

Form 20-F Addex Therapeutics Ltd. For: Dec 31
 Tweet
Tweet Share
Share
Use these links to rapidly review the document
TABLE OF CONTENTS
INDEX TO FINANCIAL STATEMENTS
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 20-F
(Mark One)
- o
- REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934
OR
- ý
- ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2019
OR
- o
- TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from to
OR
- o
- SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
Date of event requiring this shell company report
Commission File Number 001-39179
Addex Therapeutics Ltd
(Exact name of registrant as specified in its charter and translation of registrant's name into English)
Switzerland
(Jurisdiction of incorporation or organization)
Chemin des Mines 9, CH- 1202 Geneva, Switzerland
(Address of principal executive offices)
Tim Dyer
Chief Executive Officer Addex Therapeutics Ltd Chemin des Mines 9,
CH- 1202 Geneva, Switzerland
Tel: + 41 22 884 1555
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act.
| Title of each class Trading Symbol Name of each exchange on which registered | ||||
| American Depositary Shares, each representing 6 ordinary shares, CHF 1.00 per share |
ADXN | The Nasdaq Stock Market LLC | ||
Shares, par value CHF 1.00 per share* |
||||
- *
- Not for trading, but only in connection with the registration of the American Depositary Shares.
Securities registered or to be registered pursuant to Section 12(g) of the Act. None
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act. None
Indicate the number of outstanding shares of each of the issuer's classes of capital or common stock as of the close of the period covered by the annual report.
Shares, par value CHF 1.00 per share: 32,848,635 shares outstanding as of December 31, 2019
American Depositary Shares (each representing 6 ordinary shares): none outstanding as of December 31, 2019
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. o Yes ý No
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934. o Yes ý No
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. ý Yes o No
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). ý Yes o No
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or an emerging growth company. See the definitions of "large accelerated filer," "accelerated filer," and "emerging growth company" in Rule 12b-2 of the Exchange Act.
| Large accelerated filer o | Accelerated filer o | Non-accelerated filer ý | Emerging growth company ý |
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. o
- †
- The term "new or revised financial accounting standard" refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012.
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
| U.S. GAAP o | International Financial Reporting Standards as issued by the International Accounting Standards Board ý |
Other o |
If "Other" has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow. o Item 17 o Item 18
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). o Yes ý No
1
2
Unless otherwise indicated or the context otherwise requires, all references in this Annual Report on Form 20-F to the terms "Addex," "Addex Therapeutics," "Addex Therapeutics Ltd," "the company," "we," "us" and "our" refer to Addex Therapeutics Ltd together with its subsidiaries.
We own trademarks for Addex Therapeutics in Switzerland. We also have trademarks for AddeLite and ProxyLite in relation to our screening technologies in the United States, Switzerland and the People's Republic of China and, in the case of ProxyLite, the EU. All other trade names, trademarks and service marks of other companies appearing in this Annual Report on Form 20-F are the property of their respective holders. Solely for convenience, the trademarks and trade names in this Annual Report on Form 20-F may be referred to without the ® and ™ symbols, but such references should not be construed as any indicator that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto. We do not intend to use or display other companies' trademarks and trade names to imply a relationship with, or endorsement or sponsorship of us by, any other companies.
Our reporting currency is the Swiss franc. The exchange rate between the Swiss franc and the U.S. dollar as of April 17, 2020 was $1.0342 per CHF 1.0. We present our consolidated financial statements in accordance with International Financial Reporting Standards, or IFRS, as adopted by the International Accounting Standards Board, or IASB. Readers of this Annual Report on Form 20-F should note that there may be certain differences between the presentation of our financial position, results of operations and cash flows under IFRS and U.S. generally accepted accounting principles.
The terms "dollar," "USD" or "$" refer to U.S. dollars and the terms "Swiss Francs" or "CHF" refer to the legal currency of Switzerland.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 20-F contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, that are based on our management's beliefs and assumptions and on information currently available to our management. All statements other than present and historical facts and conditions contained in this Annual Report on Form 20-F, including statements regarding our future results of operations and financial positions, business strategy, plans and our objectives for future operations, are forward-looking statements. When used in this Annual Report on Form 20-F, the words "anticipate," "believe," "continue" "could," "estimate," "expect," "intend," "may," "might," "ongoing," "objective," "plan," "potential," "predict," "should," "will" and "would," or the negative of these and similar expressions identify forward-looking statements. Forward-looking statements include, but are not limited to, statements about:
- •
- the development of our product candidates, including statements regarding the timing of initiation, completion and the outcome of pre-clinical
studies or clinical trials and related preparatory work, the period during which the results of the studies or trials will become available and our research and development programs with respect to
our product candidates;
- •
- our ability to obtain and maintain regulatory approval of our product candidates in the indications for which we plan to develop them, and any
related restrictions, limitations or warnings in the label of an approved drug or therapy;
- •
- our plans to collaborate, or statements regarding the ongoing collaborations, with partner companies;
- •
- our plans to research, develop, manufacture and commercialize our product candidates;
- •
- the timing of our regulatory filings for our product candidates;
3
- •
- the size and growth potential of the markets for our product candidates;
- •
- our ability to raise additional capital;
- •
- our commercialization, marketing and manufacturing capabilities and strategy;
- •
- our expectations regarding our ability to obtain and maintain intellectual property;
- •
- our ability to attract and retain qualified employees and key personnel;
- •
- our ability to contract with third-party suppliers and manufacturers and their ability to perform adequately;
- •
- how long we will qualify as an emerging growth company or a foreign private issuer;
- •
- our estimates regarding future revenue, expenses and needs for additional financing;
- •
- our belief that our existing cash, cash equivalents and marketable securities as of December 31, 2019 will be sufficient to fund our
operating expenses and capital expenditure requirements through at least December 2021;
- •
- regulatory developments in the United States and foreign countries; and
- •
- other risks and uncertainties, including those listed in this section of this Annual Report on Form 20-F titled "Item 3.D—Risk Factors."
You should refer to the section of this Annual Report on Form 20-F titled "Item 3.D—Risk Factors" for a discussion of important factors that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. As a result of these factors, we cannot assure you that the forward-looking statements in this Annual Report on Form 20-F will prove to be accurate. Furthermore, if our forward- looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame or at all. We undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law.
You should read this Annual Report on Form 20-F and the documents that we reference in this Annual Report on Form 20-F and have filed as exhibits to this Annual Report on Form 20-F completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of our forward-looking statements by these cautionary statements.
This Annual Report on Form 20-F contains market data and industry forecasts that were obtained from industry publications. These data involve a number of assumptions and limitations, and you are cautioned not to give undue weight to such estimates. We have not independently verified any third-party information. While we believe the market position, market opportunity and market size information included in this Annual Report on Form 20-F is generally reliable, such information is inherently imprecise.
This Annual Report on Form 20-F contains estimates, projections and other information concerning our industry, our business and the markets for our product candidates. Information that is based on estimates, forecasts, projections, market research or similar methodologies is inherently subject to uncertainties, and actual events or circumstances may differ materially from events and circumstances that are assumed in this information. Unless otherwise expressly stated, we obtained this industry, business, market and other data from our own internal estimates and research as well as from reports, research surveys, studies and similar data prepared by market research firms and other third parties such as investment banking analysts, industry, medical and general publications, government data and similar sources. In addition, assumptions and estimates of our and our industry's future performance are necessarily subject to a high degree of uncertainty and risk due to a variety of factors, including those described in the section titled "Item 3.D—Risk Factors. "These and other factors could cause our future performance to differ materially from our assumptions and estimates.
4
Item 1. Identity of Directors, Senior Management and Advisers.
Not applicable.
Item 2. Offer Statistics and Expected Timetable.
Not applicable.
- A.
- Selected Financial Data
We have derived the consolidated statements of operations data for the years ended December 31, 2019, 2018 and 2017 presented below and the consolidated balance sheet data as of December 31, 2019, 2018 presented below from our audited consolidated financial statements appearing elsewhere in this Annual Report on Form 20-F. The consolidated balance sheet data as of December 31, 2017 has been derived from our audited consolidated financial statements not included in this Annual Report on Form 20-F. Our consolidated financial statements have been prepared in accordance with International Financial Reporting Standards, or IFRS, as adopted by the International Accounting Standards Board, or IASB. You should read this data together with our consolidated financial statements and related notes included elsewhere in this Annual Report on Form 20-F and the information under the caption "Management's Discussion and Analysis of Financial Condition and Results of Operations." Our historical results are not necessarily indicative of our future results. The exchange rate between the Swiss franc and the U.S. dollar as of April 17, 2020 was $1.0342 per CHF 1.0.
| |
For the years ended December 31, |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2019 | 2018 | 2017 | |||||||
| |
(CHF in thousands, except share and per share data) |
|||||||||
Consolidated statement of operations data: |
||||||||||
Revenue from contract with customer |
2,763 | 6,044 | — | |||||||
Other income |
71 | 659 | 500 | |||||||
Operating costs |
||||||||||
Research and development |
(12,454 | ) | (4,919 | ) | (2,629 | ) | ||||
General and administration |
(4,984 | ) | (3,209 | ) | (1,106 | ) | ||||
Total operating costs |
(17,438 | ) | (8,128 | ) | (3,735 | ) | ||||
Operating income / (loss) |
(14,604 |
) |
(1,425 |
) |
(3,235 |
) |
||||
Finance income |
37 |
— |
— |
|||||||
Finance costs |
(214 | ) | (220 | ) | (45 | ) | ||||
Net loss before tax |
(14,781 | ) | (1,645 | ) | (3,280 | ) | ||||
Income tax expense |
— | — | — | |||||||
Net income / (loss) for the period |
(14,781 | ) | (1,645 | ) | (3,280 | ) | ||||
Weighted average shares outstanding |
26,428,269 |
23,293,237 |
12,941,439 |
|||||||
Number of shares outstanding |
32,848,635 | 28,564,031 | 15,384,988 | |||||||
Income / (loss) per share for income / (loss) attributable to the ordinary equity holders of the company: |
||||||||||
Basic income / (loss) per share |
(0.56 | ) | (0.07 | ) | (0.25 | ) | ||||
Diluted income / (loss) per share(1) |
(0.56 | ) | (0.07 | ) | (0.25 | ) | ||||
5
| |
As of December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2019 | 2018 | 2017 | |||||||
Consolidated balance sheet data: |
||||||||||
Cash and cash equivalents |
31,537 | 41,670 | 2,579 | |||||||
Working capital(2) |
26,708 | 39,817 | 1,576 | |||||||
Total assets |
33,029 | 42,214 | 3,063 | |||||||
Debt |
— | — | — | |||||||
Total liabilities |
7,505 | 2,973 | 1,721 | |||||||
Share capital |
32,849 | 28,564 | 15,385 | |||||||
Accumulated losses |
(300,847 | ) | (286,067 | ) | (284,422 | ) | ||||
Total equity |
25,524 | 39,241 | 1,343 | |||||||
- (1)
- See
Note 23 to our audited consolidated financial statements appearing elsewhere in this Annual Report on Form 20-F for a description of the method
used to compute diluted net (loss)/income per share.
- (2)
- We define working capital as current assets less current liabilities.
- B.
- Capitalization and Indebtedness
Not applicable.
- C.
- Reasons for the Offer and Use of Proceeds
Not applicable.
- D.
- Risk Factors
Our business faces significant risks. You should carefully consider all of the information set forth in this Annual Report on Form 20-F and in our other filings with the United States Securities and Exchange Commission, or the SEC, including the following risk factors which we face and which are faced by our industry. Our business, financial condition, results of operations and growth prospects could be materially adversely affected by any of these risks. This report also contains forward-looking statements that involve risks and uncertainties. Our results could materially differ from those anticipated in these forward-looking statements, as a result of certain factors including the risks described below and elsewhere in this Annual Report and our other SEC filings. See "Special Note Regarding Forward-Looking Statements" above.
Risks Related to Our Business
We will need significant amounts of additional new capital to fund our continued development activities.
As of December 31, 2019, we had CHF 31.5 million of cash and cash equivalents. Our monthly spending levels vary based on new and ongoing development and corporate activities. Currently, on a going concern basis, we expect to be able to finance our operations through at least December 2021, unless we are able to raise new funds. Accordingly, we intend to primarily focus our resources on continuing to investigate dipraglurant, an mGlu5 negative allosteric modulator, for use in Parkinson's disease and dystonia and corporate development activities aimed at securing resources from investors, partners and grant providers to advance our other clinical and preclinical programs, as well as our allosteric modulator discovery platform.
Our budgeted external costs for the development plans described above and further detailed in the section entitled "Item 4—"Information on the Company" are based on discussions with contract research organizations and other external suppliers, and for some of these external costs we have not entered into any agreements or other arrangements that would establish or guarantee the costs of these
6
programs. There is a risk that these development plans could be more costly than we anticipate, including as a result of unanticipated delays.
Although we believe that we will have sufficient resources to fund our intended operations through at least December 2021, we cannot assure you of this and our ability to finance our operations and pursue our intended development plans beyond that date which will depend on our ability to generate additional funding through further partnerships or grants and amounts we may raise through further financings such as additional equity offerings. If our development plans are not successful, we may not be able to generate additional funding through partnerships or grants, or raise further financing through equity offerings or otherwise, or we may only be able to do so on terms that are not favorable to our shareholders.
To the extent that we raise additional capital through the issuance of shares or other securities convertible into shares, our existing shareholders will be diluted. Future issuances of such securities, or the perception that such sales may occur, could adversely affect the trading price of our shares and impair our ability to raise capital through future offerings of shares or other equity securities. No prediction can be made as to the effect, if any, that future sales of shares or the availability of shares for future sales will have on the trading price of our shares.
We cannot guarantee that we will have sufficient funds available in the future to develop and commercialize our current or future drug candidates.
We have limited sources of revenue and will need substantial additional capital to develop and commercialize our product candidates. We may be unable to raise additional capital when needed, or at all, which would force us to reduce or discontinue operations. We do not expect to realize meaningful revenue from product sales, milestone payments or royalties in the foreseeable future, if at all. Our revenue sources are, and, we believe, will remain, extremely limited until and unless our product candidates are clinically tested, approved for commercialization and successfully marketed. To date, we have primarily financed our operations through the sale of securities, milestone payments from partners and grants from foundations and governmental agencies. Our ability to raise additional funds will depend on financial, economic and other factors, many of which are beyond our control. Under Swiss law, shareholders have certain preemptive rights to subscribe for newly issued securities in proportion to the nominal value of shares held. These preemptive rights, unless waived, may cause delays and uncertainties in any future equity offering, including in pricing, number of shares offered and dilutive effects, which discourage investment in our securities. We can provide no assurance that we can obtain access to sufficient funds when needed. If we fail to obtain additional funds at acceptable terms when needed, we may have to delay, reduce or terminate our research and development programs, limit strategic opportunities or be forced to cease operations, which may adversely affect our business, financial condition, results of operations and prospects.
We have a history of net losses and negative cash flows, and we expect that such losses will continue for the foreseeable future and that we may never achieve or maintain profitability.
Since we began operations in 2002, we have not had product revenue and our expenses have substantially exceeded our revenue, resulting in continuing operating losses and an accumulated deficit of CHF 300.9 million at December 31, 2019. For the year ended December 31, 2019, we incurred a net loss of CHF 14.8 million. These losses have resulted principally from costs incurred in research and development of our drug candidates and general and administrative expense.
We expect to continue to incur significant operating losses in the foreseeable future, primarily due to the cost of our research and development programs, preclinical studies and clinical trials and the regulatory approval process for drug candidates. The amount of future losses is uncertain and our ability to achieve profitability, if ever, will depend on, among other things, us or partners successfully
7
developing drug candidates, obtaining regulatory approval to market and commercialize drug candidates, manufacturing any approved products on commercially reasonable terms, establishing a sales and marketing organization or suitable third party alternatives for any approved product and raising sufficient funds to finance our activities. If we and/or our partners are unable to develop and commercialize one or more of our drug candidates or if sales revenue from any drug candidate that receives approval is insufficient, we will not achieve profitability, which could have a material adverse effect on our business, financial condition, results of operations and prospects.
We are a development-stage company working with novel approaches to therapeutics, which may not be successful.
We have devoted our resources to the discovery and development of allosteric modulators for neurological diseases. Since inception, we have focused on building a drug discovery platform, including a knowledge-based library and proprietary biological screening tools as well as a portfolio of drug candidates. Discovery and development of allosteric modulators involves novel approaches to therapeutics. We are subject to the risks of failure inherent in the development of product candidates based on new technologies. There is little precedent for the successful commercialization of products based on our technologies, and there are a number of technological challenges that we must overcome to complete most of our development efforts. If we are not successful in development, it will have a material adverse effect on our business, financial condition, results of operations and prospects.
We have no products on the market and we may never generate revenue from the sale or licensing of product candidates.
Our ability to achieve and sustain profitability depends on obtaining regulatory approvals for and successfully commercializing our product candidates, either alone or with third parties, such as our partner for ADX71149, Janssen, and our partner for GABAB PAM, Indivior. Currently, none of our product candidates has been approved for marketing and commercialization or is in Phase 3 trials. We cannot guarantee that any of our product candidates will be successfully tested, approved by the U.S. Food and Drug Administration, or FDA, the European Medicines Agency, or EMA, Swissmedic, Swiss Agency for Therapeutic Products, or any other regulatory agency or marketed and commercialized at any time in the foreseeable future or at all. If approval is obtained for a product candidate, we cannot assure you that we will be able to generate or sustain revenue from any sales due to factors such as whether the product is manufactured at a competitive cost or accepted in the market, as well as general and industry-specific local and international economic pressures. With our strategy to focus on allosteric modulator development, these risks continue to be significant and may increase to the extent the space becomes more competitive or less favored in the commercial marketplace. Our focus on rare disease indications with the potential for orphan drug designation limits the size of the patient population for even an approved product, unless approval is expanded for use beyond a particular rare disease. Because of the inherently small patient population for treatment of a rare disease, an approved product with orphan drug designation for which pricing is not approved or accepted in the market at an appropriate level may not generate enough revenue to offset costs of development, manufacturing, marketing and commercialization despite any benefits received from the designation, such as market exclusivity, assistance in clinical trial design, a reduction in user fees or tax credits related to development expense, and our business may be adversely affected.
We have been granted U.S. Orphan Drug Designation for dipraglurant for PD-LID and may seek Orphan Drug Designation for other product candidates, and we may be unable to maintain the benefits associated with Orphan Drug Designation, including the potential for market exclusivity.
Regulatory authorities in some jurisdictions, including the United States and Europe, may designate drugs and therapeutic biologics for relatively small patient populations as orphan drugs.
8
Under the Orphan Drug Act, the FDA may designate a drug as an orphan drug if it is intended to treat a rare disease or condition, which is generally defined as a patient population of fewer than 200,000 individuals annually in the United States, or a patient population greater than 200,000 in the United States where there is no reasonable expectation that the cost of developing the drug will be recovered from sales in the United States. In the United States, Orphan Drug Designation entitles a party to financial incentives such as opportunities for grant funding toward clinical trial costs, tax advantages and user-fee waivers. In addition, if a product that has Orphan Drug Designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications, including a full NDA, to market the same product for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity or where the manufacturer is unable to assure sufficient product quantity.
We have obtained Orphan Drug Designation for dipraglurant and if we may be able to obtain Orphan Drug Designation for any of our future product candidates in specific indications, we may not be the first to obtain marketing approval for dipraglurant or any other such product candidates for the orphan-designated indication due to the uncertainties associated with developing pharmaceutical products. In addition, exclusive marketing rights in the United States may be limited if we seek approval for an indication broader than the orphan-designated indication or may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition.
Further, even if we obtain orphan drug exclusivity in the U.S. for a product, that exclusivity may not effectively protect the product from competition because different drugs or therapeutic biologics with different active moieties can be approved for the same condition. Even after an orphan product is approved, the FDA can subsequently approve the same drug or therapeutic biologic with the same active moiety for the same condition if the FDA concludes that the later drug or therapeutic biologic is safer, more effective or makes a major contribution to patient care. In Europe, we could be prevented from marketing our products if a similar medicinal product is granted orphan drug designation for the same indications that we are pursuing. Once authorized, with a limited number of exceptions, neither the competent authorities of the EU member states, the EMA, or the European Commission are permitted to accept applications or grant marketing authorization for other similar medicinal products with the same therapeutic indication. Marketing authorization could also be granted to a similar medicinal product with the same orphan indication if the latter product is safer, more effective or otherwise clinically superior to the original orphan medicinal product. Orphan Drug Designation neither shortens the development time or regulatory review time of a drug or therapeutic biologic nor gives the drug or therapeutic biologic any advantage in the regulatory review or approval process. In addition, while we may seek Orphan Drug Designation for our future product candidates, we may never receive such designations.
The future of our business and operations depends on the success of our allosteric modulator development programs, including our most advanced proprietary product candidate, dipraglurant.
We are substantially dependent on the success of our current lead drug candidate, dipraglurant, which we are developing ourselves. In March 2012, we announced the completion of a Phase 2a clinical trial in the United States and Europe with dipraglurant for the treatment of PD-LID. Though the development so far has produced positive results, further development and commercialization for the treatment of PD-LID or other disease indications may not be successful or may experience additional significant delays and setbacks. For example, we are undertaking significant risk in planning a pivotal development program for dipraglurant for the treatment of PD-LID, without having conducted any additional exploratory clinical trials beyond the Phase 2a proof of concept clinical trial; our last clinical
9
trial of dipraglurant in PD-LID was completed eight years ago and the diagnosis and standard of care for PD-LID may have changed in the interim, including standards for marketing authorization. We believe that a failure to develop our most advanced drug candidates, or to do so in a timely manner, would not only harm those programs but also industry and investor confidence in our other programs and could have a material adverse effect on our business, financial condition, results of operations and prospects.
Our dependence on Janssen to develop and commercialize ADX71149 and Indivior to develop and commercialize GABAB PAM exposes us to significant risks.
Our collaboration with Janssen, Indivior and any future partner, may not be scientifically, clinically or commercially successful. We are dependent upon Janssen and Indivior, and may be dependent upon any other partners with which we may collaborate in the future, to perform and fund development activities, including clinical testing, regulatory filings and the manufacture and marketing of products. Under our collaboration and license agreements with our partners, our partners have sole responsibility for the financing and development of selected compounds through preclinical and clinical trials, as well as registration procedures and commercialization, if any, in the United States, Japan, the United Kingdom, Germany, France, Spain and Italy. Our partners have authority over all aspects of the development of selected compounds and may develop or commercialize third-party compounds with a different mechanism of action for identical use. Our role on the joint development committee formed under the collaboration and license agreement is advisory and we do not have authority to determine or veto actions. Our partners may take independent action concerning product development, marketing strategies, manufacturing and supply issues and rights relating to intellectual property. Thus, the success of ADX71149 and GABAB PAM for the treatment of CNS and related diseases currently depends entirely upon the efforts of Janssen and Indivior, respectively. Janssen and Indivior each have significant discretion in determining the efforts and resources it applies to the development and, if approval is obtained, commercialization and marketing of ADX71149 and GABAB PAM, respectively. Janssen and Indivior may not be effective in obtaining approvals in their respective fields of use, marketing any approved products or arranging for any necessary sublicense, supply, manufacturing or distribution relationships, or our partners may change their strategic focus or pursue alternative technologies in a manner that results in reduced or delayed revenue to us. Our partners have a variety of marketed products and their own corporate objectives may not be consistent with our best interests. Changes of this nature might also occur if our partners are acquired or experience changes in management. In any future disagreement with us, our partners will have significantly greater financial and managerial resources on which to draw. Any disagreement could lead to lengthy and expensive litigation or other dispute resolution proceedings as well as extensive financial and operational consequences to us and have a material adverse effect on our business, financial conditions, results of operations and prospects
Our failure to collaborate successfully with partners may delay, impair or prevent the development or commercialization of our drug candidates.
Our business strategy requires us to enter into various forms of collaboration arrangements with other companies, licensors or licensees to research, develop and commercialize our drug candidates. We are unlikely to be able to enter into new collaborative arrangements, with respect to the drug candidates we are currently developing internally, until we complete at least the next stage of their respective development activities. We cannot assure you that we will be able to maintain our existing collaborations with Janssen and Indivior, negotiate collaboration arrangements in the future on acceptable terms with first choice partners, if at all, or that any such collaboration arrangements will be successful. To the extent that we are not able to maintain or establish such arrangements, we would be forced to seek alternatives, including undertaking drug development and commercialization activities on our own, which would increase our capital requirements and could require us to limit the scope of
10
some or all of our other research and development activities. Under a collaboration agreement, we are likely to have limited influence over the future development or commercialization of the relevant drug candidates. Such development or commercialization may depend significantly on the efforts and activities of the collaborator. Under the terms of an agreement, a collaborator may have significant discretion in determining the efforts and resources it dedicates to the collaboration, which may change over time depending on the collaborator's overall strategic priorities. The suspension or termination of our collaboration arrangements, the failure of our collaboration arrangements to be successful or the delay in the development or commercialization of drug candidates pursuant to collaborations could have a material adverse effect on our business, financial condition, results of operations and prospects.
Our future success depends on our ability to retain key employees, consultants and advisors and to attract, retain and motivate qualified personnel.
We are highly dependent on members of our executive team. The loss of the services of any of them may adversely impact the achievement of our objectives.
Recruiting and retaining qualified employees, consultants and advisors for our business, including scientific and technical personnel, is also critical to our success. Competition for skilled personnel is intense and the turnover rate can be high. We may not be able to attract and retain personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies and academic institutions for skilled individuals. In addition, failure to succeed in preclinical studies, clinical trials or applications for marketing approval may make it more challenging to recruit and retain qualified personnel. The inability to recruit, or loss of services of certain executives, key employees, consultants or advisors, may impede the progress of our research, development and commercialization objectives and have a material adverse effect on our business, financial condition, results of operations and prospects.
Furthermore, a number of our key staff reside in California, which in the past has experienced both severe earthquakes and wildfires. Disruptions to the services provided by our staff based in California due to earthquakes, wildfires or other natural disasters could delay or disrupt our business and operations.
If third parties on which we depend to conduct our clinical trials do not perform as contractually required, fail to satisfy regulatory or legal requirements or miss expected deadlines, our clinical development programs could be delayed and otherwise adversely affected.
We rely on third party clinical investigators, contract research organizations, or CROs, clinical data management organizations, medical institutions and consultants to design, conduct, supervise and monitor preclinical studies and clinical trials in relation to our product candidates. Because we rely on third parties and do not have the ability to conduct clinical trials independently, we have less control over the timing, quality and other aspects of clinical trials than we would if we conducted them on our own. These investigators, CROs and consultants are not our employees and we have limited control over the amount of time and resources that they dedicate to our programs. These third parties may have contractual relationships with other entities, some of which may be our competitors, which may draw time and resources from our programs. If we cannot contract with acceptable third parties on commercially reasonable terms, or at all, or if these third parties do not carry out their contractual duties, satisfy legal and regulatory requirements for the conduct of clinical trials or meet expected deadlines, our clinical development program could be delayed or otherwise adversely affected. In all events, we are responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trial. The FDA requires us to comply with good clinical practices for conducting, recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. Our reliance on third parties that we do not control does not relieve us of
11
these responsibilities and requirements. The third parties with which we contract might not be diligent, careful or timely in conducting our clinical trials, as a result of which the clinical trials could be delayed or unsuccessful. Any such event could have a material adverse effect on our business, financial condition, results of operations and prospects.
Because we rely on third party manufacturing and supply partners, our clinical development supplies and other materials may become limited or interrupted or may not be of satisfactory quantity.
We rely on third party manufacturing and supply partners for our research and development, preclinical studies and clinical trials. We currently do not have in-house facilities to manufacture our research and development, preclinical and clinical drug supplies. In the event that any of our suppliers, for research and development, or preclinical studies or clinical trials, fail to perform their respective obligations in terms of quality, timing or otherwise, or if our supply of such components or other materials become limited or interrupted for other reasons, we may not be able to develop or market our drug candidates on a timely and cost-competitive basis, if at all, which may have a material adverse effect on our business, financial condition, results of operations and prospects. There can be no assurance that our supply of research and development, preclinical and clinical development drugs and other materials will not be limited, interrupted, restricted in certain geographic regions or of satisfactory quality. If the suppliers that currently manufacture our clinical drug supplies cannot continue to do so, we can provide no assurance that we will be able to obtain alternative components and materials from other manufacturers of acceptable quality, or on terms or in quantities acceptable to us, or that we will not require additional components and other materials to manufacture or use our drug candidates. In addition, suppliers need to meet applicable manufacturing requirements and undergo rigorous facility and process validation tests required by regulatory authorities in order to comply with applicable regulatory standards, such as current Good Manufacturing Practices, or cGMP. We cannot provide assurance that our suppliers will comply with such requirements.
Our product candidates may not successfully obtain regulatory approval.
Even if we are able to initiate Phase 3 clinical trials and they are completed, there can be no assurance that we will receive approval from the FDA, the EMA, Swissmedic, Swiss Agency for Therapeutic Products, or any other relevant government agencies. Any approval, if any, may be delayed or may be obtained on restrictive terms. This may occur if a drug candidate does not show acceptable safety and efficacy in preclinical studies and clinical trials or otherwise does not meet applicable regulatory standards for approval or the drug candidate does not prove as effective as, or does not offer therapeutic or other improvements over, existing or future drugs used to treat the same or similar illness or conditions. Failure by us or a partner to obtain approval for products candidates could have a material adverse effect on our business, financial condition, results of operations and prospects.
Our drug candidates must prove their efficacy and safety in rigorous clinical testing that is expensive, time-consuming and may be delayed, suspended or terminated at any time.
Drug approval requires extensive, time consuming and expensive clinical testing to demonstrate safety, tolerability and efficacy of a drug and meet other regulatory standards for authorization to market and commercialize. The development of innovative drugs is inherently risky and the utility and success of a drug will depend on its efficacy and side effect profile for the target patient population. Preclinical studies and clinical trials are long, expensive and uncertain processes. Successful results obtained in preclinical studies and early clinical trials may not be predictive of results in later clinical trials and do not ensure that later preclinical studies or clinical trials will be successful. Clinical trials may be delayed, suspended or terminated as a result of many factors, many of which are or may be beyond our control, such as:
- •
- suspension or termination of clinical trials by regulators, institutional review boards or data safety monitoring boards;
12
- •
- termination due to safety issues or lack of efficacy of the drug tested;
- •
- a collaboration partner's termination of an arrangement with us or inadequate dedication of financial or other resources towards
- •
- development under an arrangement with us;
- •
- inability to enter into adequate collaboration arrangements to complete the development or commercialization and manufacturing of our drug
candidates;
- •
- insufficient availability of a drug product in accordance with cGMP quality; or
- •
- slower than expected enrollment of patients or lack of compliance by patients.
We or a partner may be required to conduct clinical trials or other testing of drug candidates beyond those currently contemplated, in particular, if the currently contemplated trials fail to complete successfully or if the results of those trials or tests are negative or inconclusive. It may take us several years to complete this testing, if at all, and failure can occur at any stage of the process, which could delay, increase costs associated with or prevent approval or commercialization of a drug candidate. Even after approval, if any, a drug may be shown to be unsafe or not have its purported effect. As a result, we or a partner may be required to conduct additional trials or studies, be subject to fines, suspension or withdrawal of approval, drug recalls, product seizures, operating restrictions or criminal prosecution. In all such cases, our anticipated development or commercialization timelines may not be met, which could have a material adverse effect on our business, financial condition, results of operations and prospects.
We face competition from entities that have developed or may develop similar or different product candidates aimed at the indications on which we are focusing.
The development and commercialization of drugs is highly competitive. We compete with a variety of multinational pharmaceutical companies and specialized pharmaceutical companies, including products approved for marketing and/or product candidates under development, for each of the product candidates and each of the indications for which we are developing our product candidates. Competitor firms include Adamas Pharmaceuticals, Avanir Pharmaceuticals, Inc., Eli Lilly and Company, F. Hoffmann-La Roche Ltd, Heptares Therapeutics Ltd, Indivior (as certain to product candidates outside the scope of our collaboration with Indivior), Lundbeck Pharmaceuticals Ltd, Medytox Korea Co., Ltd., Merck & Co. Inc., Neuraltus Pharmaceuticals, Inc., Newron Pharmaceuticals, Inc. and Novartis Pharma AG, as well as technology being developed at universities and other research institutions. Many of our competitors have significantly greater financial, technical, manufacturing, marketing, sales and supply resources or experience than we have. Our competitors have developed, are developing or will develop drug candidates and processes that will compete with our drug candidates. Competitors may enjoy a significant competitive advantage if they are able to achieve patent protection, obtain marketing authorizations and commence commercial sales of their drugs before us. Competing drugs could present superior treatment alternatives for our targeted indications, including by being more effective, safer or convenient, and even make our drug candidates or know-how obsolete before we reach the market. In addition, competitors may sell drugs below the price level at which appropriate return for our investment in drug development is possible. As a result of these factors, we may be unable to successfully develop commercially feasible drugs and our commercial opportunities may be reduced or eliminated, and we may not be able to successfully compete. This would have a material adverse effect on our business, financial condition, results of operations and prospects.
13
We may fail to obtain, maintain or enforce licenses, patents and proprietary technology.
Our success depends in part on our ability to obtain patent protection for our drug candidates and processes, preserve our trade secrets and other proprietary rights and to defend and enforce our rights against infringement in Europe, the United States and other countries. If we are unable to do so, our drugs, technologies and know-how may not provide us with a competitive advantage. The validity and breadth of claims in patent applications involve complex legal and factual questions and, therefore, involve uncertainty. We own 13 U.S. and 238 foreign patents and a number of pending patent applications that cover various aspects of our technologies. No assurance can be given that patents based on pending patent applications or any future patent applications will be issued. We may need to refine or narrow our claims. Due to their broad scope, some of our generic compound claims may not be patentable. Other of our patent applications may not be granted if third parties have earlier filed applications for inventions covered by our pending patent applications. The scope of any patent protection we are able to obtain may not provide us with sufficient protection against competing drugs or provide competitive advantages to us. Any of the patents that have been or may be issued to us may be held invalid or unenforceable if subsequently challenged by competitors or other third parties. Furthermore, there can be no assurance that others have not developed or will not develop similar drugs, duplicate any of our drugs or design around any patents that have been or may be issued to us. Any of our granted, valid and enforceable patents will provide protection for only a limited period of time. We cannot assure that we will obtain any extensions of patent protection that are sometimes offered if certain clinical development extension application deadlines are met or that we will be successful in seeking any method of use patent. If a method of use patent is granted but product patents are not granted or expire, third parties would be able to develop products using the method in indications not covered by the method of use patent.
We may be restricted in our development and any commercialization activities by third-party patents and patent applications.
Our commercial success depends on our ability to operate without infringing third-party patents and other intellectual property or market exclusivity rights. If we are not able to do so, we may be subject to infringement actions. We may not be aware of all patents and patent applications that may impact our ability to make, use or sell our product candidates. Other parties may have filed, or may file in the future, patent applications covering compounds or drug candidates that are similar to ours. Any such event could have a material adverse effect on our business, financial condition, results of operations and prospects. In addition, because patent applications can take many years to issue and are not published for a period of time ranging on the jurisdictions in which we applied for registration, there may be applications currently pending, unknown to us, which may later result in patents that our drug candidates or technology may infringe. Any conflicts arising from the patent rights of others could significantly reduce the scope of our patents and limit our ability to obtain meaningful patent protection. We may be required to obtain licenses to those patents or to develop or obtain alternative technology. We may not be able to obtain any such licenses on acceptable terms, or at all. Any failure to obtain such licenses could delay or prevent us from pursuing the development or commercialization, if any, of our product candidates.
We may fail to protect our intellectual property rights, including trade secrets and know-how.
Our success depends on our ability to obtain and enforce intellectual property rights, including trade secrets and non-patentable know-how related to our allosteric modulator platform. We seek to protect or secure this intellectual property, in part, by entering confidentiality agreements with and receiving assignments from our employees, consultants, suppliers, licensees, funding partners and other contractual partners and advisers. We may not always be able to obtain these agreements or assignments. Even if we obtain these agreements or assignments, there can be no assurance that they
14
will effectively protect our intellectual property rights or prevent improper use or disclosure of confidential information or that they will not be breached. We may not have adequate remedies for any breach of these agreements or assignments, or our trade secrets or non-patentable know-how may otherwise become known or be independently developed by competitors. In addition, these agreements or assignments may conflict with, or be subject to, the rights of third parties with which our employees, consultants, suppliers, licensees, funding partners or other contractual partners or advisers had previous employment, consulting or other relationships. Any such event could have a material adverse effect on our business, financial condition, results of operations and prospects.
We may have to defend against or initiate lawsuits to protect our intellectual property rights.
In the future, third parties with patent claims that overlap with our intended activities may decide to sue us for monetary damages or to prevent us from manufacturing, selling or developing our drug candidates. We could also become subject to claims that we or our employees have inadvertently or otherwise used or disclosed intellectual property, trade secrets or other proprietary information of an employee's former employer, particularly if such employer is a university or pharmaceutical company. Additionally, to protect our patent rights, we may decide to initiate lawsuits against third parties. Defending against or initiating such claims, which typically go on for years before a legal judgment or settlement is obtained, would involve significant effort and expense and could divert management's attention from the operation of our business. Any such proceedings could involve prior art and put our patents at risk of being invalidated or interpreted narrowly and our pending patent applications at risk of not being issued. In addition, there is a risk that some of our confidential information could be compromised by disclosure in such proceedings and provide competitors with access to our proprietary information. Further, the outcome of any such proceedings may be unfavorable to us. If the manufacture, use or sale of any of our drug candidates infringes the patents, or violates other proprietary rights, of third parties, a court or settlement agreement may require us to pay actual damages and, potentially, penalties, including the other party's attorney's fees, which may be substantial. We could also be required to cease the development, manufacture, use and sale of drugs that infringe the patent rights of others, to expend significant resources to redesign our technology so that it does not infringe the patent rights of others, to develop or acquire non-infringing technology, which may not be possible, or to obtain licenses to the infringed intellectual property, which may not be available to us on acceptable terms or at all. We cannot guarantee that we will have sufficient financial or other resources to protect intellectual property significant to the development of our product candidates.
Even if a product candidate receives regulatory approval, lack of market acceptance may prevent us from generating revenue from commercialization of the product.
Even if a product candidate is approved, if we or a partner are not successful in commercializing the product, we will not generate revenue from sales. Revenue generated from an approved product depends on its successful commercialization. Many factors may impede successful commercialization, many of which are or may be beyond our or a partner's control. These factors include the proprietary rights of third parties, including our competitors, the failure of a product to prove effective as, or offer therapeutic or other improvements over, existing or future drugs used to treat the same or similar conditions or the inability of a product to gain acceptance by patients, the medical community or third-party payers, such as insurance companies or government reimbursement programs, or the inability of produce a product in commercial quantities at an acceptable cost, or at all. Even if our drug development is successful and marketing authorization has been obtained, our ability, or our partners' ability, to generate significant revenue will depend on the acceptance of our drugs by physicians, patients, third-party payers and the medical community. We cannot assure you that we or our partners will achieve market acceptance of our drug candidates or generate revenue once we or our partners obtain marketing authorization. The market acceptance of any of our drug candidates depends on a
15
number of factors, including the continued demonstration of efficacy and safety in commercial use, cost-effectiveness, convenience and ease of administration, competition, marketing and distribution support, the scope of the approved uses and labeling requirements, prevalence and severity of any side effects, and adequate government or other third-party coverage or reimbursement for the cost of the drug. To the extent competitors are able to commercialize competing drugs before our drugs have achieved market approval and acceptance, we may have difficulty gaining market acceptance if physicians, patients, third- party payers and the medical community have grown accustomed to use of the competing drugs, whether or not such competing drugs are more effective or have other advantages over our drug.
Any commercialization efforts by us will require us to develop sales, marketing and distribution capabilities internally or through arrangements with third parties.
Sales, marketing and distribution capabilities are key elements of a successful commercialization strategy, none of which we currently have internally. If any of our product candidates are approved, we intend to market the product either directly or through other strategic alliances and distribution arrangements with third parties. To commercialize our drugs, we will need to enter into new collaborations with third parties or develop our own marketing and sales force with technical expertise and supporting distribution capability. If we decide to market our products directly, we will need to commit significant financial and managerial resources to develop a marketing and sales force with technical expertise and with supporting distribution, administration and compliance capabilities. If we rely on third parties with such capabilities to market our products, we will need to establish and maintain partnership arrangements, and there can be no assurance that we will be able to enter into third-party marketing or distribution arrangements on acceptable terms or at all. To the extent that we do enter into such arrangements, we will be dependent on our marketing and distribution partners. In entering into third-party marketing or distribution arrangements, we expect to incur significant additional expense and there can be no assurance that such third parties will establish adequate sales and distribution capabilities or be successful in gaining market acceptance for our products and services. Any factors preventing or limiting the market acceptance of our drug candidates could have a material adverse effect on our business, financial condition, results of operations and prospects. There can be no assurance that we will be able to build up our own marketing and sales organization, to attract and maintain established collaboration partners for the third-party commercialization of our drug candidates, to enter into agreements on acceptable terms for sales and marketing, if at all, or that any such collaboration arrangements will be successful. As a consequence, we would be forced to seek alternatives, redirect our resources or have to limit the scope of our research and development activities in other fields and thereby delay the launch and sales of any or all of our drug candidates, or raise new funds. Accordingly, this could have a material adverse effect on our business, financial condition, results of operations and prospects.
We may become exposed to costly and damaging liability claims and may not be able to maintain sufficient liability insurance to cover these claims.
Our business with pharmaceutical drugs entails a potential risk of substantial liability for damages, including drug liability and environmental liability, which are inherent in the development, testing and manufacturing of our drug candidates. It is always possible that a drug, even after marketing authorization, may exhibit unforeseen failures or adverse side effects. We can provide no assurance that sufficient insurance coverage will be available to us at acceptable terms, or at all, for any damages or costs in connection with any liability claims. Liability lawsuits are costly and time consuming and may divert management's attention from their normal responsibilities. If any of our drugs were to fail or produce adverse side effects, substantial uninsured losses could result, which could have a material adverse effect on our business, financial condition, results of operations and prospects. Even where drug failures or side effects are not so serious as to warrant withdrawing the drug from the market or
16
liability in damages, they may reduce the drug's competitiveness or adversely affect our reputation, which could have a material adverse effect on our business, financial condition, results of operations and prospects.
We and our partners are subject to significant government regulation, including marketing authorization requirements, which could increase the cost of developing our drug candidates or delay, prevent or limit the commercialization of our drug candidates.
We and our partners are subject to extensive and rigorous governmental regulation and the applicable regulatory requirements are subject to change. Our and our partner's research and development, preclinical studies and clinical trials, manufacturing, safety, efficacy, record-keeping, labeling, marketing, sales and distribution of our drug candidates are regulated by the EMA, the FDA, Swissmedic, Swiss Agency for Therapeutic Products, and other government agencies in countries where we are testing or intend to test and market our drug candidates. Before a clinical trial can begin, we and our partners must obtain approval from the competent national authority in the country where the trial is planned to be conducted. A favorable opinion from a competent ethics committee or an independent institutional review board on the clinical trial application is also needed. We cannot assure we or our partners will obtain authorization for further testing of drug candidates already in clinical trials or for human clinical trials of any or all of our other candidates currently in research or pre-clinical development. We, and our partners or regulatory authorities may suspend or terminate clinical trials at any time if it is thought that the participants are being exposed to unacceptable health risks. It may take us or our collaborators several years to complete this testing, and failure can occur at any stage of the process.
The governmental regulation of development of drug candidates extends beyond clinical trials to approvals required for their sale and monitoring after sale, including safety reporting requirements, regulatory oversight of drug promotion and marketing and cGMP. A failure by us or our partners to obtain marketing authorization or a delay in obtaining and maintaining approval could damage our reputation and adversely affect the marketing of our drugs and our ability to generate revenue, which could have a material adverse effect on our business, financial condition, results of operations and prospects. In addition, marketing authorizations, if granted, may not include all uses for which we may seek to market a drug, thereby limiting the potential market for the drug. Moreover, even after marketing authorization is obtained, a marketed drug, its manufacturer and its manufacturing facilities are subject to continual review and periodic inspections by the relevant authorities. Consequently, any discovery of previously unknown problems with an approved drug, manufacturer or manufacturing facilities may result in restrictions on the drug or manufacturer, including a requirement to withdraw the drug from the market. In addition, regulatory requirements are evolving in a manner that cannot be predicted. Changes in existing regulations of EMA, FDA, Swissmedic, Swiss Agency for Therapeutic Products or other regulations or the adoption of new regulations could prevent us from obtaining or maintaining, or affect the timing of, future marketing authorizations. Changes in regulatory policy during the period of development of a drug or regulatory review may result in delays or rejections of approvals of the drug candidates. Any change in the regulations governing us could have a material adverse effect on our business, financial condition, results of operations and prospects.
Current healthcare laws and regulations and future legislative or regulatory changes to the healthcare system may affect our ability to sell any drugs we may develop.
Healthcare laws are subject to change, which may affect our ability to sell any product candidates for which we receive marketing and commercialization approval. In the U.S., an important potential market for our drug candidates, there have been a number of legislative and regulatory changes to the healthcare system that could affect our future results of operations. In particular, there have been and
17
continue to be a number of initiatives at the U.S. federal and state levels that seek to reduce healthcare costs.
Individual states in the United States have become increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access, and marketing cost disclosure and transparency measures, and designed to encourage importation from other countries and bulk purchasing. Legally-mandated price controls on payment amounts by third-party payers or other restrictions could harm our business, results of operations, financial condition and prospects. In addition, regional healthcare authorities and individual hospitals in the United States are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs. This could reduce ultimate demand for our products or put pressure on our product pricing, which could negatively affect our business, results of operations, financial condition and prospects.
In addition, given recent federal and state government initiatives directed at lowering the total cost of healthcare, Congress and state legislatures will likely continue to focus on healthcare reform, the cost of prescription drugs and biologics and the reform of the Medicare and Medicaid programs. While we cannot predict the full outcome of any such legislation, it may result in decreased reimbursement for drugs and biologics, which may further exacerbate industry-wide pressure to reduce prescription drug prices. This could harm our ability to generate revenue. In addition, legislation has been introduced in Congress that, if enacted, would permit more widespread importation or re-importation of pharmaceutical products from foreign countries into the United States, including from countries where the products are sold at lower prices than we might sell our products in the United States. Such legislation, or similar regulatory changes, could put competitive pressure on our ability to profitably price our products, which, in turn, could adversely affect our business, results of operations, financial condition and prospects. It is also possible that other legislative proposals having similar effects will be adopted.
Certain European countries utilize reference pricing to control the prices of drugs. Use of reference pricing may increase, which could restrict the sales potential for many new drugs unless the drug can be significantly differentiated from existing drugs.
Additional governmental and regulatory proposals and health care reforms are possible. However, we are unable to forecast what additional legislation or regulation relating to the health care industry or third-party reimbursement may be enacted in the future, or what effect such legislation or regulation would have on our business. Our business could be harmed by other health care reforms that may be erected or adopted in the future, and in particular this could have a material adverse effect on the amounts that private payers will pay for drugs. As a consequence, we may not be able to realize an appropriate return on our investment in research and development and generate revenue sufficient to attain profitability, even if our drugs are approved for marketing. This could have a material adverse effect on our business, financial condition, results of operations and prospects.
The availability and level of third-party reimbursement for our potential drugs will be uncertain, and it may be difficult to obtain or maintain expected price levels.
Our or a partner's ability successfully to commercialize our drug candidates and to attract strategic partners for our drug candidates or future drugs will depend in part on price levels and on the extent to which reimbursement for the costs of treatment with these drug candidates will be available from government health administration authorities, private health insurers and other third-party payers, as well as government health care programs. Governments and other third-party payers are increasingly attempting to contain health care costs, in part by challenging the price of medical drugs and services or by restricting the eligibility for reimbursement. Health care cost pressure could lead to pricing
18
pressure which could adversely affect pricing of dipraglurant, ADX71149, GABAB PAM and our other potential drugs. Seeking third-party reimbursement is a time-consuming and costly process, which will require us and our partners to provide scientific and clinical support for the use of each of our drug candidates to each third-party payer separately. Significant uncertainty exists as to the payment status of newly approved medical drugs. The unavailability or inadequacy of third-party reimbursement, or legislation controlling treatments or prices, could have an adverse effect on the price level and consequently the market acceptance of our drug candidates and may have a material adverse effect on our results or operations, financial condition and prospects.
Any non-compliance by us with the environmental, health and safety laws and regulations that we are subject to could result in fines, suspension of drugs research and development or cessation of our operations or civil liability.
We are subject to a variety of health, safety and environmental laws and regulations in the jurisdictions in which we operate, particularly in our research and development activities, as well as in our pre-clinical studies. These laws and regulations govern, among other things, the use, storage, handling and discharge or disposal of hazardous materials, chemicals and compounds, including wastewater discharge, air emissions and waste management, where we operate. Our research and development programs involve the controlled use of hazardous materials, chemical and biological materials and controlled pre-clinical animal studies. Although we believe that we hold all permits currently required to operate our business and otherwise comply with current laws and regulations, any failure by us to comply with present or future laws and regulations could result in fines, suspension of research and development or cessation of our operations. We, like many of our competitors, have incurred, and will continue to incur, capital and operating expenditures and other costs in the ordinary course of our business in complying with such laws and regulations in most of the jurisdictions in which we operate. We do not currently anticipate any material additional capital expenditures in respect of such regulations outside of the ordinary course of our business. However, the risk of environmental liability is inherent in our business and there can be no assurance that additional material costs of complying with environmental regulations will not arise in the future. Our research and manufacturing activities involve the use of hazardous materials. Although we believe that our safety procedures for handling and disposing of hazardous materials (including medical and biological waste) comply with relevant laws and regulations, we cannot eliminate the risk of accidental or manmade contamination, injury or damage from these materials. In the event of an accident or environmental discharge, we may be held liable for any resulting damages. We cannot assure you that the amount of our insurance coverage will be sufficient to satisfy any such damages. As a result, any such accident could have a material adverse effect on our business, financial condition, results of operation and prospects. In addition, changes to existing or future laws and regulations may result in the imposition on us of significant additional environmental, health and safety compliance costs.
We are exposed to currency fluctuation risks and other financial risks.
For the year ended December 31, 2019, approximately 58% and 98% of our costs and revenue, respectively, were denominated in currencies other than the Swiss franc. As a result, our business is affected by fluctuations in foreign exchange rates between the Swiss franc and other currencies, particularly U.S. dollars, the Euro and the British pound. A significant amount of our costs are denominated in currencies other than Swiss francs as we source supplies, research and development, consulting and other services in several countries other than Switzerland. On the revenue side, a significant amount relates to currencies other than Swiss francs. The research grants from The Michael J. Fox Foundation for Parkinson's Research are paid in U.S dollars, whereas under our agreement with Janssen, all milestone payments and royalties payable by Janssen to us are denominated in Euros. Furthermore, under our agreement with Indivior, all research funding, milestones payments and royalties payable by Indivior to us are denominated in U.S dollars. Since our
19
reporting currency is the Swiss francs, we convert financial line items into Swiss francs at the applicable foreign exchange rates. As our business grows, we expect that a significant part of our revenue, including milestone payments and royalties, and of our costs, including costs for clinical trials, will be denominated in U.S. dollars, the Euro or the British pound. Unfavorable fluctuations in the value of the Swiss franc compared to these other currencies could have a material adverse effect on our business, financial condition, results of operations and prospects.
Our current operations are concentrated in one location and any events affecting this location may have material adverse consequences.
Our current operations are located in our facilities situated in Geneva, Switzerland. Any unplanned event, such as flood, fire, explosion, earthquake or other accidents that result in us being unable to fully utilize the facilities, may have a material adverse effect on our ability to operate our business, particularly on a daily basis, and have significant negative consequences on our financial and operating conditions. Loss of access to these facilities may result in increased costs, delays in the development of our product candidates or interruption of our business operations. As part of our risk management policy, we maintain insurance coverage at levels we believe are appropriate for our business. However, in the event of an accident at these facilities, we cannot assure you that the amounts of insurance will be sufficient to satisfy any damages and losses. If our facilities are unable to operate because of an accident or for any other reason, even for a short period of time, any or all of our research and development programs may be harmed. Any business interruption may have a material adverse effect on our business, financial position, results of operations and prospects.
We are subject to risks related to data privacy concerns, cyber security breaches and failure to comply with privacy regulations and security requirements relating to data.
In the ordinary course of our business we come to possess sensitive personal data, including information from clinical trials, and health data obtained in connection with reporting of adverse events. We are subject to data protection laws, privacy requirements and other regulatory restrictions in the various jurisdictions in which we operate.
Our failure to keep apprised of, and comply with, privacy, data use and security laws, standards and regulations, including, for instance, unauthorized disclosure of, or access to, data, could result in the suspension or revocation of our approvals or registrations, the limitation, suspension or termination of services or the imposition of administrative, civil or criminal penalties, including fines which may be as high as €20 million or 4% of our annual worldwide revenue (whichever is greater) for serious infringements of the EU General Data Protection Regulation that became effective in May 2018. In addition, we may obtain health information from third parties in the United States (including research institutions from which we may obtain clinical trial data) that are subject to privacy and security requirements under the Health Insurance Portability and Accountability Act of 1996, as amended, or HIPAA. Depending on the facts and circumstances, we could be subject to criminal penalties, including if we knowingly obtain, use, or disclose individually identifiable health information maintained by a HIPAA-covered entity in a manner that is not authorized or permitted by HIPAA. In addition, such failure or non-compliance may cause existing or potential partners, including hospitals, physicians and patients to cease interacting with us, and could damage our reputation and brand. In addition, to the extent more restrictive laws, rules or security requirements relating to business and personal data are adopted in the future in the various jurisdictions in which we operate, such changes could have an adverse impact on our business by increasing our costs or imposing restrictions on our business processes. Accordingly, our failure to keep apprised of, and comply with, privacy, data use and security laws, standards and regulations could have a material adverse effect on our reputation, business, financial condition, results and prospects. Our financial exposure to any actual or alleged breach of
20
such regulations or standards may either not be insured against or not fully covered through our current insurance.
Cyber security attacks on our servers, information systems and databases, or the third party servers, information systems and databases on which our information is stored, could compromise the security of our data or could cause interruptions in the operations of our businesses. Notwithstanding safeguards, cyber security breaches, internal security breaches, physical security breaches or other unauthorized or accidental access to our servers, other information systems or databases could result in tampering with, or the theft or publication of, sensitive information or the deletion or modification of data, or could otherwise cause interruptions in our operations.
The tampering with, disruption to, or the theft or publication of, sensitive information or the deletion or modification of records held either in our systems or the systems of others to which we have access, could subject us to increased costs and exposure to litigation. The loss of confidential information could result in the payment of damages and reputational harm and have a material adverse effect on our business, financial condition, results and prospects.
Our financial exposure from the items referenced above could either not be insured against or not fully covered through any insurance that we maintain and could have a material adverse effect on our business, financial condition, results and prospects.
The global pandemic caused by COVID-19 could materially and adversely impact our business and clinical trials, including potentially delaying the initiation of our planned Phase 2b/3 pivotal clinical trial of dipraglurant in PD-LID patients.
In early 2020, a coronavirus disease (COVID-19) pandemic developed globally resulting in a significant number of infections and negative effects on economic activity. Since then, the pandemic has, among other, caused various emergency measures to be applied by various countries around the world (including the United States as well as our country of incorporation, Switzerland) and has brought along substantial volatility in financial markets both globally and in Switzerland. While COVID-19 is still spreading and the final implications of the pandemic are difficult to estimate at this stage, it is clear that it will affect the lives of a large portion of the global population and cause significant effects. At this time, the pandemic has caused states of emergency to be declared in various countries, travel restrictions imposed globally, quarantines established in certain jurisdictions and various institutions and companies being closed. The Company is actively monitoring the situation and is taking any necessary measures to respond to the situation in cooperation with the various stakeholders.
The Company has suspended the initiation of its planned placebo-controlled Phase 2b/3 pivotal clinical trial of dipraglurant in PD-LID patients. The Company decided to suspend the trial based on the inability of planned clinical trial sites in the United States to initiate the trial in full compliance with the Company's planned clinical trial procedures including with respect to data reporting, data monitoring, and the recommendations of various health authorities that the infirm patients who would participate in the trial not risk being exposed to COVID-19 at clinical trial sites. Such sites have been and may continue to be required to focus their limited resources on matters unrelated to our planned clinical trial, thereby decreasing availability, in whole or in part, for services to our planned clinical trial. The Company will not be able to initiate the trial until these risks have been significantly reduced or remediated. Although the Company believes, based on current projections of the pandemic, that it will be able to initiate the trial in the second half of 2020, the duration of the COVID-19 crisis is uncertain and, if the enumerated risks are not addressed, the Company may have to adjust its expectations as to trial initiation, including potentially initiating the trial in 2021, in order to accommodate the foregoing factors. In addition, the COVID-19 pandemic may affect the operations of the FDA and other health authorities, which could result in delays of reviews and approvals, including
21
with respect to dipraglurant and our other product candidates. Any such delays could increase the cost of our planned clinical trial and increase the uncertainty of receiving approval from the FDA for dipraglurant in PD-LID patients.
Depending on the duration of the COVID-19 crisis and continued negative impact on global economic activity, the Company may have to take additional measures that will have a negative impact on the Company business continuity and may experience certain liquidity restraints as well as incur impairments on its assets. The exact impact on the Company's activities in 2020 and thereafter cannot be reasonably predicted. However, based on the risk mitigation measures undertaken, the Company concluded that there is no material uncertainty that may cast a significant doubt upon the Company's ability to continue as a going concern.
Risks Related to our ADSs and Shares and our Prospective Nasdaq Listing
The market price for our shares and ADSs may be highly volatile and could decline significantly.
Our securities have a relatively small public float and may be less liquid and more volatile than securities of companies with broader public ownership. Factors affecting the market price of the securities, many of which are beyond our control, include:
- •
- low daily trading volume of our securities on the SIX Swiss Exchange and on Nasdaq;
- •
- announcements by us and developments that impact our financial results, business and partners;
- •
- fluctuations in our financial position or operating results;
- •
- changes in our business strategy and operations;
- •
- changes in our senior management team or Board of Directors;
- •
- changes in the recommendations of securities analysts regarding us or our industry;
- •
- investor need for liquidity;
- •
- investor assessment of the valuation of us and our competitors;
- •
- fluctuations in interest rates;
- •
- price and volume of the markets where our securities trade; and
- •
- future offerings of our securities.
In addition, securities markets in general have from time to time, and in particular in recent years, experienced significant price and volume fluctuations. Such fluctuations, as well as the economic environment as a whole, can have a substantial negative effect on the market price of our securities, regardless of our operating results or our financial position. Any such broad market fluctuations may adversely affect the trading price of our securities.
Compared to prior periods, we expect to incur increased costs as a result of operating as a company with securities listed in the United States in addition to Switzerland, and our senior management will be required to devote substantial time to new compliance initiatives and corporate governance practices.
ADSs representing our ordinary shares were listed on Nasdaq on January 29, 2020. As a company with securities listed in the United States in addition to Switzerland, and particularly after we no longer qualify as an emerging growth company, we expect to incur significant legal, accounting, and other expenses that we did not incur in previous periods. The Sarbanes-Oxley Act of 2002, the Dodd-Frank Wall Street Reform and Consumer Protection Act, the listing requirements of Nasdaq and other applicable securities rules and regulations impose various requirements on non-U.S. reporting public companies, including the establishment and maintenance of effective disclosure and financial controls
22
and corporate governance practices. Our senior management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations will increase our legal and financial compliance costs and will make some activities more time-consuming and costly.
Pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, or Section 404, we will be required to furnish a report by our senior management on our internal control over financial reporting beginning with our second annual report to be filed with the SEC. However, while we remain an emerging growth company, we will not be required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. To prepare for eventual compliance with Section 404, we will need to continue to dedicate internal resources, potentially engage outside consultants and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented, and implement a continuous reporting and improvement process for internal control over financial reporting. We anticipate that the process to document and evaluate our internal control over financial reporting will be both costly and challenging.
An active market may not develop in which investors can resell such ADSs.
Although our shares have traded on SIX since 2007, we cannot predict the extent to which an active market for ADSs representing our shares will develop or be sustained on Nasdaq, or how the development of such a market might affect the market price for our shares on SIX. The price at which ADSs representing our shares trade on Nasdaq may or may not be correlated with the price at which our shares trade on SIX.
Fluctuations in the exchange rate between the U.S. dollar and the Swiss franc may increase the risk of holding the ADSs.
Our share price is quoted on SIX in Swiss francs, while the ADSs trade on Nasdaq in U.S. dollars. Fluctuations in the exchange rate between the U.S. dollar and the Swiss franc may result in temporary differences between the value of the ADSs and the value of our shares, which may result in heavy trading by investors seeking to exploit such differences. In addition, as a result of fluctuations in the exchange rate between the U.S. dollar and the Swiss franc, the U.S. dollar equivalent of the proceeds that a holder of the ADSs would receive upon the sale in Switzerland of any shares withdrawn from the depositary receipts facility, and the U.S. dollar equivalent of any cash dividends paid in Swiss francs on our shares represented by the ADSs, could also decline.
Future sales, or the possibility of future sales, of a substantial number of ADSs representing our shares or our shares could adversely affect the price of such securities.
Future sales of a substantial number of ADSs representing our shares or our shares, or the perception that such sales will occur, could cause a decline in the market price of ADSs representing our shares and our shares. As of December 31, 2019, we had 32,848,645 shares issued and outstanding. There were no ADSs representing our shares outstanding as of such date. All of our outstanding shares are freely tradeable on SIX. In addition, other than shares held by our affiliates, all such shares are able to be deposited with the depositary in exchange for ADSs representing such shares, which ADSs will be freely tradeable. If holders sell substantial amounts of ADSs or shares in the respective public markets therefor, or if the market perceives that such sales may occur, the market price of ADSs representing our shares and our shares and our ability to raise capital through an issue of equity securities in the future could be adversely affected.
23
We have never paid dividends on our share capital, and we do not anticipate paying cash dividends in the foreseeable future.
We have never declared or paid cash dividends on our share capital. We do not anticipate paying cash dividends on our registered Shares in the foreseeable future. We currently intend to retain all available funds and any future earnings to fund the development and growth of our business. Any future determination to declare cash dividends will be made at the discretion of our Board of Directors, subject to compliance with applicable laws and covenants under current or future credit facilities, which may restrict or limit our ability to pay dividends and will depend on our financial condition, operating results, capital requirements, distributable profits and/or distributable reserves from capital contributions, general business conditions and other factors that our Board of Directors may deem relevant. As a result, capital appreciation, if any, of our securities will be your sole source of gain for the foreseeable future.
The exercise of equity incentive instruments granted under our equity incentive plan could dilute our share capital.
Pursuant to our existing equity incentive plan, ESCs with subscription rights to purchase shares, employee stock option plan, or ESOP, and warrants may be exercisable at prices below the market price of our shares at the time of exercise. To the extent that these instruments are exercised in the future, holders of our registered shares will be diluted. As of December 31, 2019, there were 11,906,248 shares reserved for issuance pursuant to subscription rights outstanding under our existing equity incentive plan, including 198,750 shares reserved for ESCs, 5,540,600 shares reserved for the ESOP, 6,166,898 shares reserved for warrants.
Holders of ADSs may not have the same voting rights as the holders of our shares and may not receive voting materials in time to be able to exercise their right to vote.
Except as described in Exhibit 15.2, holders of ADSs representing our shares are not able to exercise voting rights attaching to the underlying shares on an individual basis. Holders of ADSs representing our shares have appointed the depositary or its nominee as their representative to exercise the voting rights attaching to the shares underlying such ADSs. Holders of ADSs representing our shares may not receive voting materials in time to instruct the depositary to vote. The depositary may not be liable for any failure to carry out any instructions to vote, for the manner in which any vote is cast or for the effect of any such vote. As a result, holders of ADSs representing our shares may not be able to exercise voting rights and may lack recourse if such ADSs are not voted as requested. In addition, holders of ADSs representing our shares will not be able to call a shareholders' meeting.
Holders of ADSs representing our shares may not receive distributions on our shares underlying our ADSs or any value for them if it is illegal or impractical to make them available to such holders.
The depositary for ADSs representing our shares has agreed to pay to holders of such ADSs cash dividends or other distributions that it or the custodian receives on our shares after deducting its fees and expenses. Holders of ADSs representing our shares will receive these distributions in proportion to the number of our shares underlying their ADSs. However, in accordance with the limitations set forth in the deposit agreement, it may be unlawful or impractical for the depositary to make a distribution available to holders of ADSs representing our shares. We have no obligation to take any other action to permit the distribution of ADSs representing our shares, shares themselves, rights or anything else to holders of ADSs representing our shares. This means that holders of ADSs representing our shares may not receive any distributions that we make on our shares or any value from them if it is unlawful or impractical to make such distributions available to holders. These restrictions may negatively impact the trading value of ADSs representing our shares.
24
Holders of ADSs may be subject to limitations on transfer of their ADSs.
ADSs are transferable on the books of the depositary. However, the depositary may close its transfer books at any time or from time to time when it deems expedient in connection with the performance of its duties. In addition, the depositary may refuse to deliver, transfer, or register transfers of ADSs generally when our books or the books of the depositary are closed, or at any time if we or the depositary deems it advisable to do so because of any requirement of law or of any government or governmental body, or under any provision of the deposit agreement, or for any other reason in accordance with the terms of the deposit agreement.
ADSs holders may not be entitled to a jury trial with respect to claims arising under the deposit agreement, which could augur less favorable results to the plaintiff(s) in any such action.
The deposit agreement governing the ADSs representing our shares provides that holders and beneficial owners of ADSs irrevocably waive the right to a trial by jury in any legal proceeding arising out of or relating to the deposit agreement or the ADSs, including claims under federal securities laws, against us or the depositary to the fullest extent permitted by applicable law. If this jury trial waiver provision is prohibited by applicable law, an action could nevertheless proceed under the terms of the deposit agreement with a jury trial. To our knowledge, the enforceability of a jury trial waiver under the federal securities laws has not been finally adjudicated by a federal court. However, we believe that a jury trial waiver provision is generally enforceable under the laws of the State of New York, which govern the deposit agreement, by a court of the State of New York or a federal court, which have non-exclusive jurisdiction over matters arising under the deposit agreement, applying such law. In determining whether to enforce a jury trial waiver provision, New York courts and federal courts will consider whether the visibility of the jury trial waiver provision within the agreement is sufficiently prominent such that a party has knowingly waived any right to trial by jury. We believe that this is the case with respect to the deposit agreement and the ADSs. In addition, New York courts will not enforce a jury trial waiver provision in order to bar a viable setoff or counterclaim sounding in fraud or one which is based upon a creditor's negligence in failing to liquidate collateral upon a guarantor's demand, or in the case of an intentional tort claim (as opposed to a contract dispute), none of which we believe are applicable in the case of the deposit agreement or the ADSs. No condition, stipulation or provision of the deposit agreement or ADSs serves as a waiver by any holder or beneficial owner of ADSs or by us or the depositary of compliance with any provision of the federal securities laws. If you or any other holder or beneficial owner of ADSs brings a claim against us or the depositary in connection with matters arising under the deposit agreement or the ADSs, you or such other holder or beneficial owner may not be entitled to a jury trial with respect to such claims, which may have the effect of limiting and discouraging lawsuits against us and / or the depositary. If a lawsuit is brought against us and / or the depositary under the deposit agreement, it may be heard only by a judge or justice of the applicable trial court, which would be conducted according to different civil procedures and may augur different results than a trial by jury would have had, including results that could be less favorable to the plaintiff(s) in any such action, depending on, among other things, the nature of the claims, the judge or justice hearing such claims, and the venue of the hearing.
The rights accruing to holders of our shares may differ from the rights typically accruing to shareholders of a U.S. corporation.
We are organized under the law of Switzerland. The rights of holders of shares are governed by the laws of Switzerland and by our Articles of Association. These rights differ in certain respects from the rights of shareholders in typical U.S. corporations. See the sections entitled "Description of Share Capital and Articles of Association—Differences in Corporate Law" and "Description of Share Capital and Articles of Association—Articles of Association—Other Swiss Law Considerations" in the prospectus included in the Registration Statement on From F-1 (333-235554) that we filed with the
25
SEC on January 28, 2020, which are incorporated by reference herein for a description of the principal differences between the provisions of Swiss law applicable to us and, for example, the Delaware General Corporation Law relating to shareholders' rights and protections.
Claims of U.S. civil liabilities may not be enforceable against us.
We are incorporated under the law of Switzerland. Certain of our directors reside outside the United States. As a result, it may not be possible for investors to effect service of process within the United States upon such persons or to enforce judgments obtained in U.S. courts against them or us, including judgments predicated upon the civil liability provisions of the U.S. federal securities laws. The United States and Switzerland do not currently have a treaty providing for recognition and enforcement of judgments (other than arbitration awards) in civil and commercial matters. Consequently, a final judgment for payment given by a court in the United States, whether or not predicated solely upon U.S. securities laws, would not automatically be recognized or enforceable in Switzerland. In addition, uncertainty exists as to whether Swiss courts would entertain original actions brought in Switzerland against us or our directors predicated upon the securities laws of the United States or any state in the United States. Any final and conclusive monetary judgment for a definite sum obtained against us in U.S. courts would be treated by the courts of Switzerland. Whether these requirements are met in respect of a judgment based upon the civil liability provisions of the U.S. securities laws, including whether the award of monetary damages under such laws would constitute a penalty, is an issue for the court making such decision. If a Swiss court gives judgment for the sum payable under a U.S. judgment, the Swiss judgment will be enforceable by methods generally available for this purpose. These methods generally permit the Swiss court discretion to prescribe the manner of enforcement. As a result, U.S. investors may not be able to enforce against us or our certain of our directors, or certain experts named herein who are residents of Switzerland or countries other than the United States, any judgments obtained in U.S. courts in civil and commercial matters, including judgments under the U.S. federal securities laws.
We are organized under the laws of Switzerland and our jurisdiction of incorporation is Plan-les-Ouates, Geneva, Switzerland. Moreover, a number of our directors and executive officers and a number of directors of each of our subsidiaries are not residents of the United States, and all or a substantial portion of the assets of such persons are located outside the United States. As a result, it may not be possible for investors to effect service of process within the United States upon us or upon such persons or to enforce against them judgments obtained in U.S. courts, including judgments in actions predicated upon the civil liability provisions of the federal securities laws of the United States. We have been advised by our Swiss counsel that there is doubt as to the enforceability in Switzerland of original actions, or in actions for enforcement of judgments of U.S. courts, of civil liabilities to the extent predicated upon the federal and state securities laws of the United States. Original actions against persons in Switzerland based solely upon the U.S. federal or state securities laws are governed, among other things, by the principles set forth in the Swiss Federal Act on International Private Law of 1987, as amended, or PILA. This statute provides that the application of provisions of non-Swiss law by the courts in Switzerland shall be precluded if the result was incompatible with Swiss public policy. Also, mandatory provisions of Swiss law may be applicable regardless of any other law that would otherwise apply.
Switzerland and the United States do not have a treaty providing for reciprocal recognition of and enforcement of judgments in civil and commercial matters. The recognition and enforcement of a judgment of the courts of the United States in Switzerland is governed by the principles set forth in the PILA. This statute provides in principle that a judgment rendered by a non-Swiss court may be enforced in Switzerland only if:
- •
- the non-Swiss court had jurisdiction pursuant to the PILA;
26
- •
- the judgment of such non-Swiss court has become final and non-appealable;
- •
- the judgment does not contravene Swiss public policy;
- •
- the court procedures and the service of documents leading to the judgment were in accordance with the due process of law; and
- •
- no proceeding involving the same position and the same subject matter was first brought in Switzerland, or adjudicated in Switzerland, or was earlier adjudicated in a third state and this decision is recognizable in Switzerland.
We currently qualify as a foreign private issuer and, as a result, we are not subject to U.S. proxy rules and are subject to reporting obligations under the Exchange Act, that, to some extent, are more lenient and less frequent than those of a U.S. domestic public company.
Because we qualify as a foreign private issuer under the Exchange Act, we are exempt from certain provisions of the Exchange Act that are applicable to U.S. domestic public companies, including (i) the sections of the Exchange Act regulating the solicitation of proxies, consents, or authorizations in respect of a security registered under the Exchange Act; (ii) the sections of the Exchange Act requiring insiders to file public reports of their stock ownership and trading activities and liability for insiders who profit from trades made in a short period of time; and (iii) the rules under the Exchange Act requiring the filing with the SEC of quarterly reports on Form 10-Q containing unaudited financial and other specified information or current reports on Form 8-K upon the occurrence of specified significant events. In addition, foreign private issuers are not required to file their annual report on Form 20-F until 120 days after the end of each fiscal year, while U.S. domestic issuers that are accelerated filers are required to file their annual report on Form 10-K within 75 days after the end of each fiscal year. Foreign private issuers also are exempt from Regulation Fair Disclosure, aimed at preventing issuers from making selective disclosures of material information. As a result of the above, you may not have the same protections afforded to shareholders of companies that are not foreign private issuers.
As a foreign private issuer and as permitted by the listing requirements of Nasdaq, we follow certain Swiss corporate governance rules instead of certain corporate governance requirements of Nasdaq.
As a foreign private issuer, we follow certain of our home country corporate governance rules instead of certain corporate governance requirements of Nasdaq. For example, we are exempt from Nasdaq regulations that require a listed U.S. company to:
- •
- have a majority of the board of directors consist of independent directors as such term is defined by Nasdaq;
- •
- have nominating and compensations committees that are fully independent, as defined by Nasdaq;
- •
- solicit proxies and provide proxy statements for all shareholder meetings; and seek shareholder approval for the implementation of certain equity compensation plans and issuances of shares.
For an overview of our corporate governance principles, including those which comply with certain of the requirements above, see the section entitled "Description of Share Capital and Articles of Association" in the prospectus included in the Registration Statement on From F-1 (333-235554) that we filed with the SEC on January 28, 2020. In accordance with our Nasdaq listing, our Audit Committee is required to comply with the provisions of Section 301 of the Sarbanes-Oxley Act of 2002 and Rule 10A-3 of the Exchange Act, both of which also are applicable to Nasdaq-listed U.S. companies. To the extent we follow Swiss corporate governance practices instead of Nasdaq governance requirements applicable to domestic issuers, you may not have the same protections afforded to shareholders of companies that are subject to these Nasdaq requirements.
27
We may lose our foreign private issuer status, which would then require us to comply with the Exchange Act's domestic reporting regime and Nasdaq's corporate governance requirements applicable to a domestic issuer, and cause us to incur significant incremental legal, accounting and other expenses.
Although we currently qualify as a foreign private issuer, in order to maintain this status, either (a) a majority of our shares, including shares represented by ADSs, must be either directly or indirectly owned of record by non-residents of the United States or (b)(i) a majority of our executive officers or directors must not be U.S. citizens or residents, (ii) more than 50 percent of our assets must be located outside of the United States and (iii) our business must be administered principally outside of the United States. If we lose our status as a foreign private issuer, we would be required to comply with the Exchange Act reporting and other requirements applicable to U.S. domestic issuers, which are more detailed and extensive than the requirements for foreign private issuers. We would also be required to make changes in our corporate governance practices in accordance with various SEC and Nasdaq rules. The regulatory and compliance costs to us under U.S. securities laws if we are required to comply with the reporting requirements applicable to a U.S. domestic issuer will be significantly higher than the costs that we would incur as a foreign private issuer. As a result, we expect that the loss of foreign private issuer status would increase our legal and financial compliance costs and would make some activities highly time consuming and costly.
We are an "emerging growth company," and we cannot be certain if the reduced reporting requirements applicable to "emerging growth companies" will make ADSs representing our shares or our shares less attractive to investors.
We are an "emerging growth company" as defined in the JOBS Act. For as long as we continue to be an emerging growth company, we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies, including not being required to comply with the auditor attestation requirements of Section 404, exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. We may take advantage of these exemptions until we are no longer an emerging growth company. We could be an emerging growth company for up to five years, although circumstances could cause us to lose that status earlier, including if the aggregate market value of ADSs representing our shares and our shares held by non-affiliates exceeds $700 million as of any June 30 (the end of our second fiscal quarter) before that time, in which case we would no longer be an emerging growth company as of the following December 31 (our fiscal year-end). We cannot predict if investors will find ADSs representing our shares or our shares less attractive because we may rely on these exemptions. If some investors find such securities less attractive as a result, there may be a less active trading market for ADSs representing our shares or our shares and the price of such securities may be more volatile.
If we fail to maintain an effective system of internal control over financial reporting, we may not be able to accurately report our financial results or prevent fraud. As a result, shareholders could lose confidence in our financial and other public reporting, which would harm our business and the trading price of ADSs representing our shares or our shares.
Effective internal controls over financial reporting are necessary for us to provide reliable financial reports and, together with adequate disclosure controls and procedures, are designed to prevent fraud. Any failure to implement required new or improved controls, or difficulties encountered in their implementation could cause us to fail to meet our reporting obligations. In addition, any testing by us conducted in connection with Section 404, or any subsequent testing by our independent registered public accounting firm, may reveal deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses or that may require prospective or retroactive changes to our financial statements or identify other areas for further attention or improvement. Inadequate internal
28
controls could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of ADSs representing our shares or our shares.
Management will be required to assess the effectiveness of our internal controls annually. However, for as long as we are an "emerging growth company" under the JOBS Act, our independent registered public accounting firm will not be required to attest to the effectiveness of our internal controls over financial reporting pursuant to Section 404. An independent assessment of the effectiveness of our internal controls could detect problems that our management's assessment might not. Undetected material weaknesses in our internal controls could lead to financial statement restatements requiring us to incur the expense of remediation and could also result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our financial statements.
If securities or industry analysts do not publish research, or publish inaccurate or unfavorable research, about our business, the price of ADSs representing our shares or our shares and the trading volume thereof could decline.
The trading market for ADSs representing our shares and our shares will depend in part on the research and reports that securities or industry analysts publish about us or our business. Since we did not undertake a primary offering of ADSs representing our shares in connection with the listing of ADSs on Nasdaq, we do not anticipate that many or any industry analysts in the United States will publish such research and reports in the United States about our shares or ADSs. If no or too few securities or industry analysts commence or continue coverage on us, the trading price for ADSs representing our shares and our shares could be affected. If one or more of the analysts who may eventually cover us downgrade such ADSs or shares or publish inaccurate or unfavorable research about our business, the trading price of ADSs representing our shares or our shares would likely decline. If one or more of these analysts cease coverage of us or fail to publish reports on us regularly, demand for ADSs representing our shares or our shares could decrease, which might cause the price of such securities and the trading volume thereof to decline.
If we are a passive foreign investment company, or PFIC, for U.S. federal income tax purposes, the consequences to U.S. holders of our shares or ADSs representing our shares may be adverse.
Based on our analysis of our income, assets, activities and market capitalization for our taxable year ended December 31, 2018, we believe that we were classified as a "passive foreign investment company," or PFIC, for our taxable year ended December 31, 2019. We intend to annually provide U.S. Holders, upon request, a "PFIC Annual Information Statement", with the information required to allow U.S. Holders to make a "qualified electing fund" election, or "QEF Election" for United States federal income tax purposes. The application of the PFIC rules is subject to uncertainty in several respects, and therefore, no assurances can be provided with respect to our PFIC status for our taxable year ended December 31, 2018 or with regard to our PFIC status in the past or in the future. Under the Code, a non-U.S. company will be considered a PFIC for any taxable year in which (1) 75% or more of its gross income consists of passive income or (2) 50% or more of the average quarterly value of its assets consists of assets that produce, or are held for the production of, passive income. For purposes of these tests, passive income includes dividends, interest, gains from the sale or exchange of investment property and certain rents and royalties. In addition, for purposes of the above calculations, a non-U.S. corporation that directly or indirectly owns at least 25% by value of the shares of another corporation is treated as if it held its proportionate share of the assets and received directly its proportionate share of the income of such other corporation. If we are a PFIC for any taxable year during which a U.S. Holder (as defined below under "Material Income Tax Considerations—Material U.S. Federal Income Tax Considerations for U.S. Holders") holds our shares or ADSs representing our shares, we will continue to be treated as a PFIC with respect to such U.S. Holder in all succeeding
29
years during which the U.S. Holder owns the shares or ADSs representing our shares, regardless of whether we continue to meet the PFIC test described above, unless the U.S. Holder makes a specified election once we cease to be a PFIC. If we are classified as a PFIC for any taxable year during which a U.S. Holder holds our shares or ADSs representing our shares, the U.S. Holder may be subject to adverse tax consequences regardless of whether we continue to qualify as a PFIC, including ineligibility for any preferred tax rates on capital gains or on actual or deemed dividends, interest charges on certain taxes treated as deferred, and additional reporting requirements. For further discussion of the PFIC rules and the adverse U.S. federal income tax consequences in the event we are classified as a PFIC, see "Item 10.E—Taxation."
If a United States person is treated as owning at least 10% of our shares, such holder may be subject to adverse U.S. federal income tax consequences.
If a U.S. Holder is treated as owning, directly, indirectly or constructively, at least 10% of the value or voting power of our shares (directly or in the form of ADSs representing our shares), such U.S. Holder may be treated as a "United States shareholder" with respect to each "controlled foreign corporation" in our corporate group, if any. If such group includes one or more U.S. subsidiaries, certain of our non-U.S. subsidiaries could be treated as controlled foreign corporations, regardless of whether we are treated as a controlled foreign corporation. A United States shareholder of a controlled foreign corporation may be required to annually report and include in its U.S. taxable income its pro rata share of "Subpart F income," "global intangible low-taxed income" and investments in U.S. property by controlled foreign corporations, regardless of whether we make any distributions. An individual that is a United States shareholder with respect to a controlled foreign corporation generally would not be allowed certain tax deductions or foreign tax credits that would be allowed to a United States shareholder that is a U.S. corporation. Failure to comply with these reporting obligations may subject a United States shareholder to significant monetary penalties and may prevent the statute of limitations with respect to such shareholder's U.S. federal income tax return for the year for which reporting was due from starting. We cannot provide any assurances that we will assist our investors in determining whether any of our non-U.S. subsidiaries are treated as a controlled foreign corporation or whether such investor is treated as a United States shareholder with respect to any of such controlled foreign corporations. Further, we cannot provide any assurances that we will furnish to any United States shareholder information that may be necessary to comply with the reporting and tax paying obligations described in this risk factor. U.S. Holders should consult their tax advisors regarding the potential application of these rules to their investment in our shares or ADSs representing our shares.
Tax authorities may disagree with our positions and conclusions regarding certain tax positions, resulting in unanticipated costs, taxes or non-realization of expected benefits.
A tax authority may disagree with tax positions that we have taken, which could result in increased tax liabilities. A tax authority could assert that we are subject to tax in a jurisdiction where we believe we have not established a taxable connection, often referred to as a "permanent establishment" under international tax treaties, and such an assertion, if successful, could increase our expected tax liability in one or more jurisdictions. A tax authority may take the position that material income tax liabilities, interest and penalties are payable by us, in which case, we expect that we might contest such assessment. Contesting such an assessment may be lengthy and costly and if we were unsuccessful in disputing the assessment, the result could increase our anticipated effective tax rate.
Item 4. Information on the Company.
A. History and Development of the Company
Our legal and commercial name is Addex Therapeutics Ltd. We are a Swiss limited company organized under the laws of Switzerland. We were founded in 2002 with an indefinite duration. We are
30
currently registered in Geneva, Switzerland. Our principal executive offices are located at Chemin des Mines 9, CH-1202 Geneva, Switzerland. Our telephone number is +41 884 1555. Investors should contact us for any inquiries through the address and telephone number of our principal executive office, or via our U.S. office at 650 California Street in San Francisco, CA, telephone number +1 (415) 429-2591. We maintain a website at www.addextherapeutics.com. The reference to our website is an inactive textual reference only and the information contained in, or that can be accessed through, our web site is not a part of this Annual Report on Form 20-F. The SEC maintains an Internet site that contains reports and other information that we file electronically with the SEC at http:// www.sec.gov.
B. Business Overview
We are a clinical-stage pharmaceutical company focused on the development and commercialization of an emerging class of novel orally available small molecule drugs known as allosteric modulators. Allosteric modulators target a specific receptor or protein and alter the effect of the body's own signaling molecules on their target through a novel mechanism of action. These innovative small molecule drug candidates offer several potential advantages over conventional non-allosteric molecules and may offer an improved therapeutic approach to existing drug treatments. To date, our research and development efforts have been primarily focused on building a portfolio of proprietary drug candidates based on our allosteric modulator development capability. The allosteric modulator principle has broad applicability across a wide range of biological targets and therapeutic areas, but our primary focus is on G-protein coupled receptors, or GPCR, targets implicated in neurological diseases, where we believe there is a clear medical need for new therapeutic approaches.
Using our allosteric modulator discovery capabilities, we have developed a pipeline of proprietary clinical and preclinical stage drug candidates. We or our partners are developing these clinical and preclinical stage proprietary drug candidates for diseases for which there are no approved therapies or where improved therapies are needed. These include PD-LID, non-parkinsonian dystonia, addiction, epilepsy, Charcot-Marie-Tooth type 1A neuropathy, or CMT1A, and other neurodegenerative diseases. Some of these indications are classified as rare diseases that may allow for orphan drug designation by regulatory agencies in major commercial markets, such as the United States, Europe and Japan. Orphan drug designation may entitle the recipient to benefits, in the jurisdiction granting the designation, such as market exclusivity following approval and assistance in clinical trial design, a reduction in user fees or tax credits related to development expenses.
We are developing our lead drug candidate, dipraglurant, as an mGlu5 NAM, for the treatment of PD-LID. We are planning to initiate a placebo-controlled Phase 2b/3 pivotal clinical trial of dipraglurant in PD-LID patients in the second half of 2020, pending removal of governmental restrictions and lessening of the impact of the global coronavirus pandemic on the U.S. healthcare system, which delayed our previously anticipated initiation in the first quarter of 2020. The study will be conducted at approximately 50 sites in the United States and will target enrolment of approximately 140 patients. We have received orphan drug designation from the United States Food and Drug Administration, or FDA, for dipraglurant in PD-LID and expect to report topline results in the second quarter of 2022. In parallel, dipraglurant's therapeutic use in dystonia is being investigated in preclinical models. We are also conducting a research program under our strategic partnership with Indivior UK Limited, or Indivior, to discover novel orally available GABAB PAMs. We are currently in late lead optimization and expect to deliver a drug candidate by the end of 2020 for Indivior to develop for addiction. Under terms of the agreement with Indivior, we have the right to select drug candidates for development in certain exclusive indications outside of addiction. We plan to develop our selected drug candidate in CMT1A, an indication that has been clinically validated with baclofen, an orthosteric agonist of GABAB.
Allosteric modulators have broad applicability for many clinically validated GPCR targets which are implicated in multiple therapeutic indications. We intend to continue to leverage our scientific
31
expertise in allosteric modulation and our proprietary technology platform to discover novel drug candidates for the treatment of neurological diseases.
Based on our expertise in allosteric modulation, our goal is to build a leading neuroscience company focused on conditions where current treatment options are limited and where unmet medical needs exist. Our business strategy includes the possibility of entering into collaborative arrangements with third parties to complete the development and commercialization of our proprietary drug candidates, such as our partnership with Janssen Pharmaceuticals, Inc., or Janssen, a subsidiary of Johnson & Johnson, for ADX71149 and our strategic partnership with Indivior for GABAB PAM. We cannot forecast with any degree of certainty which proprietary products or indications, if any, will be subject to future collaborative arrangements, in whole or in part, and how such arrangements would affect our development plan or capital requirements. To date, we have secured grants and other funding from: The Michael J. Fox Foundation for Parkinson's Research, or MJFF, for the development of dipraglurant for the treatment of PD-LID; the National Institute of Drug Abuse, or NIDA, to generate important data on the role of GABAB in addiction; the Swiss Innovation Agency, or Innosuisse, to advance our understanding of the role of our drug candidates in neurodegenerative and psychiatric diseases; and the Eurostars Joint Programme, or Eurostars to identify novel drug candidates on mGlu7 NAM for PTSD. As we advance our clinical and preclinical programs, we will continue to apply for subsidies, grants and government or agency sponsored studies that could offset or reduce our development costs.
The development and commercialization of drugs is highly competitive. We compete with a variety of multinational pharmaceutical companies and specialized pharmaceutical companies, including products approved for marketing and/or product candidates under development, for each of the product candidates and each of the indications for which we are developing. Furthermore, Government authorities in the United States, at the federal, state and local levels, and in other countries, extensively regulate, among other things, the research, development, testing, manufacture, packaging, storage, recordkeeping, labeling, advertising, promotion, distribution, marketing, import and export of pharmaceutical products, such as those we are developing. The processes for obtaining regulatory approvals in the United States and in foreign countries, along with subsequent compliance with applicable statutes and regulations, require the expenditure of substantial time and financial resources.
Research and Development Portfolio
Using our allosteric modulator platform and drug discovery and development expertise, we have established a portfolio of clinical and preclinical programs, both internally and with partners.
Internally Developed Product Candidates
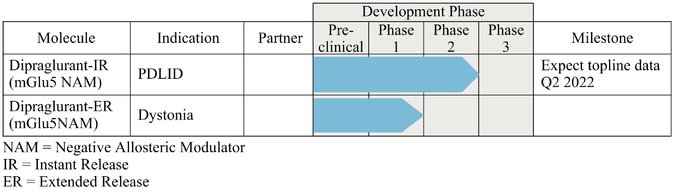
Dipraglurant, for the treatment of levodopa-induced dyskinesia associated with Parkinson's disease. We are developing dipraglurant as a novel orally available mGlu5 NAM for the treatment of PD-LID.
32
PD-LID is a disease with significant commercial opportunity as improved therapies are needed. We believe that, subject to regulatory approval, dipraglurant may offer an innovative and differentiated treatment approach from existing therapies. In a 28 day Phase 2a placebo-controlled clinical trial, conducted in the United States and Europe, in patients with PD-LID, dipraglurant met its primary end point, was generally well tolerated and no clinically significant abnormalities of safety monitoring parameters occurred. In addition, at Day 1 and Day 14, dipraglurant showed statistically significant effects on PD-LID clinical symptoms, as measured using mAIMs. However, an increasing placebo response resulted in the effect of dipraglurant on PD-LID clinical symptoms not showing statistical significance at Day 28. We have substantially completed the preparation of dipraglurant to start registration studies in PD-LID and expect to initiate a Phase 2b/3 placebo-controlled pivotal clinical trial of dipraglurant in PD-LID patients in the second half of 2020, pending removal of governmental restrictions and lessening of the impact of the global coronavirus pandemic on the U.S. healthcare system, which delayed our previously anticipated initiation in the first quarter of 2020. The study will be conducted at approximately 50 sites in the United States and will target enrolment of approximately 140 patients. We have also received orphan drug designation from the US FDA for dipraglurant in PD-LID and expect to report topline results in the second quarter of 2022.
Dipraglurant, for the treatment of non-Parkinsonian dystonia. We are developing an extended release formulation of dipraglurant as a novel orally available NAM for the treatment of dystonia. At the appropriate time, we expect to be able to start a Phase 2a proof of concept clinical trial. There are many types of dystonias that present a commercial opportunity for dipraglurant. We believe that subject to regulatory approval, dipraglurant may offer an innovative and differentiated treatment approach for multiple types of dystonia. We are currently completing preclinical evaluations of dipraglurant in preclinical models of dystonia.
Externally Developed Out-licensed Product Candidate
ADX71149 (mGlu2 PAM) for the treatment of epilepsy. Our partnered drug candidate, ADX71149 is a novel orally active mGlu2 PAM. Our partner, Janssen, has completed Phase 1 and two Phase 2a clinical trials in schizophrenia and anxious depression, respectively, and is currently evaluating future development in epilepsy. Under our agreement with Janssen, Janssen is responsible for financing the development and commercialization, if any, of ADX71149.
Material Internal Research Programs

GABAB PAM for the treatment of addiction. Our partner, Indivior, has licensed worldwide rights to our GABAB PAM program and is responsible for all development, manufacture and commercialization of any selected GABAB PAM drug candidates. Under the agreement, we are responsible for executing
33
a research program funded by Indivior to discover novel drug candidates. Indivior has the right to select GABAB PAM drug candidates from our research program. We expect to deliver drug candidates for selection in 2020. We believe that addiction is an indication with a significant commercial opportunity. Existing therapies often do not provide effective control of symptoms or have side effects that discourage adherence. Subject to regulatory approval, we believe that GABAB PAM may offer an innovative and differentiated treatment approach from existing therapies and may provide benefit to patients.
GABAB PAM for the treatment of CMT1A. Our license agreement with Indivior provides for a funded research program, under which we have the right to select drug candidates for exclusive development in certain indications outside of addiction, including CMT1A, a rare disease indication. We plan to pursue orphan drug designation for a selected drug candidate for CMT1A. We believe an oral small molecule GABAB PAM with a once-a-day dosing and without the adherence-limiting side effects of baclofen, which is currently used off label, could bring benefit to patients and consequently present a strong commercial opportunity for us.
mGlu7 NAM for the treatment of PTSD: We are developing mGlu7 NAMs as a novel orally available treatment to reduce fear memory in PTSD. This is a disorder that can lead to intense fear and anxiety. Current medication is unspecific and ineffective, with a number of side effects. By selectively targeting mGlu7 with NAMs, the brain circuitries involved in fear and anxiety can be more precisely modulated, potentially resulting in a more focused response and fewer side effects than current therapeutic approaches. Subject to regulatory approval, we believe our mGlu7 NAM may offer an innovative and differentiated treatment approach from existing therapies. We are in late lead optimization and a consortium led by us has been awarded a €4.85 million grant from the Eurostars Joint Programme to advance the program to drug candidate stage.
Early Stage Internal Research Programs
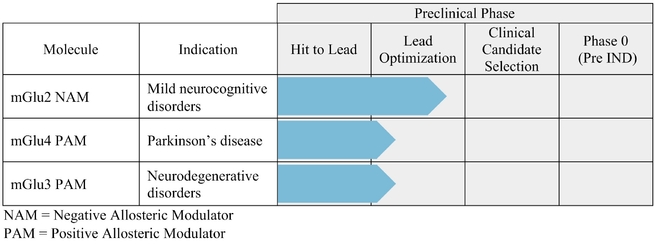
mGlu2 NAM for the treatment of mNCD: We are developing mGlu2 NAM as a novel orally available treatment for mNCD associated with Alzheimer's disease, Parkinson's disease and depressive disorders. We are currently optimizing multiple chemical series of highly selective mGlu2 NAMs offering advanced compounds at the late stage of lead optimization.
mGlu4 PAM for the treatment of Parkinson's disease: We are developing mGlu4 PAM as a novel orally available treatment for Parkinson's disease. We are currently optimizing multiple chemical series of highly selective mGlu4 PAMs with compounds in early lead optimization.
34
mGlu3 PAM for the treatment of neurodegenerative disorders: We are developing mGlu3 PAM as a novel orally available treatment for neurodegenerative disorders. We are currently optimizing multiple chemical series of highly selective mGlu3 PAMs, with compounds in early lead optimization.
Introduction to Allosteric Modulation
Disease and the Role of Proteins
Proteins are complex biological molecules that have many structural and functional roles in the body. They are critical components in the lines of communication between the cells of the body known as signaling pathways. It is now recognized that signaling pathways are altered in many disease states through changes in the function of essential proteins underlying the series of cellular events required for normal biological activity. Most drug treatments are focused on modifying these biological signaling pathways by altering the activity profile of selected proteins suspected to play a key role in the manifestation of a particular disease. The major proteins targeted in drug discovery include membrane-bound receptors, such as G-protein coupled receptors, or GPCRs, or ionotropic (ion channels) receptors and enzymes.
GPCRs as Drug Targets
GPCRs are the largest family of integral membrane receptors, accounting for approximately 3-4% of the human genome. GPCRs have evolved to recognize a range of endogenous stimuli and act to transmit messages encoded in stimuli from the exterior to the interior of the cell. The ubiquitous cell surface distribution of GPCRs and their involvement in virtually all biological processes have made GPCRs extremely attractive targets for drug development. In fact, most currently marketed drugs act on GPCRs, emphasizing their importance for drug development.
Conventional Approaches to GPCR Drug Discovery
The drug discovery process involves the design of molecules that interact with a target with high specificity and efficacy. Traditional approaches to drug discovery focus on mimicking or inhibiting the actions of the endogenous activator for a target receptor. Conventionally, this has been done by the design and chemical synthesis of small molecule agonists (activators) or antagonists (inhibitors) that act in a competitive manner through interaction with the same binding site as the endogenous activator.
Competitive agonists and antagonists must have a sufficiently high affinity for the target receptor to displace the endogenous activator and must be maintained at a sufficiently high concentration in the region of the receptor in order to exert an effect. Under these conditions, agonists will induce an activated state and antagonists will induce an inactivated state, and in both states, receptors will not be responsive to natural fluctuations in the levels of endogenous activator, thereby interfering with normal physiological signaling.
Although this approach has historically yielded a number of blockbuster drugs, such as Clopidogrel, or Plavix, Salmeterol, or Serevent, and Aripiprazole, or Abilify, significant challenges remain with respect to the continued development of therapeutically useful GPCR competitive agonists or antagonists due to either lack of receptor selectivity or undesirable side effects.
Allosteric Modulators as GPCR Drugs
In contrast to competitive orthosteric compounds, allosteric modulators of GPCRs interact with binding sites that are topographically distinct from the binding site of the endogenous activator, and therefore do not compete with the endogenous activator. This means that allosteric modulators do not activate or inhibit receptors on their own, but only in the presence of an endogenous activator do they enhance (positively modulate) or inhibit (negatively modulate) the natural physiological activity of the
35
receptor. Consequently, allosteric modulators offer the possibility to preserve normal physiological receptor function while controlling pathologic activity caused by over- or under-activation of an endogenous receptor.
We believe that by applying this non-competitive allosteric modulator approach, we may be able to produce efficacious drug candidates with potentially beneficial properties:
- •
- Novel drug class: Allosteric modulators are a novel class of orally
available small molecule drug candidates with chemical structures unrelated to that of competitive agonist or antagonist drugs and, as such, represent drug candidates with a high potential for
composition of matter patent protection.
- •
- Superior receptor sub-type selectivity: The binding site for an endogenous
activator is in general, highly conserved within a GPCR family and achieving receptor subtype selectivity within a family has not always been possible for competitive agonists. The best
examples of this are the muscarinic acetylcholine and the metabotropic glutamate receptor families, for which developing competitive, sub-type selective agonists has not been successful thus far. In
contrast, allosteric modulator binding sites, being independent of endogenous stimuli, have evolved with a much higher structural diversity than endogenous activator binding sites and consequently
offer the potential for the synthesis of drug candidates with much greater sub-type selectivity.
- •
- Ability to discover small molecule drugs for a greater number of GPCR
targets: Several GPCR targets are currently thought to be beyond the reach of conventional competitive drug discovery approaches due to
the complexity of the interaction of the endogenous activator with the receptor; including, for example certain peptides, high molecular weight hormones and lipids. For these targets, the allosteric
modulator approach represents a novel pathway to develop orally active activator or inhibitor small molecules.
- •
- Ability to re-address well characterized and clinically validated GPCR targets where the pharmaceutical industry has
exhausted competitive compound drug discovery approaches: Allosteric modulator drug candidates offer a promising way to revisit these targets,
providing novel small molecules while capitalizing on the existing knowledge on well-validated GPCR targets.
- •
- Improved safety: Allosteric modulators control pathologic activity while
preserving natural physiological signaling activity due to their lack of effect in the absence of the endogenous activator. In addition, they show no or less tolerance to their effect when chronically
administered, thereby not requiring increased dosing as is often the case with competitive compounds. Together with their superior selectivity, these allosteric modulator compounds have the potential
for improved safety compared to their competitive analogs.
- •
- Clinical use in combination: Given that allosteric modulators target different binding sites to conventional agonists or antagonists, allosteric modulator drugs may find clinical utility in combination therapies for certain clinical indications.
Orally available brain penetrant small molecule drug candidates that are highly selective for their therapeutic targets and interact with their target in a modulatory manner preserving natural physiological signaling are particularly suitable for neurological diseases.
Our Platform and Competitive Positioning in Allosteric Modulation
We believe that we have a recognized expertise in allosteric modulator R&D. Since our inception in 2002, we have focused exclusively on allosteric modulation drug discovery and development. We have engaged leading experts in the field of allosteric modulation who have years of experience in both industry and academia, including Robert Lutjens, our head of discovery biology and Jean-Philippe Rocher, our head of discovery chemistry.
36
We have established a unique chemical library of more than 70,000 allosteric modulator compounds, in addition to highly specialized biological systems that are required for the identification and screening of high affinity, orally active small molecule allosteric modulators. Combined with our allosteric modulator library, these high-throughput detection systems have enabled us to build what we believe to be the largest clinical and pre-clinical portfolio of proprietary allosteric modulator compounds.
Dipraglurant, an mGlu5 NAM, is our lead development compound. It is in late-stage development and scheduled to start a placebo-controlled Phase 2b/3 pivotal clinical trial in PD-LID patients in the second half of 2020, pending removal of governmental restrictions and lessening of the impact of the global coronavirus pandemic on the U.S. healthcare system, which delayed our previously anticipated initiation in the first quarter of 2020. Topline results are expected in the second quarter of 2022. ADX71149, a mGlu2 PAM licensed to Janssen, has completed Phase 1 and two Phase 2a clinical trials demonstrating good tolerability. Its further development is being evaluated by our partner, Janssen, with a key focus on evaluating it for the treatment of epilepsy. Furthermore, we believe the depth of our in-house discovered portfolio further demonstrates our expertise in the allosteric modulation field.
Our Strategy
Based on our expertise in allosteric modulation, our goal is to build a leading neuroscience company focused on conditions where current treatment options are limited and where unmet medical needs exist. Since our inception, we have raised an aggregate of CHF 325 million in equity financing and generated an aggregate of CHF 57 million of revenues, which we have used to build our proprietary allosteric modulator technology platform and our pipeline of drug products and candidates. We have secured financing from leading U.S. healthcare investors, including New Enterprise Associates, New Leaf Venture Partners and CAM Capital. The key elements of our strategy include the following:
- •
- Continue to advance dipraglurant for the treatment of PD-LID. We are
planning to initiate a placebo-controlled Phase 2b/3 pivotal clinical trial of dipraglurant in PD-LID patients in the second half of 2020, pending removal of governmental restrictions and
lessening of the impact of the global coronavirus pandemic on the U.S. healthcare system, which delayed our previously anticipated initiation in the first quarter of 2020. The study will be conducted
at approximately 50 sites in the United States and will target enrollment of approximately 140 patients. We expect to report data in the second quarter of 2022.
- •
- Pursue additional indications for dipraglurant. We believe dipraglurant has
applications outside of PD-LID including non-parkinsonian dystonia. We are currently evaluating dipraglurant in preclinical models of dystonia and will evaluate pursing the development of dipraglurant
in this and other indications.
- •
- Continue to advance our GABAB PAM research programs. We are
currently executing a research program funded by Indivior to discover novel GABAB PAM drug candidates. We are currently in late lead optimization and expect to deliver drug candidates in
2020 for our partner, Indivior, to develop for addiction and for Addex's internal program focused on CMT1A.
- •
- Continue to advance our preclinical programs. We are currently conducting
lead optimization activities on a number of highly innovative programs and aim to select drug candidates for at least one program in 2020.
- •
- Develop a targeted commercialization strategy for dipraglurant. We invented dipraglurant and therefore own all commercial rights. As we advance dipraglurant through its registration program for PD-LID toward regulatory approval, we will evaluate options for commercialization. These options may include building our own internal sales force, entering into a joint marketing
37
- •
- Leverage our expertise and experience in allosteric modulation. We are
focused on developing allosteric modulators discovered from our proprietary discovery technology platform. We will continue to invest in our platform and pursue novel drug targets where we believe our
platform can provide novel drug candidates where an unmet medical need exists.
- •
- Pursue collaborative arrangement with other pharmaceutical companies. We may seek collaborative arrangements with third parties to advance the development and commercialization of our drug candidates.
arrangement with another pharmaceutical company, or out-licensing certain rights to another pharmaceutical company.
Our Strengths
Our current strategic focus is the development of certain proprietary drug candidates in our existing portfolio. We believe that we have a number of competitive advantages that distinguish us from our competitors.
- •
- Robust in-house discovered pipeline with lead product candidate advancing into pivotal registration clinical
trials. We have established an in-house discovered pipeline of eight clinical and preclinical proprietary programs. Internally, these programs
include dipraglurant for the treatment of PD-LID and dystonia, GABAB PAM for the treatment of CMT1A. Partnered programs include ADX71149 for the treatment of epilepsy which is licensed to
Janssen and GABAB PAM for the treatment of addiction which is licensed to Indivior. We have also received orphan drug designation from the US FDA for dipraglurant in PD-LID and we expect
to initiate a placebo-controlled Phase 2b/3 pivotal clinical trial of dipraglurant in PD-LID patients in the second half of 2020, pending removal of governmental restrictions and lessening of
the impact of the global coronavirus pandemic on the U.S. healthcare system, which delayed our previously anticipated initiation in the first quarter of 2020.
- •
- High value partnerships. In January 2018, we entered into a strategic
partnership and license agreement with Indivior for the discovery, development and commercialization of novel GABAB PAM compounds for the treatment of addiction and other neurological
diseases. Under the agreement, Indivior has sole responsibility for, including the financing of, development of selected compounds under the agreement through preclinical and clinical trials, as well
as registration procedures and commercialization, if any. We are responsible for executing a research program funded by Indivior to discover novel GABAB PAM drug candidates. Under the
terms of the agreement, we are eligible for a minimum of $4 million in research funding and payment of $330 million on successful achievement of pre-specified clinical, regulatory and
commercial milestones and tiered royalties on the level of net sales from high single digits up to low double-digit. In December 2004, we entered into a collaboration and license agreement with
Janssen for the discovery, development and commercialization of novel mGlu2 PAM compounds for the treatment neurological diseases. ADX71149 is one of the drug candidates discovered and selected for
development by Janssen and under the agreement Janssen has sole responsibility for, including the financing of, development of selected compounds under the agreement through preclinical and clinical
trials, as well as registration procedures and commercialization, if any. Under the terms of the agreement, we are eligible for payments of $109 million upon successful achievement of
pre-specified clinical and regulatory milestones and a flat low double-digit royalty on net sales.
- •
- Global leadership in an emerging drug class. Allosteric modulators are an emerging class of oral small molecule therapeutics that have the potential to become drug candidates for a number of disease indications. We believe that our expertise, unique knowledge-based library and proprietary biological screening tools make us a leader in allosteric modulation-based drug
38
- •
- Focus on rare diseases with high unmet medical needs. We plan to focus on
therapeutic indications with substantial unmet or undermet medical needs and commercial potential, including indications considered rare diseases for the purpose of orphan drug designation by the FDA.
If we acquire orphan designation for a drug candidate, the designation would provide benefits such as market exclusivity after approval, and potential reductions in user fees or tax credits related to
development expenses. In addition, as we advance drug products through clinical development, we will continue to pursue appropriate regulatory pathways available to us (such as Breakthrough Therapy
Designation, Fast Track or Accelerated Approval and Priority Review) to expedite our programs and accelerate the timeline to an approval.
- •
- Experienced team. Our team has extensive global experience and its members are recognized experts in their respective fields. We seek to leverage the complementary skill sets of our team members in our approach to drug discovery and development and draw on their prior experience gained at leading international companies, and pharmaceutical and biotech companies. Dr. Roger Mills, an experienced chief medical officer and head of development, joined us in 2016 to lead our clinical development organization and in particular the, development of dipraglurant in PD-LID. Dr. Mills and his wider team have significant experience and a successful track record of developing drugs for Parkinson's disease patients.
discovery and development and that we have the potential to develop patentable, novel, highly differentiated oral small molecules for clinically validated targets considered "undruggable" or beyond the reach of conventional drug discovery approaches.
Our Internally Developed Product Candidates
Dipraglurant
Dipraglurant is a selective, orally active small molecule drug product which acts as an mGlu5 NAM. We discovered dipraglurant and hold composition of matter and polymorph patents granted in the United States and Europe. Dipraglurant is selective for mGlu5 and does not have significant activity or binding affinity to other mGlu or other CNS receptors, such as serotonin, GABA or dopamine receptors. There are currently no drugs of this class on the market.
Dipraglurant for the treatment of Parkinson's disease levodopa induced dyskinesia (PD-LID)
We have conducted a Phase 2a proof of concept clinical trial of dipraglurant in PD-LID, in which dipraglurant met its primary end point, was generally well tolerated. We expect to initiate a placebo-controlled Phase 2b/3 pivotal clinical trial of dipraglurant in PD-LID patients in the second half of 2020, pending removal of governmental restrictions and lessening of the impact of the global coronavirus pandemic on the U.S. healthcare system, which delayed our previously anticipated initiation in the first quarter of 2020. This registration clinical trial will be powered at 90%, the industry norm with the objective of demonstrating benefits on clinical symptoms in PD-LID patients over a 12-week treatment duration. The term "power" of a study is a measure of the probability that a clinical trial is a meaningful test of the hypotheses (or primary endpoints) that the clinical trial is designed to test. The study will be conducted at approximately 50 sites in the United States and will target enrolment of approximately 140 patients.
Parkinson's disease is a progressive neurodegenerative disease that results in the loss of dopaminergic neurons in the substantia nigra, or SN. One consequence of the depletion of dopamine in this disease is a series of movement disorders, including bradykinesia, akinesia, tremor, gait disorders and problems with balance. Early in the course of the disease, these motor symptoms of Parkinson's disease are effectively treated by dopamine replacement with the use of levodopa or dopamine D2 receptor agonists or monoamine oxidase B inhibitors. However, as the disease progresses, these agents become less effective in controlling motor symptoms and PD-LID often emerges.
39
PD-LID is involuntary movement that may affect any body area, including the face, trunk or limbs. Oral levodopa is currently the most effective treatment available for motor symptoms associated with Parkinson's disease. However, long term levodopa use is often associated with the development of dyskinesia, which may be as disabling as the symptoms of Parkinson's disease.
Dyskinesias are comprised principally of two types of movement: chorea, which is a rapid uncontrolled movement, and dystonia, which is a slow, often painful, writhing movement.
Even though levodopa provides more effective motor symptom control than other currently available therapies, physicians tend to delay use of levodopa use for as long as possible, using dopamine agonists or monoamine oxidase B inhibitors in the early stages of the disease, due to the inexorable onset of dyskinesia onset with levodopa use. Dopamine agonists and monoamine oxidase B inhibitors become less effective as Parkinson's disease progresses and are associated with dose limiting side effects, including Impulse Control Disorders, or ICDs, such as pathological addictions to gambling, shopping, eating or sex.
The occurrence of PD-LID is linked to the neurodegenerative process of PD and is not solely related to the duration of dopamine replacement therapy. For instance, in severe advanced stage Parkinson's disease patients, dyskinesia can be provoked after a first high dose of levodopa. Chronic or high dose dopamine replacement treatments alone do not lead to dyskinesia, but may lower the threshold for dyskinesia occurrence following dosing, as neurodegeneration progresses. Efforts to reduce the use of high doses of levodopa or dopamine agonists, by using more frequent lower doses or extended release formulations, can improve dyskinesias but may be at the expense of optimal motor function. In the later stages of Parkinson's disease, the patient and physician have to juggle good motor symptom control against the occurrence of levodopa-induced dyskinesia.
If dyskinesia could be effectively treated, or even delayed or eliminated, it is likely that doctors would use levodopa earlier in the treatment of Parkinson's disease. Currently available therapies, such as amantadine and Deep Brain Stimulation, or DBS surgery, often have limited effectiveness or tolerance in patients. The response of patients varies widely to amantadine, which in its generic form is commonly used off label to treat dyskinesia. Typically, amantadine only works for some, if any, dyskinesias suffered by a patient. Amantadine often has side effects which may limit its use, and some patients do not tolerate it at all. Some of the more common side effects of amantadine include blurred vision, digestive issues, postural hypotension, dizziness, falls, ankle oedema, drowsiness, trouble sleeping, depression and psychotic symptoms. DBS surgery, a non-pharmacological treatment strategy, is used primarily for patients whose symptoms cannot be satisfactorily controlled with medications. Patients experience varied results with DBS surgery, and even patients who experience better motor symptom control with DBS surgery may have continued symptoms of dyskinesia. Further, many patients are unwilling to undergo DBS surgery, since it is a costly, invasive surgical procedure that could result in complications.
40
Figure 1: Pharmacokinetic profile of Dipraglurant and L-Dopa in Humans
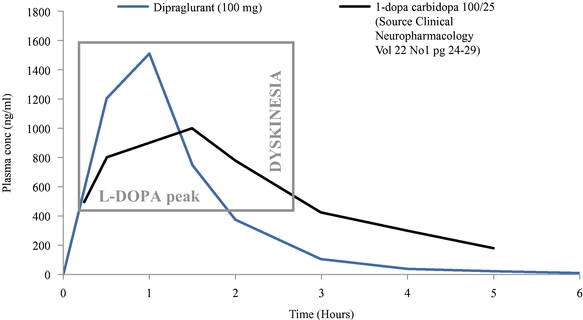
The rapid absorption and relatively short half-life of dipraglurant in the current immediate-release, or IR, formulation are thought to be well suited for use in LID. The pharmacokinetic, or PK, profile of dipraglurant (IR) mirrors that of levodopa with peak plasma concentration occurring around the same time as that of levodopa and the duration of plasma exposure covering that of the "On" period (the time when levodopa is having its effect, Figure 1). Dyskinesia occurs predominantly during the period immediately following levodopa dosing; troublesome peak-dose dyskinesia (dyskinesia which severely interferes with the patient's daily activity) usually appears as levodopa reaches Cmax and parallels the period of maximal clinical benefit. Based on its similar PK profile, dipraglurant is expected to optimally inhibit the abnormal glutamate stimulation during peak levodopa dose, while releasing the receptor during normal activity. Furthermore, dipraglurant will wash out between doses, releasing the mGlu5 receptor when not required, i.e. when levodopa has worn off.
Dipraglurant completed Phase 2a PD-LID clinical trial
We evaluated the efficacy, safety and tolerability of dipraglurant at 50 and 100 mg in a Phase 2a proof-of-concept four weeks, randomized, double-blind, placebo-controlled, parallel-group out-patient clinical trial in 76 patients with Parkinson's disease (dipraglurant n = 52, placebo n = 24) with moderate or severe LID. The study was conducted in 25 centers in the United States, France, Germany and Austria. The severity of LID was evaluated by both clinicians and the patients using the modified Abnormal Involuntary Movement Scale, or mAIMS, patient diaries and the patient global impression of change, or PGIC, and the clinician global impression of change, or CGIC, for both dyskinesia and motor symptoms of Parkinson's disease. Motor symptoms of Parkinson's disease were assessed using the Unified Parkinson Disease Rating Scale, or UPDRS.
The Phase 2a proof of concept clinical trial of dipraglurant in PD-LID met its primary end point, was generally well tolerated and no clinically significant abnormalities of safety monitoring parameters occurred. In addition, dipraglurant showed statistically significant effects on PD-LID as measured using mAIMS at Day 1 and Day 14. However, an increasing placebo response resulted in the effect of dipraglurant on PD-LID symptoms not showing statistical significance at Day 28.
41
Figure 2: Dipraglurant Phase 2a PD-LID clinical trial design
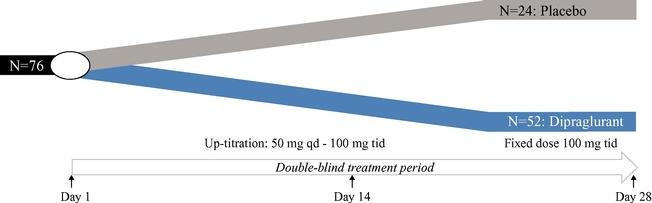
Figure 3: Dipraglurant Phase 2a PD-LID clinical trial dosing schedule
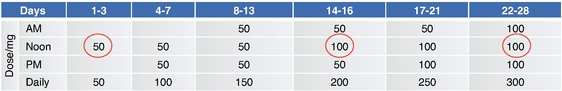
Patients were randomized to receive either active or placebo treatment at a 2:1 ratio. A blinded dose-titration regimen was employed over the first 3 weeks of treatment. Following the initial intake of a single capsule containing 50 mg or matching placebo, dose and dosing frequency were progressively escalated up to the target regimen of 100 mg tid (total daily dose of 300 mg) beginning on Day 22; the up-titration scheme was customizable based on tolerability. Dyskinesia was measured using the mAIMS at the mid-day does of dipraglurant and L-dopa on Day 1, 14 and 28. Due to the short half-life of dipraglurant the acute effect of 50mg dose was measured on Day 1 and the acute effect of 100mg dose was measure on Day 14 and Day 28.
Outcome Measures
The primary objective of the study was safety and tolerability. This was assessed based on vital signs, physical and neurological examination, electrocardiogram, or ECG, laboratory tests, and AE monitoring.
Secondary efficacy outcome measures included the mAIMS, UPDRS and patient diaries.
The main efficacy endpoint was severity of dyskinesia determined with mAIMS for dyskinesia in face, neck, trunk, and each of the upper and lower limbs. At screening, patients had to specify a dose of levodopa between 10.30 am and 3.30 pm that was regularly associated with moderate to severe dyskinesia. As severity of dyskinesia generally correlates with the plasma concentration of levodopa (peak dose dyskinesia) and as Tmax of dipraglurant is comparable, study medication was to be taken within 15 minutes prior to levodopa. Dyskinesia following this midday dose was assessed by the mAIMS on Days 0, 1, 14 and 28.
42
Figure 4: MMRM analysis of the effect of dipraglurant on the peak mAIMS score reported as reduction from baseline
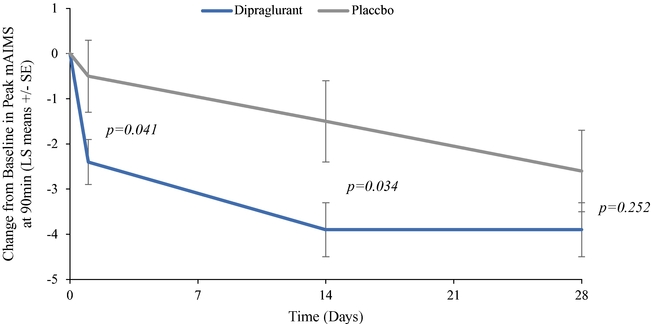
The above figure shows the effect of dipraglurant on dyskinesia as measured using the mAIMS at Day 1, 14 and 28. Dipraglurant reduced dyskinesia compared to placebo at all visits over the 28 days and showed statistically significant effects at Day 1 and 14 as well as improvements of ³30% at Day 14 which were sustained through Day 28. The level of improvement with dipraglurant at each time point and Day 28 was about twice that of placebo. However, an increasing placebo response resulted in the effect of dipraglurant on PD-LID symptoms not showing statistical significance at Day 28. We believe the dose titration technique employed along with the 2:1 randomization of active to placebo, as well as the fact that placebo-mitigating techniques were not deployed in the study, contributed to the placebo response.
43
Figure 5: Dipraglurant cumulative % of PD-LID patients showing > 30% change of peak mAIMS from baseline
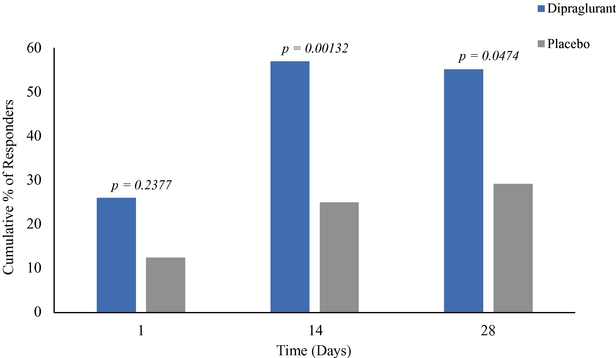
Responders were defined as those patients reaching at least 30% improvement in peak mAIMS scores compared to baseline. At all visits, the percentage of responders was higher in the dipraglurant-treated group than in the placebo group and, on Days 14 and 28, exceeded 50% and showed a statistically significant difference over placebo.
Figure 6: Clinician rated global impression of change in LID patients after administration of dipraglurant and placebo
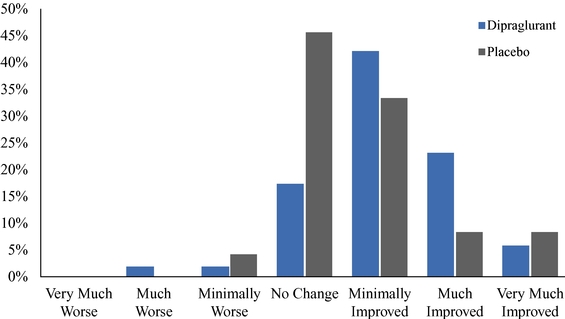
44
There was a significant improvement in the CGI-C as determined by the investigators. Figure 6 summarizes the results across each score level and for each treatment group at Day 28. The CGIC for improvement of dyskinesia was assessed by the investigating physician at Day 28 compared to baseline. The dipraglurant group improved by 71.2% versus 49.9% for the placebo group at Day 28 (p < 0.05).
Other Secondary Measures of Efficacy
Dipraglurant did not worsen motor control
UPDRS Part III scores in the post-levodopa dosing period did not differ between treatment groups. There was no evidence that dipraglurant led to increased parkinsonism or "Off" periods. This was an expected result given the mechanism of action, but was important to establish, as it confirms that the anti-dyskinetic benefits observed with treatment did not come at the cost of worsened motor control.
Patient reported effects on PD motor symptoms and dyskinesia—diary data
Mean daily "Off" time tended to decrease in each treatment group in weeks 1, 2 and 3, but at week 4, "Off" time decreased by about 50 minutes per day in the dipraglurant group versus an increase of about 9 minutes in the placebo group. Although, the week 4 difference was not statistically significant, or NSS (the study was not powered to detect such changes), it remains interesting and suggests the potential for additional benefits of treatment in this patient population.
Figure 7. Pattern of motor complications of dipraglurant group patients over the course of a day, as reported in patients' diaries—baseline versus week 4
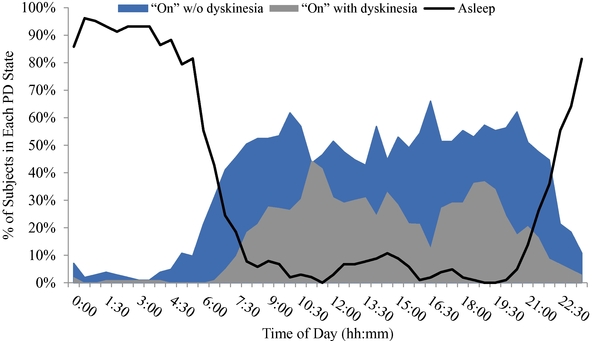
45
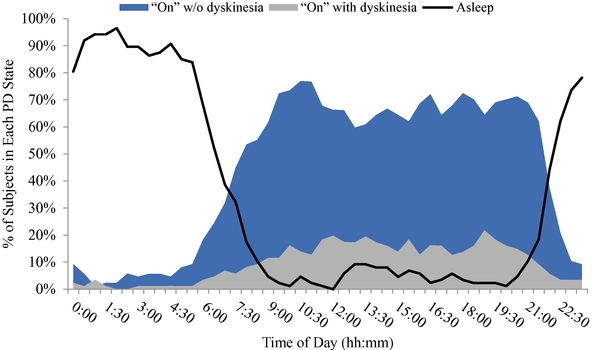
The mean daily percentage of time in "On" with no dyskinesia increased in both treatment groups. At week 4, the dipraglurant group showed a 2.28 hours increase compared to 1.78 hours for the placebo group (NSS).
Overall, there were more patients in the dipraglurant group who reported "On" time without (w/o) dyskinesia and fewer patients reporting "On" time with dyskinesia (Figure 7). In this analysis, dipraglurant treatment improved the percentage of patients in "On" time without LID from morning to late afternoon.
Safety and Tolerability Data
This first study in PD patients met its primary objective and generally demonstrated tolerability for dipraglurant taken at doses of up to 100 mg tid for 4 weeks. The types of adverse events, or AEs, observed in PD patients up to 75 years of age were generally similar to those seen in healthy normal subjects. Fifty-two patients were exposed to the study drug vs. 24 patients on placebo.
The overall incidence of treatment emergent adverse events, or TEAEs, for the 4-week treatment period was 88.5% for the dipraglurant group compared to 75% for placebo. As doses increased over the titration period, an increase in AE incidence was observed. In Weeks 1 and 2, AE incidence was 53.8% for patients receiving dipraglurant 50 mg vs. 58.3% for placebo and, in Weeks 3 and 4, it was 73.1% for dipraglurant 100 mg vs. 62.5% for placebo. In both treatment groups, AEs occurred most commonly in the central nervous system (51.9% dipraglurant vs. 45.8% placebo).
The only significantly increased event compared to placebo was nausea. Although dyskinesia or worsening of dyskinesia was reported more frequently for dipraglurant than for placebo (n=11 vs. 3; 21.2% vs. 12.5%), there was no pattern to these reports and most were transient. In addition, 3 of the 11 patients who reported worsening dyskinesia in the treated group did so only in the follow-up period (i.e. when not taking the drug), thus the dyskinesia recurred only after therapy had stopped. Therefore, the adjusted AE% was similar, 15.3% for dipraglurant vs. 12.5% for placebo.
Two patients withdrew due to TEAEs. Both were in the dipraglurant group at the 100 mg dose level.
46
- •
- One case concerned a 68-year-old man who experienced severe nausea, dizziness, and anxiety after he had been treated with dipraglurant for
3 weeks. At the time of AE, the patient was receiving 100 mg tid. After 3 days, the medication was discontinued and the AEs resolved without sequelae.
- •
- The other case concerned a 69-year-old man who experienced several AEs. On treatment Day 6, when he was taking 50 mg bid, he experienced moderate dyskinesia and fell. On Day 8, he experienced mild nausea and continued to experience moderate dyskinesia and sweating. The patient continued with the protocol defined dose escalation and started the 100 mg midday dose on Day 14. On Day 17, study medication was discontinued. By Day 21, nausea had resolved, whereas dyskinesia and sweating had not.
Only one severe AE occurred in the dipraglurant group, and, as it occurred 2 days after the end of treatment, it was considered unrelated to study medication.
Clinical laboratory tests in the study (biochemistry, hematology and urinalysis) did not show any relevant differences, neither over time nor between groups. There were no clinically significant ECG changes that occurred in the study in patients receiving dipraglurant.
Dipraglurant clinical development plan—registration program in PD-LID
The pivotal program for LID currently comprises two placebo-controlled studies that are intended to support registration. The 100 mg tid dose of dipraglurant will be evaluated versus placebo in both studies, as it showed a significant reduction in LID symptoms at Day 1 and Day 14, in the earlier in the earlier POC clinical trial, the drug was also generally well tolerated and no clinically significant abnormalities of safety monitoring parameters occurred. Following successful completion of the first Phase 2b/3 pivotal clinical trial, an End of Phase 2, or EOP2, meeting with the FDA will be planned to obtain agreement on the final design of a confirmatory Phase 3 clinical trial and any remaining elements of the registration program.
Dipraglurant Phase 2b/3 pivotal clinical trial in PD-LID patients—Study 301
We have substantially completed preparation to start registration studies in PD-LID and expect to initiate a double-blind, placebo-controlled, parallel group design Phase 2b/3 pivotal clinical trial of dipraglurant in PD-LID patients in the second half of 2020, pending removal of governmental restrictions and lessening of the impact of the global coronavirus pandemic on the U.S. healthcare system, which delayed our previously anticipated initiation in the first quarter of 2020. The study will be conducted at approximately 50 sites in the United States and will target enrollment of approximately 140 patients. Furthermore, the primary endpoint will be efficacy in reducing PD-LID measured as the change over time in Unified Dyskinesia Rating Scale, or UDysRS at week 12 compared to baseline. We expect to report data in the second quarter of 2022.
47
Figure 8: Design of dipraglurant PD-LID Phase 2b/3 study 301 clinical trial

The study will enroll patients 30-85 years of age with PD and clinically relevant LID. At screening, LID severity will be assessed to determine eligibility using the UDysRS and the Movement Disorder Society Unified Parkinson's Rating Scales (MDS-UPDRS) in order to ensure that only patients with moderate to severe LID are included in the study. During the screening period, patients will receive Brief Psychosocial Therapy adapted for dyskinesia, or BPST-Dys. This is a structured non-pharmacological intervention, directed by trained site-staff, BPST-Dys has been demonstrated to reduce anxiety associated with neurodegenerative disease including PD. The aim is to ensure that any anxiety overlay is reduced before patients enter the randomized period of the study such that the baseline symptom severity is more reflective of the condition itself. At the baseline visit, the severity of LID symptoms will be reassessed using the UDysRS, the MDS-UPDRS and PD diary entries to confirm that the patient continues to meet the study entry criteria. Subjects who have improved during the screening period and do not meet the inclusion criteria at their baseline visit will be excluded prior to randomization. Although this may result in a higher screen-failure rate, it is intended to reduce placebo response and ensure that patients randomized to study require pharmacologic treatment for their condition.
All PD medications will be required to be stable prior to screening and remain so during the course of the study.
Following completion of the 12-week randomized study, patients who choose to do so will be allowed to enroll in a separate open label safety study, or OLS, if the investigator considers it to be of benefit.
Objectives and endpoints. The key objectives of the study are to evaluate the efficacy and safety of dipraglurant in reducing dyskinesia symptoms in PD patients with LID without worsening of the Parkinson's disease. The primary and secondary endpoints that are currently planned in the protocol include the following:
Primary Endpoint. Change in the Unified Dyskinesia Rating Scale (UDysRS) total score from baseline to week 12.
Key Secondary Endpoints—Efficacy
- •
- Standardized Patient Diary based assessments of "On" time without troublesome dyskinesia, as well as "Off" time.
48
- •
- Other Secondary Endpoints include the Clinician's Global Impression of Severity (CGI-S)
Safety Assessments
Safety measures will be assessed during the course of the study and during a one week follow-up period for those patients not electing to enter the open-label study.
UDysRS, the Primary Endpoint
In this study, the primary endpoint will be the UDysRS. The UDysRS was the endpoint that was used in the Gocovri pivotal studies leading to the drug's FDA approval for PD Dyskinesia. Of all the scales used for assessing dyskinesia, only the UDysRS contains anchored, clinician-evaluated measures of dyskinesia. In this scale, patients perform 4 tasks, and raters evaluate dyskinesia severity and disability for each. Although this rating measure incorporates the concepts of the mAIMS, the UDysRS has the advantage of consolidating both patient-based perceptions of disability and objective rater-based assessments of impairment and disability (Goetz et al., Mov Disord 2008).
Placebo-minimization procedures
A number of factors may have contributed to the high placebo response in the Phase 2a POC study. A number of steps will therefore be taken in the pivotal studies to curb excessive placebo response, reduce variability and therefore enable a more precise estimate of treatment effect of dipraglurant in the treatment of LID:
- •
- The UDysRS measure will be used as the primary efficacy endpoint instead of the mAIMS. The mAIMS is not as sensitive to changes in LID as the
UDysRS, as described in the 8-scale comparison study (Goetz et al., Mov Disord 2013) and it is more prone to placebo response than the UDysRS. The mAIMS
lacks the functional/objective items of the UDysRS and thus is largely subjective.
- •
- Raters will be qualified by the Movement Disorder Society to assess the UDysRS scores. In addition, a specialized rating company will review
UDysRS ratings both across the study and per rater to further ensure the quality and consistency of these assessments.
- •
- Randomized patients will be required to have dyskinesia scores at screening and baseline that reflect moderate to severe symptoms.
- •
- A non-pharmacologic intervention BPST-Dys will be incorporated into the screening period to help exclude placebo-responders from the
randomized, controlled portion of the trial.
- •
- The longer 12 week randomized treatment period should also help to mitigate placebo response, compared to the 4-week treatment duration tested in the proof-of-concept study.
Dipraglurant confirmatory Phase 3 pivotal clinical trial in PD-LID patients—Study 303
The confirmatory Phase 3 study will also be a two-arm double-blind, placebo-controlled, parallel group design comparing 100 mg dipraglurant versus placebo in PD patients with LID. Treatment will be over a 6 month period to meet FDA requirements. The Phase 3 trial will follow a similar study design to the Phase 2b/3 pivotal clinical trial. Although the treatment duration will be 24 weeks in total, the primary endpoint will remain the change in UDysRS total score from baseline to week 12. The 24 week UDysRS score will evaluate maintenance of effect as a secondary endpoint. At the completion of the study, eligible patients will have the option of rolling into the ongoing OLS study, if the investigator considers it to be of benefit.
49
Dipraglurant in PD motor symptoms
There is an increasing body of literature that suggests that inhibiting mGlu5 in the striatopallidal pathway may also improve the motor symptoms of PD and may also prevent excitotoxic damage to the substantia nigra. Dipraglurant was investigated in the haloperidol induced catalepsy (HIC) model, an animal model of Parkinson's disease. Haloperidol is an antagonist of the dopamine D2 receptor and overcoming the catalepsy (immobility) induced by haloperidol administration is suggestive of antiparkinsonian activity and may also have relevance for other movement disorders, such as tardive dyskinesia and dystonia, where reduced activity of dopamine D2 receptors is implicated. In the rat HIC model, dipraglurant reduced the amount of time rats were immobile, in a dose dependent manner. The effective plasma concentration was similar to that for the treatment of dyskinesia in the MPTP macaque and that which was seen to be effective in PD-LID patients. The suggestion of antiparkinsonian activity was also supported by observations in the Phase 2a clinical trial. In week 4 of treatment, patients reported an average "Off" time reduction of 50 minutes per day. Also, both patients and clinicians reported improvements in PD symptoms compared to placebo. Although none of these results were statistically significant, the observations were interesting and were noted by the PD experts who took part in the trials. PD motor symptom effects will continue to be evaluated in the larger pivotal clinical trials.
Dipraglurant in PD non-motor symptoms
As well as suffering from difficulty with poor and uncontrolled motor symptoms, PD patients also suffer from a wide variety of non-motor symptoms. Among these are affective disorders (e.g., anxiety, depression and anhedonia) and compulsive behavioral disorders (e.g., sex, alcohol, gambling, and shopping addiction). These compulsive behaviors are particularly linked to treatment with dopamine agonists and more specifically to those which act on the dopamine D3 receptor as well as D2 (e.g. pramipexole). Inhibition of mGlu5 is pre-clinically and clinically validated for the treatment of anxiety and depression, although no mGlu5 inhibitors are yet marketed for these indications. Also, inhibition of mGlu5 has been shown to have anti-addictive properties in a number of animal models, including cocaine self-administration in rats. These data suggest that mGlu5 inhibition may potentially have utility in treating certain non-motor symptoms of PD.
Dipraglurant for the treatment of non-Parkinsonian dystonia
Dystonia is a movement disorder that causes the muscles to contract and spasm involuntarily. There are about 500,000 people with one or other form of dystonia in North America. The involuntary muscle contractions force the body into repetitive and often twisting movements as well as awkward, irregular, sometimes painful postures. Dystonia aetiologies and symptoms are heterogeneous. There are approximately 23 forms of dystonia, and dozens of diseases and conditions include dystonia as a major symptom. Dystonia may affect a single body area (focal), multiple areas (segmental) or be generalized throughout multiple muscle groups. Further, dystonias are distinguished as either primary, with idiopathic or genetic causes, or secondary, induced by drugs or toxins. A number of types of dystonia are classified as rare, including cervical dystonia, DYT1 familial generalized dystonia or X-linked dystonia parkinsonism. It is estimated that over 30% of PD patients experience dystonia as a symptom or a complication of treatment.
Dystonia affects people of all ages and backgrounds. Dystonia causes varying degrees of disability and pain, from mild to severe. Presently, there is no cure for dystonia. Doctors often prescribe drugs for the treatment of dystonia off-label, i.e., drugs that have not been approved for the indication being treated. Since these drugs have not been approved for the treatment of dystonia, they have not undergone rigorous clinical trials for the indication.
Current therapies include oral drugs such as anticholinergic agents, dopamine receptor agonists/antagonists and baclofen. The efficacy of these drugs is marginal and side effects further limit compliance and usage. The only available indicated treatment is botulinum toxin injections, which are only suitable for focal or segmental dystonia treatment. In addition, Deep Brain Stimulation, or DBS, surgery is sometimes used for both focal and generalized refractory dystonia. However, for both of these approaches, patients are left with inadequate response, and therefore a need exists for an oral, safe and effective treatment for dystonia.
50
Initial data from the testing of dipraglurant in the MPTP macaque model of LID and a small subset of patients in the Phase 2a clinical trial of dipraglurant in patients with PD-LID suggest that dipraglurant may have a role in treating dystonia. In the MPTP macaque model of LID, dipraglurant reduced dystonia following levodopa administration to the same extent as chorea. We are currently evaluating dipraglurant in preclinical models of dystonia.
Externally Developed Out-licensed Product Candidate
ADX71149 for the treatment of Epilepsy
Epilepsy is one of the most common serious neurological disorders affecting about 65 million people globally (Thurman et al. 2011). It affects 1% of the population by age 20 and 3% of the population by age 75 (Holmes et al. 2008). Epilepsy describes a condition in which a person has recurrent seizures due to a chronic, underlying process. Epilepsy refers to a clinical phenomenon rather than a single disease entity, since there are many forms and causes. Epilepsy is defined by any of the following conditions: (1) at least two unprovoked (or reflex) seizures occurring >24 h apart; (2) one unprovoked (or reflex) seizure and a probability of further seizures similar to the general recurrence risk (at least 60%) after two unprovoked seizures, occurring over the next 10 years; (3) diagnosis of an epilepsy syndrome (Fisher et al. 2014). The synaptic vesicle protein 2A (SV2A) has been identified as a broad spectrum anti-convulsant target in models of partial and generalized epilepsy, and studies in animal models and human tissue suggest that changes in the expression of SV2A are implicated in epilepsy (Mendoza-Torreblanca et al. 2013; Kaminski et al. 2012). SV2A ligands include levetiracetam (Lynch et al. 2004), which is an anti-epileptic drug commercialized under trademark Keppra®, which is approved in Europe and the USA as a monotherapy or add-on therapy in patients diagnosed with epilepsy.
Our partnered drug candidate ADX71149 is a novel orally active mGlu2 PAM. Our partner, Janssen, has completed Phase 1 and two Phase 2a clinical trials in schizophrenia and anxious depression, respectively. Janssen has announced that ADX71149 has been extensively profiled in preclinical models of epilepsy showing both standalone activity and in combination with SV2A ligands including levetiracetam (a commercialized antiepileptic drug). Janssen has patented the combination of mGlu2 PAM with SV2A ligands for the treatment of epilepsy and is currently evaluating the development path forward for ADX71149. Epilepsy is an indication with a large commercial opportunity. Existing therapies frequently provide ineffective control of symptoms or have side effects that discourage adherence. We believe that, subject to regulatory approval, ADX71149 may provide a substantial benefit to patients. Under our agreement with Janssen, Janssen is responsible for, including the financing of, development and commercialization, if any, of ADX71149.
Material Internal Research Programs
GABAB PAM
Activation of the GABAB, receptor, a Family C class of GPCR, is clinically and commercially validated. Generic GABAB receptor agonist, baclofen, also known as chlorophenibut, is marketed for spasticity and some spinal cord injuries, and used for overactive bladder, or OAB, but is not commonly used due to a variety of side effects of the drug and its rapid clearance. Researchers have shown that baclofen is effective in reducing drug self-administration, cravings, and anxiety, and thus promotes abstinence.
Our GABAB PAM drug candidates are novel, orally available, small molecules that have demonstrated positive effects and tolerability in several preclinical rodent models of pain, anxiety, addiction and OAB and have also shown activity in a genetic model of CMT1A. A GABAB PAM differs from the generic drug baclofen in that it is a PAM rather than an orthosteric agonist at the GABAB receptor. The GABAB PAM only acts when the natural ligand (GABA) activates the receptor,
51
and therefore respecting the physiological cycle of activation. It has been proposed that PAMs produce fewer adverse effects and lead to less tolerance to effect than direct agonists (May and Christopoulos 2003; Langmead and Christopoulos 2006; Perdona et al. 2011; Urwyler 2011; Gjoni et al., 2008; Ahnaou et al).
GABAB PAM for the treatment of addiction
Our Partner, Indivior, has licensed worldwide rights to our GABAB PAM program and is responsible for all development, manufacture and commercialization of any selected GABAB PAM drug candidates. Under the agreement, we are responsible for executing a research program funded by Indivior to discover novel drug candidates. Indivior has the right to select GABAB PAM drug candidates from our research program and we expect to deliver drug candidates by the end of 2020. Addiction is an indication with a significant commercial opportunity. Existing therapies often do not provide effective control of symptoms or have side effects that discourage adherence. Subject to regulatory approval, we believe that GABAB PAM offers an innovative and differentiated treatment approach from existing therapies and may be of substantial benefit to patients.
Scientific advances have revolutionized our understanding of addiction as a chronic, relapsing disease and not a moral failure. Drug addiction is a complex illness which is characterized by intense and, at times, uncontrollable drug craving, along with compulsive drug seeking and use that persist even in the face of devastating consequences. Addiction affects multiple brain circuits, including those involved in reward and motivation, learning and memory, and inhibitory control over behavior. While a person initially chooses to take drugs, over time the effects of prolonged exposure on brain functioning compromise the ability to choose, and seeking and consuming the drug become compulsive, often eluding a person's self-control or willpower. Because drug abuse and addiction have so many dimensions and disrupt so many aspects of an individual's life, treatment is not simple. Addiction treatment must help the individual stop using drugs, maintain a drug-free lifestyle, and achieve productive functioning in the family, at work and in society. Patients typically require long-term or repeated episodes of care to achieve the ultimate goal of sustained abstinence and recovery of their lives.
GABAB PAM for the treatment of CMT1A
Our license agreement with Indivior provides for a funded research program, under which we have the right to select drug candidates for exclusive development in certain indications outside of addiction, including CMT1A, a rare disease indication. We plan to pursue orphan drug designation for a selected drug candidate in CMT1A. Subject to regulatory approval, believe an oral small molecule GABAB PAM without the adherence-limiting side effects of baclofen and with a once-a-day dosing could bring benefit to patients and consequently present a strong commercial opportunity for us. Our GABAB PAM compounds have demonstrated the potential role of this mechanism in CMT1A and we plan to complete lead optimization and select a clinical candidate for this indication.
Charcot-Marie-Tooth disease, previously classified as a subtype of muscular dystrophy, is a rare hereditary motor and sensory neuropathy, or HMSN, which causes demyelination of the peripheral nerves. The disease leads to damage or destruction to the myelin sheath covering nerve fibers. The nerve fibers most severely affected are those that stimulate movement, with the nerves in the legs being affected first and most severely. Similar symptoms may appear in the arms and hands, which may include a claw- like hand.
The disease is very impactful for patients due to the neurological pain and muscular disability. A combination of lower motor neuron-type motor deficits and sensory symptoms are observed, and paresis and muscle atrophy develop with areflexia. The chronic nature of the motor neuropathy commonly results in foot deformity, hammertoes, very high-arched feet, loss of lower leg musculature,
52
resulting in skinny calves, numbness of the foot or leg, "slapping" gait (feet hit the floor hard when walking), foot drop (inability to hold foot horizontal) and weakness of the hips, legs or feet. Involvement of the hands may follow as the disease progresses. Signs of sensory system dysfunction are common and include loss of vibration and joint position sense followed by decreased pain and temperature sensation. Onset of CMT1A occurs between age 5 and 25 years, with a prevalence of 1 in 5,000. There are no known cures for this debilitating condition. Current therapies are primarily symptomatic, such as physiotherapy.
We expect to select a drug candidate for development by the end of 2020.
mGlu7 NAM
The mGlu7 receptor is a class C GPCR and is the most highly conserved of all mGlu subtypes, exhibiting the widest distribution in the brain. It is localized pre-synaptically at a broad range of glutamatergic and GABAergic synapses and is thought to be one of the most important mGlu subtypes in regulating CNS function.
mGlu7 NAM for the treatment of PTSD
Preclinical data suggest that mGlu7 antagonism could alleviate stress-related anxiety and depressive symptoms and deficits in amygdala-dependent behaviors (fear response and conditioned taste aversion). These data are consistent with the abundant localization of mGlu7 in brain regions involved in the control of fear and emotion supporting the potential use of mGlu7 NAM for the treatment of PTSD.
PTSD is a serious anxiety disorder that can occur in people who have experienced or witnessed a traumatic event such as a natural disaster, a serious accident, a terrorist act, war/combat, rape or other violent personal assault. PTSD can occur in all people, in people of any ethnicity, nationality or culture, and any age. PTSD affects approximately 3.5 percent of U.S. adults, and an estimated one in 11 people will be diagnosed with PTSD in their lifetime. Women are twice as likely as men to have PTSD. People with PTSD have intense, disturbing thoughts and feelings related to their experience that last long after the traumatic event has ended. They may relive the event through flashbacks or nightmares; they may feel sadness, fear or anger; and they may feel detached or estranged from other people. People with PTSD may avoid situations or people that remind them of the traumatic event, and they may have strong negative reactions to something as ordinary as a loud noise or an accidental touch.
Current treatments are primarily based on psychotherapy, as medication is nonspecific and usually ineffective, often with a number of side effects.
We are developing mGlu7 NAM as a novel orally available treatment to reduce fear memory in PTSD. By selectively targeting mGlu7 with NAMs, the brain circuitries involved in fear and anxiety can be more precisely modulated, potentially resulting in higher efficacy and fewer side effects. Subject to regulatory approval, we believe the mGlu7 NAM may offer an innovative and differentiated treatment approach from existing therapies. We are in late lead optimization and a consortium led by us has been awarded a €4.85 million grant from the Eurostars Joint Programme, to advance the program to drug candidate stage.
Internal Early Stage Research Programs
mGlu2 NAM
The mGlu2 receptor, is a class C GPCR and is broadly distributed throughout the cortex as well as highly expressed in the hippocampus and perforant path. Activation of mGlu2 leads to inhibition of glutamate release in the synapse and therefore mGlu2 NAM has the potential to treat medical conditions linked to lowered glutamate levels in the brain via restoration of a normalized glutamatergic tone such as cognitive impairment in Alzheimer's disease and depression.
53
mGlu2 NAM for the treatment of mild neurocognitive disorders
mGlu2 NAM is one of the most promising experimental therapeutic strategies for the treatment of mNCD. Our mGlu2 NAM has been tested in the beta amyloid-induced memory impairment model in rodents which mimics aspects of pathophysiology and progressive memory impairment observed in Alzheimer's disease or, AD. In this study, our mGlu2 NAM dose-dependently reversed working memory impairment in the novel object recognition test, without affecting locomotor activity. Donepezil, a marketed drug currently used to treat symptoms of AD, was used as the positive control and the magnitude of the effect induced by the mGlu2 NAM was similar to that of donepezil. Preclinical research from other groups suggest not only that mGlu2 NAM might slow the progression of AD, an effect not seen with any marketed drug, but also that it may have a synergistic effect on cognition when combined with donepezil.
We are currently optimizing multiple chemical series of highly selective, orally active mGlu2 NAMs offering advanced compounds at the late stage of lead optimization.
mGlu4 PAM
The mGlu4 receptor, is a class C GPCR and is expressed primarily on presynaptic terminals, functioning as an autoreceptor or heteroceptor with its activation leading to decreases in neurotransmitter release from presynaptic terminals. The mGlu4 is uniquely distributed in key brain regions involved in multiple CNS disorders. In particular, mGlu4 is abundant in striato-pallidal synapses and on the subthalamo-nigral projections within the basal ganglia circuitry, a key area implicated in movement disorders, like Parkinson's disease.
mGlu4 PAM for the treatment of Parkinson's disease
Parkinson's disease (PD) is a progressive neurodegenerative disorder that is caused by loss of dopaminergic neurons in the basal ganglia, the brain center for movement initiation and coordination. mGlu4 receptors are strategically localized to counteract neurotransmitter imbalance and restore motor behavior in patients.
Parkinson's disease is characterized by motor symptoms such as slowness in initiating and executing movements (akinesia and bradykinesia, respectively), muscular rigidity, resting tremor, postural instability, gait dysfunction and freezing. These symptoms are caused by the degeneration of dopaminergic neurons in the substantia nigra and depletion of dopamine in the striatum. Actually, in a healthy brain, there is a balance between the direct and indirect pathways of the basal ganglia. The direct pathway stimulates the initiation of movements through an activation of the thalamocortical neurons. By contrast, the indirect pathway leads to the inhibition of the thalamocortical neurons, and subsequently inhibition of movements. These two pathways are modulated by the dopaminergic neurons from the substantia nigra, that activate the direct pathway and inhibit the indirect pathway. In PD, these dopaminergic neurons degenerate, and dopamine loss in the striatum leads to an over-stimulation of glutamate transmission at the subthalamo-nigral synapses, and then an over-inhibition of the thalamocortical neurons. The ability to carry out voluntary movements is impaired, and this leads to the motor symptoms of PD.
Current treatments are aimed at replacing dopamine or mimicking its effects by chronically administering patients with the dopamine precursor L-DOPA, inhibitors of dopamine catabolic enzymes or direct dopamine receptor agonists. Although these treatments provide good symptomatic relief in the early to middle stages of PD, they lose their efficacy as the disease progresses and their chronic administration is associated with invalidating side effects (dyskinesia, motor fluctuations, behavior disturbances). Finally, no PD therapies have demonstrated neuroprotection and delaying disease progression. Therefore, new treatments are required for PD that target the neurochemical systems downstream dopamine itself in the basal ganglia.
54
By decreasing GABAergic and glutamatergic transmission in the indirect pathway, mGlu4 activation is expected to restore the equilibrium between the direct and indirect pathways, and then to restore motor behaviors in PD. This has been demonstrated in animal models, including the MPTP monkey model. Several studies in animal models have demonstrated that this strategy is promising for the treatment of motor and non-motor symptoms of PD, as well as for disease modification. In the immune system mGlu4 has been found on dendritic cells. Emerging data implicate mGlu4 in several indications such as multiple sclerosis, Parkinson's disease, anxiety, neuropathic and inflammatory pain, schizophrenia, autism and diabetes.
We are developing mGlu4 PAM as a novel orally available treatment for PD. Subject to regulatory approval, we believe our mGlu4 PAM program may offer an innovative and differentiated treatment approach from existing therapies.
mGlu3 PAM
The mGlu3 receptor, is a class C GPCR involved in modulation of glutamatergic neurotransmission. Expression of mGlu3 receptors is high in pyramidal cells in the prefrontal cortex and neocortical regions, as well as in astrocytes and oligodendrocytes. So far, industry has only found orthosteric compounds that act as mGlu3 receptor agonists or antagonists. All of these compounds suffer from poor selectivity and have activity on other mGlu receptors, and in particular the mGlu2 receptor. Targeting the allosteric site of the mGlu3 receptor provides a unique approach to find subtype selective compounds and will allow a focused strategy to modulate specifically those pathways involving the mGlu3 receptor.
mGlu3 PAM for the treatment of neurodegeneration
Scientific evidence suggests astroglial mGlu3 receptor activation leads to neuroprotection, through modulation of glutamate excitotoxicity and glutamate transport, neurotrophin production and reduction of oxidative damage. This points to the potential utility of mGlu3 PAMs for neurodegenerative disease such as Alzheimer's or Parkinson's diseases.
We are currently optimizing multiple chemical series of highly selective, orally active mGlu3 PAMs offering compounds at the lead optimization stage.
Material agreements
Collaboration Agreement with Indivior for Development of novel GABAB PAM Compounds, including for the Treatment of Addiction and Other CNS Diseases
In January 2018, we entered into an agreement with Indivior for the discovery, development and commercialization of novel GABAB PAM compounds for the treatment of addiction and other CNS diseases. This agreement included the selected clinical candidate, ADX71441. In addition, Indivior agreed to fund a research program at Addex to discover novel GABAB PAM compounds.
Indivior has sole responsibility, including funding liability, for development of selected compounds under the agreement through preclinical and clinical trials, as well as registration procedures and commercialization, if any, worldwide. Indivior has the right to design development programs for selected compounds under the agreement. Through our participation in a joint development committee, we review, in an advisory capacity, any development programs designed by Indivior. However, Indivior has authority over all aspects of the development of such selected compounds.
Under terms of the agreement, we have granted Indivior an exclusive license to use relevant patents and know-how in relation to the development and commercialization of product candidates selected by Indivior. Subject to agreed conditions, Addex and Indivior jointly own all intellectual property rights that are jointly developed, and Addex or Indivior individually own all intellectual
55
property rights that Addex or Indivior develop individually. Addex has retained the right to select compounds from the research program for further development in areas outside the interest of Indivior including Charcot-Marie-Tooth type 1A neuropathy, or CMT1A. Under certain conditions, but subject to certain consequences, Indivior may terminate the agreement.
Under terms of the agreement, we received a non-refundable upfront fee of $5.0 million for the right to use the clinical candidate, ADX71441, including all materials and know-how related to this clinical candidate. In addition, we are eligible for payments on successful achievement of pre-specified clinical, regulatory and commercial milestones totaling $330 million. In addition, we are eligible for a royalty in the low teens on net sales of applicable products on a country-by country-basis. The term of the royalty for each licensed product in any particular country commences on such product's launch and ends on the latest of ten year anniversary of launch, expiration of certain applicable patent rights, and expiration of certain applicable marketing or data exclusivity periods. On February 14th, 2019, Indivior terminated the development of their selected compound, ADX71441.
Separately, Indivior funds research at Addex, based on a research plan to be mutually agreed between the parties, to discover novel GABAB PAM compounds. These future novel GABAB PAM compounds, if selected by Indivior, become licensed compounds. We agreed with Indivior to an initial research term and duration of two years, that can be extended by twelve-month increments and a minimum annual funding of $2 million for the Addex R&D costs incurred. Following Indivior's selection of one newly identified compound, Addex has the right to also select one additional newly identified compound. Addex is responsible for the funding of all development and commercialization costs of its selected compounds and Indivior has no rights to the Addex selected compounds. The initial two-year research term was expected to run from May 2018 to April 2020. On October 7, and December 20, 2019, Indivior agreed an additional research funding of $0.8 million, increasing the research funding by $1.6 million, for the research period.
Indivior may terminate the Agreement in its entirety or with respect to one or more countries or products upon 90 days' prior written notice prior to receipt of marketing approval for product candidates or twelve months' prior written notice after the receipt of marketing approval.
Addex may terminate the agreement if Indivior commits a material breach of the agreement and fails to cure such breach within 90 days of Addex's written notification to Indivior or fails to cure breaches to any payment obligations breach within 30 days.
Collaboration Agreement with Janssen for Development of Novel mGlu2 PAM Compounds, including ADX71149, for the Treatment of CNS and Related Diseases
On December 31, 2004, we entered into a collaboration and license agreement with Janssen Pharmaceuticals Inc., or Janssen, for the discovery, development and commercialization of novel mGlu2 PAM compounds for the treatment of CNS and related diseases. We agreed with Janssen to an initial research term and duration of two years that can be extended if parties mutually agree thereto in writing. The agreement was not extended. ADX71149 is one of the drug candidates discovered and selected for development by Janssen under the agreement. In 2012, Janssen announced completion of a Phase 2a clinical trial in Europe with ADX71149 for the treatment of schizophrenia demonstrating proof of principal in patients with negative symptoms of schizophrenia, such as apathy, social withdrawal, loss of emotional expression or sleep disorders. Janssen also conducted a Phase 2a clinical trial with ADX71149 for the treatment of patients with anxious depression, which failed to show statistically significant effects. In addition, Janssen announced in 2015 that ADX71149 demonstrated synergies with levetiracetam (a globally commercialized antiepileptic drug) in preclinical models of epilepsy.
Under our agreement with Janssen, we have granted Janssen an exclusive license to use relevant patents and know-how in relation to the development and commercialization of product candidates
56
selected by Janssen under the agreement and a non-exclusive worldwide license to conduct research on the collaboration compounds using relevant patents and know-how. Subject to certain conditions, we and Janssen own, jointly, all intellectual property rights that we and Janssen develop jointly, and we or Janssen own all intellectual property rights that we or Janssen develop individually. Under certain conditions, but subject to certain consequences, Janssen may terminate the agreement for any reason, subject to a 90-day notice period.
Janssen has sole responsibility, including funding liability, for development of selected compounds under the agreement through preclinical and clinical trials, as well as registration procedures and commercialization, if any, in the United States, Japan, the United Kingdom, Germany, France, Spain and Italy. Janssen has the right to design development programs for selected compounds under the agreement. Through our participation in a joint development committee, we review, in an advisory capacity, any development programs designed by Janssen. However, Janssen has authority over all aspects of the development of selected compounds and may develop or commercialize third-party compounds with a different mechanism of action for identical use.
Under the terms of the Janssen agreement, we received an upfront fee of CHF 4.6 million and research funding of CHF 6.4 million during the research period, which ran from 2005 to 2007. In addition, we are eligible for payments on successful achievement of pre-specified clinical and regulatory milestones. We are also eligible for a royalty in the low teens on net sales of applicable products on a country-by country-basis and on a product by product basis, for a period commencing on the date of first sale and ending upon the latest of the expiration of twelve years from the date of first sale of a product in a given country or the last to expire of our patents containing a claim covering composition of compound comprised in a product sold by Janssen, its affiliates or sublicensees in such country. We received a CHF 1.5 million milestone payment in relation to the entry of ADX71149 into Phase 1 in July 2009 and a CHF 2.6 million milestone payment in relation to the entry of ADX71149 into Phase 2 in June 2011. We are eligible for a further €109 million in success-based development and regulatory milestones and low double digit royalties on net sales.
In the event that no compounds are discovered or identified during the research period, the agreement shall terminate.
In the event that there are compounds but no mGlu2 PAM are discovered during the research period or 6 months thereafter, our and Janssen's rights and obligations to develop commercialize, pay royalties and milestones on mGlu2 PAM compounds will terminate. The contract also contains termination rights for non-defaulting parties upon breach.
Intellectual Property
Patents and Proprietary Rights
We own thirteen U.S. and 228 foreign patents and a number of pending patent applications that cover various aspects of our allosteric modulator technologies and discovery platform, including several classes of compounds which are potentially useful as modulators of mGlu5, mGlu2, mGlu4 and GABAB. More specifically, our patents and patent applications cover compounds, pharmaceutical compositions, polymorphs and uses of compounds for medical treatment.
Our patent strategy is to file patent applications on innovations and improvements to cover a majority of the major pharmaceutical markets in the world. We typically file priority applications at the United Kingdom Patent Office to establish a priority date for the generic subject matter and examples which are available at the filing date of each invention. Subsequently, we file international applications under the Patent Cooperation Treaty or PCT, with extra examples to support the scope of the claims (International Phase). After the International Phase, we file patent applications in selected countries representing potential major markets for our drug candidates (National/Regional Phase).
57
Generally, patents have a term of twenty years from the earliest priority date, assuming all maintenance fees are paid. In some instances, patent terms can be increased or decreased, depending on the laws and regulations of the country or jurisdiction that issued the patent. Wherever appropriate and legally possible, we aim at obtaining patent protection for novel molecules, composition of matter and uses for drugs and inventions originating from our research and development efforts, as well as new manufacturing and other processes and formulations. In each case, we carefully balance the value of patent protection against the advantage of keeping the know-how regarding the invention confidential. We aim to position the claims of our applications to exploit gaps in prior art.
We have two patent families covering dipraglurant as a composition of matter and its polymorphs which are useful as mGlu5 NAMs: 71 patents have been granted to us, including 4 in the United States and 67 in other international jurisdictions (Austria, Belgium, Bulgaria, Cyprus, Czech Republic, Denmark, Estonia, Finland, France, Germany, Great Britain, Greece, Hungary, Iceland, Ireland, Italy, Lithuania, Luxembourg, Monaco, Netherlands, Norway, Poland, Portugal, Romania, Slovakia, Slovenia, Spain, Sweden, Switzerland, Turkey, Armenia, Australia, Azerbaijan, Belarus, Brazil, Canada, China Hong Kong, Indonesia, India, Israel, Japan, Kazakhstan, South Korea, Kyrgyzstan, Moldova, Mexico, New Zealand, Philippines, Russia, Singapore, South Africa, Tajikistan and Turkmenistan). We also have 39 patent applications pending.
We have one patent family covering compounds which are useful as GABAB PAMs, of which 49 patents have been granted, including two in the United States and 47 in other international jurisdictions (Austria, Belgium, Bulgaria, Cyprus, Czech Republic, Denmark, Estonia, Finland, France, Germany, Great Britain, Greece, Hungary, Iceland, Ireland, Italy, Latvia, Lithuania, Luxembourg, Malta, Monaco, Netherlands, Norway, Poland, Portugal, Romania, Slovakia, Slovenia, Spain, Sweden, Switzerland, Turkey, Ukraine, Australia, Azerbaijan, Belarus, Brazil, Canada, China, India, Indonesia, Israel, Japan, Kazakhstan, New Zealand, Russia and South Africa).
Jointly with Janssen, we have 81 patents in three patent families covering compounds which are useful as mGlu2 PAMs, including ADX71149, which is explicitly exemplified and claimed as a compound and as a pharmaceutical composition, we have five patents in the United States and 86 in other international jurisdictions (Albania, Austria, Belgium, Bosnia & Herzegovina, Bulgaria, Croatia, Cyprus, Czech Republic, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Iceland, Ireland, Italy, Latvia, Lithuania, Luxembourg, North Macedonia, Turkey, Ukraine, United Kingdom, Algeria, Argentina, Australia, Bahrain, Brazil, Canada, China, Chile, Hong Kong, Israel, India, Indonesia, Japan, Kuwait, Republic of Korea, Malaysia, Mexico, New Zealand, Oman, Philippines, Qatar, Russia, Singapore, Saudi Arabia, South Africa, Vietnam, Taiwan, Thailand and United Arab Emirates).
Furthermore, we have 17 patents in two patent families covering compounds that have potential utility as mGlu4 PAMs, which include two patents granted in the United States and 15 in Europe (Belgium, France, Germany, Great Britain, Hungary, Italy, Spain and Switzerland). One family is owned by us and a second family is jointly owned by us and Merck & Co Inc. pursuant to our collaboration agreement for the development of mGlu4 PAM, which we entered into in 2007. Our granted patents have expiration dates ranging from 2025 to 2034 without extensions.
The patent positions of pharmaceutical and biotechnology companies are uncertain and involve complex legal and factual issues. There can be no assurance that patents that have been issued will be held valid and enforceable in a court of law. Even for patents that are held valid and enforceable, the legal process associated with obtaining such a judgment is time consuming and costly. Additionally, issued patents can be subject to opposition or other proceedings that can result in the revocation of the patent or maintenance of the patent in amended form, and potentially in a form that renders the patent without commercially relevant or broad coverage. Further, our competitors may be able to circumvent and otherwise design around our patents. Even if a patent is issued and enforceable,
58
because development and commercialization of pharmaceutical products can be subject to substantial delays, patents may expire early and provide only a short period of protection, if any, following the commercialization of a product covered by any of our patents. We may have to participate in interference proceedings declared by the U.S. Patent and Trademark Office, which could result in a loss of the patent or substantial cost to us.
U.S. and foreign patent rights and other proprietary rights exist that are owned by third parties and relate to pharmaceutical compositions and reagents, medical devices and equipment and methods for preparation, packaging and delivery of pharmaceutical compositions. We cannot predict with any certainty which, if any, of these rights will be considered relevant to our technology by authorities in the various jurisdictions where such rights exist, nor can we predict with certainty which, if any, of these rights will or may be asserted against us by third parties. We could incur substantial costs in defending ourselves and our partners against any such claims. Furthermore, parties making such claims may be able to obtain injunctive or other equitable relief, which could effectively block our ability to develop or commercialize some or all of our products in the U.S. and abroad and could result in the award of substantial damages. In the event of a claim of infringement, we or our partners may be required to obtain one or more licenses from third parties. There can be no assurance that we can obtain a license to any technology that we determine we need on reasonable terms, if at all, or that we could develop or otherwise obtain alternative technology. The failure to obtain licenses if needed may have a material adverse effect on our business, results of operations and financial condition.
We also rely on trade secret protection for our confidential and proprietary information. No assurance can be given that we can meaningfully protect our trade secrets. Others may independently develop substantially equivalent confidential and proprietary information or otherwise gain access to, or disclose, our trade secrets. Our success will depend in part on our ability to obtain and maintain patent protection for our drugs, preserve trade secrets, prevent third parties from infringing upon our proprietary rights and operate without infringing upon the proprietary rights of others, both in Switzerland and in other territories worldwide.
It is our policy to require our employees and consultants, outside scientific collaborators, sponsored researchers and other advisors who receive confidential information from us to execute confidentiality agreements upon the commencement of employment or consulting relationships with us. These agreements provide that all confidential information developed or made known to the individual during the course of the individual's relationship with us is to be kept confidential and not disclosed to third parties except in specific circumstances. The agreements provide that all inventions conceived by an employee shall be our property. There can be no assurance, however, that these agreements will provide meaningful protection or adequate remedies for our trade secrets in the event of unauthorized use or disclosure of such information.
Trademarks
We own trademarks for Addex Pharmaceuticals in Switzerland. We also have trademarks for AddeLite and ProxyLite in relation to our screening technologies in the United States, Switzerland and the People's Republic of China and, in the case of ProxyLite, the EU.
Competition
The development and commercialization of drugs is highly competitive. We compete with a variety of multinational pharmaceutical companies and specialized pharmaceutical companies, including products approved for marketing and/or product candidates under development, for each of the
59
product candidates and each of the indications for which we are developing our product candidates, as follows:
Dipraglurant for the treatment of PD-LID
Amantadine, Gocovri (extended release amantadine) and Deep Brain Stimulation (DBS) surgery are currently available therapies for the treatment of PD-LID. In addition, several drug candidates currently in clinical development could compete with dipraglurant for the treatment of PD-LID. Avanir Pharmaceuticals, Inc. is developing AVP-923 (NMDA antagonist), Neuraltus Pharmaceuticals, Inc. is developing NP002 (nicotine receptor agonist), Newron Pharmaceuticals, Inc. is developing safinamide (MAO-B inhibitor), Novartis Pharma AG is developing AQW051 (alpha 7 nAChR inhibitor), and Prilenia Therapeutics Inc. is developing pridopidine (D2 receptor agonist).
Dipraglurant for the treatment of dystonia
Currently available therapies include tetrabenazine (a dopamine antagonist), with a broad label for movement disorders, levodopa for levodopa responsive dystonia, botulinum toxin for focal and limb dystonia and DBS surgery. Other compounds, such as baclofen, anticholinergic drugs and benzodiazepines, are used off label or within the broad label context of treating muscle spasms. In addition, drug candidates currently in development could compete with dipraglurant for the treatment of dystonias, including MT10109 clostridium botulinum toxin, currently in development by Medytox Korea Co., Ltd. for cervical dystonia and transcranial magnetic stimulation.
GABAB PAM for the treatment of addiction
Currently available treatments of addiction include Buprenorphine (Suboxone®, Subutex®, Probuphine®, Sublocade™), naltrexone (Vivitrol®) to treat opioid addiction; bupropion (Zyban®) and varenicline (Chantix®) to treat nicotine addiction; and naltrexone (Vivitrol®), Acamprosate (Campral®), Disulfiram (Antabuse®) to treat alcohol addiction. Baclofen (a GABAB agonist) has been largely used off-label to treat alcohol abuse, and its approval is under review in France. In addition, several novel derivatives of baclofen are in clinical development and Astellas is in Phase I with ASP8062 as a novel GABAB PAM for substance-related disorders.
GABAB PAM for the treatment of CMT1A
Currently, there is no disease-modifying treatment available for CMT1A. Currently approved therapies for relief from certain symptoms of CMT1A, including musculoskeletal and neuropathic pain, include anti-inflammatory drugs, tricyclic antidepressants and anticonvulsants. In addition, Pharnext is seeking approval in the US and Europe of PXT3003, a novel oral fixed-dose combination of baclofen, naltrexone and sorbitol.
ADX71149 for the treatment of epilepsy
Currently available therapies treatment of epilepsy includes racetams such as Brivaracetam (Briviact) or Levetiracetam (Keppra); benzodiazepines such as diazepam (Valium), clonazepam (Klonopin), lorazepam (Ativan); carboxamides such as Carbamazepine (Carbatrol or Tegretol) and Eslicarbazepine (Aptiom); GABA analogs such as Gabapentin (Neurotin) or Pregabalin (Lyrica); Perampanel (Fycompa). Late stage drug candidates in development which could compete with ADX714419 include Ganaxolone, Cannabidiol (Epidiolex), Everolimus (Afinitor / Votubia), ZX008.
Government Regulation and Product Approval
Government authorities in the United States, at the federal, state and local levels, and in other countries, extensively regulate, among other things, the research, development, testing, manufacture,
60
packaging, storage, recordkeeping, labeling, advertising, promotion, distribution, marketing, import and export of pharmaceutical products, such as those we are developing. The processes for obtaining regulatory approvals in the United States and in foreign countries, along with subsequent compliance with applicable statutes and regulations, require the expenditure of substantial time and financial resources.
Marketing
United States Government Regulation
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or FDCA, and its implementing regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable United States requirements at any time during the drug development process, approval process or after approval, may subject an applicant to a variety of administrative or judicial sanctions, such as the FDA's refusal to approve pending new drug applications, or NDAs, withdrawal of an approval, imposition of a clinical hold, issuance of warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties.
The process required by the FDA before a drug may be marketed in the United States generally involves:
- •
- completion of pre-clinical laboratory tests, animal studies and formulation studies in compliance with the FDA's good laboratory practice, or
GLP, regulations, including laboratory evaluation of product chemistry, toxicity and formulation, as well as animal studies to assess potential safety and efficacy;
- •
- submission to the FDA of an Investigational New Drug, or IND, which must become effective before human clinical trials may begin;
- •
- approval by an independent institutional review board, or IRB, at each clinical site before each clinical trial may be initiated;
- •
- performance of adequate and well-controlled clinical trials in accordance with good clinical practice, or GCP, requirements to establish the
safety and
- •
- efficacy of the proposed drug for each indication;
- •
- submission to the FDA of an NDA;
- •
- satisfactory completion of an FDA advisory committee review, if applicable;
- •
- satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance
with cGMP requirements, and to assure that the facilities, methods and controls are adequate to preserve the drug's identity, strength, quality and purity; satisfactory completion of an FDA inspection
of selected clinical sites to assure compliance with GCPs and the integrity of the clinical data; payment of user fees; and
- •
- FDA review and approval of the NDA.
Clinical Trials
Prior to the initiation of clinical testing, a sponsor must submit to the FDA an IND, application to the FDA, including the results of pre-clinical studies, manufacturing information, analytical data and any available clinical data or literature. Some pre-clinical testing may continue even after the IND is
61
submitted. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to one or more proposed clinical trials and places the clinical trial on a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND may not result in the FDA allowing clinical trials to commence.
Clinical trials involve the administration of the investigational new drug to human subjects under the supervision of qualified investigators in accordance with GCP requirements, which include the requirement that all research subjects provide their informed consent in writing for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. In addition, an IRB at each institution participating in the clinical trial must review and approve the plan for any clinical trial before it commences at that institution, and the IRB must continue to oversee the clinical trial while it is being conducted. Information about certain clinical trials must be submitted within specific timeframes to the National Institutes of Health, or NIH, for public dissemination on their ClinicalTrials.gov website.
Human clinical trials are typically conducted in three sequential phases, which may overlap or be combined. In Phase 1, the drug is initially introduced into healthy human subjects or patients with the target disease or condition and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an initial indication of its effectiveness. In Phase 2, the drug typically is administered to a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. In Phase 3, the drug is administered to an expanded patient population, generally at geographically dispersed clinical trial sites, in well-controlled clinical trials to generate enough data to statistically evaluate the safety and efficacy of the product for approval, to establish the overall risk-benefit profile of the product and to provide adequate information for the labeling of the product.
Progress reports detailing the results of the clinical trials must be submitted, at least annually, to the FDA, and more frequently if certain serious adverse events occur. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, or at all. Furthermore, the FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB's requirements, or if the drug has been associated with unexpected serious harm to patients.
Marketing Approval
Assuming successful completion of the required clinical testing, the results of the pre-clinical and clinical studies, together with detailed information relating to the product's chemistry, manufacture, controls and proposed labeling, among other things, are submitted to the FDA as part of an NDA requesting approval to market the product for one or more indications. In most cases, the submission of an NDA is subject to a substantial application user fee. Under the Prescription Drug User Fee Act, or PDUFA, guidelines that are currently in effect, the FDA has a goal of ten months from the date of "filing" of a standard NDA for a new molecular entity to review and act on the submission. This review typically takes twelve months from the date the NDA is submitted to the FDA because the FDA has sixty days from submission to make a "filing" decision.
In addition, under the Pediatric Research Equity Act, certain NDAs or supplements to an NDA must contain data that are adequate to assess the safety and effectiveness of the drug for the claimed
62
indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements. Unless otherwise required by regulation, the pediatric data requirements do not apply to products with orphan designation.
The FDA also may require submission of a risk evaluation and mitigation strategy, or REMS, plan to ensure that the benefits of the drug outweigh its risks. The REMS plan could include medication guides, physician communication plans, assessment plans, and / or elements to assure safe use, such as restricted distribution methods, patient registries or other risk minimization tools.
The FDA conducts a preliminary review of all NDAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews an NDA to determine, among other things, whether the drug is safe and effective and whether the facility in which it is manufactured, processed, packaged or held meets standards designed to assure the product's continued safety, quality and purity.
The FDA may refer an application for a novel drug to an advisory committee. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA will typically inspect one or more clinical trial sites to assure compliance with GCP requirements.
The testing and approval process for an NDA requires substantial time, effort and financial resources, and takes several years to complete. Data obtained from pre-clinical and clinical testing are not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The FDA may not grant approval of an NDA on a timely basis, or at all.
After evaluating the NDA and all related information, including the advisory committee recommendation, if any, and inspection reports regarding the manufacturing facilities and clinical trial sites, the FDA may issue an approval letter, or, in some cases, a complete response letter. A complete response letter generally contains a statement of specific conditions that must be met in order to secure final approval of the NDA and may require additional clinical or pre-clinical testing in order for FDA to reconsider the application. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If and when those conditions have been met to the FDA's satisfaction, the FDA will typically issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications.
Even if the FDA approves a product, it may limit the approved indications for use of the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies, including Phase 4 clinical trials, be conducted to further assess a drug's safety after approval, require testing and surveillance programs to monitor the product after
63
commercialization, or impose other conditions, including distribution and use restrictions or other risk management mechanisms under a REMS, which can materially affect the potential market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-marketing studies or surveillance programs. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes, and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Orange Book Listing
In seeking approval for a drug through an NDA, applicants are required to list with the FDA certain patents whose claims cover the applicant's product. Upon approval of an NDA, each of the patents listed in the application for the drug is then published in the FDA's Approved Drug Products with Therapeutic Equivalence Evaluations, known as the Orange Book. Any applicant who files an Abbreviated New Drug Application, or ANDA, seeking approval of a generic equivalent version of a drug listed in the Orange Book referencing a drug listed in the Orange Book must certify, for each patent listed in the Orange Book for the referenced drug, to the FDA that (1) no patent information on the drug product that is the subject of the application has been submitted to the FDA, (2) such patent has expired, (3) the date on which such patent expires or (4) such patent is invalid or will not be infringed upon by the manufacture, use or sale of the drug product for which the application is submitted. The fourth certification described above is known as a paragraph IV certification. A notice of the paragraph IV certification must be provided to each owner of the patent that is the subject of the certification and to the holder of the approved NDA to which the ANDA. The applicant may also elect to submit a "section viii" statement certifying that its proposed label does not contain (or carves out) any language regarding the patented method-of-use rather than certify to a listed method-of-use patent. This section viii statement does not require notice to the patent holder or NDA owner. There might also be no relevant patent certification.
If the reference NDA holder and patent owners assert a patent challenge directed to one of the Orange Book listed patents within 45 days of the receipt of the paragraph IV certification notice, the FDA is prohibited from approving the application until the earlier of 30 months from the receipt of the paragraph IV certification expiration of the patent, settlement of the lawsuit, or a decision in the infringement case that is favorable to the applicant. Even if the 45 days expire, a patent infringement lawsuit can be brought and could delay market entry, but it would not extend the FDA-related 30-month stay of approval.
Post-Approval Requirements
Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications, manufacturing changes or other labeling claims, are subject to further testing requirements and prior FDA review and approval. There also are continuing annual user fee requirements for any marketed products and the establishments at which such products are manufactured, as well as application fees for supplemental applications with clinical data.
Even if the FDA approves a product, it may limit the approved indications for use of the product, require that contraindications, warnings or precautions be included in the product labeling, including a boxed warning, require that post-approval studies, including Phase 4 clinical trials, be conducted to further assess a drug's safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution restrictions or other risk management mechanisms under a REMS, which can materially affect the potential market and
64
profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-marketing studies or surveillance programs.
In addition, drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies, and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with cGMP requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon the sponsor and any third party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in mandatory revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
- •
- restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls;
- •
- fines, warning letters or holds on post-approval clinical trials;
- •
- refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product approvals;
- •
- product seizure or detention, or refusal to permit the import or export of products; or
- •
- injunctions or the imposition of civil or criminal penalties.
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label, although physicians, in the practice of medicine, may prescribe approved drugs for unapproved indications. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to liability.
In addition, the distribution of prescription pharmaceutical products is subject to the Prescription Drug Marketing Act, or PDMA, which regulates the distribution of drugs and drug samples at the federal level, and sets minimum standards for the registration and regulation of drug distributors by the states. Both the PDMA and state laws limit the distribution of prescription pharmaceutical product samples and impose requirements to ensure accountability in distribution.
Federal and State Fraud and Abuse, Data Privacy and Security, and Transparency Laws and Regulations
In addition to FDA restrictions on marketing of pharmaceutical products, federal and state healthcare laws and regulations restrict business practices in the biopharmaceutical industry. These laws may impact, among other things, our current and future business operations, including our clinical research activities, and proposed sales, marketing and education programs and constrain the business or financial arrangements and relationships with healthcare providers and other parties through which we market, sell and distribute our products for which we obtain marketing approval. These laws include
65
anti-kickback and false claims laws and regulations, data privacy and security, and transparency laws and regulations, including, without limitation, those laws described below.
The federal Anti-Kickback Statute prohibits, among other things, individuals or entities from knowingly and willfully offering, paying, soliciting or receiving remuneration, directly or indirectly, overtly or covertly, in cash or in kind to induce or in return for purchasing, leasing, ordering or arranging for or recommending the purchase, lease or order of any item or service reimbursable under Medicare, Medicaid or other federal healthcare programs. The term "remuneration" has been broadly interpreted to include anything of value. The federal Anti-Kickback Statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers and formulary managers on the other hand. Although there are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, the exceptions and safe harbors are drawn narrowly. Practices that involve remuneration that may be alleged to be intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exception or safe harbor.
The reach of the federal Anti-Kickback Statute was also broadened by the Patient Protection and Affordable Care Act of 2010, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively the ACA, which, among other things, amended the intent requirement of the federal Anti-Kickback Statute such that a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it in order to have committed a violation. In addition, the ACA provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act and the civil monetary penalties statute.
The federal civil and criminal false claims laws, including the False Claims Act, which prohibit, among other things, any individual or entity from knowingly presenting, or causing to be presented, a false claim for payment to the federal government or knowingly making, using or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government. A claim includes "any request or demand" for money or property presented to the U.S. government. Several pharmaceutical and other healthcare companies have been prosecuted under these laws for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. Other companies have been prosecuted for causing false claims to be submitted because of the companies' marketing of products for unapproved, and thus non-reimbursable, uses.
The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, created additional federal criminal statutes that prohibit, among other things, knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private third party payors and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and their respective implementing regulations, impose certain requirements relating to the privacy, security and transmission of individually identifiable health information without appropriate authorization on certain health plans, healthcare clearinghouses and certain healthcare providers, known as covered entities, and their respective business associates, independent contractors that perform certain services involving the use or disclosure of individually identifiable health information. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce HIPAA and seek attorneys' fees and costs associated with pursuing federal civil actions.
66
The federal Physician Payments Sunshine Act requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children's Health Insurance Program, with specific exceptions, to report annually to the Centers for Medicare & Medicaid Services, or CMS, information related to payments or other transfers of value made to physicians and teaching hospitals, and applicable manufacturers and applicable group purchasing organizations to report annually to CMS ownership and investment interests held by physicians and their immediate family members.
We may also be subject to state and foreign law equivalents of each of the above federal laws; state laws that require manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; state laws that require pharmaceutical companies to comply with the pharmaceutical industry's voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or that otherwise restrict payments that may be made to healthcare providers; state and local laws that require the registration of pharmaceutical sales representatives; as well as state and foreign laws that govern the privacy and security of health information in some circumstances, many of which differ from each other in substantial ways and often are not preempted by HIPAA, thus complicating compliance efforts.
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. Because of the breadth of these laws and the narrowness of the statutory exceptions and regulatory safe harbors available, it is possible that some of our business activities could be subject to challenge under one or more of such laws. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to substantial civil, criminal and administrative penalties, damages, fines, disgorgement, imprisonment, exclusion from participating in government funded healthcare programs, such as Medicare and Medicaid, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, contractual damages, reputational harm and the curtailment or restructuring of our operations. To the extent that any of our products are sold in a foreign country, we may be subject to similar foreign laws and regulations, which may include, for instance, applicable post-marketing requirements, including safety surveillance, anti-fraud and abuse laws and implementation of corporate compliance programs and reporting of payments or transfers of value to healthcare professionals.
Coverage and Reimbursement
Market acceptance and sales of any drug products depend in part on the extent to which reimbursement for drug products will be available from third party payors, including government health administration authorities, managed care organizations and other private health insurers. third party payors decide which drug products they will pay for and establish reimbursement levels. third party payors often rely upon Medicare coverage policy and payment limitations in setting their own coverage and reimbursement policies. However, decisions regarding the extent of coverage and amount of reimbursement to be provided for drug products are made on a payor-by-payor basis. One payor's determination to provide coverage for a drug product does not assure that other payors will also provide coverage, and adequate reimbursement, for the drug product. Additionally, a third party payor's decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Each payor determines whether or not it will provide coverage for a drug product, what amount it will pay the manufacturer for the therapy, and on what tier of its formulary it will be placed. The position on a payor's list of covered drugs, or formulary, generally determines the co-payment that a patient will need to make to obtain the drug product and can
67
strongly influence the adoption of such drug product by patients and physicians. Patients who are prescribed drug products for their conditions and providers prescribing such drug products generally rely on third party payors to reimburse all or part of the associated costs. Patients are unlikely to use a drug product unless coverage is provided and reimbursement is adequate to cover a substantial portion of the cost of the drug product.
Reimbursement by a third party payor may depend upon a number of factors, including the third party payor's determination that a drug product is neither experimental nor investigational, safe, effective, and medically necessary, appropriate for the specific patient, cost-effective, supported by peer-reviewed medical journals and included in clinical practice guidelines.
Third party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular drug products. Even if reimbursement is available, the level of reimbursement is unpredictable. Inadequate coverage and reimbursement can impact the demand for, or the price of, drug products. If coverage and adequate reimbursement are not available, or are available only to limited levels, drug products may not be successfully commercialized. Further, adequate third party payor reimbursement may not be available to enable price levels sufficient to realize appropriate returns on investment in drug product development.
In addition, the federal government and state legislatures have continued to implement cost containment programs, including price controls and restrictions on coverage and reimbursement. To contain costs, governmental healthcare programs and third party payors are increasingly challenging the price, scrutinizing the medical necessity and reviewing the cost-effectiveness of drug products.
Impact of Healthcare Reform on our Business
In the United States and some foreign jurisdictions, there have been, and continue to be, several legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of drug product candidates, restrict or regulate post-approval activities, and affect the profitable sale of drug product candidates.
Among policy makers and payors in the United States and elsewhere, there is substantial interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and / or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been affected by major legislative initiatives. In March 2010, the ACA was passed, which substantially changed the way healthcare is financed by both the government and private insurers, and greatly impacts the U.S. pharmaceutical industry. The ACA, among other things: (i) increased the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program and extends the rebate program to individuals enrolled in Medicaid managed care organizations; (ii) established an annual, nondeductible fee on any entity that manufactures or imports certain specified branded prescription drugs and biologic agents apportioned among these entities according to their market share in some government healthcare programs; (iii) expanded the availability of lower pricing under the 340B drug pricing program by adding new entities to the program; (iv) increased the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program, to 23.1% and 13% of the average manufacturer price for most branded and generic drugs, respectively and capped the total rebate amount for innovator drugs at 100% of the Average Manufacturer Price, or AMP; (v) expanded the eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for individuals with income at or below 133% of the federal poverty level, thereby potentially increasing manufacturers' Medicaid rebate liability; (vi) established a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% (and 70%, commencing January 1, 2019) point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a
68
condition for the manufacturer's outpatient drugs to be covered under Medicare Part D; (vii) created a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; and (viii) established a Center for Medicare Innovation at CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug.
Some of the provisions of the ACA have yet to be implemented, and there have been judicial and Congressional challenges to certain aspects of the ACA, as well as recent efforts by the Trump administration to repeal or replace certain aspects of the ACA. Since January 2017, President Trump has signed two Executive Orders and other directives designed to delay the implementation of certain provisions of the ACA or otherwise circumvent some of the requirements for health insurance mandated by the ACA. Concurrently, Congress has considered legislation that would repeal or repeal and replace all or part of the ACA. While Congress has not passed comprehensive repeal legislation, two bills affecting the implementation of certain taxes under the ACA have been signed into law. On December 22, 2017, new legislation was signed into law (H.R. 1, "An Act to provide for reconciliation pursuant to titles II and V of the concurrent resolution on the budget for fiscal year 2018," or the Tax Cuts and Jobs Act) that significantly revised the U.S. Internal Revenue Code of 1986, as amended, or the Code. The Tax Cuts and Jobs Act includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the ACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the "individual mandate". Additionally, on January 22, 2018, President Trump signed a continuing resolution on appropriations for fiscal year 2018 that delayed the implementation of certain ACA-mandated fees, including the so-called "Cadillac" tax on certain high cost employer-sponsored insurance plans, the annual fee imposed on certain health insurance providers based on market share, and the medical device excise tax on non-exempt medical devices. Further, the Bipartisan Budget Act of 2018, or the BBA, among other things, amends the ACA, effective January 1, 2019, to close the coverage gap in most Medicare drug plans, commonly referred to as the "donut hole".
Other legislative changes have been proposed and adopted since the ACA was enacted. These changes include aggregate reductions to Medicare payments to providers of 2% per fiscal year pursuant to the Budget Control Act of 2011, which began in 2013, and due to subsequent legislative amendments to the statute, including the BBA, will remain in effect through 2027 unless additional Congressional action is taken. The American Taxpayer Relief Act of 2012, among other things, further reduced Medicare payments to several providers, including hospitals and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These new laws may result in additional reductions in Medicare and other healthcare funding, which could have an adverse effect on customers for our drug candidates, if approved, and, accordingly, our financial operations.
Additionally, there has been heightened governmental scrutiny in the United States of pharmaceutical pricing practices in light of the rising cost of prescription drugs and biologics. Such scrutiny has resulted in several recent congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for products. At the federal level, the Trump administration's budget proposal for fiscal year 2019 contains further drug price control measures that could be enacted during the 2019 budget process or in other future legislation, including, for example, measures to permit Medicare Part D plans to negotiate the price of certain drugs under Medicare Part B, to allow some states to negotiate drug prices under Medicaid, and to eliminate cost sharing for generic drugs for low-income patients. While any proposed measures will require authorization through additional legislation to become effective, Congress and the Trump administration have each indicated that it will continue to seek new legislative and / or administrative measures to control drug costs. At the state
69
level, legislatures are increasingly passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
Manufacturing and Supply
We rely on third party manufacturing and supply partners for our supply for, preclinical studies and clinical trials. We currently do not have in-house facilities to manufacture our research and development, preclinical and clinical drug supplies.
Legal Proceedings
From time to time, we may become involved in litigation or other legal proceedings relating to claims arising from the ordinary course of business. There are currently no claims or actions pending against us that, in the opinion of our management, are likely to have a material adverse effect on our results of operations, financial condition or cash flows.
C. Organizational Structure
The following diagram illustrates our corporate structure:
D. Property, Plants and Equipment.
Our registered office is located in 12, Chemin des Aulx, Plan-les-Ouates, Geneva, Switzerland. Our headquarters are located in Campus Biotech, Chemin des Mines 9, Geneva, Switzerland, and consist of approximately 500 square meters of office and lab space, which houses our in-house R&D function. We also have one office in San Francisco, California, United States for our subsidiary Addex Pharmaceuticals Inc. We may require additional space and facilities as our business expands.
Item 4A. Unresolved Staff Comments.
Not applicable.
Item 5. Operating and Financial Review and Prospects.
You should read the following discussion in conjunction with our audited consolidated financial statements, including the related notes thereto, included in this Annual Report on Form 20-F. In addition to historical information, this discussion contains forward-looking statements that involve risks and uncertainties. You should read the sections of this Annual Report on Form 20-F entitled "Item 3D—Risk
70
Factors" and "Special Note Regarding Forward-Looking Statements" for a discussion of the factors that could cause our actual results to differ materially from our expectations.
Overview
We are a clinical-stage pharmaceutical company focused on the development and commercialization of an emerging class of novel orally available small molecule drugs known as allosteric modulators. Allosteric modulators target a specific receptor or protein and alter the effect of the body's own signaling molecules on their target through a novel mechanism of action. These innovative small molecule drug candidates offer several potential advantages over conventional non-allosteric molecules and may offer an improved therapeutic approach to existing drug treatments. To date, our research and development efforts have been primarily focused on building a portfolio of proprietary candidates based on our allosteric modulator development capability. The allosteric modulator principle has broad applicability across a wide range of biological targets and therapeutic areas, but our primary focus is on G-protein coupled receptors, or GPCR, targets implicated in neurological diseases, where we believe there is a clear medical need for new therapeutic approaches.
Using our allosteric modulator discovery capabilities, we have developed a pipeline of proprietary clinical and preclinical stage drug candidates. We or our partners are developing these clinical and preclinical stage proprietary drug candidates for diseases for which there are no approved therapies or where improved therapies are needed. These include levodopa induced dyskinesia associated with Parkinson's disease, non-parkinsonian dystonia, addiction, epilepsy, Charcot-Marie-Tooth type 1A neuropathy, or CMT1A, and other neurodegenerative diseases. Some of these indications are classified as rare diseases that may allow for orphan drug designation by regulatory agencies in major commercial markets, such as the United States, Europe and Japan. Orphan drug designation may entitle the recipient to benefits in the jurisdiction granting the designation, such as market exclusivity following approval and assistance in clinical trial design, a reduction in user fees or tax credits related to development expense.
Our Partner, Janssen Pharmaceuticals Inc., or Janssen, has licensed worldwide rights to our second clinical program, ADX71149 (mGlu2 PAM), and is responsible for development, manufacture and commercialization. Janssen has completed two Phase 2 studies in schizophrenia and anxious depression generating mixed results. Janssen has conducted several preclinical studies in epilepsy and continues to evaluate the program in other neurological disorders.
Our Partner, Indivior PLC, or Indivior, has licensed worldwide rights to our GABAB PAM, program and is responsible for all development, manufacture and commercialization of any selected GABAB PAM drug candidate. Under the agreement, we are responsible for executing a research program funded by Indivior to discovery novel GABAB PAM drug candidates. Indivior's primary therapeutic focus is addiction and under the agreement we have the right to select certain drug candidates for future independent development in certain exclusive indications including CMT1A. The program is currently in late lead optimization.
In addition, we are conducting a number of early stage research programs including mGlu7 NAM, mGlu2 NAM, mGlu4 PAM and mGlu3 PAM.
We were founded in May 2002 and completed our initial public offering of ordinary shares on the Swiss SIX Exchange in May 2007. On January 29, 2020, we listed American Depositary Shares (ADSs) representing our ordinary shares on the Nasdaq Stock Market following the United States Securities and Exchange Commission (SEC) having declared our registration statements on Forms F1 and F6 effective. Our operations to date have included organizing and staffing our company, raising capital, out-licensing rights to our mGlu2 PAM and GABAB PAM programs and conducting preclinical studies and clinical trials. To date, we have generated CHF 57 million of revenue from the sale of license rights to certain of our research programs. We have historically financed our operations mainly through the
71
sale of equity. As of February 29, 2020, we had raised an aggregate of CHF 325 million of gross proceeds.
We have never been profitable and had incurred significant net losses in each period since our inception. Our net losses were CHF 14.8 million, CHF 1.7 million and CHF 3.3 million for years ended December 31, 2019, 2018 and 2017, respectively. As of December 31, 2019, we had accumulated losses of CHF 300.8 million. We expect to continue to incur significant expenses and operating losses for the foreseeable future. We anticipate that our expenses will increase significantly in connection with our ongoing activities as we:
- •
- continue to invest in the research and development of our allosteric modulator discovery platform and pipeline, and specifically in connection
with our Phase 2b/3 clinical trial of dipraglurant for the treatment of PD-LID and any additional clinical trials that we may conduct for product candidates;
- •
- hire additional research and development, and general and administrative personnel;
- •
- maintain, expand and protect our intellectual property portfolio;
- •
- identify and in-license or acquire additional product candidates; and
- •
- incur additional costs associated with operating as a public company in the United States.
We will need substantial additional funding to support our operating activities as we advance our research and product candidates through clinical development, seek regulatory approval and prepare for, and if any of our product candidates are approved, proceed to commercialization. Adequate funding may not be available to us on acceptable terms, or at all.
We have no manufacturing facilities, and all of our manufacturing activities are contracted out to third parties. Additionally, we currently utilize third-party clinical research organizations, or CROs, to carry out our clinical development and trials. We do not yet have a sales organization.
License Agreement with Indivior
In January 2018, we entered into an agreement with Indivior for the discovery, development and commercialization of novel GABAB PAM compounds for the treatment of addiction and other CNS diseases. This agreement included the selected clinical candidate, ADX71441. In addition, Indivior agreed to fund a research program at Addex to discover novel GABAB PAM compounds.
Indivior has sole responsibility, including funding liability, for development of selected compounds under the agreement through preclinical and clinical trials, as well as registration procedures and commercialization, if any, worldwide. Indivior has the right to design development programs for selected compounds under the agreement. Through our participation in a joint development committee, we review, in an advisory capacity, any development programs designed by Indivior. However, Indivior has authority over all aspects of the development of such selected compounds.
Under terms of the agreement, we have granted Indivior an exclusive license to use relevant patents and know-how in relation to the development and commercialization of product candidates selected by Indivior. Subject to agreed conditions, Addex and Indivior jointly own all intellectual property rights that are jointly developed, and Addex or Indivior individually own all intellectual property rights that Addex or Indivior develop individually. Addex has retained the right to select compounds from the research program for further development in areas outside the interest of Indivior including Charcot-Marie-Tooth type 1A neuropathy, or CMT1A. Under certain conditions, but subject to certain consequences, Indivior may terminate the agreement.
Under terms of the agreement, we received a non-refundable upfront fee of $5.0 million for the right to use the clinical candidate, ADX71441, including all materials and know-how related to this
72
clinical candidate. In addition, we are eligible for payments on successful achievement of pre-specified clinical, regulatory and commercial milestones totaling $330 million, and royalties on net sales of mid-single digits to low teens double-digit. On February 14th, 2019, Indivior terminated the development of their selected compound, ADX71441.
Separately, Indivior funds research at Addex, based on a research plan to be mutually agreed between the parties, to discover novel GABAB PAM compounds. These future novel GABAB PAM compounds, if selected by Indivior, become licensed compounds. We agreed with Indivior to an initial research term of two years, that can be extended by twelve-month increments and a minimum annual funding of $2 million for the Addex R&D costs incurred. Following Indivior's selection of one newly identified compound, Addex has the right to also select one additional newly identified compound. Addex is responsible for the funding of all development and commercialization costs of its selected compounds and Indivior has no rights to the Addex selected compounds. The initial two-year research term was expected to run from May 2018 to April 2020. In 2019, Indivior agreed an additional research funding of $1.6 million, for the research period.
The contract contains two distinct material promises and performance obligations: (1) the selected compound ADX71441 which falls within the definition of a licensed compound, whose rights of use and benefits thereon was transferred in January 2018 and, (2) the research services to be conducted by Addex and funded by Indivior to discover novel GABAB PAM compounds for clinical development that may be discovered over the research term of the agreement and selected by Indivior.
License Agreement with Janssen
Under our agreement with Janssen Pharmaceuticals Inc. (formerly known as Ortho-McNeil-Janssen Pharmaceuticals Inc), or Janssen, we granted Janssen an exclusive license to use relevant patents and know-how in relation to the development and commercialization of product candidates selected by Janssen under the agreement and a non-exclusive worldwide license to conduct research on the collaboration compounds using relevant patents and know-how. Subject to certain conditions, we and they agreed to own, jointly, all intellectual property rights that we develop jointly and, individually, all intellectual property rights that either party develops individually. Under certain conditions, but subject to certain consequences, Janssen may terminate the agreement for any reason, subject to a 90-day notice period.
Janssen has sole responsibility, including funding liability, for development of selected compounds under the agreement through preclinical and clinical trials, as well as registration procedures and commercialization, if any, in the United States, Japan, the United Kingdom, Germany, France, Spain and Italy. Janssen has the right to design development programs for selected compounds under the agreement. Through our participation in a joint development committee, we review, in an advisory capacity, any development programs designed by Janssen. However, Janssen has authority over all aspects of the development of selected compounds and may develop or commercialize third-party compounds.
Under the terms of the Janssen agreement, we received an upfront fee of CHF 4.6 million and research funding of CHF 6.4 million during the research period, which ran from 2005 to 2007. In addition, we are eligible for payments on successful achievement of pre-specified clinical and regulatory milestones and a low double-digit royalty on net sales. We received a CHF 1.5 million milestone payment in relation to the entry of ADX71149 into Phase 1 in July 2009 and a CHF 2.6 million milestone payment in relation to the entry of ADX71149 into Phase 2 in June 2011. We are eligible for a further €109 million in success-based development and regulatory milestones and low double digit royalties on net sales.
73
Components of Results of Operations
Revenue
From the beginning of January 2017 through December 2019, we recognized CHF 8.8 million as revenue primarily under our license agreement with Indivior. We do not have approval to market or commercialize any of our product candidates, we have never generated revenue from the sale of products and we do not expect to generate any revenue from product sales for the foreseeable future. Prior to approval of a product candidate, we will seek to generate revenue from a combination of license fees, milestone payments in connection with collaborative or strategic relationships, royalties resulting from the licensing of our drug candidates and payments from sponsored research and development activities.
Revenue from collaborative arrangements comprises the fair value for the sale of products and services, net of value-added tax, rebates and discounts. Revenue from the rendering of services is recognized in the accounting period in which the services are rendered, by reference to completion of the specific transaction assessed on the basis of the actual service provided as a proportion of the total service to be provided. Revenue from collaborative arrangements may include the receipt of non-refundable license fees, milestone payments, and research and development payments. When we have continuing performance obligations under the terms of the arrangements, non-refundable fees and payments are recognized as revenue by reference to the completion of the performance obligation and the economic substance of the agreement.
Our revenue has varied, and we expect revenue to continue to vary, substantially from year to year, depending on the structure and timing of milestone events, as well as our development and commercialization strategies and those of our collaboration partners for our product candidates. We, therefore, believe that historical period to period comparisons are not meaningful and should not be relied upon as an indicator of our future revenue and performance potential.
Other Income
From the beginning of January 2017 through December 2019, we recognized CHF 1.2 million as other income primarily relating to grants from The Michael J. Fox Foundation for Parkinson's Research, or MJFF, relating to certain clinical activities related to dipraglurant development in Parkinson's disease levodopa-induced dyskinesia, or PD-LID, and TrKB PAM discovery activities.
Grants are recognized at their fair value where there is reasonable assurance that the grant will be received and that we will comply with all associated conditions. Grants relating to costs are recognized as other income in the statement of income over the period necessary to match them with the costs that they are intended to compensate.
Operating Expenses
Research and Development Costs
From the beginning of January 2017 through December 2019, we incurred CHF 20 million in research and development costs. They consist mainly of direct research costs, which include: costs associated with the use of contract research organizations, or CROs, and consultants hired to assist in our research and development activities, personnel costs, share-based compensation for our employees and consultants, costs related to regulatory affairs and intellectual property, as well as depreciation for assets used in research and development activities.
We typically use our employee, consultant and infrastructure resources across our research and development programs. We track by program the directly attributable costs from CROs and consultants.
74
The following table provides a breakdown of our outsourced research and development costs that are directly attributable to the specified programs for the years ended December 31, 2019, 2018 and 2017:
| |
For the years ended December 31, |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2019 | 2018 | 2017 | |||||||
| |
(CHF in thousands) |
|||||||||
Dipraglurant—PD-LID |
7,044 | 1,416 | 816 | |||||||
GABAB PAM |
1,516 | 477 | — | |||||||
Other discovery programs |
658 | 486 | 264 | |||||||
| | | | | | | | | | | |
Total outsourced research and development costs |
9,218 | 2,379 | 1,080 | |||||||
| | | | | | | | | | | |
We expect our research and development costs will increase for the foreseeable future as we seek to advance the development of our programs. At this time, we cannot reasonably estimate or know the nature, timing and estimated costs of the efforts that will be necessary to complete the development of our product candidates. We are also unable to predict when, if ever, material net cash inflows will commence from sales of our product candidates. This is due to the numerous risks and uncertainties associated with developing such product candidates, including:
- •
- uncertainty to discover efficacious clinical candidate;
- •
- uncertainty to be able to efficiently manufacture and distribute drug products;
- •
- competitor intellectual property restraining our freedom to operate;
- •
- the number of patients and sites required for clinical trials;
- •
- the length of time required to enroll patients, run the clinical study and analyze the results; and
- •
- the results of our clinical trials.
In addition, the probability of success for any of our product candidates will depend on numerous factors, including competition, manufacturing capability and commercial viability. A change in the outcome of any of these variables with respect to the development of any of our product candidates would significantly change the costs, timing and viability associated with the development of that product candidate.
General and Administrative Costs
General and administrative costs consist primarily of personnel costs, including salaries, benefits and share-based compensation cost for our employees as well as corporate facility costs not otherwise included in research and development expenses, legal fees related to corporate matters and fees for accounting and financial or tax consulting services.
We anticipate that our general and administrative costs will increase in the future to support continued research and development activities. We also anticipate that we will incur increased accounting, audit, legal, compliance and director and officer insurance costs, as well as investor and public relations expenses, associated with being listed on the Nasdaq stock market.
Finance Result, Net
Finance result net, consists mainly of currency exchange differences, interest expenses relating to the negative interest rates on Swiss franc cash deposits since January 2018 partially offset by positive interest income on USD bank deposits and short term deposits since the year ended December 31, 2019.
75
Taxation
We are subject to corporate taxation in Switzerland. We are also entitled under Swiss laws to carry forward any losses incurred for a period of seven years and can offset our losses carried forward against future taxes. As of December 31, 2019, we had tax loss carry forwards totaling CHF 190 million of which CHF 16 million will expire by the end of 2020. Deferred income taxes are not recognized as we do not believe it is probable that we will generate sufficient taxable profits to utilize these tax loss carry forwards.
Analysis of Results of Operations
The following table presents our consolidated results of operations for the fiscal years 2019, 2018 and 2017.
| |
For the years ended December 31, |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2019 | 2018 | 2017 | |||||||
| |
(CHF in thousands) |
|||||||||
Revenue |
2,763 | 6,044 | — | |||||||
Other Income |
71 | 659 | 500 | |||||||
Research and development costs |
(12,454 | ) | (4,919 | ) | (2,629 | ) | ||||
General and administrative costs |
(4,984 | ) | (3,209 | ) | (1,106 | ) | ||||
Operating loss |
(14,604 | ) | (1,425 | ) | (3,235 | ) | ||||
Finance income |
37 | — | — | |||||||
Finance expense |
(213 | ) | (220 | ) | (45 | ) | ||||
| | | | | | | | | | | |
Net loss |
(14,780 | ) | (1,645 | ) | (3,280 | ) | ||||
| | | | | | | | | | | |
Year Ended December 31, 2019 Compared to Year Ended December 31, 2018
Revenue
The following table sets forth our revenue in 2019 and 2018.
| |
For the years ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2019 | 2018 | |||||
| |
(CHF in thousands) |
||||||
Fees from sale of license rights |
— | 4,876 | |||||
Collaborative research funding |
2,763 | 1,168 | |||||
| | | | | | | | |
Total |
2,763 | 6,044 | |||||
| | | | | | | | |
Revenue decreased by CHF 3.3 million in 2019 compared to 2018 primarily due to the absence of the USD 5.0 million (CHF 4.8 million) upfront payment received from Indivior in January 2018 partially offset by the increase in collaborative research funding from Indivior.
76
Other Income
The following table sets forth the other income in 2019 and 2018.
| |
For the years ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2019 | 2018 | |||||
| |
(CHF in thousands) |
||||||
Research grants |
49 | 609 | |||||
Other service income |
22 | 50 | |||||
| | | | | | | | |
Total |
71 | 659 | |||||
| | | | | | | | |
Other income decreased by CHF 0.6 million in 2019 compared to 2018 primarily due to the absence of MJFF research grants recognized in 2018. In 2019, research grants relate to amounts recognized under our Eurostars/Innosuisse grant award. Other service income relates to consulting services performed by our finance and administration department.
Research and Development Expenses
The following table sets forth our research and development expenses in 2019 and 2018.
| |
For the years ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2019 | 2018 | |||||
| |
(CHF in thousands) |
||||||
Dipraglurant—PD-LID |
7,044 | 1,416 | |||||
GABAB PAM |
1,516 | 477 | |||||
Other discovery programs |
658 | 486 | |||||
| | | | | | | | |
Subtotal outsourced R&D per program |
9,218 | 2,379 | |||||
| | | | | | | | |
Staff costs |
1,956 | 1,307 | |||||
Depreciation and amortization |
264 | 2 | |||||
Professional fees |
153 | 494 | |||||
Laboratory consumables |
230 | 144 | |||||
Patent maintenance and registration costs |
268 | 262 | |||||
Operating leases |
— | 134 | |||||
Short-term leases |
27 | — | |||||
Other operating expenses |
338 | 197 | |||||
| | | | | | | | |
Subtotal unallocated R&D expenses |
3,236 | 2,540 | |||||
| | | | | | | | |
Total |
12,454 | 4,919 | |||||
| | | | | | | | |
Our research and development costs increased by CHF 7.5 million in 2019 compared to 2018 primarily due to CHF 5.6 million of increased outsourced R&D expenses related to our dipraglurant PD-LID program and CHF 1.0 million related to our GABAB PAM program.
In addition to increased directly attributable CRO and consulting costs we significantly increased our headcount in 2019 resulting in a CHF 0.6 million increase in staff costs.
The increase in depreciation and amortization relates to the recognition of the right-of-use assets of long-term operating leases in accordance with IFRS16 effective from January 1, 2019.
77
General and Administrative Costs
The following table sets forth our general and administrative costs in 2019 and 2018.
| |
For the years ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2019 | 2018 | |||||
| |
(CHF in thousands) |
||||||
Staff costs |
2,333 | 918 | |||||
Depreciation and amortization |
69 | 1 | |||||
Professional fees |
1,932 | 1,809 | |||||
Operating leases |
— | 45 | |||||
Other operating costs |
650 | 436 | |||||
| | | | | | | | |
Total |
4,984 | 3,209 | |||||
| | | | | | | | |
General and administrative costs increased by CHF 1.8 million in 2019 compared to 2018 primarily due to the increase of headcount and costs related to preparing the listing of ADSs on the Nasdaq Stock Market.
The increase in depreciation and amortization relates to the recognition of the right-of-use assets of long-term operating leases in accordance with IFRS16 effective from January 1, 2019.
Other operating costs increased in 2019 due to the travels, insurance premiums and associated listing costs.
Finance Result, Net
Finance result net, remained stable in 2019 compared to 2018 and primarily relates to currency exchange differences, interest expenses relating to the negative interest rates on Swiss franc cash deposits since January 2018 partially offset by positive interest income on USD bank deposits and short term deposits since the year ended December 31, 2019.
Year Ended December 31, 2018 Compared to Year Ended December 31, 2017
Revenue
The following table sets forth our revenue in 2018 and 2017.
| |
For the years ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2018 | 2017 | |||||
| |
(CHF in thousands) |
||||||
Fees from sale of license rights |
4,876 | — | |||||
Collaborative research funding |
1,168 | — | |||||
| | | | | | | | |
Total |
6,044 | — | |||||
| | | | | | | | |
Revenue increased to CHF 6.0 million in 2018 compared to zero in 2017 due to amounts received under our license and research agreement with Indivior related to our GABAB PAM program.
78
Other Income
The following table sets forth the other income in 2018 and 2017.
| |
For the years ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2018 | 2017 | |||||
| |
(CHF in thousands) |
||||||
Research grants |
609 | 465 | |||||
Other service income |
50 | 35 | |||||
| | | | | | | | |
Total |
659 | 500 | |||||
| | | | | | | | |
Other income increased by CHF 0.2 million in 2018 compared to 2017 primarily due to increased income recognized from MJFF research grants. The MJFF research grants fund certain activities related to our dipraglurant PD-LID program and TrKB PAM discovery program. Other service income relates to consulting services performed by our finance and administration department.
Research and Development Expenses
The following table sets forth our research and development expenses in 2018 and 2017.
| |
For the years ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2018 | 2017 | |||||
| |
(CHF in thousands) |
||||||
Dipraglurant—PD-LID |
1,416 | 816 | |||||
GABAB PAM |
477 | — | |||||
Other discovery programs |
486 | 264 | |||||
| | | | | | | | |
Subtotal outsourced R&D per program |
2,379 | 1,080 | |||||
| | | | | | | | |
Staff costs |
1,307 | 491 | |||||
Depreciation and amortization |
2 | 12 | |||||
Professional fees |
494 | 566 | |||||
Laboratory consumables |
144 | 30 | |||||
Patent maintenance and registration costs |
262 | 180 | |||||
Operating leases |
134 | 75 | |||||
Short-term leases |
— | — | |||||
Other operating expenses |
197 | 195 | |||||
| | | | | | | | |
Subtotal unallocated R&D expenses |
2,540 | 1,549 | |||||
| | | | | | | | |
Total |
4,919 | 2,629 | |||||
| | | | | | | | |
Research and development expenses increased by CHF 2.3 million in 2018 compared to 2017 primarily due to CHF 0.6 million increase in the costs of outsourced development activities related to our dipraglurant PD-LID program and CHF 0.5 million increase in the costs of outsourced research activities related to our GABAB PAM program and to a lesser extent our mGlu4 PAM program. In addition to increased directly attributable CRO and consulting costs we significantly increased our headcount in 2018 resulting in a CHF 0.8 million increase in staff costs.
79
General and Administrative Costs
The following table sets forth our general and administrative costs in 2018 and 2017.
| |
For the years ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2018 | 2017 | |||||
| |
(CHF in thousands) |
||||||
Staff costs |
918 | 260 | |||||
Depreciation and amortization |
1 | 4 | |||||
Professional fees |
1,809 | 543 | |||||
Operating leases |
45 | 22 | |||||
Other operating costs |
436 | 277 | |||||
| | | | | | | | |
Total |
3,209 | 1,106 | |||||
| | | | | | | | |
General and administrative costs increased by CHF 2.1 million in 2018 compared to 2017 primarily due to grants of equity incentive units to employees, consultants and board members which in turn resulted in an increase in share-based compensation being recorded in professional fees and staff costs.
Finance Result, Net
Finance result, net increased to a net expense of CHF 0.2 million in 2018 compared to virtually zero in 2017 primarily due to currency exchange differences and interest expenses related to the negative interest rate charged on Swiss francs cash deposits.
Liquidity and Capital Resources
Since our inception, we have generated CHF 57 million of revenue and have incurred net losses and negative cash flows from our operations. We have funded our operations primarily through the sale of equity. From inception through December 31, 2019, we raised an aggregate of CHF 325 million of gross proceeds from the sale of equity. As of December, 31, 2019, we had CHF 31.5 million in cash and cash equivalents.
Our primary uses of cash are to fund operating expenses, primarily research and development expenditures. Cash used to fund operating expenses is impacted by the timing of when we pay these expenses, as reflected in the changes in our outstanding accounts payable and accrued expenses. We currently have no ongoing material financing commitments, such as lines of credit or guarantees.
We expect our expenses to increase in connection with our ongoing activities, particularly as we continue to advance our portfolio of product candidates, initiate further clinical trials and seek marketing approval for our product candidates. In addition, if we obtain marketing approval for any of our product candidates, we expect to incur significant commercialization expenses related to program sales, marketing, manufacturing and distribution to the extent that such sales, marketing and distribution are not the responsibility of potential collaborators. Furthermore, in connection with our recent listing of ADS on the Nasdaq, we expect to incur additional costs associated with operating as a public company in the United States. Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. If we are unable to raise capital when needed or on attractive terms, we would be forced to delay, reduce or eliminate our research and development programs or future commercialization efforts.
We expect our existing cash and cash equivalents will enable us to fund our operating expenses and capital expenditure requirements for at least the next 12 months. We have based this estimate on
80
assumptions that may prove to be wrong, and we could utilize our available capital resources sooner than we currently expect. Our future capital requirements will depend on many factors, including:
- •
- the scope, progress, results and costs of our ongoing and planned preclinical studies and clinical trials for dipraglurant;
- •
- the timing and amount of milestone and royalty payments we may receive under our license agreements;
- •
- the extent to which we in-license or acquire other product candidates and technologies;
- •
- the number and development requirements of other product candidates that we may pursue;
- •
- the costs, timing and outcome of regulatory review of our product candidates;
- •
- the costs associated with building out our Swiss and U.S. operations; and
- •
- the costs and timing of future commercialization activities, including drug manufacturing, marketing, sales and distribution, for any of our product candidates for which we receive marketing approval.
Identifying potential product candidates and conducting preclinical studies and clinical trials is a time-consuming, expensive and uncertain process that takes many years to complete, and we may never generate the necessary data or results required to obtain marketing approval and achieve product sales. In addition, our product candidates, if approved, may not achieve commercial success. Our revenue, if any, will be derived from sales of products that we do not expect to be commercially available for many years, if at all.
Until such time, if ever, as we can generate substantial product revenue, we may finance our cash needs through a combination of equity offerings, debt financings, collaborations, strategic alliances and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms of any additional securities may include liquidation or other preferences that adversely affect your rights as a shareholder. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends.
If we raise funds through additional collaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or to grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
81
The following table shows a summary of our cash flows for the periods indicated:
| |
For the years ended December 31, |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2019 | 2018 | 2017 | |||||||
| |
(CHF in thousands) |
|||||||||
Cash and cash equivalents at the beginning of the period |
41,670 | 2,579 | 1,410 | |||||||
Net cash flows from / (used in) operating activities |
(9,482 | ) | 1,752 | (2,140 | ) | |||||
Net cash flows from / (used in) investing activities |
(43 | ) | (57 | ) | (1 | ) | ||||
Net cash flows from / (used in) financing activities |
(464 | ) | 37,385 | 3,355 | ||||||
| | | | | | | | | | | |
Increase/(Decrease) in cash and cash equivalents |
(9,989 | ) | 39,080 | 1,214 | ||||||
| | | | | | | | | | | |
Effect of the exchange rates |
(144 | ) | 11 | (45 | ) | |||||
| | | | | | | | | | | |
Cash and cash equivalents at end of period |
31,537 | 41,670 | 2,579 | |||||||
| | | | | | | | | | | |
Operating Activities
Net cash flows from or used in operating activities consist of the net loss adjusted for changes in working capital, that are current assets and current liabilities, and for non-cash items such as depreciation and the value of share-based services.
During the year ended December 31, 2019, operating activities used CHF 9.5 million of cash primarily due to our net loss of CHF 14.8 million adjusted for changes in net working capital of CHF 2.9 million and non-cash items of CHF 2.1 million. Non-cash items relate mainly to the value of share-based services. Changes in working capital relate mainly to an increases of CHF 1.1 million and CHF0.9 million in payables and accruals, respectively that are primarily related to our dipraglurant PD-LID program and professional service fees related to our recent listing of ADSs on the Nasdaq stock market, as well as CHF 0.7 million of increased contract liabilities related to our funded research contract with Indivior.
During the year ended December 31, 2018, operating activities generated positive cash flows of CHF 1.8 million primarily due to the revenue of CHF 6.0 million from the licensing and research agreement with Indivior that limited the consolidated net loss to CHF 1.7 million and non-cash items of CHF 2.4 million primarily consisting of the value of share-based services. Changes in net working capital of CHF 0.7 million primarily include a CHF 1.1 million increase in payables and accruals related to dipraglurant manufacturing and PD-LID clinical trial preparation costs off-set by a decrease of CHF 0.4 million in deferred income, primarily related to the recognition of research grants from MJFF.
During the year ended December 31, 2017, operating activities used CHF 2.1 million of cash primarily as the result of our net loss of CHF 3.3 million, as adjusted for the positive change in net working capital of CHF 0.3 million and non-cash items of CHF 0.8 million relating mainly to the value of share-based services.
Investing Activities
Net cash used in investing activities consist primarily of investments in computer and laboratory equipment, security rental deposits related to laboratory and office space and purchase of our own shares.
During the year ended December 31, 2019, net cash used in investing activities primarily related to investments in security rental deposits for our US office and to a lesser extent computer and laboratory equipment.
82
During the year ended December 31, 2018, net cash used in investing activities primarily related to investments in security rental deposits related to laboratory and office space, and to a lesser extent computer and laboratory equipment.
During the year ended December 31, 2017, net cash used in investing activities primarily related to investments in computer and laboratory equipment.
Financing Activities
Net cash flows from financing activities consists of proceeds from the sale of equity securities, whilst net cash flows used in financing activities primarily relates to the principal element of lease payments under IFRS 16 and interest expenses on Swiss francs cash deposits.
During the year ended December 31, 2019, net cash flows used in financing activities primarily related to the principal element of lease payments and associated interest expense resulting from the adoption of IFRS16, effective from January 1, 2019.
During the year ended December 31, 2018, net cash from financing activities primarily related to proceeds from the sale of equity securities.
During the year ended December 31, 2017, net cash from financing activities primarily related to proceeds from the sale of treasury shares.
Lease liabilities and commitments
The maturities for lease payments in relation to operating lease under IFRS 16 as of December 31, 2019 are as follows:
| |
Less than 1 Year |
1 to 3 Years |
3 to 5 Years |
More than 5 Years |
Total cash out flows |
Carrying amount liabilities |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
(CHF in thousands) |
||||||||||||||||||
Lease liabilities |
393 | 164 | 18 | — | 576 | 550 | |||||||||||||
Lease liabilities relate to the rent of laboratories, equipment, offices and related spaces used by the Group. There are no cancellable operating lease commitments over 5 years.
We enter into contracts in the normal course of business with CROs for clinical trials, preclinical studies, manufacturing and other services and products for operating purposes. These contracts generally provide for termination upon notice, and therefore we believe that our non-cancelable obligations under these agreements are not material.
Off-Balance Sheet Arrangements
For the years ended December 31, 2019, 2018 and 2017, we did not have, and we do not currently have, any off-balance sheet arrangements, as defined in the rules and regulations of the U.S. Securities and Exchange Commission.
Outstanding Debt
We do not engage in trading activities involving non-exchange traded contracts nor do we currently have any debt outstanding. We therefore believe that we are not materially exposed to any financing, liquidity, market or credit risk that could arise if we had engaged in these relationships.
83
Critical Accounting Policies and Significant Judgments and Estimates
Our management's discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which we have prepared in accordance with International Financial Reporting Standards, or IFRS. The preparation of our consolidated financial statements requires us to make estimates, assumptions and judgments that affect the reported amounts of assets, liabilities, costs and expenses. We base our estimates and assumptions on historical experience and other factors that we believe to be reasonable under the circumstances. We evaluate our estimates and assumptions on an ongoing basis. Our actual results may differ from these estimates.
While our significant accounting policies are described in more detail in Note 4 to our consolidated financial statements appearing elsewhere in this prospectus. We believe the following accounting policies to be most critical to understanding our historical financial performance as they relate to the more significant areas involving management's judgments and estimates.
Revenue recognition
Under IFRS 15, we recognize as revenue our non-refundable license fees, milestone, research activities and royalties when our customer obtains control of promised services, in an amount that reflects the consideration which we expect to receive in exchange for those rendered services. To assess revenue recognition for arrangements that we determine are within the scope of IFRS 15, we perform the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) we satisfy a performance obligation. We only apply the five-step model to contracts when it is probable that we will collect the consideration we are entitled to in exchange for services we transfer to the customer.
At contract inception, once the contract is determined to be within the scope of IFRS 15, we assess the services promised within each contract and determine those that are performance obligations and assess whether each promised service is distinct. We use the most likely method to estimate any variable consideration and include such consideration in the amount of the transaction price based on an estimated stand-alone selling price. Revenue is recognized for the respective performance obligation when (or as) the performance obligation is satisfied.
Other income
We recognized grants at their fair value when we have the reasonable assurance that they will be received, and all conditions will be complied with. Grants are recognized in the accounting period as the costs they intend to compensate are incurred.
Recognition of Research and Development Costs
We recognize expenses incurred in carrying out our research and development activities in line with our best estimation of the stage of completion of each separately contracted study or activity. This includes the calculation of research and development accruals at the end of each period to account for expenditure that has been incurred. This requires us to estimate the full costs to complete each study or activity and to estimate the current stage of completion. There have been no material adjustments to estimates based on the actual costs incurred for the periods presented. In all cases, we expense the full cost of each study or activity by the time the final study report or, where applicable, product, has been received.
We will recognize an internally-generated intangible asset arising from our development activities only when an asset is created that can be identified, it is probable that the asset created will generate future economic benefits and the development cost of the asset can be measured reliably. We have
84
determined that regulatory and marketing approvals are the earliest points at which the probable threshold for the creation of an internally generated intangible asset can be achieved. We therefore expense all research and development expenditure incurred prior to achieving such approvals as it is incurred. None of our product candidates have yet received regulatory and marketing approvals.
Share-Based Compensation
We measure and recognize compensation expense for all equity incentive units based on the estimated fair value of the award on the grant date. We only grant equity incentive units to our employees, key consultants and board members. The fair value is recognized as expense, net of estimated forfeitures, over the requisite service period, which is generally the vesting period of the respective award.
The fair value at the grant date of the equity incentive units is determined using either an option pricing method that uses a Black-Scholes model or a binomial valuation model. In establishing these models, a number of assumptions are made by management. The fair value per share for our shares is the closing price of our shares as reported by the Swiss SIX exchange on the applicable grant date. A number of assumptions on the volatility of the underlying shares and on the risk free rate are made in these models.
Employee Benefits
We maintain a pension plan for all employees in Switzerland that is maintained through payments to a legally independent collective foundation. This pension plan qualifies under IFRS as a defined benefit pension plan. There are no pension plans for the subsidiaries in the United States.
The liability recognized in the balance sheet in respect of defined benefit pension plans is the present value of the defined benefit obligation at the end of the reporting period less the fair value of plan assets. The defined benefit obligation is calculated annually by an independent actuary, using the projected unit credit method. The present value of the defined benefit obligation is determined by discounting the estimated future cash outflows using interest rates of high-quality corporate bonds that are denominated in the currency in which the benefits will be paid, and that have terms to maturity approximating to the terms of the related pension obligation.
Actuarial gains and losses arising from experience adjustments and changes in actuarial assumptions are charged or credited to equity in other comprehensive income in the period in which they arise. Past-service costs are recognized immediately in the consolidated statement of comprehensive loss.
Recent Accounting Pronouncements
See Note 2.2 to our consolidated financial statements included elsewhere in this Annual Report on Form 20-F for a description of recent accounting pronouncements applicable to our consolidated financial statements.
Qualitative and Quantitative Disclosures about Financial Risks
We operate primarily in Switzerland, Europe and in the United States and are therefore exposed to market risk, which represents the risk of loss that may impact our financial position due to adverse changes in financial market prices and rates.
As of December 31, 2019, we had CHF 31.5 million of cash and cash equivalents and we had no debt.
85
JOBS Act Transition Period
Subject to certain conditions, as an emerging growth company, we may rely on certain of these exemptions under the Jumpstart Our Business Startups Act of 2012, or the JOBS Act, including without limitation, (1) providing an auditor's attestation report on our system of internal controls over financial reporting pursuant to Section 404(b) of the Sarbanes-Oxley Act and (2) complying with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor's report providing additional information about the audit and the financial statements, known as the auditor discussion and analysis. We will remain an emerging growth company until the earlier to occur of (1) the last day of the fiscal year (a) following the fifth anniversary of the completion of this offering, (b) in which we have total annual gross revenues of at least $1.07 billion or (c) in which we are deemed to be a "large accelerated filer" under the rules of the U.S. Securities and Exchange Commission, which means the market value of our common shares that is held by non-affiliates exceeds $700 million as of the prior June 30, and (2) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.
Item 6. Directors, Senior Management and Employees.
A. Directors and Senior Management.
The following table sets forth information regarding our executive officers and directors, including their ages, as of December 31, 2019. Our directors are appointed for one-year terms, which expire on the occasion of each annual general meeting. Accordingly, the terms of the directors set forth below will expire on the date of our 2020 annual general meeting of shareholders.
Name
|
Age | Position(s) | |||
|---|---|---|---|---|---|
| Executive Officers: | |||||
| Tim Dyer | 51 | Chief Executive Officer and Director | |||
| Roger Mills | 62 | Chief Medical Officer and Director | |||
| Robert Lütjens | 51 | Head of Discovery—Biology | |||
| Jean-Philippe Rocher | 60 | Head of Discovery—Chemistry | |||
| Lénaic Teyssédou(3) | 34 | Head of Finance | |||
Non-Employee Directors: |
|||||
| Vincent Lawton(1)(2) | 70 | Chairman | |||
| Raymond Hill(1) | 74 | Director | |||
| Isaac Manke(2) | 42 | Director | |||
| Jake Nunn(2) | 49 | Director | |||
- (1)
- Member
of the Compensation Committee
- (2)
- Member
of the Audit Committee
- (3)
- Mr. Teyssédou is not a member of the Executive Management
Executive Officers
Tim Dyer, Co-Founder, Director and Chief Executive Officer: Since co-founding Addex in 2002, Mr. Dyer has played a pivotal role in building the Addex Group, raising CHF 300 million of capital, including Addex IPO and negotiating licensing agreements with pharmaceutical industry partners that generated more than CHF 57 million in cash inflows. Prior to founding Addex, he spent 10 years with Price Waterhouse, or PW, and PricewaterhouseCoopers, or PwC, in the UK and Switzerland as part of the audit and business advisory group. At PwC in Switzerland, Mr. Dyer's responsibilities included
86
managing the service delivery to a diverse portfolio of clients including high growth start-up companies, international financial institutions and venture capital and investment companies. Mr. Dyer has extensive experience in finance, corporate development, business operations and the building of start-up companies. He is a UK Chartered Accountant and holds a BSc (Hons) in Biochemistry and Pharmacology from the University of Southampton, UK.
Roger Mills, Director and Chief Medical Officer: Dr. Mills brings more than 30 years of biopharmaceutical industry experience at both large global pharmaceutical companies and smaller biotechnology companies, including Acadia Pharmaceuticals, Pfizer, Gilead Sciences, Abbott Laboratories and The Wellcome Foundation, across a spectrum of disease areas. His extensive track record includes managing drug development programs from Investigational New Drug Application preparation through to post-marketing and OTC products, including NUPLAZIDM for the treatment of Parkinson's Disease Psychosis, as well as regulatory affairs and business development activities. Most recently, Dr. Mills was with Acadia Pharmaceuticals for nine years, serving as Executive Vice President, Development and Chief Medical Officer. In this role, he oversaw the largest ever international phase III program in Parkinson's Disease Psychosis, and led its New Drug Application submission to the US Food and Drug Administration (FDA) for NUPLAZID, which was subsequently approved and remains the first and only medication approved by the FDA in this indication. Dr. Mills currently serves as a Visiting Professor at the Centre for Age Related Diseases, Institute of Psychiatry, Psychology and Neuroscience, King's College London. He received his medical degree from Imperial College, Charing Cross Hospital Medical School, London, United Kingdom.
Robert Lütjens, Head of Discovery—Biology: Dr. Lütjens is responsible for all biology activities and has extensive experience in drug discovery. He established the biology capabilities and built the Company's small molecule allosteric modulator biology platform. He played a pivotal role in the success of both internal and partnered programs, including the discovery of dipraglurant and ADX71149, both of which progressed into phase II clinical development. Prior to joining Addex at inception in 2002, Dr. Lütjens completed a postdoctoral fellowship in the Department of Neuropharmacology at the Scripps Research Institute, in La Jolla, CA, where he focused on understanding molecular changes involved in addiction disorders. Dr. Lütjens obtained his degrees in Biology from the University of Geneva, his master's at the Swiss Institute for Experimental Cancer Research and his Ph.D. thesis at the Glaxo Institute for Molecular Biology in Geneva and the Institute for Cellular Biology and Morphology in Lausanne. Dr. Lu¨tjens is co-author of over 30 peer-reviewed publications and patents.
Jean-Philippe Rocher, Head of Discovery—Chemistry: Dr. Rocher is responsible for all chemistry activities and has extensive experience in drug discovery. He returns to Addex from Pierre Fabre where he was Director of CNS Programs from March 2014 to May 2018. Joining Addex at its inception in 2002, Dr. Rocher established the company's chemistry capabilities and built its small molecule allosteric modulator chemistry platform. He played a pivotal role in the success of both internal and partnered programs, including the discovery of dipraglurant and ADX71149, both of which progressed into phase II clinical development. Prior to joining Addex, Dr. Rocher was director of chemistry at Devgen NV (Gent, Belgium), senior research scientist for GlaxoSmithKline KK (Tsukuba, Japan), scientific project leader in CNS at Mitsubishi Tanabe (Yokohama, Japan) and Head of Drug Discovery Unit for Battelle (Geneva, Switzerland). He started his career as a research scientist in the dermatology research center of Galderma (Sophia-Antipolis, France) following a PhD in medicinal chemistry and Pharm D at the Faculty of Pharmacy of Lyon (France). He is a co-author of more than 40 research publications and patents.
Lénaic Teyssédou, Head of Finance: Mr. Teyssédou has served as our Head of Finance since June 2017 and is responsbile for finances and administration at Addex. Prior to joining the company, Mr. Teyssédou worked for four years in the transaction support department of French audit company,
87
where he was responsible for the acquisition due diligence of various clients, including major financial institutions. Mr. Teyssédou is a French Chartered Accountant and holds two Master degrees in Finance and Management from EM Strasbourg Business School, France.
Non-Employee Directors
Vincent Lawton, Chairman of the Board of Directors: Professor Lawton was Vice President Merck Europe and Managing Director of MSD UK until he stepped down in 2006, after 26 years' service internationally for Merck & Co Inc. He was appointed CBE (Commander of the British Empire) by the Queen of England for services to the Pharmaceutical Industry. During his tenure, MSD UK achieved sustained commercial success, launching many new medicines to the market in a wide range of therapeutic areas, becoming the fastest growing company in the market over a number of years. He worked in commercial, research and senior management roles in France, the US and Canada, Spain and throughout Europe. As President of the UK Industry Association, the ABPI, he negotiated industry pricing, worked with Government bodies to help establish the UK globally as a leading center of clinical research. He served on the board of the UK regulatory authority (MHRA) from 2008 to 2015. He is a Senior Strategy Advisor for Imperial College Department of Medicine, University of London and serves as a consultant to a number of leading healthcare organizations. He studied Psychology at the University of London and holds an undergraduate degree and PhD.
Raymond Hill, Director: Raymond Hill was previously a member of the Board of Directors from the Annual General Meetings of 2008 until 2012. Currently Visiting Professor of Pharmacology at Imperial College in London, and Non-Executive Director of Avilex (Denmark), Asceneuron (Switzerland) and was NED of Orexo AB (Sweden) from 2008 to 2019. Prior to his retirement, he was Executive Director, Licensing and External Research at Merck/MSD in Europe (2002 - 2008); Executive Director, Pharmacology (1990-2002) at the Merck Neuroscience Research Centre and had oversight responsibility for Neuroscience research at the Banyu Research Labs in Tsukuba, Japan (1997-2002). At Merck, he chaired a number of discovery project teams including those responsible for the marketed products Maxalt® and Emend®. Dr. Hill received his academic training (BPharm PhD) at the University of London. He was awarded an Honorary DSc by the University of Bradford in 2004 and was elected to Fellowship of the Academy of Medical Sciences in 2005. He was a lecturer in Pharmacology at the University of Bristol School of Medicine from 1974 to 1983 and supervisor in Pharmacology at Downing College, University of Cambridge from 1983 to 1988. He joined the pharmaceutical industry in 1983 as Head of Biology and founder member of the Park Davis Research Unit at Cambridge. In 1988, he joined SK&F (United Kingdom) as Group Director of Pharmacology and in 1990 moved to Merck. He is a past Council Member of the UK Academy of Medical Sciences and President Emeritus of the British Pharmacological Society. He is a Visiting Professor at the University of Bristol and was a member of the UK Government Advisory Council on the Misuse of Drugs from 2010 to 2019. He continues to serve on the ACMD Working Group on the Medicinal Uses of Cannabis and is a member of the Royal Pharmaceutical Society Science and Research Board.
Isaac Manke, Director: Dr. Manke has more than 15 years of experience in the life science industry as an investor, research analyst, consultant and scientist. Isaac joined New Leaf Venture Partners, or NLV, in 2009 and was promoted to Partner from 2014. Isaac's investment activities with NLV started with a focus on venture investments in the biopharmaceutical sector. During his time at NLV, he led the firm's public investment activities initially with the public portfolio within NLV-II, and from 2014 through 2019, had day-to-day management and oversight responsibility for the NLV Biopharma Opportunities Funds. Isaac has been a board member or observer for several companies, including the boards of True North Therapeutics (acquired by Bioverativ) and Karos Pharmaceuticals (acquired by an undisclosed company). Prior to joining NLV, Isaac was an Associate in the Global Biotechnology Equity Research group at Sanford C. Bernstein. Previously, Isaac worked as an Associate in the Biotechnology Equity Research group at Deutsche Bank and was a Senior Analyst at Health
88
Advances, a biopharmaceutical and medical device strategy consulting firm. Isaac received a B.A. in Biology and a B.A. in Chemistry at Minnesota State University (Moorhead), and a Ph.D. in Biophysical Chemistry and Molecular Structure at the Massachusetts Institute of Technology, or MIT. Isaac's discoveries led to several publications in top journals, including Science and Cell, and were selected by Science as one of the "2003: Signaling Breakthroughs of the Year". These discoveries also resulted in four issued patents.
Jake Nunn, Director: Mr.Nunn has more than 25 years of experience in the life science industry as an investor, independent director, research analyst and investment banker. Jake is currently a venture advisor at New Enterprise Associates, or NEA, where he was a partner from 2006 to 2018, focusing on later-stage specialty pharmaceuticals, biotechnology and medical device investments and managing a number of NEA's public investments in healthcare. Jake is a Director of Oventus Medical Ltd. (ASX: OVN), Regulus Therapeutics (Nasdaq: RGLS) and Trevena, Inc. (Nasdaq: TRVN). He previously was a Director of Dermira Inc. (acquired by Eli Lilly), Hyperion Therapeutics (acquired by Horizon Pharma PLC), TriVascular (acquired by Endologix), Aciex Therapeutics (acquired by Nicox SA), Transcept Pharmaceuticals (merged with Paratek) and a board observer at Vertiflex, Inc. (acquired by Boston Scientific). Prior to NEA, Jake worked at MPM Capital as a Partner with the MPM BioEquities Fund, where he specialized in public, PIPE and mezzanine-stage life sciences investing. Previously, he was a healthcare research analyst and portfolio manager at Franklin Templeton Investments. Jake was also an investment banker with Alex. Brown & Sons. He received an MBA from the Stanford Graduate School of Business and an AB in Economics from Dartmouth College. Jake holds the Chartered Financial Analyst designation, is a member of the CFA Society of San Francisco, and recently completed the Stanford GSB Directors' Consortium executive education program.
Family Relationships
There are no family relationships among any of our executive officers or directors.
B. Compensation. Compensation of Executive Officers and Directors
Board of Directors
The compensation of the member of the Board consists of fixed and variable elements. The fixed element comprises a fixed annual monetary compensation per board term from one general meeting of shareholders to the next. The variable element comprises a monetary compensation based on board meeting attendance and equity incentive units (share options and equity sharing certificates). Our social security contributions are accrued on the fixed and variable elements. Board member social security contributions are accrued on the fair value of equity incentive units. Equity incentive units are granted based on the discretion of the Board. In addition, we reimburse members of the Board for out-of-pocket expenses incurred in relation to their services on an on-going basis upon presentation of the corresponding receipts. The most recent review of compensation for members of the Board took place on December 13, 2018.
Executive Management
The compensation of members of the Executive Management consists of fixed and variable elements. The fixed element may include a base salary or a cash retainer paid under a consulting contract. The variable element may include performance-related cash or share based bonuses, consulting fees based on chargeable hours and equity incentive units (equity sharing certificates and share options). Company contributions to pension plans, death and invalidity insurances and social security contributions are accrued on all fixed and variable element compensation that relates to an employment relationship. Both company and employee social security contributions are accrued for all shares or equity incentive unit compensation. The amount of the fixed element depends on the
89
position, responsibilities, experience and skills, and takes into account individual performance. The fixed element is reviewed at the end of each year by the Board. Any changes in the fixed elements are made effective in January of the following year. The variable elements are based on individual and company performance. The potential variable cash bonus is determined in the employment contract and in general is a percentage of the base salary. Where the Executive Manager has been engaged under a consulting contract, the variable element is based on the time spent at the contractually defined rate of remuneration. At the beginning of each year the Board decides, on the total amount of variable elements including the amount of cash and equity incentive units to be granted for the previous year based on the achievement of Company goals. Equity incentive units are granted based on the discretion of the Board. Variable cash compensation paid to Executive Managers in 2019 includes bonus and consulting fees.
Equity Incentive Plans
The purpose of our share purchase, share option and equity sharing certificate programs (refer to note 15 of the consolidated financial statements included elsewhere in this Annual Report on Form 20-F) is to provide members of the Board, Executive Management, employees and certain consultants with an opportunity to benefit from the potential appreciation in the value of our shares, thus providing an increased incentive for participants to contribute to our future success and prosperity, enhancing the value of the shares for the benefit of our shareholders and increasing our ability to attract and retain individuals of exceptional skill. In addition, these plans provide us with a mechanism to engage services for non-cash consideration. The grant of any share option or equity sharing certificate is at the discretion of the Board. Key factors considered by the Board in making grants of share options or equity sharing certificates are the amount of shareholder approved conditional capital, the benchmarking with other companies as well as individual performance. The strike price is determined by the Board and is primarily based on the closing price of our shares on the SIX Swiss Exchange on the grant date. The transfer of treasury shares under the share purchase plan to settle consulting services are based on predefined terms of the consulting contract.
Indirect benefits
We may contribute to the pension plan and maintains certain insurance for death and invalidity for the members of the Executive Management. New entrants may be eligible for reimbursement of relocation costs, compensation for lost benefits or stock granted by a previous employer, international school for children or language courses for a limited time period. No Indirect benefits have been paid to Executive Management in 2019. We have not granted any loans, credits or guarantees to members of the Board or of the Executive Management in 2019.
Measurement basis for compensation
The measurement basis for each component of compensation is described below:
- •
- Fixed Cash compensation, variable cash compensation and shares acquired under the share purchase plan: accrual basis;
- •
- Equity incentive units: fair value at the grant date in accordance with IFRS 2 valuation methodology; and
- •
- Employers' social security: accrual basis except for equity incentive units where the notional amount is calculated based on the fair value at grant date.
90
Compensation of the Board of Directors in 2019
| |
|
Variable compensation (Value in CHF—unaudited) |
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
2019
|
Fixed cash compensation |
cash attendance |
number of equity incentive units |
value of equity incentive units |
Total 2019 |
|||||||||||
Vincent Lawton |
25,858 | 25,858 | — | — | 51,716 | |||||||||||
Raymond Hill |
15,341 | 15,341 | — | — | 30,682 | |||||||||||
Tim Dyer |
— | — | — | — | — | |||||||||||
Roger Mills |
— | — | — | — | — | |||||||||||
Jake Nunn |
13,284 | 13,284 | — | — | 26,568 | |||||||||||
Isaac Manke |
10,627 | 10,627 | — | — | 21,254 | |||||||||||
| | | | | | | | | | | | | | | | | |
Total |
65,110 | 65,110 | 0 | 0 | 130,220 | |||||||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Compensation to the Executive Management in 2019
| |
|
Variable compensation (Value in CHF—unaudited) |
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
2019
|
Fixed cash compensation |
Cash(3) | number of equity incentive units(2) |
value of shares(2) |
Total 2019 |
|||||||||||
Total Executive Management(1) |
890,350 | 303,287 | 356,605 | 402,363 | 1,596,000 | |||||||||||
| | | | | | | | | | | | | | | | | |
- (1)
- The
highest paid member of Executive Management in 2019 was the CEO, Tim Dyer, who received CHF 429,268 of fixed cash compensation, CHF 74,719 of
variable cash compensation and 243,506 equity incentive units. The value of equity incentive units including accrued social charges amounted to CHF 205,258.
- (2)
- Equity
incentive units include shares awarded for consulting services under the share purchase plan and share options granted under our share option plan.
- (3)
- Executive managers have been engaged under consulting contracts which include hourly and daily rates with a monthly cap.
C. Board Practices.
Composition of Our Board of Directors
Our board of directors is currently composed of six members. As a foreign private issuer, under the listing requirements and rules of Nasdaq, we are not required to have independent directors on our board of directors, except that our audit committee is required to consist fully of independent directors, subject to certain phase-in schedules. However, our board of directors has determined that Vincent Lawton and Jake Nunn do not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of director and that each of these directors is "independent" as that term is defined under Nasdaq rules. There are no family relationships among any of our directors or senior management.
Committees of Our Board of Directors
The audit committee, which consists of Vincent Lawton, Isaac Manke and Jake Nunn, assists the board of directors in overseeing our accounting and financial reporting processes. Vincent Lawton serves as chairman of the audit committee. The audit committee consists exclusively of members of our
91
board who are financially literate, and Jake Nunn is considered an "audit committee financial expert" as defined by applicable SEC rules and has the requisite financial sophistication as defined under the applicable Nasdaq rules and regulations. Our board of directors has determined that Vincent Lawton, Isaac Manke and Jake Nunn are independent directors under Nasdaq listing rules and under Rule 10A-3 under the Exchange Act. The audit committee is governed by a charter that complies with Nasdaq rules. The audit committee's responsibilities include:
- •
- to review and assess the effectiveness of the statutory auditors and the group auditors, in particular their independence from Addex. In
connection therewith, it reviews in particular additional assignments given by the Company or its subsidiaries. It may issue binding regulations or directives in connection with such additional
assignments;
- •
- to review and assess the scope and plan of the audit, the examination process and the results of the audit and to examine whether the
recommendations issued by the auditors have been implemented by management;
- •
- to review the auditors' reports, to discuss their contents with the auditors and with the management;
- •
- to approve the terms and conditions of the engagement of the auditors;
- •
- to review the effectiveness of the internal audit function, its professional qualifications, resources and independence and its cooperation
with external audit;
- •
- to approve the annual internal audit concept and the annual internal audit report, including the responses of the management thereto
- •
- to assess the risk assessment established by the management and the proposed measures to reduce risks;
- •
- to assess the state of compliance with norms within Addex;
- •
- to review in cooperation with the auditors, the CEO and Head of Finance whether the accounting principles and the financial control mechanism
of Addex and its subsidiaries are appropriate in view of our size and complexity;
- •
- to review the annual and interim statutory and consolidated financial statements intended for publication. It should discuss these with the CEO
and the Head of Finance and, separately, with the head of external audit;
- •
- to make a proposal to the Board with respect to these annual and interim statutory and consolidated financial statements; and
- •
- the responsibility for approving the annual financial statements remains with the Board.
Compensation Committee
The compensation committee, which consists of Vincent Lawton and Raymond Hill, advises the board in determining executive compensation. The remuneration committee's responsibilities include:
- •
- to review and assess on a regular basis the remuneration system of Addex (including the management incentive plans) and to make proposals in
connection thereto to the Board;
- •
- to recommend the terms of employment, in particular the remuneration package, of the CEO and to make proposals in relation to the remuneration
of Directors;
- •
- to recommend upon proposal of the CEO the terms of employment, in particular the remuneration package, of employees reporting directly to the CEO as well as review matters
92
related to the compensation of other top managers, as well as the general employee compensation, benefit policies and HR practices of Addex; and
D. Employees.
We had 18.50 full-time employees and consultants as of December 31, 2019. None of our employees are represented by any collective bargaining unit. We believe that we maintain good relations with our employees.
E. Share Ownership.
For information regarding the share ownership of our directors and executive officers, see "Item 6.B-Compensation" and "Item 7.A-Major Shareholders."
Item 7. Major Shareholders and Related Party Transactions
A. Major shareholders.
The following table sets forth information with respect to the beneficial ownership of our shares as of December 31, 2019 for:
- •
- each beneficial owner of 5% or more of our outstanding shares;
- •
- each of our directors and executive officers; and
- •
- all of our directors and executive officers as a group.
Beneficial ownership is determined in accordance with the rules of the SEC. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities and include shares issuable upon the exercise of options that are immediately exercisable or exercisable within 60 days of December 31, 2019. Percentage ownership calculations are based on 32,848,635 shares outstanding as of December 31, 2019. 2019 plus, consistent with SEC rules on disclosure of beneficial ownership, shares that each security holder has the ability to acquire within 60 days of December 31, 2019, due to outstanding equity interests becoming vested or exercisable. The percentage of shares beneficially owned before offering shown on the table reflect these incremental shares that a security holder has the ability to acquire within the time frame noted. Except as otherwise indicated in the table below, addresses of the
93
directors and executive officers are c/o Addex Therapeutics Ltd, Chemin des Mines 9, 1202 Geneva, Switzerland.
| |
Number of shares beneficially owned |
Percentage of shares beneficially owned |
||
|---|---|---|---|---|
5% or Greater Shareholders: |
||||
Addex Pharma SA(1) |
6,415,002 | 19.5% | ||
Growth Equity Opportunities Fund IV, LLC(2) |
6,624,600 | 19.0% | ||
New Leaf Biopharma Opportunities I, L.P.(3) |
2,316,293 | 6.9% | ||
CDK Associates, LLC(4) |
2,316,293 | 6.9% | ||
Credit Suisse Funds AG(5) |
2,130,964 | 6.4% | ||
Executive Officers and Directors: |
|
|||
Tim Dyer(6) |
2,244,891 | 6.5% | ||
Vincent Lawton |
* | * | ||
Raymond Hill |
* | * | ||
Isaac Manke |
* | * | ||
Roger Mills |
* | * | ||
Jake Nunn |
* | * | ||
Robert Lütjens |
* | * | ||
Jean-Philippe Rocher |
* | * | ||
Lénaïc Teyssédou |
* | * | ||
All current directors and executive officers as a group (9 persons) |
3,467,513 | 9.7% |
- *
- Represents
beneficial ownership of less than one percent.
- (1)
- Consists
of 6,353,549 shares and 61,453 shares issuable upon exercise of outstanding warrants directly held by Addex Pharma SA that is wholly owned by Addex
Therapeutics Ltd. Tim Dyer, Chief Executive Officer of the Group is the sole board member of Addex Pharma SA and one of the board members of Addex Therapeutics Ltd. Tim Dyer makes
investment decisions on behalf of Addex Pharma SA based on instructions from the board of directors of Addex Therapeutics Ltd. A full list of members of the board of directors of Addex
therapeutics Ltd is disclosed elsewhere in this Annual Report on Form 20-F. The address of Addex Pharma SA is Chemin des Mines 9, 1202 Geneva, Switzerland.
- (2)
- Consist
of 4,568,690 shares and 2,055,910 shares issuable upon exercise of outstanding warrants directly held by Growth Equity Opportunities Fund IV LLC ("GEO
IV") that is wholly owned by New Enterprise Associates 15, L.P ("NEA 15"). The shares directly held by GEO IV are indirectly held by NEA 15, whose general partner is NEA Partners 15, L.P ("NEA
Partners 15"). The general partner of NEA Partners 15 is NEA 15 GP, LLC ("NEA 15 LLC"). The individual Managers of NEA 15 LLC, or collectively, the NEA 15 Managers are
Peter J. Barris, Forest Baskett, Anthony A. Florence Jr., Joshua Makower, Mohamad Makhzoumi, David M.Mott, Scott D.Sandell and Peter W.Sonsini. GEO IV, NEA 15, NEA Partners 15, NEA 15 LLC and
the NEA 15 Managers share voting and dispositive power with regard to our securities held directly by GEO IV. The address of Growth Equity Opportunities Fund IV LLC is c/o New Enterprise
Associate 15 L.P, Timonium, MD 21093.
- (3)
- Consist of 1,597,444 shares and 718,849 shares issuable upon exercise of outstanding warrants directly held by New Leaf Biopharma Opportunities I, L.P and indirectly held by
94
New Leaf Venture Management III LLC. The individual Managers of New Leaf Venture Management III LLC are Ronald Hunt and Vijay Lathi. The address of New Leaf Biopharma Opportunities I, L.P is c/o Corporation Trust Company/Center, 1209 Orange Street, Wilmington, DE 19801.
- (4)
- Consist
of 1,597,444 shares and 718,849 shares issuable upon exercise of outstanding warrants directly held by CDK Associates LLC and indirectly held by Bruce
Kovner who can exercise the voting rights and investment control at his own discretion. The address of CDK Associates LLC is Princeton, New Jersey 08540.
- (5)
- Consist
of 1,455,964 shares and 675,000 shares issuable upon the exercise of warrants indirectly held by Credit Suisse Funds AG with voting power whilst Credit
Suisse Asset Management AG, has investing power. The collective investment scheme CS (CH) Small Cap Switzerland directly holds 1,175,125 shares and 675,000 shares issuable upon exercise of warrants.
The individual Managers of Credit Suisse Funds AG and Credit Suisse Asset Management AG are Simon Go¨tschmann, Thomas Scha¨rer, Emil Stark and Gabriele Wyss. The
address of Credit Suisse Funds AG is Kalandergasse 4, 8045 Zurich, Switzerland.
- (6)
- The 2,244,891 shares consist of 435,192 shares and 1,809,699 shares issuable upon the exercise of 1,795,323 outstanding options and 14,376 outstanding warrants.
B. Related Party Transactions.
Since January 1, 2018, we have engaged in the following transactions with our directors, executive officers and holders of more than 5% of our outstanding voting securities and their affiliates, which we refer to as our related-parties.
2018 Private Placement
On March 28, 2018, we issued an aggregate of 13,037,577 of our shares, 136,561 of which were recorded as treasury shares, and warrants to purchase up to 5,866,898 of our shares. The price per share plus warrant to purchase 0.45 of a share was CHF 3.13. Each warrant has an exercise price per ordinary share of CHF 3.43 and a term of seven years. We utilized the net proceeds of the private placement of units primarily to advance our drug development programs and for general corporate purposes. The table below summarizes the issuance of such shares and warrants that were issued to members of our board of directors, our executive officers or holders of more than 5% of our shares.
| |
Shares purchased |
Shares issuable upon exercise of warrants purchased |
|||||
|---|---|---|---|---|---|---|---|
5% or Greater Shareholders |
|||||||
Growth Equity Opportunities Fund IV, LLC(1) |
4,568,690 | 2,055,910 | |||||
New Leaf Biopharma Opportunities I, L.P.(2) |
1,597,444 | 718,849 | |||||
CDK Associates, LLC(3) |
1,597,444 | 718,849 | |||||
Credit Suisse Funds AG(4) |
1,500,000 | 675,000 | |||||
Executive Officers and Directors |
|||||||
Tim Dyer(5) |
31,948 | 14,376 | |||||
- (1)
- Growth Equity Opportunities Fund IV, wholly owned by New Enterprise Associate LP, is a holder of more than 5% of our shares and has appointed Jake Nunn as Board Member
95
- (2)
- New
Leaf Biopharma Opportunities I, L.P, owned by New Leaf Venture Management III LLC, is a holder of more than 5% of our shares and has appointed Isaac Manke
as Board Member
- (3)
- CDK
Associates LLC, owned by Bruce Kovner, is a holder of more than 5% of our shares.
- (4)
- Credit
Suisse Funds AG indirectly holds more than 5% of our shares, through collective investment schemes whose CS (CH) Small Cap Switzerland directly holds more
than 5%.
- (5)
- Mr. Tim Dyer is a holder of more than 5% of our shares and is a board member.
In connection with the foregoing private placement of securities, we entered into a registration rights agreement with the purchasers of the securities, pursuant to which we granted such purchasers the right to have their ordinary shares and ordinary shares issuable upon the exercise of warrants purchased in the private placement registered with the SEC for resale in the United States. All such shares were registered for resale on our Registration Statement on Form F-1 (no. 333-235554 that was declared effective on January 29, 2020.
Director and Executive Officer Compensation
See "Item 6.B-Compensation of Directors and Executive Officers" for information regarding compensation of directors and executive officers.
We have entered into indemnification agreements with each of our directors and executive officers. These agreements require us to indemnify our directors and executive officers to the fullest extent permitted by law.
Tim Dyer, our CEO, provided his services to us through TMD Advisory Ltd liab. Co, or TMDA, until November 1, 2018. He is now employed by us. TMDA rented administrative offices to us for CHF 27 thousand in 2018 and nil in 2019. We also performed accounting services for TMDA for CHF 50 thousand in 2018 and nil in 2019.
Related-Party Transactions Policy
We have adopted a related-party transaction policy that sets forth our procedures for the identification, review, consideration and approval or ratification of related-party transactions. For purposes of our policy only, a related-party transaction is a transaction, arrangement or relationship, or any series of similar transactions, arrangements or relationships, in which we and any related parties are, were or will be participants, which are not in the ordinary course of business, (2) at arms' length and (3) in which the amount involved exceeds CHF 120,000. Transactions involving compensation for services provided to us as an employee or director are not covered by this policy. For purposes of this policy, a related party is any executive officer, director (or nominee for director) or beneficial owner of more than 5% of any class of our voting securities, including any of their immediate family members and any entity owned or controlled by such persons.
Under the policy, if a transaction has been identified as a related-party transaction, including any transaction that was not a related-party transaction when originally consummated or any transaction that was not initially identified as a related-party transaction prior to consummation, our management must present information regarding the related-party transaction to our board of directors for review, consideration and approval. The presentation must include a description of, among other things, the material facts, the interests, direct and indirect, of the related parties, the benefits to us of the transaction and whether the transaction is on terms that are comparable to the terms available to or from, as the case may be, an unrelated third party or to or from employees generally. Under the policy, we will collect information that we deem reasonably necessary from each director, executive officer and,
96
to the extent feasible, significant shareholder to enable us to identify any existing or potential related-party transactions and to effectuate the terms of the policy. In addition, our board of directors has adopted a Code of Business Conduct and Ethics, under which our employees and directors will have an affirmative responsibility to disclose any transaction or relationship that reasonably could be expected to give rise to a conflict of interest. In considering related-party transactions, our audit committee, or other independent body of our board of directors, will take into account the relevant available facts and circumstances including:
- •
- the risks, costs and benefits to us;
- •
- the impact on a director's independence in the event that the related party is a director, immediate family member of a director or an entity
with which a director is affiliated;
- •
- the availability of other sources for comparable services or products; and
- •
- the terms available to or from, as the case may be, unrelated third parties or to or from employees generally.
The policy requires that, in determining whether to approve, ratify or reject a related-party transaction, our audit committee, or other independent body of our board of directors, must consider, in light of known circumstances, whether the transaction is in, or is not inconsistent with, our best interests and those of our shareholders, as our audit committee, or other independent body of our board of directors, determines in the good faith exercise of its discretion.
C. Interests of Experts and Counsel.
Not applicable.
A. Consolidated Statements and Other Financial Information. Consolidated Financial Statements
Our consolidated financial statements are appended at the end of this Annual Report, starting at page F-1, and are incorporated herein by reference.
Dividend Distribution Policy
We have never paid a dividend, and we do not anticipate paying dividends in the foreseeable future. We intend to retain all available funds and any future earnings to fund the development and expansion of our business.
Under Swiss law, any dividend must be proposed by our board of directors and approved by a shareholders' meeting. In addition, our auditors must confirm that the dividend proposal of our board of directors conforms to Swiss statutory law and our articles of association. A Swiss corporation may pay dividends only if it has sufficient distributable profits brought forward from the previous business years ("Gewinnvortrag") or if it has distributable reserves ("frei verfügbare Reserven"), each as evidenced by its audited standalone statutory balance sheet prepared pursuant to Swiss law and after allocations to reserves required by Swiss law and its articles of association have been deducted. Distributable reserves are generally booked either as "free reserves" ("freie Reserven") or as "reserve from capital contributions" ("Reserven aus Kapitaleinlagen"). Distributions out of issued share capital, which is the aggregate nominal value of a corporation's issued shares, may be made only by way of a share capital reduction. See "Description of Share Capital and Articles of Association" in the prospectus included in the Registration Statement on From F-1 (333-235554) that we filed with the SEC on January 27, 2020.
97
B. Significant Changes.
On January 29, 2020, we listed American Depositary Shares (ADSs) representing our ordinary shares on the Nasdaq Stock Market following the United States Securities and Exchange Commission (SEC) having declared our registration statements on Forms F1 and F6 effective. The ADSs are listed for trading on Nasdaq under the symbol "ADXN". Addex has not registered any new issuance of securities and its shares will continue to be admitted to trading on SIX Swiss Exchange. Reference is made to Note 26 to the consolidated financial statements of our financial statements included elsewhere in this Annual Report on Form 20-F.
Item 9. The Offer and Listing.
A. Offer and Listing Details.
Our shares have been listed on Nasdaq Capital Market, or Nasdaq, under the symbol "ADXN" since January 29, 2020, and on the SIX Swiss Exchange, or SIX, under the symbol "ADXN" since January 29, 2020. Prior to May 22, 2007 , there was no public trading market for shares.
B. Plan of Distribution.
Not applicable.
C. Markets.
Our shares have been listed on Nasdaq Capital Market, or Nasdaq, under the symbol "ADXN" since January 29, 2020, and on the SIX Swiss Exchange, or SIX, under the symbol "ADXN" since January 29, 2020. Prior to May 22, 2007 , there was no public trading market for shares.
D. Selling Shareholders.
Not applicable.
E. Dilution.
Not applicable.
F. Expenses of the Issue.
Not applicable.
Item 10. Additional Information.
A. Share Capital.
Not applicable.
B. Memorandum and Articles of Association.
See the information set forth in the prospectus included in the Registration Statement on From F-1 (333-235554) that we filed with the SEC on January 27, 2020 under the captions "Description of Share Capital and Articles of Association," "Comparison of Swiss Law and Delaware Law," Description of American Depositary Shares," and "Service of Process and Enforcement of Judgments," which is incorporated herein by reference.
98
C. Material Contracts.
Collaboration Agreement with Indivior for Development of novel GABAB PAM Compounds, including for the Treatment of Addiction and Other CNS Diseases
In January 2018, we entered into an agreement with Indivior for the discovery, development and commercialization of novel GABAB PAM compounds for the treatment of addiction and other CNS diseases. This agreement included the selected clinical candidate, ADX71441. In addition, Indivior agreed to fund a research program at Addex to discover novel GABAB PAM compounds.
Indivior has sole responsibility, including funding liability, for development of selected compounds under the agreement through preclinical and clinical trials, as well as registration procedures and commercialization, if any, worldwide. Indivior has the right to design development programs for selected compounds under the agreement. Through our participation in a joint development committee, we review, in an advisory capacity, any development programs designed by Indivior. However, Indivior has authority over all aspects of the development of such selected compounds.
Under terms of the agreement, we have granted Indivior an exclusive license to use relevant patents and know-how in relation to the development and commercialization of product candidates selected by Indivior. Subject to agreed conditions, Addex and Indivior jointly own all intellectual property rights that are jointly developed, and Addex or Indivior individually own all intellectual property rights that Addex or Indivior develop individually. Addex has retained the right to select compounds from the research program for further development in areas outside the interest of Indivior including Charcot-Marie-Tooth type 1A neuropathy, or CMT1A. Under certain conditions, but subject to certain consequences, Indivior may terminate the agreement.
Under terms of the agreement, we received a non-refundable upfront fee of $5.0 million for the right to use the clinical candidate, ADX71441, including all materials and know-how related to this clinical candidate. In addition, we are eligible for payments on successful achievement of pre-specified clinical, regulatory and commercial milestones totaling $330 million. In addition, we are eligible for a royalty in the low teens on net sales of applicable products on a country-by country-basis. The term of the royalty for each licensed product in any particular country commences on such product's launch and ends on the latest of ten year anniversary of launch, expiration of certain applicable patent rights, and expiration of certain applicable marketing or data exclusivity periods. On February 14th, 2019, Indivior terminated the development of their selected compound, ADX71441.
Separately, Indivior funds research at Addex, based on a research plan to be mutually agreed between the parties, to discover novel GABAB PAM compounds. These future novel GABAB PAM compounds, if selected by Indivior, become licensed compounds. We agreed with Indivior to an initial research term and duration of two years, that can be extended by twelve-month increments and a minimum annual funding of $2 million for the Addex R&D costs incurred. Following Indivior's selection of one newly identified compound, Addex has the right to also select one additional newly identified compound. Addex is responsible for the funding of all development and commercialization costs of its selected compounds and Indivior has no rights to the Addex selected compounds. The initial two-year research term was expected to run from May 2018 to April 2020. On October 7, and December 20, 2019, Indivior agreed an additional research funding of $0.8 million, increasing the research funding by $1.6 million, for the research period.
Indivior may terminate the Agreement in its entirety or with respect to one or more countries or products upon 90 days' prior written notice prior to receipt of marketing approval for product candidates or twelve months' prior written notice after the receipt of marketing approval.
Addex may terminate the agreement if Indivior commits a material breach of the agreement and fails to cure such breach within 90 days of Addex's written notification to Indivior or fails to cure breaches to any payment obligations breach within 30 days.
99
Collaboration Agreement with Janssen for Development of Novel mGlu2 PAM Compounds, including ADX71149, for the Treatment of CNS and Related Diseases
On December 31, 2004, we entered into a collaboration and license agreement with Janssen Pharmaceuticals Inc., or Janssen, for the discovery, development and commercialization of novel mGlu2 PAM compounds for the treatment of CNS and related diseases. We agreed with Janssen to an initial research term and duration of two years that can be extended if parties mutually agree thereto in writing. The agreement was not extended. ADX71149 is one of the drug candidates discovered and selected for development by Janssen under the agreement. In 2012, Janssen announced completion of a Phase 2a clinical trial in Europe with ADX71149 for the treatment of schizophrenia demonstrating proof of principal in patients with negative symptoms of schizophrenia, such as apathy, social withdrawal, loss of emotional expression or sleep disorders. Janssen also conducted a Phase 2a clinical trial with ADX71149 for the treatment of patients with anxious depression, which failed to show statistically significant effects. In addition, Janssen announced in 2015 that ADX71149 demonstrated synergies with levetiracetam (a globally commercialized antiepileptic drug) in preclinical models of epilepsy.
Under our agreement with Janssen, we have granted Janssen an exclusive license to use relevant patents and know-how in relation to the development and commercialization of product candidates selected by Janssen under the agreement and a non-exclusive worldwide license to conduct research on the collaboration compounds using relevant patents and know-how. Subject to certain conditions, we and Janssen own, jointly, all intellectual property rights that we and Janssen develop jointly, and we or Janssen own all intellectual property rights that we or Janssen develop individually. Under certain conditions, but subject to certain consequences, Janssen may terminate the agreement for any reason, subject to a 90-day notice period.
Janssen has sole responsibility, including funding liability, for development of selected compounds under the agreement through preclinical and clinical trials, as well as registration procedures and commercialization, if any, in the United States, Japan, the United Kingdom, Germany, France, Spain and Italy. Janssen has the right to design development programs for selected compounds under the agreement. Through our participation in a joint development committee, we review, in an advisory capacity, any development programs designed by Janssen. However, Janssen has authority over all aspects of the development of selected compounds and may develop or commercialize third-party compounds with a different mechanism of action for identical use.
Under the terms of the Janssen agreement, we received an upfront fee of CHF 4.6 million and research funding of CHF 6.4 million during the research period, which ran from 2005 to 2007. In addition, we are eligible for payments on successful achievement of pre-specified clinical and regulatory milestones. We are also eligible for a royalty in the low teens on net sales of applicable products on a country-by country-basis and on a product by product basis, for a period commencing on the date of first sale and ending upon the latest of the expiration of twelve years from the date of first sale of a product in a given country or the last to expire of our patents containing a claim covering composition of compound comprised in a product sold by Janssen, its affiliates or sublicensees in such country. We received a CHF 1.5 million milestone payment in relation to the entry of ADX71149 into Phase 1 in July 2009 and a CHF 2.6 million milestone payment in relation to the entry of ADX71149 into Phase 2 in June 2011. We are eligible for a further €109 million in success-based development and regulatory milestones and low double digit royalties on net sales.
In the event that no compounds are discovered or identified during the research period, the agreement shall terminate.
In the event that there are compounds but no mGlu2 PAM are discovered during the research period or 6 months thereafter, our and Janssen's rights and obligations to develop commercialize, pay
100
royalties and milestones on mGlu2 PAM compounds will terminate. The contract also contains termination rights for non-defaulting parties upon breach.
For additional information on our material contracts, please see "Item 4. Information on the Company," Item 6. Directors, Senior Management and Employees," and "Item 7.B. Related Party Transactions" of this Annual Report on 20-F.
D. Exchange Controls.
There are no Swiss governmental laws, decrees or regulations that restrict, in a manner material to us, the export or import of capital, including any foreign exchange controls, or that generally affect the remittance of dividends or other payments to non-residents or non-citizens of Switzerland who hold our shares.
E. Taxation.
The following is a description of the material U.S. federal income tax consequences to the U.S. Holders described below of acquiring, owning and disposing of our shares or ADSs representing our shares. It is not a comprehensive description of all tax considerations that may be relevant to a particular person's decision to acquire securities. This discussion applies only to U.S. Holders that are initial purchasers of our ADSs representing our shares and that will hold such ADSs as a capital asset for tax purposes (generally, property held for investment). In addition, it does not describe all of the tax consequences that may be relevant in light of a U.S. Holder's particular circumstances, including state and local tax consequences, estate tax consequences, alternative minimum tax consequences, the potential application of the Medicare contribution tax, and tax consequences applicable to U.S. Holders subject to special rules, such as:
- •
- banks, insurance companies, and certain other financial institutions;
- •
- U.S. expatriates and certain former citizens or long-term residents of the United States;
- •
- dealers or traders in securities who use a mark-to-market method of tax accounting;
- •
- persons holding our shares or ADSs representing our shares as part of a hedging transaction, "straddle," wash sale, conversion transaction or
integrated transaction or persons entering into a constructive sale with respect to our shares or ADSs representing our shares;
- •
- persons whose "functional currency" for U.S. federal income tax purposes is not the U.S. dollar;
- •
- brokers, dealers or traders in securities, commodities or currencies;
- •
- tax-exempt entities or government organizations;
- •
- S corporations, partnerships, or other entities or arrangements classified as partnerships for U.S. federal income tax purposes;
- •
- regulated investment companies or real estate investment trusts;
- •
- persons who acquired our shares or ADSs representing our shares pursuant to the exercise of any employee stock option or otherwise as
compensation;
- •
- persons required under Section 451(b) of the Code to conform to the timing of income accruals with respect to our ADSs representing our
shares or the shares represented by such ADSs;
- •
- persons that own or are deemed to own (including by attribution) ten percent or more of our shares by voting power or value; and
- •
- persons holding our shares or ADSs representing our shares in connection with a trade or business, permanent establishment, or fixed base outside the United States.
101
Holders of our shares or ADSs representing our shares who fall within one of the categories above are advised to consult their tax advisor regarding the specific tax consequences which may apply to their particular situation.
If an entity that is classified as a partnership for U.S. federal income tax purposes holds our shares or ADSs representing our shares, the U.S. federal income tax treatment of a partner will generally depend on the status of the partner and the activities of the partnership. Partnerships holding our shares or ADSs representing our shares and partners in such partnerships are encouraged to consult their tax advisers as to the particular U.S. federal income tax consequences of holding and disposing of such shares or ADSs representing our shares.
The discussion is based on the Internal Revenue Code of 1986, as amended, or the Code, administrative pronouncements, judicial decisions, final, temporary and proposed Treasury Regulations, and the Convention between the United States of America and the Swiss Confederation for the Avoidance of Double Taxation with Respect to Taxes on Income, signed on October 2, 1996, as amended and currently in force, or the U.S.-Swiss Tax Treaty, in each case, as in effect and available on the date hereof. All of the foregoing are subject to change, which change could apply retroactively, and to differing interpretations, all of which could affect the tax considerations described below. There can be no assurances that the U.S. Internal Revenue Service, or the IRS, will not take a position concerning the tax consequences of the acquisition, ownership and disposition of our shares or ADSs representing our shares or that such a position would not be sustained by a court. We have not obtained, nor do we intend to obtain, a ruling with respect to the U.S. federal income tax considerations in the purchase, ownership or disposition of our shares or ADSs representing our shares. Accordingly, holders should consult their own tax advisors concerning the U.S. federal, state, local and non-U.S. tax consequences of acquiring, owning and disposing of our shares or ADSs representing our shares in their particular circumstances.
A "U.S. Holder" is a holder who, for U.S. federal income tax purposes, is a beneficial owner of our shares or ADSs representing our shares who is eligible for the benefits of the U.S.-Swiss Tax Treaty and is:
- •
- a citizen or individual resident of the United States;
- •
- a corporation, or other entity taxable as a corporation, created or organized in or under the laws of the United States, any state therein or
the District of Columbia;
- •
- an estate the income of which is subject to U.S. federal income taxation regardless of its source; or
- •
- a trust if (1) a U.S. court is able to exercise primary supervision over the administration of the trust and one or more U.S. persons have authority to control all substantial decisions of the trust or (2) the trust has a valid election to be treated as a U.S. person under applicable U.S. Treasury Regulations.
U.S. Holders are encouraged to consult their tax advisers concerning the U.S. federal, state, local and foreign tax consequences of owning and disposing of our shares or ADSs representing our shares in their particular circumstances.
The discussion below assumes that the representations contained in the deposit agreement are true and that the obligations in the deposit agreement and any related agreement will be complied with in accordance with their terms. Generally, a holder of an ADS should be treated for U.S. federal income tax purposes as holding the shares represented by the ADS. Accordingly, no gain or loss will be recognized upon an exchange of ADSs representing our shares for shares. The U.S. Treasury has expressed concerns that intermediaries in the chain of ownership between the holder of an ADS and the issuer of the security underlying the ADS may be taking actions that are inconsistent with the
102
beneficial ownership of the underlying security. Accordingly the creditability of foreign taxes, if any, as described below, could be affected by actions taken by intermediaries in the chain of ownership between the holders of ADSs representing our shares and our company if, as a result of such actions, the holders of ADSs representing our shares are not properly treated as beneficial owners of the underlying shares.
Passive Foreign Investment Company Rules
Based on our analysis of our income, assets, activities and market capitalization for our taxable year ended December 31, 2019, we believe that we are classified as a "passive foreign investment company," or PFIC, for our taxable year ended December 31, 2019. The application of the PFIC rules is subject to uncertainty in several respects, and therefore, no assurances can be provided with respect to our PFIC status for our taxable year ended December 31, 2019 or with regard to our PFIC status in the past or in the future. If we are classified as a PFIC in any taxable year, a U.S. Holder will be subject to special rules generally intended to reduce or eliminate any benefits from the deferral of U.S. federal income tax that a U.S. Holder could derive from investing in a non-U.S. company that does not distribute all of its earnings on a current basis.
A non-U.S. corporation will be classified as a PFIC for any taxable year in which, after applying certain look-through rules with respect to the income and assets of its subsidiaries, either:
- •
- at least 75% of its gross income is "passive income"; or
- •
- at least 50% of its gross assets (determined on the basis of a quarterly average) is attributable to assets that produce "passive income" or are held for the production of "passive income."
For this purpose, cash is a passive asset and passive income generally includes dividends, interest, royalties and rents (other than royalties and rents derived in the active conduct of a trade or business and not derived from a related person). We will be treated as owning our proportionate share of the assets and earning our proportionate share of the income of any other corporation, the equity of which we own, directly or indirectly, 25% or more (by value).
The determination of whether we are a PFIC is a fact-intensive determination made on an annual basis and the applicable law is subject to varying interpretation. If we are a PFIC for any taxable year during which a U.S. Holder holds our shares or ADSs representing our shares, such U.S. Holder will be subject to special tax rules discussed below and could suffer adverse tax consequences.
A separate determination must be made after the close of each taxable year as to whether we are a PFIC for that year. As a result, our PFIC status may change from year to year. The total value of our assets for purposes of the asset test generally will be calculated using the market price of our shares or ADSs representing our shares, which may fluctuate considerably. Fluctuations in the market price of the shares or ADSs representing our shares may result in our being a PFIC for any taxable year. Because of the uncertainties involved in establishing our PFIC status, our United States tax counsel expresses no opinion regarding our PFIC status.
If we are classified as a PFIC in any year with respect to which a U.S. Holder owns our shares or ADSs representing our shares, we will continue to be treated as a PFIC with respect to such U.S. Holder in all succeeding years during which the U.S. Holder owns the shares or ADSs representing our shares, regardless of whether we continue to meet the tests described above unless (i) we cease to be a PFIC and the U.S. Holder has made a "deemed sale" election under the PFIC rules, or (ii) the U.S. Holder makes a "QEF Election" (defined below) or is eligible to make and makes a mark-to-market election (as described below), with respect to all taxable years during such U.S. Holder's holding period in which we are a PFIC. If the "deemed sale" election is made, a U.S. Holder will be deemed to have sold the shares or ADSs representing our shares the U.S. Holder holds at their fair market value as of the date of such deemed sale and any gain from such deemed sale would be subject to the rules
103
described below. After the deemed sale election, so long as we do not become a PFIC in a subsequent taxable year, the U.S. Holder's shares or ADSs representing our shares with respect to which such election was made will not be treated as shares in a PFIC and the U.S. Holder will not be subject to the rules described below with respect to any "excess distribution" the U.S. Holder receives from us or any gain from an actual sale or other disposition of the shares or ADSs representing our shares. U.S. Holders should consult their tax advisors as to the possibility and consequences of making a deemed sale election if such election becomes available.
For each taxable year we are treated as a PFIC with respect to U.S. Holders, U.S. Holders will be subject to special tax rules with respect to any "excess distribution" such U.S. Holder receives and any gain such U.S. Holder recognizes from a sale or other disposition (including a pledge) of our shares or ADSs representing our shares, unless (i) such U.S. Holder makes a QEF Election or (ii) our shares or ADSs representing our shares constitute "marketable" securities, and such U.S. Holder makes a mark-to-market election as discussed below. Distributions a U.S. Holder receives in a taxable year that are greater than 125% of the average annual distributions a U.S. Holder received during the shorter of the three preceding taxable years or the U.S. Holder's holding period for the shares or ADSs representing our shares will be treated as an excess distribution. Under these special tax rules:
- •
- the excess distribution or gain will be allocated ratably over a U.S. Holder's holding period for the shares or ADSs representing our shares;
- •
- the amount allocated to the current taxable year, and any taxable year prior to the first taxable year in which we became a PFIC, will be
treated as ordinary income; and
- •
- the amount allocated to each other year will be subject to the highest tax rate in effect for that year and the interest charge generally applicable to underpayments of tax will be imposed on the resulting tax attributable to each such year.
The tax liability for amounts allocated to years prior to the year of disposition or "excess distribution" cannot be offset by any net operating losses for such years, and gains (but not losses) realized on the sale of the shares or ADSs representing our shares cannot be treated as capital gains, even if a U.S. Holder holds the shares or ADSs representing our shares as capital assets. In addition, dividend distributions made to you will not qualify for the lower rates of taxation applicable to qualified dividends discussed below under "Taxation of Distributions."
If we are a PFIC, a U.S. Holder will generally be subject to similar rules with respect to distributions we receive from, and our dispositions of the stock of, any of our direct or indirect subsidiaries that also are PFICs, as if such distributions were indirectly received by, and / or dispositions were indirectly carried out by, such U.S. Holder. U.S. Holders should consult their tax advisors regarding the application of the PFIC rules to our subsidiaries.
U.S. Holders can avoid the interest charge on excess distributions or gain relating to our shares or ADSs representing our shares by making a mark-to-market election with respect to the shares or ADSs representing our shares, provided that the shares or ADSs representing our shares are "marketable." Shares or ADSs representing our shares will be marketable if they are "regularly traded" on certain U.S. stock exchanges or on a foreign stock exchange that meets certain conditions. For these purposes, the shares or ADSs representing our shares will be considered regularly traded during any calendar year during which they are traded on, other than in de minimis quantities, at least 15 days during each calendar quarter. Any trades that have as their principal purpose meeting this requirement will be disregarded. Our ADSs representing our shares will be listed on Nasdaq, which is a qualified exchange for these purposes. Consequently, if our ADSs representing our shares remain listed on Nasdaq and are regularly traded, and you are a U.S. Holder of ADSs representing our shares, we expect the mark-to-market election would be available to you if we are a PFIC. Each U.S. Holder should consult its tax advisor as to the whether a mark-to-market election is available or advisable with respect to the
104
shares or ADSs representing our shares. It should be noted that it is intended that only our ADSs representing our shares and not our shares will be listed on Nasdaq. Consequently, our shares may not be marketable if SIX Swiss Exchange (where our shares are listed) does not meet the applicable requirements. U.S. Holders should consult their tax advisors regarding the availability of the mark-to-market election for shares that are not represented by ADSs.
A U.S. Holder that makes a mark-to-market election must include in ordinary income for each year an amount equal to the excess, if any, of the fair market value of our shares or ADSs representing our shares at the close of the taxable year over the U.S. Holder's adjusted tax basis in the shares or ADSs representing our shares. An electing holder may also claim an ordinary loss deduction for the excess, if any, of the U.S. Holder's adjusted basis in the shares or ADSs representing our shares over the fair market value of the shares or ADSs representing our shares at the close of the taxable year, but this deduction is allowable only to the extent of any net mark-to-market gains for prior years. Gains from an actual sale or other disposition of the shares or ADSs representing our shares will be treated as ordinary income, and any losses incurred on a sale or other disposition of the shares will be treated as an ordinary loss to the extent of any net mark-to-market gains for prior years. Once made, the election cannot be revoked without the consent of the IRS unless the shares or ADSs representing our shares cease to be marketable.
However, a mark-to-market election generally cannot be made for equity interests in any lower-tier PFICs that we own, unless shares of such lower-tier PFIC are themselves "marketable." As a result, even if a U.S. Holder validly makes a mark-to-market election with respect to our shares or ADSs representing our shares, the U.S. Holder may continue to be subject to the PFIC rules (described above) with respect to its indirect interest in any of our investments that are treated as an equity interest in a PFIC for U.S. federal income tax purposes. U.S. Holders should consult their tax advisors as to the availability and desirability of a mark-to-market election, as well as the impact of such election on interests in any lower-tier PFICs.
Alternatively, a U.S. Holder can make an election, if we provide the necessary information, to treat us and each lower-tier PFIC as a qualified electing fund, or a QEF Election, in the first taxable year we (and our relevant subsidiaries) are treated as a PFIC with respect to the U.S. Holder. If such election remains in place while we and any lower-tier PFIC subsidiaries are PFICs, we and our subsidiaries will not be treated as PFICs with respect to such U.S. Holder. A U.S. Holder must make the QEF Election for us and for each of our subsidiaries that is a PFIC by attaching a separate properly completed IRS Form 8621 for each such PFIC to the U.S. Holder's timely filed U.S. federal income tax return. If we are a PFIC for our taxable year ending December 31, 2019, or any subsequent taxable years, we expect to provide U.S. Holders, upon request, a "PFIC Annual Information Statement", with the information required to allow U.S. Holders to make a QEF Election for United States federal income tax purposes.
If a U.S. Holder makes a QEF Election with respect to a PFIC in lieu of the tax consequences described above, the U.S. Holder will be currently taxable on its pro rata share of the PFIC's ordinary earnings and net capital gain (at ordinary income and capital gain rates, respectively) for each taxable year that the entity is classified as a PFIC. If a U.S. Holder makes a QEF Election with respect to us, any distributions paid by us out of our earnings and profits that were previously included in the U.S. Holder's income under the QEF Election would not be taxable to the holder. A U.S. Holder will increase its tax basis in its shares or ADSs representing our shares by an amount equal to any income included under the QEF Election and will decrease its tax basis by any amount distributed on the shares or ADSs representing our shares that is not included in the holder's income. In addition, a U.S. Holder will recognize capital gain or loss on the disposition of shares or ADSs representing our shares in an amount equal to the difference between the amount realized and the holder's adjusted tax basis in the shares or ADSs representing our shares. U.S. Holders should note that if they make QEF Elections with respect to us and lower-tier PFICs, they may be required to pay U.S. federal income tax
105
with respect to their shares or ADSs representing our shares for any taxable year significantly in excess of any cash distributions (which may be zero) received on the shares or ADSs representing our shares for such taxable year. U.S. Holders should consult their tax advisors regarding making QEF Elections in their particular circumstances. If a U.S. Holder does not make and maintain a QEF election for the U.S. Holder's entire holding period for the shares or ADSs representing our shares by making the election for the first year in which the U.S. Holder owns the shares or ADSs representing our shares, the U.S. Holder will be subject to the adverse PFIC rules discussed above unless the U.S. Holder can properly make a 'purging election' with respect to the shares or ADS representing our shares in connection with the U.S. Holder's QEF Election. A purging election may require the U.S. Holder to recognize taxable gain on the U.S. Holder's shares or ADSs representing our shares. No purging election is necessary for a U.S. Holder that timely makes a QEF election for the first year in which the U.S. Holder acquired our shares or ADSs representing our shares.
The U.S. federal income tax rules relating to PFICs are complex. Prospective U.S. investors are urged to consult their own tax advisors with respect to the acquisition, ownership and disposition of our shares or ADSs representing our shares, the consequences to them of an investment in a PFIC, any elections available with respect to our shares or ADSs representing our shares and the IRS information reporting obligations with respect to the acquisition, ownership and disposition of our shares or ADSs representing our shares.
Taxation of Distributions
Subject to the discussion above under "Passive Foreign Investment Company Rules," distributions paid on our shares or ADSs representing our shares will generally be treated as dividends to the extent paid out of our current or accumulated earnings and profits (as determined under U.S. federal income tax principles). Because we may not calculate our earnings and profits under U.S. federal income tax principles, we expect that distributions generally will be reported to U.S. Holders as dividends. Subject to applicable limitations, dividends paid to certain non-corporate U.S. Holders may be taxable at preferential rates applicable to "qualified dividend income" if we are a "qualified foreign corporation" and certain other requirements (discussed below) are met. A non-U.S. corporation (other than a corporation that is classified as a PFIC for the taxable year in which the dividend is paid or the preceding taxable year) generally will be considered to be a qualified foreign corporation (a) if it is eligible for the benefits of a comprehensive tax treaty with the United States which the Secretary of Treasury of the United States determines is satisfactory for purposes of this provision and which includes an exchange of information provision, or (b) with respect to any dividend it pays on ADSs representing our shares which are readily tradable on an established securities market in the United States. We have applied to list our ADSs representing our shares on Nasdaq, which is an established securities market in the United States, and we expect the ADSs representing our shares to be readily tradable on Nasdaq. There can be no assurance that the ADSs representing our shares will be considered readily tradable on an established securities market in the United States in later years. The Company, which is incorporated under the laws of Switzerland, believes that it qualifies as a resident of Switzerland for purposes of, and is eligible for the benefits of, the U.S.-Swiss Tax Treaty, although there can be no assurance in this regard. Further, the IRS has determined that the U.S.-Swiss Tax Treaty is satisfactory for purposes of the qualified dividend rules and that it includes an exchange-of-information program. Therefore, subject to the discussion under "Passive Foreign Investment Company Rules," above, such dividends will generally be "qualified dividend income" in the hands of individual U.S. Holders, provided that a holding period requirement (more than 60 days of ownership, without protection from the risk of loss, during the 121-day period beginning 60 days before the ex-dividend date) and certain other requirements are met. The dividends will not be eligible for the dividends-received deduction generally allowed to corporate U.S. holders.
106
However, the qualified dividend income treatment will not apply if we are treated as a PFIC. In addition, the amount of the dividend will be treated as foreign-source dividend income to U.S. Holders and will not be eligible for the dividends-received deduction generally available to U.S. corporations under the Code. Dividends will generally be included in a U.S. Holder's income on the date of the U.S. Holder's receipt of the dividend. The amount of any dividend income paid in foreign currency will be the U.S. dollar amount calculated by reference to the exchange rate in effect on the date of actual or constructive receipt, regardless of whether the payment is in fact converted into U.S. dollars. If the dividend is converted into U.S. dollars on the date of receipt, a U.S. Holder should not be required to recognize foreign currency gain or loss in respect of the dividend income. A U.S. Holder may have foreign currency gain or loss if the dividend is converted into U.S. dollars after the date of receipt. Such gain or loss would generally be treated as U.S.-source ordinary income or loss. The amount of any distribution of property other than cash generally will be the fair market value of such property on the date of distribution.
A U.S. Holder generally may claim the amount of any Swiss withholding tax as either a deduction from gross income or a credit against its U.S. federal income tax liability. The foreign tax credit is subject to numerous complex limitations that must be determined and applied on an individual basis. Generally, the credit cannot exceed the proportionate share of a U.S. Holder's U.S. federal income tax liability that such U.S. Holder's taxable income bears to such U.S. Holder's worldwide taxable income. In applying this limitation, a U.S. Holder's various items of income and deduction must be classified, under complex rules, as either "foreign source" or "U.S. source." In addition, the creditability of foreign taxes could be affected by actions taken by intermediaries in the chain of ownership between the holders of our shares or ADSs representing our shares and our company if, as a result of such actions, the holders of our shares or ADSs representing our shares are not properly treated as beneficial owners of the underlying shares. Each U.S. Holder should consult its own tax advisors regarding the foreign tax credit rules.
Sale or Other Taxable Disposition of Shares and ADSs Representing Our Shares
Subject to the discussion above under "Passive Foreign Investment Company Rules," gain or loss realized on the sale or other taxable disposition of our shares or ADSs representing our shares will be capital gain or loss, and will be long-term capital gain or loss if the U.S. Holder held the shares or ADSs representing our shares for more than one year. The amount of the gain or loss will equal the difference between the U.S. Holder's adjusted tax basis in the shares or ADSs representing our shares disposed of and the amount realized on the disposition, in each case as determined in U.S. dollars. The adjusted tax basis in our shares or ADSs representing our shares generally will be equal to the cost of such shares or ADSs. This gain or loss will generally be U.S.-source gain or loss for foreign tax credit purposes. The deductibility of capital losses is subject to limitations.
If the consideration received by a U.S. Holder is not paid in U.S. dollars, the amount realized will be the U.S. dollar value of the payment received determined by reference to the spot rate of exchange on the date of the sale or other disposition. However, if the shares or ADSs representing our shares are treated as traded on an "established securities market" and you are either a cash basis taxpayer or an accrual basis taxpayer that has made a special election (which must be applied consistently from year to year and cannot be changed without the consent of the IRS), you will determine the U.S. dollar value of the amount realized in a non-U.S. dollar currency by translating the amount received at the spot rate of exchange on the settlement date of the sale. If you are an accrual basis taxpayer that is not eligible to or does not elect to determine the amount realized using the spot rate on the settlement date, you will recognize foreign currency gain or loss to the extent of any difference between the U.S. dollar amount realized on the date of sale or disposition and the U.S. dollar value of the currency received at the spot rate on the settlement date.
107
Information Reporting and Backup Withholding
U.S. Holders generally will be subject to information reporting requirements with respect to dividends on our shares or ADSs representing our shares and on the proceeds from the sale, exchange or disposition of our shares or ADSs representing our shares that are paid within the United States or through certain U.S.-related financial intermediaries, unless the U.S. Holder is an "exempt recipient." In addition, U.S. Holders may be subject to backup withholding on such payments, unless the U.S. Holder provides a correct taxpayer identification number and a duly executed IRS Form W-9 or otherwise establishes an exemption. Backup withholding is not an additional tax, and the amount of any backup withholding from a payment to a U.S. Holder will be allowed as a credit against a U.S. Holder's U.S. federal income tax liability and may entitle such holder to a refund, provided that the required information is timely furnished to the IRS.
Certain Reporting Requirements
U.S. Holders paying more than $100,000 for our shares or ADSs representing our shares generally may be required to file IRS Form 926 reporting the payment of the offer price for our shares or ADSs representing our shares to us. Substantial penalties may be imposed upon a U.S. Holder that fails to comply. Each U.S. Holder should consult its own tax advisor as to the possible obligation to file IRS Form 926.
Information with Respect to Foreign Financial Assets
Certain U.S. Holders who are individuals (and, under regulations, certain entities) may be required to report information relating to our shares or ADSs representing our shares, subject to certain exceptions (including an exception for shares or ADSs representing our shares held in accounts maintained by certain U.S. financial institutions). Such U.S. Holders who fail to timely furnish the required information may be subject to a penalty. Additionally, if a U.S. Holder does not file the required information, the statute of limitations with respect to tax returns of the U.S. Holder to which the information relates may not close until three years after such information is filed. U.S. Holders should consult their tax advisers regarding their reporting obligations with respect to their ownership and disposition of our shares or ADSs representing our shares.
Swiss Tax Considerations
Swiss Federal, Cantonal and Communal Individual Income Tax and Corporate Income Tax
Non-Resident Shareholders
Holders of or shares or ADSs representing our shares who are not resident in Switzerland for tax purposes, and who, during the relevant taxation year, have not engaged in a trade or business carried on through a permanent establishment or fixed place of business situated in Switzerland for tax purposes (all such shareholders are hereinafter referred to as the "Non-Resident Shareholders"), will not be subject to any Swiss federal, cantonal and communal income tax on dividends and similar cash or in-kind distributions on ADSs representing our shares (including dividends on liquidation proceeds and stock dividends) (hereinafter referred to as the "Dividends"), distributions based upon a capital reduction (Nennwertrückzahlungen) or paid out of reserves from capital contributions (Reserven aus Kapitaleinlagen) on shares underlying the ADSs, or capital gains realized on the sale or other disposition of ADSs (see, however, "Swiss Federal Withholding Tax" below for a summary of Swiss federal withholding tax on Dividends).
108
Resident Private Shareholders
Swiss resident individuals who hold their ADSs as private assets all such shareholders are hereinafter referred to as the "Resident Private Shareholders") are required to include Dividends, but not distributions based upon a capital reduction (Nennwertrückzahlungen) or paid out of reserves from capital contributions (Reserven aus Kapitaleinlagen) of the shares underlying the ADSs, in their personal income tax return and are subject to Swiss federal, cantonal and communal income tax on any net taxable income for the relevant taxation period, including the Dividends, but not the distributions based upon a capital reduction (Nennwertrückzahlungen) or paid out of reserves from capital contributions (Reserven aus Kapitaleinlagen). Capital gains resulting from the sale or other dispositions of ADSs are not subject to Swiss federal, cantonal and communal income tax, and conversely, capital losses are not tax-deductible for Resident Private Shareholders. See "Domestic Commercial Shareholders" below for a summary of the taxation treatment applicable to Swiss resident individuals, who, for income tax purposes, are classified as "professional securities dealers".
(C) Domestic Commercial Shareholders
Corporate and individual shareholders who are resident in Switzerland for tax purposes and corporate and individual shareholder who are not resident in Switzerland, and who, in each case, hold their ADSs as part of a trade or business carried on in Switzerland, in the case of corporate and individual shareholders not resident in Switzerland, through a permanent establishment or fixed place of business situated, for tax purposes, in Switzerland, are required to recognize Dividends, distributions based upon a capital reduction (Nennwertrückzahlungen) or paid out of reserves from capital contributions (Reserven aus Kapitaleinlagen) received on shares underlying the ADSs and capital gains or losses realized on the sale or other disposition of ADSs in their income statement for the relevant taxation period and are subject to Swiss federal, cantonal and communal individual or corporate income tax, as the case may be, on any net taxable earnings for such taxation period. The same taxation treatment also applies to Swiss-resident private individuals who, for income tax purposes, are classified as "professional securities dealers" for reasons of, inter alia, frequent dealing, or leveraged investments in ADSs and other securities (the shareholders referred to in this paragraph), hereinafter for the purposes of this section, as the "Domestic Commercial Shareholders"). Domestic Commercial Shareholders who are corporate taxpayers may be eligible for dividend relief (Beteiligungsabzug) in respect of Dividends and distributions based upon a capital reduction (Nennwertrückzahlungen) or paid out of reserves from capital contributions (Reserven aus Kapitaleinlagen) if the shares underlying the ADSs held by them as part of a Swiss business have an aggregate market value of at least CHF 1 million.
Swiss Cantonal and Communal Private Wealth Tax and Capital Tax
Non-Resident Shareholders
Non-Resident Shareholders are not subject to Swiss cantonal and communal private wealth tax or capital tax.
Resident Private Shareholders and Domestic Commercial Shareholders
Resident Private Shareholders and Domestic Commercial Shareholders who are individuals are required to report their ADSs as part of private wealth or their Swiss business assets, as the case may be, and will be subject to Swiss cantonal and communal private wealth tax on any net taxable wealth (including the ADSs), in the case of Domestic Commercial Shareholders to the extent the aggregate taxable wealth is allocated in Switzerland. Domestic Commercial Shareholders who are corporate taxpayers are subject to Swiss cantonal and communal capital tax on taxable capital to the extent the aggregate taxable capital is allocated to Switzerland.
109
Swiss Federal Withholding Tax
Dividends that the Company pays on the shares underlying the ADSs are subject to Swiss Federal withholding tax (Verrechnungssteuer) at a rate of 35% on the gross amount of the Dividend. The Company is required to withhold the Swiss federal withholding tax from the Dividend and remit it to the Swiss Federal Tax Administration. Distributions based upon a capital reduction (Nennwertrückzahlungen) or paid out of reserves from capital contributions (Reserven aus Kapitaleinlagen) are not subject to Swiss federal withholding tax.
The Swiss federal withholding tax on a Dividend will be refundable in full to a Resident Private Shareholder and to a Domestic Commercial Shareholder, who, in each case, inter alia, as a condition to refund, duly reports the Dividend in his or her individual income tax return as income or recognizes the Dividends in its income statement as earnings, as applicable.
A Non-Resident Shareholder may be entitled to a partial refund of the Swiss federal withholding tax on Dividend if the country of his or her residence for tax purposes has entered into a bilateral treaty for the avoidance of double taxation with Switzerland and the conditions of such treaty are met. Such shareholders should be aware that the procedures for claiming tax treaty benefits (and the time required for obtaining a refund) might be different from country to country. For example, a shareholder who is resident of the U.S. for the purposes of the bilateral treaty between the U.S. and Switzerland is eligible for a refund of the amount of the withholding tax in excess of the 15% treaty rate, provided such shareholder: (i) qualifies for benefits under this treaty and qualifies as beneficial owner of the Dividends; (ii) hold, directly or indirectly, less than 10% of the voting stock of the Company; (iii) does not qualify as a pension scheme or retirement arrangement for the purpose of the bilateral treaty; and (iv) does not conduct business through a permanent establishment or fixed base in Switzerland to which the ADSs are attributable. Such an eligible U.S. shareholder may apply for a refund of the amount of the withholding tax in excess of the 15% treaty rate. The applicable refund request form may be filed with the Swiss Federal Tax Administration following receipt of the dividend and the relevant deduction certificate, however no later than December 31 of the third year following the calendar year in which the dividend was payable.
Swiss Federal Stamp Taxes
Any dealings in the ADSs, where a bank or another securities dealer in Switzerland, as defined in the Swiss Federal Stamp Tax Act, acts as intermediary or is a party to the transaction, are, subject to certain exemptions provided for in the Swiss Federal Stamp Tax Act, subject to Swiss securities turnover tax at an aggregate tax rate of up to 0.3% of the consideration paid for such ADSs.
International Automatic Exchange of Information in Tax Matters
On November 19, 2014, Switzerland signed the Multilateral Competent Authority Agreement, which is based on article 6 of the OECD/Council of Europe administrative assistance convention and is intended to ensure the uniform implementation of automatic exchange of information (the "AEOI"). The Federal Act on the International Automatic Exchange of Information in Tax Matters (the "AEOI Act") entered into force on January 1, 2017. The AEOI Act is the legal basis for the implementation of the AEOI standard in Switzerland.
The AEOI is being introduced in Switzerland through bilateral agreements or multilateral agreements. The agreements have, and will be, concluded on the basis of guaranteed reciprocity, compliance with the principle of specialty (i.e., the information exchanged may only be used to assess and levy taxes (and for criminal tax proceedings)) and adequate data protection.
Based on such multilateral agreements and bilateral agreements and the implementing laws of Switzerland, Switzerland exchanges data in respect of financial assets, including the Shares, held in, and
110
income derived thereon and credited to, accounts or deposits with a paying agent in Switzerland for the benefit of individuals resident in a EU member state or in a treaty state.
Swiss Facilitation of the Implementation of the U.S. Foreign Account Tax Compliance Act
Switzerland has concluded an intergovernmental agreement with the U.S. to facilitate the implementation of FATCA. The agreement ensures that the accounts held by U.S. persons with Swiss financial institutions are disclosed to the U.S. tax authorities either with the consent of the account holder or by means of group requests within the scope of administrative assistance. Information will not be transferred automatically in the absence of consent, and instead will be exchanged only within the scope of administrative assistance on the basis of the double taxation agreement between the U.S. and Switzerland. On October 8, 2014, the Swiss Federal Council approved a mandate for negotiations with the U.S. on changing the current direct-notification-based regime to a regime where the relevant information is sent to the Swiss Federal Tax Administration, which in turn provides the information to the U.S. tax authorities.
F. Dividends and Paying Agents.
Not applicable.
G. Statement by Experts.
Not applicable.
H. Documents on Display.
We are subject to the information reporting requirements of the Exchange Act applicable to foreign private issuers and under those requirements will file reports with the SEC. Those reports may be inspected without charge at the locations described below. As a foreign private issuer, we are exempt from the rules under the Exchange Act related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we are not required under the Exchange Act to file periodic reports and financial statements with the SEC as frequently or as promptly as United States companies whose securities are registered under the Exchange Act.
We maintain a corporate website at www.addextherapeutics.com. We intend to post our Annual Report on Form 20-F on our website promptly following it being filed with the SEC. Information contained on, or that can be accessed through, our website does not constitute a part of this Annual Report on Form 20-F. We have included our website address in this Annual Report on Form 20-F solely as an inactive textual reference. The Securities and Exchange Commission maintains a website (www.sec.gov) that contains reports, proxy and information statements and other information regarding registrants, such as Addex, that file electronically with the SEC.
I. Subsidiary Information.
Not required.
Item 11. Quantitative and Qualitative Disclosures About Market Risk.
We operate primarily in Switzerland, Europe and in the United States and are therefore exposed to market risk, which represents the risk of loss that may impact our financial position due to adverse changes in financial market prices and rates. As of December 31, 2019, we had CHF 31.5 million of cash and cash equivalents and we had no debt.
111
Interest Rate Risk
Our cash is held in readily available checking and money market accounts. As of December 31, 2019, Swiss francs balance represented 64% of the cash and cash equivalents of the company. Since January 1, 2018, Swiss banks have partially re-invoiced to their clients a part of the negative interests on Swiss franc deposit that they pay to the Swiss National Bank. For the year ended December 31, 2019 the effective negative interest rate on Swiss francs cash deposits paid by Addex was limited to—0.45%. As a result, a change in market interest rates would not have had any significant impact on our financial position or results of operations. As of December 31, 2019, we had no debt and, therefore, no material interest rate risk exposure.
Foreign Currency Exchange Risk
We operate primarily in Switzerland, the United States and Europe more broadly and our functional currency is the Swiss franc, and as a result, we are exposed to (1) transactional foreign exchange risk when we or a subsidiary enter into a transaction in a currency other than our or its functional currency and (2) translational foreign exchange risk when we translate financial statements of our foreign subsidiaries from their functional currency into Swiss francs.
Transactional Risk
Our expenses are generally denominated in the currencies of the countries where the relevant transaction takes place, which is primarily in Switzerland, the United States, and to a lesser extent in the Euro-zone countries and United Kingdom. Transactions in foreign currencies of our Swiss company are recorded in Swiss francs at the applicable exchange rate on the date of the relevant transaction. Our results of operations and cash flows are, therefore, subject to fluctuations due to changes in foreign currency exchange rates and may be adversely affected in the future due to changes in foreign currency exchange rates.
Translational Risk
Because our reporting currency is the Swiss franc, or CHF, we may be exposed to translation risk when the income statements of our subsidiaries located in countries outside Switzerland are converted into Swiss francs using the average exchange rate for the period, and whilst revenues and costs are unchanged in local currency, changes in exchange rates may lead to effects on the converted balances of revenue, costs and the result in Swiss francs .
To date, our risk management policy is to economically hedge 100% of anticipated transactions in each major currency for the subsequent 12 months. As our operations grow, we will continue to reassess our approach to manage our risk relating to fluctuations in foreign currency rates.
Capital Risk
We are not regulated and not subject to specific capital requirements, however, we aim to be compliant with the specific needs of the Swiss law. To ensure that statutory capital requirements are met, we monitor capital periodically on an interim basis as well as annually. From time to time, we may take appropriate measures or propose capital increases at the shareholders' meeting to ensure the necessary capital remains intact.
Item 12. Description of Securities Other than Equity Securities.
A. Debt Securities.
Not applicable.
112
B. Warrants and Rights.
Not applicable.
C. Other Securities.
Not applicable.
D. American Depositary Shares.
Fees and Charges
ADS holders are required to pay the following fees under the terms of the deposit agreement governing the terms of the ADS:
| Service | Fee | |
|---|---|---|
• Issuance of ADSs (e.g., an issuance of ADS upon a deposit of shares, upon a change in the ADS(s)-to-shares ratio, or for any other reason), excluding ADS issuances as a result of distributions of shares) |
• Up to U.S. 5¢ per ADS issued |
|
• Cancellation of ADSs (e.g., a cancellation of ADSs for delivery of deposited property, upon a change in the ADS(s)-to-shares ratio, or for any other reason) |
• Up to U.S. 5¢ per ADS cancelled |
|
• Distribution of cash dividends or other cash distributions (e.g., upon a sale of rights and other entitlements) |
• Up to U.S. 5¢ per ADS held |
|
• Distribution of ADSs pursuant to (i) stock dividends or other free stock distributions, or (ii) exercise of rights to purchase additional ADSs |
• Up to U.S. 5¢ per ADS held |
|
• Distribution of securities other than ADSs or rights to purchase additional ADSs (e.g., upon a spin-off) |
• Up to U.S. 5¢ per ADS held |
|
• ADS Services |
• Up to U.S. 5¢ per ADS held on the applicable record date(s) established by the depositary |
|
• Registration of ADS transfers (e.g., upon a registration of the transfer of registered ownership of ADSs, upon a transfer of ADSs into DTC and vice versa, or for any other reason) |
• Up to U.S. 5¢ per ADS (or fraction thereof) transferred |
|
• Conversion of ADSs of one series for ADSs of another series (e.g., upon conversion of Partial Entitlement ADSs for Full Entitlement ADSs, or upon conversion of Restricted ADSs (each as defined in the Deposit Agreement) into freely transferable ADSs, and vice versa). |
• Up to U.S. 5¢ per ADS (or fraction thereof) converted |
113
ADS holders are also be responsible for paying certain charges such as:
- •
- taxes (including applicable interest and penalties) and other governmental charges;
- •
- the registration fees as may from time to time be in effect for the registration of shares on the share register and applicable to transfers of
shares to or from the name of the custodian, the depositary or any nominees upon the making of deposits and withdrawals, respectively;
- •
- certain cable, telex and facsimile transmission and delivery expenses;
- •
- the fees, expenses, spreads, taxes and other charges of the depositary and/or service providers (which may be a division, branch or affiliate
of the depositary) in the conversion of foreign currency;
- •
- the reasonable and customary out-of-pocket fees and expenses incurred by the depositary in connection with compliance with exchange control
regulations and other regulatory requirements applicable to shares, ADSs and ADRs; and
- •
- the fees, charges, costs and expenses incurred by the depositary, the custodian, or any nominee in connection with the ADR program.
ADS fees and charges for (i) the issuance of ADSs, and (ii) the cancellation of ADSs are charged to the person for whom the ADSs are issued (in the case of ADS issuances) and to the person for whom ADSs are cancelled (in the case of ADS cancellations). In the case of ADSs issued by the depositary into DTC, the ADS issuance and cancellation fees and charges may be deducted from distributions made through DTC, and may be charged to the DTC participant(s) receiving the ADSs being issued or the DTC participant(s) holding the ADSs being cancelled, as the case may be, on behalf of the beneficial owner(s) and will be charged by the DTC participant(s) to the account of the applicable beneficial owner(s) in accordance with the procedures and practices of the DTC participants as in effect at the time. ADS fees and charges in respect of distributions and the ADS service fee are charged to the holders as of the applicable ADS record date. In the case of distributions of cash, the amount of the applicable ADS fees and charges is deducted from the funds being distributed. In the case of (i) distributions other than cash and (ii) the ADS service fee, holders as of the ADS record date will be invoiced for the amount of the ADS fees and charges and such ADS fees and charges may be deducted from distributions made to holders of ADSs. For ADSs held through DTC, the ADS fees and charges for distributions other than cash and the ADS service fee may be deducted from distributions made through DTC, and may be charged to the DTC participants in accordance with the procedures and practices prescribed by DTC and the DTC participants in turn charge the amount of such ADS fees and charges to the beneficial owners for whom they hold ADSs. In the case of (i) registration of ADS transfers, the ADS transfer fee will be payable by the ADS Holder whose ADSs are being transferred or by the person to whom the ADSs are transferred, and (ii) conversion of ADSs of one series for ADSs of another series, the ADS conversion fee will be payable by the Holder whose ADSs are converted or by the person to whom the converted ADSs are delivered.
In the event of refusal to pay the depositary fees, the depositary may, under the terms of the deposit agreement, refuse the requested service until payment is received or may set off the amount of the depositary fees from any distribution to be made to the ADS holder.
Note that the fees and charges that ADS holders may be required to pay may vary over time and may be changed by us and by the depositary. ADS holders will receive prior notice of such changes. The depositary may reimburse us for certain expenses incurred by us in respect of the ADR program, by making available a portion of the ADS fees charged in respect of the ADR program or otherwise, upon such terms and conditions as we and the depositary agree from time to time.
114
Item 13. Defaults, Dividend Arrearages and Delinquencies.
Not applicable.
Item 14. Material Modifications to the Rights of Security Holders and Use of Proceeds.
Not applicable.
Item 15. Controls and Procedures. Disclosure Controls and Procedures
Our Chief Executive Officer (principal executive officer) and Head of Finance (principal financial officer), after evaluating the effectiveness of our disclosure controls and procedures (as defined in Rule 13a-15(e) of the Exchange Act) as of December 31, 2019, have concluded that, as of such date, our disclosure controls and procedures were effective and ensured that information required to be disclosed by us in reports that we file or submit under the Exchange Act is accumulated and communicated to our management, including our Chief Executive Officer (principal executive officer) and Head of Finance (principal financial officer), to allow timely decisions regarding required disclosure and is recorded, processed, summarized and reported within the time periods specified by the SEC's rules and forms.
Management's Annual Report on Internal Control over Financial Reporting
This Annual Report does not include a report of management's assessment regarding internal control over financial reporting due to a transition period established by rules of the SEC for newly public companies.
Attestation Report of the Registered Public Accounting Firm
This Annual Report does not include an attestation report of our registered public accounting firm. For as long as we are an "emerging growth company" under the JOBS Act, our independent registered public accounting firm will not be required to attest to the effectiveness of our internal controls over financial reporting pursuant to Section 404.
Changes in Internal Control Over Financial Reporting
There has been no change in our internal control over financial reporting (as defined in Rule 13a-15(f) under the Exchange Act) that occurred during the period covered by this Annual Report that has materially affected, or is reasonably likely to materially affect, internal control over financial reporting.
Not applicable.
Item 16A. Audit Committee Financial Expert.
Our board of directors has determined that Jake Nunn is an "audit committee financial expert" as defined by SEC rules and has the requisite financial sophistication under the applicable rules and regulations of the Nasdaq Stock Market. Mr Nunn is independent as such term is defined in Rule 10A-3 under the Exchange Act and under the listing standards of the Nasdaq Stock Market.
115
Item 16B. Code of Business Conduct and Ethics.
We have adopted a Code of Business Conduct and Ethics, or the Code of Conduct, that is applicable to all of our employees, executive officers and directors. A copy of the Code of Conduct is available on our website at www.addextherapeuticscom.
Item 16C. Principal Accountant Fees and Services.
PricewaterhouseCoopers SA, or PwC, has served as our independent registered public accounting firm for 2019 and 2018. Our accountants billed the following fees to us for professional services in each of those fiscal years:
| |
Year ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2019 | 2018 | |||||
| |
(in thousands) |
||||||
Audit Fees |
782 | 105 | |||||
Audit-Related Fees |
3 | 9 | |||||
Tax Fees |
— | 10 | |||||
Other Fees |
— | — | |||||
| | | | | | | | |
Total |
785 | 124 | |||||
| | | | | | | | |
"Audit Fees" consist of fees billed for the annual audit of our consolidated financial statements, and the statutory audit of our consolidated and stand-alone financial statements. Audit Fees also include services that only our independent external auditor can reasonably provide, such as the review of documents filed with the U.S. stock exchange.
"Audit-Related Fees" consist of fees billed for assurance and related services that are reasonably related to the performance of the audit or review of its financial statements or that are traditionally performed by the external auditor, and mainly include services such as comfort letters issued in connection with securities offerings, due diligence and agreed-upon or expanded audit procedures.
"Tax Fees" consist of tax consultations, such as advice in connection with employees' taxation arising from share-based compensation.
"Other Fees" consist of advisory services relating to the adoption or application of IFRS.
Audit and Non-Audit Services Pre-Approval Policy
To ensure the independence and objectivity of our external auditors, the provision of all non-audit services by the external auditors are pre- approved by our audit committee.
Item 16D. Exemptions from the Listing Standards for Audit Committees.
Not applicable.
Item 16E. Purchases of Equity Securities by the Issuer and Affiliated Purchasers.
Not applicable.
Item 16F. Change in Registrant's Certifying Accountant.
Not applicable.
116
Item 16G. Corporate Governance.
Summary of Significant Corporate Governance Differences From Nasdaq Listing Standards
As a "foreign private issuer," as defined by the SEC, we are permitted to follow home country corporate governance practices, instead of certain corporate governance practices required by Nasdaq for U.S. domestic issuers. While we follow most Nasdaq corporate governance rules, we follow Swiss corporate governance practices in lieu of Nasdaq corporate governance rules as follows:
- •
- We do not intend to comply with Nasdaq Rule 5605(b)(1), which requires that our board of directors be comprised of a majority of
independent directors. Such requirements are not required under the laws of Switzerland. However, we do make an assessment of the independence of our directors under Swiss corporate governance
practices and have concluded that the majority of our directors are independent based upon those standards.
- •
- We do not intend to follow Nasdaq Rule 5605(d)(1) regarding the compensation committee charter or Nasdaq Rule 5605(d)(2)
regarding compensation committee composition. Such requirements are not required under the laws of Switzerland. However, we do maintain a remuneration committee in line with Swiss law.
- •
- We do not intend to follow Nasdaq Rule 5605(e)(1)(A) with respect to having director nominees selected by independent directors
constituting a majority of our board's independent directors in a vote in which only independent directors participate or Nasdaq Rule 5605(e)(2) regarding the adoption of a formal written
charter or board resolution, as applicable, addressing the nominations process. Such requirements are not required under the laws of Switzerland.
- •
- We do not intend to follow Nasdaq Rule 5620(c) regarding quorum requirements applicable to meetings of shareholders. Such quorum requirements are not required under the laws of Switzerland.
We must comply with Nasdaq Rule 5640 Notification of Noncompliance and Rule 5640 Voting Rights. Further, we must have an audit committee that satisfies Rule 5605(c)(3), which addresses audit committee responsibilities and authority, and that consists of committee members that meet the independence requirements of Rule 5605(c)(2)(A)(ii), subject to a transition period for newly public companies.
Because we are a foreign private issuer, our directors and senior management are not subject to short-swing profit and insider trading reporting obligations under Section 16 of the Exchange Act. They and our other shareholders will, however, be subject to the obligations to report changes in share ownership under Section 13 of the Exchange Act and related SEC Rules.
Item 16H. Mine Safety Disclosure.
Not applicable.
117
Item 17. Financial Statements.
See pages F-1 through F-42 of this Annual Report on Form 20-F.
Item 18. Financial Statements.
Not applicable.
List of exhibits:
118
- †
- Indicates
a management contract or any compensatory plan, contract or arrangement.
- #
- Confidential treatment has been granted from the Securities and Exchange Commission as to certain portions of this document.
119
The Registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this Annual Report on its behalf.
| Addex Therapeutics Ltd. | ||||||
/s/ Tim Dyer |
/s/ Lénaic Teyssédou |
|||||
| Name: | Tim Dyer | Name: | Lénaic Teyssédou | |||
| Title: | Chief Executive Officer | Title: | Head of Finance | |||
Switzerland
Dated: April 24, 2020
120
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Shareholders of Addex Therapeutics Ltd
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Addex Therapeutics Ltd and its subsidiaries (the "Company") as of December 31, 2019 and 2018, and the related consolidated statements of loss, comprehensive loss, changes in equity and cash flows for each of the three years in the period ended December 31, 2019, including the related notes (collectively referred to as the "consolidated financial statements"). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2019 and 2018, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2019 in conformity with International Financial Reporting Standards as issued by the International Accounting Standards Board.
Change in Accounting Principle
As discussed in Note 2.2 to the consolidated financial statements, the Company changed the manner in which it accounts for leases in 2019.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits of these consolidated financial statements in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/
PricewaterhouseCoopers SA
Geneva, Switzerland
April 24, 2020
We have served as the Company's auditor since 2002.
F-2
| |
|
As at December, 31 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| |
Notes | 2019 | 2018 | ||||||
| |
Amounts in Swiss francs |
||||||||
ASSETS |
|||||||||
Current assets |
|
||||||||
Cash and cash equivalents |
6 | 31,536,803 | 41,670,158 | ||||||
Other financial assets |
7 | 13,968 | 7,983 | ||||||
Receivables |
7 | 118,028 | 273,016 | ||||||
Prepayments |
7 | 720,063 | 199,410 | ||||||
| | | | | | | | | | |
Total current assets |
32,388,862 | 42,150,567 | |||||||
| | | | | | | | | | |
Non-current assets |
|||||||||
Right-of-use assets |
8 | 543,340 | — | ||||||
Property, plant and equipment |
9 | 27,626 | 8,868 | ||||||
Non-current financial assets |
10 | 68,911 | 54,404 | ||||||
| | | | | | | | | | |
Total non-current assets |
639,877 | 63,272 | |||||||
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
Total assets |
33,028,739 | 42,213,839 | |||||||
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
LIABILITIES AND EQUITY |
|||||||||
Current liabilities |
|||||||||
Current lease liabilities |
12 | 373,025 | — | ||||||
Payables and accruals |
11 | 4,196,411 | 2,121,084 | ||||||
Contract liability |
16 | 945,737 | 212,744 | ||||||
Deferred income |
13 | 165,389 | — | ||||||
| | | | | | | | | | |
Total current liabilities |
5,680,562 | 2,333,828 | |||||||
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
Non-current liabilities |
|||||||||
Non-current lease liabilities |
12 | 177,220 | — | ||||||
Retirement benefits obligations |
21 | 1,481,738 | 639,351 | ||||||
Deferred income |
13 | 165,390 | — | ||||||
| | | | | | | | | | |
Total non-current liabilities |
1,824,348 | 639,351 | |||||||
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
Equity |
|||||||||
Share capital |
14 | 32,848,635 | 28,564,031 | ||||||
Share premium |
14 | 286,375,977 | 286,476,912 | ||||||
Reserves |
7,146,506 | 10,266,402 | |||||||
Accumulated deficit |
(300,847,289 | ) | (286,066,685 | ) | |||||
| | | | | | | | | | |
Total equity |
25,523,829 | 39,240,660 | |||||||
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
Total liabilities and equity |
33,028,739 | 42,213,839 | |||||||
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
The accompanying notes form an integral part of these consolidated financial statements.
F-3
Consolidated Statements of Loss
| |
Notes | December 31, 2019 |
December 31, 2018 |
December 31, 2017 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
|
Amounts in Swiss francs |
||||||||||
Revenue from contract with customer |
16 | 2,762,830 | 6,043,855 | — | ||||||||
Other income |
17 | 70,835 | 658,818 | 499,894 | ||||||||
Operating costs |
||||||||||||
Research and development |
(12,453,876 | ) | (4,918,793 | ) | (2,628,901 | ) | ||||||
General and administration |
(4,983,946 | ) | (3,208,505 | ) | (1,106,049 | ) | ||||||
| | | | | | | | | | | | | |
Total operating costs |
18 | (17,437,822 | ) | (8,127,298 | ) | (3,734,950 | ) | |||||
| | | | | | | | | | | | | |
Operating loss |
(14,604,157 | ) | (1,424,625 | ) | (3,235,056 | ) | ||||||
| | | | | | | | | | | | | |
Finance income |
36,874 | — | — | |||||||||
Finance expense |
(213,321 | ) | (220,173 | ) | (45,350 | ) | ||||||
| | | | | | | | | | | | | |
Finance costs |
22 | (176,447 | ) | (220,173 | ) | (45,350 | ) | |||||
| | | | | | | | | | | | | |
Net loss before tax |
(14,780,604 | ) | (1,644,798 | ) | (3,280,406 | ) | ||||||
Income tax expense |
20 | — | — | — | ||||||||
| | | | | | | | | | | | | |
Net loss for the year |
(14,780,604 | ) | (1,644,798 | ) | (3,280,406 | ) | ||||||
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
Basic and diluted loss per share for loss attributable to the ordinary equity holders of the Company |
23 | (0.56 | ) | (0.07 | ) | (0.25 | ) | |||||
The accompanying notes form an integral part of these consolidated financial statements.
F-4
Consolidated Statements of Comprehensive Loss
| |
Notes | December 31, 2019 | December 31, 2018 | December 31, 2017 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
|
Amounts in Swiss francs |
|
|||||||||
Net loss for the year |
(14,780,604 | ) | (1,644,798 | ) | (3,280,406 | ) | ||||||
Other comprehensive loss |
|
|||||||||||
Items that will never be reclassified to the statement of income: |
||||||||||||
Remeasurements of retirement benefits obligations |
21 | (745,855 | ) | (375,479 | ) | (9,909 | ) | |||||
Items that may be classified subsequently to the statement of income |
||||||||||||
Exchange difference on translation of foreign operations differences |
(838 | ) | (181 | ) | (871 | ) | ||||||
| | | | | | | | | | | | | |
Other comprehensive loss for the year, net of tax |
(746,693 | ) | (375,660 | ) | (10,780 | ) | ||||||
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
Total comprehensive loss for the year |
(15,527,297 | ) | (2,020,458 | ) | (3,291,186 | ) | ||||||
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
The accompanying notes form an integral part of these consolidated financial statements.
F-5
Consolidated Statements of Changes in Equity
| |
Share Capital |
Share Premium |
Treasury Shares Reserve |
Foreign Currency Translation Reserve |
Other Reserves |
Accumulated Deficit |
Total | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Amounts in Swiss Francs |
|||||||||||||||||||||
January 1, 2017 |
13,454,553 | 263,100,700 | (1,953,067 | ) | (651,271 | ) | 7,409,158 | (281,141,481 | ) | 218,592 | ||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | |
Net loss for the year |
— | — | — | — | — | (3,280,406 | ) | (3,280,406 | ) | |||||||||||||
Other comprehensive loss |
— | — | — | (871 | ) | (9,909 | ) | — | (10,780 | ) | ||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | |
Total comprehensive loss |
— | — | — | (871 | ) | (9,909 | ) | (3,280,406 | ) | (3,291,186 | ) | |||||||||||
Issue of shares |
1,930,435 | — | — | — | — | — | 1,930,435 | |||||||||||||||
Share capital issuance costs |
— | (25,573 | ) | — | — | — | — | (25,573 | ) | |||||||||||||
Value of share-based services |
— | — | — | — | 800,188 | — | 800,188 | |||||||||||||||
Movement in treasury shares: |
||||||||||||||||||||||
Capital increase |
— | — | (1,930,435 | ) | — | — | — | (1,930,435 | ) | |||||||||||||
Sale of shares to investors |
— | 1,647,645 | 1,617,523 | — | — | — | 3,265,168 | |||||||||||||||
Net sales under liquidity agreement |
— | — | 6,006 | 6,006 | ||||||||||||||||||
Exercise of ESC |
— | — | 108,000 | — | — | — | 108,000 | |||||||||||||||
Settlement of supplier invoices |
— | 129,236 | 132,096 | — | — | — | 261,332 | |||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | |
January 1, 2018 |
15,384,988 | 264,852,008 | (2,019,877 | ) | (652,142 | ) | 8,199,437 | (284,421,887 | ) | 1,342,527 | ||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | |
Net loss for the year |
— | — | — | — | — | (1,644,798 | ) | (1,644,798 | ) | |||||||||||||
Other comprehensive loss |
— | — | — | (181 | ) | (375,479 | ) | — | (375,660 | ) | ||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | |
Total comprehensive loss |
— | — | — | (181 | ) | (375,479 | ) | (1,644,798 | ) | (2,020,458 | ) | |||||||||||
Issue of shares |
13,179,043 | 24,461,056 | — | — | — | — | 37,640,099 | |||||||||||||||
Share capital issuance costs |
— | (2,963,415 | ) | — | — | — | — | (2,963,415 | ) | |||||||||||||
Value of share-based services |
— | — | — | — | 2,298,933 | — | 2,298,933 | |||||||||||||||
Value of Warrants |
— | — | — | — | 3,308,982 | — | 3,308,982 | |||||||||||||||
Movement in treasury Shares: |
||||||||||||||||||||||
Capital increase |
— | — | (568,902 | ) | — | — | — | (568,902 | ) | |||||||||||||
Settlement of suppliers invoices |
— | 120,908 | 87,176 | — | — | — | 208,084 | |||||||||||||||
Net purchases under liquidity agreement |
— | 6,355 | (11,545 | ) | — | — | — | (5,190 | ) | |||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | |
January 1, 2019 |
28,564,031 | 286,476,912 | (2,513,148 | ) | (652,323 | ) | 13,431,873 | (286,066,685 | ) | 39,240,660 | ||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | |
Net loss for the year |
— | — | — | — | — | (14,780,604 | ) | (14,780,604 | ) | |||||||||||||
Other comprehensive loss |
— | — | — | (838 | ) | (745,855 | ) | — | (746,693 | ) | ||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | |
Total comprehensive loss |
— | — | — | (838 | ) | (745,855 | ) | (14,780,604 | ) | (15,527,297 | ) | |||||||||||
Issue of shares |
4,284,604 | — | — | — | — | — | 4,284,604 | |||||||||||||||
Share capital issuance costs |
— | (170,411 | ) | — | — | — | — | (170,411 | ) | |||||||||||||
Value of share-based services |
— | — | — | — | 1,685,965 | — | 1,685,965 | |||||||||||||||
Movement on warrants |
— | — | (288 | ) | — | — | — | (288 | ) | |||||||||||||
Movement in treasury shares: |
||||||||||||||||||||||
Capital increase |
— | — | (4,284,604 | ) | — | — | — | (4,284,604 | ) | |||||||||||||
Settlement of suppliers invoices |
92,604 | 196,610 | — | — | — | 289,214 | ||||||||||||||||
Net sales under liquidity agreement |
— | (23,128 | ) | 29,114 | — | — | — | 5,986 | ||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | |
December 31, 2019 |
32,848,635 | 286,375,977 | (6,572,316 | ) | (653,161 | ) | 14,371,983 | (300,847,289 | ) | 25,523,829 | ||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | |
The accompanying notes form an integral part of these consolidated financial statements.
F-6
Consolidated Statements of Cash Flows
| |
Notes | December 31, 2019 |
December 31, 2018 |
December 31, 2017 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
|
Amounts in Swiss francs |
||||||||||
Net loss for the year |
(14,780,604 | ) | (1,644,798 | ) | (3,280,406 | ) | ||||||
Adjustments for: |
||||||||||||
Depreciation |
8 and 9 | 333,844 | 2,937 | 15,249 | ||||||||
Value of share-based services |
15 | 1,685,965 | 2,298,933 | 800,188 | ||||||||
Pension costs |
21 | 96,532 | 20,008 | 19,520 | ||||||||
Finance costs |
234,663 | 123,840 | 45,471 | |||||||||
Decrease / (increase) in other financial assets |
(5,985 | ) | 3,308 | (4,992 | ) | |||||||
Decrease / (increase) in receivables |
154,988 | (77,134 | ) | (83,159 | ) | |||||||
(Increase) in prepayments |
(520,653 | ) | (40,487 | ) | (137,488 | ) | ||||||
Increase / (decrease) in payables and accruals |
1,966,160 | 1,083,315 | (212,131 | ) | ||||||||
Increase in contract liability |
732,993 | 212,744 | — | |||||||||
Increase / (decrease) in deferred income |
330,779 | (439,022 | ) | 439,022 | ||||||||
Services paid in shares |
289,214 | 208,085 | 258,903 | |||||||||
| | | | | | | | | | | | | |
Net cash (used in) / from operating activities |
(9,482,104 | ) | 1,751,729 | (2,139,823 | ) | |||||||
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
Cash flows from investing activities |
||||||||||||
Purchase of property, plant and equipment |
9 | (28,459 | ) | (9,054 | ) | (697 | ) | |||||
Purchase of non-current financial assets |
10 | (14,795 | ) | (47,317 | ) | — | ||||||
| | | | | | | | | | | | | |
Net cash used in investing activities |
(43,254 | ) | (56,371 | ) | (697 | ) | ||||||
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
Cash flows from financing activities |
||||||||||||
Proceeds from issue of shares—capital increase |
14 | — | 40,488,180 | — | ||||||||
Costs paid on issue of shares |
— | (2,963,415 | ) | (25,573 | ) | |||||||
Costs paid on issue of shares subscribed by the Group |
(61,244 | ) | — | — | ||||||||
Sale/ (purchase) of treasury shares |
5,986 | (5,373 | ) | 3,380,747 | ||||||||
Principal element of lease payments |
(316,793 | ) | — | — | ||||||||
Interests received |
36,874 | — | — | |||||||||
Interests paid |
22 | (128,518 | ) | (134,307 | ) | (171 | ) | |||||
| | | | | | | | | | | | | |
Net cash (used in) / from financing activities |
(463,695 | ) | 37,385,085 | 3,355,003 | ||||||||
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
(Decrease) / Increase in cash and cash equivalents |
(9,989,053 | ) | 39,080,443 | 1,214,483 | ||||||||
Cash and cash equivalents at beginning of the year |
6 | 41,670,158 | 2,579,248 | 1,410,065 | ||||||||
Exchange difference on cash and cash equivalents |
(144,302 | ) | 10,467 | (45,300 | ) | |||||||
| | | | | | | | | | | | | |
Cash and cash equivalents at end of the year |
6 | 31,536,803 | 41,670,158 | 2,579,248 | ||||||||
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
The accompanying notes form an integral part of these consolidated financial statements.
F-7
Notes to the Consolidated Financial Statements
(Amounts in Swiss francs)
1. General information
Addex Therapeutics Ltd (the "Company"), formerly Addex Pharmaceuticals Ltd, and its subsidiaries (together, the "Group") are a clinical stage pharmaceutical group applying its leading allosteric modulator drug discovery platform to discovery and development small-molecule pharmaceutical products, with an initial focus on central nervous system disorders.
The Company is a Swiss stockholding corporation domiciled c/o Addex Pharma SA, Chemin des Aulx 12, CH-1228 Plan-les-Ouates, Geneva, Switzerland and the parent company of Addex Pharma SA, Addex Pharmaceuticals France SAS and Addex Pharmaceuticals Inc., company created on May 29, 2019 registered in Delaware with its principal business location in San Francisco, California, United States. Its registered shares are traded at the SIX, Swiss Exchange, under the ticker symbol ADXN. On January 29, 2020, the Group listed on the Nasdaq Stock Market, American Depositary Shares (ADSs) under the symbol "ADX", without a new issuance of securities. ADSs represents ordinary shares that continues to be admitted to trading on SIX Swiss Exchange.
These consolidated financial statements have been approved for issuance by the Board of Directors on April 7, 2020.
2. Summary of significant accounting policies
The principal accounting policies applied in the preparation of these consolidated financial statements are set out below. These policies have been consistently applied to all the years presented, unless otherwise stated.
2.1 Basis of preparation
The consolidated financial statements of Addex Therapeutics Ltd have been prepared in accordance with International Financial Reporting Standards (IFRS) as issued by the International Accounting Standards Board ("IASB"), and under the historical cost convention.
The preparation of financial statements in conformity with IFRS requires the use of certain critical accounting estimates. It also requires management to exercise its judgment in the process of applying the Group's accounting policies. The areas involving a higher degree of judgment or complexity, or areas where assumptions and estimates are significant to the consolidated financial statements are disclosed in note 4 "Critical accounting estimates and judgements".
Due to rounding, numbers presented throughout these consolidated financial statements may not add up precisely to the totals provided. All ratios and variances are calculated using the underlying amount rather than the presented rounded amount.
Where necessary, comparative figures have been revised to confirm with the current year 2019 presentation.
2.2 Standards and interpretations published by the IASB
New standards adopted by the Group
IFRS 16
IFRS 16 provides a single lessee accounting model, requiring lessees to recognise assets and liabilities for all leases and lessors to confirm the continuation of classifying leases as operating or
F-8
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
2. Summary of significant accounting policies (Continued)
finance. The Group is not a lessor and is only impacted by the standard from a lessee point of view. The standard replaces IAS 17, under which operating lease payments were charged to the income statement on a straight-line basis over the term of the contract.
The Group has adopted IFRS16 Leases from its mandatory adoption date of January 1, 2019 and applied the cumulative catch-up approach without restating the comparative amounts for the year prior to first adoption.
In applying IFRS 16 for the first time, the group has used the following practical expedients permitted by the standard:
- •
- applying a single discount rate to a portfolio of leases with reasonably similar characteristics
- •
- relying on previous assessments on whether leases are onerous as an alternative to performing an impairment review—there were no
onerous contracts as at 1 January 2019
- •
- accounting for operating leases with a remaining lease term of less than 12 months as at 1January 2019 as short-term leases
- •
- excluding initial direct costs for the measurement of the right-of-use asset at the date of initial application, and
- •
- using hindsight in determining the lease term where the contract contains options to extend or terminate the lease.
The group has also elected not to reassess whether a contract is, or contains a lease at the date of initial application. Instead, for contracts entered into before the transition date the group relied on its assessment made applying IAS 17 and Interpretation 4 Determining whether an Arrangement contains a Lease.
On January 1, 2019, the Group recognized right-of-use asset and lease liabilities for CHF 544,510 that were previously classified as operating leases under the principles of IAS 17 Leases. The lease liabilities were measured at the present value of the remaining lease payments, discounted using a weighted average incremental borrowing rate of 4.70%.
F-9
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
2. Summary of significant accounting policies (Continued)
The following table presents the reconciliation between the non-cancellable operating lease commitments reported as of December 31, 2018 and the lease liabilities recognized on January 1, 2019:
| |
Total | |||
|---|---|---|---|---|
Operating lease commitments disclosed as at December 31, 2018 |
272,498 | |||
Adjustments as a result of different treatment of extension and termination options |
297,721 | |||
| | | | | |
Total lease commitments as at December 31, 2018, |
570,219 | |||
| | | | | |
| | | | | |
| | | | | |
Discount using the incremental borrowing rate at the date of the initial application |
(24,015 | ) | ||
Gross liabilities as per January 1, 2019 under IFRS 16 |
546,204 | |||
| | | | | |
| | | | | |
| | | | | |
Short term leases |
(1,694 | ) | ||
| | | | | |
Lease liability recognized as at January 1, 2019 |
544,510 | |||
| | | | | |
| | | | | |
| | | | | |
Of which are: |
||||
Current lease liabilities |
303,627 | |||
Non-current lease liabilities |
240,883 | |||
Lease liability recognized as at December 31, 2019 |
||||
Current lease liabilities |
373,025 |
|||
Non-current lease liabilities |
177,220 | |||
Right-of-use assets were measured at the amount equal to the lease liability, adjusted by the amount of any prepaid or accrued lease payments relating to that lease recognized in the balance sheet as at December 31, 2018. The right-of-use assets relate to the following types of assets:
| |
December 31, 2019 |
January 1, 2019 |
|||||
|---|---|---|---|---|---|---|---|
Properties |
496,126 | 483,350 | |||||
Equipment |
47,214 | 61,160 | |||||
| | | | | | | | |
Total right-of-use assets |
543,340 | 544,510 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The adoption of IFRS 16 Leases did not have a material impact on the Group's net loss after tax or on the Group's loss per share.
Other standards
The following new standards, amendments to standards and interpretations which are mandatory for the financial periods beginning on January 1, 2019 did not have any material impact on the consolidated financial statements:
Plan Amendments, Curtailment or Settlement: Amendments to IAS 19 Employee Benefits relates to accounting for plan amendments, curtailments and settlements where changes are made to pension plans. The amendments require an entity: (1) to use updated assumptions to determine current service cost and net interest for the remainder of the period after a plan amendment, curtailment or
F-10
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
2. Summary of significant accounting policies (Continued)
settlement; and (2) to recognise in profit or loss as part of past service cost, or a gain or loss on settlement, any reduction in a surplus, even if that surplus was not previously recognised because of the impact of the asset ceiling. On this basis, the Group has completed its assessment and determined this did not have a material impact on the consolidated financial statements.
There are no other amendments or new standards relevant for the Group for the financial period beginning on January 1 2019 .
New standards and interpretations not yet adopted
There are new standards, amendments to standards and interpretations that are not yet effective, which have been deemed by the Group as currently not relevant, hence are not listed or discussed further here.
2.3 Consolidation
Subsidiaries are all entities over which the Group has control. The Group controls an entity when the Group is exposed to, or has rights to, variable returns from its involvement with the entity and has the ability to affect those returns through its power over the entity. Subsidiaries are fully consolidated from the date on which control is transferred to the Group. They are de-consolidated from the date that control ceases.
Inter-company transactions, balances and unrealized gains on transactions between Group companies are eliminated. Unrealized losses are also eliminated unless the transaction provides evidence of an impairment of the asset transferred. Accounting policies of subsidiaries have been changed where necessary to ensure consistency with the policies adopted by the Group. The reporting date of all Group companies is December 31.
2.4 Segment reporting
The Group operates in one segment, which is the discovery, development and commercialization of small-molecule pharmaceutical products. A single management team that reports to the chief executive officer comprehensively manages the entire business. The chief operating decision-maker is the Chief Executive Officer who reviews the statement of operations of the Group on a consolidated basis, makes decisions and manages the operations of the Group as a single operating segment. The Group's activities are not affected by any significant seasonal effect. Revenue is attributable to the Company's country of domicile, Switzerland.
2.5 Foreign currency transactions
Functional and presentation currency
Items included in the financial statements of each of the Group's entities are measured using the currency of the primary economic environment in which the entity operates ("the functional currency"). The consolidated financial statements are presented in Swiss francs, which is the Group's presentation currency.
F-11
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
2. Summary of significant accounting policies (Continued)
Transactions and balances
Foreign currency transactions are translated into the functional currency using the exchange rates prevailing at the dates of the transactions or valuation where items are re-measured. Foreign exchange gains and losses resulting from the settlement of such transactions and from the translation at year-end exchange rates of monetary assets and liabilities denominated in foreign currencies are recognized in the statement of income.
Foreign exchange gains and losses that relate to borrowings and cash and cash equivalents are presented in the statement of income within 'finance cost'.
Group companies
The results and financial position of the Group's subsidiary that has a functional currency different from the presentation currency are translated into the presentation currency as follows:
- •
- assets and liabilities for each balance sheet presented are translated at the closing rate at the date of that balance sheet;
- •
- income and expenses for each statement of income are translated at the average exchange rate; and
- •
- all resulting exchange differences are recognized in other comprehensive income.
2.6 Property, plant and equipment
Property, plant and equipment are stated at historical cost less accumulated depreciation, and impairment (if any). Historical cost includes expenditure that is directly attributable to the acquisition of the item. Subsequent costs are included in the asset's carrying amount or recognized as a separate asset, as appropriate, only when it is probable that future economic benefits associated with the item will flow to the Group and the cost of the item can be measured reliably. All other repairs and maintenance are charged to the statement of income during the financial period in which they are incurred. Depreciation is calculated using the straight-line method to allocate their cost to their residual values over their estimated useful lives as follows:
Computer equipment |
3 years | |
Laboratory equipment |
4 years | |
Furniture and fixtures |
5 years | |
Chemical library |
5 years |
The assets' residual values and useful lives are reviewed, and adjusted if appropriate, at each balance sheet date. An asset's carrying amount is written down immediately to its recoverable amount if the asset's carrying amount is greater than its estimated recoverable amount (see note 2.7). Gains and losses on disposals are determined by comparing proceeds with the carrying amount, and are included in the statement of income.
2.7 Impairment of non-financial assets
Assets that are subject to depreciation or amortization are reviewed for impairment annually, and whenever events or changes in circumstances indicate that the carrying amount may not be recoverable.
F-12
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
2. Summary of significant accounting policies (Continued)
An impairment loss is recognized for the amount by which the asset's carrying amount exceeds its recoverable amount. The recoverable amount is the higher of an asset's fair value less costs to sell and value in use. For the purposes of assessing impairment, assets are grouped at the lowest levels for which there are separately identifiable cash flows (cash generating units). Prior impairment of non-financial assets other than goodwill is reviewed for possible reversal at each reporting date.
2.8 Financial assets
The Group has one category of financial assets, namely "receivables". Receivables are non-derivative financial assets with fixed or determinable payments that are not quoted in an active market. These assets are held for collection of contractual cash flows which represent solely the payment of principal and interest. They arise when the Group provides money, goods or services directly to a debtor with no intention of trading the receivable. They are included in current assets, except for maturities greater than 12 months after the balance sheet date, which are classified as non-current assets. Receivables are included in other current assets in the balance sheet (see note 7).
Receivables are initially measured at fair value and subsequently measured at amortized cost. The amortized cost of a financial asset is the amount at which the financial asset is measured at initial recognition minus the principal repayments, plus the cumulative amortisation using the effective interest method of any difference between that initial amount and the maturity amount, adjusted for any loss allowance. The gross carrying amount of a financial asset is the amortized cost of a financial asset before adjusting for any loss allowance. Receivables are derecognized when settled.
The Company classifies a contract asset as a receivable when the Company's right to consideration is unconditional. If the Company transfers control of goods or services to a customer before the customer pays consideration, the Company records either a contract asset or a receivable depending on the nature of the Company's right to consideration for its performance. Contract assets and contract liabilities arising from the same contract are netted and presented as either a single net contract asset or net contract liability.
Impairment of financial assets
The Group recognizes a loss allowance for expected credit losses on lease receivables, trade receivables, contract assets, financial guarantee contracts investments, in debt instruments that are measured at amortized cost. The amount of expected credit losses is updated at each reporting date to reflect changes in credit risk since initial recognition of the respective financial instrument.
The Group always recognizes lifetime ECL (expected credit losses) for trade receivables, contract assets and lease receivables where applicable. The expected credit losses on these financial assets are estimated using a provision matrix based on the Group's historical credit loss experience, adjusted for factors that are specific to the debtors, general economic conditions and an assessment of both the current as well as the forecast direction of conditions at the reporting date, including time value of money where appropriate.
Lifetime ECL represents the expected credit losses that will result from all possible default events over the expected life of a financial instrument. In contrast, 12-month ECL represents the portion of lifetime ECL that is expected to result from default events on a financial instrument that are possible within 12 months after the reporting date.
F-13
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
2. Summary of significant accounting policies (Continued)
2.9 Cash and cash equivalents
Cash and cash equivalents include cash on hand, deposits held at call with banks and other short-term highly liquid investments with original maturities of three months or less. They are both readily convertible to known amounts of cash and so near their maturity that they present insignificant risk of changes in value because of changes in interest rates. Any bank overdrafts are not netted against cash and cash equivalents but are shown as part of current liabilities on the consolidated balance sheet.
2.10 Share capital
Shares are classified as equity. Incremental costs directly attributable to the issue of new shares are shown as a deduction, net of tax, from the proceeds.
Where any Group company purchases the Company's equity share capital (treasury shares), the consideration paid, including any directly attributable incremental cost (net of income taxes) is recorded as a deduction from equity attributable to the Company's equity holders as a treasury share reserve until the shares are cancelled, reissued or disposed of. When such shares are subsequently sold or reissued, any consideration received, net of any directly attributable incremental transaction costs and the related income tax effect, the nominal amount is reversed from the treasury share reserve, with any remaining difference to the total transaction value being recognized in share premium.
The Company has entered into a liquidity contract where an independent broker buys and sells the Company's shares held in the broker's custody. Such shares are presented in the treasury share reserve.
The Company also uses treasury shares to partially settle services rendered by third and related parties. When shares are issued for this purpose, the nominal share value is recognized as a treasury share reserve and the value above par is presented as a share premium.
2.11 Equity instruments
Equity instruments issued by the Group are recorded at the fair value of the proceeds received, net of direct issuance costs.
2.12 Trade payables
Trade payables are recognized initially at fair value and subsequently measured at amortized cost using the effective interest method. All payables have a contract maturity within 1 year.
2.13 Grants
Grants are not recognized until there is reasonable assurance that the Group will comply with the conditions attaching to them and that the grants will be received. Grants are recognized in profit or loss on a systematic basis over the periods in which the Group recognizes as expenses the related costs for which the grants are intended to compensate. Specifically, grants whose primary conditions is that the Group should undertake specific research activities within a defined period of time, are recognized as deferred income in the consolidated statement of financial position and transferred to the statement of profit or loss on a systematic and rationale basis over the defined timeframe.
F-14
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
2. Summary of significant accounting policies (Continued)
2.14 Deferred income tax
Deferred income tax is recorded in full, using the liability method, on temporary differences arising between the tax bases of assets and liabilities and their carrying amounts in the consolidated financial statements. However, if the deferred income tax arises from initial recognition of an asset or liability in a transaction other than a business combination that at the time of the transaction affects neither accounting nor taxable profit or loss, it is not accounted for. Deferred income tax is determined using tax rates and laws that have been enacted or substantively enacted by the balance sheet date and are expected to apply when the related deferred income tax asset is realized or the deferred income tax liability is settled.
Deferred income tax assets are recognized to the extent that it is probable that future taxable profit will be available against which the temporary differences can be utilized.
Deferred income tax is recorded on temporary differences arising on investments in subsidiaries, except where the Group and it is probable that the temporary difference will not reverse in the foreseeable future.
Potential deferred income tax assets from tax loss carry forwards exceed deferred tax liabilities. Deferred income tax assets from tax loss carry forwards are initially recognized to the extent that there are suitable deferred income tax liabilities, then to the extent that the realization of the related tax benefit through future taxable profits is probable.
2.15 Pension obligations
The Group operates one pension scheme. The scheme is generally funded through payments to insurance companies or trustee-administered funds, determined by periodic actuarial calculations. The Group has defined benefit plans. A defined benefit plan is a pension plan that defines an amount of pension benefit that an employee will receive on retirement, usually dependent on one or more factors such as age, years of service and compensation. Actuarial gains and losses arising from experience adjustments and changes in actuarial assumptions are recognized immediately in other comprehensive income and past-service costs are recognized immediately in the statement of income.
The liability recognized in the balance sheet in respect of defined benefit pension plans is the defined benefit obligation at the balance sheet date minus the fair value of the plan assets. The defined benefit obligation is calculated at least annually by an independent actuary using the projected unit credit method. The present value of the defined obligation is determined by discounting the estimated future cash outflows using interest rates of high-quality corporate bonds that are denominated in the currency in which the benefits will be paid, and that have terms to maturity approximating to the terms of the related pension liability.
2.16 Share-based compensation
The Group operates an equity sharing certificates' equity incentive plan, a share option plan, and a share purchase plan. The Group also from time to time grants warrants to brokers and investors. The fair value of the services received in exchange for the grant or transfer of equity sharing certificates, options, shares or warrants is recognized in the Consolidated Financial Statements over the period for which the services are received. The total amount to be recognized over the vesting period is determined by reference to the fair value of the equity incentive unit granted or transferred. The fair
F-15
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
2. Summary of significant accounting policies (Continued)
value of instruments granted includes any market performance conditions and excludes the impact of any service and non-market performance vesting conditions. Service and non-market performance conditions are included in assumptions about the number of equity incentive units that are expected to vest.
At each balance sheet date, the Group revises its estimates for the number of equity incentive units that are expected to vest. It recognizes the impact of the revision to original estimates, if any, in the statement of income, with a corresponding adjustment to equity.
The proceeds received net of any directly attributable transaction costs are credited to share capital (nominal value) and share premium when the equity incentive units are exercised.
2.17 Revenue recognition
The Group recognizes revenue from the license of intellectual property and providing research and development services:
License of intellectual property
If the license to the Group's intellectual property is determined to be distinct from the other performance obligations identified in the arrangement, the Group recognizes revenues when the license conveys a right of use, or there is a right of access to the underlying intellectual property. For licenses that are sold in conjunction with a related service, the Group uses judgment to assess the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied over time or at a point in time. If the performance obligation is settled over time, the Group determines the appropriate method of measuring progress for purposes of recognizing license revenue. The Group evaluates the measure of progress each reporting period and, if necessary, adjusts the measure of performance and related revenue recognition.
Research and development services
The Group has an arrangement with its partner that includes deploying its full-time employees for research and development activities. The Group assesses if these research and development activities are considered distinct in the context of the respective contract and, if so, they are accounted for as a separate performance obligation. This revenue is recorded within "Revenue from contract with customer" over time as the activities are performed.
Contract balances
The Group receives payments and determines credit terms from its customers for its various performance obligations based on billing schedules established in each contract. The actual timing of the income recognition, billings and cash collections may result in other current receivables, accrued revenue (contract assets), and deferred revenue (contract liabilities) being recorded on the balance sheets. Amounts are recorded as other current receivables when the Group's right to consideration is unconditional. The Group does not assess whether a contract has a significant financing component if the expectation at contract inception is such that the period between payment by the customer and the transfer of the promised goods or services to the customer will be one year or less.
F-16
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
2. Summary of significant accounting policies (Continued)
Under IFRS 15, the Group recognizes as revenue our non-refundable license fees, milestone, research activities and royalties when our customer obtains control of promised services, in an amount that reflects the consideration which we expect to receive in exchange for those rendered services. To assess revenue recognition for arrangements that we determine are within the scope of IFRS 15, we perform the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) we satisfy a performance obligation. We only apply the five-step model to contracts when it is probable that we will collect the consideration we are entitled to in exchange for services we transfer to the customer. At contract inception, once the contract is determined to be within the scope of IFRS 15, we assess the services promised within each contract and determine those that are performance obligations and assess whether each promised service is distinct. We use the most likely method to estimate any variable consideration and include such consideration in the amount of the transaction price based on an estimated stand-alone selling price. Revenue is recognized for the respective performance obligation when (or as) the performance obligation is satisfied.
2.18 Finance income and expense
Interest received or paid on cash and cash equivalents are classified in the statement of cash flows under financing activities.
2.19 Leases
The Group assesses whether a contract is or contains a lease, at inception of the contract. The Group recognizes a right-of-use asset and a corresponding lease liability with respect to all lease arrangements in which it is the lessee, except for short-term leases (defined as leases with a lease term of 12 months or less) and leases of low value assets (less than USD 5 thousand). For these leases, the Group recognizes the lease payments as an operating expense on a straight-line basis over the term of the lease unless another systematic basis is more representative of the time pattern in which economic benefits from the leased assets are consumed.
The lease liability is initially measured at the present value of the lease payments as from the commencement date of the lease until the expected termination date. In determining the lease term, management consider all facts and circumstances that create an economic incentive to exercise an extension option, or not to exercise a termination option. Extension option are only considered if the lease is reasonably certain to be extended. The assessment of reasonable certainty is only revised if a significant event or a significant change in circumstances, that is within the control of the lessees, occurs. The lease payments are discounted by using the rate implicit in the lease. If this rate cannot be readily determined, the Group uses its incremental borrowing rate, being the rate that the individual lessee would have to pay to borrow the funds necessary to obtain an asset of similar value to the right-of-use asset in a similar economic environment with similar terms, security and conditions. The lease liability is presented as a separate line in the consolidated statement of financial position. The interest expense is presented in the line finance expenses in the consolidated statement of income.
The right-of-use assets comprise the initial measurement of the corresponding lease liability, lease payments made at or before the commencement day, less any lease incentives received and any initial direct costs. They are subsequently measured at cost less accumulated depreciation and impairment
F-17
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
2. Summary of significant accounting policies (Continued)
losses. They are depreciated over the shorter period of lease term and useful life of the underlying asset. If a lease transfers ownership of the underlying asset or the cost of the right-of-use asset reflects that the Group expects to exercise a purchase option, the related right-of-use asset is depreciated over the useful life of the underlying asset. The depreciation starts at the commencement date of the lease. The right-of-use assets are presented as a separate line in the consolidated statement of financial position.
All lease payments on leases are presented as part of the cash flow from financing activities, except for the short-term and low value leases cash flows, which are booked under operating activities.
2.20 Research and development
Research and development costs are expensed as incurred. Costs incurred on development projects are recognized as intangible assets when the following criteria are fulfilled:
- •
- it is technically feasible to complete the intangible asset so that it will be available for use or sale;
- •
- management intends to complete the intangible asset and use or sell it;
- •
- there is an ability to use or sell the intangible asset;
- •
- it can be demonstrated how the intangible asset will generate probable future economic benefits;
- •
- adequate technical, financial and other resources to complete the development and to use or sell the intangible asset are available; and
- •
- the expenditure attributable to the intangible asset during its development can be reliably measured.
In the opinion of management, due to uncertainties inherent in the development of the Group's products, the criteria for development costs to be recognized as an asset, as prescribed by IAS 38, "Intangible Assets", are not met.
3. Financial risk management
3.1 Financial risk factors
The Group's activities expose it to a variety of financial risks: market risk, credit risk, liquidity risk and capital risk. The Group's overall risk management program focuses on the unpredictability of financial markets and seeks to minimize potential adverse effects on the Group's financial performance. Risk management is carried out by the Group's finance department (Group Finance) under the policies approved by the Board. Group Finance identifies, evaluates and in some instances economically hedges financial risks in close co-operation with the Group's operating units. The Board provides written guidance for overall risk management, as well as written policies covering specific areas, such as foreign exchange risk, interest-rate risk, use of derivative financial instruments and non-derivative financial instruments, credit risk and investing excess liquidity.
F-18
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
3. Financial risk management (Continued)
Market risk and foreign exchange risk
The Group operates internationally and is exposed to foreign exchange risk arising from various exposures, primarily with respect to the Euro, US dollar and UK pound. Foreign exchange risk arises from future commercial transactions, recognized assets and liabilities and net investments in foreign operations. To manage foreign exchange risk Group Finance maintains foreign currency cash balances to cover anticipated future requirements. The Group's risk management policy is to economically hedge 50% to 100% of anticipated transactions in each major currency for the subsequent 12 months. The Group has a subsidiary in France and in United States of America, whose net assets are exposed to foreign currency translation risk. In 2019, a 10% increase or decrease in the EUR/CHF exchange rate would have resulted in a CHF 19,920 (respectively CHF 52,398 in 2018 and CHF 11,144 in 2017) decrease or increase in net income and shareholders' equity as at December 31, 2019, a 10% increase or decrease in the GBP/CHF exchange rate would have resulted in a CHF 12,489 (respectively CHF 15,965 in 2018 and CHF 3,791 in 2017) decrease or increase in net income and shareholders' equity as at December 31, 2019 and a 10% increase or decrease in the USD/CHF exchange rate would have resulted in a CHF 972,596 (respectively CHF 1,224,506 in 2018 and CHF 86,326 in 2017) increase or decrease in net income and shareholders' equity as at December 31, 2019. Movements in other currencies would not have had a material impact. The Group is not exposed to equity price risk or commodity price risk as it does not invest in these classes of investment.
Interest rate risk
The Group's exposure to interest rate fluctuations is limited because the Group has no interest-bearing indebtedness. The Company's Swiss franc cash holdings are subject to negative interest rates at certain thresholds defined by its bank counterparties. A 10% increase or decrease in the interest rates charged by the counterparties would not have had a material impact on the net income for the period.
Credit risk
Credit risk is managed on a Group basis. Credit risk arises from cash and cash equivalents and deposits with banks, as well as credit exposures to collaboration partners. The Group has a limited number of collaboration partners and consequently has a significant concentration of credit risk. The Group has policies in place to ensure that credit exposure is kept to a minimum and significant concentrations of credit risk are only granted for short periods of time to high credit quality partners. The Group's policy is to invest funds in low risk investments including interest bearing deposits. For banks and financial institutions, only independently rated parties with a minimum rating of "A" are accepted (see note 6).
Liquidity risk
The Group's principal source of liquidity is its cash reserves which are obtained through the sale of new shares and to a lesser extent the sale of its research and development stage products. Group Finance monitors rolling forecasts of the Group's liquidity requirements to ensure it has sufficient cash to meet operational needs. The ability of the Group to maintain adequate cash reserves to sustain its activities in the medium term is highly dependent on the Group's ability to raise further funds from the licensing of its development stage products and the sale of new shares. Consequently, the Group is exposed to significant liquidity risk (see note 4).
F-19
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
3. Financial risk management (Continued)
3.2 Capital risk management
The Group is not regulated and not subject to specific capital requirements. The amount of equity depends on the Group's funding needs and statutory capital requirements. The Group monitors capital periodically on an interim and annual basis. From time to time, the Group may take appropriate measures or propose capital increases to its shareholders to ensure the necessary capital remains intact. The Group did not have any short-term or long-term debt outstanding as of December 31, 2019 and 2018.
3.3 Fair value estimation
The nominal value less estimated credit adjustments of trade receivables and payables are assumed to approximate to their fair values due to the short-term maturity of these instruments and are held at their amortized cost in accordance with IFRS 9. The fair value of other financial assets and liabilities for disclosure purposes is estimated by discounting the future contractual cash flows at the current market interest rate that is available to the Group for similar financial instruments.
4. Critical accounting estimates and judgments
The Group makes estimates and assumptions concerning the future. These estimates and judgments are continually evaluated and are based on historical experience and other factors, including expectations of future events that are believed to be reasonable under the circumstances. The resulting accounting estimates will, by definition, seldom equal the related actual results. The estimates and assumptions that have a significant risk of causing a material adjustment to the carrying amounts of assets and liabilities or may have had a significant impact on the reported results are disclosed below:
Going concern
The Group's accounts are prepared on a going concern basis. To date, the Group has financed its cash requirements primarily from share issuances and licensing certain of its research and development stage products. The Group is a development-stage enterprise and is exposed to all the risks inherent in establishing a business. The Group maintains detailed financial forecasts and monitors actual results on a regular basis so that measures can be taken to ensure the Group remains solvent.
Revenue recognition
Revenue is primarily from fees related to licenses, milestones and research services. Given the complexity of the relevant agreements, judgements are required to identify distinct performance obligations; allocate the transaction price to these performance obligations and determine when the performance obligations are met. In particular, the Group's judgement over the estimated stand alone selling price which is used to allocate the transaction price to the performance obligations is disclosed in note 16.
Grants
Grants are recorded at their fair value when there is reasonable assurance that they will be received and recognized as income when the group has satisfied the underlying grant conditions. In
F-20
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
4. Critical accounting estimates and judgments (Continued)
certain circumstances, grant income may be recognized before explicit grantor acknowledgement that the conditions have been met.
Accrued research and development costs
The Group records accrued expenses for estimated costs of research and development activities conducted by third party service providers. The Group records accrued expenses for estimated costs of research and development activities based upon the estimated amount of services provided but not yet invoiced, and these costs are included in accrued expenses on the balance sheets and within research and development expenses in the statements of loss. These costs are a significant component of research and development expenses. Accrued expenses for these costs are recorded based on the estimated amount of work completed in accordance with agreements established with these third parties.
To date, the Group has not experienced significant changes in the estimates of accrued research and development expenses after a reporting period. However, due to the nature of estimates, the Group may be required to make changes to the estimates in the future as it becomes aware of additional information about the status or conduct of its research activities.
Research and development costs
The Group recognizes expenditure incurred in carrying out its research and development activities, including development supplies, until it becomes probable that future economic benefits will flow to the Group, which results in recognizing such costs as intangible assets, involving a certain degree of judgement. Currently, such development supplies are associated with pre-clinical and clinical trials of specific products that do not have any demonstrated technical feasibility.
Deferred taxes
Deferred tax is the tax expected to be payable or recoverable on differences between the carrying amounts of assets and liabilities in the financial statements and the corresponding tax bases used in the computation of taxable profit and is accounted for using the liability method. Deferred tax liabilities are generally recognized for all taxable temporary differences and deferred tax assets are recognized to the extent that it is probable that taxable profits will be available against which deductible temporary differences can be utilized. Probability over those tax benefits are assessed by Management on the basis of business projections on each relevant entity.
The carrying amount of deferred tax assets is reviewed at each reporting date and reduced to the extent that it is no longer probable that sufficient taxable profits will be available to allow all or part of the asset to be recovered.
Deferred tax is calculated at the tax rates that are expected to apply in the period when the liability is settled or the asset is realized based on tax laws and rates that have been enacted or substantively enacted at the reporting date.
The measurement of deferred tax liabilities and assets reflects the tax consequences that would follow from the manner in which the Group expects, at the end of the reporting period, to recover or settle the carrying amount of its assets and liabilities.
F-21
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
4. Critical accounting estimates and judgments (Continued)
Deferred tax is recognized in profit or loss, except when they relate to items that are recognized in other comprehensive income or directly in equity, in which case, the current and deferred tax are also recognized in other comprehensive income or directly in equity respectively.
Share-based compensation
The Group recognizes an expense for share-based compensation based on the valuation of equity incentive units using binomial and Black-Scholes valuation models. A number of assumptions on the volatility of the underlying shares and on the risk-free rate are made in these models. Should the assumptions and estimates underlying the fair value of these instruments vary significantly from management's estimates, then the share-based compensation expense would be materially different from the amounts recognized. Had these assumptions been modified within their feasible ranges, i.e. a 10% increase or decrease in the volatility assumption and a risk-free rate of 0.5 or zero, and the Group calculated the share-based compensation based on the higher and lower values of these ranges, share-based compensation expense in 2019 would have been CHF 1,239,680 or CHF 2,023,158, respectively CHF 1,696,301 or CHF 2,762,285 in 2018 and CHF 711,856 or CHF 911,946 in 2017. This is compared to the amount recognized as an expense in 2019 of CHF 1,685,965 (respectively CHF 2,298,933 in 2018 and CHF 800,188 in 2017). Additional information is disclosed in note 15.
Pension obligations
The present value of the pension obligations depends on a number of assumptions that are determined on an actuarial basis such as discount rates, future salary and pension increases, and mortality rates. Any changes in these assumptions will impact the carrying amount of pension obligations. The Group determines the appropriate discount rate at the beginning of each year. This is the interest rate that should be used to determine the present value of estimated future cash outflows expected to be required to settle the pension obligations. In determining the appropriate discount rate, the Group considers the interest rates of high-quality corporate bonds that are denominated in the currency in which the benefits will be paid, and that have terms to maturity approximating the terms of the related pension liability. Other key assumptions for pension obligations are based in part on current market conditions. Additional information is disclosed in note 21.
5. Segment information
Management has identified one single operating segment, related to the discovery, development and commercialization of small-molecule pharmaceutical products.
Information about products, services and major customers
External income of the Group for the years ended December 31, 2019 and 2018 is derived from the business of discovery, development and commercialization of pharmaceutical products. Income was earned from the sale of license rights, rendering of research services to a pharmaceutical company and grants earned.
Information about geographical areas
External income is exclusively recorded in the Swiss operating company.
F-22
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
5. Segment information (Continued)
Analysis of revenue from contract with customer and other income by nature is detailed as follows:
| |
2019 | 2018 | 2017 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Fees from sale of license rights |
— | 4,876,000 | — | |||||||
Collaborative research funding |
2,762,830 | 1,167,855 | — | |||||||
Grants earned |
49,405 | 609,212 | 464,916 | |||||||
Other service income |
21,430 | 49,606 | 34,978 | |||||||
| | | | | | | | | | | |
Total |
2,833,665 | 6,702,673 | 499,894 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Analysis of revenue from contract with customer and other income by major counterparties is detailed as follows:
| |
2019 | 2018 | 2017 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Indivior PLC |
2,762,830 | 6,043,855 | — | |||||||
The Michael J. Fox Foundation |
— | 609,212 | 464,916 | |||||||
Eurostars (Innosuisse) |
49,405 | — | — | |||||||
Other counterparties |
21,430 | 49,606 | 34,978 | |||||||
| | | | | | | | | | | |
Total |
2,833,665 | 6,702,673 | 499,894 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
For more detail, refer to note 16, "Revenue from contract with customer" and note 17 "Other Income".
The geographical allocation of long-lived assets is detailed as follows:
| |
December 31, 2019 |
December 31, 2018 |
|||||
|---|---|---|---|---|---|---|---|
Switzerland |
498,066 | 62,866 | |||||
United States of America |
141,420 | — | |||||
France |
391 | 406 | |||||
| | | | | | | | |
Total |
639,877 | 63,272 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The geographical analysis of operating costs is as follows:
| |
2019 | 2018 | 2017 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Switzerland |
17,409,808 | 8,119,953 | 3,719,191 | |||||||
United States of America |
21,214 | — | — | |||||||
France |
6,800 | 7,345 | 15,759 | |||||||
| | | | | | | | | | | |
Total operating costs (note 18) |
17,437,822 | 8,127,298 | 3,734,950 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
There was capital expenditure of CHF 28,459 in 2019 and CHF 9,054 in 2018.
F-23
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
6. Cash and cash equivalents
| |
December 31, 2019 |
December 31, 2018 |
|||||
|---|---|---|---|---|---|---|---|
Cash at bank and on hand |
26,889,923 | 41,670,158 | |||||
Short term deposits in USD |
4,646,880 | — | |||||
| | | | | | | | |
Total cash and cash equivalents |
31,536,803 | 41,670,158 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Split by currency:
| |
December 31, 2019 |
December 31, 2018 |
|||||
|---|---|---|---|---|---|---|---|
CHF |
64.31 | % | 72.33 | % | |||
USD |
35.03 | % | 26.87 | % | |||
EUR |
0.26 | % | 0.51 | % | |||
GBP |
0.40 | % | 0.29 | % | |||
| | | | | | | | |
Total |
100.00 | % | 100.00 | % | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The Group pays interests on CHF cash and cash equivalents and earns interests on USD cash and cash equivalents. The Group invest its cash balances into a variety of current and deposit accounts in two different Swiss banks to limit negative interests. In addition, the Group invests a portion of its USD cash in line with its treasury guidelines.
All cash and cash equivalents were held either at bank or on hand as at December 31, 2019 and December 31, 2018.
Credit quality of cash and cash equivalents
The table below shows the cash and cash equivalents by credit rating of the major counterparties:
| |
December 31, 2019 |
December 31, 2018 |
|||||
|---|---|---|---|---|---|---|---|
External credit rating of counterparty |
|||||||
P-1 / A-1 |
31,536,646 | 41,670,040 | |||||
Cash on hand |
157 | 118 | |||||
| | | | | | | | |
Total cash and cash equivalents |
31,536,803 | 41,670,158 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
External credit ratings of counterparties were obtained from Moody's (P-1) or Standard & Poor's (A-1), respectively.
F-24
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
7. Other current assets
| |
December 31, 2019 |
December 31, 2018 |
|||||
|---|---|---|---|---|---|---|---|
Other financial assets |
13,968 | 7,983 | |||||
Receivables |
118,028 | 273,016 | |||||
Prepayments |
720,063 | 199,410 | |||||
| | | | | | | | |
Total other current assets |
852,059 | 480,409 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The Group applies the IFRS 9 simplified approach to measure expected credit losses, using lifetime expected loss allowance for all trade receivables and contract assets. As of December 31, 2019, the receivables comprise of five non-governmental debtors whose combined outstanding balances are CHF 88,075 (two non-governmental debtors for CHF 115,949 as of December 31, 2018). The Group has considered these customers to have a low risk of default based on historic loss rates and forward-looking information on macroeconomic factors affecting the ability of the customers to settle the receivables. As a result, excepted loss allowance has been deemed as nil as of December 31, 2019 (2018: nil).
The increase in prepayments primarily relates to advance payments relating to R&D service contracts.
8. Right-of-use assets
| |
Properties | Equipment | Total | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
At December 31, 2019 |
||||||||||
Opening net book amount |
— | — | — | |||||||
Adoption of IFRS16 as at January 1, 2019 |
483,350 | 61,160 | 544,510 | |||||||
Additions |
308,987 | 13,541 | 322,528 | |||||||
Depreciation charge |
(296,656 | ) | (27,487 | ) | (324,143 | ) | ||||
Exchange differences |
445 | — | 445 | |||||||
| | | | | | | | | | | |
Closing net book amount |
496,126 | 47,214 | 543,340 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
At December 31, 2019 |
||||||||||
Cost |
792,337 | 74,701 | 867,038 | |||||||
Accumulated depreciation |
(296,211 | ) | (27,487 | ) | (323,698 | ) | ||||
| | | | | | | | | | | |
Net book value |
496,126 | 47,214 | 543,340 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
The Group recorded a depreciation charge in 2019 of CHF 259,940 as part of research and development expenses and CHF 64,203 as part of general and administration expenses.
The maturity analysis of lease liabilities is presented under note 12.
F-25
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
9. Property, plant and equipment
| |
Equipment | Furniture & fixtures |
Chemical Library |
Total | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Year ended December 31, 2018 |
|||||||||||||
Opening net book amount |
2,464 | — | 287 | 2,751 | |||||||||
Additions |
9,054 | — | — | 9,054 | |||||||||
Depreciation charge |
(2,650 | ) | — | (287 | ) | (2,937 | ) | ||||||
| | | | | | | | | | | | | | |
Closing net book amount |
8,868 | — | — | 8,868 | |||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
At December 31, 2018 |
|||||||||||||
Cost |
1,594,405 | 7,564 | 1,207,165 | 2,809,134 | |||||||||
Accumulated depreciation |
(1,585,537 | ) | (7,564 | ) | (1,207,165 | ) | (2,800,266 | ) | |||||
| | | | | | | | | | | | | | |
Net book value |
8,868 | — | — | 8,868 | |||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Year ended December 31, 2019 |
|||||||||||||
Opening net book amount |
8,868 | — | — | 8,868 | |||||||||
Additions |
28,459 | — | — | 28,459 | |||||||||
Depreciation charge |
(9,701 | ) | — | — | (9,701 | ) | |||||||
| | | | | | | | | | | | | | |
Closing net book amount |
27,626 | — | — | 27,626 | |||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
At December 31, 2019 |
|||||||||||||
Cost |
1,622,865 | 7,564 | 1,207,165 | 2,837,594 | |||||||||
Accumulated depreciation |
(1,595,239 | ) | (7,564 | ) | (1,207,165 | ) | (2,809,968 | ) | |||||
| | | | | | | | | | | | | | |
Net book value |
27,626 | — | — | 27,626 | |||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
The Group recorded a depreciation charge in 2019 of CHF 4,732 (2018: CHF 2,068) as part of research and development expenses and CHF 4,969 (2018: CHF 869) as part of general and administration expenses.
10. Non-current financial assets
| |
December 31, 2019 |
December 31, 2018 |
|||||
|---|---|---|---|---|---|---|---|
Security rental deposits |
68,911 | 54,404 | |||||
| | | | | | | | |
Total non-current financial assets |
68,911 | 54,404 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Security rental deposits relate to laboratory and office space which has increased during 2019 due to the new company in United States. The applicable interest rate to such deposits is immaterial, and therefore, the value approximates amortized cost.
F-26
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
11. Payables and accruals
| |
December 31, 2019 |
December 31, 2018 |
|||||
|---|---|---|---|---|---|---|---|
Trade payables |
2,216,147 | 1,148,801 | |||||
Social security and other taxes |
107,415 | 14,921 | |||||
Accrued expenses |
1,872,849 | 957,362 | |||||
| | | | | | | | |
Total payables and accruals |
4,196,411 | 2,121,084 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
All payables mature within 3 months. Accrued expenses relate primarily to amounts accrued under R&D service contracts and professional fees. At December 31, 2019, amounts have increased in line with increased R&D activities. The carrying amounts of trade payables do not materially differ from their fair values, due to their short-term nature.
12. Lease liabilities
The maturities for lease liabilities as of December 31, 2019 are as follows:
| |
Less than 1 Year |
1 to 5 Years |
More than 5 Years |
Total cash out flows |
Carrying amount liabilities |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Lease liabilities |
392,954 | 182,664 | — | 575,618 | 550,245 | |||||||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Lease liabilities relate to the rent of laboratories, equipment, offices and related spaces used by the Group.
The net debt is detailed as follows:
| |
Leases | Cash and cash equivalents |
Other financial assets |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
Net debt as at January 1, 2018 |
— | 2,579,248 | 11,291 | |||||||
Cash flows |
— | 39,080,443 | (3,308 | ) | ||||||
Acquisition—Finance leases and operating Leases |
— | — | — | |||||||
Foreign exchange differences |
— | 10,467 | — | |||||||
Net debt as at December 31, 2018 |
— | 41,670,158 | 7,983 | |||||||
Recognized on adoption of IFRS 16 |
(544,510 | ) | — | — | ||||||
| | | | | | | | | | | |
Total |
(544,510 | ) | 41,670,158 | 7,983 | ||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Cash flows |
316,348 | (9,989,053 | ) | 5,955 | ||||||
Acquisition—Leases |
(322,528 | ) | — | — | ||||||
Foreign exchange differences |
445 | (144,302 | ) | — | ||||||
Net debt as at December 31, 2019 |
(550,245 | ) | 31,536,803 | 13,938 | ||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
F-27
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
13. Deferred income
The Group expects the deferred income to be recognized as follows:
| |
December 31, 2019 |
December 31, 2018 |
|||||
|---|---|---|---|---|---|---|---|
Expected income recognition in year one after the balance sheet date |
165,389 | — | |||||
Expected income recognition in year two after the balance sheet date |
165,390 | — | |||||
| | | | | | | | |
Total deferred income |
330,779 | — | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The deferred income relates to a grant from Eurostars/Innosuisse. See note 17 "other income" for further information related to the Eurostars/Innosuisse project.
14. Share capital
| |
Number of shares | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
Common shares |
Treasury shares |
Total | |||||||
Balance at January 1, 2018 |
15,384,988 | (1,964,973 | ) | 13,420,015 | ||||||
Issue of shares—capital increase |
13,179,043 | (278,027 | ) | 12,901,016 | ||||||
Settlement of suppliers' invoices |
— | 87,176 | 87,176 | |||||||
Net purchase of treasury shares |
— | (2,652 | ) | (2,652 | ) | |||||
| | | | | | | | | | | |
Balance at December 31, 2018 |
28,564,031 | (2,158,476 | ) | 26,405,555 | ||||||
| | | | | | | | | | | |
Issue of shares—capital increase |
4,284,604 | (4,284,604 | ) | — | ||||||
Settlement of suppliers' invoices |
— | 196,610 | 196,610 | |||||||
Net sale of treasury shares |
— | 2,983 | 2,983 | |||||||
| | | | | | | | | | | |
Balance at December 31, 2019 |
32,848,635 | (6,243,487 | ) | 26,605,148 | ||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
The Company maintains a liquidity contract with Kepler Capital Markets SA ("Kepler"). Under the agreement, the Group has provided Kepler with cash and shares to enable them to buy and sell the Company's shares. At December 31, 2019, 27,925 (2018: 30,908) treasury shares are recorded under this agreement in the treasury share reserve and CHF 13,968 (2018: CHF 7,983) is recorded in other financial assets.
At December 31, 2019, the total issued share capital is CHF 32,848,635 (2018: CHF 28,564,031), consisting of 32,848,635 shares (2018: 28,564,031). All shares have a nominal value of CHF 1.
On May 17, 2019, the Company issued 4,284,604 new shares from the authorized capital to its 100% owned subsidiary, Addex Pharma SA at CHF 1. These shares are held as treasury shares.
On March 28, 2018, the Company increased its share capital by issuing 13,037,577 new shares with a nominal value of CHF 1 each at an issue price of CHF 3.13 per share. Of these new shares, 12,901,016 were placed with investors raising CHF 40.4 million of gross proceeds and the remaining 136,561 new shares were recorded as treasury shares at the issue price of CHF 427,436. Each new share received a 7-year warrant to purchase 0.45 of a share at a price of CHF 3.43 per share. A total of 5,866,898 warrants were granted of which 5,806,882 to investors. The fair value of each of the warrants
F-28
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
14. Share capital (Continued)
issued to investors is CHF 0.56, and has been calculated using the Black-Scholes valuation model and recorded in equity as a cost of the capital increase, with a volatility of 37.15% and an annual risk free rate of 0.13%. The total value of the warrants granted to investors amounts to CHF 3,308,982.
On March 16, 2018, the Group issued 141,466 new shares from the conditional capital to its 100% owned subsidiary, Addex Pharma SA at CHF 1. These shares have been issued to replenish the treasury share reserve, which had previously been used to settle the exercise of share options.
For the fiscal year ended December 31, 2019, the Group used 196,610 treasury shares (2018: 87,176) to purchase services from consultants including 113,099 (2018: 37,824) shares for Roger Mills, the Group's Chief Medical Officer (2018: 32,362 shares for Tim Dyer, the Group's Chief Executive Officer). The total value of consulting services settled in shares was CHF 289,214 (2018: CHF 208,084). Under a liquidity agreement, the Group recorded net sales of treasury shares of CHF 5,986 (2018: net purchases of CHF 5,190).
15. Share-based compensation
The total share-based compensation expense recognized in the statement of loss for equity incentive units granted to directors, executives, employees, consultants and investors has been recorded under the following headings:
| |
2019 | 2018 | 2017 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Research and development |
433,536 | 880,982 | 511,789 | |||||||
General and administration |
1,252,429 | 1,417,951 | 288,399 | |||||||
| | | | | | | | | | | |
Total share-based compensation |
1,685,965 | 2,298,933 | 800,188 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Analysis of share-based compensation by equity incentive plan is detailed as follows:
| |
2019 | 2018 | 2017 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Equity sharing certificate plan |
37,776 | 77,336 | 28,588 | |||||||
Share purchase plan |
45,593 | 38,296 | 34,821 | |||||||
Share option plans |
1,602,596 | 2,183,301 | 736,779 | |||||||
| | | | | | | | | | | |
Total share-based compensation |
1,685,965 | 2,298,933 | 800,188 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Equity Sharing Certificate Equity Incentive Plan
On June 1, 2010, the Company established an equity incentive plan based on equity sharing certificates (ESCs) to provide incentives to directors, executives, employees and consultants of the Group. Each ESC provides the holder (i) a right to subscribe for 1,000 shares in the Company, and (ii) a right to liquidation proceeds equivalent to that of shareholders. All rights of the ESCs expire after their defined exercise period with the ownership of the ESCs reverting to the Group. ESCs granted are subject to certain vesting conditions based on service period defined in each grant agreement. The holder of vested ESCs has the right to subscribe to shares at the subscription price if the underlying share price has reached the floor price. The floor and subscription price are defined by the Board of Directors in each grant agreement at the time of issuance. In the event of a change in
F-29
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
15. Share-based compensation (Continued)
control, all ESCs are automatically vested. The Group has no legal or constructive obligation to repurchase or settle ESCs in cash.
Movements in the number of share subscription rights attached to the ESCs outstanding are as follows:
| |
2019 | 2018 | 2017 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
At January 1 |
265,600 | 275,933 | 354,433 | |||||||
Granted |
— | — | 108,000 | |||||||
Expired |
(66,850 | ) | (10,333 | ) | (78,500 | ) | ||||
Exercised |
— | — | (108,000 | ) | ||||||
| | | | | | | | | | | |
At December 31 |
198,750 | 265,600 | 275,933 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
At December 31, 2019, of the outstanding 198,750 subscription rights (respectively 2018: 265,600 and 2017: 275,933) attached to the ESCs, 144,750 were exercisable (respectively 2018: 184,600 and 2017: 128,533). On April 1, 2019, the exercise period of 90,750 vested ESCs has been extended for 5 years. Include in share-based compensation for the year 2019, CHF 8,667 relates to the fair value adjustment for exercise period extensions of vested options.
The outstanding subscription rights as at December 31, 2019 and 2018 have the following expiry dates, subscription prices and floor prices:
At December 31, 2019
| |
Subscription prices / floor prices (CHF) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Expiry date
|
1.00 / 2.30 | 2.00 / 2.30 | Total | |||||||
2024 |
90,750 | — | 90,750 | |||||||
2027 |
— | 108,000 | 108,000 | |||||||
| | | | | | | | | | | |
Total subscription rights |
90,750 | 108,000 | 198,750 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
At December 31, 2018
| |
Subscription prices / floor prices (CHF) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Expiry date
|
1.00 / 2.30 | 2.00 / 2.30 | Total | |||||||
2019 |
151,600 | — | 151,600 | |||||||
2020 |
6,000 | — | 6,000 | |||||||
2027 |
— | 108,000 | 108,000 | |||||||
| | | | | | | | | | | |
Total subscription rights |
157,600 | 108,000 | 265,600 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
F-30
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
15. Share-based compensation (Continued)
At December 31, 2017
| |
Subscription prices / floor prices (CHF) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Expiry date
|
1.00 / 2.30 | 2.00 / 2.30 | 5.00 /10.00 | 7.00 / 14.00 | Total | |||||||||||
2018 |
— | — | 8,000 | 2,333 | 10,333 | |||||||||||
2019 |
151,600 | — | — | — | 151,600 | |||||||||||
2020 |
6,000 | — | — | — | 6,000 | |||||||||||
2027 |
— | 108,000 | — | — | 108,000 | |||||||||||
| | | | | | | | | | | | | | | | | |
Total subscription rights |
157,600 | 108,000 | 8,000 | 2,333 | 275,933 | |||||||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Share option plans
The Company established a share option plan to provide incentives to directors, executives, employees and consultants of the Group.
During 2019, the Group granted the following options with vesting over 4 years and a 10-year exercise period as follow:
| |
Number | Exercise price |
Expiry date |
|||||
|---|---|---|---|---|---|---|---|---|
January 1, 2019 |
243,506 | 2.25 | December 31, 2028 | |||||
July 1, 2019 |
187,189 | 1.50 | June 30, 2029 | |||||
October 1, 2019 |
30,000 | 1.80 | September 30, 2029 | |||||
| | | | | | | | | |
Total 2019 |
460,695 | |||||||
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
On June 1, 2018 the Group granted 2,467,584 options at an exercise price of CHF 3, with vesting rights over 4 years and a 10-year exercise period.
Movements in the number of options outstanding are as follows:
| |
2019 | 2018 | 2017 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
At January 1 |
5,128,680 | 2,661,096 | 779,813 | |||||||
Granted |
460,695 | 2,467,584 | 1,901,283 | |||||||
Expired |
(48,775 | ) | — | (20,000 | ) | |||||
| | | | | | | | | | | |
At December 31 |
5,540,600 | 5,128,680 | 2,661,096 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
At December 31, 2019, of the outstanding 5,540,600 share options (respectively 2018: 5,128,680 and 2017:2,661,096), 2,811,825 were exercisable (respectively 2018: 1,736,764 and 2017:773,489).
On April 1, 2019, the exercise period of 506,351 vested options has been extended for 5 years and share-based compensation related to the fair value adjustment for the exercise period extensions of CHF 75,331 has been recognized in 2019.
F-31
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
15. Share-based compensation (Continued)
The outstanding share options as at December 31, 2019 and 2018 have the following expiry dates:
At December 31, 2019
| |
Exercises prices (CHF) | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Expiry date
|
1.00 | 1.50 | 1.80 | 2.00 | 2.08 | 2.25 | 3.00 | Total | |||||||||||||||||
2020 |
— | — | — | 49,687 | — | — | — | 49,687 | |||||||||||||||||
2021 |
— | — | — | 105,000 | 50,000 | — | — | 155,000 | |||||||||||||||||
2024 |
— | — | — | 506,351 | — | — | — | 506,351 | |||||||||||||||||
2027 |
292,261 | — | — | 1,609,022 | — | — | — | 1,901,283 | |||||||||||||||||
2028 |
— | — | — | — | — | 243,506 | 2,467,584 | 2,711,090 | |||||||||||||||||
2029 |
— | 187,189 | 30,000 | — | — | — | — | 217,189 | |||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | |
Total |
292,261 | 187,189 | 30,000 | 2,270,060 | 50,000 | 243,506 | 2,467,584 | 5,540,600 | |||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | |
At December 31, 2018
| |
Exercises prices (CHF) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Expiry date
|
1.00 | 2.00 | 2.08 | 3.00 | Total | |||||||||||
2019 |
— | 555,126 | — | — | 555,126 | |||||||||||
2020 |
— | 49,687 | — | — | 49,687 | |||||||||||
2021 |
— | 105,000 | 50,000 | — | 155,000 | |||||||||||
2027 |
292,261 | 1,609,022 | — | — | 1,901,283 | |||||||||||
2028 |
— | — | — | 2,467,584 | 2,467,584 | |||||||||||
| | | | | | | | | | | | | | | | | |
Total |
292,261 | 2,318,835 | 50,000 | 2,467,584 | 5,128,680 | |||||||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
At December 31, 2017
| |
Exercises prices (CHF) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Expiry date
|
1.00 | 2.00 | 2.08 | 3.00 | Total | |||||||||||
2019 |
— | 555,126 | — | — | 555,126 | |||||||||||
2020 |
— | 49,687 | — | — | 49,687 | |||||||||||
2021 |
— | 105,000 | 50,000 | — | 155,000 | |||||||||||
2027 |
292,261 | 1,609,022 | — | — | 1,901,283 | |||||||||||
| | | | | | | | | | | | | | | | | |
Total |
292,261 | 2,318,835 | 50,000 | — | 2,661,096 | |||||||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
F-32
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
15. Share-based compensation (Continued)
The weighted average fair value of share options granted during 2019 determined using a Black-Scholes model was CHF 0.68 (2018: CHF 1.03). The significant inputs to the model were:
| |
2019 | 2018 | 2017 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Weighted average share price per share at the grant date |
CHF 1.93 | CHF 2.94 | CHF 2.27 | |||||||
Weighted average strike price per share |
CHF 1.92 | CHF 3.00 | CHF 1.85 | |||||||
Weighted average volatility |
36.45 | % | 36.86 | % | 43.00 | % | ||||
Dividend yield |
— | — | — | |||||||
Weighted average annual risk free rate / annual risk-free rate |
0.13 | % | 0.13 | % | 0.13 | % | ||||
Share purchase plan
The Group established a share purchase plan under which services are settled for shares. Under the plan directors, executives, employees and consultants may receive fully paid ordinary shares from the Group's treasury share reserve for services rendered. During 2019, 196,610 shares (respectively 2018: 87,176 and 2017: 132,096) were transferred to settle CHF 289,214 (respectively 2018: CHF 208,085 and 2017: CHF 258,903) of consulting fees.
16. Revenue from contract with customer
License & research agreement with Indivior PLC
On January 2, 2018, the Group entered into an agreement with Indivior PLC (Indivior) for the discovery, development and commercialization of novel GABAB PAM compounds for the treatment of addiction and other CNS diseases. This agreement included the selected clinical candidate, ADX71441. In addition, Indivior agreed to fund a research program at the Group to discover novel GABAB PAM compounds.
The contract contains two distinct material promises and performance obligations: (1) the selected compound ADX71441 which falls within the definition of a licensed compound, whose rights of use and benefits thereon was transferred in January 2018 and, (2) the research services to be conducted by the Group and funded by Indivior to discover novel GABAB PAM compounds for clinical development that may be discovered over the research term of the agreement and selected by Indivior.
Indivior has sole responsibility, including funding liability, for development of selected compounds under the agreement through preclinical and clinical trials, as well as registration procedures and commercialization, if any, worldwide. Indivior has the right to design development programs for selected compounds under the agreement. Through the Group's participation in a joint development committee, the Group reviews, in an advisory capacity, any development programs designed by Indivior. However, Indivior has authority over all aspects of the development of such selected compounds.
Under terms of the agreement, the Group granted Indivior an exclusive license to use relevant patents and know-how in relation to the development and commercialization of product candidates selected by Indivior. Subject to agreed conditions, the Group and Indivior jointly own all intellectual property rights that are jointly developed and the Group or Indivior individually own all intellectual property rights that the Group or Indivior develop individually. The Group has retained the right to
F-33
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
16. Revenue from contract with customer (Continued)
select compounds from the research program for further development in areas outside the interest of Indivior including Charcot-Marie-Tooth type 1A neuropathy, or CMT1A. Under certain conditions, but subject to certain consequences, Indivior may terminate the agreement.
The Group received in January 2018, a non-refundable upfront fee of USD 5.0 million for the right to use the clinical candidate, ADX71441, including all materials and know-how related to this clinical candidate. In addition, the Group is eligible for payments on successful achievement of pre-specified clinical, regulatory and commercial milestones totaling USD 330 million and royalties on net sales of mid-single digits to low teens double-digit.
On February 14, 2019, Indivior terminated the development of their selected compound, ADX71441. Separately, Indivior funds research at the Group, based on a research plan to be mutually agreed between the parties, to discover novel GABAB PAM compounds. These future novel GABAB PAM compounds, if selected by Indivior, become licensed compounds. The Group agreed with Indivior to an initial research term of two years, that can be extended by twelve-month increments and a minimum annual funding of USD 2 million for the Group's R&D costs incurred. Following Indivior's selection of one newly identified compound, the Group has the right to also select one additional newly identified compound. The Group is responsible for the funding of all development and commercialization costs of its selected compounds and Indivior has no rights to the Group's selected compounds. The initial two-year research term was expected to run from May 2018 to April 2020. On October 7, 2019 and December 20, 2019, Indivior agreed an additional research funding of USD 0.8 million, increasing the research funding by USD 1.6 million, for the research period.
The research activities started on May 1, 2018. For the year ended December 31, 2019, the Group recognized CHF 2.8 million as revenue (2018: CHF 1.2 million) and recorded CHF 0.9 million as contract liability (2018: CHF 0.2 million). In 2018, the non-refundable upfront fees of USD 5 million (CHF 4.9 million) for the right of use the clinical candidate ADX71441 was recorded as revenue.
Janssen Pharmaceuticals Inc. (formerly Ortho-McNeil-Janssen Pharmaceuticals Inc).
On December 31, 2004, the Group entered into a research collaboration and license agreement with Janssen Pharmaceuticals Inc. (JPI). In accordance with this agreement, JPI has acquired an exclusive worldwide license to develop mGluR2PAM compounds for the treatment of human health. The Group is eligible to receive up to EUR 109 million in success-based development and regulatory milestone, and low double digit royalties on net sales. The Group considers these various milestones to be variable consideration as they are contingent upon achieving uncertain, future development stages and net sales. For this reason the Group considers the achievement of the various milestones as binary events that will be recognized as revenue upon occurrence. No amounts have been recognized under this agreement in 2019 and 2018.
F-34
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
17. Other income
Under grant agreements with Eurostars/Innosuisse and Michael J.Fox Foundation, the Group is required to complete specific research activities within a defined period of time. The Group's funding is fixed and received, based on the satisfactory completion of these agreed research activities and incurring the related costs.
Eurostars/Innosuisse
In 2019, the Group received CHF 380,184 from Eurostars/Innosuisse that partially funds the two coming years of the project named DISARM FEAR. It aims at developing small-molecule negative allosteric modulators (NAMs) that targets the metabotropic glutamate receptor 7 (mGlu7) as a potential treatment to reduce fear memory in post-traumatic stress disorder (PTSD).
Michael J.Fox Foundation for Parkinson's Research (MJFF)
Until 2018, the Group has received USD 1.8 Million from MJFF for the funding of clinical testing of Dipraglurant for the treatment of Parkinson's disease levodopa-induced dyskinesia (PD-LID) and TrKB PAM discovery activities.
For the year ended December 31, 2019 the Group recognized as other income CHF 49,405 related to the funding from Eurostars/Innosuisse. For the years ended December 31, 2018 and 2017 the Group respectively recognized CHF 609,212 and CHF 464,916 in other income from Michael J.Fox Foundation. The Grants are deferred and recognized as other income in the statement of loss over the period according to when the Group has satisfied the underlying grant conditions. As of December 31, 2019 the Group recognized CHF 330,079 from Eurostars/Innosuisse as deferred income, including CHF 165,389 on short term (less than one year) and CHF 165,390 (more than one year), in accordance with the budget for the use of the grant received. As of December 31, 2018, deferred income were nil.
In 2019, the Group has additionally recognized as other income CHF 21,430 from IT consultancy agreements.
18. Operating costs
| |
2019 | 2018 | 2017 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Staff costs (note 19) |
4,288,815 | 2,224,206 | 751,277 | |||||||
Depreciation |
333,844 | 2,938 | 15,249 | |||||||
External research and development costs |
9,350,667 | 2,368,457 | 841,308 | |||||||
Laboratory consumables |
230,097 | 144,169 | 29,764 | |||||||
Patent maintenance and registration costs |
268,143 | 261,954 | 180,125 | |||||||
Professional fees |
1,951,661 | 2,313,722 | 1,347,913 | |||||||
Operating leases |
— | 179,102 | 96,889 | |||||||
Short term leases |
26,150 | — | — | |||||||
Other operating costs |
988,445 | 632,750 | 472,425 | |||||||
| | | | | | | | | | | |
Total operating costs |
17,437,822 | 8,127,298 | 3,734,950 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Operating expenses have increased significantly during the year due to expanded R&D activities. Professional fees primarily relate to legal, auditors, G&A consultants and board members.
F-35
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
19. Staff costs
| |
2019 | 2018 | 2017 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Wages and salaries |
2,438,448 | 1,273,382 | 544,912 | |||||||
Social charges and insurances |
243,232 | 112,524 | 59,749 | |||||||
Value of share-based services (note 15) |
1,310,888 | 719,374 | 83,459 | |||||||
Retirement benefit expenses (note 21) |
296,247 | 118,926 | 63,157 | |||||||
| | | | | | | | | | | |
Total staff cost |
4,288,815 | 2,224,206 | 751,277 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
20. Taxes
| |
December 31, 2019 |
December 31, 2018 |
December 31, 2017 |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
Loss before tax |
14,780,604 | 1,644,798 | 3,280,406 | |||||||
| | | | | | | | | | | |
Tax calculated at a tax rate of 23.4% |
3,458,661 | 384,883 | 767,615 | |||||||
Effect of different tax rates in other countries |
(1,660 | ) | (1,719 | ) | (3,687 | ) | ||||
Expenses charged against equity |
(39,876 | ) | (693,439 | ) | (5,984 | ) | ||||
Expenses not deductible for tax purposes |
(418,356 | ) | (542,632 | ) | (191,812 | ) | ||||
| | | | | | | | | | | |
Total tax losses not recognized as deferred tax asset |
(2,998,769 | ) | 852,907 | (566,132 | ) | |||||
| | | | | | | | | | | |
Income tax expense |
— | — | — | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
The Group has revised the 2018 and 2017 tax rate and disclosure to include the additional cantonal and communal taxes that are applicable in order to be consistent with 2019.
The Group has decided not to recognize any deferred income tax assets at December 31, 2019, 2018 or 2017. The key factors which have influenced Management in arriving at this evaluation are the fact that the Group has not yet a history of making profits and product development remains at an early stage.
The amounts of deferred income tax assets that arise from sources other than tax loss carry forwards and the amounts of deferred income tax liabilities are insignificant in comparison to the unrecognized tax loss carry forwards.
F-36
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
20. Taxes (Continued)
The tax losses carry forwards of the Group and their respective expiring dates are as follows:
| |
December 31, 2019 |
December 31, 2018 |
December 31, 2017 |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
2018 |
— | — | 28,861,010 | |||||||
2019 |
— | 27,481,171 | 28,287,766 | |||||||
2020 |
15,982,220 | 15,982,220 | 15,982,220 | |||||||
2021 |
1,224,210 | 1,224,210 | 1,224,210 | |||||||
2022 |
3,540,541 | 3,540,541 | 3,540,541 | |||||||
2023 |
141,425,567 | 3,309,636 | 3,309,636 | |||||||
2024 |
290,949 | 290,949 | 1,125,258 | |||||||
2025 |
3,586,490 | 3,586,490 | — | |||||||
2026 |
23,467,858 | — | — | |||||||
| | | | | | | | | | | |
Total unrecorded tax losses carry forwards |
189,517,835 | 55,415,217 | 82,330,641 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
As of December 31, 2019, the unrecorded taxes losses carried forwards raised to CHF 189,517,835, because the swiss tax administration accepted to renew CHF 138 million tax losses, on July 18, 2019.
21. Retirement benefits obligations
Apart from the social security plans fixed by the law, the Group sponsors an independent pension plan. The Group has contracted with Swiss Life based in Lausanne for the provision of occupational benefits. All benefits in accordance with the regulations are reinsured in their entirety with Swiss Life within the framework of the corresponding contract. This pension solution fully reinsures the risks of disability, death and longevity with Swiss Life. The latter invests the vested pension capital and provides a 100% capital and interest guarantee. The pension plan is entitled to an annual bonus from Swiss Life comprising the effective savings, risk and cost results. Although, as is the case with many Swiss pension plans, the amount of ultimate pension benefit is not defined, certain legal obligations of the plan create constructive obligations on the employer to pay further contributions to fund an eventual deficit; this results in the plan nevertheless being accounted for as a defined benefit plan. All employees are covered by this plan, which is a defined benefit plan. Retirement benefits are based on contributions, computed as a percentage of salary, adjusted for the age of the employee and shared approximately 45% / 55% by employee and employer. In addition to retirement benefits, the plans provide death and long-term disability benefits to its employees. Liabilities and assets are revised every year by an independent actuary. Assets are held in the insurance company. In accordance with IAS 19 (revised), plan assets have been estimated at fair market values and liabilities have been calculated according to the "projected unit credit" method. The Group recorded a pension benefit charge in 2019 of CHF 296,247 (2018: CHF 118,926) as part of staff costs.
F-37
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
21. Retirement benefits obligations (Continued)
Employment benefit obligations
The amounts recognized in the balance sheet are determined as follows:
| |
2019 | 2018 | |||||
|---|---|---|---|---|---|---|---|
Defined benefit obligation |
(8,583,214 | ) | (7,060,278 | ) | |||
Fair value of plan assets |
7,101,476 | 6,420,927 | |||||
| | | | | | | | |
Funded status |
(1,481,738 | ) | (639,351 | ) | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The amounts recognized in the statements of loss are as follows:
| |
2019 | 2018 | |||||
|---|---|---|---|---|---|---|---|
Current service cost |
(286,515 | ) | (115,146 | ) | |||
Interest cost |
(81,829 | ) | (37,903 | ) | |||
Interest income |
72,097 | 34,123 | |||||
| | | | | | | | |
Company pension cost (note 19) |
(296,247 | ) | (118,926 | ) | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The movement in the defined benefit obligations during the year is as follows:
| |
2019 | 2018 | |||||
|---|---|---|---|---|---|---|---|
Defined benefit obligation at beginning of year |
(7,060,278 | ) | (3,607,276 | ) | |||
Service cost |
(286,515 | ) | (115,146 | ) | |||
Interest cost |
(81,829 | ) | (37,903 | ) | |||
Employee contribution |
(166,150 | ) | (84,096 | ) | |||
Actuarial gain / (loss) arising from changes in financial assumptions |
(875,960 | ) | 197,291 | ||||
Actuarial gain / (loss) arising from changes in demographic assumptions |
91,212 | — | |||||
Actuarial gain / (loss) on experience adjustment |
(263,491 | ) | (573,684 | ) | |||
Benefits paid/ (deposited) |
59,797 | (2,839,464 | ) | ||||
| | | | | | | | |
Defined benefit obligations at end of year |
(8,583,214 | ) | (7,060,278 | ) | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The movements in the fair value of plan assets during the year are as follows:
| |
2019 | 2018 | |||||
|---|---|---|---|---|---|---|---|
Fair value of plan assets at beginning of year |
6,420,927 | 3,363,412 | |||||
Interest income |
72,097 | 34,123 | |||||
Employees' contributions |
166,150 | 84,096 | |||||
Company contribution |
199,715 | 98,918 | |||||
Plan assets gains |
302,384 | 914 | |||||
Benefits (paid)/ deposited |
(59,797 | ) | 2,839,464 | ||||
| | | | | | | | |
Fair value of plan assets at end of year |
7,101,476 | 6,420,927 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
F-38
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
21. Retirement benefits obligations (Continued)
The
principal actuarial assumptions used were as follows:
| |
December 31, 2019 |
December 31, 2018 |
||
|---|---|---|---|---|
Discount rate |
0.20% | 0.90% | ||
Mortality tables |
BVG2015 GT | BVG2015 GT | ||
Salary growth rate |
1.00% | 1.00% | ||
Pension growth rate |
0.00% | 0.00% |
The discount rate and the life expectancy were identified as significant actuarial assumptions for the Swiss pension plan. The following impacts on the defined benefit obligation are to be expected:
- •
- 0.25% increase or decrease in the discount rate would lead to a decrease of 4.47% (2018: 4.38%) or an increase of 5.22% (2018: 4.74%) in the
defined benefit obligation of the Swiss pension plan;
- •
- 0.25% increase or decrease in the interest rate on retirement savings capital would lead to an increase of 0.59% (2018: 1.19%) or a decrease of
0.53% (2018: 1.16%) in the defined benefit obligation of the Swiss pension plan;
- •
- 0.25% increase or decrease in salaries would lead to an increase of 0.03% (2018: 0.18%) or a decrease of 0.02% (2018: 0.18%) in the defined
benefit obligation of the Swiss pension plan.
- •
- +/-1 year in the life expectancy would lead to an increase of 1.86% (2018: 1.55%) or a decrease of 1.92% (2018: 1.56%) in the defined benefit obligation of the Swiss pension plan.
The above sensitivity analyses are based on a change in an assumption while holding all other assumptions constant. In practice, this is unlikely to occur, and changes in some of the assumptions may be correlated. When calculating the sensitivity of the defined benefit obligations to significant actuarial assumptions the same method (present value of the defined benefit obligation calculated with the projected unit credit method at the end of the reporting period) has been applied as when calculating the pension liability recognized within the consolidated balance sheets.
The methods and types of assumptions used in preparing the sensitivity analysis did not change compared to the prior period.
The estimated Group contributions to pension plans for the financial year 2020 amounts to CHF 213,000.
The following table shows the funding of the defined benefit pensions and actuarial adjustments on plan liabilities:
| |
2019 | 2018 | |||||
|---|---|---|---|---|---|---|---|
Present value of defines benefit obligation |
(8,583,214 | ) | (7,060,278 | ) | |||
Fair value of plan assets |
7,101,476 | 6,420,927 | |||||
| | | | | | | | |
Deficit in the plan |
(1,481,738 | ) | (639,351 | ) | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Experience adjustment |
(1,048,239 | ) | (376,393 | ) | |||
Actuarial gains on plan assets |
302,384 | 914 | |||||
F-39
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
21. Retirement benefits obligations (Continued)
The following table shows the estimated benefit payments for the next ten years where the number of employees remains constant:
2020 |
318,000 | |||
2021 |
308,000 | |||
2022 |
301,000 | |||
2023 |
298,000 | |||
2024 |
655,000 | |||
2025 - 2029 |
1,456,000 |
22. Finance costs
| |
2019 | 2018 | 2017 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Interests income |
36,874 | — | — | |||||||
Interests expense on leases |
(22,603 | ) | — | — | ||||||
Interests cost |
(105,915 | ) | (134,307 | ) | (171 | ) | ||||
Foreign exchange losses |
(84,803 | ) | (85,866 | ) | (45,179 | ) | ||||
| | | | | | | | | | | |
Finance costs |
(176,447 | ) | (220,173 | ) | (45,350 | ) | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
23. Loss per share
Basic and diluted loss per share is calculated by dividing the loss attributable to equity holders of the Company by the weighted average number of shares in issue during the year excluding shares purchased by the Group and held as treasury shares.
| |
2019 | 2018 | 2017 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Loss attributable to equity holders of the Company |
(14,780,604 | ) | (1,644,798 | ) | (3,280,406 | ) | ||||
Weighted average number of shares in issue |
26,428,269 | 23,293,237 | 12,941,439 | |||||||
| | | | | | | | | | | |
Basic and diluted loss per share |
(0.56 | ) | (0.07 | ) | (0.25 | ) | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
The Company has three categories of dilutive potential shares as at December 31, 2019, 2018 and 2017: equity sharing certificates (ESCs), share options and warrants. As of December 31, 2019, 2018 and 2017, equity sharing certificates, share options and warrants have been ignored in the calculation of the loss per share, as they would be antidilutive.
The total number of dilutive instruments as of December 31, 2019 is 11,906,248 (Respectively 2018: 11,561,178 and 2017: 3,237,029) which consists of 198,750 ESCs (respectively 2018: 265,600 and 2017: 275,933), 5,540,600 ESOP (respectively 2018: 5,128,680 and 2017: 2,661,096) and 6,166,898 warrants (respectively 2018: 6,166,898 and 2017: 300,000). These options could potentially dilute basic earnings per share in the future.
F-40
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
24. Commitments and contingencies
Operating leases commitments
Commitments for operating lease as of December 31, 2018 are as follows:
| |
Less than 1 Year |
1 to 5 Years |
More than 5 Years |
Total cash out flows |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Operating lease commitments |
142,298 | 130,200 | — | 272,498 | |||||||||
Operating lease commitments consist mainly of rental contracts for laboratories, offices and related spaces used by the Group. There are no commitments over 5 years.
Capital commitments
As at December 31, 2019 and 2018, the Group has no contracted capital expenditure.
Contingencies
As part of the ordinary course of business, the Group is subject to contingent liabilities in respect of certain litigation. Currently, there is no outstanding litigation.
25. Related party transactions
Related parties include members of the Board of Directors and the Executive Management of the Group. The following transactions were carried out with related parties:
Key management compensation
| |
2019 | 2018 | 2017 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Salaries, other short-term employee benefits and post-employment benefits |
1,156,427 | 522,163 | 133,180 | |||||||
Consulting fees |
364,535 | 577,078 | 737,685 | |||||||
Share-based compensation |
1,434,190 | 2,019,430 | 595,835 | |||||||
| | | | | | | | | | | |
|
2,955,152 | 3,118,671 | 1,466,700 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
The Group has revised the 2018 share-based compensation reported in this note to reflect actual shared based compensation expense recorded in the consolidated statements of loss of the year, rather than the total fair value at the grant date, as previously disclosed in 2018.
Salaries, other short-term employee benefits and post-employment benefits relate to members of the Board of Directors and Executive Management who are employed by the Group. In 2019, consulting fees relate solely to Roger Mills, a member of the Executive Management who delivers his services to the Group under a consulting contract. In 2018, consulting fees are related to Roger Mills as well as Tim Dyer who delivered his consulting services through TMD Advisory Ltd ("TMDA") until October 31, 2018. TMDA invoiced the Group for the rent of administrative premises, CHF 26,682 in 2018 (CHF 22,536 in 2017 and nil in 2019), whilst the Group invoiced accounting services to TMDA of CHF 49,606 in 2018 (CHF 34,978 in 2017 and nil in 2019) recorded in other income. The Group has a net payable to the Board of Directors and Executive Management of CHF 176,089 at December 31,
F-41
Notes to the Consolidated Financial Statements (Continued)
(Amounts in Swiss francs)
25. Related party transactions (Continued)
2019 (2018: CHF 169,486) and a net receivable of CHF 6,087 at December 31, 2017. In addition, the Group has a net payable to TMDA of CHF 116,994 at December 31, 2018 (2017: 176,640) for consulting services and a net receivable of CHF 82,589 (2017: 82,589) for invoiced administrative services.
26. Events after the balance sheet date
On January 29, 2020 the Group listed American Depositary Shares (ADSs) representing its ordinary shares on the Nasdaq Stock Market and the United States Securities and Exchange Commission (SEC) declared its registration statement on Form F1 and F6 becoming effective. The ADSs are listed for trading on Nasdaq under the symbol "ADXN". Addex has not registered any new issuance of securities and its shares will continue to be admitted to trading on SIX Swiss Exchange.
In early 2020 a coronavirus disease (COVID-19) pandemic developed globally resulting in a significant number of infections and negative effects on economic activity. The Group is actively monitoring the situation and is taking any necessary measures to respond to the situation in cooperation with the various stakeholders. As of the date of approving these financial statements, the Group has suspended the initiation of a placebo-controlled Phase 2b/3 pivotal clinical trial of dipraglurant in PD-LID patients. Depending on the duration of the COVID-19 crisis and continued negative impact on global economic activity, the Group may have to take additional measures that will have a negative impact on the Groups business continuity and may experience certain liquidity restraints as well as incur impairments on its assets. The exact impact on the Group's activities in 2020 and thereafter cannot be reasonably predicted. However, based on the risk mitigation measures undertaken, the Group concluded that there is no material uncertainty that may cast a significant doubt upon the Group's ability to continue as a going concern.
F-42
Certification by the Principal Executive Officer pursuant to
Securities Exchange Act Rules 13a-14(a) and 15d-14(a)
as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002
I, Tim Dyer, certify that:
1. I have reviewed this annual report on Form 20-F of Addex Therapeutics Ltd;
2. Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
3. Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the company as of, and for, the periods presented in this report;
4. The company’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) for the company and have:
(a) Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the company, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
(b) Evaluated the effectiveness of the company’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
(c) Disclosed in this report any change in the company’s internal control over financial reporting that occurred during the period covered by the annual report that has materially affected, or is reasonably likely to materially affect, the company’s internal control over financial reporting; and
5. The company’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the company’s auditors and the audit committee of the company’s board of directors (or persons performing the equivalent functions):
(a) All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the company’s ability to record, process, summarize and report financial information; and
(b) Any fraud, whether or not material, that involves management or other employees who have a significant role in the company’s internal control over financial reporting.
|
Date: April 24, 2020 |
| |
|
|
| |
|
/s/ Tim Dyer |
| |
|
Name: |
Tim Dyer |
|
|
Title: |
Chief Executive Officer |
|
|
|
(Principal Executive Officer) |
|
Certification by the Principal Financial Officer pursuant to
Securities Exchange Act Rules 13a-14(a) and 15d-14(a)
as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002
I, Lénaïc Teyssédou, certify that:
1. I have reviewed this annual report on Form 20-F of Addex Therapeutics Ltd;
2. Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
3. Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the company as of, and for, the periods presented in this report;
4. The company’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) for the company and have:
(a) Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the company, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
(b) Evaluated the effectiveness of the company’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
(c) Disclosed in this report any change in the company’s internal control over financial reporting that occurred during the period covered by the annual report that has materially affected, or is reasonably likely to materially affect, the company’s internal control over financial reporting; and
5. The company’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the company’s auditors and the audit committee of the company’s board of directors (or persons performing the equivalent functions):
(a) All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the company’s ability to record, process, summarize and report financial information; and
(b) Any fraud, whether or not material, that involves management or other employees who have a significant role in the company’s internal control over financial reporting.
|
Date: April 24, 2020 |
| |
|
|
| |
|
/s/ Lénaïc Teyssédou |
| |
|
Name: |
Lénaïc Teyssédou |
|
|
Title: |
Head of Finance |
|
|
|
(Principal Financial Officer) |
|
Certification by the Principal Executive Officer pursuant to
18 U.S.C. Section 1350, as adopted pursuant to
Section 906 of the Sarbanes-Oxley Act of 2002
In connection with the Annual Report of Addex Therapeutics Ltd (the “Company”) on Form 20-F for the fiscal year ended December 31, 2019 as filed with the Securities and Exchange Commission on the date hereof (the “Report”), I, Tim Dyer, Chief Executive Officer of the Company, certify, pursuant to 18 U.S.C. § 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, that to my knowledge:
(1) The Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended; and
(2) The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
|
Date: April 24, 2020 |
| |
|
|
| |
|
/s/ Tim Dyer |
| |
|
Name: |
Tim Dyer |
|
|
Title: |
Chief Executive Officer |
|
|
|
(Principal Executive Officer) |
|
Certification by the Principal Financial Officer pursuant to
18 U.S.C. Section 1350, as adopted pursuant to
Section 906 of the Sarbanes-Oxley Act of 2002
In connection with the Annual Report of Addex Therapeutics Ltd (the “Company”) on Form 20-F for the fiscal year ended December 31, 2019 as filed with the Securities and Exchange Commission on the date hereof (the “Report”), I, Lénaïc Teyssédou, Head of Finance of the Company, certify, pursuant to 18 U.S.C. § 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, that to my knowledge:
(1) The Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended; and
(2) The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
|
Date: April, 24, 2020 |
| |
|
|
| |
|
/s/ Lénaïc Teyssédou |
| |
|
Name: |
Lénaïc Teyssédou |
|
|
Title: |
Head of Finance |
|
|
|
(Principal Financial Officer) |
|
Serious News for Serious Traders! Try StreetInsider.com Premium Free!
You May Also Be Interested In
- Celebrate Motherhood in Style: JTV® Shares Top Gifts for Mother's Day
- InternationalReserve Offers Attractive Investment Alternatives as Inflation Hedge
- National Council on Aging Hosts Age+Action Conference May 6-8, 2024
Create E-mail Alert Related Categories
SEC FilingsSign up for StreetInsider Free!
Receive full access to all new and archived articles, unlimited portfolio tracking, e-mail alerts, custom newswires and RSS feeds - and more!
