive a refund pursuant to a double taxation treaty or the aforementioned option for foreign corporations, the shareholder has to submit a completed form for refund (available at the Federal Central Office for Taxes (http://www.bzst.de) as well as at the German embassies and consulates) together with a withholding tax certificate (Kapitalertragsteuerbescheinigung) issued by the institution that withheld the tax.
The exemption from withholding tax in accordance with the EU Parent Subsidiary Directive or a double tax treaty and the aforementioned options for a refund of the withholding tax (with or without protection under a double taxation treaty) depend on whether certain additional prerequisites (in particular so-called substance requirements) are fulfilled. The applicable withholding tax relief will only be granted if the preconditions of the German anti avoidance rules (so called Directive Override or Treaty Override), in particular Section 50d, paragraph 3, German Income Tax Act (Einkommensteuergesetz) are fulfilled.
The aforementioned reductions of (or exemptions from) withholding tax are further restricted if (i) the applicable double taxation treaty provides for a tax reduction resulting in an applicable tax rate of less than 15% and (ii) the shareholder is not a corporation that directly holds at least 10% in the equity capital of the Company and is subject to tax on its income and profits in its state of residence without being exempt. In this case, the reduction of (or exemption from) withholding tax is subject to the following three cumulative prerequisites: (i) the shareholder must qualify as beneficial owner of the shares in the Company for a minimum holding period of 45 consecutive days occurring within a period of 45 days prior and 45 days after the due date of the dividends, (ii) the shareholder has to bear at least 70% of the change in value risk related to the shares in the Company during the minimum
156
holding period without being directly or indirectly hedged and (iii) the shareholder must not be required to fully or largely compensate directly or indirectly the dividends to third parties. However, these further prerequisites do not apply if the shareholder has been the beneficial owner of the shares in the Company for at least one uninterrupted year upon receipt of the dividends.
For individual or corporate shareholders tax resident outside Germany not holding the shares through a permanent establishment (Betriebsstätte) in Germany or as business assets (Betriebsvermögen) for which a permanent representative (ständiger Vertreter) has been appointed in Germany, the remaining and paid withholding tax (if any) is final (i.e., not refundable) and settles the shareholder's limited tax liability in Germany. For individual or corporate shareholders tax resident in Germany (that are, for example, shareholders whose residence, domicile, registered office or place of management is located in Germany) holding their shares as business assets, as well as for shareholders tax resident outside of Germany holding their shares through a permanent establishment in Germany or as business assets for which a permanent representative has been appointed in Germany, the withholding tax withheld (including solidarity surcharge) can be credited against the shareholder's personal income tax or corporate income tax liability in Germany. Any withholding tax (including solidarity surcharge) in excess of such tax liability is refunded. For individual shareholders tax resident in Germany holding the Company's shares as private assets, the withholding tax is a final tax (Abgeltungsteuer), subject to the exceptions described in the following section.
Pursuant to special rules on the restriction of withholding tax credit, the credit of withholding tax is subject to the following three cumulative prerequisites: (i) the shareholder must qualify as beneficial owner of the shares in the Company for a minimum holding period of 45 consecutive days occurring within a period of 45 days prior and 45 days after the due date of the dividends, (ii) the shareholder has to bear at least 70% of the change in value risk related to the shares in the Company during the minimum holding period without being directly or indirectly hedged and (iii) the shareholder must not be required to fully or largely compensate directly or indirectly the dividends to third parties. Absent the fulfillment of all of the three prerequisites, three-fifths of the withholding tax imposed on the dividends must not be credited against the shareholder's (corporate) income tax liability, but may, upon application, be deducted from the shareholder's tax base for the relevant assessment period. A shareholder that has received gross dividends without any deduction of withholding tax due to a tax exemption without qualifying for a full tax credit has to notify the competent local tax office accordingly and has to make a payment in the amount of the omitted withholding tax deduction. The special rules on the restriction of withholding tax credit do not apply to a shareholder whose overall dividend earnings within an assessment period do not exceed € 20,000 or that has been the beneficial owner of the shares in the Company for at least one uninterrupted year upon receipt of the dividends.
Taxation of dividend income of shareholders tax resident in Germany holding the Company's shares as private assets
For individual shareholders (individuals) resident in Germany holding the Company's shares as private assets, dividends are subject to a flat tax rate which is satisfied by the withholding tax actually withheld (Abgeltungsteuer). Accordingly, dividend income will be taxed at a flat tax rate of 25% plus 5.5% solidarity surcharge thereon (in total 26.375%) and church tax (Kirchensteuer) in case the shareholder is subject to church tax because of his individual circumstances. An automatic procedure for deduction of church tax by way of withholding will apply to shareholders being subject to church tax unless the shareholder has filed a blocking notice (Sperrvermerk) with the German Federal Tax Office (details related to the computation of the concrete tax rate including church tax are to be discussed with the individual tax adviser of the relevant shareholder). Except for an annual lump sum savings allowance (Sparer-Pauschbetrag) of up to €801 (for individual filers) or up to € 1,602 (for married couples and for partners in accordance with the registered partnership law (Gesetz über die Eingetragene
157
Lebenspartnerschaft) filing jointly), private individual shareholders will not be entitled to deduct expenses incurred in connection with the capital investment from their dividend income.
The income tax owed for the dividend income is satisfied by the withholding tax withheld by the Dividend Paying Agent. However, if the flat tax results in a higher tax burden as opposed to the private shareholder's individual tax rate, the private shareholder can opt for taxation at his individual personal income tax rate. In that case, the final withholding tax will be credited against the income tax. However, pursuant to the German tax authorities and a court ruling, private shareholders are nevertheless not entitled to deduct expenses incurred in connection with the capital investment from their income. The option can be exercised only for all capital income from capital investments received in the relevant assessment period uniformly, and married couples as well as partners in accordance with the registered partnership law filing jointly may only jointly exercise the option.
Exceptions from the flat tax rate (satisfied by withholding at source) (Abgeltungsteuer) may apply—that is, only upon application—for shareholders who have a shareholding of at least 25% in a company and for shareholders who have a shareholding of at least 1% in the Company and work for a company in a professional capacity. In such a case, the same rules apply as for sole proprietors holding the shares as business assets. See "—Taxation of dividend income of shareholders tax resident in Germany holding the Company's shares as business assets—Sole proprietors."
Taxation of dividend income of shareholders tax resident in Germany holding the Company's shares as business assets
If a shareholder holds the Company's shares as business assets, the taxation of the dividend income depends on whether the respective shareholder is a corporation, a sole proprietor or a partnership.
Corporations
Dividend income of corporate shareholders is exempt from corporate income tax, provided that the incorporated entity holds a direct participation of at least 10% in the share capital of a company at the beginning of the calendar year in which the dividends are paid. The acquisition of a participation of at least 10% in the course of a calendar year is deemed to have occurred at the beginning of such calendar year for the purpose of this rule. Participations in the share capital of the Company which a corporate shareholder holds through a partnership, including co-entrepreneurships (Mitunternehmerschaften), are attributable to such corporate shareholder only on a pro rata basis at the ratio of the interest share of the corporate shareholder in the assets of the relevant partnership. However, 5% of the tax exempt dividends are deemed to be non-deductible business expenses for tax purposes and therefore are subject to corporate income tax (plus solidarity surcharge) and trade tax, i.e., tax exemption of 95%. Business expenses incurred in connection with the dividends received are entirely tax-deductible.
For trade tax purposes the entire dividend income is subject to trade tax (i.e., the tax-exempt dividends must be added back when determining the trade taxable income), unless the corporation shareholder holds at least 15% of the Company's registered share capital at the beginning of the relevant tax assessment period (Erhebungszeitraum). In case of an indirect participation via a partnership please refer to the section "Partnerships" below.
If the shareholding is below 10% in the share capital, dividends are taxable at the applicable corporate income tax rate of 15% plus 5.5% solidarity surcharge thereon and trade tax (the rate of which depends on the municipalities the corporate shareholder resides in).
Special regulations apply which abolish the 95% tax exemption if the Company's shares are held as trading portfolio assets in the meaning of Section 340e of the German commercial code
158
(Handelsgesetzbuch) by (i) a credit institution (Kreditinstitut), (ii) a financial service institution (Finanzdienstleistungsinstitut) or (iii) a financial enterprise within the meaning of the German Banking Act (Kreditwesengesetz), in case more than 50% of the shares of such financial enterprise are held directly or indirectly by a credit institution or a financial service institution, as well as by a life insurance company, a health insurance company or a pension fund in case the shares are attributable to the capital investments, resulting in fully taxable income.
Sole proprietors
For sole proprietors (individuals) resident in Germany holding shares as business assets dividends are subject to the partial income rule (Teileinkünfteverfahren). Accordingly, only (i) 60% of the dividend income will be taxed at his/her individual personal income tax rate plus 5.5% solidarity surcharge thereon and church tax (if applicable) and (ii) 60% of the business expenses related to the dividend income are deductible for tax purposes. In addition, the dividend income is entirely subject to trade tax if the shares are held as business assets of a permanent establishment in Germany within the meaning of the German Trade Tax Act (Gewerbesteuergesetz), unless the shareholder holds at least 15% of the Company's registered share capital at the beginning of the relevant assessment period. The trade tax levied will be eligible for credit against the shareholder's personal income tax liability based on the applicable municipal trade tax rate and the individual tax situation of the shareholder.
Partnerships
In case shares are held by a partnership, the partnership itself is not subject to corporate income tax or personal income tax. In this regard, corporate income tax or personal income tax (and church tax, if applicable) as well as solidarity surcharge, are levied only at the level of the partner with respect to their relevant part of the profit and depending on their individual circumstances.
If the partner is a corporation, the dividend income will be subject to corporate income tax plus solidarity surcharge. See "—Corporations."
If the partner is a sole proprietor (individual), the dividend income will be subject to the partial income rule. See "—Sole Proprietors."
The dividend income is subject to trade tax at the level of the partnership (provided that the partnership is liable to trade tax), unless the partnership holds at least 15% of a company's registered share capital at the beginning of the relevant assessment period, in which case the dividend income is exempt from trade tax. There are no explicit statutory provisions concerning the taxation of dividends with regard to a corporate shareholder of the partnership. However, trade tax will be levied on 5% of the dividends to the extent they are attributable to the shares of such corporate partners to whom at least 10% of the shares of the Company are attributable on a look-through basis, since such portion of the dividends will be deemed to be non-deductible business expenses.
If a partner is an individual, depending on the applicable municipal trade tax rate and the individual tax situation, the trade tax paid at the level of the partnership is partly or entirely be credited against the partner's personal income tax liability.
In case of a corporation being a partner, special regulations will apply with respect to trading portfolio assets of credit institutions, financial service institutions or financial enterprises within the meaning of the German Banking Act (Kreditwesengesetz) or life insurance companies, health insurance companies or pension funds. See "—Corporations."
Thus, the actual trade tax charge, if any, at the level of the partnership depends on the shareholding quota of the partnership and the nature of the partners (e.g., individual or corporation).
159
Taxation of dividend income of shareholders tax resident outside of Germany
For foreign individual or corporate shareholders tax resident outside of Germany not holding the shares through a permanent establishment in Germany or as business assets for which a permanent representative has been appointed in Germany, the deducted withholding tax (possibly reduced by way of a tax relief under a double tax treaty or domestic tax law, such as in connection with the EU Parent Subsidiary Directive) is final (that is, not refundable) and settles the shareholder's limited tax liability in Germany, unless the shareholder is entitled to apply for a withholding tax refund or exemption.
In contrast, individual or corporate shareholders tax resident outside of Germany holding the Company's shares through a permanent establishment in Germany or as business assets for which a permanent representative has been appointed in Germany are subject to the same rules as applicable (and described above) to shareholders resident in Germany holding the shares as business assets. The withholding tax withheld (including solidarity surcharge) is credited against the shareholder's personal income tax or corporate income tax liability in Germany.
Taxation of Capital Gains
Withholding tax on capital gains
Capital gains realized on the disposal of shares are only subject to withholding tax if a German branch of a German or foreign credit or financial institution, a German securities trading Company or a German securities trading bank stores or administrates or carries out the sale of the shares and pays or credits the capital gains. In those cases, the institution (and not the company) is required to deduct the withholding tax at the time of payment for the account of the shareholder and has to pay the withholding tax to the competent tax authority. In case the shares in Centogene N.V. are held (i) as business assets by a sole proprietor, a partnership or a corporation and such shares are attributable to a German business or (ii) in case of a corporation being subject to unlimited corporate income tax liability in Germany, the capital gains are not subject to withholding tax. In case of clause (i), the withholding tax exemption is subject to the condition that the paying agent has been notified by the beneficiary (Gläubiger) that the capital gains are exempt from withholding tax. The respective notification has to be filed by using the officially prescribed form.
Taxation of capital gains realized by shareholders tax resident in Germany holding shares as private assets
For individual shareholders (individuals) resident in Germany holding shares as private assets, capital gains realized on the disposal of shares are subject to final withholding tax. Accordingly, capital gains will be taxed at a flat tax rate of 25% plus a 5.5% solidarity surcharge thereon (in total 26.375%) and church tax, in case the shareholder is subject to church tax because of his individual circumstances. An automatic procedure for deduction of church tax by way of withholding will apply to shareholders being subject to church tax unless the shareholder has filed a blocking notice (Sperrvermerk) with the German Federal Tax Office (details related to the computation of the concrete tax rate including church tax are to be discussed with the individual tax adviser of the relevant shareholder). The taxable capital gain is calculated by deducting the acquisition costs of the shares and the expenses directly related to the disposal from the proceeds of the disposal. Apart from that, except for an annual lump sum savings allowance (Sparer-Pauschbetrag) of up to €801 (for individual filers) or up to €1,602 (for married couples and for partners in accordance with the registered partnership law (Gesetz über die Eingetragene Lebenspartnerschaft) filing jointly), private individual shareholders will not be entitled to deduct expenses incurred in connection with the capital investment from their capital gain.
In case the flat tax results in a higher tax burden as opposed to the private shareholder's individual tax rate, the private shareholder can opt for taxation at his individual personal income tax rate. In that case, the withholding tax (including solidarity surcharge) withheld will be credited against the income
160
tax. However, pursuant to the German tax authorities the private shareholders are nevertheless not entitled to deduct expenses incurred in connection with the capital investment from their income. The option can be exercised only for all capital income from capital investments received in the relevant assessment period uniformly, and married couples as well as for partners in accordance with the registered partnership law filing jointly may only jointly exercise the option.
Capital losses arising from the sale of the shares can only be offset against other capital gains resulting from the disposition of the shares or shares in other stock corporations during the same calendar year. Offsetting of overall losses with other income (such as business or rental income) and other capital income is not possible. Such losses are to be carried forward and to be offset against positive capital gains deriving from the sale of shares in stock corporations in future years.
The final withholding tax will not apply if the seller of the shares or in case of gratuitous transfer, its legal predecessor has held, directly or indirectly, at least 1% of the Company's registered share capital at any time during the five years prior to the disposal. In that case capital gains are subject to the partial income rule. Accordingly, only (i) 60% of the capital gains will be taxed at his individual personal income tax rate plus a 5.5% solidarity surcharge thereon and church tax (if applicable) and (ii) 60% of the business expenses related to the capital gains are deductible for tax purposes. The withholding tax withheld (including solidarity surcharge) will be credited against the shareholder's personal income tax liability in Germany.
Taxation of capital gains realized by shareholders tax resident in Germany holding the Company's shares as business assets
If a shareholder holds shares as business assets, the taxation of capital gains realized on the disposal of such shares depends on whether the respective shareholder is a corporation, a sole proprietor or a partnership:
Corporations
Capital gains realized on the disposal of shares by a corporate shareholder are generally exempt from corporate income tax and trade tax. However, 5% of the tax-exempt capital gains are deemed to be non-deductible business expenses for tax purposes and therefore are subject to corporate income tax (plus solidarity surcharge) and trade tax, i.e., tax exemption of 95%. Business expenses incurred in connection with the capital gains are entirely tax-deductible.
Capital losses incurred upon the disposal of shares or other impairments of the share value are not tax-deductible. A reduction of profit is also defined as any losses incurred in connection with a loan or security in the event the loan or the security is granted by a shareholder or by a related party thereto or by a third person with the right of recourse against the before-mentioned persons, and the shareholder holds directly or indirectly more than 25% of the company's registered share capital.
Special regulations apply if the shares are held as trading portfolio assets by a credit institution, a financial service institution or a financial enterprise within the meaning of the German Banking Act (Kreditwesengesetz) as well as by a life insurance company, a health insurance company or a pension fund. See "—Corporations."
Sole Proprietors
If the shares are held by a sole proprietor, capital gains realized on the disposal of the shares are subject to the partial income rule. Accordingly, only (i) 60% of the capital gains will be taxed at his/her individual personal income tax rate plus a 5.5% solidarity surcharge thereon and church tax (if applicable) and (ii) 60% of the business expenses related to the dividend income are deductible for tax purposes. In addition, 60% of the capital gains are subject to trade tax if the shares are held as
161
business assets of a permanent establishment in Germany within the meaning of the German Trade Tax Act (Gewerbesteuergesetz). The trade tax levied, depending on the applicable municipal trade tax rate and the individual tax situation, is partly or entirely credited against the shareholder's personal income tax liability.
Partnerships
In case the shares are held by a partnership, the partnership itself is not subject to corporate income tax or personal income tax as well as a solidarity surcharge (and church tax) since partnerships qualify as transparent for German tax purposes. In this regard, corporate income tax or personal income tax as well as a solidarity surcharge (and church tax, if applicable) are levied only at the level of the partner with respect to their relevant part of the profit and depending on their individual circumstances.
If the partner is a corporation, the capital gains will be subject to corporate income tax plus a solidarity surcharge. See "—Corporations." Trade tax will be levied additionally at the level of the partner insofar as the relevant profit of the partnership is not subject to trade tax at the level of the partnership. However, with respect to both corporate income and trade tax, the 95% exemption rule as described above applies.
If the partner is a sole proprietor (individual), the capital gains are subject to the partial income rule. See "—Sole Proprietors."
In addition, if the partnership is liable to trade tax, 60% of the capital gains are subject to trade tax at the level of the partnership, to the extent the partners are individuals, and 5% of the capital gains are subject to trade tax, to the extent the partners are corporations. However, if a partner is an individual, depending on the applicable municipal trade tax rate and the individual tax situation, the trade tax paid at the level of the partnership is credited against the partner's personal income tax liability.
With regard to corporate partners, special regulations apply if they are held as trading portfolio assets by credit institutions, financial service institutions or financial enterprises within the meaning of the German Banking Act or life insurance companies, health insurance companies or pension funds, as described above.
Taxation of capital gains realized by shareholders tax resident outside of Germany
Capital gains realized on the disposal of the shares by a shareholder tax resident outside of Germany are subject to German taxation provided that (i) the Company's shares are held as business assets of a permanent establishment or as business assets for which a permanent representative has been appointed in Germany, or (ii) the shareholder or, in case of a gratuitous transfer, its legal predecessor has held, directly or indirectly, at least 1% of the company's shares capital at any time during a five-year period prior to the disposal. In these cases, capital gains are generally subject to the same rules as described above for shareholders resident in Germany. However, in case the shares are not attributable to a German permanent establishment or permanent representative the 5% taxation (see "—Corporations—Taxation of capital gains realized by shareholders tax resident in Germany holding the Company's shares as business assets") shall not apply and the capital gains are fully exempt from German tax.
However, except for the cases referred to in clause (i) above, some of the double tax treaties concluded with Germany provide for a full exemption from German taxation.
162
Inheritance and Gift Tax
The transfer of the Company's shares to another person by way of succession or donation is subject to German inheritance and gift tax (Erbschaft- und Schenkungsteuer) if:
(i) the decedent, the donor, the heir, the donee or any other beneficiary has his/her/its residence, domicile, registered office or place of management in Germany at the time of the transfer, or is a German citizen who has not stayed abroad for more than five consecutive years without having a residence in Germany; or
(ii) (irrespective of the personal circumstances) the shares are held by the decedent or donor as business assets for which a permanent establishment in Germany is maintained or a permanent representative is appointed in Germany; or
(iii) (irrespective of the personal circumstances) at least 10% of the shares are held, directly or indirectly by, the decedent or person making the gift, himself or together with a related party in terms of Section 6 Foreign Tax Act.
Special regulations apply to qualified German citizens who maintain neither a residence nor their domicile in Germany but in a low tax jurisdiction, and to former German citizens, also resulting in inheritance and gift tax. The few double tax treaties on inheritance and gift tax which Germany has entered into provide that German inheritance and gift tax is levied only in case of (i) and, with certain restrictions, in case of (ii).
Other Taxes
No German capital transfer tax (Kapitalverkehrsteuer), value-added tax (Umsatzsteuer), stamp duty (Stempelgebühr) or similar taxes are levied when acquiring, holding or transferring the Company's shares. No value-added tax will be levied unless the shareholder validly opts for it. Net wealth tax (Vermögensteuer) is currently not levied in Germany.
On January 22, 2013, the Council of the European Union approved the resolution of the ministers of finance from eleven EU member states (including Germany) to introduce a Financial Transaction Tax ("FTT") within the framework of enhanced cooperation. On February 14, 2013, the European Commission published a proposal for a Council Directive implementing enhanced cooperation in the area of financial transaction tax. The plan focuses on levying a tax of 0.1% (0.01% for derivatives) on the purchase and sale of financial instruments.
A joint statement issued by 10 of the 11 participating EU member states in October 2016 reaffirmed the intention to introduce FTT. However, at the moment not many details are available. Thus, it is not known to what extent the elements of the European Commission's proposal outlined in the preceding paragraph will be followed in relation to the taxation of shares. The FTT proposal remains subject to negotiation between the participating Member States and is subject to political discussion. It may, therefore, be altered prior to the implementation, the timing of which remains unclear. Additional EU member states may decide to participate.
Prospective holders of the shares are advised to seek their own professional advice in relation to FTT.
F. Dividends and Paying Agents
Not applicable.
G. Statement by Experts
Not applicable.
163
H. Documents on Display
We are subject to the informational requirements of the Exchange Act. Accordingly, we are required to file reports and other information with the SEC, including annual reports on Form 20-F and reports on Form 6-K. The SEC maintains an internet website that contains reports and other information about issuers, like us, that file electronically with the SEC. The address of that website is www.sec.gov.
I. Subsidiary Information
Not applicable.
Item 11. Quantitative and Qualitative Disclosures About Market Risk
In the ordinary course of our business activities, we are exposed to various risks that are beyond our control, including credit risk, liquidity risk, market and sales risk and currency risk.
Credit Risk
Our default risk generally arises from trade and other receivables and is influenced mainly by the characteristics of individual international customers, as well as deposits with banks.
Our customers in the pharmaceutical segment are mainly pharmaceutical companies which are usually listed companies, or strongly financed by private equity funds. Our customers in the diagnostics segment are mainly hospitals, labs and physicians. 75% of the revenues from our diagnostics segment are generated from customers who have had business relationships with us since 2017. To avoid defaults, we may request prepayment when entering into new business relationships with physicians.
In addition to the macroeconomic situation generally, the development of international healthcare markets is a key economic factor affecting our business. These markets are closely monitored by our sales and other staff. The maximum default risk for trade receivables as of December 31, 2018 and 2019 by geographical region was as follows:
| |
As of December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2018 | 2019 | |||||
| |
(€ in thousands) |
||||||
Europe |
2,900 | 3,311 | |||||
Middle East |
7,766 | 10,470 | |||||
North America |
1,074 | 4,156 | |||||
Latin America |
604 | 811 | |||||
Other Regions |
190 | 180 | |||||
| | | | | | | | |
Total trade receivables, gross |
12,534 | 18,928 | |||||
Expected credit loss |
1,633 | 2,335 | |||||
| | | | | | | | |
Total trade receivables, net |
10,901 | 16,593 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Overdue trade receivables in the Middle East region mainly relate to major customers from the diagnostics segment. The trade receivables due from the top 10 diagnostics customers in the MENA region as of December 31, 2019 represent over 85% of overdue balances for the Middle East. These customers are mainly government hospitals administered by the Ministry of Health in the respective countries as well as distributors and, based on our past experience, they normally require a longer period to settle outstanding trade receivables.
164
Liquidity Risk
We are exposed to liquidity risk in both the availability of finance and the repayment of borrowings, as we may not be in a position to meet our financial liabilities as contractually agreed by providing cash or other financial assets. We manage our liquidity in order to ensure that sufficient cash and cash equivalents are available for us to meet our payment obligations when these fall due, without incurring unacceptable losses or damaging our reputation.
We strive to maintain cash and cash equivalents at a level above that of the expected cash outflows for financial liabilities (apart from trade payables) during the next 60 days.
We completed our initial public offering in November 2019. As at December 31, 2019, we had cash and cash equivalent of €41,095 thousand. We assessed the concentration of risk and concluded it to be low.
In addition to the cash and cash equivalent available as of December 31, 2019, we also have access to other sources of funding, including the amount of expected cash inflows from trade and other receivables. We also have secured credit lines of €3,500 thousand that bear interest at 3.33% to 4.50%, of which €2,636 thousand was utilized as of December 31, 2019 and €1,915 thousand was utilized as of December 31, 2018.
Market and Sales Risk
Market risks primarily arise from changing reimbursement structures as well as pricing pressure and pressure to innovate in the highly dynamic market environment of genetic diagnostic testing. We monitor developments very closely through our local sales teams and their reporting structures. We also closely monitor our individual segmental results by conducting periodic analyses of our competitive landscape, with the aim of enabling rapid pricing and product enhancements, if necessary.
Currency Risk
We are also exposed to currency risks where contracts are concluded in foreign currencies. The vast majority of the products and services we provide, however, are invoiced in Euros. Our main functional currencies other than the euro are the U.S. dollar, the Indian rupee (the "INR") and the Arab Emirates Dirham (the "AED"), as shown in the table below.
| |
As of December 31, 2019 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
USD | INR | AED | |||||||
| |
(€ in thousands) |
|||||||||
Trade receivables |
4,275 | 36 | (1 | ) | ||||||
Trade payables and other liabilities |
(2,801 | ) | (98 | ) | (15 | ) | ||||
| | | | | | | | | | | |
Net risk statement of financial position |
1,474 | (62 | ) | (16 | ) | |||||
The impact on our earnings before tax for the years ended December 31, 2018 and 2019 or equity as of December 31, 2018 and 2019 of a 10% change in the U.S. dollar exchange rate as compared to the euro would not be material. In the future, we expect our exposure to the U.S. dollar to increase over time as our business grows. The impact on our earnings before tax for the years ended December 31, 2018 and 2019 or equity as of December 31, 2018 and 2019 of a 10% change in the INR and AED would not be material.
165
Item 12. Description of Securities Other Than Equity Securities
A. Debt Securities
Not applicable.
B. Warrants and Rights
Not applicable.
C. Other Securities
Not applicable.
D. American Depositary Shares
Not applicable.
166
Item 13. Defaults, Dividend Arrearages and Delinquencies
None.
Item 14. Material Modifications to the Rights Of Security Holders and Use Of Proceeds
A. Material Modifications to Instruments
Not applicable.
B. Material Modifications to Rights
Not applicable.
C. Withdrawal or Substitution of Assets
Not applicable.
D. Changes in Trustees or Paying Agents
Not applicable.
E. Use of Proceeds
On November 6, 2019, our registration statement on Form F-1 (File No. 333-234177), as amended, was declared effective by the SEC for our initial public offering, pursuant to which we offered and sold a total of 4,000,000 of our common shares, €0.12 nominal value per share, at a public offering price of $14.00 per share. SVB Leerink LLC and Evercore Group L.L.C acted as joint book-running managers, and Robert W. Baird & Co. Incorporated and BTIG, LLC. acted as co-managers. The offering was completed on November 12, 2019.
The total expenses incurred in connection with our initial public offering were approximately $9.4 million (€8.4 million), which included $3.9 million (€3.5 million) in underwriting discounts and commissions and approximately $5.5 million (€4.9 million) in other costs and expenses. None of the transaction expenses included payments to directors or officers of our company or their associates, persons owning more than 10% or more of our equity securities or our affiliates. None of the net proceeds from our initial public offering were paid, directly or indirectly, to any of our directors or officers or their associates, persons owning 10% or more of our equity securities or our affiliates.
We received net proceeds of approximately $46.6 million (€41.9 million) from our initial public offering. For the period from the effective date of the F-1 registration statement to the year ended December 31, 2019, we used the net proceeds from our initial public offering as follows:
- •
- approximately $1.8 million (€1.6 million) for research and development under our pharmaceutical segment,
including the development and clinical validation of biomarkers, as well as for growth of our partnership opportunities through sales and marketing investments;
- •
- approximately $2.0 million (€1.8 million) for the development of our knowledge-driven information platform,
including investments in new information technology, artificial intelligence and other software solutions that improve our processes and enhance our data documentation, and for the development of
solutions driving precision medicine based treatments; and
- •
- the remainder for working capital and other general corporate purposes.
167
Item 15. Controls and Procedures
A. Disclosure Controls and Procedures
As required by Rule 13a-15 under the Exchange Act, management, including our chief executive officer and our chief financial officer, has evaluated the effectiveness of our disclosure controls and procedures as of the end of the period covered by this report. Disclosure controls and procedures refer to controls and other procedures designed to ensure that information required to be disclosed in the reports we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. Disclosure controls and procedures include, without limitations, controls and procedures designed to ensure that information required to be disclosed by us in our reports that we file or submit under the Exchange Act is accumulated and communicated to management, including our principal executive and principal financial officers, or persons performing similar functions, as appropriate to allow timely decisions regarding our required disclosures.
Based on the foregoing, our chief executive officer and our chief financial officer have concluded that, as of December 31, 2019, our disclosure controls and procedures were not effective due to the material weakness in our internal control over financial reporting primarily related to a lack of effective review controls over judgmental and complex areas of the financial statement close process and a lack of routine financial statement close process controls.
B. Management's Annual Report on Internal Control Over Financial Reporting
This Annual Report does not include a report of management's assessment regarding internal control over financial reporting due to a transition period established by rules of the SEC for newly public companies.
C. Attestation Report of the Registered Public Accounting Firm
This Annual Report does not include an attestation report of our independent registered public accounting firm regarding internal control over financial reporting due to a transition period established by rules of the SEC for newly public companies.
D. Changes in Internal Control Over Financial Reporting
This Annual Report does not include disclosure of changes in control over financial reporting due to a transition period established by the rules of the SEC for newly public companies.
Item 16A. Audit Committee Financial Expert
Our supervisory board has determined that Berndt Modig and Peer Schatz each satisfy the "independence" requirements set forth in Rule 10A-3 under the Exchange Act and Berndt Modig qualifies as an "audit committee financial expert," as such term is defined in the rules of the SEC. For more information see "Item 6. Directors, Senior Management and Employees—C. Board Practices—Committees—Audit Committee."
We adopted a Code of Business Conduct and Ethics (the "Code") applicable to our management board, our supervisory board, and company personnel, which is a code of ethics as defined in Item 16B of Form 20-F promulgated by the SEC. The full text of the Code can be found on our website at www.centogene.com. Information contained on, or that can be accessed through, our website does not
168
constitute a part of this Annual Report and is not incorporated by reference herein. If we make any amendment to the Code or grant any waivers, including any implicit waiver, from a provision of the Code, we will disclose the nature of such amendment or waiver on our website to the extent required by the rules and regulations of the SEC.
Item 16C. Principal Accountant Fees and Services
Our financial statements have been prepared in accordance with IFRS and are audited by Ernst & Young GmbH Wirtschaftsprüfungsgesellschaft ("Ernst & Young"), our independent registered public accounting firm registered with the Public Company Accounting Oversight Board in the United States.
Ernst & Young has served as our independent registered public accounting firm for each of the three years ended December 31, 2017, 2018 and 2019, for which audited financial statements appear in this Annual Report.
| |
For the year ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2018 | 2019 | |||||
| |
(in thousands) |
||||||
Audit |
1,281 | 496 | |||||
Audit-related fees |
— | — | |||||
Tax fees |
— | — | |||||
All other fees |
— | — | |||||
| | | | | | | | |
Total |
1,281 | 496 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Audit fees relate to (i) audit services, (ii) certain procedures on our quarterly results, (iii) services related to our statutory and regulatory filings for certain of our subsidiaries, including Centogene AG and (iv) fees in connection with the issuance of a comfort letter for our initial public offering in November 2019 and fees for other services related thereto.
Audit Committee's Pre-Approval Policies and Procedures
The audit committee is responsible for the appointment, compensation, retention and oversight of the work of our independent registered public accounting firm.
The Audit Committee evaluates the qualifications, independence and performance of the independent registered public accounting firm as well as pre-approves and reviews the audit and non-audit services to be performed by the independent registered public accounting firm. The external audit plan and fees for professional audit services and other services rendered by Ernst & Young for the years ended December 31, 2019 and 2018 were approved by the Audit Committee.
The Audit Committee monitors compliance with the German, Dutch and U.S. rules on non-audit services provided by an independent registered public accounting firm.
Audit Work Performed by Other Than Principal Accountant if Greater than 50%
Not applicable.
Item 16D. Exemptions from the Listing Standards for Audit Committees
We are relying on the exemption under Rule 10A-3(b)(1)(iv)(A)(2) of the Exchange Act, which exempts a minority of the members of the audit committee from the independence requirements for one year from the effective date of the registration statement, filed in connection with the initial public offering. Such reliance does not adversely affect the ability of the audit committee to act independently and to satisfy the other requirements of Rule 10A-3.
169
Item 16E. Purchases of Equity Securities by the Issuer and Affiliated Purchasers
Neither we nor any affiliated purchaser has purchased any shares or other units of any class of our equity securities registered pursuant to Section 12 of the Exchange Act during the fiscal year ended December 31, 2019.
Item 16F. Change in Registrant's Certifying Accountant
Not applicable.
Item 16G. Corporate Governance
As a "foreign private issuer", as defined by the SEC, we are permitted to rely on home country governance requirements and certain exemptions thereunder rather than on the corporate governance requirements of Nasdaq. For example, in accordance with Dutch law and generally accepted business practices, our Articles of Association do not provide quorum requirements generally applicable to general meetings of shareholders. To this extent, our practice varies from the requirement of Nasdaq Listing Rule 5620(c), which requires an issuer to provide in its bylaws for a generally applicable quorum, and that such quorum may not be less than one-third of the outstanding voting shares. Although we must provide shareholders with an agenda and other relevant documents for the general meeting of shareholders, Dutch law does not have a regulatory regime for the solicitation of proxies and the solicitation of proxies is not a generally accepted business practice in the Netherlands, thus our practice varies from the requirement of Nasdaq Listing Rule 5620(b). As permitted by the listing requirements of Nasdaq, we have also opted out of the requirements of Nasdaq Listing Rule 5605(d), which requires, among other things, an issuer to have a compensation committee that consists entirely of independent directors, Nasdaq Listing Rule 5605(e), which requires independent director oversight of director nominations, and Nasdaq Listing Rule 5605(b)(2), which requires an issuer to have a majority of independent directors on its board. We are also relying on the phase-in rules of the SEC and Nasdaq with respect to the independence of our audit committee. These rules require that a majority of our directors must be independent and all members of our audit committee must meet the independence standard for audit committee members within a period of one year after the SEC has declared the F-1 registration statement effective. In addition, we have opted out of shareholder approval requirements, as included in the Nasdaq Listing Rules, for the issuance of securities in connection with certain events such as the acquisition of shares or assets of another company, the establishment of or amendments to equity-based compensation plans for employees, a change of control of the Company and certain private placements. To this extent, our practice varies from the requirements of Nasdaq Rule 5635, which generally requires an issuer to obtain shareholder approval for the issuance of securities in connection with such events.
Item 16H. Mine Safety Disclosure
Not applicable.
Miscellaneous. Disclosure under Iran Threat Reduction and Syria Human Rights Act of 2012 (ITRA)
Section 219 of the Iran Threat Reduction and Syria Human Rights Act of 2012 added Section 13(r) to the Exchange Act. Section 13(r) requires an issuer to disclose in its annual or quarterly reports, as applicable, whether it or any of its affiliates knowingly engaged in certain activities, transactions or dealings relating to Iran or with designated natural persons or entities sanctioned under programs relating to terrorism or the proliferation of weapons of mass destruction. Disclosure is required even where the activities, transactions or dealings are conducted outside the US by non-US affiliates in compliance with applicable law, and whether or not the activities are sanctionable under US law.
170
As of the date of this Annual Report, we are not aware of any activity, transaction or dealing by us during the financial year ended December 31, 2019 that is disclosable under Section 219 of the Iran Threat Reduction and Syria Human Rights Act of 2012 and Section 13(r) of the Exchange Act, except as set forth below. This information is to the best of our knowledge.
Centogene N.V., which is not a U.S. person and is not owned or controlled by U.S. persons, does not have any branches or subsidiaries based in Iran. However, we have contracts with several laboratories and one distributor in Iran through which we provide diagnostic tests to patients in Iran, primarily NIPT for pregnant women. These laboratories and our third-party distributor are not owned or controlled by the Iranian government and we do not have any agreements, commercial arrangements, or other contracts with the Iranian government. Moreover, neither our distributor nor we have entered into any arrangements with or sold any products to persons included on the Specially Designated Nationals and Blocked Persons List maintained by the U.S. Department of the Treasury's Office of Foreign Asset Control.
In the year ended December 31, 2019, our gross revenues from activities in Iran amounted to €1,277 thousand, which represented approximately 2.6% of our total gross revenues. We did not generate any net profits for the year ended December 31, 2019 from authorities in Iran.
We do not receive information regarding the identity of our distributor's downstream customers and intermediaries in Iran, and it is possible that these parties include entities, such as government-owned hospitals and pharmacies, that are owned directly or indirectly by the Iranian government or by persons or entities sanctioned in connection with terrorism or proliferation activities. As a result, we cannot establish the proportion of gross revenue or sales potentially attributable to entities affiliated with the Iranian government or parties sanctioned for disclosable activities.
We believe that our business with Iranian parties is conducted in compliance with all applicable sanctions and export controls and that such activities, which involve providing genetic testing services to patients, are not sanctionable under U.S. secondary sanctions targeting Iran. For further information, see "Item 3. Key Information—D. Risk Factors—Transactions involving Iran or other countries or parties that are targets of U.S. or other economic sanctions could expose us to certain risks and may lead some potential customers and investors to avoid doing business with us or investing in our securities."
As of the date of this Annual Report, our management does not anticipate any change in our activities in Iran that would result in a material impact on Centogene.
171
We have responded to Item 18 in lieu of this item.
Our audited consolidated financial statements are included in this Annual Report beginning at Page F-1.
172
173
- *
- Filed
herewith.
- **
- Furnished
herewith.
- †
- Certain information has been excluded from the exhibit because it both (i) is not material and (ii) would likely cause competitive harm to the Company if publicly disclosed.
174
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this Annual Report on its behalf.
| CENTOGENE N.V. | ||||
By: |
/s/ ARNDT ROLFS Name: Arndt Rolfs Title: Chief Executive Officer |
|||
Date: April 23, 2020
F-1
Report of Independent Registered Public Accounting Firm
To the Shareholders and the Supervisory Board of Centogene N.V.
Opinion on the Financial Statements
We have audited the accompanying consolidated statements of financial position of Centogene N.V. (the Company) as of December 31, 2018 and 2019, the related consolidated statements of comprehensive loss, changes in equity and cash flows for each of the three years in the period ended December 31, 2019, and the related notes (collectively referred to as the "consolidated financial statements"). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2018 and 2019, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2019, in conformity with International Financial Reporting Standards as issued by the International Accounting Standards Board.
Retrospective Application of Equity Transaction
As discussed in Note 17 to the consolidated financial statements, the Company has retrospectively applied the effects of the Corporate Reorganization to the Issued capital and Capital reserve in the consolidated statement of financial position as of December 31, 2018, as if the transaction had occurred from the earliest period presented.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
| /s/ INGO RÖDERS | /s/ CHRISTIAN PATZELT | |
| Wirtschaftsprüfer | Wirtschaftsprüfer | |
| (German Public Auditor) | (German Public Auditor) |
Ernst & Young GmbH Wirtschaftsprüfungsgesellschaft
We
have served as the Company's auditor since 2010.
Berlin, Germany
April 23, 2020
F-2
Centogene N.V.
Consolidated statements of comprehensive loss
for the years ended December 31, 2017, 2018 and 2019
(in EUR k)
| |
Note | 2017 | 2018 | 2019 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Revenue |
7 | 31,689 | 40,478 | 48,780 | |||||||||
Cost of sales |
14,939 | 19,941 | 26,005 | ||||||||||
| | | | | | | | | | | | | | |
Gross profit |
16,750 | 20,537 | 22,775 | ||||||||||
Research and development expenses |
6,396 | 6,300 | 9,590 | ||||||||||
General administrative expenses |
9,498 | 18,610 | 23,160 | ||||||||||
Selling expenses |
5,897 | 7,474 | 9,254 | ||||||||||
Other operating income |
8.1 | 1,043 | 2,306 | 3,781 | |||||||||
Other operating expenses |
8.2 | 457 | 1,065 | 2,036 | |||||||||
Real Estate Transfer Tax |
13.1 | — | — | 1,200 | |||||||||
| | | | | | | | | | | | | | |
Operating loss |
(4,455 | ) | (10,606 | ) | (18,684 | ) | |||||||
Interest and similar income |
14 | 33 | 16 | ||||||||||
Interest and similar expenses |
1,021 | 1,075 | 2,029 | ||||||||||
| | | | | | | | | | | | | | |
Financial costs, net |
8.3 | (1,007 | ) | (1,042 | ) | (2,013 | ) | ||||||
Loss before taxes |
(5,462 | ) | (11,648 | ) | (20,697 | ) | |||||||
Income taxes expenses/(benefits) |
9 | 14 | (310 | ) | 158 | ||||||||
| | | | | | | | | | | | | | |
Loss for the year |
(5,476 | ) | (11,338 | ) | (20,855 | ) | |||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Other comprehensive income, all attributable to equity holders of the parent |
10 | (8 | ) | 16 | |||||||||
Total comprehensive loss |
(5,466 | ) | (11,346 | ) | (20,839 | ) | |||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Attributable to: |
|||||||||||||
Equity holders of the parent |
(5,351 | ) | (10,971 | ) | (20,658 | ) | |||||||
Non-controlling interests |
(115 | ) | (375 | ) | (181 | ) | |||||||
| | | | | | | | | | | | | | |
|
(5,466 | ) | (11,346 | ) | (20,839 | ) | |||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Loss per share—Basic and diluted (in EUR) |
(0.4 | ) | (0.8 | ) | (1.3 | ) | |||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
The accompanying notes form an integral part of these consolidated financial statements
F-3
Centogene N.V.
Consolidated statements of financial position
as at December 31, 2018 and 2019
(in EUR k)
Assets
|
Note | Dec 31, 2018 | Dec 31, 2019 | ||||||
|---|---|---|---|---|---|---|---|---|---|
Non-current assets |
|||||||||
Intangible assets |
11 | 8,795 | 14,145 | ||||||
Property, plant and equipment |
12 | 39,115 | 8,376 | ||||||
Right-of-use assets |
13 | — | 24,932 | ||||||
Other assets |
15 | 1,948 | |||||||
| | | | | | | | | | |
|
47,910 | 49,401 | |||||||
Current assets |
|
||||||||
Inventories |
14 | 1,346 | 1,809 | ||||||
Trade receivables |
15 | 10,901 | 16,593 | ||||||
Other assets |
15 | 7,295 | 8,612 | ||||||
Cash and cash equivalents |
16 | 9,222 | 41,095 | ||||||
| | | | | | | | | | |
|
28,764 | 68,109 | |||||||
| | | | | | | | | | |
|
76,674 | 117,510 | |||||||
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
Equity and liabilities
|
|||||||||
Equity |
|||||||||
Issued capital |
17 | 1,903 | 2,383 | ||||||
Capital reserve |
17 | 45,342 | 98,099 | ||||||
Retained earnings and other reserves |
(19,964 | ) | (40,622 | ) | |||||
Non-controlling interests |
(757 | ) | (938 | ) | |||||
| | | | | | | | | | |
|
26,524 | 58,922 | |||||||
Non-current liabilities |
|
||||||||
Non-current loans |
19 | 12,915 | 1,578 | ||||||
Lease liabilities |
19 | 1,712 | 18,069 | ||||||
Deferred tax liabilities |
— | — | |||||||
Other liabilities |
19.2 | 11,240 | 9,941 | ||||||
| | | | | | | | | | |
|
25,867 | 29,588 | |||||||
Current liabilities |
|||||||||
Investment subsidies |
19.2 | 794 | 1,348 | ||||||
Current loans |
19.1 | 3,702 | 3,688 | ||||||
Lease liabilities |
19.1 | 1,350 | 3,635 | ||||||
Liabilities from income taxes |
10 | — | |||||||
Trade payables |
19.2 | 5,429 | 8,554 | ||||||
Other liabilities |
19.2, 20 | 12,998 | 11,775 | ||||||
| | | | | | | | | | |
|
24,283 | 29,000 | |||||||
| | | | | | | | | | |
|
76,674 | 117,510 | |||||||
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
The accompanying notes form an integral part of these consolidated financial statements
F-4
Centogene N.V
Consolidated statements of cash flows
for the years ended December 31, 2017, 2018 and 2019
(in EUR k)
| |
Note | 2017 | 2018 | 2019 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
Operating activities |
||||||||||||
Loss before taxes |
(5,462 | ) | (11,648 | ) | (20,697 | ) | ||||||
Adjustments to reconcile earnings to cash flow from operating activities |
||||||||||||
Amortization and depreciation |
11,12,13 | 3,237 | 5,175 | 6,579 | ||||||||
Interest income |
8.3 | (14 | ) | (33 | ) | (16 | ) | |||||
Interest expense |
8.3 | 1,021 | 1,075 | 2,029 | ||||||||
Loss/(gain) on the disposal of non-current assets |
(60 | ) | — | (532 | ) | |||||||
Share-based payment expenses |
20 | 894 | 5,521 | 6,418 | ||||||||
Real estate transfer tax expenses |
13.1 | — | — | 1,200 | ||||||||
Other non-cash items |
(32 | ) | (966 | ) | (1,856 | ) | ||||||
Changes in operating assets and liabilities: |
|
|||||||||||
Inventories |
14 | (412 | ) | (567 | ) | (463 | ) | |||||
Trade receivables |
15 | (2,430 | ) | (3,909 | ) | (5,692 | ) | |||||
Other assets |
15 | 314 | (919 | ) | (1,169 | ) | ||||||
Trade payables |
19.2 | (728 | ) | 140 | 3,125 | |||||||
Other liabilities |
(664 | ) | 1,554 | 3,299 | ||||||||
Cash flow used in operating activities |
(4,336 | ) | (4,577 | ) | (7,775 | ) | ||||||
| | | | | | | | | | | | | |
Investing activities |
||||||||||||
Cash paid for investments in intangible assets |
11 | (2,471 | ) | (3,059 | ) | (7,280 | ) | |||||
Cash paid for investments in property, plant and equipment and right-of -use assets |
12,13 | (15,564 | ) | (8,710 | ) | (296 | ) | |||||
Grants received for investment in property, plant and equipment |
19.2 | 6,802 | 3,042 | 793 | ||||||||
Grants refunded related to disposed property, plant and equipment |
13.1, 19.2 | — | — | (358 | ) | |||||||
Cash received from disposals of property, plant and equipment |
13.1 | 65 | — | 21,300 | ||||||||
Interest received |
14 | 33 | 16 | |||||||||
Cash flow (used in)/from investing activities |
(11,154 | ) | (8,694 | ) | 14,175 | |||||||
| | | | | | | | | | | | | |
Financing activities |
||||||||||||
Cash received from equity contributions, net |
17 | 19,034 | 20,073 | 41,899 | ||||||||
Cash received from loans |
19, 21.2 | 9,990 | 3,631 | 721 | ||||||||
Cash repayments of loans |
19, 21.2 | (8,749 | ) | (2,851 | ) | (12,072 | ) | |||||
Cash repayments of leases liabilities |
19, 21.2 | (1,580 | ) | (442 | ) | (3,046 | ) | |||||
Interest paid |
8.3 | (1,013 | ) | (1,075 | ) | (2,029 | ) | |||||
Cash flow from financing activities |
17,682 | 19,336 | 25,473 | |||||||||
| | | | | | | | | | | | | |
Changes in cash and cash equivalents |
2,192 | 6,065 | 31,873 | |||||||||
Cash and cash equivalents at the beginning of the period |
965 | 3,157 | 9,222 | |||||||||
| | | | | | | | | | | | | |
Cash and cash equivalents at the end of the period |
3,157 | 9,222 | 41,095 | |||||||||
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
The accompanying notes form an integral part of these consolidated financial statements
F-5
Centogene N.V.
Consolidated statements of changes in equity
for the years ended December 31, 2017, 2018 and 2019
| |
|
Attributable to the owners of the parent | |
|
||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
in EUR k
|
Note | Issued capital |
Capital reserve |
Currency translation reserve |
Retained earnings |
Total | Non- controlling interests |
Total equity |
||||||||||||||||
As of January 1, 2017 |
1 | 1,274 | 5,370 | (18 | ) | (3,624 | ) | 3,002 | (267 | ) | 2,735 | |||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | |
Loss for the year |
— |
— |
— |
(5,361 |
) |
(5,361 |
) |
(115 |
) |
(5,476 |
) |
|||||||||||||
Other comprehensive income |
— | — | 10 | — | 10 | — | 10 | |||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | |
Total comprehensive loss |
— | — | 10 | (5,361 | ) | (5,351 | ) | (115 | ) | (5,466 | ) | |||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | |
Share-based payments |
51 |
— |
51 |
— |
51 |
|||||||||||||||||||
Issuance of shares |
272 | 19,178 | — | — | 19,450 | — | 19,450 | |||||||||||||||||
Transaction cost |
— | (416 | ) | — | — | (416 | ) | — | (416 | ) | ||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | |
As of December 31, 2017 |
1,546 |
24,183 |
(8 |
) |
(8,985 |
) |
16,736 |
(382 |
) |
16,354 |
||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | |
| |
|
Attributable to the owners of the parent | |
|
||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
in EUR k
|
Note | Issued capital |
Capital reserve |
Currency translation reserve |
Retained earnings |
Total | Non- controlling interests |
Total equity |
||||||||||||||||
As of January 1, 2018 |
1 | 1,546 | 24,183 | (8 | ) | (8,985 | ) | 16,736 | (382 | ) | 16,354 | |||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | |
Loss for the year |
— |
— |
— |
(10,963 |
) |
(10,963 |
) |
(375 |
) |
(11,338 |
) |
|||||||||||||
Other comprehensive loss |
— | — | (8 | ) | — | (8 | ) | — | (8 | ) | ||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | |
Total comprehensive loss |
— | — | (8 | ) | (10,963 | ) | (10,971 | ) | (375 | ) | (11,346 | ) | ||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | |
Share-based payments |
1,443 |
— |
1,443 |
— |
1,443 |
|||||||||||||||||||
Issuance of shares |
357 | 19,716 | — | — | 20,073 | — | 20,073 | |||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | |
As of December 31, 2018 |
1,903 |
45,342 |
(16 |
) |
(19,948 |
) |
27,281 |
(757 |
) |
26,524 |
||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | |
| |
|
Attributable to the owners of the parent | |
|
||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
in EUR k
|
Note | Issued capital |
Capital reserve |
Currency translation reserve |
Retained earnings |
Total | Non- controlling interests |
Total equity |
||||||||||||||||
As of January 1, 2019 |
1 | 1,903 | 45,342 | (16 | ) | (19,948 | ) | 27,281 | (757 | ) | 26,524 | |||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | |
Loss for the year |
— |
— |
— |
(20,674 |
) |
(20,674 |
) |
(181 |
) |
(20,855 |
) |
|||||||||||||
Other comprehensive loss |
— | — | 16 | — | 16 | — | 16 | |||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | |
Total comprehensive loss |
— | — | 16 | (20,674 | ) | (20,658 | ) | (181 | ) | (20,839 | ) | |||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | |
Issuance of shares at IPO |
17 |
480 |
46,318 |
— |
— |
46,798 |
— |
46,798 |
||||||||||||||||
Transaction costs |
17 | — | (4,899 | ) | — | — | (4,899 | ) | — | (4,899 | ) | |||||||||||||
Share-based payments |
20 | — | 1,300 | — | — | 1,300 | — | 1,300 | ||||||||||||||||
Share-based payments -modification at IPO |
20(ii) | 10,038 | — | — | 10,038 | — | 10,038 | |||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | |
As of December 31, 2019 |
2,383 |
98,099 |
— |
(40,622 |
) |
59,860 |
(938 |
) |
58,922 |
|||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | |
The accompanying notes form an integral part of these consolidated financial statements
F-6
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019
1 General company information
Centogene N.V. ("the Company") and its subsidiaries focus on rare diseases that transforms real-world clinical and genetic data into actionable information for patients, physicians and pharmaceutical companies. The mission of the Company is to bring rationality to treatment decisions and to accelerate the development of new orphan drugs by using our knowledge of the global rare disease market, including epidemiological and clinical data and innovative biomarkers.
On November 7, 2019, the Company completed an initial public offering ("IPO") and is listed on Nasdaq Global Market under stock code "CNTG". 4 million common shares with a nominal value of EUR 0.12 per share were sold at a public offering price of USD 14 per share (i.e. EUR 12.58 per share), for aggregate net offering proceeds, after deducting underwriting discounts and commissions, of EUR 47 million.
In connection with the IPO, the Company underwent a corporate reorganization and Centogene N.V. became the parent holding company with 100% interest in Centogene AG since November 12, 2019. Centogene N.V. is a public company with limited liabilities incorporated in the Netherlands, with registered office located at Am Strande 7 in 18055 Rostock, Germany and trade register number 72822872.
Prior to the reorganization, Centogene AG was the parent holding company of the Group and was owned by individual common shareholders as well as institutional investors holding preference shares. As part of the reorganization, Centogene B.V. was formed and the common shares and preference shares of Centogene AG owned by individual shareholders and institutional investors were exchanged to common shares of Centogene B.V. As a result, Centogene B.V. became the holding company of Centogene AG, while the individual shareholders and institutional investors had a 100% shareholding in Centogene B.V. Effective with the IPO, Centogene B.V. changed its legal form and became Centogene N.V. and common shares of Centogene B.V. were converted to common shares of Centogene N.V.
2 Basis of preparation
The corporate reorganization, as described above, is considered a continuation of the Centogene group resulting in no change in the carrying values of assets or liabilities. As a result, the financial statements for periods prior to the IPO and the corporate reorganization are the financial statements of Centogene AG as the predecessor to the Company for accounting and reporting purposes. Unless otherwise specified, "the Company" refers to Centogene N.V., and Centogene AG throughout the remainder of these notes, while "the Group" refers to Centogene N.V., Centogene AG and its subsidiaries.
The consolidated financial statements of the Group were prepared in accordance with International Financial Reporting Standards ("IFRS") as issued by the International Accounting Standards Board (the "IASB"). The accounting policies used in the fiscal year 2019 generally correspond to the policies applied by Centogene AG in the prior year, except for certain amendments to the standards which are effective for annual periods beginning on or after January 1, 2019 (see note 3). In addition, certain prior period information has been reclassified to conform with current year presentation (see note 17).
These consolidated financial statements are presented in euro, which is the Group's functional currency. Unless otherwise specified, all financial information presented in euro is rounded to the nearest thousand (EUR k) in line with customary commercial practice.
F-7
Notes to the consolidated financial statements as of December 31, 2018 and 2019
and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
3 Effects of new accounting standards
(a) New standards adopted by the Group as of January 1, 2019
IFRS 16 Leases
IFRS 16 supersedes IAS 17 Leases, IFRIC 4 Determining whether an Arrangement contains a Lease, SIC-15 Operating Leases-Incentives and SIC-27 Evaluating the Substance of Transactions Involving the Legal Form of a Lease. The standard sets out the principles for the recognition, measurement, presentation and disclosure of leases and requires lessees to account for most leases under a single on-balance sheet model.
The Group has lease contracts for land and buildings, offices as well as various items of plant, machinery, motor vehicles and other equipment. Prior to the adoption of IFRS 16, the Group classified each of its leases (as lessee) at the inception date as either a finance lease or an operating lease. A lease was classified as a finance lease if it transferred substantially all of the risks and rewards incidental to ownership of the leased asset to the Group; otherwise it was classified as an operating lease. Finance leases were capitalized at the commencement of the lease at the inception date fair value of the leased property or, if lower, at the present value of the minimum lease payments. Lease payments were apportioned between interest (recognized as finance costs) and reduction of the lease liability. For operating leases, the leased property was not capitalized, and the lease payments were recognized as rent expense in the statement of profit or loss on a straight-line basis over the lease term. Any prepaid rent and accrued rent were recognized under Prepayments and Trade and other payables, respectively.
The Group adopted IFRS 16 as of January 1, 2019, using the modified retrospective method of adoption. The Group elected to use the transition practical expedient allowing the standard to be applied only to contracts that were previously identified as leases applying IAS 17 and IFRIC 4 at the date of initial application. The Group also elected to use the recognition exemptions for lease contracts that, at the commencement date, have a lease term of 12 months or less and do not contain a purchase option ('short-term leases'), and lease contracts for which the underlying asset is of low value ('low-value assets').
Upon adoption of IFRS 16, the Group applied a single recognition and measurement approach for all leases under which it is the lessee, except for short-term leases and leases of low-value assets. The Group recognized lease liabilities to make lease payments and right-of-use assets representing the right to use the underlying assets.
In accordance with the modified retrospective method of adoption, the Group did not change the initial carrying amounts of recognized assets and liabilities at the date of initial application for leases previously classified as finance leases (i.e., the right-of-use assets and lease liabilities equal the lease assets and liabilities recognized under IAS 17) and did not restate its comparative figures but recognized the cumulative effect of adopting IFRS 16 as an adjustment to equity at the beginning of the current period.
F-8
Notes to the consolidated financial statements as of December 31, 2018 and 2019
and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
3 Effects of new accounting standards (Continued)
The effect of adoption of IFRS 16 is as follows:
Impact on the statement of financial position (increase/(decrease)) as at January 1, 2019:
| |
in EUR k | |||
|---|---|---|---|---|
Assets |
||||
Property, plant and equipment |
(5,364 | ) | ||
Right-of-use assets |
5,767 | |||
| | | | | |
Total assets |
403 | |||
| | | | | |
| | | | | |
| | | | | |
Liabilities |
||||
Lease liabilities—Current |
93 | |||
Lease liabilities—Non-Current |
310 | |||
| | | | | |
Total liabilities |
403 | |||
| | | | | |
| | | | | |
| | | | | |
- •
- Right-of-use assets of EUR 5,767k, were recognized and presented separately in the consolidated statement of financial position. This includes
the lease assets recognized previously under finance leases of EUR 5,364k, that were reclassified from Property, plant and equipment.
- •
- Additional lease liabilities of EUR 403k, were recognized.
IFRIC Interpretation 23 Uncertainty over Income Tax Treatment (the "Interpretation")
The Interpretation addresses the accounting for income taxes when tax treatments involve uncertainty that affects the application of IAS 12 and does not apply to taxes or levies outside the scope of IAS 12, nor does it specifically include requirements relating to interest and penalties associated with uncertain tax treatments. The Interpretation specifically addresses the following:
- –
- Whether an entity considers uncertain tax treatments separately;
- –
- The assumptions an entity makes about the examination of tax treatments by taxation authorities;
- –
- How an entity determines taxable profit (tax loss), tax bases, unused tax losses, unused tax credits and tax rates; and
- –
- How an entity considers changes in facts and circumstances.
The Group determines whether to consider each uncertain tax treatment separately or together with one or more other uncertain tax treatments and uses the approach that better predicts the resolution of the uncertainty.
Upon adoption of the Interpretation, the Group considered whether it has any uncertain tax positions. The Group determined, based on its review, the tax loss positions of both the Company and the subsidiaries and no outstanding income tax liabilities, the Interpretation did not have an impact on the consolidated financial statements.
(b) New standards not yet effective
Furthermore, the new and amended standards and interpretations that are issued, but not yet effective, up to the date of issuance of the Group's financial statements are disclosed below. The
F-9
Notes to the consolidated financial statements as of December 31, 2018 and 2019
and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
3 Effects of new accounting standards (Continued)
Group intends to adopt these new and amended standards and interpretations when they become effective.
Amendments to IAS 1 and IAS 8: Definition of Material
In October 2018, the IASB issued amendments to IAS 1 Presentation of Financial Statements and IAS 8 Accounting Policies, Changes in Accounting Estimates and Errors to align the definition of 'material' across the standards and to clarify certain aspects of the definition. The new definition states that, 'Information is material if omitting, misstating or obscuring it could reasonably be expected to influence decisions that the primary users of general purpose financial statements make on the basis of those financial statements, which provide financial information about a specific reporting entity.'
The amendments to the definition of material is not expected to have a significant impact on the Group's consolidated financial statements.
Amendments to IFRS 3: Definition of a Business
In October 2018, the IASB issued amendments to the definition of a business in IFRS 3 Business Combinations to help entities determine whether an acquired set of activities and assets is a business or not. They clarify the minimum requirements for a business, remove the assessment of whether market participants are capable of replacing any missing elements, add guidance to help entities assess whether an acquired process is substantive, narrow the definitions of a business and of outputs, and introduce an optional fair value concentration test. New illustrative examples were provided along with the amendments.
Since the amendments apply prospectively to transactions or other events that occur on or after the date of first application, the Group will not be affected by these amendments on the date of transition.
4 Basis of consolidation
As discussed in note 1 and note 2, as a result of the corporate reorganization, Centogene N.V. consolidates Centogene AG and Centogene AG is considered to be the predecessor to Centogene N.V. for accounting and reporting purposes.
The basis of consolidation includes the entities over which Centogene N.V. has control within the meaning of IFRS 10 Consolidated Financial Statements. According to IFRS 10, Centogene N.V. has control of an investee when it has direct or indirect power over the investee, exposure, or rights to variable returns from its involvement with the investee and the ability to use its power over the investee to affect those returns. Control is established when it is possible to influence operating and financial policies of the investee, typically with a share in the voting rights or shareholding of more than 50% in the investee. An entity is included in the Group's basis of consolidation from the point in time when the Group obtains control of the entity and ceases when the Group loses control of the subsidiary. Assets, liabilities, income and expenses of a subsidiary acquired or disposed of during the year are included in the consolidated financial statements from the date the Group gains control until the date the Group ceases to control the subsidiary.
F-10
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
4 Basis of consolidation (Continued)
Profit or loss and each component of other comprehensive income are attributed to the equity holders of the parent of the Group and to the non-controlling interests, even if this results in the non-controlling interests having a deficit balance. All intra-group assets and liabilities, equity, income, expenses and cash flows relating to transactions between members of the Group are eliminated in full upon consolidation.
If the Group loses control over a subsidiary, it derecognizes the related assets, liabilities, non-controlling interest and other components of equity, while any resultant gain or loss is recognized in profit or loss.
5 Significant accounting policies
The Group applied the following accounting policies consistently for all of the periods presented in these consolidated financial statements.
(a) Foreign currency and currency translation
The Group's consolidated financial statements are presented based on the parent company's functional currency. For each entity, the Group determines the functional currency and items included in the financial statements of each entity are measured using that functional currency. The Group uses the direct method of consolidation and on disposal of a foreign operation, the gain or loss that is reclassified to profit or loss reflects the amount that arises from using this method.
Transactions in foreign currency are translated into the respective entity's functional currency at the spot rate prevailing on the date of the transaction.
The functional currency of each entity is the respective local currency, since the entities carry out their business activities independently from a financial, economic and organizational perspective.
Monetary assets and liabilities denominated in foreign currency are translated to the functional currency using the closing rate at the reporting date. Currency translation differences are recognized immediately through profit or loss. Non-monetary items denominated in a foreign currency that are measured at historical cost are not translated at the reporting date.
On consolidation, the assets and liabilities of foreign operations are translated into euros using the closing rate on the reporting date. Income and expenses of foreign operations are translated using the exchange rate prevailing on the date of the transaction or the annual average exchange rate. Equity is translated using historical rates until the entity is removed from the Group's basis of consolidation. Any resulting currency translation differences are recorded in other comprehensive income and recognized under the currency translation reserve in equity if the exchange difference is not allocable to the non-controlling interests.
The exchange rates used are presented in the following table:
| |
Average rate | Closing rate | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2017 | 2018 | 2019 | Dec 31, 2017 | Dec 31, 2018 | Dec 31, 2019 | |||||||||||||
USD (EUR 1) |
1.1297 | 1.1779 | 1.1191 | 1.1993 | 1.1419 | 1.1234 | |||||||||||||
AED (EUR 1) |
4.1549 | 4.2713 | 4.0985 | 4.3874 | 4.1396 | 4.0795 | |||||||||||||
INR (EUR 1) |
73.5324 | 79.3177 | 78.7980 | 76.6055 | 78.5156 | 80.187 | |||||||||||||
F-11
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
5 Significant accounting policies (Continued)
(b) Revenues from contracts with customers
The Group provides pharmaceutical solutions and diagnostic tests enabled by its knowledge and interpretation-based platform. Revenue from contracts with customers is recognized when control of the goods or services are transferred to the customer at an amount that reflects the consideration to which the Group expects to be entitled in exchange for those goods or services, usually on delivery of the goods.
- (i)
- Pharmaceutical segment
- –
- Revenue from early patient recruitment and identification, epidemiological insights, biomarker discovery and patient monitoring is based on fee
per sample, milestone fees and fixed fees. The revenues from these solutions are recognized over time using an input method based on the work rendered in order to measure progress towards complete
satisfaction of the services.
- –
- Revenue from the licensing of intellectual property for an unlimited period, usually in the structure of an upfront fee, is recognized at a
point in time, when the right (or license) to use intellectual property is conveyed.
- –
- Revenues from the licensing of intellectual property for a certain period, being a right to access such intellectual property as defined in
IFRS 15, is recognized over time over the licensing period.
- –
- Revenue from the sale of CentoCards is recognized at a point in time when the control of the CentoCards has transferred to the customer, which
typically occurs on delivery.
The Group's contracts with customers relate to a variety of solutions provided to the Group's pharmaceutical partners in order to accelerate their development of treatments for rare diseases, including early patient recruitment and identification, epidemiological insights, biomarker discovery and patient monitoring. The collaboration agreements are structured on a fee per sample basis, milestone basis, fixed fee basis, royalty basis or a combination of these. In addition, some of the Group's contracts with its pharmaceutical partners also include sales of CentoCards for the collection of biological samples from patients.
The performance obligations in Pharmaceutical segment can either be satisfied over time or at a point in time depending on the structure of the collaborations, which are determined based on nature of the service provided, as detailed below.
- (ii)
- Diagnostics segment
Revenues from the Group's diagnostics segment are typically generated from genetic sequencing and diagnostics services that the Group provides to clients, who are typically physicians, laboratories or hospitals, either directly or through distributors. Revenues are based on a negotiated price per test or on the basis of agreements to provide certain testing volumes over defined periods. The Group has concluded that the services rendered in the diagnostics segment comprise one performance obligation.
The performance obligation in the Diagnostics segment is recognized over time, using an input method to measure progress towards complete satisfaction of the service. In order to measure progress, the Group uses a standardized process which measures progress to completion by stages,
F-12
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
5 Significant accounting policies (Continued)
consisting of (i) a preparation stage, (ii) a clarification stage, (iii) a sequencing stage, and (iv) an output stage. The percentages attributed to those stages are indicative of the cost incurred in performing the respective stage in relation to total cost.
The Group has disaggregated revenue recognized from contracts with customers into categories that depict how the nature, amount, timing and uncertainty of revenue and cash flows are affected by economic factors. The Group has also disclosed information about the relationship between the disclosure of disaggregated revenue and the revenue information disclosed for each reportable segment. See note 7 for the disclosure on disaggregated revenue.
Contract balances
- (i)
- Contract assets
A contract asset is the right to consideration in exchange for goods or services transferred to the customer. If the Group satisfies a performance obligation by transferring goods or services to a customer before the customer pays consideration or before payment is due, a contract asset is recognized for the earned consideration that is conditional. Contract assets are subject to impairment assessment, refer to accounting policies of impairment of financial assets in note 5(n) "Financial instruments".
- (ii)
- Trade receivables
A receivable represents the Group's right to an amount of consideration that is unconditional (i.e., only the passage of time is required before payment of the consideration is due). Refer to accounting policies of impairment of financial assets in note 5(n) "Financial instruments".
- (iii)
- Contract liabilities
A contract liability is the obligation to transfer goods or services to a customer for which the Group has received consideration or an amount of consideration is due from the customer (whichever is earlier). If a customer pays consideration before the Group transfers goods or services to the customer, a contract liability is recognized when the payment is made or the payment is due (whichever is earlier). Contract liabilities are recognized as revenue when the Group performs under the contract.
(c) Finance income and finance costs
Interest income and expenses are recognized in the period which they relate to through profit or loss using the effective interest rate method.
(d) Current versus non-current classification
The Group presents assets and liabilities in the statement of financial position based on current/non-current classification. An asset is current when it is:
- –
- Expected to be realized or intended to be sold or consumed in the normal operating cycle
- –
- Held primarily for the purpose of trading
- –
- Expected to be realized within twelve months after the reporting period; or
F-13
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
5 Significant accounting policies (Continued)
- –
- Cash or cash equivalent unless restricted from being exchanged or used to settle a liability for at least twelve months after the reporting period
All other assets are classified as non-current.
A liability is current when:
- –
- It is expected to be settled in the normal operating cycle
- –
- It is held primarily for the purpose of trading
- –
- It is due to be settled within twelve months after the reporting period; or
- –
- There is no unconditional right to defer the settlement of the liability for at least twelve months after the reporting period
The terms of the liability that could, at the option of the counterparty, result in its settlement by the issue of equity instruments do not affect its classification.
The Group classifies all other liabilities as non-current.
Deferred tax assets and liabilities are classified as non-current assets and liabilities
(e) Intangible assets
Research and development
Expenses for research activities are recognized through profit or loss in the period in which they are incurred.
Development expenditures on an individual project are recognized as an intangible asset from the date the Group can demonstrate:
- –
- the product or process is technically and commercially feasible so that the asset will be available for use or sale
- –
- the Group has the intention and its ability and intention to use or sell the asset
- –
- a future economic benefit is probable
- –
- the Group has sufficient resources to complete the development and
- –
- the development costs can be measured reliably.
The Group's research and development activities mainly relate to development of biomarkers and IT driven solutions. With respect to biomarkers, the development stage is usually considered to be achieved when the target validation process is completed and commercialization is probable. With respect to IT driven solutions, the development stage is considered to be achieved upon the completion of the Group's internal validation test. Before such dates, any development costs are recognized in profit or loss and may not be subsequently capitalized.
Capitalized development costs are recognized at cost less accumulated amortization and any accumulated impairment losses. They are only amortized as from the date the asset is ready for its intended use, which in the case of biomarkers is normally at the time the patent application for such
F-14
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
5 Significant accounting policies (Continued)
biomarker is made. Amortization expense commences when the assets ready to be put in use, and is recorded in cost of sales and research and development expenses.
Capitalized development costs which are still under development are tested for impairment annually and when circumstances indicate that the carrying value may be impaired.
Other intangible assets
Other intangible assets purchased by the Group with finite useful lives are recognized at cost less accumulated amortization and any accumulated impairment losses. Subsequent expenditure is only capitalized if it increases the future economic benefits of the respective asset.
Intangible assets are amortized over their estimated useful life using the straight-line method and assessed for impairment whenever there is an indication that the intangible asset may be impaired.
The estimated useful lives are as follows:
- –
- Software, patents and trademarks: 3-7 years
- –
- Capitalized development costs: 7 years
The useful lives and depreciation methods are reviewed annually to ensure that the methods and periods of depreciation are consistent with the expected economic benefit from the asset.
(f) Property, plant and equipment
Property, plant and equipment are carried at cost less any accumulated depreciation and any accumulated impairment losses.
The cost of property, plant and equipment comprises its purchase price including customs duties and non-refundable acquisition taxes, and proportionate VAT not deductible from input tax as well as any directly attributable costs of bringing the asset to its working condition and location for its intended use.
Subsequent expenditure is only capitalized if it is probable that the future economic benefits associated with the expenditure will flow to the Group.
Depreciation is calculated over the estimated useful life using the straight-line method. The Group has assessed that none of its property, plant and equipment has a residual value. The estimated useful lives of significant property, plant and equipment are as follows:
- –
- Freehold land is not depreciated
- –
- Buildings: 33 years and
- –
- Plant and other equipment, furniture and fixtures: 2-15 years
An item of property, plant and equipment is derecognized upon disposal or when no future economic benefits are expected from its use or disposal. Any gain or loss arising on derecognition of the asset (calculated as the difference between the net disposal proceeds and the carrying amount of the asset) is included in the statement of comprehensive loss when the asset is derecognized.
F-15
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
5 Significant accounting policies (Continued)
The depreciation methods, useful lives and residual values are reviewed, and adjusted prospectively if appropriate, as of each reporting date.
Assets under construction are reported at cost and are allocated to property, plant and equipment until they are completed and put into operational use, from which point onwards they are depreciated.
(g) Leases
Before January 1, 2019 prior to adoption of IFRS 16, the accounting treatment of leases depended on if key risk and rewards of ownerships of the assets under leases were transferred. Assets that are held by the Group under a lease that transfers the key risks and rewards of ownership to the Group are classified as finance leases. The leased asset is initially measured at the lower of fair value and the present value of the minimum lease payments. After initial recognition, the asset is carried in accordance with applicable accounting policy for the asset.
Finance lease payments are apportioned between finance costs and the reduction of the outstanding liability. The finance costs are allocated to each period during the lease term so as to produce a constant periodic rate of interest on the remaining balance of the liability.
Assets from other leases are classified as operating leases and the respective lease expenses are recognised in profit or loss on a straight-line basis over the lease term.
Since January 1, 2019, the Group adopted IFRS 16 and assesses at contract inception whether a contract is, or contains, a lease. That is, if the contract conveys the right to control the use of an identified asset for a period of time in exchange for a consideration.
Group as a lessee
The Group applies a single recognition and measurement approach for all leases, except for short-term leases and leases of low-value assets. The Group recognizes lease liabilities to make lease payments and right-of-use assets representing the right to use the underlying assets.
- (i)
- Right-of-use assets
The Group recognizes right-of-use assets at the commencement date of the lease (i.e., the date the underlying asset is available for use). Right-of-use assets are measured at cost, less any accumulated depreciation and impairment losses, and adjusted for any remeasurement of lease liabilities. The cost of right-of-use assets includes the amount of lease liabilities recognized, initial direct costs incurred, and lease payments made at or before the commencement date less any lease incentives received. Unless the Group is reasonably certain to obtain ownership of the leased asset at the end of the lease term, the recognized right-of-use assets are depreciated on a straight-line basis over the shorter of its lease term and the estimated useful lives, as follows:
- –
- Buildings: 33 years
- –
- Offices: 4 - 12 years and
- –
- Plant and other equipment, furniture and fixtures: 2-15 years
If ownership of the leased asset transfers to the Group at the end of the lease term or the cost reflects the exercise of a purchase option, depreciation is calculated using the estimated useful life of the asset.
The right-of-use assets are also subject to impairment. Refer to accounting policies of impairment of financial assets in note 5(n) "Financial instruments".
F-16
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
5 Significant accounting policies (Continued)
- (ii)
- Lease liabilities
At the commencement date of the lease, the Group recognizes lease liabilities measured at the present value of lease payments to be made over the lease term. The lease payments include fixed payments (including in-substance fixed payments) less any lease incentives receivable, variable lease payments that depend on an index or a rate, and amounts expected to be paid under residual value guarantees. The lease payments also include the exercise price of a purchase option reasonably certain to be exercised by the Group and payments of penalties for leases reasonably certain to be terminated. The variable lease payments that do not depend on an index or a rate are recognized as expenses in the period during which the event or condition that triggers the payment occurs.
In calculating the present value of lease payments, the Group uses the incremental borrowing rate at the lease commencement date if the interest rate implicit in the lease is not readily determinable. After the commencement date, the amount of lease liabilities is increased to reflect the accretion of interest and reduced for the lease payments made. In addition, the carrying amount of lease liabilities is remeasured if there is a modification, a change in the lease term, a change in the in-substance fixed lease payments or a change in the assessment to purchase the underlying asset.
- (iii)
- Short-term leases and leases of low-value assets
The Group applies the short-term lease recognition exemption to its short-term leases of machinery and equipment (i.e., those leases that have a lease term of 12 months or less from the commencement date and do not contain a purchase option). It also applies the lease of low-value assets recognition exemption to leases of office equipment that are considered of low value (i.e., below EUR 5k). Lease payments on short-term leases and leases of low-value assets are recognized as expenses on a straight-line basis over the lease term.
- (iv)
- Sale and leaseback transactions
The Group applies IFRS 15 for determining if the transfer of an asset to the buyer (lessor) is to be accounted for as a sale of assets. After the sale of assets is concluded, the Group measures the right-of-use assets arising from the leaseback at the proportion of the previous carrying value of the asset that relates to the right of use retained by the Group. Accordingly, the Group recognizes only the amount of any gain or loss that relates to the rights transferred to the buyer (lessor).
If the fair value of the consideration for the sale of an asset does not equal the fair value of the asset, or if the payments for the leases are not at market rates, the Group makes the following adjustments to measure the sale proceeds at fair value:
- •
- any below-market terms shall be accounted for as a prepayment of lease payments
- •
- any above-market terms shall be accounted for as additional financing provided by the buyer-lessor to the seller-lessee
(h) Impairment of non-financial assets
Property, plant and equipment and intangible assets are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Whenever the carrying amount of an asset exceeds its recoverable amount, an impairment loss is
F-17
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
5 Significant accounting policies (Continued)
recognized in profit or loss. The recoverable amount is measured as the higher of fair value less costs to sell and value in use. Recoverable amounts are estimated either for individual assets or, if an individual asset does not generate cash flows independently of other assets, for the whole cash-generating unit.
- (i)
- Inventories
Inventories are measured at the lower of cost and net realizable value. Inventories are recognized at cost based on the first in first out (FIFO) method.
Net realizable value is the estimated selling price in the ordinary course of business, less estimated costs of completion and the estimated costs necessary to make the sale.
- (j)
- Government grants
Government grants are recognized where there is reasonable assurance that the grant will be received and all attached conditions will be complied with. Grants that are intended to compensate the Group for expenses incurred are recognized through profit or loss on a systematic basis over the periods in which expenses are recognized.
Government grants which relate to an asset are initially recognized as deferred income at nominal amounts. They are subsequently released to profit or loss on a systematic basis over the expected useful life of the related asset.
The release of deferred income related to either type of grant is presented as other operating income (see note 8).
(k) Share-based payments
Plan recipients (including senior executives and certain member of Supervisory Board) of the Group receive remuneration in the form of share-based payments, whereby the recipients render services as consideration for equity instruments (equity-settled transactions) or settled in cash (cash-settled transactions).
Equity settled transactions
The cost of equity-settled transactions is determined by the fair value of the granted options when the grant is made, using a Black-Scholes Model, with further details given in note 20.
The cost is recognized in employee benefits expense (see note 8.4) or other relevant expenses, together with a corresponding increase in equity (capital reserves), over the period in which the service conditions are fulfilled (the vesting period). The cumulative expense recognized for equity-settled transactions at each reporting date until the vesting date reflects the extent to which the vesting period has expired and the Group's best estimate of the number of equity instruments that will ultimately vest. The expense or credit in profit or loss for a period represents the movement in cumulative expense recognized as at the beginning and end of that period.
Cash-settled transactions
A liability is recognized for the fair value of cash-settled transactions. The fair value is measured initially and at each reporting date up to and including the settlement date, with changes in fair value
F-18
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
5 Significant accounting policies (Continued)
recognized in employee benefits expense (see note 8.4). The fair value per option is determined using the Black-Scholes model, further details of which are given in note 20. The fair value per option is then multiplied by the Group's best estimate of the number of awards expected to vest and the portion of the expired vesting period (period in which the service conditions are fulfilled). The cumulative amount of expense recognized will be equal to the cash that is paid on settlement.
Service and non-market performance conditions are not taken into account when determining the grant date fair value of awards, but the likelihood of the conditions being met is assessed as part of the Group's best estimate of the number of equity instruments that will ultimately vest. Market performance conditions are reflected within the grant date fair value. Any other conditions attached to an award, but without an associated service requirement, are considered to be non-vesting conditions. Non-vesting conditions are reflected in the fair value of an award and lead to an immediate expensing of an award unless there are also service and/or performance conditions.
No expense is recognized for awards that do not ultimately vest because non-market performance and/or service conditions have not been met. Where awards include a market or non-vesting condition, the transactions are treated as vested irrespective of whether the market or non-vesting condition is satisfied, provided that all other performance and/or service conditions are satisfied.
If the terms and conditions of a cash-settled share-based payment transaction are modified with the result that it becomes an equity-settled share-based payment transaction, the transaction is accounted for as such from the date of the modification. Specifically, the equity-settled share-based payment transaction is measured by reference to the fair value of the equity instruments granted at the modification date and recognized in equity. The liability for the cash-settled share-based payment transaction as at the modification date is derecognized on that date. Any difference between the carrying amount of the liability derecognized and the amount of equity recognised on the modification date is recognised immediately in profit or loss.
(l) Provisions
A provision is recognized when the Group has a present obligation (legal, contractual or constructive) as a result of a past event, it is probable that an outflow of resources embodying economic benefits will be required to settle the obligation and a reliable estimate can be made of the amount of the obligation. When the Group expects some or all of a provision to be reimbursed, for example, under an insurance contract, the reimbursement misrecognized as a separate asset, but only when the reimbursement is virtually certain. The expense relating to a provision is presented in the profit or loss net of any reimbursement. Provisions are reviewed at each reporting date and adjusted to reflect the current best estimate.
If the requirements for recognizing a provision are not satisfied, the corresponding obligations are recorded as contingent liabilities unless the possibility of an outflow of resources embodying economic benefits is remote.
(m) Income taxes
Tax expense comprises current and deferred taxes. Current taxes and deferred taxes are recognized through profit or loss apart from deferred taxes related to items recognized outside profit or loss, in
F-19
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
5 Significant accounting policies (Continued)
which case it is recognized in correlation to the underlying transaction either directly in equity or in other comprehensive income.
Current income tax assets and liabilities are measured at the amount expected to be recovered from or paid to the taxation authorities. The tax rates and tax laws used to compute the amount are those that are enacted or substantively enacted at the reporting date in the countries where the Group operates and generates taxable income.
Deferred taxes are set up for temporary differences between the carrying amounts of assets and liabilities for group financial reporting purposes at the reporting date and the amounts used for tax purposes. Deferred tax liabilities are recognized for all taxable temporary differences, except:
- –
- temporary differences arising from the initial recognition of assets or liabilities in the course of a business transaction that is not a
business combination and does not affect either the accounting profit or the taxable profit or loss
- –
- temporary differences associated with investments in subsidiaries if the Group controls the timing of the reversal of the temporary differences, and it is probable that the differences will not reverse in the foreseeable future.
The carrying amount of deferred tax assets is reviewed at each reporting date and reduced to the extent that it is no longer probable that sufficient taxable profit will be available to allow all or part of the deferred tax asset to be utilized. Unrecognized deferred tax assets are re-assessed at each reporting date and are recognized to the extent that it has become probable that future taxable profits will allow the deferred tax asset to be recovered.
Deferred tax assets and liabilities are measured at the tax rates that are expected to apply in the year when the asset is realized or the liability is settled, based on tax rates that have been enacted or substantively enacted at the reporting date.
Deferred tax assets and deferred tax liabilities are offset against each other if certain conditions are met.
(n) Financial instruments
- (i)
- Financial assets
The Group's financial assets principally consist of those accounted for as receivables and contract assets.
Receivables and contract assets
Receivables, including contract assets, are non-derivative financial assets with fixed or determinable payments that are not quoted in an active market. Contract assets and trade receivables that do not contain a significant financing component or for which the Group has applied the practical expedient are measured at the transaction price determined under IFRS 15. Refer to the accounting policies in note 5(b) "Revenues from contracts with customers".
F-20
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
5 Significant accounting policies (Continued)
After initial recognition, Receivables and contract assets are subsequently carried at amortized cost using the effective interest rate method less any impairment losses. Gains and losses are recognized in the profit or loss for the period when the assets are derecognized or impaired.
Derecognition
A financial asset or a part of a financial asset is derecognized when the Group no longer has the contractual rights to the asset or the right to receive cash flows from the asset have expired.
Impairment
The Group recognizes an allowance for expected credit losses (ECLs). ECLs are based on the difference between the contractual cash flows due in accordance with the contract and all the cash flows that the Group expects to receive, discounted at an approximation of the original effective interest rate.
The Group applies a simplified approach in calculating ECLs. Therefore, the Group does not track changes in credit risk, but instead recognizes a loss allowance based on lifetime ECLs at each reporting date. The Group has established a provision matrix that is based on its historical credit loss experience, adjusted for forward-looking factors specific to the debtors and the economic environment.
The Group considers a financial asset in default when contractual payments are 180 days past due. However, in certain cases, the Group may also consider a financial asset to be in default when internal or external information indicates that the Group is unlikely to receive the outstanding contractual amounts in full before taking into account any credit enhancements held by the Group. A financial asset is written off when there is no reasonable expectation of recovering the contractual cash flows.
Further disclosures relating to impairment of trade receivables, including contract assets, are in note 21.
- (ii)
- Financial liabilities
All financial liabilities are recognized initially at fair value and, in the case of loans and borrowings and payables, net of directly attributable transaction costs.
The Group's financial liabilities include trade and other payables (include contract liabilities), as well as loans and borrowings including bank overdrafts.
Loans and borrowings
Loans and borrowings are initially recognized at fair value and subsequently measured at amortized cost using the effective interest rate method, taking into account any principal repayments and any discount or premium on acquisition and including transaction costs and fees that are an integral part of the effective interest rate.
Gains or losses are recognized through profit or loss at the time the liabilities are derecognized or disposed of.
F-21
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
5 Significant accounting policies (Continued)
Derecognition
A financial liability is derecognized when the obligation underlying the liability is discharged, canceled or expires.
When an existing financial liability is replaced by another from the same lender on substantially different terms, or the terms of an existing liability are substantially modified, such an exchange or modification is treated as the derecognition of the original liability and the recognition of a new liability. The difference in the respective carrying amounts is recognized through profit or loss.
(o) Cash and cash equivalents
Cash and cash equivalents comprise cash on hand and bank balances, including short-term, highly liquid investments that can be quickly converted into cash amounts. These have original maturities of three months or less and are subject to a low risk of fluctuation in value.
6 Accounting judgments and estimates
The preparation of the consolidated financial statements requires the management board to make judgments, estimates and assumptions that affect the application of accounting policies and the reported amounts of revenues, expenses, assets and liabilities, and the accompanying disclosures. Actual results could differ from those estimates. Estimates and underlying assumptions are reviewed on an ongoing basis and revisions of estimates are recorded prospectively.
6.1 Judgments
Development costs
Development costs are recognized in accordance with the accounting policy for certain internally generated assets. The Group's research and development activities mainly relate to development of biomarkers and IT driven solutions. With respect to biomarkers, the development stage is usually considered to be achieved when the target validation process is completed and commercialization is probable. With respect to IT driven solutions, the development stage is considered to be achieved upon the completion of the Group's internal validation test. Before such date, any development costs are recognized in profit or loss and may not be subsequently capitalized. As of December 31, 2019, the carrying amount of capitalized development costs was EUR 14,145k (2018: EUR 8,795k). This amount includes investments in the development of biomarkers and IT driven solutions (e.g., the Group's CentoMD database and CentoPortal online platform).
Provision for expected credit losses of trade receivables and contract assets
The Group uses a provision matrix to calculate ECLs for trade receivables and contract assets. The provision rates are based on days past due for groupings of various customer segments that have similar loss patterns (e.g. by segment, geography, customer type and rating).
The provision matrix is initially based on the Group's historical observed default rates. The Group will calibrate the matrix to adjust the historical credit loss experience with forward-looking information. For instance, if forecasted economic conditions (i.e., gross domestic product) are expected to deteriorate over the next year which can lead to an increased number of defaults in the manufacturing
F-22
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
6 Accounting judgments and estimates (Continued)
sector, the historical default rates are adjusted. At every reporting date, the historical observed default rates are updated and changes in the forward-looking estimates are analyzed.
The assessment of the correlation between historical observed default rates, forecasted economic conditions and ECLs is a significant estimate. The amount of ECLs is sensitive to changes in circumstances and of forecasted economic conditions. The Group's historical credit loss experience and forecast of economic conditions may also not be representative of customer's actual default in the future. The information about the ECLs on the Group's trade receivables and contract assets is disclosed in note 21.
Deferred tax asset on loss carryforwards
The tax losses carried forward do not expire. In the light of the Company's loss history, the recognition of deferred taxes for tax losses carried forward and deductible temporary differences is limited to the future reversal of existing taxable temporary differences.
6.2 Assumptions and estimation uncertainties
Information concerning assumptions and estimation uncertainty that have a significant risk of causing a material adjustment to the fiscal year ended on December 31, 2019 are presented in the following disclosures. The Group based its assumptions and estimates on parameters available when the consolidated financial statements were prepared. Existing circumstances and assumptions about future developments, however, may change due to market changes or circumstances arising that are beyond the control of the Group. Such changes are reflected in the assumptions when they occur.
Share-based payments
Estimating fair value for share-based payment transactions requires a determination of the most appropriate valuation model, which depends on the terms and conditions of the grant. The Group measures the fair value of cash-settled transactions with employees (including senior executives) using the Black-Scholes model to determine the liability incurred at the date of grant, as well as at the end of each reporting period, until the date of settlement (including cancellation and replacement), with any changes in fair value recognised in profit or loss. This requires a reassessment of the estimates used at the end of each reporting period. No cash-settled share-based transactions were outstanding as of December 31, 2019.
For the measurement of the fair value of equity-settled transactions at the grant date (including those issued to replace the cash-settled transactions), the Group also uses the Black-Scholes model. The fair value at grant date of equity-settled transactions is not updated at the end of each reporting period.
Valuation of Share Options
The Black-Scholes option pricing model requires the input of subjective assumptions, including assumptions about the expected life of share-based awards and share price volatility. As a company only recently listed on NASDAQ stock exchange, the Group's share price does not have sufficient historical information to be used as a reference, and therefore subjective inputs were included when estimating the fair value of our common shares to be used in the Black-Scholes option pricing model.
F-23
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
6 Accounting judgments and estimates (Continued)
In addition, our management concluded a volatility of 70% (2018: 70%; 2017: 60%) an appropriate assumption used for the valuation of our share options, considering the historical volatility of other comparable publicly traded companies.
The Group intends to continue to consistently apply this methodology using the same comparable companies until a sufficient amount of historical information regarding the volatility of our own share price as a public company becomes available.
The assumptions and models used for estimating fair value for share-based payment transactions are disclosed in note 20.
7 Segment information and revenue from contracts with customers
For management purposes, the Group is organized into business units based on its products and services and has two reportable segments, as follows:
- •
- Pharmaceutical segment: This
segment provides a variety of solutions to our pharmaceutical partners, including target discovery, early patient recruitment and identification, epidemiological insights, biomarker discovery and
patient monitoring, in order to accelerate their development of treatments for rare diseases; and
- •
- Diagnostics segment: This segment provides genetic sequencing and diagnostics services to our clients, who are typically physicians, laboratories or hospitals, either directly or through distributors.
Residual operating activities of the Group are reported as 'Corporate'. These include the group functions for communications, human resources, finance (including treasury and taxes), legal, research and development and other supporting activities.
The management board is the Chief Operating Decision Maker and monitors the operating results of its business units separately for the purpose of making decisions about resource allocation and performance assessment. Segment performance is evaluated based on segment results and is measured with reference to the Adjusted EBITDA, which is operating loss presented in the consolidated statements of comprehensive loss, adjusted for corporate expenses, depreciation and amortization as well as share-based payment expenses.
| |
2017 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
in EUR k
|
Pharmaceutical | Diagnostics | Corporate | Total | |||||||||
Rendering of services |
12,326 | 17,758 | — | 30,084 | |||||||||
Sales of goods |
1,605 | — | — | 1,605 | |||||||||
Revenues from external customers |
13,931 | 17,758 | — | 31,689 | |||||||||
Adjusted EBITDA |
10,870 |
2,552 |
(13,746 |
) |
(324 |
) |
|||||||
Capital Expenditures |
1,464 |
607 |
15,964 |
18,035 |
|||||||||
Additions to property, plant and equipment |
241 | 607 | 14,716 | 15,564 | |||||||||
Additions to intangible assets |
1,223 | — | 1,248 | 2,471 | |||||||||
Other segment information |
|||||||||||||
Depreciation and amortization |
793 | 1,311 | 1,133 | 3,237 | |||||||||
Research and development expenses |
35 | — | 6,361 | 6,396 | |||||||||
F-24
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
7 Segment information and revenue from contracts with customers (Continued)
| |
2018 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
in EUR k
|
Pharmaceutical | Diagnostics | Corporate | Total | |||||||||
Rendering of services |
16,077 | 23,171 | — | 39,248 | |||||||||
Sales of goods |
1,230 | — | — | 1,230 | |||||||||
Revenues from external customers |
17,307 | 23,171 | — | 40,478 | |||||||||
Recognized over time |
12,077 |
23,171 |
— |
35,248 |
|||||||||
Recognized at a point in time |
5,230 | — | — | 5,230 | |||||||||
Revenues from external customers |
17,307 | 23,171 | — | 40,478 | |||||||||
Adjusted EBITDA |
13,641 |
2,285 |
(15,836 |
) |
90 |
||||||||
Capital Expenditures |
|||||||||||||
Additions to property, plant and equipment |
1,225 | 1,917 | 5,568 | 8,710 | |||||||||
Additions to intangible assets |
1,948 | — | 1,111 | 3,059 | |||||||||
Other segment information |
|||||||||||||
Depreciation and amortization |
1,222 | 1,838 | 2,115 | 5,175 | |||||||||
Research and development expenses |
334 | — | 5,966 | 6,300 | |||||||||
| |
2019 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
in EUR k
|
Pharmaceutical | Diagnostics | Corporate | Total | |||||||||
Rendering of services |
19,089 | 27,258 | — | 46,347 | |||||||||
Sales of goods |
2,433 | — | — | 2,433 | |||||||||
Revenues from external customers |
21,522 | 27,258 | — | 48,780 | |||||||||
Recognized over time |
17,159 |
27,258 |
— |
44,417 |
|||||||||
Recognized at a point in time |
4,363 | — | — | 4,363 | |||||||||
Revenues from external customers |
21,522 | 27,258 | — | 48,780 | |||||||||
Adjusted EBITDA |
14,956 |
2,306 |
(22,949 |
) |
(5,687 |
) |
|||||||
Capital Expenditures |
|||||||||||||
Additions to property, plant and equipment and right-of-use assets |
1,362 | 1,998 | 17,908 | 21,268 | |||||||||
Additions to intangible assets |
3,603 | — | 3,677 | 7,280 | |||||||||
Other segment information |
|||||||||||||
Depreciation and amortization |
1,308 | 2,032 | 3,239 | 6,579 | |||||||||
Research and development expenses |
— | — | 9,590 | 9,590 | |||||||||
Adjustments
Corporate expenses, depreciation and amortization, interest and similar income and expenses, as well as share-based payment expenses are not allocated to individual segments as the underlying instruments are managed on a group basis. Current taxes and deferred taxes are allocated to Corporate as they are also managed on a group basis.
Corporate expenses for the year ended December 31, 2019 also included expenses incurred in relation to the IPO as described in note 1 of EUR 1,092k (2018: EUR nil ;2017: EUR nil) (see
F-25
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
7 Segment information and revenue from contracts with customers (Continued)
note 8.2) as well as real estate transfer tax of EUR 1,200k (2018: EUR nil ;2017: EUR nil) related to an intercompany sale of land and building (see note 13.1).
Capital expenditure consists of additions of property, plant and equipment, right-of-use assets and intangible assets.
Reconciliation of segment Adjusted EBITDA to Group Loss for the Period
| |
2017 | 2018 | 2019 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Reportable segment Adjusted EBITDA |
13,422 | 15,926 | 17,262 | |||||||
Corporate expenses |
(13,746 | ) | (15,836 | ) | (22,949 | ) | ||||
|
(324 | ) | 90 | (5,687 | ) | |||||
Share-based payment expenses |
(894 | ) | (5,521 | ) | (6,418 | ) | ||||
Depreciation and amortization |
(3,237 | ) | (5,175 | ) | (6,579 | ) | ||||
Operating loss |
(4,455 | ) | (10,606 | ) | (18,684 | ) | ||||
Financial costs, net |
(1,007 | ) | (1,042 | ) | (2,013 | ) | ||||
Income taxes |
(14 | ) | 310 | (158 | ) | |||||
Loss for the year |
(5,476 | ) | (11,338 | ) | (20,855 | ) | ||||
Geographical information
| |
2017 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
in EUR k
|
Pharmaceutical | Diagnostics | Total | |||||||
Revenues |
||||||||||
Europe |
493 | 5,183 | 5,676 | |||||||
- Germany* |
— | — | — | |||||||
Middle East |
— | 8,846 | 8,846 | |||||||
- Saudi Arabia# |
— | 4,926 | 4,926 | |||||||
North America |
13,438 | 1,459 | 14,897 | |||||||
- United States# |
13,438 | 44 | 13,482 | |||||||
Latin America |
— | 1,474 | 1,474 | |||||||
Asia Pacific |
— | 796 | 796 | |||||||
Total |
13,931 | 17,758 | 31,689 | |||||||
F-26
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
7 Segment information and revenue from contracts with customers (Continued)
| |
2018 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
in EUR k
|
Pharmaceutical | Diagnostics | Total | |||||||
Revenues |
||||||||||
Europe |
654 | 6,196 | 6,850 | |||||||
- Germany* |
654 | 407 | 1,061 | |||||||
Middle East |
— | 12,401 | 12,401 | |||||||
- Saudi Arabia# |
— | 5,475 | 5,475 | |||||||
North America |
16,653 | 1,460 | 18,113 | |||||||
- United States# |
16,653 | 643 | 17,296 | |||||||
Latin America |
— | 2,185 | 2,185 | |||||||
Asia Pacific |
— | 929 | 929 | |||||||
Total |
17,307 | 23,171 | 40,478 | |||||||
| |
2019 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
in EUR k
|
Pharmaceutical | Diagnostics | Total | |||||||
Revenues |
||||||||||
Europe |
381 | 7,066 | 7,447 | |||||||
- Germany* |
233 | 275 | 508 | |||||||
- Netherlands** |
— | 25 | 25 | |||||||
Middle East |
122 | 13,977 | 14,099 | |||||||
- Saudi Arabia# |
— | 7,417 | 7,417 | |||||||
North America |
20,896 | 2,380 | 23,276 | |||||||
- United States# |
20,896 | 1,882 | 22,778 | |||||||
Latin America |
123 | 2,864 | 2,987 | |||||||
Asia Pacific |
— | 971 | 971 | |||||||
Total |
21,522 | 27,258 | 48,780 | |||||||
- *
- country
of the incorporation of Centogene AG
- **
- country
of the incorporation of Centogene N.V.
- #
- countries contributing more than 10% of the Group's total consolidated revenues for the respective year ended December 31, 2017, 2018 and 2019
We collaborated with the majority of our pharmaceutical partners on a worldwide basis in 2017, 2018 and 2019. In addition, in cases where our pharmaceutical partners are developing a new rare disease treatment, it is generally anticipated that the final approved treatment will be made available globally. As a result, we allocate the revenues of our pharmaceutical segment by geographical region by reference to the location where each pharmaceutical partner mainly operates, which is based on the region from which most of their revenues are generated. The allocation of revenues in our diagnostics segment is based on the location of each customer.
During the year ended December 31, 2019, revenues from one pharmaceutical partner represented 24.3% of the Group's total revenues (2018: 27.3%; 2017: 37.7%)
During the year ended December 31, 2019, Centogene entered into several collaborations with pharmaceutical partners, of which upfront fees totaling EUR 1,930k were received. Such upfront
F-27
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
7 Segment information and revenue from contracts with customers (Continued)
payments were recognized as revenues during the year as they represented the transaction price allocated to the one-off transfer of the Group's intellectual property—provision of epidemiological insights of relevant rare diseases and relevant data. For the year ended December 31, 2018, upfront payments totaling EUR 4,000k were received and recognized as revenues during the period as they represented the transaction price to be allocated to the grant of licences which are distinct and qualify as a licence to use such intellectual property for an unlimited period or for the time specified in the agreements. No such revenues were recognized for the year ended December 31, 2017.
Non-current assets of the Group consist of right-of-use assets (under IFRS 16), property, plant and equipment (including finance leases already capitalized under IAS17 for prior years), as well as intangible assets. All of such assets are located in Germany, which is the country of the business address of the Centogene AG, except for property, plant and equipment of EUR 286k (2018: EUR 718k) and right-of-use assets of EUR 1,042k (2018: EUR nil), which are located in the United States.
Contract balances
in EUR k
|
Dec 31, 2018 | Dec 31, 2019 | |||||
|---|---|---|---|---|---|---|---|
Trade receivables (note 15) |
8,572 | 12,709 | |||||
Contract assets (note 15) |
2,329 | 3,884 | |||||
Contract liabilities (note 19.2) |
297 | 3,748 | |||||
The contract assets primarily relate to the Group's rights to consideration for work completed but not billed at the reporting date on the tests for the diagnostics segment, with the satisfaction of the respective performance obligation measured by reference to stages in a standardized process. The contract assets also include work performed for pharmaceutical partners which are based on milestone fees. In 2019, EUR 8k (2018: EUR 2k) was recognised as provision for expected credit losses on contract assets. The contract assets are transferred to receivables when the rights become unconditional. This usually occurs when the Group issues an invoice to the customer.
The contract liabilities primarily relate to the advance consideration received from pharmaceutical partners for which revenue is recognized over time, and consideration from sales of CentoCards which have not yet been delivered. Within contract liabilities, EUR 1,430k relates to the aggregate amount of transaction price allocated to performance obligations that are either unsatisfied or partially unsatisfied as of December 31, 2019 (2018: EUR nil), among which EUR 230k will be recognized as revenues within one year upon completion of related patient recruitment and identification activities, and the remaining EUR 1,200k will be recognized over three year in accordance to the licensing period.
The amount of EUR 230k included in contract liabilities as of December 31, 2018 has been recognized as revenues in 2019 (2018: EUR 464k).
F-28
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
8 Other income and expenses
8.1 Other operating income
in EUR k
|
2017 | 2018 | 2019 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Government grants |
637 | 1,611 | 2,641 | |||||||
Gain on disposal of property, plant and equipment |
— | — | 532 | |||||||
Exchange rate gains |
159 | 147 | 314 | |||||||
Income from the reversal of provisions |
— | 309 | 89 | |||||||
Others |
247 | 239 | 205 | |||||||
Total other operating income |
1,043 | 2,306 | 3,781 | |||||||
Government grants contain performance-based grants to subsidize research, development and innovation in the state of Mecklenburg-Western Pomerania from funds granted by the European Regional Development Fund. Furthermore, government grants contain the release of deferred income from investment related grants.
In July 2019, the Group entered into a sale and leaseback transaction. According to which, the Group sold the Rostock headquarters building to a third party and then leased the building from the third party for a period of 12 years at a fixed rate per month with the option to extend. The sale of the Rostock headquarters resulted in a gain of EUR 532k and is recognized in the current period (See note 13.1).
8.2 Other operating expenses
in EUR k
|
2017 | 2018 | 2019 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Currency losses |
84 | 250 | 192 | |||||||
Provision for expected credit losses (note 21.2) |
367 | 792 | 752 | |||||||
Other |
6 | 23 | 1,092 | |||||||
Total other operating expenses |
457 | 1,065 | 2,036 | |||||||
Other operating expenses for the year ended December 31, 2019 included costs incurred related to the IPO charged to profit and loss of EUR 1,092k (2018: EUR nil; 2017: EUR nil).
8.3 Financial costs, net
in EUR k
|
2017 | 2018 | 2019 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Interest expenses from loans |
(827 | ) | (922 | ) | (1,690 | ) | ||||
Unwinding of the discount on lease liabilities |
(194 | ) | (153 | ) | (339 | ) | ||||
Interest income from loans and receivables |
14 | 33 | 16 | |||||||
Total |
(1,007 | ) | (1,042 | ) | (2,013 | ) | ||||
F-29
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
8 Other income and expenses (Continued)
8.4 Employee benefits expense
in EUR k
|
2017 | 2018 | 2019 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Wages and salaries |
13,505 | 17,965 | 23,854 | |||||||
Social security contributions |
2,144 | 2,492 | 3,212 | |||||||
Share-based payments |
894 | 5,521 | 5,714 | |||||||
Termination benefits |
35 | 56 | 63 | |||||||
Total |
16,578 | 26,034 | 32,843 | |||||||
Social security contributions include contributions to state pension scheme of EUR 1,136k (2018: EUR 1,046k; 2017: EUR 987k) as defined contribution plan expenses.
9 Income taxes
Taxes recognized through profit or loss:
in EUR k
|
2017 | 2018 | 2019 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Current tax expenses |
(23 | ) | (87 | ) | (158 | ) | ||||
Current year |
(27 | ) | (87 | ) | (1 | ) | ||||
Adjustments for prior periods |
4 | — | (157 | ) | ||||||
Deferred tax (expense)/ income |
(9 | ) | 397 | — | ||||||
Temporary differences |
31 | 527 | (514 | ) | ||||||
Tax losses |
(22 | ) | (130 | ) | 514 | |||||
Total income tax (expenses)/ benefit |
(14 | ) | 310 | (158 | ) | |||||
No income taxes were recognized directly in other comprehensive income for the years ended December 31, 2017, 2018 and 2019.
A reconciliation of the effective tax rate to the Group's statutory rate of 31.1% for each of the years ended December 31, 2017, 2018 and 2019 is presented in the table below.
in EUR k
|
2017 | 2018 | 2019 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Loss before tax |
(5,462 | ) | (11,648 | ) | (20,697 | ) | ||||
Taxes on the basis of the Company's domestic tax rate |
1,701 |
3,623 |
6,445 |
|||||||
Tax rate effect of foreign tax jurisdictions |
228 | 406 | 412 | |||||||
Non-deductible expenses |
(78 | ) | (105 | ) | (441 | ) | ||||
Current year losses for which no deferred tax assets were recognized |
(1,842 | ) | (3,528 | ) | (6,416 | ) | ||||
Tax income related to prior years |
4 | — | (157 | ) | ||||||
Other effects |
(27 | ) | (86 | ) | (1 | ) | ||||
Income tax (expenses)/ benefit |
(14 | ) | 310 | (158 | ) | |||||
The domestic tax rate of 31.1% is composed of the corporate income tax rate of 15%, the solidarity surcharge of 5.5% of this corporate income tax, as well as trade tax of 15.3%. The tax rate effects from foreign tax jurisdictions are primarily attributable to the tax-exempt profit of a Group subsidiary located in Dubai.
F-30
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
9 Income taxes (Continued)
Tax losses carryforwards for which no deferred tax assets were recognized amount to EUR 41,570k in Germany (2018: EUR 21,728k; 2017: EUR 9,994k) and to EUR 505k in other countries (2018: EUR 788k; 2017: EUR 790k).
Tax losses carried forward in Germany do not expire. Foreign tax losses carried forward may be restricted. In the light of the Group's loss history, the recognition of deferred taxes for tax losses carried forward and deductible temporary differences was limited to the future reversal of existing taxable temporary differences.
For temporary differences associated with investments in the amount of EUR 4,360k (2018: EUR 3,049k; 2017: EUR 1,791), no deferred tax liability has been recognized because the Company is able to control the timing of the reversal and it is probable that the difference will not reverse in the foreseeable future.
The below table shows a breakdown of deferred taxes in the Group's statement of financial position.
| |
December 31, 2018 | December 31, 2019 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
in EUR k
|
Deferred tax assets |
Deferred tax liabilities |
Deferred tax assets |
Deferred tax liabilities |
|||||||||
Intangible assets |
— | (2,053 | ) | — | (3,013 | ) | |||||||
Property, plant and equipment |
— | — | — | (156 | ) | ||||||||
Other assets (costs relating IPO) |
— | (807 | ) | — | — | ||||||||
Measurement of service contracts |
— | (125 | ) | — | (173 | ) | |||||||
Share-based payments |
2,208 | — | — | — | |||||||||
Government grants |
— | — | 2,051 | — | |||||||||
Unused tax losses |
777 | — | 1,291 | — | |||||||||
Sum |
2,985 | (2,985 | ) | 3,342 | (3,342 | ) | |||||||
Offset |
(2,985 | ) | 2,985 | (3,342 | ) | 3,342 | |||||||
Deferred Taxes |
— | — | — | — | |||||||||
10 Loss Per Share
Basic loss per share is calculated by dividing loss for the period attributable to equity holders of the Group by the weighted average number of shares outstanding during the same period, adjusted for the effect of the corporate reorganization as discussed in Note 1 and applied retrospectively to all prior periods presented. The weighted average number of outstanding shares for the year ended December 31, 2019, after adjusted for the effect of the corporate reorganization were 16,409,285 (2018: 14,112,841; 2017: 12,065,714).
For the periods included in these financial statements, the share options are not included in the diluted loss per share calculation as the Company was loss-making in all these periods. Due to the anti-dilutive nature of the outstanding options, basic and diluted loss per share is equal.
F-31
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
Assets
11 Intangible assets
Reconciliation of carrying amounts
in EUR k
|
Internally generated/ acquired biomarkers |
Internally developed database |
Purchased rights, licenses, software |
Total | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Acquisition and production cost |
|||||||||||||
As of Jan 1, 2018 |
5,812 | 2,804 | 2,193 | 10,809 | |||||||||
| | | | | | | | | | | | | | |
Additions |
1,321 | 561 | 1,177 | 3,059 | |||||||||
| | | | | | | | | | | | | | |
As of Dec 31, 2018 |
7,133 | 3,365 | 3,370 | 13,868 | |||||||||
| | | | | | | | | | | | | | |
Reclass from property, plant and equipment |
386 | — | — | 386 | |||||||||
Reclass* |
900 | 758 | (1,658 | ) | — | ||||||||
Additions |
3,603 | 2,379 | 1,298 | 7,280 | |||||||||
| | | | | | | | | | | | | | |
As of Dec 31, 2019 |
12,022 | 6,502 | 3,010 | 21,534 | |||||||||
| | | | | | | | | | | | | | |
Accumulated amortization and impairment |
|||||||||||||
As of Jan 1, 2018 |
1,782 | 534 | 1,014 | 3,330 | |||||||||
| | | | | | | | | | | | | | |
Amortization |
878 | 513 | 352 | 1,743 | |||||||||
| | | | | | | | | | | | | | |
As of Dec 31, 2018 |
2,660 | 1,047 | 1,366 | 5,073 | |||||||||
| | | | | | | | | | | | | | |
Amortization |
1,171 | 723 | 422 | 2,316 | |||||||||
| | | | | | | | | | | | | | |
As of Dec 31, 2019 |
3,831 | 1,770 | 1,788 | 7,389 | |||||||||
| | | | | | | | | | | | | | |
Carrying amounts |
|||||||||||||
As of Dec 31, 2018 |
4,473 | 2,318 | 2,004 | 8,795 | |||||||||
| | | | | | | | | | | | | | |
As of Dec 31, 2019 |
8,191 | 4,732 | 1,222 | 14,145 | |||||||||
| | | | | | | | | | | | | | |
- *
- The reclassification of EUR 1,658k from purchased rights, licenses, software represented purchased rights related to certain biomarkers in prior years, as well as expenses incurred for certain licenses and softwares which were used in the process of developing internal IT driven solutions. Reclassification is made to allow more transparent presentation considering this is more in line with each sub-group of intangible assets.
Development costs and amortization
Internally generated intangible assets include capitalized development costs for biomarkers and IT driven solutions such as CentoPortal and the CentoMD mutation database (see notes 5 and 6 regarding measurement).
The amortization of patents, trademarks and development costs is expensed and recorded under "cost of sales" to the extent the related intangible is used in generating revenue and recorded in research and development expenses to the extent the related intangibles are used for R&D purposes.
F-32
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
12 Property, plant and equipment
Please refer to the following table for the development from January 1, 2018 to December 31, 2019:
in EUR k
|
Land | Buildings | Plant | Other equipment, furniture and fixtures |
Assets under construction |
Total | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Acquisition and production cost |
|||||||||||||||||||
As of Jan 1, 2018 |
2,149 | — | 12,869 | 3,501 | 20,678 | 39,197 | |||||||||||||
Additions |
— | — | 3,142 | 1,154 | 4,414 | 8,710 | |||||||||||||
Reclass |
— | 24,891 | — | 201 | (25,092 | ) | — | ||||||||||||
| | | | | | | | | | | | | | | | | | | | |
As of Dec 31, 2018 |
2,149 | 24,891 | 16,011 | 4,856 | — | 47,907 | |||||||||||||
| | | | | | | | | | | | | | | | | | | | |
Reclass to right-of-use assets* |
(6,303 | ) | (32 | ) | — | (6,335 | ) | ||||||||||||
Reclass to intangible assets (note 11) |
— | — | — | (386 | ) | (386 | ) | ||||||||||||
Additions |
— | — | 22 | 274 | — | 296 | |||||||||||||
Disposal (note 13.1) |
(2,149 | ) | (21,637 | ) | — | — | — | (23,786 | ) | ||||||||||
| | | | | | | | | | | | | | | | | | | | |
As of Dec 31, 2019 |
— | 3,254 | 9,730 | 4,712 | — | 17,696 | |||||||||||||
| | | | | | | | | | | | | | | | | | | | |
Accumulated depreciation and impairment |
|||||||||||||||||||
As of Jan 1, 2018 |
— | — | 4,089 | 1,271 | — | 5,360 | |||||||||||||
Depreciation |
— | 612 | 2,089 | 731 | — | 3,432 | |||||||||||||
| | | | | | | | | | | | | | | | | | | | |
As of Dec 31, 2018 |
— | 612 | 6,178 | 2,002 | — | 8,792 | |||||||||||||
| | | | | | | | | | | | | | | | | | | | |
Reclass to right-of-use assets* |
(963 | ) | (8 | ) | (971 | ) | |||||||||||||
Depreciation |
— | 737 | 940 | 779 | — | 2,456 | |||||||||||||
Disposal (note 13.1) |
— | (957 | ) | — | — | — | (957 | ) | |||||||||||
| | | | | | | | | | | | | | | | | | | | |
As of Dec 31, 2019 |
— | 392 | 6,155 | 2,773 | — | 9,320 | |||||||||||||
| | | | | | | | | | | | | | | | | | | | |
Carrying amounts |
|||||||||||||||||||
As of Dec 31, 2018 |
2,149 | 24,279 | 9,833 | 2,854 | — | 39,115 | |||||||||||||
| | | | | | | | | | | | | | | | | | | | |
As of Dec 31, 2019 |
— | 2,862 | 3,575 | 1,939 | — | 8,376 | |||||||||||||
| | | | | | | | | | | | | | | | | | | | |
- *
- The reclass to right-of-use assets of EUR 5,364k represented the carrying amount of assets previously classified as finance leases under IAS 17 and recognized as right-of-use assets upon the adoption of IFRS 16. As disclosed in note 3(a), the Group has applied a modified retrospective method of adoption and did not change the initial carrying amounts of recognized assets and liabilities at the date of initial application for leases previously classified as finance leases.
Assets under construction
The Group progressed and completed the construction of a new laboratory and headquarters in Rostock in 2018. Additions to assets under construction during the year ended December 31, 2018 were EUR 4,414k and assets under construction totaling EUR 25,092k were transferred to Plant and Buildings upon completion.
F-33
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
12 Property, plant and equipment (Continued)
Security
The Syndicated Loan Facility of the Group was secured by a land charge in the amount of EUR 19,910k and by the assignment of certain laboratory equipment as of December 31, 2018 (see note 19), which were replaced by security over short-term deposits of EUR 1,500k in December 2019. (see note 16).
13 Leases
The Group has lease contracts for land and buildings and offices in Germany and the United States, as well as various items of plant, machinery, motor vehicles and other equipment used in its operations. Leases for land and buildings is related to the sale and leaseback transaction of the Rostock headquarters building (see note 13.1) with a lease term of 12 years, while the lease terms of offices in Berlin and Boston, Massachusetts are 12 years and 4 years respectively. Leases of plant and machinery and other equipment generally have lease terms between 2 and 4 years, while motor vehicles generally have lease terms of 3 years. The Group's obligations under its leases are secured by the lessor's title to the leased assets. Generally, the Group is restricted from subleasing the leased assets. In addition, a bank guarantee of EUR 3,000k (which is secured by cash deposit of EUR 1,500k) and rental deposits of EUR 257k are required to be maintained for the leases of Rostock headquarters building and Berlin offices until the expiry or termination of the leases. Leases of certain plant and machineries were also secured with rental deposits of EUR 191k.
The lease contracts of Rostock headquarters building and office in Boston, Massachusetts include extension options. These options are negotiated by management to provide flexibility in managing the leased-asset portfolio and align with the Group's business needs. The lease of Rostock headquarters building allows the Group to extend the rental contract twice, each for a period of 6 years, after the expiration of agreement in September 2031 with rental payments of EUR 1,400k per annum. Such extension option was not included in the right-of-use assets and lease liabilities, as it is not reasonably certain that such extension option will be exercised. The lease of office in Boston, Massachusetts provides for an extension option for a two-year period after the expiry of contract on June 30, 2020. The cashflow resulting from the exercise of such extension option was included in the lease term. None of the lease contracts contain termination options.
The Group also has certain leases of motor vehicles and premises with lease terms of 12 months or less and leases of office equipment with low value. The Group applies the 'short-term lease' and 'lease of low-value assets' recognition exemptions for these leases.
F-34
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
13 Leases (Continued)
Set out below are the carrying amounts of right-of-use assets and movements during the period:
In EUR k
|
Building* | Offices | Plant and equipment |
Other equipment |
Motor Vehicles |
Total | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
As at January 1, 2019 |
— | 391 | 5,340 | 24 | 12 | 5,767 | |||||||||||||
Additions |
13,456 | 4,288 | 2,824 | 386 | 18 | 20,972 | |||||||||||||
Depreciation expenses |
(330 | ) | (272 | ) | (1,175 | ) | (20 | ) | (10 | ) | (1,807 | ) | |||||||
| | | | | | | | | | | | | | | | | | | | |
As at December 31, 2019 |
13,126 | 4,407 | 6,989 | 390 | 20 | 24,932 | |||||||||||||
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
- *
- As the lease of land and buildings are made through one contract, all the related right-of-use assets are allocated to Buildings.
Set out below are the carrying amounts of lease liabilities and the movements during the period:
in EUR k
|
Lease liabilities |
|||
|---|---|---|---|---|
As at January 1, 2019 |
3,465 | |||
Additions |
20,946 | |||
Interest expenses |
339 | |||
Payments |
(3,046 | ) | ||
| | | | | |
As at December 31, 2019 |
21,704 | |||
| | | | | |
| | | | | |
| | | | | |
Current |
3,635 | |||
Non-current |
18,069 | |||
The maturity analysis of lease liabilities is disclosed in note 21.
The following are the amounts recognised in profit or loss:
in EUR k
|
2019 | |||
|---|---|---|---|---|
Depreciation expense of right-of-use assets |
1,807 | |||
Interest expenses on lease liabilities |
339 | |||
Rent expenses—short-term leases |
185 | |||
Rent expense—leases of low-value assets |
25 | |||
| | | | | |
Total amounts recognized in profit or loss |
2,356 | |||
| | | | | |
The Group had total cash outflows for leases of EUR 3,256k in 2019. All additions to right-of-use assets and lease liabilities in 2019 were non-cash in nature. The future cash outflows relating to non-cancellable short-term leases and leases of low-value assets, are disclosed in note 24.
13.1 Sale and Leaseback transaction
In June 2019, in preparation for a sale and leaseback transaction, Centogene AG sold its land and building (the Rostock headquarters building) with a carrying value of EUR 22,829k to a subsidiary of the Group. Such intercompany transaction resulted in a real estate transfer tax expense of EUR 1,200k and was recognized in the year ended December 31, 2019.
F-35
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
13 Leases (Continued)
In July 2019, the Group concluded the sale and leaseback transaction, according to which, the Company sold the Rostock headquarters building to a third party for EUR 24,000k, representing the fair value of the building as of June 30, 2019. The Group then leased the building from the third party for a period of 12 years at a fixed rate per month with the option to extend.
The consideration received was used to repay the loans related to the construction of the building of EUR 10,776k (see note 19), plus additional interest of EUR 1,159k. In addition, part of the consideration was used to pay for the rental deposits of the lease of EUR 3,000k. In November 2019, the Group has arranged a bank guarantee to replace the rental deposits to the lessor, while a cash deposit of EUR 1,500k was used to secure the bank guarantee. In addition, government grants received which related to the purchase of land amounting to EUR 358k were refunded to the relevant authority subsequent to the transaction (note 19.2).
The transaction was recorded according to IFRS 16, resulting in a gain on disposal of fixed assets of EUR 532k (see note 8.1), a decrease in property, plant and equipment of approximately EUR 22,829k, an increase of right-of-use assets of approximately EUR 13,456k (see note 13) and an increase in lease liabilities of approximately EUR 14,091k (see note 19).
14 Inventories
in EUR k
|
Dec 31, 2018 | Dec 31, 2019 | |||||
|---|---|---|---|---|---|---|---|
Raw materials, consumables and supplies |
1,323 | 1,644 | |||||
Finished goods and merchandise |
23 | 165 | |||||
Inventories |
1,346 | 1,809 | |||||
In the year ended December 31, 2019, raw materials, consumables and changes in inventories of finished goods and work in process recorded as expenses under "cost of sales" came to EUR 11,285k (2018: EUR 9,473k; 2017: EUR 6,588k).
15 Trade and other receivables and other assets
in EUR k
|
Dec 31, 2018 | Dec 31, 2019 | |||||
|---|---|---|---|---|---|---|---|
Non-current |
|||||||
Other assets—Rental deposits |
— | 1,948 | |||||
|
— | 1,948 | |||||
Current |
|||||||
Trade receivables |
8,572 | 12,709 | |||||
Contract assets |
2,329 | 3,884 | |||||
Receivables due from shareholders |
2,170 | 2,766 | |||||
Other assets |
5,125 | 5,846 | |||||
|
18,196 | 25,205 | |||||
| | | | | | | | |
Total non-current and current trade and other receivables and other assets |
18,196 | 27,153 | |||||
Trade receivables are non-interest bearing and are generally due in 30 to 90 days. In general, portfolio-based bad debt allowances are recognized on trade receivables and contract assets (see note 21.2).
F-36
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
15 Trade and other receivables and other assets (Continued)
As of December 31, 2018, the Group's trade receivables and contract assets were designated as collateral in respect of existing Loan agreements (see note 19.1). In December 2019, the loan agreements were revised and secured by short-term deposits with a carrying amount of EUR 1,500k (note 16) and the security interests in the trade receivables and contract assets were released accordingly.
Other assets
Other assets include VAT receivables of EUR 1,311k (2018: EUR 1,317k), prepaid expenses of EUR 3,481k (2018: EUR 476k) as well as receivables from grants of EUR 409k (2018: EUR 489k). As of December 31, 2018, other assets also included costs relating to the initial public offering of EUR 2,591k, which were offset against capital reserve upon the completion of the transaction in November 2019.
16 Cash and short-term deposits
As of December 31, 2019, the Group has pledged its short-term deposits with carrying amount of EUR 1,500k (2018: EUR nil) and EUR 2,500k (2018: EUR 1,500k) respectively, to fulfil collateral requirements in respect of existing secured bank loan and overdraft facility up to EUR 2,500k. Subsequent to the year end, the Group has pledged its short-term deposits of EUR 500k related to another overdraft facility up to EUR 500k. See note 19 for further details.
The restriction applying to the collateral may be terminated at any time subject to the full amount of the relevant bank loans and the overdrafts being repaid.
Equity and liabilities
17 Equity
On October 29, 2019, the general meeting of shareholders of the Company resolved to approve the corporate reorganization and to execute the Deed of Conversion. The reorganization was effected by the following procedures:
- 1.
- Existing
shareholders of common shares and preferred shares of Centogene AG subscribed for new common shares in Centogene B.V., and in return transferred their
respective shares (both common and preferred) in Centogene AG to Centogene B.V. as a contribution in kind. The exchange ratios for the common and preferred shares of Centogene AG to common
shares of Centogene B.V. were as follows:
- –
- Preferred shares of Centogene AG were exchanged for common shares of Centogene B.V. (share split) on a 1.00 to 89.6125
basis
- –
- Common shares of Centogene AG were exchanged for common shares of Centogene B.V. (share split) on a 1.00 to 33.2238 basis
- 2.
- the legal form of Centogene B.V. were converted from a Dutch private company with limited liability to a Dutch public company, Centogene N.V. and the articles of association of Centogene N.V. became effective. The conversion took place by means of the execution of a
F-37
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
17 Equity (Continued)
notarial deed of conversion and amendment, and resulted in a name change from Centogene B.V. to Centogene N.V.
The corporate reorganization was fully completed on November 12, 2019. All share, per-share and related information presented in the financial statements and corresponding disclosure notes have been retrospectively adjusted, where applicable, to reflect the impact of the share split resulting from the reorganization.
Issued capital and capital reserve
Common Shares
As of December 31, 2018, 15,861,340 common shares of Centogene N.V. with a nominal value of EUR 0.12 (converted from 230,445 common shares with a conversion ratio of 33.2238 and 91,562 preferred shares with a conversion ratio of 89.6125, both with a nominal value of EUR 1.00), were issued and fully paid up.
The preferred shares were issued to certain investors to fund the Company's development activities. The preferred shares each had one voting right per share and did not contain a redemption feature or a contractual right to fixed dividends. The preferred shareholders were entitled to a disproportionate share of the net assets of the Company in case of certain exit events, including IPO, which was reflected by the different conversion ratios (share split) for common and preferred shares of Centogene AG to Centogene B.V.
As a result of the IPO, all issued and paid-in preferred shares were converted to common shares, based on the conversion ratio above which reflected the return to investors as agreed in the relevant investment agreements. As of December 31, 2019, the authorized but unissued common share capital amounted to EUR 7,097k.
in thousands of shares
|
2019 | |||
|---|---|---|---|---|
Common shares issued as a result of corporate reorganization |
15,861 | |||
Issued at IPO |
4,000 | |||
Common shares issued as of Dec 31, fully paid |
19,861 | |||
in thousands of shares
|
as of Dec 31, 2019 |
|||
|---|---|---|---|---|
Authorized common shares of EUR 0.12 each |
79,000 | |||
The holders of common shares are entitled to the Company's approved dividends and other distributions as may be declared from time to time by the Company, and is entitled to vote per share on all matters to be voted at the Company's annual general meetings.
Capital reserve
As of December 31, 2019, capital reserve included a share premium of EUR 90,297k, being amounts contributed by shareholders at the issuance of shares in excess of the par value of the shares issued, net of any transaction costs incurred for the share issuance. During the year ended December 31, 2019, transaction costs related to IPO, that are directly attributable to issuing the new shares, of EUR 4,899k were recorded against capital reserve. Of this amount, a total of EUR 328k was
F-38
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
17 Equity (Continued)
paid in the year ended December 31, 2018, and EUR 565k was not yet paid and included in other liabilities as of December 31, 2019 (2018: EUR 1,695k) (see note 19.2).
The capital reserve consists of the share premium account and amounts recorded in respect of share-based payments. For additional information on the share-based payments, please refer to note 20.
18 Capital management
The Group's objective when managing capital are to safeguard the Company's ability to continue as a going concern and finance all necessary sustainable developments, so that it can continue to provide returns for shareholders and benefits for other stakeholders. In particular, care is taken and an optimal capital structure is tried to achieve to reduce the cost of capital. With the IPO in November 2019, the Group also put more attention on achieving a healthy capital base to increase the confidence of investors and the capital market.
During the years ended December 31, 2017 and 2018, the Group has deployed debt capital for the development of the Rostock headquarters building, and compliance with certain financial covenants were required under the bank loan agreements. Accordingly, the Group also monitored the compliance with these covenants as part of the capital management. The financial covenants related to the bank loans were removed in December 2019 upon additional short-term cash deposits of EUR 1,500k were provided as security.
The Group manages its capital structure and makes adjustments in light of changes in economic conditions and the risk characteristics of its activities. To maintain or adjust the capital structure, the Group may adjust the return to shareholders, issue new shares, or pay additional interests to reduce debt.
19 Financial liabilities
19.1 Interest-bearing loans
in EUR k
|
Dec 31, 2018 | Dec 31, 2019 | |||||
|---|---|---|---|---|---|---|---|
Non-current liabilities |
|||||||
Non-current portion of secured bank loans |
12,055 | 968 | |||||
Municipal loans |
860 | 610 | |||||
| | | | | | | | |
Total non-current loans |
12,915 | 1,578 | |||||
Lease liabilities |
1,712 | 18,069 | |||||
| | | | | | | | |
Total non-current liabilities |
14,627 | 19,647 | |||||
Current liabilities |
|||||||
Current portion of secured bank loans |
1,787 | 802 | |||||
Bank overdrafts |
1,915 | 2,636 | |||||
Municipal loans |
— | 250 | |||||
| | | | | | | | |
Total current loans |
3,702 | 3,688 | |||||
Current portion of lease liabilities |
1,350 | 3,635 | |||||
| | | | | | | | |
Total current liabilities |
5,052 | 7,323 | |||||
| | | | | | | | |
Total non-current and current liabilities |
19,679 | 26,970 | |||||
F-39
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
19 Financial liabilities (Continued)
Financial covenants applied to secured bank loans which stipulate quarterly targets for the company's solvency ratio and net debt ratio as well as covenants related to revenue and EBITDA for the year ended December 31, 2018. The Group obtained formal waivers from the lenders for such covenants for the year ended December 31, 2018. Therefore the secured bank loans were disclosed as current and non-current liabilities based on the contractual maturity of such loans.
As of December 31, 2018, the secured bank loans were also secured by trade receivables (including contract assets) with a carrying amount of EUR 10,901k (see note 15). In addition, they were also secured by a land charge in the amount of EUR 19,910k and by assignment of certain laboratory equipment (see note 12). In December 2019, after a majority of outstanding bank loans were repaid using the consideration received from the sale and leaseback transaction (see note 13.1), short-term cash deposits of EUR 1,500k were used to secure the remaining bank loans outstanding (see note 16), and in exchange, the requirement of compliance with financial covenants were removed, and all collaterals over trade receivables (including contract assets) and certain laboratory equipment, as well as land charge, were released.
The following table is based on the original terms and conditions:
Conditions and statement of liabilities
The outstanding loans as of December 31, 2019 and 2018 have the following conditions:
| |
|
|
|
Dec 31, 2018 | Dec 31, 2019 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
in EUR k
|
Currency | Nominal interest rate | Maturity | Nominal amount |
Carrying amount |
Nominal amount |
Carrying amount |
|||||||||||||
Secured bank loan |
EUR | 3.50% | 2016-19 | 6 | 6 | — | — | |||||||||||||
Secured bank loan |
EUR | 2.50% | 2017-25 | 5,633 | 5,633 | — | — | |||||||||||||
Secured bank loan |
EUR | 2.50% | 2017-25 | 5,633 | 5,633 | — | — | |||||||||||||
Secured bank loan |
EUR | 3.95% | 2017-25 | 2,570 | 2,570 | 1,770 | 1,770 | |||||||||||||
Municipal loan |
EUR | 8.25%; plus 1.5% profit-related; 0.75% on losses |
2020-21 | 500 | 500 | 500 | 500 | |||||||||||||
Municipal loan |
EUR | 8%; plus 1.5% profit-related; 0.75% on losses |
2022 | 360 | 360 | 360 | 360 | |||||||||||||
Bank overdrafts |
EUR | 4.46% | Rollover | — | — | 476 | 476 | |||||||||||||
Bank overdrafts |
EUR | 3.75% | Rollover | 1,915 | 1,915 | 2,160 | 2,160 | |||||||||||||
Lease liabilities |
EUR | 3.5%*,5.4%-8.9% | 2017-31 | 3,062 | 3,062 | 21,704 | 21,704 | |||||||||||||
| | | | | | | | | | | | | | | | | | | | | |
Total interest-bearing financial liabilities |
19,679 | 19,679 | 26,970 | 26,970 | ||||||||||||||||
- *
- represents the incremental borrowing rate of the Group at the commencement of the leases
The bank overdrafts of EUR 2,160k as of December 31, 2019 (2018: 1,915k) were secured by short-term deposits with a carrying amount of EUR 2,500k (2018: EUR 1,500k) (see note 16). The bank overdrafts of EUR 476k (2018: EUR nil) were secured by guarantees provided by certain of the Company's shareholders, which were released by providing security over a short-term deposit with a carrying amount of EUR 500k subsequent to the year ended December 31, 2019.
The municipal loan due to MBMV (Mittelständische Bürgschaftsbank Mecklenburg-Vorpommern) of EUR 860k (2018: EUR 860k) with a remaining term between 2-3 years and an interest rate of 8.25%/8% is also secured by guarantees provided by the Group's shareholders. Subsequent to the year
F-40
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
19 Financial liabilities (Continued)
end, the municipal loans were repaid in full in February 2020 and the shareholder guarantees were released accordingly.
19.2 Trade payables and other liabilities
in EUR k
|
Dec 31, 2018 | Dec 31, 2019 | |||||
|---|---|---|---|---|---|---|---|
Trade payables |
5,429 | 8,554 | |||||
Government grants (deferred income) |
12,034 | 11,289 | |||||
Liability for Virtual Stock Option Program |
7,093 | 2,769 | |||||
Contract liabilities |
297 | 3,748 | |||||
Others |
5,618 | 5,258 | |||||
Trade payables and other liabilities |
30,471 | 31,618 | |||||
Non-current |
11,240 | 9,941 | |||||
Current |
19,231 | 21,677 | |||||
Government grants mainly include investment-related government grants. These were received for the purchase of certain items of property, plant and equipment for the research and development facilities in Mecklenburg-Western Pomerania, including the Rostock facility. The grants were issued in the form of investment subsidies as part of the joint federal and state program, "Verbesserung der regionalen Wirtschaftsstruktur" (improvement of the regional economic structure) in connection with funds from the European Regional Development Fund. Additional grants received during the year ended December 31, 2019 relating to the purchase of certain items of property, plant and equipment amounted to EUR 793k (2018: EUR 3,042k). Subsequent to the sale and leaseback transaction, investment-related government grant received in prior years of EUR 358k relating to purchase of land was refunded to the authority (note 13.1).
In addition, other liabilities include personnel-related liabilities for vacation and bonuses totaling EUR 2,264k (2018: EUR 1,955k) as well as liabilities for wage and church tax of EUR 376k (2018: EUR 307k). Other liabilities also include costs relating to IPO of EUR 565k (2018: EUR 1,695k) (see note 17).
20 Share-based payments
At December 31, 2018 and 2019, the Group had the following share-based payment arrangements.
- (i)
- Virtual share option program 2016 (Cash-settled)
On July 1, 2016, the Group established a virtual share option program ("2016 VSOP") under Centogene AG that entitles the management board to grant virtual share options to individuals, in regard to services they provide and their continuous commitment to the Group. The 2016 VSOP allowed the management board to grant up to 1,000,000 virtual options, representing 5% of the original 205,000 shares of Centogene AG which are issued and owned by the original shareholders. The share options are subject to service conditions.
The completion of IPO in November 2019 was defined as one of the "exit events" in the 2016 VSOP program. Accordingly, all options granted under 2016 VSOP were vested immediately in full. In addition, holders of vested options are entitled to receive a direct cash payment from the Company according to the calculation as stipulated in the program, which is determined based on the IPO price
F-41
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
20 Share-based payments (Continued)
of the shares of Centogene N.V. and the exercise prices of the vested options. As of December 31, 2019, all options under 2016 VSOP were considered vested and exercised and a liability with an carrying amount of EUR 2,768k (2018: EUR 2,170k) was recorded.
The payment to the option holders will be reimbursed by the original shareholders to the Company at the same time as the obligation to pay the options holders arises. A respective receivable against shareholders was recorded (see note 15). As this is a shareholder transaction, the respective receivable against shareholders was recorded against equity (capital reserve).
| |
2018 | 2019 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Number | WAEP | Number | WAEP | |||||||||
Outstanding at January 1 |
802,283 | 3.22 | 802,283 | 3.22 | |||||||||
Exercised during the year |
— | — | (802,283 | ) | 3.45 | ||||||||
Outstanding at December 31 |
802,283 | 3.22 | — | — | |||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Vested at December 31 |
756,083 | 2.74 | — | — | |||||||||
Exercisable at December 31 |
— | — | — | — | |||||||||
The weighted average remaining contractual life for the share options outstanding as at December 31, 2018 was seven years. The range of exercise prices for options outstanding as of December 31, 2018 was EUR 1.0 to EUR 6.0. The intrinsic value of the options vested as of December 31, 2018 was EUR 2,169k.
- (ii)
- Virtual share option program 2017 (Cash-settled)
In 2017, the Group established an additional virtual share option program ("2017 VSOP") that entitled the management board to grant virtual share options to individuals, in regard to services they provide and their continuous commitment to the Group. The 2017 VSOP allowed the management board to grant up to 29,560 virtual options, representing approximately 10% of the total shares of Centogene AG which were then issued and anticipated to be issued after additional investment by the investors. Under this program, holders of vested options were entitled to receive a direct cash payment from the Company, which is determined based on the exit price of the Company's shares, upon the occurrence of any of the "exit events" as defined in share option program. The vesting period shall be three years commencing on the day of grant, where one-third of the granted options shall be vested at the end of each year of grant. Upon an exit event, the vesting of any unvested awards will be accelerated.
As part of the corporate reorganization, in connection with the IPO (see note 1), a transfer agreement was entered into between the holders of the 2017 VSOP, Centogene AG and the Company in November 2019, according to which, the 2017 VSOP was terminated, and the option holders were
F-42
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
20 Share-based payments (Continued)
instead granted new share options of the Centogene N.V., determined based on the IPO price of the shares of Centogene N.V. and the number of options granted (see note 20(iii)).
| |
2018 | 2019 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Number | WAEP | Number | WAEP | |||||||||
Outstanding at January 1 |
4,318 | 1.0 | 10,496 | 1.0 | |||||||||
Granted during the year |
6,178 | 1.0 | 5,878 | 1.0 | |||||||||
Cancelled during the year |
— | — | (16,374 | ) | 1.0 | ||||||||
Replacement awards granted during the year (note 20(iii)) |
— | — | 805,308 | 0.12 | |||||||||
| | | | | | | | | | | | | | |
Outstanding at December 31 |
10,496 | 1.0 | 805,308 | 0.12 | |||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Vested at December 31 |
5,040 | 1.0 | 805,308 | 0.12 | |||||||||
Exercisable at December 31 |
— | — | — | — | |||||||||
The weighted average remaining contractual life for the share options outstanding as at December 31, 2018 was eight years and the weighted average fair value of options outstanding was EUR 540.3. The exercise price for options outstanding as of December 31, 2018 was EUR 1. The intrinsic value of the options vested as of December 31, 2018 was EUR 2,722k.
Upon the completion of the IPO, the liability under 2017 VSOP was calculated based on cash payment entitled to by the holders of options from the Company as stipulated in the program.
The cancellation of 2017 VSOP and the grant of new share options of Centogene N.V. was accounted for as a modification under IFRS 2. The total accumulated liability prior to the modification date of EUR 10,038k was reclassified to the capital reserve, since the new share options of Centogene N.V. are classified as an equity-settled share-based payment (see note 20(iii) below).
- (iii)
- Equity share option - Replacement (ESOP 2017)
As discussed in note 20(ii), share options of Centogene N.V. were issued to the holders of options originally granted under 2017 VSOP as part of the corporate reorganization to replace the cash-settled share-based program.
The number of options granted to each holder was based on the number of options granted to them under 2017 VSOP and the IPO price of Centogene N.V. Accordingly, 805,308 new share options were granted pursuant to the Centogene N.V. Long-term Incentive Plan, with each option representing one common share of Centogene N.V., and an exercise price equal to the nominal value of the share of Centogene N.V., which is EUR 0.12.
The options were considered vested upon the completion of the IPO, but are not exercisable in the first 180 days subsequent to the listing (lock-up period).
The contractual life for the share options as at December 31, 2019 is ten years and the weighted average fair value of options outstanding was EUR 12.46.
The fair value of share options issued under ESOP 2017 are equity-settled and the fair value of the options were recognized in equity under capital reserve on the date of grant.
F-43
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
20 Share-based payments (Continued)
- (iv)
- Equity share option 2019 (ESOP 2019)
In 2019, an agreement was entered into between the Company and an individual of the Supervisory Board. According to this agreement, a total of 396,522 options, each option representing one common share, were granted pursuant to the Centogene N.V. Long-term Incentive Plan to the individual Supervisory Board member with exercise price equaling to the IPO price, which is EUR 12.58 per option, on the date of the IPO of the Company. The vesting period shall be three years commencing on the day of grant, where one-third of the granted options shall be vested at the end of each year of grant, and the first year ending on March 31, 2020.
The contractual life for the share options as at December 31, 2019 is ten years and the weighted average fair value of options outstanding was EUR 9.08. The share options issued under "ESOP 2019" will be equity-settled and the fair value of the options were recognized in equity under capital reserve, based on the fair value on the date of grant, and will be charged to profit or loss over the vesting period.
Valuation of Options
Virtual share option program 2016 and 2017
The fair values of the 2016 VSOP upon its exercise and 2017 VSOP upon its cancellation in 2019 were based on the cash payment entitled to by the holders of the virtual options, which were calculated according to the formulae as stipulated in the respective programs. The cash payment is with reference to the share price of Centogene N.V. at the date of IPO.
For the year ended December 31, 2018, the fair values of 2016 VSOP and 2017 VSOP were calculated based on the enterprise value of the Company, which is determined by discounting the future cash flows to be generated by the Company according to the cash flow projection, and using the Black-Scholes option pricing model. The cash flow projection included specific estimates for ten years and a terminal growth rate of 2%. The discount rate applied of 15%, was a post-tax measure estimated based on the historical industry average weighted average cost of capital, with a possible debt leveraging of 0%-5% at a market interest rate of 6%.
The values assigned to the key assumptions represent management's assessment of future trends in the relevant industries and have been based on historical data from both external and internal sources.
The key assumptions used to derive the option value are set out below:
| |
2018 | |||
|---|---|---|---|---|
Volatility (%) |
70 | |||
Risk-free interest rate (%) |
(0.8 | ) | ||
Dividend yield (%) |
0 | |||
Option term (years) |
0.4 | |||
Equity share option 2017 and Equity share option 2019
The fair values of ESOP 2017 and ESOP 2019 were estimated at the date of grant using the Black-Scholes option pricing model, taking into account the terms and conditions on which the share options were granted. It takes into account historical and expected dividends, and the share price volatility of the other public company in the relevant industries to predict the share performance. There are no cash settlement alternatives for either the option holders or the Company.
F-44
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
20 Share-based payments (Continued)
The key assumptions used to derive the option value are set out below:
| |
2019 | ||||||
|---|---|---|---|---|---|---|---|
| |
ESOP 2017 | ESOP 2019 | |||||
Exercise price (EUR) |
0.12 | 12.58 | |||||
Share price at grant date (EUR) |
12.58 | 12.58 | |||||
Volatility (%) |
70 | 70 | |||||
Risk-free interest rate (%) |
(0.7 | ) | (0.7 | ) | |||
Dividend yield (%) |
0 | 0 | |||||
Option term (years) |
10 | 10 | |||||
The expense recognized for the above share-based payment transactions during the year is shown in the following table:
| |
2018 | 2019 | |||||
|---|---|---|---|---|---|---|---|
Expenses arising from cash-settled share-based payment transactions |
5,521 | 5,714 | |||||
- 2016 VSOP |
1,442 | 596 | |||||
- 2017 VSOP (2019: including modification gain) |
4,079 | 5,118 | |||||
Expenses arising from equity-settled share-based payment transactions |
— | 704 | |||||
| | | | | | | | |
Total expenses arising from share-based payment transactions |
5,521 | 6,418 | |||||
Financial instruments
21 Financial instruments—fair values and risk management
21.1 Classifications and fair values
The carrying values of the Group's financial assets and financial liabilities approximate their fair value.
21.2 Financial risk management
The Group is exposed to the following risks from the use of financial instruments:
- –
- Credit risk
- –
- Liquidity risk
- –
- Currency risk
Credit risk
Credit risk is the risk that a counterparty will not meet its obligations under a financial instrument or customer contract, leading to a financial loss. The Group is exposed to credit risk from its operating activities (primarily trade receivables) and from its financing activities, including deposits with banks and financial institutions and foreign exchange transactions. The carrying amount of the financial assets corresponds to the maximum default risk.
F-45
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
21 Financial instruments—fair values and risk management (Continued)
Trade receivables and contract assets
The Group utilizes a receivables management system that closely manages open items of major customers. The Group's customers in the pharmaceutical segment are mainly pharmaceutical companies which are usually listed companies, or strongly financed by private equity funds. The Group's customers in the diagnostics segment are mainly hospitals, labs and physicians, of which approximately 75% of the revenues from Diagnostics segment were generated from customers who have had business relationships with the Group at least since 2017. To avoid default, the Company may request prepayment for new business with physicians.
In addition to the macroeconomic situation generally, the development of international healthcare markets is a key economic factor in assessing the default risk related to trade receivables and contract assets. These markets are closely monitored by the Group.
An impairment analysis is performed at each reporting date using a provision matrix to measure expected credit losses. The provision rates are based on days past due for groupings of various customer segments with similar loss patterns (i.e. by customers from different segment; customers from different geographical region and customer type). The calculation reflects the probability weighted outcome, the time value of money and reasonable and supportable information that is available at the reporting date about past events, current conditions and forecasts of future economic conditions. The maximum exposure to credit risk at the reporting date is the carrying value of each class of financial assets disclosed in note 15. The Group does not hold collateral as security and does not request letters of credit or other forms of credit insurance. The Group evaluates the concentration of risk with respect to trade receivables and contract assets and recorded credit losses reflecting the expected lifetime loss, based on different types of customers.
Considering the major exposure to the credit risk arising from the diagnostics segment, the Group focused its impairment analysis on the trade receivables due from customers in the diagnostic segment, in particular the MENA and Europe regions as they represent the majority of that segment's revenue. In addition to applying the provision matrix, the Group performed an individual customer analysis on major debtors, with reference to the past history (such as sales and collection in the previous periods) and the assessment of their current financial condition and other relevant factors and evaluated if additional specific impairment losses would be necessary.
F-46
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
21 Financial instruments—fair values and risk management (Continued)
Set out below is the information regarding the credit risk exposure of the Group's trade receivables and contract assets using a provision matrix
| |
As of December 31, 2018 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
in EUR k
|
Total Gross amount |
Not past due |
Past due 1 –30 days |
Past due 31 - 90 days |
Past due by more than 90 days |
|||||||||||
Middle East |
7,766 | 3,065 | 401 | 1,560 | 2,740 | |||||||||||
Europe |
2,900 | 2,052 | 356 | 240 | 252 | |||||||||||
Latin America |
604 | 415 | 81 | 74 | 34 | |||||||||||
North America |
1,074 | 728 | 230 | 79 | 37 | |||||||||||
Asia Pacific |
190 | 175 | 10 | — | 5 | |||||||||||
| | | | | | | | | | | | | | | | | |
Total |
12,534 | 6,435 | 1,078 | 1,953 | 3,068 | |||||||||||
Expected credit loss rate |
13.0 | % | 0.1 | % | 0.5 | % | 2 | % | 51.6 | % | ||||||
Expected credit loss |
1,633 | 6 | 5 | 39 | 1,583 | |||||||||||
| |
As of December 31, 2019 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
in EUR k
|
Total Gross amount |
Not past due |
Past due 1 - 30 days |
Past due 31 - 90 days |
Past due by more than 90 days |
|||||||||||
Middle East |
10,470 | 3,956 | 721 | 1,411 | 4,382 | |||||||||||
Europe |
3,311 | 2,476 | 268 | 222 | 345 | |||||||||||
Latin America |
811 | 611 | 53 | 42 | 105 | |||||||||||
North America |
4,156 | 3,908 | 53 | 24 | 171 | |||||||||||
Asia Pacific |
180 | 151 | 18 | 9 | 2 | |||||||||||
| | | | | | | | | | | | | | | | | |
Total |
18,928 | 11,102 | 1,113 | 1,708 | 5,005 | |||||||||||
Expected credit loss rate |
12.3 | % | 0.3 | % | 1.0 | % | 1.2 | % | 45.4 | % | ||||||
Expected credit loss |
2,335 | 31 | 11 | 21 | 2,272 | |||||||||||
Overdue trade receivables in MENA region mainly related to the major customers from diagnostics segment. The trade receivables due from top 10 diagnostics customers in MENA region as of December 31, 2019 represented over 85% of overdue balances for Middle East. These customers are mainly government hospitals administered by Ministry of Health in the respective countries as well as distributors. Based on our past experience, they normally require a longer period to settle the outstanding trade receivables.
Set out below is the movement in the allowance for expected credit losses of trade receivables and contract assets:
in EUR k
|
2018 | 2019 | |||||
|---|---|---|---|---|---|---|---|
As of January 1 |
841 | 1,633 | |||||
Provision for expected credit losses (note 8.2) |
792 | 752 | |||||
Write-off |
— | (30 | ) | ||||
As of December 31 |
1,633 | 2,355 | |||||
F-47
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
21 Financial instruments—fair values and risk management (Continued)
Cash and cash equivalents
As of December 31, 2019, the Group held cash and cash equivalents of EUR 41,095k (2018: EUR 9,222k). This total, therefore, also represents the maximum default risk with regard to these assets. The cash and cash equivalents are deposited at banks or financial institutions that have a rating of BAA to AA.
Liquidity risk
The liquidity risk is the risk of the Group possibly not being in a position to meet its financial liabilities as contractually agreed by providing cash or other financial assets.
The Group's objective is to maintain a balance between continuity of funding and flexibility through the use of bank overdrafts and lease contracts.
Managing liquidity within the Group is intended to ensure that - as far as possible - sufficient cash and cash equivalents are always available to meet payment obligations when these fall due, in both normal and challenging conditions, without incurring unacceptable losses or damaging the Group's reputation.
The Group strives to maintain cash and cash equivalents at a level above that of the expected cash outflows for financial liabilities (apart from trade payables) during the next 60 days. As at December 31, 2019, approximately 27.2% of the Group's interest-bearing liabilities is mature in less than one year (2018: 25.7%) based on the carrying value of borrowings reflected in the financial statements. The increase in the ratio is due to repayment of non-current bank loans in 2019 upon the completion of sale and leaseback transaction.
The Company has recently completed the IPO in November 2019. As at December 31, 2019, the Group had cash and cash equivalent of EUR 41,095k (2018: EUR 9,222k), the Group assessed the concentration of risk and concluded it to be low.
In addition to the cash and cash equivalent available as of December 31, 2019, the Group also has access to other sources of funding, including the amount of expected cash inflows from trade and other receivables. As of December 31, 2019, the expected cash inflows from trade and other receivables within two months amounts to EUR 6,644k (2018: EUR 3,830k), which would be similar to the amount of trade payables due as of then. As at December 31, 2019, the Group has secured credit lines for totaling EUR 3,500k. These bear interest of 3.33% - 4.50% (2018: EUR 4,000k; 3.33% - 4.50%). EUR 2,636k were utilized as of December 31, 2019 (2018: EUR 1,915k).
The table below presents the remaining contractual terms of the financial liabilities on the reporting date, including estimated interest payments. The figures are undiscounted gross amounts,
F-48
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
21 Financial instruments—fair values and risk management (Continued)
including estimated interest payments and interest on undrawn loan funds, but without showing the impact of offsetting.
| |
Contractually agreed cash flows | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Dec 31, 2018 in EUR k |
Carrying amount |
Total | Less than 2 months |
2 to 12 months |
1 to 5 years |
More than 5 years |
|||||||||||||
Bank overdrafts |
1,915 | 1,915 | 1,915 | — | — | — | |||||||||||||
Secured bank loans |
13,842 | 15,985 | 236 | 1,965 | 5,808 | 7,976 | |||||||||||||
Lease liabilities |
3,062 | 3,234 | 239 | 1,196 | 1,799 | — | |||||||||||||
Municipal Loans |
860 | 1,273 | — | — | — | 1,273 | |||||||||||||
Trade payables |
5,429 | 5,429 | 3,920 | 1,509 | — | — | |||||||||||||
| | | | | | | | | | | | | | | | | | | | |
|
25,108 | 27,836 | 6,310 | 4,670 | 7,607 | 9,249 | |||||||||||||
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| |
Contractually agreed cash flows | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Dec 31, 2019 in EUR k |
Carrying amount |
Total | Less than 2 months |
2 to 12 months |
1 to 5 years |
More than 5 years |
|||||||||||||
Bank overdrafts |
2,636 | 2,636 | 2,636 | — | — | — | |||||||||||||
Secured bank loans |
1,770 | 1,866 | 12 | 848 | 1,006 | — | |||||||||||||
Lease liabilities |
21,704 | 25,934 | 755 | 3,034 | 9,574 | 12,571 | |||||||||||||
Municipal Loans |
860 | 1,022 | — | 327 | 695 | — | |||||||||||||
Trade payables |
8,554 | 8,554 | 6,871 | 1,683 | — | — | |||||||||||||
| | | | | | | | | | | | | | | | | | | | |
|
35,524 | 40,012 | 10,274 | 5,892 | 11,275 | 12,571 | |||||||||||||
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
Reconciliation of liabilities arising from financing activities
| |
|
|
Non-cash changes | |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
in EUR k
|
Jan 1, 2018 | Cash flows | Additions | Changes in maturity |
Dec 31, 2018 | |||||||||||
Non-current financial liabilities |
3,851 | (1,373 | ) | 856 | 11,293 | 14,627 | ||||||||||
Non-current portion of secured bank loans |
— | — | — | 12,055 | 12,055 | |||||||||||
Municipal loans |
2,000 | (1,140 | ) | — | — | 860 | ||||||||||
Non-current lease liabilities |
1,851 | (233 | ) | 856 | (762 | ) | 1,712 | |||||||||
Current financial liabilities |
15,490 | 267 | 588 | (11,293 | ) | 5,052 | ||||||||||
Current portion of secured bank loans |
13,837 | 5 | — | (12,055 | ) | 1,787 | ||||||||||
Bank overdrafts |
— | 1,915 | — | — | 1,915 | |||||||||||
Current leases liabilities |
1,653 | (1,653 | ) | 588 | 762 | 1,350 | ||||||||||
| | | | | | | | | | | | | | | | | |
Total |
19,341 | (1,106 | ) | 1,444 | — | 19,679 | ||||||||||
F-49
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
21 Financial instruments—fair values and risk management (Continued)
| |
|
|
Non-cash changes | |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
in EUR k
|
Jan 1, 2019 | Cash flows | Additions | Changes in maturity |
Dec 31, 2019 | |||||||||||
Non-current financial liabilities |
14,627 | (12,783 | ) | 18,910 | (1,107 | ) | 19,647 | |||||||||
Non-current portion of secured bank loans |
12,055 | (11,087 | ) | — | — | 968 | ||||||||||
Municipal loans |
860 | — | — | (250 | ) | 610 | ||||||||||
Non-current lease liabilities |
1,712 | (1,696 | ) | 18,910 | (857 | ) | 18,069 | |||||||||
Current financial liabilities |
5,052 |
(1,614 |
) |
2,778 |
1,107 |
7,323 |
||||||||||
Current portion of secured bank loans |
1,787 | (985 | ) | — | — | 802 | ||||||||||
Bank overdrafts |
1,915 | 721 | — | — | 2,636 | |||||||||||
Municipal loans |
— | — | — | 250 | 250 | |||||||||||
Current leases liabilities |
1,350 | (1,350 | ) | 2,778 | 857 | 3,635 | ||||||||||
Total |
19,679 | (14,397 | ) | 21,688 | — | 26,970 | ||||||||||
Currency risk
The Group is exposed to currency risk in cases where contracts are concluded in foreign currencies. The vast majority of goods delivered and services the Company provided, including those for international customers, are invoiced in euro.
The main functional currencies of group companies are the euro, USD, the Indian rupee and the Arab Emirates Dirham. The following table presents the net foreign currency exposure of the Group as at December 31, 2018 and 2019.
| |
Dec 31, 2018 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
in EUR k
|
USD | INR | AED | |||||||
Trade receivables |
1,674 | 65 | 4 | |||||||
Trade payables and other liabilities |
(2,193 | ) | (2 | ) | (5 | ) | ||||
Net exposure _ |
(519 | ) | 63 | (1 | ) | |||||
| |
Dec 31, 2019 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
in EUR k
|
USD | INR | AED | |||||||
Trade receivables |
4,275 | 36 | (1 | ) | ||||||
Trade payables and other liabilities |
(2,801 | ) | (98 | ) | (15 | ) | ||||
Net exposure |
1,474 | (62 | ) | (16 | ) | |||||
Sensitivity analysis relating to changes in exchange rates:
Given the exposure to foreign currencies as presented above, the impact to the Group's earnings before tax or equity from a 10% change in the US dollar exchange rate would not be material.
F-50
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
22 List of subsidiaries
The major subsidiaries of the Group are listed below.
| |
|
Equity interests (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|
Name
|
Country in which primary activities are pursued |
Dec 31, 2018 | Dec 31, 2019 | ||||||
Centogene AG |
Germany | 100 | 100 | ||||||
Centogene IP GmbH |
Germany | 100 | 100 | ||||||
Centogene Shared Service GmbH |
Germany | 100 | 100 | ||||||
Centogene Fzllc, Dubai |
Dubai | 100 | 100 | ||||||
Ludewig Wasserbau GmbH* |
Germany | 100 | 0 | ||||||
Centogene US LLC, Burlington, USA |
USA | 100 | 100 | ||||||
Centogene GmbH, Vienna |
Austria | 90 | 90 | ||||||
Centogene India Pvt. Ltd |
India | 51 | 51 | ||||||
LPC GmbH |
Germany | 51 | 51 | ||||||
- *
- The 100% interest in Ludewig Wasserbau GmbH was sold as part of the sale and leaseback transaction during the year ended December 31, 2019. Please refer to note 13.1 for details.
23 Non-controlling interests
The table below shows information on each subsidiary of the Group with material, non-controlling interests before intercompany eliminations.
| |
Centogene India Pvt. Ltd |
LPC GmbH | |||||
|---|---|---|---|---|---|---|---|
Dec 31, 2019
|
49% | 49% | |||||
in EUR k
|
|||||||
Net assets/(liabilities) |
(1,263 | ) | (546 | ) | |||
Carrying amount of non-controlling interests |
(619 | ) | (268 | ) | |||
Revenue |
687 | 0 | |||||
Profit/(loss) |
(311 | ) | (56 | ) | |||
Profit/loss allocated to non-controlling interests |
(152 | ) | (27 | ) | |||
24 Commitments
Future payments for non-cancellable leases
The Group has various non-cancellable lease contracts of office equipment and storage spaces which had a lease term of less than 12 months or were related to leases of low-value assets, and therefore the short-term lease recognition exemption was applied to these contracts. The future lease payments for these non-cancellable lease contracts are EUR 72k within one year (2018: EUR 197k) and EUR 36k within five years (2018: 55k).
Future payment obligations
During 2019, the Group concluded agreements with suppliers, for goods and services to be provided in 2020 with a total payment obligation of around EUR 802k (2018: EUR 1,013k).
F-51
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
25 Related parties
During the year ended December 31, 2019, the Group had the following related party transactions:
Transaction with shareholders
Based on a shareholder agreement from January 2016 the payment to the option holders of the VSOP 2016 will be reimbursed by the original shareholders to the Company at the same time when the obligation to pay the options holders arises. A respective receivable against shareholders was recorded (refer to note 15). The shareholder agreement has a term till December 31, 2023.
Transactions with members of management in key positions
Remuneration of members of key management
in EUR k
|
2017 | 2018 | 2019 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Short-term employee benefits |
1,843 | 2,354 | 3,313 | |||||||
Post-employment pension and medical benefits |
10 | 10 | 10 | |||||||
Share-based payment transactions |
530 | 2,893 | 3,395 | |||||||
| | | | | | | | | | | |
Total compensation to key management |
2,383 | 5,257 | 6,718 | |||||||
440,475 share options were granted under ESOP 2017 to key management personnel, allowing to purchase common shares of the Company, as a result of the replacement of previous cash-settled share-based transaction (see note 20). The options are fully vested and exercisable after a lock-up period of 6 months. The exercise price of the share options is EUR 0.12, and the options expire in 2029. There are no pension commitments for members of the management board.
The supervisory board received remuneration for its activities of EUR 499k in the reporting year (2018: EUR 341k; 2017: EUR 160k). In addition, as disclosed in note 20, an individual of the supervisory board received share options from the Company upon completion of IPO. Share-based payment expenses of EUR 704k (2018: EUR nil; 2017: EUR nil) was charged to profit and loss for the year ended December 31, 2019.
For the year ended December 31, 2019, consultant fees totaling EUR 152k (2018: EUR nil; 2017: EUR nil) was charged to profit or loss in relation to corporate strategy services provided by a member of the supervisory board. For the years ended December 31, 2018 and 2017, there were also consulting fees of EUR 64k and EUR 490k respectively relating to services provided by a member of the supervisory board and an entity controlled by a member of the supervisory board.
26 Contingent liabilities
In May 2016, the Company was informed in writing by the Universitair Medisch Centrum Utrecht ("UMCU") that a claim had been initiated against UMCU regarding a prenatal diagnostic test that the Company conducted at their request which failed to identify a specific mutation present in a patient. On November 8, 2018, the UMCU and Neon Underwriting Limited formally filed a legal claim in the local court in Rostock, Germany against the Company alleging that the Company's negligence in performing the test resulted in the misdiagnosis of the patient. UMCU is seeking recovery for compensatory damages as a result of the alleged misdiagnosis. By court order of November 8, 2018, the Regional Court of Rostock set the amount in dispute at EUR 880k.
F-52
Notes to the consolidated financial statements as of December 31, 2018 and 2019 and
for the three years ended December 31, 2017, 2018 and 2019 (Continued)
26 Contingent liabilities (Continued)
The Company intends to rigorously defend its position and considers that it is not probable the legal claim towards the Company will be successful and as a result has not recognized a provision for this claim as of December 31, 2019. In addition, in case a settlement would be required, the Company believes that the corresponding liability will be fully covered by the respective insurance coverage.
27 Subsequent Events
- (a)
- Repayment of municipal loans and release of shareholders' guarantees—As disclosed in note 19.1, subsequent to the year ended December 31, 2019, the Company has repaid the outstanding municipal loan in full of EUR 860k in February 2020. In addition, a short-term deposit of EUR 500k was provided to another bank to secure the overdraft facility, of which EUR 476k (2018: EUR nil) was utilized as of December 31, 2019.
- (b)
- Impact of COVID-19—The COVID-19 pandemic, which began in December 2019 in China and has spread worldwide, has caused many governments to implement measures to slow the spread of the outbreak through quarantines, travel restrictions, heightened border scrutiny and other measures. The Company has taken a series of actions aimed at safeguarding the Company's employees and business associates, including implementing a work-from-home policy for employees except for those related to our laboratory operations. These disruptions could result in increased costs of execution of operational plans or may negatively impact our business due to its negative impact on the global economy.
After the above transactions, all guarantees provided by certain shareholders in relation to the Group's financial liabilities and facilities were released.
As part of the Company's initiative to help local, national and international authorities in their efforts to diagnose cases of COVID-19, the Company has commenced testing for COVID-19 in March 2020. In April 2020, in order to increase testing capacity, the Company acquired laboratory facilities and equipment for a total consideration of EUR 1,800 thousand and leased laboratory space in Hamburg, Germany. The lease is charged at a fixed rate and covers a fixed period of five years, with an option to extend. Such lease contract is accounted for under IFRS 16 and accordingly right-of-use assets and lease liabilities of approximately EUR 450 thousand will be recognized.
Although the provision of testing for the COVID-19 virus is anticipated to generate additional revenues to the Company, the impact of the pandemic to the global economy, international trade and business activities may also have a negative impact to its operating results, and therefore the Company is unable to provide a reasonable estimate of the related financial impact at this time.
These consolidated financial statements were approved by management on April 23, 2020.
F-53
DESCRIPTION OF THE RIGHTS OF EACH CLASS OF SECURITIES REGISTERED UNDER SECTION 12 OF THE SECURITIES EXCHANGE ACT OF 1934 AS OF DECEMBER 31, 2019.
As of the date of the Annual Report on Form 20-F of which this Exhibit 2.2 is a part, Centogene N.V. (the “Company”, “we”, “us” or “our”) has only one class of securities registered under Section 12 of the Securities Exchange Act of 1934, as amended: the Company’s registered common shares.
The following description of our common shares is a summary and does not purport to be complete. It is subject to and qualified in its entirety by reference to our articles of association, which are incorporated by reference as an exhibit to this Annual Report on Form 20-F of which this Exhibit 2.2 is a part.
As of April 23, 2020, our authorized share capital was €9,480,000, consisting of 79,000,000 common shares, par value €0.12 per share, of which we had 19,861,340 common shares outstanding.
Common Shares
The following summarizes the main rights of holders of our common shares:
· each holder of common shares is entitled to one vote per share on all matters to be voted on by shareholders generally, including the election of managing directors and supervisory directors;
· there are no cumulative voting rights;
· the holders of our common shares are entitled to dividends and other distributions as may be declared from time to time by us out of funds legally available for that purpose, if any;
· upon our liquidation, dissolution or winding-up, the holders of common shares will be entitled to share ratably in the distribution of all of our assets remaining available for distribution after satisfaction of all our liabilities; and
· the holders of common shares have preemptive rights in case of share issuances or the grant or rights to subscribe for shares, except if such rights are limited or excluded by the corporate body authorized to do so and except in such cases as provided by Dutch law and our articles of association.
The Company may not make calls on shareholders in excess of the aggregate nominal value of the shares a shareholder has subscribed for.
Limitations on the Rights to Own Securities
Our common shares may be issued to individuals, corporations, trusts, estates of deceased individuals, partnerships and unincorporated associations of persons. Our articles of association contain no limitation on the rights to own our shares and no limitation on the rights of nonresidents of the Netherlands or foreign shareholders to hold or exercise voting rights.
Shareholders’ Meetings
General meetings of shareholders may be held in Amsterdam, Arnhem, Assen, The Hague, Haarlem, ‘s-Hertogenbosch, Groningen, Leeuwarden, Lelystad, Maastricht, Middelburg, Rotterdam, Schiphol (Haarlemmermeer), Utrecht or Zwolle, all in the Netherlands. The annual general meeting of shareholders must be held within six months of the end of each financial year. Additional extraordinary general meetings of shareholders may also be held, whenever considered appropriate by the management board or the supervisory board and shall be held within three months after our management board has considered it to be likely that our equity has decreased to an amount equal to or lower than half of its paid up and called up share capital, in order to discuss the measures to be taken if so required.
Pursuant to Dutch law, one or more shareholders or others with meeting rights under Dutch law who jointly represent at least one-tenth of the issued share capital may request us to convene a general meeting, setting out in detail the matters to be discussed. If we have not taken the steps necessary to ensure that such meeting can be held within six weeks after the request, the requesting party/parties may, on their application, be authorized by the competent Dutch court in preliminary relief proceedings to convene a general meeting of shareholders. The court shall disallow the application if it does not appear that the applicants have previously requested our management board and our supervisory board to convene a general meeting and neither our management board nor our supervisory board have taken the necessary steps so that the general meeting could be held within six weeks after the request.
General meetings of shareholders can be convened by a notice, which shall include an agenda stating the items to be discussed as well as other information as required by Dutch law, including for the annual general meeting of shareholders, among other things, the adoption of the annual accounts, appropriation of our profits and proposals relating to the composition of the management board and supervisory board, including the filling of any vacancies in such bodies. In addition, the agenda shall include such items as have been included therein by the management board or the supervisory board. The agenda shall also include such items requested by one or more shareholders, or others with meeting rights under Dutch law, representing at least 3% of the issued share capital. Requests must be made in writing or by electronic means and received by us at least 60 days before the day of the convocation of the meeting. No resolutions shall be adopted on items other than those that have been included in the agenda.
In accordance with the Dutch Corporate Governance Code (the “DCGC”) and our articles of association, shareholders having the right to put an item on the agenda under the rules described above shall exercise such right only after consulting the management board in that respect. If one or more shareholders intend to request that an item be put on the agenda that may result in a change in the company’s strategy (for example, the removal of managing directors or supervisory directors), the management board must be given the opportunity to invoke a reasonable period to respond to such intention. Such period shall not exceed 180 days (or such other period as may be stipulated for such purpose by Dutch law and/or the DCGC from time to time). If invoked, the management board must use such response period for further deliberation and constructive consultation, in any event with the shareholders(s) concerned, and shall explore the alternatives. At the end of the response time, the management board shall report on this consultation and the exploration of alternatives to the general meeting of shareholders. This shall be supervised by our supervisory board. The response period may be invoked only once for any given general meeting of shareholders and shall not apply: (a) in respect of a matter for which a response period has been previously invoked; or (b) if a shareholder holds at least 75% of the company’s issued share capital as a consequence of a successful public bid. The response period may also be invoked in response to shareholders or others with meeting rights under Dutch law requesting that a general meeting of shareholders be convened, as described above.
The general meeting is presided over by the chairman of the supervisory board. If no chairman has been elected or if he or she is not present at the meeting, the general meeting shall be presided over by another supervisory director present at the meeting. If no supervisory director is present, the meeting shall be presided over by our CEO. If no CEO has been elected or if he or she is not present at the meeting, the general meeting shall be presided over by another managing director present at the meeting. If no managing director is present at the meeting, the general meeting shall be presided over by any other person appointed by the general meeting. In each case, the person who should chair the general meeting pursuant to the rules described above may appoint another person to chair the general meeting instead. Managing directors and supervisory directors may always attend a general meeting of shareholders. In these meetings, they have an advisory vote. The chairman of the meeting may decide at his or her discretion to admit other persons to the meeting.
All shareholders and others with meeting rights under Dutch law are authorized to attend the general meeting of shareholders, to address the meeting and, in so far as they have such right, to vote.
Each common share confers the right on the holder to cast one vote at the general meeting of shareholders. Shareholders may vote by proxy. No votes may be cast at a general meeting of shareholders on shares held by us or our subsidiaries or on shares for which we or our subsidiaries hold depositary receipts. Nonetheless, the holders of a right of use and enjoyment (vruchtgebruik) and the holders of a right of pledge (pandrecht) in respect of shares held by us or our subsidiaries in our share capital are not excluded from the right to vote on such shares, if the right of use and enjoyment (vruchtgebruik) or the right of pledge (pandrecht) was granted prior to the time such shares were
acquired by us or any of our subsidiaries. Neither we nor any of our subsidiaries may cast votes in respect of a share on which we or such subsidiary holds a right of use and enjoyment (vruchtgebruik) or a right of pledge (pandrecht). Shares which are not entitled to voting rights pursuant to the preceding sentences will not be taken into account for the purpose of determining the number of shareholders that vote and that are present or represented, or the amount of the share capital that is provided or that is represented at a general meeting of shareholders.
Decisions of the general meeting of shareholders are taken by a simple majority of votes cast, except where Dutch law or our articles of association provide for a qualified majority or unanimity. Shareholders’ rights as described in the Company’s articles of association may only be affected for as far the changed rights still comply with mandatory Dutch law. Furthermore, an amendment to the Company’s articles of association requires a resolution of the general meeting of the shareholders and a notarial deed in giving effect to the amendment.
Dividends and Other Distributions
Dividends
We may only make distributions to our shareholders if our shareholders’ equity (eigen vermogen) exceeds the sum of the paid-up and called-up share capital plus any reserves required by Dutch law or by our articles of association. Under our articles of association, the management board may decide that all or part of the profits are carried to reserves. After reservation by the management board of any profit, the remaining profit will be at the disposal of the general meeting of shareholders for distribution, subject to restrictions of Dutch law and approval by our supervisory board.
We only make a distribution of dividends to our shareholders after the adoption of our annual accounts demonstrating that such distribution is legally permitted. The management board is permitted, subject to certain requirements, to declare interim dividends without the approval of the general meeting of shareholders, but only with the approval of the supervisory board.
Dividends and other distributions shall be made payable not later than the date determined by the management board. Claims to dividends and other distributions not made within five years from the date that such dividends or distributions became payable, will lapse and any such amounts will be considered to have been forfeited to us (verjaring).
We have not adopted a dividend policy with respect to future dividends. Subject the restrictions described above, any dividend policy (if we were to adopt one) will depend on many factors, such as our results of operations, financial condition, cash requirements, prospects and other factors deemed relevant by our management board and supervisory board.
Exchange Controls
Under Dutch law, there are no exchange controls applicable to the transfer to persons outside of the Netherlands of dividends or other distributions with respect to, or of the proceeds from the sale of, shares of a Dutch company, subject to applicable restrictions under sanctions and measures, including those concerning export control, pursuant to European Union regulations, the Sanctions Act 1977 (Sanctiewet 1977) or other legislation, applicable anti-boycott regulations and similar rules.
Squeeze-Out Procedures
Pursuant to Section 2:92a of the Dutch Civil Code, a shareholder who holds at least 95% of our issued share capital for his own account, alone or together with group companies, may initiate proceedings against the other shareholders jointly for the transfer of their shares to such shareholder. The proceedings are held before the Enterprise Chamber of the Amsterdam Court of Appeal, or the Enterprise Chamber (Ondernemingskamer), and can be instituted by means of a writ of summons served upon each of the other shareholders in accordance with the provisions of the Dutch Code of Civil Procedure (Wetboek van Burgerlijke Rechtsvordering). The Enterprise Chamber may grant the claim for squeeze-out in relation to the other shareholders and will determine the price to be paid for the shares, if necessary, after appointment of one or three experts who will offer an opinion to the Enterprise Chamber on the value to be paid for the shares of the other shareholders. Once the order to transfer becomes final
before the Enterprise Chamber, the person acquiring the shares shall give written notice of the date and place of payment and the price to the holders of the shares to be acquired whose addresses are known to him. Unless the addresses of all of them are known to the acquiring person, such person is required to publish the same in a daily newspaper with a national circulation.
Dissolution and Liquidation
Under our articles of association, we may be dissolved by a resolution of the general meeting of shareholders, subject to a proposal of the management board approved by our supervisory board. In the event of a dissolution, the liquidation shall be effected by the management board, under supervision of our supervisory board, unless the general meeting decides otherwise. To the extent that any assets remain after payment of all debts, those assets shall be distributed to the holders of common shares.
Certain information has been excluded from this Exhibit 4.5 because it is both (i) not material and (ii) would likely cause competitive harm if publicly disclosed. [*****] indicates that information has been redacted.
Execution Version
2019 AMENDMENT No. 3
TO GLOBAL MASTER SERVICES AGREEMENT AND TO SUPPLY AGREEMENT
This amendment (“2019 Amendment No. 3”) to the Global Master Services Agreement dated 1 January 2015 (“MSA”) and the Supply Agreement dated 1 January 2016 (“Supply Agreement”), each as amended by amendment agreements dated 23 December 2015, 8 March 2016, 3 May 2017, 2 July 2018, 19 July 2017 and 15 July 2019 (the “Amendment Agreements”), and side letters dated 7 June 2016, 24 November 2016, 9 August 2017, 24 August 2017, 8 September 2017, 2 July 2018, 19 December 2018, 29 May 2019 and 18 January 2019 (the “Side Letters”), is entered into as of 10 December 2019 by and between:
(1) SHIRE INTERNATIONAL GMBH, a Swiss limited liability company having its registered office at Zahlerweg 10, 6301 Zug, Switzerland (“Shire”);
(2) SHIRE PHARMACEUTICALS IRELAND LTD, an Irish limited liability company having its registered office at Block 2 & 3 Miesian Plaza, 50 -58 Baggot Street Lower, Dublin 2, Ireland (“SPIL”); and
(3) CENTOGENE AG, a German stock corporation incorporated under the laws of the Federal Republic of Germany with principal office in Rostock, registered with the district court (‘Amtsgericht’) in Rostock under HRB 13225 and having a business address at Am Strande 7, 18055 Rostock, Germany (“Centogene”).
PREAMBLE
(A) Under the MSA, Centogene agreed to provide certain diagnostic testing services relating to dried-blood-spot cards for the purpose of identifying patients suffering from lysosomal storage disorders and other rare diseases to Shire and its affiliates.
(B) Under the Supply Agreement, Centogene agreed to develop, manufacture and supply DBS test kits for use in certain required countries on the basis of Centogene’s existing DBS test kits (ii) manufacture new DBS test kits and (iii) supply SPIL and its affiliates with new DBS test kits.
(C) On January 8, 2019 Takeda Pharmaceutical Company Limited acquired Shire plc.
(D) Shire, SPIL and Centogene now wish to extend the term of the MSA and Supply Agreement until 31 March 2021 and amend certain financial and other aspects of their cooperation going forward, on the understanding that any further supplies of products and services beyond 31 March 2021 required by the Takeda group of companies will be subject to further agreement between Takeda and Centogene.
NOW, THEREFORE, Shire, Centogene and SPIL, intending to be legally bound, hereby agree to amend the MSA and Supply Agreement (as amended by the Amendment Agreements and Side Letters) as follows:
1 DEFINITIONS
In this 2019 Amendment No. 3, any capitalized terms shall have the meaning set forth in the MSA or the Supply Agreement (as amended by the Amendment Agreements and Side Letters), unless a term is specifically defined under this 2019 Amendment No. 3.
2 AMENDMENTS TO MSA
2.1. Shire and Centogene agree to replace Section 3.3 of the MSA with the following:
3.3 Timeframes. Centogene shall process submitted Samples, perform Diagnostic Tests and notify the Physician or Third Party laboratory that the Diagnostic Results are available for download through Centogene’s CentoPortal within ten (10) Business Days after receipt of the relevant Sample, provided the Test Kits and/or Testing Request Forms submitted to Centogene contain sufficient testing material and all information required by the Test Kits and/or Testing Request Forms (e.g. patient consent, sender information, target, clinically relevant information as regards to New Test Kits, etc.).
2.2 Shire and Centogene agree to amend Section 3.6 of the MSA with respect to the calendar year 2020 and first calendar quarter of 2021 as follows:
Subject to the below, Shire shall make the following payments to Centogene for the performance of an unlimited and uncapped number of Diagnostic Tests for Morbus Fabry, Morbus Gaucher, Morbus Hunter, MPS1, MPS2, MPS3, MPS4, MPS6 and MPS7 in the calendar year 2020 and first calendar quarter of 2021 respectively (the “Flat Fee”):
2020: €[*****] ([*****] Euro)
Q1 2021: €[*****] ([*****] Euro)
With respect to the calendar year 2020 and first calendar quarter of 2021, the Flat Fee shall include all costs and charges incurred by Centogene with respect to diagnostic testing under the MSA (as amended and supplemented by the Amendment Agreements and Side Letters). Centogene hereby expressly waives any additional payment claims under the MSA, including without limitation:
(i) all Excess Diagnostic Tests or other charges for additional diagnostic tests under Section 3.6(c);
(ii) all Additional Processing Fees under Section 3.2(a) and 3.6(d);
(iii) manual processing fees for processing whole blood (EDTA) samples from:
(a) Ukraine and Kazakhstan, under Section 2.1 of the Side Letter dated 24 August 2017; and
(b) Serbia, under Section 2.1 of the Side Letter dated 29 May 2019.
(iv) all Quality Validation Testing Fees under Section 2(a) of the Side Letter dated 2 July 2018;
(v) all shipment and importation costs, duties and licensing fees, including costs of managing importation licences;
(vi) all reporting costs under Section 3 of 2019 Amendment No. 2; and
(vii) all costs relating to hospital testing in Germany pursuant to Side Letters dated 7 June 2016, 9 August 2017 and 18 January 2019.
Centogene shall invoice Shire for €[*****] ([*****] Euro), being [*****] per cent ([*****]%) of the Flat Fee for the calendar year 2020, upon signature of this 2019 Amendment No. 3 and Shire shall make payment of such invoice within 30 days of receipt.
The remainder of the Flat Fee shall be invoiced on a pro rata monthly basis in advance on the first Business Day of each calendar month i.e. €[*****] per month in 2020 and €[*****] per month in Q1 2021. Shire shall make payment within ninety (90) calendar days of receipt of Centogene’s invoice.
Notwithstanding the concept of the Flat Fee, if Centogene performs less than [*****] Diagnostic Tests in 2020 or [*****] in Q1 2021, reconciliation will take place in the subsequent calendar year or quarter (as the case may be). At the start of such subsequent calendar year or quarter, Centogene will issue a credit note against which future Shire or other Shire affiliate invoices may be offset, for an amount equal to the difference between the annual flat fee and the value of the actual tests performed, invoiced at the prices set forth in Exhibit 1 of the MSA (Diagnostic Tests and Prices).
If Shire wishes Centogene to process any Third Party DBS test kits that have not previously been validated for use in Centogene’s systems, it shall notify Centogene and provide details of the required processing. Centogene shall within ten Business Days after receipt of Shire’s notification provide Shire with an estimate of the timeframe and cost of validation, such cost not to exceed €[*****]. If Shire approves the estimate Centogene shall validate the Third Party DBS test kits within the agreed timeframe and cost.
2.3 Shire and Centogene agree to add a new Section 5.5 to the MSA, as follows:
5.5 Support. Centogene shall commit sufficient project management resource to ensure swift internal communication and coordination within Centogene so that it meets the requirements of, and fully discharges its obligations under, this Agreement.
2.4 Shire and Centogene agree to replace Sections 12.1 and 12.2 of the MSA with the following:
12.1 Term. This Agreement shall come into force at the Effective Date and shall remain in full force until 31 March 2021, unless terminated in accordance with Section 12.3 et seq.
12.2 [Not used]
3 SUPPLY AGREEMENT
3,1 SPIL and Centogene agree to add the following to Section 3.4 of the Supply Agreement:
Notwithstanding the foregoing, Centogene will develop Contract DBS Test Kits in accordance with the following timings:
(a) for any of the following changes to the Contract DBS Test Kits, Centogene shall provide the first technical file to Shire within [*****] ([*****]) days after the effective date of Shire’s Development Order:
1. translation into local languages that are not currently offered by Centogene;
2. changes to filter paper material and/or physical dimensions specifications;
3. changes to outer card material and/or physical dimensions specifications;
4. changes to art work layout impacting where the information will be presented on the Centogene card;
5. changes to art work layout and or language content specific to the instructions for use or, to the extent solely requested by Shire and unrelated from requirements resulting from an initial review or a Yearly Review, the informed consent form.
(b) for all other changes to or re-development of such artwork Centogene shall provide artwork specifications to Shire within [*****] ([*****]) Business Days after the effective date of Shire’s Development Order;
(c) Centogene shall incorporate Shire’s input into any artwork specifications within [*****] ([*****]) Business Days of receipt of Shire’s comments, if, when and insofar as comments were accepted by Centogene, provided that:
(i) any discussions between the Parties concerning Shire’s comments will extend the timeline; and
(ii) both Parties reply to questions and/or provide comments on the input of the respective other Party within [*****] ([*****]) Business Days of receipt; and
(d) Centogene shall submit the technical file to the respective Regulatory Authorities for Regulatory Approval within [*****] ([*****]) month of Shire’s approval of such artwork file.
3.2 SPIL and Centogene agree to replace Sections 8.2 to 8.7 of the Supply Agreement with the following:
8.2 Supply. Within [*****] ([*****]) days after the end of a calendar month, Centogene shall invoice Shire for the Contract DBS Test Kits Delivered in the preceding calendar month.
8.3 Kit Development. Within [*****] ([*****]) days after the end of a calendar month, Centogene shall invoice Shire for the Kit Development activities completed in the preceding calendar month, based in the Kit Development fees as defined in Exhibit 3.
8.4 Third Party Expenditures to be borne by Shire. Centogene shall bear all costs for the use of Third Party consultants, unless Shire Inc. participates in the costs as set forth under this Section 8.4. Only upon provision of a detailed cost estimate and subject to prior written approval by Shire International GmbH, actual and documented costs by Centogene for use of Third Party regulatory service providers to obtain Regulatory Approval for any country will be reimbursed by Shire Inc, subject to a maximum of:
(a) €[*****] per country in respect of any ‘Tier 1’ countries (namely CE Mark countries, FDA, MEDSAP countries, Israel, Canada, Australia, New Zealand); and
(b) €[*****] per country in respect of any other (‘Tier 2’) countries.
Third Party expenditures shall be invoiced immediately after payment of such expenditures by Centogene to Shire Inc, provided that Centogene presents originals of the Third Party invoices.
8.5 [not used]
8.6 [not used]
8.7 Payment Term. Payment will be made within ninety (90) days of receipt of invoice.
3.3 SPIL and Centogene agree to replace Exhibit 3 of the Supply Agreement in its entirely with a new Exhibit 3, attached to this 2019 Amendment No. 3 as Appendix 1.
3.4 SPIL and Centogene agree to amend Section 9.6 and to add a new Section 9.7 to the Supply Agreement, as follows:
9.6 No Exclusivity. Nothing in this Agreement shall prevent Shire from obtaining its demand for DBS test kits, which are not processed by Centogene under the Global Master Services Agreement, from any Third Party. Shire intends and agrees to use reasonable efforts to use solely Centogene’s CentoCards as its preferred sample collection kit in countries where Contract DBS Test Kits are processed by Centogene under the Global Master Services Agreement.
9.7 Support. Centogene shall commit sufficient project management resource to ensure swift internal communication and coordination within Centogene so that it meets the requirements of, and fully discharges its obligations under, this Agreement.
4 2018 SIDE LETTER
4.1 Shire and Centogene agree to extend the terms of the Side Letter to the MSA on the testing of expired contract DBS Test kits dated 2 July 2018 until 31 March 2021, provided however that Shire shall have the right to terminate that Side Letter upon thirty (30) days prior written notice to Centogene.
5 OTHER TERMS
5.1 All other terms and conditions as set forth in the MSA and the Supply Agreement (As amended by the Amendment Agreements and Side Letters) shall remain in full force and effect.
|
Place: |
Zug, Switzerland |
|
Place: |
Berlin | ||
|
|
|
|
|
| ||
|
Date: |
10/12/19 |
|
Date: |
11.12.2019 | ||
|
|
|
|
|
| ||
|
for and on behalf of |
|
for and on behalf of | ||||
|
Shire International GmbH |
|
Centogene AG | ||||
|
|
|
| ||||
|
/s/ Remco Lemarcq |
|
/s/ Prof. Dr. Arndt Rolfs | ||||
|
|
|
|
|
| ||
|
Name: |
Remco Lemarcq |
|
Name: |
Prof. Dr. Arndt Rolfs | ||
|
|
|
|
|
| ||
|
Title: |
Proxy Holder |
|
Title: |
CEO | ||
|
|
|
| ||||
|
|
|
| ||||
|
Place: |
Dublin, Ireland |
|
| |||
|
|
|
|
| |||
|
Date: |
10/12/2019 |
|
| |||
|
for and on behalf of Shire Pharmaceuticals Ireland Ltd |
|
| ||||
|
|
|
| ||||
|
/s/ Denis Ahern |
|
| ||||
|
|
|
|
| |||
|
Name: |
Denis Ahern |
|
| |||
|
|
|
|
| |||
|
Title: |
Director |
|
| |||
APPENDIX 1
Exhibit 3 - Prices
Development
Kit Development: [*****] € for each new Kit Development project or project to modify Existing DBS Test Kits which require translation into local languages that are not currently offered by Centogene. [*****] € for each new Kit Development project or each project to modify Existing DBS Test Kits which do not require such translations.
Regulatory
Actual and documented expenditures by Centogene for use of certain Third Party consultants as described in Section 8.4 of the Agreement.
Centogene will present copies of the Third Party invoices.
Supply
Price per Contract DBS Test Kits (finally assembled filter card, incl. protective covering, envelope, instructions and consent): €[*****]
The above price is inclusive of legal fees, annual QM fees, storage fees and quality control for production fees.
CERTIFICATION BY THE CHIEF EXECUTIVE OFFICER PURSUANT TO
SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, Arndt Rolfs, certify that:
1. I have reviewed this Annual Report on Form 20-F of Centogene N.V.;
2. Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
3. Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the company as of, and for, the periods presented in this report;
4. The company’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) for the company and have:
a. Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the company, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
b. Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
c. Evaluated the effectiveness of the company’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
d. Disclosed in this report any change in the company’s internal control over financial reporting that occurred during the period covered by the annual report that has materially affected, or is reasonably likely to materially affect, the company’s internal control over financial reporting.
5. The company’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the company’s auditors and the audit committee of the company’s board of directors (or persons performing the equivalent functions):
a. All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the company’s ability to record, process, summarize and report financial information; and
b. Any fraud, whether or not material, that involves management or other employees who have a significant role in the company’s internal control over financial reporting.
Date: April 23, 2020
|
/s/ Arndt Rolfs |
|
|
Arndt Rolfs |
|
|
Chief Executive Officer |
|
CERTIFICATION BY CHIEF FINANCIAL OFFICER PURSUANT TO
SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, Richard Stoffelen, certify that:
1. I have reviewed this Annual Report on Form 20-F of Centogene N.V.;
2. Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
3. Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the company as of, and for, the periods presented in this report;
4. The company’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) for the company and have:
a. Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the company, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
b. Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
c. Evaluated the effectiveness of the company’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
d. Disclosed in this report any change in the company’s internal control over financial reporting that occurred during the period covered by the annual report that has materially affected, or is reasonably likely to materially affect, the company’s internal control over financial reporting.
5. The company’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the company’s auditors and the audit committee of the company’s board of directors (or persons performing the equivalent functions):
a. All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the company’s ability to record, process, summarize and report financial information; and
b. Any fraud, whether or not material, that involves management or other employees who have a significant role in the company’s internal control over financial reporting.
Date: April 23, 2020
|
/s/ Richard Stoffelen |
|
|
Richard Stoffelen |
|
|
Chief Financial Officer |
|
CERTIFICATION BY THE CHIEF EXECUTIVE OFFICER PURSUANT TO
18 U.S.C. SECTION 1350, AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the Annual Report on Form 20-F of Centogene N.V. (the “Company”) for the fiscal year ended December 31, 2019 (the “Report”), I, Arndt Rolfs, certify pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, that, to the best of my knowledge:
1. the Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934; and
2. the information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
Date: April 23, 2020
|
/s/ Arndt Rolfs |
|
|
Arndt Rolfs |
|
|
Chief Executive Officer |
|
CERTIFICATION BY THE CHIEF FINANCIAL OFFICER PURSUANT TO
18 U.S.C. SECTION 1350, AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the Annual Report on Form 20-F of Centogene N.V. (the “Company”) for the fiscal year ended December 31, 2019 (the “Report”), I, Richard Stoffelen, certify pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, that, to the best of my knowledge:
1. the Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934; and
2. the information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
Date: April 23, 2020
|
/s/ Richard Stoffelen |
|
|
Richard Stoffelen |
|
|
Chief Financial Officer |
|
Consent of Independent Registered Public Accounting Firm
We consent to the incorporation by reference in the Registration Statement (Form S-8 No. 333-234551) pertaining to the Centogene N.V. Long-Term Incentive Plan of our report dated April 23, 2020, with respect to the consolidated financial statements of Centogene N.V. included in this Annual Report (Form 20-F) for the year ended December 31, 2019.
|
/s/ Ingo Röders |
|
/s/ Christian Patzelt |
|
Wirtschaftsprüfer |
|
Wirtschaftsprüfer |
|
(German Public Auditor) |
|
(German Public Auditor) |
Ernst & Young GmbH Wirtschaftsprüfungsgesellschaft
Berlin, Germany
April 23, 2020
Serious News for Serious Traders! Try StreetInsider.com Premium Free!
You May Also Be Interested In
- 7-DAY DEADLINE ALERT: The Schall Law Firm Encourages Investors in Lantronix, Inc. with Losses to Contact the Firm
- Unforgettable Experiences Await: Secure Your Spot at NPE2024
- Hybrid Train Market Size Expected to Grow US$ 38.48 Billion by 2031, Driven by Flexibility and Versatility, Government support and incentives
Create E-mail Alert Related Categories
SEC FilingsSign up for StreetInsider Free!
Receive full access to all new and archived articles, unlimited portfolio tracking, e-mail alerts, custom newswires and RSS feeds - and more!
