

Form S-1/A Jaguar Health, Inc.
 Tweet
Tweet Share
Share
Use these links to rapidly review the document
TABLE OF CONTENTS
As filed with the Securities and Exchange Commission on July 5, 2019.
Registration No. 333-231399
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
Amendment No. 1
to
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
JAGUAR HEALTH, INC.
(Exact name of registrant as specified in its charter)
| Delaware (State or other jurisdiction of incorporation or organization) |
2834 (Primary Standard Industrial Classification Code Number) |
46-2956775 (I.R.S. Employer Identification Number) |
201 Mission Street, Suite 2375
San Francisco, California 94105
(415) 371-8300
(Address, including zip code, and telephone number, including
area code, of registrant's principal executive office)
Lisa A. Conte
Chief Executive Officer and President
Jaguar Health, Inc.
201 Mission Street, Suite 2375
San Francisco, California 94105
(415) 371-8300
(Name, address, including zip code, and telephone number, including area code, of agent for service)
| Copies to: | ||
Donald C. Reinke, Esq. Reed Smith LLP 101 Second Street, Suite 1800 San Francisco, California 94105 (415) 543-8700 |
Richard A. Friedman, Esq. Stephen A. Cohen, Esq. Sheppard, Mullin, Richter & Hampton LLP 30 Rockefeller Plaza New York, NY 10112 (212) 653-8700 |
|
Approximate date of commencement of proposed sale to the public:
As soon as practicable after this registration statement is declared effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933 check the following box: ý
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of "large accelerated filer," "accelerated filer," "smaller reporting company," and "emerging growth company" in Rule 12b-2 of the Exchange Act.
| Large accelerated filer o | Accelerated filer o | Non-accelerated filer o |
Smaller reporting company ý Emerging growth company ý |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act. ý
CALCULATION OF REGISTRATION FEE
|
||||
| Title of Each Class of Securities to be Registered(1) |
Proposed Maximum Aggregate Offering Price(1) |
Amount of Registration Fee |
||
|---|---|---|---|---|
Class A Units consisting of: |
$12,000,000(4) | $1,454.40 | ||
(i) Shares of common stock, par value $0.0001 per share(2) |
||||
(ii) Warrants to purchase common stock(2) |
||||
Class B Units consisting of: |
$12,000,000(4) | $1,454.40 | ||
(i) Series B Convertible Preferred Stock, par value $0.0001 per share |
||||
(ii) Common stock issuable upon conversion of Series B Preferred Stock(2)(3) |
||||
(iii) Warrants to purchase common stock(2) |
||||
Common Stock issuable upon exercise of warrants(2) |
$60,000,000 | $7,272.00 | ||
Total |
$84,000,000 | $10,180.80(5) | ||
|
||||
- (1)
- Estimated
solely for the purpose of calculating the amount of the registration fee pursuant to Rule 457(o) of the Securities Act of 1933, as amended. Includes
the offering price of any additional securities that the underwriter has the option to purchase.
- (2)
- Pursuant
to Rule 416, the securities being registered hereunder include such indeterminate number of additional securities as may be issued after the date
hereof as a result of stock splits, stock dividends or similar transactions.
- (3)
- No
fee pursuant to Rule 457(i) under the Securities Act of 1933, as amended.
- (4)
- The
Registrant is issuing Class A Units and/or Class B Units up to $12 million in the aggregate.
- (5)
- The Registrant previously paid $1,212 in connection with the initial filing of this Registration Statement on May 10, 2019.
The Registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the registration statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
The information in this prospectus is not complete and may be changed. We may not sell these securities until the registration statement relating to these securities filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED JULY 5, 2019
PRELIMINARY PROSPECTUS

JAGUAR HEALTH, INC.
2,521,008 Class A Units, consisting of Common Stock and Warrants or
12,000 Class B Units consisting of Series B Convertible Preferred Stock and Warrants
(and 2,521,008 shares of common stock underlying shares of Series B Convertible Preferred Stock and 5,042,016 shares of Common Stock underlying Warrants)
We are offering 2,521,008 Class A Units, with each Class A Unit consisting of (i) one share of common stock, par value $0.0001 per share, (ii) one warrant to purchase one share of common stock that expires on the earlier of (a) 12 months from the date of issuance and (b) 30 calendar days following the public announcement of Positive Interim Results (as defined on page 63) related to the diarrhea results from the HALT-D Investigator Initiated Trial (referred to as the "Series 1 warrants"), and (iii) one warrant to purchase one share of our common stock that expires on the first date on the earlier of (a) 5 years from the date of issuance and (b) 30 calendar days following the public announcement by the Company that a pivotal trial using crofelemer (Mytesi, or the same or similar product with a different name) for the treatment of cancer therapy related diarrhea has met its primary endpoint in accordance with the protocol (referred to as the "Series 2 warrants" and, together with the Series 1 warrant and the shares of common stock underlying the Series 1 warrants and Series 2 warrants, the "Class A Units") at an assumed public offering price of $4.76 per Class A Unit, which was the closing price of our common stock on June 27, 2019. The Series 1 warrants have an exercise price that is 125% of the assumed public offering price of the Class A Units. The Series 2 warrants have an exercise price that is 125% of the assumed offering price of the Class A Units.
We are also offering to those purchasers whose purchase of common stock in this offering would result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock following the consummation of this offering, the opportunity to purchase, if they so choose, in lieu of the shares of common stock that would result in ownership in excess of 4.99% (or, at the election of the purchaser, 9.99%), 12,000 Class B Units. Each Class B Unit will consist of one share of Series B Convertible Preferred Stock, with a stated value of $1,000 (the "Series B Preferred Stock") and convertible into shares of common stock at the public offering price of the Class A Units, together with the equivalent number of warrants as would have been issued to such purchaser of Class B Units if they had purchased Class A Units (together with the shares of common stock underlying such warrants, the "Class B Units" and, together with the Class A Units, the "Units") at an assumed public offering price of $1,000 per Class B Unit. For each Class B Unit we sell, the number of Class A Units we are offering will be decreased by $1,000 divided by the Class A Unit offering price. Because we will issue warrants as part of each Class A Unit or Class B Unit, the number of warrants sold in this offering will not change as a result of a change in the mix of the Class A Units and Class B Units sold.
The Class A Units and Class B Units have no stand-alone rights and will not be certificated or issued as stand-alone securities and the shares of common stock, Series B Preferred Stock and warrants comprising such Units are immediately separable and will be issued separately in this offering.
The underwriters have the option to purchase additional shares of common stock and/or warrants to purchase shares of common stock solely to cover over- allotments, if any, at the price to the public less the underwriting discounts and commissions. The over-allotment option may be used to purchase shares of common stock, or warrants, or any combination thereof, as determined by the underwriters, but such purchases cannot exceed an aggregate of 15% of the number of shares of common stock (including the number of shares of common stock issuable upon conversion of shares of Series B Preferred Stock) and warrants sold in the primary offering. The over-allotment option is exercisable for 45 days from the date of this prospectus.
Our Common Stock is listed on the Nasdaq Capital Market under the symbol "JAGX." On June 27, 2019, the last reported sale price of our common stock on the Nasdaq Capital Market was $4.76 per share. Certain investors, including certain of our officers, directors and existing investors and their affiliated entities, have agreed to purchase an aggregate of approximately $3.55 million of securities in this offering at the public offering price and on the same terms as the other purchasers in this offering. The underwriters will receive the same underwriting discounts and commissions on any securities purchased by these entities as they will on any other securities sold to the public in this offering.
Investing in our Common Stock involves a high degree of risk. Before deciding whether to invest in our securities, you should consider carefully the risks that we have described on page 12 of this prospectus under the caption "Risk Factors" and in the documents incorporated by reference into this prospectus and in any amendments or supplements to this prospectus.
|
||||||
| |
Per Class A Unit |
Per Class B Unit |
Total |
|||
|---|---|---|---|---|---|---|
Public offering price |
$ | $ | $ | |||
Underwriting discounts and commissions(1)(2) |
$ | $ | $ | |||
Proceeds, before expenses, to us |
$ | $ | $ | |||
|
||||||
- (1)
- See
"Underwriting" beginning on page 132 of this prospectus for a description of compensation payable to the underwriter.
- (2)
- We have granted a 45-day option to the underwriters to purchase additional shares of common stock and/or warrants to purchase shares of common stock (up to 15% of the number of shares of common stock and warrants sold in the primary offering) solely to cover over-allotments, if any.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The underwriter expects to deliver the shares and warrants against payment therefor on or about , 2019.
Sole Book-Running Manager
Ladenburg Thalmann
, 2019
We have not, and the underwriter has not, authorized anyone to provide any information or to make any representations other than those contained in this prospectus or in any free writing prospectus prepared by or on behalf of us or to which we have referred you. We take no responsibility for, and can provide no assurance as to the reliability of, any other information that others may give you. This prospectus is an offer to sell only the shares offered hereby, but only under the circumstances and in the jurisdictions where it is lawful to do so. The information contained in this prospectus or in any applicable free writing prospectus is current only as of its date, regardless of its time of delivery or any sale of shares of our common stock. Our business, financial condition, results of operations and prospects may have changed since that date. We are not, and the underwriter is not, making an offer of these securities in any jurisdiction where such offer is not permitted.
For investors outside the United States: we have not, and the underwriter has not, done anything that would permit this offering or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the United States. Persons outside the United States who come into possession of this prospectus must inform themselves about, and observe any restrictions relating to, the offering of securities and the distribution of this prospectus outside the United States.
Jaguar Health, our logo, Napo Pharmaceuticals, Mytesi, Canalevia, Equilevia and Neonorm are our trademarks that are used in this prospectus. This prospectus also includes trademarks, tradenames and service marks that are the property of other organizations. Solely for convenience, trademarks and tradenames referred to in this prospectus appear without the ©, ® or ™ symbols, but those references are not intended to indicate that we will not assert, to the fullest extent under applicable law, our rights or that the applicable owner will not assert its rights, to these trademarks and tradenames.
2
This summary highlights information contained elsewhere in this prospectus. This summary does not contain all of the information you should consider before investing in our common stock. You should read this entire prospectus carefully, especially the section in this prospectus titled "Risk Factors" appearing elsewhere in this prospectus the information incorporated by reference herein, including our financial statements and notes in our annual report on Form 10-K for 2018 and quarterly reports on Form 10-Q, before making an investment decision.
As used in this prospectus, references to "Jaguar," "we," "us" or "our" refer to Jaguar Health, Inc.
Overview
We are a commercial stage pharmaceutical company focused on developing and commercializing novel, sustainably derived gastrointestinal products on a global basis. Our wholly-owned subsidiary, Napo Pharmaceuticals, Inc. ("Napo"), focuses on developing and commercializing proprietary human gastrointestinal pharmaceuticals for the global marketplace from plants used traditionally in rainforest areas. Our Mytesi (crofelemer) product is a first-in-class anti-secretory agent, approved by the U.S. Food and Drug Administration ("FDA") for the symptomatic relief of noninfectious diarrhea in adults with HIV/AIDS on antiretroviral therapy.
Jaguar was founded in San Francisco, California as a Delaware corporation on June 6, 2013 to develop and commercialize animal health products. Effective as of December 31, 2013, Jaguar was a wholly-owned subsidiary of Napo, and, until May 13, 2015, Jaguar was a majority-owned subsidiary of Napo. On July 31, 2017, the merger of Jaguar Animal Health, Inc. and Napo became effective, at which point Jaguar Animal Health's name changed to Jaguar Health, Inc. and Napo began operating as a wholly-owned subsidiary of Jaguar focused on human health and the ongoing commercialization of, and development of follow-on indications for Mytesi. Most of the activities of the Company are now focused on the commercialization of Mytesi and development of follow-on indications for crofelemer and a second-generation anti-secretory product, lechlemer. In the field of animal health, we have limited activities which are focused on developing and commercializing first-in-class gastrointestinal products for dogs, dairy calves, foals, and high value horses.
We believe Jaguar is poised to realize a number of synergistic, value adding benefits—an expanded pipeline of potential blockbuster human follow-on indications of crofelemer, and a second-generation anti-secretory agent—upon which to build global partnerships. As previously announced, Jaguar, through Napo, now holds extensive global rights for Mytesi, and crofelemer manufacturing may occur at two FDA-inspected and approved facilities, including a newly constructed, multimillion-dollar commercial manufacturing facility. Additionally, several of the drug product candidates in Jaguar's Mytesi pipeline are backed by strong Phase 2 and proof of concept evidence from completed human clinical trials.
Mytesi is a novel, first-in-class anti-secretory agent which has a basic normalizing effect locally on the gut, and this mechanism of action has the potential to benefit multiple gastrointestinal disorders. Mytesi is in development for multiple possible follow-on indications, including cancer therapy-related diarrhea (CTD); orphan-drug indications for infants and children with congenital diarrheal disorders (CDDs) and short bowel syndrome (SBS); supportive care for inflammatory bowel disease (IBD); irritable bowel syndrome (IBS); and for idiopathic/functional diarrhea. In addition, a second-generation proprietary anti-secretory agent, lechlemer, is in development for cholera. Mytesi has received orphan-drug designation for pediatric SBS.
3
Recent Developments
Nasdaq Compliance
On June 21, 2019, the Company received a letter from The Nasdaq Stock Market that the Company has regained compliance with Listing Rule 5450(a)(1), which requires the Company's common stock to maintain a minimum bid price of $1.00 per share. The notification letter confirmed that the minimum bid price deficiency had been cured and that the Company was in compliance with all applicable listing standards. Nasdaq had previously notified the Company of its non-compliance with Listing Rule 5450(a)(1) on November 14, 2018, following 30 consecutive business days for which the closing bid price of the Company's common stock did not meet the $1.00 per share minimum requirement.
Kingdon Debt Refinancing
On May 28, 2019, Jaguar entered into a guaranty and suretyship agreement, pursuant to which it guaranteed payment and performance of all obligations of Napo, arising under and in connection with approximately $10.5 million outstanding aggregate amount of convertible promissory notes issued by Napo pursuant to the Amended and Restated Note Purchase Agreement, dated March 31, 2017, by and between Napo, Kingdon Associates, M. Kingdon Offshore Master Fund L.P., Kingdon Family Partnership, L.P., and Kingdon Credit Mater Fund L.P. (collectively, the "Existing Notes"). The Existing Notes bear interest at a rate of 10% per annum and mature on December 31, 2019.
On May 28, 2019, Jaguar and Napo (collectively, the "Borrower") entered into an exchange agreement with Chicago Venture Partners, L.P. ("CVP"), the holder of the Existing Notes, pursuant to which CVP exchanged the Existing Notes for a secured promissory note in the original principal amount of $10,535,900.42 ("Exchange Note 1") and a secured promissory note in the original principal amount of $2,296,926.16 ("Exchange Note 2" and together with Exchange Note 1, the "Exchange Notes"). The Exchange Notes bear interest at the rate of 10% per annum and mature on December 31, 2020. The outstanding balance of Exchange Note 2 is equal to the exchange fee that Jaguar agreed to pay CVP in consideration of certain accommodations granted to Jaguar and Napo, including but not limited to the extension of the maturity dates of the Existing Notes and the legal and other fees incurred by CVP in connection with the effectuation of the transactions contemplated under the Exchange Agreement. Through June 27, 2019, Jaguar has issued 754,838 shares of Common Stock to CVP in exchange for a reduction of approximately $4.8 million of the Exchange Notes. The shares of Common Stock that were exchanged for portions of the Exchange Notes were issued in reliance on the exemption from registration provided under Section 3(a)(9) of the Securities Act.
CVP also entered into security agreements with Jaguar (the "Jaguar Security Agreement") and Napo (the "Napo Security Agreement", and together with the Jaguar Security Agreement, the "Security Agreements"), pursuant to which CVP will receive (i) a security interest in substantially all of the Company's assets as security for the Company's obligations under Exchange Note 2 and (ii) a security interest in substantially all of Napo's assets as security for Napo's obligations under Exchange Note 1 and Exchange Note 2. Notwithstanding the foregoing, (a) the amount owing under Exchange Note 2 will not be considered part of the obligations secured by the Napo Security Agreement until such time as Jaguar receives permission from a third party and (b) the security interest granted under the Jaguar Security Agreement will be automatically terminated and released upon Jaguar's receipt of a waiver from such third party.
Reverse Stock Split
On June 3, 2019, we filed an amendment to our Third Amended and Restated Certificate of Incorporation to effect on June 7, 2019, a 1-for-70 reverse split of our voting common stock.
4
Accordingly, all of the stock figures and related market, conversion and exercise prices in this prospectus have been adjusted to reflect the reverse split.
Notes and Warrants Issuance
On March 18, 2019, Jaguar began entering into securities purchase agreements (each, a "Securities Purchase Agreement") with selected accredited investors (the "Investors" and each, an "Investor") pursuant to which Jaguar will sell promissory notes ("Bridge Notes") to such Investors. The initial offering closed on March 18, 2019, and as of June 27, 2019, $5,050,000.00 aggregate principal amount of Bridge Notes were issued in offerings and the proceeds from such offerings were paid to the Company.
The Bridge Notes bear interest at the rate of 12% per annum and mature on July 31, 2019 (the "Maturity Date"). Each Bridge Note is subject to a right to purchase by Sagard Capital Partners, L.P. and its affiliates (collectively, "Sagard"), pursuant to which Sagard may elect, within 5 business days of providing notice thereof to the holder of such Bridge Note, to purchase all or any portion of such Bridge Note and all accrued interest thereon.
At the time of entering into a Securities Purchase Agreement, an Investor may elect to purchase either a Bridge Note that is subject to a mandatory exchange provision (each a "125% Coverage Note") or a Bridge Note that is not subject to a mandatory exchange provision but is otherwise substantially the same as the 125% Coverage Note (each, a "75% Coverage Note"). The mandatory exchange provision in the 125% Coverage Notes provides that, at the Company's option upon the consummation of an underwritten public offering by the Company on or before the Maturity Date (the "Public Offering") of the Company's common stock ("Common Stock"), the principal amount of the 125% Coverage Notes plus any unaccrued interest thereon will be mandatorily exchanged into shares of Common Stock (the "Exchange Shares") at a price equal to the per share price at which the Company issues Common Stock in the Public Offering (the "Exchange Price"), subject to adjustment for reorganization, recapitalization, non-cash dividend, stock split, reverse stock split or other similar transaction. Upon such exchange, the 125% Coverage Notes would be deemed repaid and terminated.
As an inducement to enter into a Securities Purchase Agreement, (i) each holder of 75% Coverage Notes will receive a 5-year warrant (the "75% Coverage Warrant") to purchase shares of Common Stock (the "Bridge Warrant Shares") in an amount equal to 75% of the principal amount of such holder's 75% Coverage Note divided by the Exercise Price (as defined below) and (ii) each holder of 125% Coverage Notes will receive a 5-year warrant (the "125% Coverage Warrant" and, together with the 75% Coverage Warrant, the "Bridge Warrants") to purchase Bridge Warrant Shares in an amount equal to 125% of the principal amount of such holder's 125% Coverage Note divided by the Exercise Price. The exercise price for the 75% Coverage Warrant and 125% Coverage Warrant is the price per share at which the Company issues Common Stock in the Public Offering, provided that if the Company has not consummated a Public Offering by the Maturity Date, then the exercise price will be equal to the closing sales price of the shares of Common Stock on the Maturity Date, in each case subject to adjustment for reclassification of the Common Stock, non-cash dividend, stock split, reverse stock split or other similar transaction (the "Exercise Price").
Amendment to the Series A Preferred Stock
On March 14, 2019, Jaguar, with the written consent of the sole holder of Jaguar's issued and outstanding Series A convertible participating preferred stock ("Series A Preferred Stock"), filed a Certificate of Amendment (the "Amendment") to the Certificate of Designation of Series A Convertible Participating Preferred Stock of Jaguar (as amended, the "Series A Certificate of Designation") with the Secretary of State of the State of Delaware to (a) adjust the conversion price of the shares of Series A Preferred Stock from $194.25 per share to $19.425 per share, provided that with
5
respect to the right to vote on an as-converted basis with holders of the Company's Common Stock, holders of Series A Preferred Stock will not be entitled to vote on any matter presented to the stockholders of the Company to the extent that such vote would be in violation of Nasdaq Listing Rule 5640, and (b) adjust the 30-day volume-weighted average price ("VWAP") threshold applicable to the Company's optional redemption right and the preferred stockholders' mandatory redemption right from $1050.00 to $105.00. The Amendment became effective upon filing with the Secretary of the State of Delaware.
CVP Note Exchanges
In January through June 2019, Jaguar entered into exchange agreements with Chicago Venture Partners L.P. ("CVP"), pursuant to which the Company issued 1,150,808 shares of Common Stock in the aggregate to CVP in exchange for a reduction of approximately $11.2 million in the principal amount of the CVP Notes and Exchange Notes. As of June 26, 2019, all of the CVP Notes have been eliminated and approximately $8.0 aggregate principal amount of the Exchange Notes remains outstanding. The shares of Common Stock that were exchanged for portions of the secured promissory notes were issued in reliance on the exemption from registration provided under Section 3(a)(9) of the Securities Act.
Risks Related to Our Business
Our business, and our ability to execute our business strategy, is subject to a number of risks as more fully described in the section titled "Risk Factors." These risks include, among others, the following:
- •
- We have a limited operating history, have not yet generated any material revenues, expect to continue to incur significant research and
development and other expenses, and may never become profitable. Our independent registered public accounting firm has expressed substantial doubt about our ability to continue as a going concern.
- •
- We have generated limited revenue from operations and may need to raise additional capital to achieve our goals.
- •
- We are substantially dependent on the success of our current lead prescription drug product candidate, Mytesi, and cannot be certain that
necessary approvals will be received for planned Mytesi follow-on indications or Canalevia or that these follow on indications will be successfully commercialized, either by us or any of our partners.
- •
- The results of earlier studies may not be predictive of the results of our pivotal trials or other future studies, and we may be unable to
obtain any necessary regulatory approvals for our existing or future prescription drug product candidates under applicable regulatory requirements.
- •
- Development of prescription drug products, and, to a lesser extent, non-prescription products, for the human health and animal health market is
inherently expensive, time-consuming and uncertain, and any delay or discontinuance of our current or future pivotal trials, or dosage or formulation studies, would harm our business and prospects.
- •
- Even if we obtain any required regulatory approvals for our current or future prescription drug product candidates, they may never achieve
market acceptance or commercial success.
- •
- We are dependent upon contract manufacturers for supplies of our current prescription drug product candidates and non-prescription products and intend to rely on contract manufacturers for commercial quantities of any of our commercialized products.
6
- •
- If we are not successful in identifying, developing and commercializing additional prescription drug product candidates and non-prescription
products, our ability to expand our business and achieve our strategic objectives may be impaired.
- •
- We have a material weakness in our internal control over financial reporting related to staff turnover in our accounting department. We did not maintain a sufficient complement of internal personnel with appropriate knowledge, experience and/or training commensurate with our financial reporting requirements. If we fail to remediate the material weakness, or experience any additional material weaknesses in the future or otherwise fail to maintain an effective system of internal controls in the future, we may not be able to accurately report our financial condition or results of operations which may adversely affect investor confidence in us and, as a result, the value of our common stock.
Corporate Information
We were incorporated in the State of Delaware on June 6, 2013. Our principal executive offices are located at 201 Mission Street, Suite 2375, San Francisco, CA 94015 and our telephone number is (415) 371-8300. Our website address is www.jaguar.health. The information contained on, or that can be accessed through, our website is not part of this prospectus. Our Common Stock is listed on the NASDAQ Capital Market and trades under the symbol "JAGX." On July 31, 2017, we completed the acquisition of Napo (the "Merger") pursuant to the Agreement and Plan of Merger, dated March 31, 2017, by and among the Company, Napo, Napo Acquisition Corporation, and Napo's representative (the "Merger Agreement").
Emerging Growth Company Information
We are an "emerging growth company," as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act, and we may take advantage of certain exemptions and relief from various reporting requirements that are applicable to other public companies that are not "emerging growth companies." In particular, while we are an "emerging growth company" (i) we will not be required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act, (ii) we will be subject to reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements and (iii) we will not be required to hold nonbinding advisory votes on executive compensation or stockholder approval of any golden parachute payments not previously approved. In addition, the JOBS Act provides that an emerging growth company can delay its adoption of any new or revised accounting standards, but we have irrevocably elected not to avail ourselves of this exemption and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies. In addition, investors may find our common stock less attractive if we rely on the exemptions and relief granted by the JOBS Act. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may decline and/or become more volatile.
We may remain an "emerging growth company" until as late as December 31, 2020 (the fiscal year-end following the fifth anniversary of the closing of our initial public offering, which occurred on May 18, 2015), although we may cease to be an "emerging growth company" earlier under certain circumstances, including (i) if the market value of our common stock that is held by non-affiliates exceeds $700.0 million as of any June 30, in which case we would cease to be an "emerging growth company" as of December 31 of such year, (ii) if our gross revenue exceeds $1.07 billion in any fiscal year or (iii) if we issue more than $1.0 billion of non-convertible debt over a three-year period.
7
Securities offered |
2,521,008 Class A Units, comprising of one share of our common stock, a Series 1 warrant to purchase one share of our common stock, and a Series 2 warrant to purchase one share of our common stock (together with the shares of common stock underlying such warrants). We are also offering 12,000 Class B Units, comprising of one share of Series B Preferred Stock, with a stated value of $1,000 and convertible into shares of common stock at the public offering price of the Class A Units, together with the equivalent number of warrants as would have been issued to such purchaser of Class B Units if they had purchased Class A Units (together with the shares of common stock underlying such shares of Series B Preferred Stock and such warrants) to those purchasers, if any, whose Purchase of Class A Units in this offering would otherwise result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% (or at the election of the purchaser, 9.99%) of our outstanding common stock immediately following the consummation of this offering. For each Class B Unit we sell, the number of Class A Units we are offering will be decreased by $1,000 divided by the Class A Unit offering price. Because we will issue warrants as part of each Class A Unit or Class B Unit, the number of warrants sold in this offering will not change as a result of a change in the mix of the Class A Units and Class B Units sold. | |
Assumed Public offering price per Class A Unit |
$4.76 per Class A Unit |
|
Assumed Public offering price per Class B Unit |
$1,000 per Class B Unit |
|
Over-allotment option |
The underwriters have the option to purchase up to (i) 378,151 additional shares of common stock and/or Series B Convertible Preferred Stock, (ii) additional Series 1 warrants to purchase up to 378,151 additional shares of common stock and/or (iii) additional Series 2 warrants to purchase up to 378,151 additional shares of common stock solely to cover over-allotments, if any, at the price to the public less the underwriting discounts and commissions. The over- allotment option may be used to purchase shares of common stock, Series B Convertible Preferred Stock or warrants, or any combination thereof, as determined by the underwriters, but such purchases cannot exceed an aggregate of 15% of the number of shares of common stock (including the number of shares of common stock issuable upon conversion of shares of Series B Preferred Stock) and warrants sold in the primary offering. The over-allotment option is exercisable for 45 days from the date of this prospectus. |
8
Series 1 Warrant |
The Series 1 warrants will be exercisable beginning on the date of issuance and expire on the earlier of (1) 12 months from the date of issuance and (2) 30 calendar days following the public announcement of Positive Interim Results (as defined on page 63) related to the diarrhea results from the HALT-D Investigator Initiated Trial, at an initial exercise price per share that is equal to 125% of the public offering price of the Class A Units, subject to appropriate adjustment in the event of recapitalization events, stock dividends, stock splits, stock combinations, reclassifications, reorganizations or similar events affecting our common stock. |
|
Series 2 Warrant |
The Series 2 warrants will be exercisable beginning on the date of issuance and expire on the earlier of (1) 5 years from the date of issuance and (2) 30 calendar days following the public announcement by the Company that a pivotal trial using crofelemer (Mytesi, or the same or similar product with a different name) for the treatment of cancer therapy related diarrhea has met its primary endpoint in accordance with the protocol, at an initial exercise price per share that is equal to 125% of the public offering price of the Class A Units, subject to appropriate adjustment in the event of recapitalization events, stock dividends, stock splits, stock combinations, reclassifications, reorganizations or similar events affecting our common stock. |
|
|
The Series 2 warrants are callable by us in certain circumstances. Subject to certain exceptions, in the event that the Series 2 warrants are outstanding, following the date that is 180 days after the closing date, (i) the volume weighted average price of our common stock for each of 30 consecutive trading days (the "Measurement Period"), which Measurement Period commences after the date that is 180 days after the closing date, exceeds 300% of the initial exercise price (subject to adjustment for forward and reverse stock splits, recapitalizations, stock dividends and similar transactions), (ii) the average daily trading volume for such Measurement Period exceeds $500,000 per trading day and (iii) the holder is not in possession of any information that constitutes or might constitute, material non-public information which was provided by the Company, and subject to the beneficial ownership limitation for the warrants described further under "Description of Securities We are Offering—Warrants—Exercise of Warrants," then we may, within one trading day of the end of such Measurement Period, upon notice (a "Call Notice"), call for cancellation of all or any portion of the Series 2 warrants for which a notice of exercise has not yet been delivered (a "Call") for consideration equal to $0.0001 per share. Any portion of a Series 2 warrant subject to such Call Notice for which a notice of exercise shall not have been received by the Call Date (as hereinafter defined) will be canceled at 6:30 p.m. (New York City time) on the tenth trading day after the date the Call Notice is received by the Holder (such date and time, the "Call Date"). Our right to call the Series 2 warrants shall be exercised ratably among the holders based on the outstanding Series 2 warrants. |
9
|
The Series 1 warrants and Series 2 warrants are collectively referred to herein as the "warrants" The forms of warrant are filed as an exhibit to the registration statement of which this prospectus forms a part. |
|
Series B Preferred Stock |
Each share of Series B Preferred Stock is convertible at any time at the holder's option into one share of common stock. Notwithstanding the foregoing, we shall not effect any conversion of Series B Preferred Stock, with certain exceptions, to the extent that, after giving effect to an attempted conversion, the holder of shares of Series B Preferred Stock (together with such holder's affiliates, and any persons acting as a group together with such holder or any of such holder's affiliates) would beneficially own a number of shares of our common stock in excess of 4.99% (or, at the election of the purchaser prior to the date of issuance, 9.99%) of the shares of our common stock then outstanding after giving effect to such conversion. For additional information, see "Description of Securities We Are Offering—Series B Preferred Stock" for a discussion of the terms of the Series B Preferred Stock. |
|
Common stock outstanding before this offering as of July 3, 2019 |
1,837,763 |
|
Common stock to be outstanding immediately after this offering |
9,400,772 |
|
No listing of Series B Preferred Stock or warrants |
There is no established public trading market for the warrants or Series B Preferred Stock, and we do not expect an active trading market to develop. We do not intend to list the warrants or the Series B Preferred Stock on any securities exchange or other trading market. Without an active trading market, the liquidity of the warrants and the Series B Preferred Stock will be limited. |
|
Registered Securities |
This prospectus also relates to the offering of the shares issuable upon conversion of the Series B Preferred Stock and upon exercise of the warrants included in the Units. |
|
Use of Proceeds |
We estimate that the net proceeds to us from this offering will be approximately $10.2 million, based on an assumed offering price of $4.76 for each Class A Unit and $1,000 for each Class B Unit, after deducting the underwriting discounts and commissions and estimated offering expenses payable by us. We intend to use approximately $5.2 million of the net proceeds from this offering for debt repayment (including repayment of $1,047,718 to certain officers, directors and their respective affiliates) and the remainder for general corporate purposes, including working capital and to pay a certain investor as consideration for their consent and waiver in refinancing an outstanding loan. See "Use of Proceeds" for additional information. |
10
Risk factors |
You should read the "Risk Factors" section of this prospectus and in the documents incorporated by reference in this prospectus for a discussion of factors to consider before deciding to invest in our Common Stock. |
|
NASDAQ Capital Market symbol |
"JAGX" |
We have two classes of common stock: (i) voting common stock, par value $0.0001 per share, and (ii) non-voting common stock, par value $0.0001 per share. The shares offered by us in this offering are voting common stock.
The number of shares of our common stock to be outstanding after this offering is based on 1,799,381 shares of our voting common stock and 40,301,237 shares of our non-voting common stock (which non-voting shares are convertible into 38,382 shares of voting common stock) outstanding as of July 3, 2019, and excludes the following:
- •
- 1,168,592 shares of common stock issuable upon exercise of the Bridge Warrants issuable pursuant to the Securities Purchase Agreement entered
into beginning on March 18, 2019 by and among the Company and selected accredited investors assuming an exercise price equal to the assumed public offering price per Class A Unit of
$4.76;
- •
- 473,565 shares of common stock issuable upon conversion of outstanding preferred stock as of July 3, 2019, with a weighted-average
conversion price of $19.425 per share;
- •
- 39,307 shares of voting common stock issuable upon exercise of outstanding options as of July 3, 2019, with a weighted average exercise
price of $412.94 per share;
- •
- 2,984 shares of common stock issuable upon exercise of outstanding inducement options as of March 31, 2019 with a weighted-average
exercise price of $122.49 per share;
- •
- 11,558 shares of voting common stock reserved for future issuance under the 2014 Stock Incentive Plan, which includes shares of common stock
that will be issuable upon exercise of options that we expect to grant on the closing date of this offering;
- •
- 71,821 shares of voting common stock issuable upon exercise of warrants outstanding as of July 3, 2019, with a weighted average exercise
price of $90.00 per share;
- •
- 5,613 shares of voting common stock issuable upon vesting of outstanding restricted stock unit awards, or RSUs, as of July 3, 2019; and
11
Investing in our common stock involves a high degree of risk. You should carefully consider the risks described below, as well as the other information contained in or incorporated by reference in this prospectus, including our financial statements and the related notes and "Management's Discussion and Analysis of Financial Condition and Results of Operations" in our Annual Report on Form 10-K for the fiscal year ended December 31, 2018, as updated in our Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2019, before deciding whether to invest in our Common Stock. The occurrence of any of the events or developments described below could harm our business, financial condition, results of operations and prospects. In such an event, the market price of our Common Stock could decline, and you may lose all or part of your investment. Additional risks and uncertainties not presently known to us or that we currently deem immaterial also may harm our business, financial condition, results of operations and prospects.
Risks Related to Our Business
We have a limited operating history, expect to incur further losses as we grow and may be unable to achieve or sustain profitability. Our independent registered public accounting firm has expressed substantial doubt about our ability to continue as a going concern.
Since formation in June 2013, our operations have been primarily limited to the research and development of our animal prescription drug product candidate, Canalevia, to treat various forms of diarrhea in dogs, our non-prescription product, Neonorm Calf, to help dairies and calf farms proactively retain fluid in calves, the ongoing commercialization of Neonorm Foal, our antidiarrheal for newborn horses, and Equilevia, our non-prescription, personalized, premium product for total gut health in high-performance equine athletes. Since the consummation of the Merger on July 31, 2017, our operations have also been heavily focused on research, development and the ongoing commercialization of our lead prescription drug product candidate, Mytesi, which is approved by the U.S. FDA for the symptomatic relief of noninfectious diarrhea in adults with HIV/AIDS on antiretroviral therapy. As a result, we have limited meaningful historical operations upon which to evaluate our business and prospects and have not yet demonstrated an ability to broadly commercialize any of our animal health products, obtain any required marketing approval for any of our animal prescription drug product candidates or successfully overcome the risks and uncertainties frequently encountered by companies in emerging fields such as the animal health industry or the gastrointestinal health industry in general. We also have not generated any material revenue to date, and expect to continue to incur significant research and development and other expenses. Our net loss and comprehensive loss for the year ended December 31, 2018 was $32.1 million. As of December 31, 2018, we had total stockholders' equity of $5.4 million. We expect to continue to incur losses for the foreseeable future, which will increase significantly from historical levels as we expand our product development activities, seek necessary approvals for our human and veterinary drug product candidates, conduct species-specific formulation studies for our non-prescription products and increase commercialization activities. Even if we succeed in developing and broadly commercializing one or more of our products or product candidates, we expect to continue to incur losses for the foreseeable future, and we may never become profitable. If we fail to achieve or maintain profitability, then we may be unable to continue our operations at planned levels and be forced to reduce or cease operations.
As more fully discussed in Note 1 to our financial statements, we believe there is substantial doubt about our ability to continue as a going concern as we do not currently have sufficient cash resources to fund our operations through March 31, 2020, or one year from the filing date of our Form 10-K. Our financial statements do not include any adjustments that may result from the outcome of this uncertainty. If we are unable to continue as a viable entity, our stockholders may lose their entire investment.
12
We currently generate limited revenue from the sale of products and may never become profitable.
We are a pharmaceuticals company focused on the development and commercialization of novel, sustainably derived gastrointestinal products for human prescription use on a global basis. Napo, our wholly-owned subsidiary, began the commercial pre-launch activities of our first FDA-approved product, Mytesi, in February 2017. Accordingly, we have only generated limited revenue from product sales. In order to commercialize our other prescription drug product candidates, we must receive regulatory approval from the FDA in the United States and other regulatory agencies in various jurisdictions. Other than Mytesi, we have not yet received any regulatory approvals for our prescription drug product candidates. Accordingly, until and unless we receive any necessary regulatory approvals, we cannot market or sell our products in many regions. Moreover, even if we receive the necessary approvals, we may not be successful in generating revenue from sales of our products as we do not have any meaningful experience marketing or distributing our products. Accordingly, we may never generate any material revenue from our operations.
We expect to incur significant additional costs as we continue commercialization efforts for current prescription drug candidates or other product candidates, and undertake the clinical trials necessary to obtain any necessary regulatory approvals, which will increase our losses.
Napo commenced sales of Mytesi for adults with HIV/AIDS on antiretroviral therapy in September 2016. We will need to continue to invest in developing our internal and third-party sales and distribution network and outreach efforts to key opinion leaders in the gastrointestinal health industry, including physicians as applicable.
We are actively identifying additional products for development and commercialization, and will continue to expend substantial resources for the foreseeable future to develop Mytesi and lechlemer. These expenditures will include costs associated with:
- •
- identifying additional potential prescription drug product candidates and non-prescription products;
- •
- formulation studies;
- •
- conducting pilot, pivotal and toxicology studies;
- •
- completing other research and development activities;
- •
- payments to technology licensors;
- •
- maintaining our intellectual property;
- •
- obtaining necessary regulatory approvals;
- •
- establishing commercial supply capabilities; and
- •
- sales, marketing and distribution of our commercialized products.
We also may incur unanticipated costs in connection with developing and commercializing our products. Because the outcome of our development activities and commercialization efforts is inherently uncertain, the actual amounts necessary to successfully complete the development and commercialization of our current or future products and product candidates may be greater than we anticipate.
Because we anticipate incurring significant costs for the foreseeable future, if we are not successful in broadly commercializing any of our current or future products or product candidates or raising additional funding to pursue our research and development efforts, we may never realize the benefit of our development efforts and our business may be harmed.
13
We will need to raise substantial additional capital in the future in the event that we conduct clinical trials for new indications and we may be unable to raise such funds when needed and on acceptable terms, which would force us to delay, limit, reduce or terminate one or more of our product development programs.
We are forecasting continued losses and negative cash flows as we continue to fund our operating and marketing activities and research and development programs, and we will not have sufficient cash on hand to fund our operating plan through July 31, 2019 and to complete the development of all the current products in our pipeline, or any additional products we may identify. We will need to seek additional funds sooner than planned through public or private equity or debt financings or other sources such as strategic collaborations. Any such financings or collaborations may result in dilution to our stockholders, the imposition of debt covenants and repayment obligations or other restrictions that may harm our business or the value of our common stock. We may also seek from time to time to raise additional capital based upon favorable market conditions or strategic considerations such as potential acquisitions or potential license arrangements.
Our future capital requirements depend on many factors, including, but not limited to:
- •
- the scope, progress, results and costs of researching and developing our current and future prescription drug product candidates and
non-prescription products;
- •
- the timing of, and the costs involved in, obtaining any regulatory approvals for our current and any future products;
- •
- the number and characteristics of the products we pursue;
- •
- the cost of manufacturing our current and future products and any products we successfully commercialize;
- •
- the cost of commercialization activities for Mytesi and Canalevia, if approved, including sales, marketing and distribution costs;
- •
- the expenses needed to attract and retain skilled personnel;
- •
- the costs associated with being a public company;
- •
- our ability to establish and maintain strategic collaborations, distribution or other arrangements and the financial terms of such agreements;
and
- •
- the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing possible patent claims, including litigation costs and the outcome of any such litigation.
Additional funds may not be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available to us on a timely basis, we may be required to delay, limit, reduce or terminate one or more of our product development programs or future commercialization efforts.
We are substantially dependent on the success of our current lead prescription drug product candidate, Mytesi, and cannot be certain that necessary approvals will be received for planned Mytesi follow-on indications or that these product candidates will be successfully commercialized, either by us or any of our partners.
Other than Mytesi, we currently do not have regulatory approval for any of our prescription drug product candidates. Our current efforts are primarily focused on the ongoing commercialization of Mytesi, and development efforts related to Mytesi. With regard to Mytesi, we are focused on the commercial launch of the product in the United States as well as on development efforts related to a follow-on indication for Mytesi in CTD, an important supportive care indication for patients undergoing primary or adjuvant chemotherapy for cancer treatment. Mytesi is in development for multiple possible follow-on indications, including diarrhea related to targeted cancer therapy;
14
orphan-drug indications for infants and children with congenital diarrheal disorders and short bowel syndrome (SBS); supportive care for inflammatory bowel disease (IBD); irritable bowel syndrome (IBS); and for idiopathic/functional diarrhea. In addition, a second-generation proprietary anti-secretory agent is in development for cholera. Mytesi has received orphan-drug designation for SBS. Accordingly, our near term prospects, including our ability to generate material product revenue, obtain any new financing if needed to fund our business and operations or enter into potential strategic transactions, will depend heavily on the success of Mytesi.
Substantial time and capital resources have been previously devoted by third parties in the development of crofelemer, the active pharmaceutical ingredient, or API, in Mytesi and Canalevia, and the development of the botanical extract used in Equilevia and Neonorm. Both crofelemer and the botanical extract used in Equilevia and Neonorm were originally developed at Shaman Pharmaceuticals, Inc. ("Shaman"), by certain members of our management team, including Lisa A. Conte, our chief executive officer and president, and Steven R. King, Ph.D., our executive vice president of sustainable supply, ethnobotanical research and intellectual property and secretary. Shaman spent significant development resources before voluntarily filing for bankruptcy in 2001 pursuant to Chapter 11 of the U.S. Bankruptcy Code. The rights to crofelemer and the botanical extract used in Equilevia and Neonorm, as well as other intellectual property rights, were subsequently acquired by Napo from Shaman in 2001 pursuant to a court approved sale of assets. Ms. Conte founded Napo in 2001 and was the current interim chief executive officer of Napo and a member of Napo's board of directors prior to the Merger. While at Napo, certain members of our management team, including Ms. Conte and Dr. King, continued the development of crofelemer. In 2005, Napo entered into license agreements with Glenmark and Luye Pharma Group Limited for rights to various human indications of crofelemer in certain territories as defined in the respective license agreements with these licensees. Subsequently, after expending significant sums developing crofelemer, including trial design and on-going patient enrollment in the final pivotal Phase 3 trial for crofelemer for non-infectious diarrhea in adults with HIV/AIDS on antiretroviral therapy, in late 2008, Napo entered into a collaboration agreement with Salix Pharmaceuticals, Inc., or Salix, for development and commercialization rights to certain indications worldwide and certain rights in North America, Europe, and Japan, to crofelemer for human use. In January 2014, Jaguar entered into the Napo License Agreement pursuant to which Jaguar acquired an exclusive worldwide license to Napo's intellectual property rights and technology, including crofelemer and the botanical extract used in Equilevia and Neonorm, for all veterinary treatment uses and indications for all species of animals. In February 2014, most of the executive officers of Napo, and substantially all Napo's employees, became Jaguar's employees. Following the merger of Jaguar and Napo in July 2017, Napo became Jaguar's wholly-owned subsidiary. If we are not successful in the development and commercialization of Mytesi, our business and our prospects will be harmed.
The successful development and commercialization of Mytesi will depend on a number of factors, including the following:
- •
- our ability to demonstrate to the satisfaction of the FDA and any other regulatory bodies, the safety and efficacy of Canalevia;
- •
- our ability and that of our contract manufacturers to manufacture supplies of Mytesi and to develop, validate and maintain viable commercial
manufacturing processes that are compliant with current good manufacturing practices, or cGMP, if required;
- •
- our ability to successfully launch Mytesi, whether alone or in collaboration with others;
- •
- the availability, perceived advantages, relative cost, relative safety and relative efficacy of our prescription drug product candidates compared to alternative and competing treatments;
15
- •
- the acceptance of our prescription drug product candidates and non-prescription products as safe and effective by physicians, veterinarians,
patients, animal owners and the human and animal health community, as applicable;
- •
- our ability to achieve and maintain compliance with all regulatory requirements applicable to our business; and
- •
- our ability to obtain and enforce our intellectual property rights and obtain marketing exclusivity for our prescription drug product candidates and non-prescription products, and avoid or prevail in any third-party patent interference, patent infringement claims or administrative patent proceedings initiated by third parties or the U.S. Patent and Trademark Office ("USPTO").
Many of these factors are beyond our control. Accordingly, we may not be successful in developing or commercializing Mytesi, Neonorm, Equilevia, Canalevia or any of our other potential products. If we are unsuccessful or are significantly delayed in commercializing Mytesi, our business and prospects will be harmed and you may lose all or a portion of the value of your investment in our common stock.
If we are not successful in identifying, licensing, developing and commercializing additional product candidates and products, our ability to expand our business and achieve our strategic objectives could be impaired.
Although a substantial amount of our efforts is focused on the commercial performance of Mytesi, a key element of our strategy is to identify, develop and commercialize a portfolio of products to serve the gastrointestinal health market. Most of our potential products are based on our knowledge of medicinal plants. Our current focus is primarily on product candidates whose active pharmaceutical ingredient or botanical extract has been successfully commercialized or demonstrated to be safe and effective in human or animal trials. In some instances, we may be unable to further develop these potential products because of perceived regulatory and commercial risks. Even if we successfully identify potential products, we may still fail to yield products for development and commercialization for many reasons, including the following:
- •
- competitors may develop alternatives that render our potential products obsolete;
- •
- an outside party may develop a cure for any disease state that is the target indication for any of our planned or approved drug products;
- •
- potential products we seek to develop may be covered by third-party patents or other exclusive rights;
- •
- a potential product may on further study be shown to have harmful side effects or other characteristics that indicate it is unlikely to be
effective or otherwise does not meet applicable regulatory criteria;
- •
- a potential product may not be capable of being produced in commercial quantities at an acceptable cost, or at all; and
- •
- a potential product may not be accepted as safe and effective by physicians, veterinarians, patients, animal owners, key opinion leaders and other decision-makers in the gastrointestinal health market, as applicable.
While we are developing specific formulations, including flavors, methods of administration, new patents and other strategies with respect to our current potential products, we may be unable to prevent competitors from developing substantially similar products and bringing those products to market earlier than we can. If such competing products achieve regulatory approval and commercialization prior to our potential products, our competitive position may be impaired. If we fail to develop and successfully commercialize other potential products, our business and future prospects may be harmed and we will be more vulnerable to any problems that we encounter in developing and commercializing our current potential products.
16
Mytesi faces significant competition from other pharmaceutical companies, both for its currently approved indication and for planned follow-on indications, and our operating results will suffer if we fail to compete effectively.
The development and commercialization of products for human gastrointestinal health is highly competitive and our success depends on our ability to compete effectively with other products in the market. During the ongoing commercialization of Mytesi for its currently approved indication, and during the future commercialization of Mytesi for any planned follow-on indications, if such follow-on indications receive regulatory approval, we expect to compete with major pharmaceutical and biotechnology companies that operate in the gastrointestinal space, such as Takeda Pharmaceuticals, Allergan, Inc., Ironwood Pharmaceuticals, Inc., Synergy Pharmaceuticals Inc., Sebela Pharmaceuticals, Inc. and Salix Pharmaceuticals.
Many of our competitors and potential competitors in the human gastrointestinal space have substantially more financial, technical and human resources and greater ability to lower costs of manufacturing and sales and marketing than we do. Many also have more experience in the development, manufacture, regulation and worldwide commercialization of human gastrointestinal health products.
For these reasons, we cannot be certain that we and Mytesi can compete effectively.
We may be unable to obtain, or obtain on a timely basis, regulatory approval for our existing or future human or animal prescription drug product candidates under applicable regulatory requirements, which would harm our operating results.
The research, testing, manufacturing, labeling, approval, sale, marketing and distribution of human and animal health products are subject to extensive regulation. We are typically not permitted to market our prescription drug product candidates in the United States until we receive approval of the product from the FDA through the filing of an NDA or NADA, as applicable. To gain approval to market a prescription drug, we must provide the FDA with safety and efficacy data from pivotal trials that adequately demonstrate that our prescription drug product candidates are safe and effective for the intended indications. Likewise, to gain approval to market an animal prescription drug for a particular species, we must provide the FDA with safety and efficacy data from pivotal trials that adequately demonstrate that our prescription drug product candidates are safe and effective in the target species (e.g. dogs, cats or horses) for the intended indications. In addition, we must provide manufacturing data evidencing that we can produce our product candidates in accordance with cGMP. For the FDA, we must also provide data from toxicology studies, also called target animal safety studies, and in some cases environmental impact data. In addition to our internal activities, we will partially rely on contract research organizations ("CROs"), and other third parties to conduct our toxicology studies and for certain other product development activities. The results of toxicology studies, other initial development activities, and/or any previous studies in humans or animals conducted by us or third parties may not be predictive of future results of pivotal trials or other future studies, and failure can occur at any time during the conduct of pivotal trials and other development activities by us or our CROs. Our pivotal trials may fail to show the desired safety or efficacy of our prescription drug product candidates despite promising initial data or the results in previous human or animal studies conducted by others. Success of a prescription drug product candidate in prior animal studies, or in the treatment of humans, does not ensure success in subsequent studies. Clinical trials in humans and pivotal trials in animals sometimes fail to show a benefit even for drugs that are effective because of statistical limitations in the design of the trials or other statistical anomalies. Therefore, even if our studies and other development activities are completed as planned, the results may not be sufficient to obtain a required regulatory approval for a product candidate.
17
Regulatory authorities can delay, limit or deny approval of any of our prescription drug product candidates for many reasons, including:
- •
- if they disagree with our interpretation of data from our pivotal studies or other development efforts;
- •
- if we are unable to demonstrate to their satisfaction that our product candidate is safe and effective for the target indication and, if
applicable, in the target species;
- •
- if they require additional studies or change their approval policies or regulations;
- •
- if they do not approve of the formulation, labeling or the specifications of our current and future product candidates; and
- •
- if they fail to approve the manufacturing processes of our third-party contract manufacturers.
Further, even if we receive a required approval, such approval may be for a more limited indication than we originally requested, and the regulatory authority may not approve the labeling that we believe is necessary or desirable for successful commercialization.
Any delay or failure in obtaining any necessary regulatory approval for the intended indications of our human or animal product candidates would delay or prevent commercialization of such product candidates and would harm our business and our operating results.
The results of our earlier studies of Mytesi may not be predictive of the results in any future clinical trials and species-specific formulation studies, respectively, and we may not be successful in our efforts to develop or commercialize line extensions of Mytesi.
Our human and animal product pipeline includes a number of potential indications of Mytesi, our lead prescription product. The results of our studies and other development activities and of any previous studies in humans or animals conducted by us or third parties may not be predictive of future results of these clinical studies and formulation studies, respectively. Failure can occur at any time during the conduct of these trials and other development activities. Even if our formulation/clinical studies and other development activities are completed as planned, the results may not be sufficient to pursue a particular line extension for Mytesi. Further, even if we obtain promising results from our clinical trials or species-specific formulation studies, as applicable, we may not successfully commercialize any line extension. Because line extensions are developed for a particular market, we may not be able to leverage our experience from the commercial launch of Mytesi in new markets. If we are not successful in developing and successfully commercializing these line extension products, we may not be able to grow our revenue and our business may be harmed.
Development of prescription drug products is inherently expensive, time-consuming and uncertain, and any delay or discontinuance of our current or future pivotal trials would harm our business and prospects.
Development of prescription drug products for human and animal gastrointestinal health remains an inherently lengthy, expensive and uncertain process, and our development activities may not be successful. We do not know whether our current or planned pivotal trials for any of our product candidates will begin or conclude on time, and they may be delayed or discontinued for a variety of reasons, including if we are unable to:
- •
- address any safety concerns that arise during the course of the studies;
- •
- complete the studies due to deviations from the study protocols or the occurrence of adverse events;
- •
- add new study sites;
18
- •
- address any conflicts with new or existing laws or regulations; or
- •
- reach agreement on acceptable terms with study sites, which can be subject to extensive negotiation and may vary significantly among different sites.
Further, we may not be successful in developing new indications for Mytesi, and Neonorm may be subject to the same regulatory regime as prescription drug products in jurisdictions outside the United States. Any delays in completing our development efforts will increase our costs, delay our development efforts and approval process and jeopardize our ability to commence product sales and generate revenue. Any of these occurrences may harm our business, financial condition and prospects. In addition, factors that may cause a delay in the commencement or completion of our development efforts may also ultimately lead to the denial of regulatory approval of our product candidates which, as described above, would harm our business and prospects.
We will partially rely on third parties to conduct our development activities. If these third parties do not successfully carry out their contractual duties, we may be unable to obtain regulatory approvals or commercialize our current or future human or animal product candidates on a timely basis, or at all.
We will partially rely upon CROs to conduct our toxicology studies and for other development activities. We intend to rely on CROs to conduct one or more of our planned pivotal trials. These CROs are not our employees, and except for contractual duties and obligations, we have limited ability to control the amount or timing of resources that they devote to our programs or manage the risks associated with their activities on our behalf. We are responsible for ensuring that each of our studies is conducted in accordance with the development plans and trial protocols presented to regulatory authorities. Any deviations by our CROs may adversely affect our ability to obtain regulatory approvals, subject us to penalties or harm our credibility with regulators. The FDA and foreign regulatory authorities also require us and our CROs to comply with regulations and standards, commonly referred to as good clinical practices (GCPs), or good laboratory practices ("GLPs"), for conducting, monitoring, recording and reporting the results of our studies to ensure that the data and results are scientifically valid and accurate.
Agreements with CROs generally allow the CROs to terminate in certain circumstances with little or no advance notice. These agreements generally will require our CROs to reasonably cooperate with us at our expense for an orderly winding down of the CROs' services under the agreements. If the CROs conducting our studies do not comply with their contractual duties or obligations, or if they experience work stoppages, do not meet expected deadlines, or if the quality or accuracy of the data they obtain is compromised, we may need to secure new arrangements with alternative CROs, which could be difficult and costly. In such event, our studies also may need to be extended, delayed or terminated as a result, or may need to be repeated. If any of the foregoing were to occur, regulatory approval, if required, and commercialization of our product candidates may be delayed and we may be required to expend substantial additional resources.
Even if we obtain regulatory approval for planned follow-on indications of Mytesi, or for Canalevia or our other product candidates, they may never achieve market acceptance. Further, even if we are successful in the ongoing commercialization of Mytesi, we may not achieve commercial success.
If we obtain necessary regulatory approvals for planned follow-on indications of Mytesi or for Canalevia or our other product candidates, such products may still not achieve market acceptance and may not be commercially successful. Market acceptance of Mytesi, Canalevia, and any of our other products depends on a number of factors, including:
- •
- the safety of our products as demonstrated in our target animal studies;
- •
- the indications for which our products are approved or marketed;
19
- •
- the potential and perceived advantages over alternative treatments or products, including generic medicines and competing products currently
prescribed by physicians or veterinarians, as applicable, and, in the case of animal products, products approved for use in humans that are used extra-label in animals;
- •
- the acceptance by physicians, veterinarians, companion animal owners, as applicable, of our products as safe and effective;
- •
- the cost in relation to alternative treatments and willingness on the part of physicians, veterinarians, patients and animal owners, as
applicable, to pay for our products;
- •
- the prevalence and severity of any adverse side effects of our products;
- •
- the relative convenience and ease of administration of our products; and
- •
- the effectiveness of our sales, marketing and distribution efforts.
Any failure by Mytesi to achieve market acceptance or commercial success would harm our financial condition and results of operations.
Human and animal gastrointestinal health products are subject to unanticipated post-approval safety or efficacy concerns, which may harm our business and reputation.
The success of our commercialization efforts will depend upon the perceived safety and effectiveness of human and animal gastrointestinal health products, in general, and of our products, in particular. Unanticipated safety or efficacy concerns can subsequently arise with respect to approved prescription drug products, such as Mytesi, or non-prescription products, such as Neonorm, which may result in product recalls or withdrawals or suspension of sales, as well as product liability and other claims. Any safety or efficacy concerns, or recalls, withdrawals or suspensions of sales of our products could harm our reputation and business, regardless of whether such concerns or actions are justified.
Future federal and state legislation may result in increased exposure to product liability claims, which could result in substantial losses.
Under current federal and state laws, companion and production animals are generally considered to be the personal property of their owners and, as such, the owners' recovery for product liability claims involving their companion and production animals may be limited to the replacement value of the animal. Companion animal owners and their advocates, however, have filed lawsuits from time to time seeking non-economic damages such as pain and suffering and emotional distress for harm to their companion animals based on theories applicable to personal injuries to humans. If new legislation is passed to allow recovery for such non-economic damages, or if precedents are set allowing for such recovery, we could be exposed to increased product liability claims that could result in substantial losses to us if successful. In addition, some horses can be worth millions of dollars or more, and product liability for horses may be very high. While we currently have product liability insurance, such insurance may not be sufficient to cover any future product liability claims against us.
If we fail to retain current members of our senior management, or to identify, attract, integrate and retain additional key personnel, our business will be harmed.
Our success depends on our continued ability to attract, retain and motivate highly qualified management and scientific personnel. We are highly dependent upon our senior management, particularly Lisa A. Conte, our president and Chief Executive Officer. The loss of services of any of our key personnel would cause a disruption in our ability to develop our current or future product pipeline and commercialize our products and product candidates. Although we have offer letters with these key members of senior management, such agreements do not prohibit them from resigning at any time. For
20
example, the resignation of our former Chief Financial Officer, Charles O. Thompson, in September 2014, and the mutually agreed departure of our former Chief Veterinary Officer, Serge Martinod, D.V.M., Ph.D. in February 2015, caused us to incur additional expenses and expend resources to ensure a smooth transition with their respective successors, which diverted management attention away from executing our operational plan during this period. To help attract, retain, and motivate qualified management and other personnel, we use share-based incentive awards such as employee stock options and restricted stock units. Due to the decline in our stock price that has occurred since February 2016, a large percentage of the options held by our employees are underwater. As of December 31, 2018, approximately 0% of all outstanding options had an exercise price below the closing price of the stock on that date. As a result, the current situation provides a considerable challenge to maintaining employee motivation, as well as creating a serious threat to retention until a recovery commences. If our share-based compensation ceases to be viewed as a valuable benefit, our ability to attract, retain, and motivate qualified management and other personnel could be weakened, which could harm our results of operations and adversely affect the timing or outcomes of our current and planned studies, as well as the prospects for commercializing our products.
In addition, competition for qualified personnel in the human gastrointestinal health field is intense, because there are a limited number of individuals who are trained or experienced in the field. We will need to hire additional personnel as we expand our product development and commercialization activities. Even if we are successful in hiring qualified individuals, as we are a growing organization, we do not have a track record for integrating and retaining individuals. If we are not successful in identifying, attracting, integrating or retaining qualified personnel on acceptable terms, or at all, our business will be harmed.
We are dependent on two suppliers for the raw material used to produce the active pharmaceutical ingredient in Mytesi. The termination of either of these contracts would result in a disruption to product development and our business will be harmed.
The raw material used to manufacture Mytesi is crude plant latex (CPL), derived from the Croton lechleri tree, which is found in countries in South America, principally Peru. The ability of our contract suppliers to harvest CPL is governed by the terms of their respective agreements with local government authorities. Although CPL is available from multiple suppliers, we only have contracts with two suppliers to obtain CPL and arrange the shipment to our contract manufacturer. Accordingly, if our contract suppliers do not or are unable to comply with the terms of our respective agreements, and we are not able to negotiate new agreements with alternate suppliers on terms that we deem commercially reasonable, it may harm our business and prospects. The countries from which we obtain CPL could change their laws and regulations regarding the export of the natural products or impose or increase taxes or duties payable by exporters of such products. Restrictions could be imposed on the harvesting of the natural products or additional requirements could be implemented for the replanting and regeneration of the raw material. Such events could have a significant impact on our cost and ability to produce Mytesi and anticipated line extensions.
We are dependent upon third-party contract manufacturers, both for the supply of the active pharmaceutical ingredient in Mytesi, as well as for the supply of finished products for commercialization.
We have contracted with third parties for the formulation of API and botanical extract into finished products for our studies. We have also entered into memorandums of understanding with Indena S.p.A. for the manufacture of CPL received from our suppliers into the API in Canalevia to support our regulatory filings, as well as the botanical extract in Neonorm and agreed to negotiate a commercial supply agreement. Indena S.p.A. has never manufactured either such ingredient to commercial scale. Glenmark is the current manufacturer of crofelemer, the active API in Canalevia, for Mytesi, and the manufacturer on file for the NADA to which we have a right of reference. As
21
announced in October of 2015, we have entered an agreement with Patheon, a provider of drug development and delivery solutions, under which Patheon provides enteric-coated tablets to us for use in humans and animals. We also may contract with additional third parties for the formulation and supply of finished products, which we will use in our planned studies and commercialization efforts.
We will be dependent upon our contract manufacturers for the supply of the API in Mytesi and Canalevia. We currently have sufficient quantities of the API used in Mytesi to support our projected sales efforts for 2019. However, we will require additional quantities of API to ensure our ongoing sales efforts for 2020 and beyond. If our contract manufacturer cannot manufacture sufficient quantities of the API in a timely manner we could suffer losses due to lost sales opportunities. We currently have sufficient quantities of the botanical extract used in Neonorm and Equilevia to support planned commercialization efforts for Neonorm and Equilevia. However, we will require additional quantities of the botanical extract if our ongoing commercialization efforts for Neonorm or our ongoing commercial launch of Equilevia is successful. If we are not successful in reaching agreements with third parties on terms that we consider commercially reasonable for manufacturing and formulation of Mytesi, or if our contract manufacturer and formulator are not able to produce sufficient quantities or quality of the Mytesi API, or finished product under their agreements, it could delay our plans and harm our business prospects.
The facilities used by our third-party contractors are subject to inspections, including by the FDA, and other regulators, as applicable. We also depend on our third-party contractors to comply with cGMP. If our third-party contractors do not maintain compliance with these strict regulatory requirements, we and they will not be able to secure or maintain regulatory approval for their facilities, which would have an adverse effect on our operations. In addition, in some cases, we also are dependent on our third-party contractors to produce supplies in conformity to our specifications and maintain quality control and quality assurance practices and not to employ disqualified personnel. If the FDA or a comparable foreign regulatory authority does not approve the facilities of our third-party contractors if so required, or if it withdraws any such approval in the future, we may need to find alternative manufacturing or formulation facilities, which could result in delays in our ability to develop or commercialize our products, if at all. We and our third-party contractors also may be subject to penalties and sanctions from the FDA and other regulatory authorities for any violations of applicable regulatory requirements. The USDA and the European Medicines Agency (the "EMA"), employ different regulatory standards than the FDA, so we may require multiple manufacturing processes and facilities for the same product candidate or any approved product. We are also exposed to risk if our third-party contractors do not comply with the negotiated terms of our agreements, or if they suffer damage or destruction to their facilities or equipment.
If we are unable to establish sales capabilities on our own or through third parties, we may not be able to market and sell our current or future human products and product candidates, if approved, and generate product or other revenue.
We currently have limited sales, marketing or distribution capabilities, and prior to Napo's launch of Mytesi for the symptomatic relief of noninfectious diarrhea in adults with HIV/AIDS on antiretroviral therapy, and our launch of Neonorm for preweaned dairy calves, we had no experience in the sale, marketing and distribution of human or animal health products. There are significant risks involved in building and managing a sales organization, including our potential inability to attract, hire, retain and motivate qualified individuals, generate sufficient sales leads, provide adequate training to sales and marketing personnel and effectively oversee a geographically dispersed sales and marketing team. Any failure or delay in the development of our internal sales, marketing and distribution capabilities and entry into adequate arrangements with distributors or other partners would adversely impact the commercialization of Mytesi, and, if approved, Canalevia. If we are not successful in commercializing Mytesi, for its currently approved indication or for any potential Mytesi follow-on
22
indication, either on our own or through one or more distributors, or in generating upfront licensing or other fees, we may never generate significant revenue and may continue to incur significant losses, which would harm our financial condition and results of operations.
We will need to increase the size of our organization and may not successfully manage such growth.
As of March 31, 2019, we had 41 employees. Our ability to manage our growth effectively will require us to hire, train, retain, manage and motivate additional employees and to implement and improve our operational, financial and management systems. These demands also may require the hiring of additional senior management personnel or the development of additional expertise by our senior management personnel. If we fail to expand and enhance our operational, financial and management systems in conjunction with our potential future growth, it could harm our business and operating results.
If approved, our animal health prescription drug product candidates may be marketed in the United States only in the target animals and for the indications for which they are approved, and if we want to expand the approved animals or indications, it will need to obtain additional approvals, which may not be granted.
If our animal health prescription drug product candidates are approved by regulatory authorities, we may market or advertise them only in the specific species and for treatment of the specific indications for which they were approved, which could limit use of the products by veterinarians and animal owners. We intend to develop, promote and commercialize approved products for new animal treatment indications in the future, but we cannot be certain whether or at what additional time and expense we will be able to do so. If we do not obtain marketing approvals for new indications, our ability to expand our animal health business may be harmed.
Under the Animal Medicinal Drug Use Clarification Act of 1994, veterinarians are permitted to prescribe extra-label uses of certain approved animal drugs and approved human drugs for animals under certain conditions. While veterinarians may in the future prescribe and use human-approved products or use our products for extra-label uses, we may not promote our animal health products for extra-label uses. We note that extra-label uses are uses for which the product has not received approval. If the FDA determines that any of our marketing activities constitute promotion of an extra-label use, we could be subject to regulatory enforcement, including seizure of any misbranded or mislabeled drugs, and civil or criminal penalties, any of which could have an adverse impact on our reputation and expose us to potential liability. We will continue to spend resources ensuring that our promotional claims for our animal health products and product candidates remain compliant with applicable FDA laws and regulations, including materials we post or link to on our website. For example, in 2012, our Chief Executive Officer received an "untitled letter" from the FDA while at Napo regarding preapproval promotion statements constituting misbranding of crofelemer, which was then an investigational drug. These statements were included in archived press releases included on Napo's website. Napo was required to expend time and resources to revise its website to remove the links in order to address the concerns raised in the FDA's letter.
If our human or animal prescription drug product candidates are approved by regulatory authorities, the misuse or extra-label use of such products may harm our reputation or result in financial or other damages.
If our human or animal prescription drug product candidates are approved by regulatory authorities, there may be increased risk of product liability if physicians, veterinarians, patients, animal owners or others, as applicable, attempt to use such products extra-label, including the use of our products for indications or in species for which they have not been approved. Furthermore, the use of an approved human or animal drug for indications other than those indications for which such products have been approved may not be effective, which could harm our reputation and lead to an increased risk of litigation. If we are deemed by a governmental or regulatory agency to have engaged in the
23
promotion of any approved human or animal product for extra-label use, such agency could request that we modify our training or promotional materials and practices and we could be subject to significant fines and penalties, and the imposition of these sanctions could also affect our reputation and position within the gastrointestinal health industry. Any of these events could harm our reputation and our operating results.
We may not maintain the benefits associated with MUMS designation, including market exclusivity.
Although we have received MUMS designation for Canalevia for the treatment of CID in dogs, we may not maintain the benefits associated with MUMS designation. MUMS designation is a status similar to "orphan drug" status for human drugs. When we were granted MUMS designation for Canalevia for the indication of CID in dogs, we became eligible for incentives to support the approval or conditional approval of the designated use. This designation does not allow us to commercialize a product until such time as we obtain approval or conditional approval of the product.
Because Canalevia has received MUMS designation for the identified particular intended use, we are eligible to obtain seven years of exclusive marketing rights upon approval (or conditional approval) of Canalevia for that intended use and become eligible for grants to defray the cost of our clinical work. Each designation that is granted must be unique, i.e., only one designation can be granted for a particular API in a particular dosage form for a particular intended use. The intended use includes both the target species and the disease or condition to be treated.
At some point, we could lose MUMS designation. The basis for a lost designation can include but is not limited to, our failure to engage with due diligence in moving forward with a non-conditional approval, or a competing product has received conditional approval or approval prior to our product candidate for the same indication or species. In addition, MUMS designation may be withdrawn for a variety of reasons such as where the FDA determines that the request for designation was materially defective, or if the manufacturer is unable to assure sufficient quantity of the prescription drug product to meet the needs of animals with the rare disease or condition. If this designation is lost, it could have a negative impact on the product and us, which includes but is not limited to, market exclusivity related to MUMS designation, or eligibility for grants as a result of MUMS designation.
The market for our human products, and the gastrointestinal health market as a whole, is uncertain and may be smaller than we anticipate, which could lead to lower revenue and harm our operating results.
It is very difficult to estimate the commercial potential of any of our human products because the gastrointestinal health market continues to evolve and it is difficult to predict the market potential for our products. The market will depend on important factors such as safety and efficacy compared to other available treatments, changing standards of care, preferences of physicians, as applicable, the willingness of patients, as applicable, to pay for such products, and the availability of competitive alternatives that may emerge either during the product development process or after commercial introduction. If the market potential for our human products is less than we anticipate due to one or more of these factors, it could negatively impact our business, financial condition and results of operations. Further, the willingness of patients to pay for our products may be less than we anticipate, and may be negatively affected by overall economic conditions.
Insurance coverage for Mytesi for its current approved indication could decrease or end, or Mytesi might not receive insurance coverage for any approved follow-on indications, which could lead to lower revenue and harm our operating results.
For its current approved indication, Mytesi is currently covered by all of the top 10 commercial insurance plans, representing more than 245 million U.S. lives. In 50% of these plans it is currently on Tier 3 with no restrictions, and in 50% it is currently on Tier 3 with a prior authorization required. In
24
the top 10 Managed Medicare plans, which represent 24 million covered lives, Mytesi is currently covered on 10% of plans. Mytesi is currently covered on Medicaid in all 50 states. However, the nature or extent of coverage for Mytesi by any of these plans or programs could change or be terminated, or Mytesi might not receive insurance coverage for any approved follow-on indications. Either outcome could lead to significantly lower revenue and significantly harm our operating results.
We may engage in future acquisitions that increase our capital requirements, dilute our stockholders, cause us to incur debt or assume contingent liabilities and subject us to other risks.
We may evaluate various strategic transactions, including licensing or acquiring complementary products, technologies or businesses. Any potential acquisitions may entail numerous risks, including increased operating expenses and cash requirements, assimilation of operations and products, retention of key employees, diversion of our management's attention and uncertainties in our ability to maintain key business relationships of the acquired entities. In addition, if we undertake acquisitions, we may issue dilutive securities, assume or incur debt obligations, incur large one-time expenses and acquire intangible assets that could result in significant future amortization expense. Moreover, we may not be able to locate suitable acquisition opportunities and this inability could impair our ability to grow or obtain access to technology or products that may be important to the development of our business.
Certain of the countries in which we plan to commercialize our products in the future are developing countries, some of which have potentially unstable political and economic climates.
We may commercialize our products in jurisdictions that are developing and emerging countries. This may expose us to the impact of political or economic upheaval, and we could be subject to unforeseen administrative or fiscal burdens. At present, we are not insured against the political and economic risks of operating in these countries. Any significant changes to the political or economic climate in any of the developing countries in which we operate or plan to sell products either now or in the future may have a substantial adverse effect on our business, financial condition, trading performance and prospects.
Fluctuations in the exchange rate of foreign currencies could result in currency transactions losses.
As we expand our operations, we expect to be exposed to risks associated with foreign currency exchange rates. We anticipate that we may commercialize Canalevia and its line extensions in jurisdictions outside the United States. As a result, we may also be further affected by fluctuations in exchange rates in the future to the extent that sales are denominated in currencies other than U.S. dollars. We do not currently employ any hedging or other strategies to minimize this risk, although we may seek to do so in the future.
There are other gastrointestinal-focused human pharmaceutical companies, and we face competition in the marketplaces in which we operate or plan to operate.
Our commercial success in the human drug arena remains dependent on maintaining or establishing a competitive position in the market for the current, approved specialty indication of Mytesi as well as for planned Mytesi follow-on indications. In the IBS-D market in particular, several competitors have commercially available products approved for our planned IBS-D indication. The availability of our competitors' products could limit the demand, and the price we are able to charge, for any drug candidate we develop. The inability to compete with existing or subsequently introduced drug candidates would have a material adverse impact on our business, financial condition and prospects.
25
Our obligations to CVP are secured by a security interest in substantially all of our veterinary related assets and substantially all of Napo's assets, so if we default on those obligations, CVP could foreclose on our assets.
Our obligations under the secured promissory notes issued to Chicago Venture Partners, L.P. ("CVP") are secured by a security interest in substantially all of our veterinary related assets and substantially all of Napo's assets, including intellectual property, as provided in the Security Agreement, dated May 28, 2019 between Jaguar and CVP, and the Security Agreement dated May 28, 2019 between Napo and CVP. As a result, if we default on our obligations under these agreements, CVP could foreclose on its security interests and liquidate some or all of these assets, which would harm our veterinary related business, financial condition and results of operations and could require us to reduce or cease operations.
Failure in our information technology systems, including by cyber attacks or other data security incidents, could significantly disrupt our operations.
Our operations depend, in part, on the continued performance of our information technology systems. Our information technology systems are potentially vulnerable to physical or electronic break-ins, computer viruses, phishing attacks and other types of disruptions. We have and continue to experience cyber attacks of varying degrees. Our security measures may also be breached due to employee error, malfeasance, system errors or other vulnerabilities. Such breach or unauthorized access or attempts by outside parties to fraudulently induce employees or users to disclose sensitive information in order to gain access to our data could result in significant legal and financial exposure, and damage to our reputation that could potentially have an adverse effect on our business. Because the techniques used to obtain unauthorized access, or sabotage systems change frequently, become more sophisticated, and often are not recognized until launched against a target, we may be unable to anticipate these techniques or to implement adequate preventative measures. Additionally, cyber attacks could also compromise trade secrets and other sensitive information and result in such information being disclosed to others and becoming less valuable, which could negatively affect our business. Although we have information technology security systems, a successful cybersecurity attack or other data security incident could result in the misappropriation and/or loss of confidential or personal information, create system interruptions, deploy malicious software that attacks our systems, or result in financial losses. It is possible that a cybersecurity attack might not be noticed for some period of time. The occurrence of a cyber security attack or incident could result in business interruptions from the disruption of our information technology systems, or negative publicity resulting in reputational damage with our shareholders and other stakeholders and/or increased costs to prevent, respond to or mitigate cybersecurity events. In addition, the unauthorized dissemination of sensitive personal information or proprietary or confidential information could expose us or other third-parties to regulatory fines or penalties, litigation and potential liability, or otherwise harm our business.
Risks Related to Intellectual Property
We cannot be certain that our patent strategy will be effective to protect against competition
Our commercial success depends in large part on obtaining and maintaining patent, trademark and trade secret protection of our human or animal products, both prescription and non-prescription, our current human or animal product candidates and any future human or animal product candidates, and their respective components, formulations, methods used to manufacture them and methods of treatment, as well as successfully defending our patents and other intellectual property rights against third-party challenges. Our ability to stop unauthorized third parties from making, using, selling, offering to sell or importing our products or our product candidates is dependent upon the extent to which we have rights under valid and enforceable patents, trade secrets and other similar intellectual property that cover these activities. The patent prosecution process is expensive and time-consuming, and we may not be able to prepare, file and prosecute all necessary or desirable patent applications at
26
a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of inventions made in the course of development and commercialization activities in time to obtain patent protection on them.
We have a portfolio of United States and foreign issued patents and pending applications related to our products and product candidates. We have three issued United States patents listed in the FDA's Orange Book for Mytesi. We plan to rely on certain of these issued patents as protection for Canalevia. The strength of patents in the field of pharmaceuticals and animal health involves complex legal and scientific questions and can be uncertain. We cannot be certain that pending applications will issue as patents. For those patents that are already issued and even if other patents do successfully issue, third parties may challenge their validity, enforceability or scope, which may result in such patents being narrowed, invalidated or held unenforceable. Furthermore, even if they are unchallenged, our patents may not adequately protect our intellectual property or prevent others from designing around their claims. If the patents we have are not maintained or their scope is significantly narrowed or if we are not able to obtain issued patents from pending applications, our business and prospects would be harmed.
The Leahy-Smith America Invents Act, patent reform legislation enacted in 2011, could increase the uncertainties and costs surrounding the prosecution of any patent applications and the enforcement or defense of any patents that issue. The Leahy-Smith Act introduced significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted, redefine prior art, may affect patent litigation, and switch the U.S. patent system from a "first-to-invent" system to a "first-to-file" system. Under a "first-to-file" system, assuming the other requirements for patentability are met, the first inventor to file a patent application generally is entitled to the patent on an invention regardless of whether another inventor had made the invention earlier. The USPTO developed regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first-to-file provisions, became effective on March 16, 2013. Among some of the other changes to the patent laws are changes that limit where a patentee may file a patent infringement suit and that provide opportunities for third parties to challenge any issued patent in the USPTO. The Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our patents and any other patents that issue, all of which could harm our business and financial condition.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance and annuity fees on any issued patent and, in certain jurisdictions, pending applications, are due to be paid to the USPTO and foreign patent agencies in several stages over the lifetime of the patent. The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we fail to maintain the patents and patent applications covering our prescription drug products, prescription drug product candidates and non-prescription products, our competitors might be able to enter the market, which would harm our business.
27
Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, which would be costly, time-consuming and, if successfully asserted against us, delay or prevent the development and commercialization of our current or future products and product candidates.
Our research, development and commercialization activities may infringe or otherwise violate or be claimed to infringe or otherwise violate patents owned or controlled by other parties. There may be patents already issued of which we are unaware that might be infringed by a product or one of our current or future prescription drug product candidates or non-prescription products. Moreover, it is also possible that patents may exist that we are aware of, but that we do not believe are relevant to our current or future prescription drug product candidates or non-prescription products, which could nevertheless be found to block our freedom to market these products. Because patent applications can take many years to issue and may be confidential for 18 months or more after filing, there may be applications now pending of which we are unaware and which may later result in issued patents that may be infringed by our current or future prescription drug product candidates or non-prescription products. We cannot be certain that our products, current or future prescription drug product candidates or non-prescription products will not infringe these or other existing or future third-party patents. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents.
To the extent we become subject to future third-party claims against us or our collaborators, we could incur substantial expenses and, if any such claims are successful, we could be liable to pay substantial damages, including treble damages and attorney's fees if we or our collaborators are found to be willfully infringing a third party's patents. If a patent infringement suit were brought against us or our collaborators, we or they could be forced to stop or delay research, development, manufacturing or sales of the human or animal prescription drug or non-prescription product that is the subject of the suit. Even if we are successful in defending such claims, infringement and other intellectual property claims can be expensive and time-consuming to litigate and divert management's attention from our business and operations. As a result of or in order to avoid potential patent infringement claims, we or our collaborators may be compelled to seek a license from a third party for which we would be required to pay license fees or royalties, or both. Moreover, these licenses may not be available on acceptable terms, or at all. Even if we or our collaborators were able to obtain such a license, the rights may be nonexclusive, which could allow our competitors access to the same intellectual property. Any of these events could harm our business and prospects.
Our proprietary position depends upon the botanical guidance of our drug approval and patents that are formulation or method-of-use patents, which do not prevent a competitor from using the same human or animal drug for another use.
Composition-of-matter patents on the API in prescription drug products are generally considered to be the strongest form of intellectual property protection because such patents provide protection without regard to any particular method of use or manufacture or formulation of the API used. The composition-of-matter patents for crofelemer, the API in Mytesi and Canalevia, have expired, and the issued patents and applications relevant to our products and product candidates cover methods of use for crofelemer and the botanical extract in Neonorm and Equilevia.
Method-of-use patents protect the use of a product for the specified method and formulation patents cover formulations of the API or botanical extract. These types of patents do not prevent a competitor from developing or marketing an identical product for an indication that is outside the scope of the patented method or from developing a different formulation that is outside the scope of the patented formulation. Moreover, with respect to method-of-use patents, even if competitors do not actively promote their product for our targeted indications or uses for which we may obtain patents, physicians may recommend that patients use our products extra-label, and veterinarians may recommend that animal owners use these products extra-label, or animal owners may do so themselves.
28
Although extra-label use may infringe or contribute to the infringement of method-of-use patents, the practice is common and such infringement is difficult to prevent or prosecute.
We may be involved in lawsuits to protect or enforce our patents, which could be expensive, time-consuming and unsuccessful, and third parties may challenge the validity or enforceability of our patents and they may be successful.
We intend to rely upon a combination of regulatory exclusivity periods, patents, trade secret protection, and confidentiality agreements to protect the intellectual property related to Mytesi, our current prescription drug product candidates, non-prescription products and our development programs.
If the breadth or strength of protection provided by any patents, patent applications or future patents we may own, license, or pursue with respect to any of our current or future product candidates or products is threatened, it could threaten our ability to commercialize any of our current or future human or animal product candidates or products. Further, if we encounter delays in our development efforts, the period of time during which we could market any of our current or future product candidates or products under any patent protection we obtain would be reduced.
Given the amount of time required for the development, testing and regulatory review of new product candidates or products, patents protecting such candidates might expire before or shortly after such product candidates or products are commercialized. The United States Patent and Trademark Office has issued a patent term extension certificate extending the term of US 7,341,744 by 1075 days under 35 USC 156. With respect to requests for patent term extensions, the applicable authorities, including the USPTO and the FDA, and any equivalent regulatory authority in other countries, may not agree with our assessment of whether such extensions are available, and may refuse to grant extensions to patents, or may grant more limited extensions than requested. If this occurs, our competitors may take advantage of our investment in development and trials by referencing our clinical and preclinical data and launch their product earlier than might otherwise be the case.
Even where laws provide protection or we are able to obtain patents, costly and time-consuming litigation may be necessary to enforce and determine the scope of our proprietary rights, and the outcome of such litigation would be uncertain. Moreover, any actions we may bring to enforce our intellectual property against our competitors could provoke them to bring counterclaims against us, and some of our competitors have substantially greater intellectual property portfolios than we have. To counter infringement or unauthorized use of any patents we may obtain, we may be required to file infringement claims, which can be expensive and time-consuming to litigate. In addition, if we or one of our future collaborators were to initiate legal proceedings against a third party to enforce a patent covering one of our products, current product candidates, or one of our future products, the defendant could counterclaim that the patent is invalid or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity or unenforceability are commonplace and challenges to validity of patents in certain foreign jurisdictions is common as well. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness, non-enablement or lack of statutory subject matter. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant material information from the USPTO, or made a materially misleading statement, during prosecution. Under the Hatch-Waxman Act, a competitor seeking to market a generic form of Mytesi before the expiration of any of the patents listed in the FDA's Orange Book for Mytesi could file an ANDA with a certification under 21 U.S.C. § 3559(j)(2)(A)(iv) that each of these patents (except for those which the ANDA filer states it will market only after its expiration) is either invalid, unenforceable or not infringed. We may assert the patents in Hatch-Waxman litigation against the party filing the ANDA to keep the competing product off of the market until the patents expire but there is a risk that we will not succeed. The party filing the ANDA may also counterclaim in the litigation that our patents are not valid or unenforceable, and the court may find one or more claims of our patents
29
invalid or unenforceable. If this occurs, a competing generic product could be marketed prior to expiration of our patents listed in the Orange Book, which would harm our business.
Third parties may also raise similar validity claims before the USPTO in post-grant proceedings such as ex parte reexaminations, inter partes review, or post-grant review, or oppositions or similar proceedings outside the United States, in parallel with litigation or even outside the context of litigation. The outcome following legal assertions of invalidity and unenforceability is unpredictable. If a defendant were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of any future patent protection on one or more of our products or our current or future product candidates. Such a loss of patent protection could harm our business. We cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution or other basis for a finding of invalidity. Litigation proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be unsuccessful, it could have an adverse effect on the price of our common stock. Finally, we may not be able to prevent, misappropriation of our trade secrets or confidential information, particularly in countries where the laws may not protect those rights as fully as in the United States.
If we are unable to prevent disclosure of our trade secrets or other confidential information to third parties, our competitive position may be impaired.
We also rely on trade secret protection and confidentiality agreements to protect proprietary know-how that is not patentable or for which we have not filed patent applications, processes for which patents are difficult to enforce and other elements of our product development processes that involve proprietary know-how, information or technology that is not covered by patents. Although we require all of our employees to assign their inventions to us, and endeavor to execute confidentiality agreements with all of our employees, consultants, advisors and any third parties who have access to our proprietary know-how, information or technology, we cannot be certain that we have executed such agreements with all parties who may have helped to develop our intellectual property or had access to our proprietary information, or that our agreements will not be breached. We cannot guarantee that our trade secrets and other confidential proprietary information will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. If we are unable to prevent disclosure of our intellectual property to third parties, we may not be able to maintain a competitive advantage in our market, which would harm our business.
Any disclosure to or misappropriation by third parties of our confidential proprietary information could enable competitors to quickly duplicate or surpass our technological achievements, and erode our competitive position in our market.
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our products.
As is the case with other human or animal pharmaceutical product companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the human and animal health industries involves both technological and legal complexity. Therefore, obtaining and enforcing patents is costly, time-consuming and inherently uncertain. In addition, the United States has recently enacted and implemented wide-ranging patent reform legislation. The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent
30
protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the federal courts, and the USPTO, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce patents that we have or that we might obtain in the future.
We may not be able to protect our intellectual property rights throughout the world, which could impair our business.
Filing, prosecuting and defending patents on human and animal drug products, product candidates and non-prescription products throughout the world would be prohibitively expensive. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we may obtain patent protection, but where patent enforcement is not as strong as that in the United States. These products may compete with our products in jurisdictions where we do not have any issued or licensed patents and any future patent claims or other intellectual property rights may not be effective or sufficient to prevent them from so competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to animal health products, which could make it difficult for us to stop the infringement of our future patents, if any, or patents we have in licensed, or marketing of competing products in violation of our proprietary rights generally. Further, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the United States. As a result, we may encounter significant problems in protecting and defending our intellectual property both in the United States and abroad. Proceedings to enforce our future patent rights, if any, in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
Our business could be harmed if we fail to obtain certain registered trademarks in the United States or in other countries.
Our registered and pending U.S. trademarks include MYTESI®, JAGUAR HEALTH®, the Jaguar Health Logo®, NAPO®, Napo Logo®, CANALEVIA, EQUILEVIA, NEONORM®, JAGUAR ANIMAL HEALTH®, and the Jaguar Animal Health Logo®. We also own registered and pending applications for the CANALEVIA mark in a number of foreign countries. During trademark registration proceedings, we may receive rejections of our trademark applications. If so, we will have an opportunity to respond, but we may be unable to overcome such rejections. In addition, the USPTO and comparable agencies in many foreign jurisdictions may permit third parties to oppose pending trademark applications and to seek to cancel registered trademarks. If opposition or cancellation proceedings are filed against any of our trademark applications or any registered trademarks, our trademarks may not survive such proceedings. Moreover, any name we propose to use with our prescription drug product candidates in the United States, including CANALEVIA, must be approved by the FDA, regardless of whether we have registered or applied to register as a trademark. The FDA typically conducts a review of proposed prescription drug product names, including an evaluation of potential for confusion with other product names. If the FDA objects to any of our proposed proprietary product names, we may be required to expend significant additional resources in an effort to identify a suitable substitute name that would qualify under applicable trademark laws, not infringe the existing rights of third parties and be acceptable to the FDA.
31
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties.
We have received confidential and proprietary information from third parties. In addition, we employ individuals who were previously employed at other biotechnology, pharmaceutical or animal health companies. We may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise improperly used or disclosed confidential information of these third parties or our employees' former employers. Litigation may be necessary to defend against any such claims. Even if we were successful in defending against any such claims, such litigation could result in substantial cost and be a distraction to our management and employees.
Even if we receive any of the required regulatory approvals for our current or future prescription drug product candidates and non-prescription products, we will be subject to ongoing obligations and continued regulatory review, which may result in significant additional expense.
If the FDA or any other regulatory body approves any of our current or future prescription drug product candidates, or if necessary, our non-prescription products, the manufacturing processes, clinical development, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for the product may be subject to extensive and ongoing regulatory requirements. These requirements could include, but are not limited to, submissions of efficacy and safety and other post-marketing information and reports, establishment registration, and product listing, compliance with new rules promulgated under the FSMA, as well as continued compliance with cGMP, GLP and GCP for any studies that we conduct post-approval. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with our contract manufacturers or manufacturing processes, or failure to comply with regulatory requirements, are reportable events to the FDA and may result in, among other things:
- •
- restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, revised labeling, or voluntary or
involuntary product recalls;
- •
- additional clinical studies, fines, warning letters or holds on target animal studies;
- •
- refusal by the FDA, or other regulators to approve pending applications or supplements to approved applications filed by us or our strategic
collaborators related to the unknown problems, or suspension or revocation of the problematic product's license approvals;
- •
- product seizure or detention, or refusal to permit the import or export of products; and
- •
- injunctions and/or the imposition of civil or criminal penalties.
The FDA or other regulatory agency's policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates or require certain changes to the labeling or additional clinical work concerning safety and efficacy of the product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability, which would harm our business. In addition, failure to comply with these regulatory requirements could result in significant penalties.
In addition, from time to time, we may enter into consulting and other financial arrangements with veterinarians, who prescribe or recommend our products, once approved. As a result, we may be subject to state, federal and foreign healthcare and/or veterinary medicine laws. If our financial
32
relationships with veterinarians are found to be in violation of such laws that apply to us, we may be subject to penalties.
Any of our current or future prescription drug product candidates or non-prescription products may cause or contribute to adverse medical events that we would be required to report to regulatory authorities and, if we fail to do so, we could be subject to sanctions that would harm our business.
If we are successful in commercializing any of our current or future prescription drug product candidates or non-prescription products, certain regulatory authorities will require that we report certain information about adverse medical events if those products may have caused or contributed to those adverse events. The timing of our obligation to report would be triggered by the date we become aware of the adverse event as well as the nature of the event. We may fail to report adverse events we become aware of within the prescribed timeframe. We may also fail to appreciate that we have become aware of a reportable adverse event, especially if such event is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of our products. If we fail to comply with our reporting obligations, the regulatory authorities could take action including, but not limited to, criminal prosecution, seizure of our products, facility inspections, removal of our products from the market, recalls of certain lots or batches, or cause a delay in approval or clearance of future products.
Legislative or regulatory reforms with respect to animal health may make it more difficult and costly for us to obtain regulatory clearance or approval of any of our current or future product candidates and to produce, market, and distribute our products after clearance or approval is obtained.
From time to time, legislation is drafted and introduced in the U.S. Congress or other jurisdictions in which we intend to operate that could significantly change the statutory provisions governing the testing, regulatory clearance or approval, manufacture, and marketing of regulated products. In addition, the FDA's regulations and guidance are often revised or reinterpreted by the FDA and such other regulators in ways that may significantly affect our business and our products and product candidates. Similar changes in laws or regulations can occur in other countries. Any new regulations or revisions or reinterpretations of existing regulations in the United States or in other countries may impose additional costs or lengthen review times of any of our current or future products and product candidates. We cannot determine what effect changes in regulations, statutes, legal interpretation or policies, when and if promulgated, enacted or adopted may have on our business in the future. Such changes could, among other things, require:
- •
- changes to manufacturing methods;
- •
- additional clinical trials or testing;
- •
- new requirements related to approval to enter the market;
- •
- recall, replacement, or discontinuance of certain products; and
- •
- additional record keeping or the development of certain regulatory required hazard identification plans.
Each of these would likely entail substantial time and cost and could harm our financial results. In addition, delays in receipt of or failure to receive regulatory clearances or approvals for any future products would harm our business, financial condition, and results of operations.
33
We believe that our non-prescription products are not subject to regulation by regulatory agencies in the United States, but there is a risk that regulatory bodies may disagree with our interpretation, or may redefine the scope of their regulatory reach in the future, which would result in additional expense and could delay or prevent the commercialization of these products.
The FDA retains jurisdiction over all animal prescription drug products however, in many instances, the Federal Trade Commission will exercise primary or concurrent jurisdiction with FDA on non-prescription products as to post marketing claims made regarding the product. On April 22, 1996, the FDA published a statement in the Federal Register, 61 FR 17706, that it believes that the Dietary Supplement and Health Education Act ("DSHEA"), does not apply to animal health supplement products, such as our non-prescription products. Accordingly, the FDA's Center for Veterinary Medicine only regulates those animal supplements that fall within the FDA's definition of an animal drug, animal food or animal feed additive. The Federal Food Drug and Cosmetic Act defines food as "articles used for food or drink for man or other animals and articles used as components of any such article." Animal foods are not subject to pre-market approval and are designed to provide a nutritive purpose to the animals that receive them. Feed additives are defined as those articles that are added to an animal's feed or water as illustrated by the guidance documents. Our non-prescription products are not added to food, are not ingredients in food nor are they added to any animal's drinking water. Therefore, our non-prescription products do not fall within the definition of a food or feed additive. In light of the pronouncement by the FDA that the DSHEA was not intended to apply to animals, the FDA seeks to regulate such supplements as food or food additives depending on the intended use of the product. The intended use is demonstrated by how the article is included in a food, or added to the animals' intake (i.e., through its drinking water). If the intended use of the product does not fall within the proscribed use making the product a food, it cannot be regulated as a food. There is no intent to make our non-prescription products a component of an animal food, either directly or indirectly. A feed additive is a product that is added to a feed for any reason including the top dressing of an already prepared feed. Some additives, such as certain forage, are deemed to be Generally Recognized as Safe, or GRAS, and therefore, not subject to a feed Additive Petition approval prior to use. However, the substances deemed GRAS are generally those that are recognized as providing nutrients as a food does. We do not believe that our non-prescription products fit within this framework either. Finally, a new animal drug refers to drugs intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in animals. Our non-prescription Neonorm Foal and Neonorm Calf products are not intended to diagnose, cure, mitigate, treat or prevent disease and therefore, do not fit within the definition of an animal drug. Additionally, because a previously marketed human formulation of the botanical extract in our non-prescription products was regulated as a human dietary supplement subject to the DSHEA (and not regulated as a drug by the FDA), we do not believe that the FDA would regulate the animal formulation used in our non-prescription products in a different manner. We do not believe that our non-prescription products fit the definition of an animal drug, food or food additive and therefore are not regulated by the FDA at this time.
However, despite many such unregulated animal supplements currently on the market, the FDA may choose in the future to exercise jurisdiction over animal supplement products in which case, we may be subject to unknown regulations thereby inhibiting our ability to launch or to continue marketing our non-prescription products. In the past, the FDA has redefined or attempted to redefine some non-prescription non-feed products as falling within the definition of drug, feed or feed additive and therefore subjected those products to the relevant regulations. We have not discussed with the FDA its belief that the FDA currently does not exercise jurisdiction over our non-prescription products. Should the FDA assert regulatory authority over our non-prescription products, we would take commercially reasonable steps to address the FDA's concerns, potentially including but not limited to, seeking registration for such products, reformulating such products to further distance such products from regulatory control, or ceasing sale of such products. Further, the Animal and Plant Health Inspection Service, an agency of the USDA, may at some point choose to exercise jurisdiction over certain
34
non-prescription products that are not intended for production animals. We do not believe we are currently subject to such regulation, but could be in the future. If the FDA or other regulatory agencies, such as the USDA, try to regulate our non-prescription products, we could be required to seek regulatory approval for our non-prescription products, which would result in additional expense and could delay or prevent the commercialization of these products.
Even if Napo receives the required regulatory approvals for Napo's current or future prescription drug product candidates and non-prescription products, Napo will be subject to ongoing obligations and continued regulatory review, which may result in significant additional expense.
If the FDA or any other regulatory body approves any of Napo's current or future prescription drug product candidates, or if necessary, Napo's non-prescription products, the manufacturing processes, clinical development, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for the product is subject to extensive and ongoing regulatory requirements. These requirements could include, but are not limited to, submissions of efficacy and safety and other post-marketing information and reports, establishment registration, and product listing, compliance with new rules promulgated under the FSMA, as well as continued compliance with cGMP, GLP and GCP for any studies that Napo conducts post-approval. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with Napo's contract manufacturers or manufacturing processes, or failure to comply with regulatory requirements, are reportable events to the FDA and may result in, among other things:
- •
- restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, revised labeling, or voluntary or
involuntary product recalls;
- •
- additional clinical studies fines, warning letters or holds on studies;
- •
- refusal by the FDA, or other regulators to approve pending applications or supplements to approved applications filed by Napo or Napo's
strategic collaborators related to the unknown problems, or suspension or revocation of the problematic product's license approvals;
- •
- product seizure or detention, or refusal to permit the import or export of products; and
- •
- injunctions or the imposition of civil or criminal penalties.
The FDA or other regulatory agency's policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of Napo's product candidates or require certain changes to the labeling or require additional clinical work concerning safety and efficacy of the product candidates. Napo cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If Napo is slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if Napo is not able to maintain regulatory compliance, Napo may lose any marketing approval that Napo may have obtained and Napo may not achieve or sustain profitability, which would harm Napo's business. In addition, failure to comply with these regulatory requirements could result in significant penalties.
In addition, from time to time, Napo may enter into consulting and other financial arrangements with physicians, who prescribe or recommend Napo's products, once approved. As a result, Napo may be subject to state, federal and foreign healthcare laws, including but not limited to anti-kickback laws. If Napo's financial relationships with physicians are found to be in violation of such laws that apply to Napo, Napo may be subject to penalties.
35
Any of Napo's current or future prescription drug product candidates or non-prescription products may cause or contribute to adverse medical events that Napo would be required to report to regulatory authorities and, if Napo fails to do so, Napo could be subject to sanctions that would harm Napo's business.
If Napo is successful in commercializing any of Napo's current or future prescription drug product candidates or non-prescription products, certain regulatory authorities will require that Napo report certain information about adverse medical events if those products may have caused or contributed to those adverse events. The timing of Napo's obligation to report would be triggered by the date Napo becomes aware of the adverse event as well as the nature of the event. Napo may fail to report adverse events Napo becomes aware of within the prescribed timeframe. Napo may also fail to appreciate that Napo has become aware of a reportable adverse event, especially if it is not reported to Napo as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of Napo's products. If Napo fails to comply with Napo's reporting obligations, the regulatory authorities could take action including, but not limited to, criminal prosecution, seizure of Napo's products, facility inspections, removal of Napo's products from the market, recalls of certain lots or batches, or cause a delay in approval or clearance of future products.
Legislative or regulatory reforms make it more difficult and costly for Napo to obtain regulatory clearance or approval of any of Napo's current or future product candidates and to produce, market, and distribute Napo's products after clearance or approval is obtained.
From time to time, legislation is drafted and introduced in the U.S. Congress or other jurisdictions in which Napo intends to operate that could significantly change the statutory provisions governing the testing, regulatory clearance or approval, manufacture, and marketing of regulated products. In addition, the FDA's regulations and guidance are often revised or reinterpreted by the FDA and such other regulators in ways that may significantly affect Napo's business and Napo's products and product candidates. Similar changes in laws or regulations can occur in other countries. Any new regulations or revisions or reinterpretations of existing regulations in the United States or in other countries may impose additional costs or lengthen review times of any of Napo's current or future products and product candidates. Napo cannot determine what effect changes in regulations, statutes, legal interpretation or policies, when and if promulgated, enacted or adopted may have on Napo's business in the future. Such changes could, among other things, require:
- •
- changes to manufacturing methods;
- •
- additional clinical trials or testing;
- •
- new requirements related to approval to enter the market;
- •
- recall, replacement, or discontinuance of certain products; and
- •
- additional record keeping or the development of certain regulatory required hazard identification plans.
Each of these would likely entail substantial time and cost and could harm Napo's financial results. In addition, delays in receipt of or failure to receive regulatory clearances or approvals for any future products would harm Napo's business, financial condition, and results of operations.
Risks Related to Securities Markets and Investment in our Securities
Our failure to meet the continued listing requirements of The Nasdaq Capital Market could result in a delisting of our common stock.
If we fail to satisfy the continued listing requirements of The NASDAQ Capital Market, such as the minimum closing bid price requirement, Nasdaq may take steps to delist our common stock.
36
The delisting of our common stock from Nasdaq may make it more difficult for us to raise capital on favorable terms in the future. Such a delisting would likely have a negative effect on the price of our common stock and would impair your ability to sell or purchase our common stock when you wish to do so. Further, if we were to be delisted from The Nasdaq Capital Market, our common stock would cease to be recognized as covered securities and we would be subject to regulation in each state in which we offer our securities.
We have a material weakness in our internal control over financial reporting related to staff turnover in our accounting department. We did not maintain a sufficient complement of internal personnel with appropriate knowledge, experience and/or training commensurate with our financial reporting requirements. If we fail to remediate the material weakness, or experience any additional material weaknesses in the future or otherwise fail to maintain an effective system of internal controls in the future, we may not be able to accurately report our financial condition or results of operations which may adversely affect investor confidence in us and, as a result, the value of our common stock.
Our management is responsible for establishing and maintaining adequate internal control over our financial reporting, as defined in Rule 13a-15(f) under the Securities Exchange Act of 1934, as amended (the "Exchange Act").
Preparing our consolidated financial statements involves a number of complex manual and automated processes, which are dependent upon individual data input or review and require significant management judgment. One or more of these elements may result in errors that may not be detected and could result in a material misstatement of our consolidated financial statements. If we fail to maintain the adequacy of our internal controls over financial reporting, our business and operating results may be harmed and we may fail to meet our financial reporting obligations. If material weaknesses in our internal control are discovered or occur, our consolidated financial statements may contain material misstatements and we could be required to restate our financial results.
In connection with our preparation of our annual financial statements for the year ended December 31, 2018, we identified a material weakness in our internal control over financial reporting related to staff turnover in our accounting department. We did not maintain a sufficient complement of internal personnel with appropriate knowledge, experience and/or training commensurate with our financial reporting requirements. We relied on outside consulting technical experts and did not maintain adequate internal qualified personnel to properly supervise and review the information provided by the outside consulting technical experts to ensure certain significant complex transactions and technical matters were properly accounted for, specifically with respect to accurately reflecting all potential accrued services on the balance sheet at December 31, 2018. In addition, we identified inadequate internal technical staffing levels and expertise to properly supervise and review the information of the outside consulting technical experts to properly apply ASC 815-40 for liability classification of certain warrants and ASC 470-50 and ASC 470-60 to properly reflect the accounting impact to multiple modifications of the Company's debt instruments. We have concluded that we must implement new or improved controls in our financial statement close process and policies in reviewing information received from our outside consulting technical experts.
We have enhanced our internal controls, processes and related documentation necessary to remediate our material weakness. We may not be able to complete our remediation, evaluation and testing in a timely fashion. If we are unable to remediate this material weakness, or if we identify one or more other material weaknesses in our internal control over financial reporting, we will continue to be unable to conclude that our internal controls are effective. If we are unable to confirm that our internal control over financial reporting is effective we could lose investor confidence in the accuracy and completeness of our financial reports, which could cause the price of our common stock to decline.
37
If our shares become subject to the penny stock rules, it would become more difficult to trade our shares.
The SEC has adopted rules that regulate broker-dealer practices in connection with transactions in penny stocks. Penny stocks are generally equity securities with a price of less than $5.00, other than securities registered on certain national securities exchanges or authorized for quotation on certain automated quotation systems, provided that current price and volume information with respect to transactions in such securities is provided by the exchange or system. If we do not retain a listing on The NASDAQ Capital Market and if the price of our common stock is less than $5.00, our common stock will be deemed a penny stock. The penny stock rules require a broker-dealer, before a transaction in a penny stock not otherwise exempt from those rules, to deliver a standardized risk disclosure document containing specified information. In addition, the penny stock rules require that before effecting any transaction in a penny stock not otherwise exempt from those rules, a broker-dealer must make a special written determination that the penny stock is a suitable investment for the purchaser and receive (i) the purchaser's written acknowledgment of the receipt of a risk disclosure statement; (ii) a written agreement to transactions involving penny stocks and (iii) a signed and dated copy of a written suitability statement. These disclosure requirements may have the effect of reducing the trading activity in the secondary market for our common stock, and therefore stockholders may have difficulty selling their shares.
The price of our common stock could be subject to volatility related or unrelated to our operations, and purchasers of our common stock could incur substantial losses.
The trading price of our common stock could be subject to wide fluctuations in response to various factors, some of which are beyond our control. These factors include those discussed previously in this "Risk Factors" section of this report and others, such as:
- •
- delays in the commercialization of Mytesi, Neonorm, Canalevia, Equilevia or our other current or future prescription drug product candidates
and non-prescription products;
- •
- any delays in, or suspension or failure of, our current and future studies;
- •
- announcements of regulatory approval or disapproval of any of our current or future product candidates or of regulatory actions affecting our
company or our industry;
- •
- manufacturing and supply issues that affect product candidate or product supply for our studies or commercialization efforts;
- •
- quarterly variations in our results of operations or those of our competitors;
- •
- changes in our earnings estimates or recommendations by securities analysts;
- •
- the payment of licensing fees or royalties in shares of our common stock;
- •
- announcements by us or our competitors of new prescription drug products or product candidates or non-prescription products, significant
contracts, commercial relationships, acquisitions or capital commitments;
- •
- announcements relating to future development or license agreements including termination of such agreements;
- •
- adverse developments with respect to our intellectual property rights or those of our principal collaborators;
- •
- commencement of litigation involving us or our competitors;
- •
- any major changes in our board of directors or management;
38
- •
- new legislation in the United States relating to the prescription, sale, distribution or pricing of gastrointestinal health products;
- •
- product liability claims, other litigation or public concern about the safety of our prescription drug product or product candidates and
non-prescription products or any such future products;
- •
- market conditions in the human or animal industry, in general, or in the gastrointestinal health sector, in particular, including performance
of our competitors; and
- •
- general economic conditions in the United States and abroad.
In addition, the stock market, in general, or the market for stocks in our industry, in particular, may experience broad market fluctuations, which may adversely affect the market price or liquidity of our common stock. Any sudden decline in the market price of our common stock could trigger securities class-action lawsuits against us. If any of our stockholders were to bring such a lawsuit against us, we could incur substantial costs defending the lawsuit and the time and attention of our management would be diverted from our business and operations. We also could be subject to damages claims if we were found to be at fault in connection with a decline in our stock price.
You may not be able to resell our common stock when you wish to sell them or at a price that you consider attractive or satisfactory.
Prior to our initial public offering in May 2015, there was no public market for shares of our common stock. The listing of our common stock on The Nasdaq Capital Market does not assure that a meaningful, consistent and liquid trading market exists. Although our common stock is listed on The Nasdaq Capital Market, trading volume in our common stock has been limited and an active trading market for our shares may never develop or be sustained. If an active market for our common stock does not develop, you may be unable to sell your shares when you wish to sell them or at a price that you consider attractive or satisfactory. The lack of an active market may also adversely affect our ability to raise capital by selling securities in the future, or impair our ability to license or acquire other product candidates, businesses or technologies using our shares as consideration.
If securities or industry analysts do not publish research or reports about our company, or if they issue adverse or misleading opinions regarding us or our stock, our stock price and trading volume could decline.
The trading market for our common stock depends in part on the research and reports that industry or financial analysts publish about us or our business. We do not influence or control the reporting of these analysts. If one or more of the analysts who do cover us downgrade or provide a negative outlook on our company or our industry, or the stock of any of our competitors, the price of our common stock could decline. If one or more of these analysts ceases coverage of our company, we could lose visibility in the market, which in turn could cause the price of our common stock to decline.
You may be diluted by conversions of outstanding non-voting common stock and Series A Preferred Stock, exchanges of our promissory notes and exercises of outstanding options and warrants.
As of March 31, 2019, we had (i) outstanding options to purchase an aggregate of 42,291 shares of our common stock at a weighted average exercise price of $412.94 per share, (ii) RSUs for 5,613 shares of voting common stock issuable upon vesting of outstanding restricted stock unit awards, (iii) warrants to purchase an aggregate of 71,821 shares of our common stock at a weighted-average exercise price of $90.00 per share and (iv) outstanding promissory notes in an aggregate principal amount of $10,446,751. As of March 31, 2019, we had 473,565 shares of common stock issuable upon conversion of outstanding shares of Series A convertible participating preferred stock ("Series A Preferred Stock"), with a conversion price of $19.425 per share.
39
The exercise of such options and warrants, conversion of the Series A Preferred Stock and exchange of the promissory notes for shares of our common stock will result in further dilution of your investment. You will experience additional dilution upon exercise of the stock options that we intend to grant to our executive officers, directors and other employees in connection with this offering, as further described in "Compensation of Directors and Executive Officers—Offering-Related Stock Option Grants to Executive Officers, Directors and Certain Employees." In addition, you may experience further dilution if we issue common stock in the future. As a result of this dilution, you may receive significantly less in net tangible book value than the full purchase price you paid for the shares in the event of liquidation.
If shares of our non-voting common stock are converted into shares of our voting common stock, your voting power will be diluted.
As of July 3, 2019, we had 1,837,763 shares of voting common stock and 40,301,237 shares of non-voting common stock (38,382 shares of voting common stock on an as converted basis) outstanding. Generally, holders of our non-voting common stock have no voting power (other than in connection with a change of control of our company) and have no right to participate in any meeting of stockholders or to have notice thereof. However, shares of our non-voting common stock that are converted into voting common stock will have all the voting rights of the voting common stock. Shares of our non-voting common stock are convertible into shares of our voting common stock on a one thousand fifty-for-one basis (i) at the option of the respective holders thereof, at any time and from time to time on or after April 1, 2018 or (ii) automatically, without any payment of additional consideration by the holder thereof, (x) upon a transfer of such shares to any person or entity that is neither an affiliate of Nantucket nor an investment fund, investment vehicle or other account, that is, directly or indirectly, managed or advised by Nantucket or any of its affiliates pursuant to a sale of such stock to a third-party for cash in accordance with the terms and condition set forth in the Investor Rights Agreement, or (y) upon the subsequent release or transfer of such shares to the registered pre-Merger legacy stockholders of Napo's outstanding shares of common stock as of July 31, 2017 (the "Napo Legacy Stockholders"). Upon conversion of any non-voting common stock, your voting power will be diluted in proportion to the decrease in your ownership of the total outstanding voting common stock.
Provisions in our charter documents and under Delaware law could discourage a takeover that stockholders may consider favorable and may lead to entrenchment of management.
Our third amended and restated certificate of incorporation and amended and restated bylaws contain provisions that could delay or prevent changes in control or changes in our management without the consent of our board of directors. These provisions to include the following:
- •
- a classified board of directors with three-year staggered terms, which may delay the ability of stockholders to change the membership of a
majority of our board of directors;
- •
- no cumulative voting in the election of directors, which limits the ability of minority stockholders to elect director candidates;
- •
- the exclusive right of our board of directors to elect a director to fill a vacancy created by the expansion of the board of directors or the
resignation, death or removal of a director, which prevents stockholders from being able to fill vacancies on our board of directors;
- •
- the ability of our board of directors to authorize the issuance of shares of preferred stock and to determine the terms of those shares, including preferences and voting rights, without stockholder approval, which could adversely affect the rights of our common stockholders or be used to deter a possible acquisition of our company;
40
- •
- the ability of our board of directors to alter our bylaws without obtaining stockholder approval;
- •
- the required approval of the holders of at least 75% of the shares entitled to vote at an election of directors to adopt, amend or repeal our
bylaws or repeal the provisions of our third amended and restated certificate of incorporation regarding the election and removal of directors;
- •
- a prohibition on stockholder action by written consent, which forces stockholder action to be taken at an annual or special meeting of our
stockholders;
- •
- the requirement that a special meeting of stockholders may be called only by the chairman of the board of directors, the chief executive
officer, the president or the board of directors, which may delay the ability of our stockholders to force consideration of a proposal or to take action, including the removal of directors; and
- •
- advance notice procedures that stockholders must comply with in order to nominate candidates to our board of directors or to propose matters to be acted upon at a stockholders' meeting, which may discourage or deter a potential acquirer from conducting a solicitation of proxies to elect the acquirer's own slate of directors or otherwise attempting to obtain control of us.
These provisions could inhibit or prevent possible transactions that some stockholders may consider attractive.
We are also subject to the anti-takeover provisions contained in Section 203 of the Delaware General Corporation Law. Under Section 203, a corporation generally may not engage in a business combination with any holder of 15% or more of its capital stock unless the holder has held the stock for three years or, among other exceptions, the board of directors has approved the transaction.
Our amended and restated bylaws designate the Court of Chancery of the State of Delaware as the sole and exclusive forum for certain actions and proceedings that may be initiated by our stockholders, which could limit our stockholders' ability to obtain a favorable judicial forum for disputes with us or our directors, officers or other employees.
Our amended and restated bylaws provide that, unless we consent in writing to an alternative forum, the Court of Chancery of the State of Delaware will be the sole and exclusive forum for (i) any derivative action or proceeding brought on our behalf, (ii) any action asserting a claim of breach of a fiduciary duty owed by any director, officer or other employee to us or our stockholders, (iii) any action asserting a claim arising pursuant to any provision of the Delaware General Corporation Law, (iv) any action asserting a claim that is governed by the internal affairs doctrine or (v) any action to interpret, apply, enforce or determine the validity of our certificate of incorporation or bylaws. Any person purchasing or otherwise acquiring any interest in any shares of our capital stock shall be deemed to have notice of and to have consented to this provision of our amended and restated bylaws. This choice-of-forum provision may limit our stockholders' ability to bring a claim in a judicial forum that they find favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits. Alternatively, if a court were to find this provision of our amended and restated bylaws inapplicable or unenforceable with respect to one or more of the specified types of actions or proceedings, we may incur additional costs associated with resolving such matters in other jurisdictions, which could harm our business and financial condition.
We do not intend to pay dividends on our common stock, and your ability to achieve a return on your investment will depend on appreciation in the market price of our common stock.
We currently intend to invest our future earnings, if any, to fund our growth and not to pay any cash dividends on our common stock. Moreover, so long as either (i) Nantucket or any of its affiliates owns any shares of our non-voting common stock or (ii) Sagard Capital Partners, L.P. or any of its affiliates owns 35% or more of the shares of our Series A Preferred Stock, we cannot pay dividends on
41
our common stock or non-voting common stock without obtaining the prior written consent of Nantucket or Sagard, respectively. Because we do not intend to pay dividends and may be required to obtain written consent if we were to do so, your ability to receive a return on your investment will depend on any future appreciation in the market price of our common stock. We cannot be certain that our common stock will appreciate in price.
Our principal stockholders own a significant percentage of our voting stock and will be able to exert significant control over matters subject to stockholder approval.
As of March 31, 2019, our executive officers, directors, holders of 5% or more of our capital stock and their respective affiliates beneficially owned in the aggregate approximately 61% of the outstanding shares of our voting common stock. As a result of their stock ownership, these stockholders may have the ability to influence our management and policies, and will be able to significantly affect the outcome of matters requiring stockholder approval such as elections of directors, amendments of our organizational documents or approvals of any merger, sale of assets or other major corporate transaction. This may prevent or discourage unsolicited acquisition proposals or offers for our common stock that you may feel are in your best interest as one of our stockholders.
The requirements of being a public company, including compliance with the reporting requirements of the Exchange Act and the requirements of the Sarbanes-Oxley Act, may strain our resources, increase our costs and distract management, and we may be unable to comply with these requirements in a timely or cost-effective manner.
Our initial public offering had a significant, transformative effect on us. Prior to our initial public offering, our business operated as a privately-held company, and we were not required to comply with public reporting, corporate governance and financial accounting practices and policies required of a publicly-traded company. As a publicly-traded company, we incur significant additional legal, accounting and other expenses compared to historical levels. In addition, new and changing laws, regulations and standards relating to corporate governance and public disclosure, including the Dodd-Frank Wall Street Reform and Consumer Protection Act and the rules and regulations thereunder, as well as under the Sarbanes-Oxley Act, the JOBS Act and the rules and regulations of the SEC and The NASDAQ Capital Market, may result in an increase in our costs and the time that our board of directors and management must devote to our compliance with these rules and regulations. These rules and regulations have substantially increased our legal and financial compliance costs and diverted management time and attention from our product development and other business activities.
The Sarbanes-Oxley Act requires, among other things, that we assess the effectiveness of our internal control over financial reporting annually and the effectiveness of our disclosure controls and procedures quarterly. In particular, Section 404 of the Sarbanes-Oxley Act, or Section 404, requires us to perform system and process evaluation and testing of our internal control over financial reporting to allow management to report on, and our independent registered public accounting firm potentially to attest to, the effectiveness of our internal control over financial reporting. We have needed to expend time and resources on documenting our internal control over financial reporting so that we are in a position to perform such evaluation when required. As an "emerging growth company," we expect to avail ourselves of the exemption from the requirement that our independent registered public accounting firm attest to the effectiveness of our internal control over financial reporting under Section 404. However, we may no longer avail ourselves of this exemption when we cease to be an "emerging growth company." When our independent registered public accounting firm is required to undertake an assessment of our internal control over financial reporting, the cost of our compliance with Section 404 will correspondingly increase. Our compliance with applicable provisions of Section 404 requires that we incur substantial accounting expense and expend significant management time on compliance-related issues as we implement additional corporate governance practices and
42
comply with reporting requirements. Moreover, if we are not able to comply with the requirements of Section 404 applicable to us in a timely manner, or if we or our independent registered public accounting firm identifies deficiencies in our internal control over financial reporting that are deemed to be material weaknesses, the market price of our stock could decline and we could be subject to sanctions or investigations by the SEC or other regulatory authorities, which would require additional financial and management resources.
We are an "emerging growth company" and we cannot be certain if the reduced disclosure requirements applicable to "emerging growth companies" will make our common stock less attractive to investors.
We are an "emerging growth company," as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act, and we may take advantage of certain exemptions and relief from various reporting requirements that are applicable to other public companies that are not "emerging growth companies." In particular, while we are an "emerging growth company" (i) we will not be required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act, (ii) we will be subject to reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements and (iii) we will not be required to hold nonbinding advisory votes on executive compensation or stockholder approval of any golden parachute payments not previously approved. In addition, the JOBS Act provides that an emerging growth company can delay its adoption of any new or revised accounting standards, but we have irrevocably elected not to avail ourselves of this exemption and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies. In addition, investors may find our common stock less attractive if we rely on the exemptions and relief granted by the JOBS Act. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may decline and/or become more volatile.
We may remain an "emerging growth company" until as late as December 31, 2020 (the fiscal year-end following the fifth anniversary of the closing of our initial public offering, which occurred on May 18, 2015), although we may cease to be an "emerging growth company" earlier under certain circumstances, including (i) if the market value of our common stock that is held by non-affiliates exceeds $700.0 million as of any June 30, in which case we would cease to be an "emerging growth company" as of December 31 of such year, (ii) if our gross revenue exceeds $1.07 billion in any fiscal year or (iii) if we issue more than $1.0 billion of non-convertible debt over a three-year period.
Issuances of shares of common stock or securities convertible into or exercisable for shares of common stock following this offering, as well as the exercise of options and warrants outstanding, will dilute your ownership interests and may adversely affect the future market price of our common stock.
The issuance of additional shares of our common stock or securities convertible into or exchangeable for our common stock could be dilutive to stockholders if they do not invest in future offerings. We intend to use the net proceeds from this offering to continue to fund the development of our business and for general corporate purposes, which may include capital expenditures and funding our working capital needs. We may seek additional capital through a combination of private and public equity offerings, debt financings, strategic partnerships and alliances and licensing arrangements, which may cause your ownership interest to be diluted.
In addition, we have a significant number of options and warrants to purchase shares of our common stock outstanding. If these securities are exercised or converted, you may incur further dilution. Moreover, to the extent that we issue additional options or warrants to purchase, or securities convertible into or exchangeable for, shares of our common stock in the future and those options, warrants or other securities are exercised, converted or exchanged, stockholders may experience further dilution.
43
We effected two reverse stock splits since January 1, 2018, which may not achieve one or more of our objectives.
We have effected two reverse stock splits since January 1, 2018, each of which has impacted the trading liquidity of the shares of our common stock. There can be no assurance that the market price per share of our common stock after a reverse stock split will remain unchanged or increase in proportion to the reduction in the number of shares of our common stock outstanding before the reverse stock split. The market price of our shares may fluctuate and potentially decline after a reverse stock split. Accordingly, the total market capitalization of our common stock after a reverse stock split may be lower than the total market capitalization before the reverse stock split. Moreover, the market price of our common stock following a reverse stock split may not exceed or remain higher than the market price prior to the reverse stock split.
Additionally, there can be no assurance that a reverse stock split will result in a per-share market price that will attract institutional investors or investment funds or that such share price will satisfy investing guidelines of institutional investors or investment funds. As a result, the trading liquidity of our common stock may not necessarily improve. Further, if a reverse stock split is effected and the market price of our common stock declines, the percentage decline may be greater than would occur in the absence of a reverse stock split.
Risks Relating to this Offering
Our management team and board of directors will have immediate and broad discretion over the use of the net proceeds from this offering and we may use the net proceeds in ways with which you disagree.
The net proceeds from this offering will be immediately available to our management to use at their discretion. We currently intend to use the net proceeds as discussed under "Use of Proceeds" in this prospectus. We have not allocated specific amounts of the net proceeds from this offering for any other purposes. Accordingly, our management and board of directors will have significant discretion and flexibility in applying the net proceeds of this offering. You will be relying on the judgment of our management and board of directors with regard to the use of these net proceeds, and you will not have the opportunity, as part of your investment decision, to assess whether the proceeds are being used appropriately. It is possible that the net proceeds will be invested in a way that does not result in a favorable, or any, return for us or our stockholders. The failure of our management to use such funds effectively could have a material adverse effect on our business, prospects, financial condition, and results of operation.
You will experience immediate and substantial dilution in the net tangible book value per share of the Common Stock in this offering.
Since the price per share of Common Stock included in the Units being offered is substantially higher than the net tangible book value per share of our Common Stock outstanding prior to this offering, you will suffer immediate and substantial dilution in the net tangible book value per share of Common Stock included in each Unit or issuable upon exercise of warrants in this offering. See the section titled "Dilution" below for a more detailed discussion of the dilution you will incur if you purchase Units in this offering.
The terms of the Series B Preferred Stock and the warrants could impede our ability to enter into certain transactions or obtain additional financing.
The terms of the Series B Preferred Stock and the warrants require us, upon the consummation of any "fundamental transaction" (as defined in "Description of Securities We Are Offering—Series B Preferred Stock"), to, among other obligations, cause any successor entity resulting from the fundamental transaction to assume all of our obligations under the Series B Preferred Stock and the
44
warrants and the associated transaction documents. In addition, holders of Series B Preferred Stock and warrants are entitled to participate in any fundamental transaction on an as-converted or as-exercised basis, which could result in the holders of our common stock receiving a lesser portion of the consideration from a fundamental transaction. The terms of the Series B Preferred Stock and the warrants could also impede our ability to enter into certain transactions or obtain additional financing in the future.
The Series B Preferred Stock and warrants will not be listed on any securities exchange and as such there will not be a public market for such securities.
There is no established public trading market for the Series B Preferred Stock or warrants, and we do not expect a market to develop. In addition, we do not intend to apply for listing of the Series B Preferred Stock or warrants on any securities exchange or trading system. Without an active market, the liquidity of the Series B Preferred Stock and warrants will be limited, and investors may be unable to liquidate their investments in the Series B Preferred Stock and warrants.
The offering price will be set by our Board of Directors and does not necessarily indicate the actual or market value of our common stock.
Our Board of Directors will approve the offering price and other terms of this offering after considering, among other things: the number of shares authorized in our certificate of incorporation; the current market price of our common stock; trading prices of our common stock over time; the volatility of our common stock; our current financial condition and the prospects for our future cash flows; the availability of and likely cost of capital of other potential sources of capital; and market and economic conditions at the time of the offering. The offering price is not intended to bear any relationship to the book value of our assets or our past operations, cash flows, losses, financial condition, net worth or any other established criteria used to value securities. The offering price may not be indicative of the fair value of the common stock.
The warrants may not have any value.
The Series 1 warrants will be exercisable beginning on the date of issuance and expire on the earlier of (1) 12 months from the date of issuance and (2) 30 calendar days following the public announcement of Positive Interim Results (as defined on page 63) related to the diarrhea results from the HALT-D Investigator Initiated Trial, at an initial exercise price per share of $5.95 (125% of the offering price per Class A Unit). The Series 2 warrants will be exercisable beginning on the date of issuance and expire on the earlier of (1) 5 years from the date of issuance and (2) 30 calendar days following the public announcement by us that a pivotal trial using crofelemer (Mytesi, or the same or similar product with a different name) for the treatment of cancer therapy related diarrhea has met its primary endpoint in accordance with the protocol, at an initial exercise price per share of $5.95 (125% of the offering price per Class A Unit).
In the event that the price of a share of our common stock does not exceed the exercise price of the warrants during the period when the warrants are exercisable, the warrants may not have any value.
A warrant does not entitle the holder to any rights as common stockholders until the holder exercises the warrant for shares of our common stock.
Until you acquire shares of our common stock upon exercise of your warrants, the warrants will not provide you any rights as a common stockholder. Upon exercise of your warrants, you will be entitled to exercise the rights of a common stockholder only as to matters for which the record date occurs on or after the exercise date.
45
There is a limited trading market for our common stock, which could make it difficult to liquidate an investment in our common stock, in a timely manner.
Our common stock is currently traded on the Nasdaq Capital Market. Because there is a limited public market for our common stock, investors may not be able to liquidate their investment whenever desired. We cannot assure that there will be an active trading market for our common stock and the lack of an active public trading market could mean that investors may be exposed to increased risk. In addition, if we failed to meet the criteria set forth in SEC regulations, various requirements would be imposed by law on broker-dealers who sell our securities to persons other than established customers and accredited investors. Consequently, such regulations may deter broker-dealers from recommending or selling our common stock, which may further affect its liquidity.
46
The following net loss per share, basic and diluted, has been derived from the audited financial statements of the Company contained in our Annual Report on Form 10-K for the fiscal year ended December 31, 2018, and our Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2019, which are incorporated by reference in this prospectus, except that the net loss per share, basic and diluted, for the three months ended March 31, 2019 and 2018 and the year ended December 31, 2018 and 2017 have been revised to reflect the 1-for-70 reverse stock split of our issued and outstanding shares of Common Stock effective on June 7, 2019, as shown below.
The historical financial information set forth below may not be indicative of our future performance and should be read together with "Management's Discussion and Analysis of Financial Condition and Results of Operations" and our historical financial statements and notes to those statements included in our Annual Report on Form 10-K for the fiscal year ended December 31, 2018, our Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2019, and any amendment or update thereto reflected in subsequent filings with the SEC, and all other annual, quarterly and other reports that we file with the SEC after the date of the initial registration statement of which this prospectus forms a part and that also are incorporated herein by reference.
| |
|
|
Three Months Ended March 31, |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Year Ended December 31, 2018 |
Year Ended December 31, 2017 |
|||||||||||
| |
2019 | 2018 | |||||||||||
Net loss per share, basic and diluted |
$ | (153.27 | ) | $ | (531.06 | ) | $ | (16.84 | ) | $ | (54.27 | ) | |
47
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus and the documents incorporated by reference into it contain forward-looking statements within the meaning of Section 27A of the Securities Act, and Section 21E of the Securities Exchange Act of 1934, as amended (the "Exchange Act"). We have made these statements in reliance on the safe harbor provisions of the Private Securities Litigation Reform Act of 1995. All statements other than statements of historical facts contained in or incorporated by reference into this prospectus, including statements regarding our future results of operations and financial position, business strategy, prospective products, product approvals, research and development costs, timing of receipt of clinical trial, field study and other study data, and likelihood of success, commercialization plans and timing, other plans and objectives of management for future operations, and future results of current and anticipated products are forward-looking statements. These statements involve known and unknown risks, uncertainties and other important factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements.
In some cases, you can identify forward-looking statements by terms such as "may," "will," "should," "expect," "plan," "aim," "anticipate," "could," "intend," "target," "project," "contemplate," "believe," "estimate," "predict," "potential" or "continue" or the negative of these terms or other similar expressions. The forward-looking statements in this prospectus are only predictions. We have based these forward-looking statements largely on our current expectations and projections about future events and financial trends that we believe may affect our business, financial condition and results of operations. These forward-looking statements speak only as of the date of this prospectus and are subject to a number of risks, uncertainties and assumptions including those listed in the "Risk Factors" incorporated by reference into this prospectus from our Annual Report on Form 10-K, as updated by subsequent reports. Forward-looking statements are subject to inherent risks and uncertainties, some of which cannot be predicted or quantified and some of which are beyond our control. The events and circumstances reflected in our forward-looking statements may not be achieved or occur and actual results could differ materially from those projected in the forward-looking statements. Moreover, we operate in a dynamic industry and economy. New risk factors and uncertainties may emerge from time to time, and it is not possible for management to predict all risk factors and uncertainties that we may face. Except as required by applicable law, we do not plan to publicly update or revise any forward-looking statements contained herein, whether as a result of any new information, future events, changed circumstances or otherwise.
Unless otherwise indicated, information contained in this prospectus concerning our industry and the markets in which we operate, including our general expectations and market position, market opportunity and market share, is based on information from our own management estimates and research, as well as from industry and general publications and research, surveys and studies conducted by third parties. Management estimates are derived from publicly available information, our knowledge of our industry and assumptions based on such information and knowledge, which we believe to be reasonable. In addition, assumptions and estimates of our and our industry's future performance are necessarily subject to a high degree of uncertainty and risk due to a variety of factors, including those described in "Risk Factors." These and other factors could cause our future performance to differ materially from our assumptions and estimates. See "Special Note Regarding Forward-Looking Statements."
48
The net proceeds from this offering will be approximately $10.2 million, or approximately $11.8 million if the underwriter exercises its option to purchase additional shares in full, based on an assumed public offering price of $4.76 per Class A Unit and $1,000 per Class B Unit, and after deducting the estimated underwriting discounts and commissions and before estimated offering expenses payable by us.
We intend to use the net proceeds from this offering as follows:
- •
- approximately $3.0 million to fund non-clinical pipeline and business development activities;
- •
- approximately $5,193,000 to repay the aggregate principal amount and related interest on the Bridge Notes (approximately $3.55 million
of which is contributed by holders of Bridge Notes participating in this offering);
- •
- $250,000 to pay Sagard in consideration for their consent and waiver to the refinancing of approximately $10.5 million outstanding
aggregate amount of convertible promissory notes issued by Napo pursuant to the Amended and Restated Note Purchase Agreement, dated March 31, 2017, by and between Napo, Kingdon Associates, M.
Kingdon Offshore Master Fund L.P., Kingdon Family Partnership, L.P., and Kingdon Credit Master Fund L.P.
- •
- the remainder for working capital and other general corporate purposes.
As noted above, we will use a portion of the net proceeds from this offering to repay the aggregate principal amount and the related interest thereon for certain of the Bridge Notes. Affiliates to be repaid include Sagard Capital Partners, L.P., in the aggregate amount of approximately $508,712; Jim Bochnowski, in the aggregate amount of $361,162; Lisa Conte, in the aggregate amount of $101,611; and Jonathan Siegel, in the aggregate amount of $76,233. These promissory notes have a simple interest rate of 12% per year. The principal and related interest on these notes is due and payable in full on July 31, 2019. We used the proceeds from the sale and issuance of these notes for working capital and other general corporate purposes.
Pending our use of the net proceeds from this offering, we intend to invest the net proceeds in a variety of capital preservation investments, including short-term, investment grade, interest bearing instruments and U.S. government securities.
This expected use of the net proceeds to us from this offering represents our intentions based upon our current plans and business conditions. The amounts and timing of our actual expenditures may vary significantly depending on numerous factors, including the progress of our research and development efforts, the status of and results from clinical trials, any collaborations that we may enter into with third parties for our programs, any unforeseen cash needs and the factors described in "Risk Factors". Accordingly, our management will have broad discretion in applying the net proceeds from this offering. An investor will not have the opportunity to evaluate the economic, financial or other information on which we base our decisions on how to use the proceeds.
Based on our current plans, we believe that our existing cash and cash equivalents, together with the net proceeds from this offering, will be sufficient to enable us to fund our operating expenses and capital expenditure requirements through to a cash flow positive situation. We have based this estimate on assumptions that may prove to be wrong, and we could use our available capital resources sooner than we currently expect. We will need to raise substantial additional funds before we can expect to commercialize additional products, if approved. We may satisfy our future cash needs through the sale of equity securities, debt financings, working capital lines of credit, corporate collaborations or license agreements, grant funding, interest income earned on invested cash balances or a combination of one or more of these sources.
49
We have never declared or paid any cash dividends on our capital stock. We intend to retain future earnings, if any, to finance the operation and expansion of our business and do not anticipate paying any cash dividends in the foreseeable future. Any future determination related to dividend policy will be made at the discretion of our board of directors after considering our financial condition, results of operations, capital requirements, business prospects and other factors the board of directors deems relevant, and subject to the restrictions contained in any future financing instruments.
50
You should read this information in conjunction with our financial statements and related notes incorporated by reference in this prospectus.
The following table sets forth our cash and cash equivalents and capitalization, as adjusted to reflect the reverse split, as of March 31, 2019, as follows:
- •
- on an actual basis;
- •
- on a pro forma basis to give effect to (i) the issuance of 882,742 shares of common stock in exchange for a reduction of approximately
$6.8 million in the amount outstanding of the CVP Notes and Exchange Notes, which issuances occurred between April 10, 2019 and June 27, 2019 and (ii) the issuance of
1,168,592 shares of common stock for warrant exercises associated with the Bridge Notes which issuances occurred between March 18, 2019 and June 26, 2019 and for which we received cash
proceeds in the aggregate amount of $5,050,000; and
- •
- on a pro forma as adjusted basis to give further effect to (i) the repayment of the Bridge Notes upon consummation of the offering and (ii) the receipt of the estimated net proceeds from the sale by us in this offering of 2,521,008 Class A Units, at the assumed public offering price of $4.76 per Class A Unit, or 12,000 Class B Units, at the public offering price of $1,000 per Class B Unit, assuming conversion of all Series B Convertible Preferred Shares included in the Class B Units, or some combination thereof, and after deducting the estimated underwriting discounts and commissions and estimated expenses payable by us.
The following table is illustrative only and will be adjusted based on the actual public offering price and other terms of this offering determined at pricing.
You should read this table in conjunction with "Selected Financial Data" included elsewhere in this prospectus and with "Management's Discussion and Analysis of Financial Condition and Results of Operations" in our Annual Report on Form 10-K for the year ended December 31, 2018, as updated in
51
our Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2019, and our financial statements and related notes incorporated by reference herein.
| |
As of March 31, 2019 (unaudited) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
Actual | Pro Forma(1) | Pro Forma, as adjusted(1) |
|||||||
| |
(in thousands) |
|||||||||
Cash and cash equivalents |
$ | 2,572 | 7,622 | 12,816 | ||||||
| | | | | | | | | | | |
Series A convertible preferred stock: $0.0001 par value, 10,000,000 shares authorized at March 31, 2019 and December 31, 2018; 78,928 shares issued and outstanding at March 31, 2019 and December 31, 2018; (redemption value and liquidation preference of $12,738,822 and $9,199,002 at March 31, 2019 and December 31, 2018, respectively); |
$ | 9,000 | 9,000 | 9,000 | ||||||
Stockholders' (deficit) equity: |
||||||||||
Common stock: $0.0001 par value, 150,000,000 shares authorized at March 31, 2019 and December 31, 2018, respectively; 848,786 and 351,473 shares issued and outstanding at March 31, 2019 and December 31, 2018, respectively |
6 |
7 |
7 |
|||||||
Common stock: non-voting: $0.0001 par value, 50,000,000 shares authorized at March 31, 2019 and December 31, 2018; 40,301,237 shares issued and outstanding at March 31, 2019 and December 31, 2018 |
4 |
— |
— |
|||||||
Additional paid-in capital |
109,641 |
115,203 |
125,446 |
|||||||
Accumulated deficit |
(102,855 |
) |
(103,571 |
) |
(103,571 |
) |
||||
| | | | | | | | | | | |
Total stockholders' equity |
$ | 6,796 | 11,632 | 21,875 | ||||||
| | | | | | | | | | | |
Total capitalization |
$ | 15,796 | $ | 20,632 | $ | 30,875 | ||||
- (1)
- The common stock includes an additional 38,382 shares of voting common stock in the "Pro Forma" and "Pro Forma, as adjusted" columns which reflects the conversion of 40,301,237 non-voting common shares as converted to a voting common share basis.
52
If you purchase shares in this offering, your interest will be diluted to the extent of the difference between the public offering price per share of common stock and warrant and the as adjusted net tangible book value per share of our common stock after this offering. Our net tangible book value as of March 31, 2019 was ($24.5) million, or ($27.61) per share of common stock (based upon 848,786 outstanding shares of common stock). "Net tangible book value" is total assets minus the sum of liabilities and intangible assets. "Net tangible book value per share" is net tangible book value divided by the total number of shares of common stock outstanding.
Our pro forma net tangible book value as of March 31, 2019 would have been ($1.9) million, or ($0.18) per share of common stock (based upon 10,501,463 outstanding shares of common stock), after giving effect to (i) the issuance of 882,742 shares of common stock in exchange for a reduction of approximately $6.8 million in the amount outstanding of the CVP Notes and Exchange Notes, which issuances occurred between April 10, 2019 and June 27, 2019 (the "Notes Exchanges") and (ii) the issuance of 1,168,592 shares of common stock for warrant exercises associated with the Bridge Notes which issuances occurred between March 18. 2019 and June 26, 2019 and for which we received cash proceeds in the aggregate amount of $5,050,000 (the "Bridge Financing").
After giving effect to (i) the pro forma adjustments described above, (ii) the repayment of the Bridge Notes upon consummation of the offering and (iii) the sale by us in this offering of 2,521,008 Class A Units, at the assumed public offering price of $4.76 per Class A Unit or 12,000 Class B Units, at the public offering price of $1,000 per Class B Unit, assuming conversion of all Series B Convertible Preferred Shares included in the Class B Units, or some combination thereof, and after deducting the estimated underwriting discounts and commissions and estimated offering expenses that we will pay, our as adjusted net tangible book value as of March 31, 2019 would have been approximately ($1.9) million, or ($0.18) per share of common stock. This amount represents an immediate increase in net tangible book value of $27.42 per share to existing stockholders and an immediate dilution of $4.94 per share to purchasers in this offering. The following table illustrates the dilution on a per share basis:
The following table illustrates this dilution:
Assumed public offering price per Class A Unit |
$ | 4.76 | |||||
Historical net tangible book value per share as of March 31, 2019 |
$ | (27.61 | ) | ||||
Pro forma increase in net tangible book value per share attributable to the Notes Exchanges and the Bridge Financing |
$ | 1.52 | |||||
Pro forma net tangible book value per share as of March 31, 2019 |
$ | (26.09 | ) | ||||
| | | | | | | | |
Increase in pro forma net tangible book value per share attributable to this offering |
$ | 25.91 | |||||
| | | | | | | | |
Pro forma as adjusted net tangible book value per share after this offering |
$ | (0.18 | ) | ||||
| | | | | | | | |
Dilution in pro forma as adjusted net tangible book value per share to new investors in this offering |
$ | 4.94 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The dilution information set forth in the table above is illustrative only and will be adjusted based on the actual public offering price and other terms of this offering determined at pricing.
If the underwriters exercise in full their over-allotment option, our pro forma as adjusted net tangible book value per share after giving effect to this offering would be approximately $(9.4) million, or $(0.98) per share, which amount represents an immediate increase in net tangible book value of $0.71 per share to existing stockholders and a dilution to new investors of $5.74 per share.
53
The number of shares of our common stock to be outstanding after this offering is based on 848,786 shares of our voting common stock and 40,307,247 (38,832 shares of voting common stock on an as converted basis) shares of our non-voting common stock outstanding as of March 31, 2019, and excludes the following:
- •
- 10 shares of common stock issuable upon exercise of the placement agent warrant;
- •
- 1,168,592 shares of common stock issuable upon exercise of the Bridge Warrants issuable pursuant to the Securities Purchase Agreement entered
into beginning on March 18, 2019 by and among the Company and selected accredited investors assuming an exercise price equal to the assumed public offering price per Class A Unit of
$4.76;
- •
- 473,565 shares of common stock issuable upon conversion of outstanding preferred stock as of March 31, 2019, with a weighted-average
conversion price of $19.425 per share;
- •
- 39,307 shares of voting common stock issuable upon exercise of outstanding options as of March 31, 2019, with a weighted average
exercise price of $412.94 per share;
- •
- 2,984 shares of common stock issuable upon exercise of outstanding inducement options as of March 31, 2019 with a weighted-average
exercise price of $122.49 per share;
- •
- 11,558 shares of voting common stock reserved for future issuance under the 2014 Stock Incentive Plan, which includes shares of common stock
that will be issuable upon exercise of options that we expect to grant on the closing date of this offering;
- •
- 71,821 shares of voting common stock issuable upon exercise of warrants outstanding as of March 31, 2019, with a weighted average
exercise price of $90.00 per share; and
- •
- 5,613 shares of voting common stock issuable upon vesting of outstanding restricted stock unit awards, or RSUs, as of March 31, 2019.
54
DESCRIPTION OF SECURITIES WE ARE OFFERING
Description of Units
We are offering 2,521,008 Class A Units, with each Class A Unit consisting of (i) one share of common stock, par value $0.0001 per share, (ii) one Series 1 warrant to purchase one share of our common stock, and (iii) one Series 2 warrant to purchase one share of our common stock at an assumed public offering price of $4.76 per Class A Unit. The Series 1 warrants have an exercise price that is 125% of the assumed public offering price of the Class A Units. The Series 2 warrants have an exercise price that is 125% of the assumed public offering price of the Class A Units.
We are also offering to those purchasers whose purchase of common stock in this offering would result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock following the consummation of this offering, the opportunity to purchase, if they so choose, in lieu of the shares of common stock that would result in ownership in excess of 4.99% (or, at the election of the purchaser, 9.99%), 12,000 Class B Units. Each Class B Unit will consist of one share of Series B Preferred Stock with a stated value of $1,000 and convertible into shares of common stock at the public offering price of the Class A Units, together with the equivalent number of warrants as would have been issued to such purchaser of Class B Units if they had purchased Class A Units, at an assumed offering price of $1,000 per Class B Unit. For each Class B Unit we sell, the number of Class A Units we are offering will be decreased by $1,000 divided by the Class A Unit public offering price. The Series 1 warrants have an exercise price that is 125% of the assumed public offering price of the Class A Units. The Series 2 warrants have an exercise price that is 125% of the assumed public offering price of the Class A Units.
The securities of which the Units are composed (the "underlying securities") are being sold in this offering only as part of the units. However, the Class A Units and Class B Units will not be certificated and the underlying securities comprising such units are immediately separable. Each underlying security purchased in this offering will be issued independent of each other underlying security and not as part of a unit. Upon issuance, each underlying security may be transferred independent of any other underlying security, subject to applicable law and transfer restrictions.
Common Stock
- •
- As of May 31, 2019, we had 1,006,742 shares of voting common stock and 40,301,237 shares of non-voting common stock (38,382 shares of
voting common stock on an as converted basis) outstanding and 5,524,926 shares of preferred stock (473,565 shares of voting common stock on an as converted basis) outstanding. See "Capitalization"
beginning on page 50 of this prospectus for a description of the exclusions pertaining to the outstanding share information.
- •
- As of May 31, 2019, we had 29 record holders of common stock.
Voting Rights
The holders of our voting common stock are entitled to one vote per share on all matters to be voted on by our stockholders. Subject to preferences that may be applicable to any outstanding shares of preferred stock, holders of common stock are entitled to receive ratably such dividends as may be declared by our board of directors out of funds legally available for that purpose. In the event of our liquidation, dissolution or winding up, the holders of common stock are entitled to share ratably in all assets remaining after the payment of liabilities, subject to the prior distribution rights of preferred stock then outstanding. Holders of common stock have no preemptive, conversion or subscription rights. There are no redemption or sinking fund provisions applicable to the common stock.
55
Dividends
Subject to preferences that may be applicable to any outstanding preferred stock, holders of common stock are entitled to receive dividends, if any, as may be declared from time to time by our board of directors out of legally available funds.
Liquidation
In the event of our liquidation, dissolution or winding up, holders of common stock will be entitled to share ratably in the net assets legally available for distribution to stockholders after the payment of all of our debts and other liabilities and the satisfaction of any liquidation preference granted to the holders of any then outstanding shares of preferred stock.
Rights and Preferences
Holders of common stock have no preemptive, conversion, subscription or other rights, and there are no redemption or sinking fund provisions applicable to the common stock. The rights, preferences, and privileges of the holders of common stock are subject to and may be adversely affected by, the rights of the holders of shares of any series of preferred stock that we may designate in the future.
Fully Paid and Nonassessable
All of our outstanding shares of common stock are, and the shares of common stock to be issued pursuant to this offering, when paid for, will be fully paid and nonassessable.
Quotation on the NASDAQ Capital Market
Our common stock is quoted on the NASDAQ Capital Market under the symbol "JAGX".
Transfer Agent
The transfer agent of our common stock is AST. Their address is 6201 15th Avenue, Brooklyn, New York, 11219.
Series B Preferred Stock
General
In connection with this offering, our board of directors will designate shares of our preferred stock as Series B Preferred Stock. The preferences and rights of the Series B Preferred Stock will be as set forth in a Certificate of Designation (the "Series B Certificate of Designation") filed as an exhibit to the registration statement of which this prospectus is a part.
Conversion
Each share of Series B Preferred Stock will be initially convertible at any time at the holder's option into the number of shares of our common stock determined by dividing the $1,000 stated value per share of the Series B Preferred Stock by an assumed conversion price of $4.76 per share, which was the closing price of our common stock on June 27, 2019. The conversion price per share will be subject to adjustment for stock splits, stock dividends, distributions, subdivisions and combinations. Notwithstanding the foregoing, the Series B Certificate of Designation will further provide that we shall not effect any conversion of the Series B Preferred Stock, with certain exceptions, to the extent that, after giving effect to an attempted conversion, the holder of Series B Preferred Stock (together with such holder's affiliates, and any persons acting as a group together with such holder or any of such holder's affiliates) would beneficially own a number of shares of Common Stock in excess of 4.99% (or,
56
at the election of the purchaser prior to the date of issuance, 9.99%) of the shares of our common stock then outstanding after giving effect to such exercise (the "Series B Preferred Stock Beneficial Ownership Limitation").
Fundamental Transaction
In the event we consummate a merger or consolidation with or into an individual or corporation, partnership, trust, incorporated or unincorporated association, joint venture, limited liability company, joint stock company, government or other entity of any kind, pursuant to which our Common Stock is effectively converted or exchanged for other securities, cash or other property, or we, directly or indirectly, in one or more related transactions effects any reclassification, reorganization or recapitalization of the Common Stock or any compulsory share exchange pursuant to which the Common Stock is effectively converted into or exchanged for other securities, cash or property (each, a "Fundamental Transaction"), then immediately prior but subject to the occurrence of such Fundamental Transaction, each outstanding share of Series B Preferred Stock will automatically convert into shares of Common Stock, without any action of or by the holders of the Series B Preferred Stock or us, at the conversion price then in effect, and the holders of Series B Preferred Stock will receive, for each conversion share, such consideration, at the same time and subject to the same terms and conditions, as the other holders of Common Stock pursuant to the terms of such Fundamental Transaction.
Liquidation Preference
In the event of a liquidation, the holders of Series B Preferred Stock will be entitled to participate on an as-converted-to-Common-Stock basis with holders of the Common Stock in any distribution of assets of the Company to the holders of the Common Stock.
Voting Rights
With certain exceptions, as described in the Series B Certificate of Designation, the Series B Preferred Stock will have no voting rights. However, as long as any shares of Series B Preferred Stock remain outstanding, the Series B Certificate of Designation will provide that we shall not, without the affirmative vote of holders of a majority of the then-outstanding shares of Series B Preferred Stock: (a) alter or change adversely the powers, preferences or rights given to the Series B Preferred Stock or alter or amend the Series B Certificate of Designation, (b) amend the Company's certificate of incorporation or other charter documents in any manner that adversely affects any rights of the Holders, (c) increase the number of authorized shares of Series B Preferred Stock or (d) effect a stock split or reverse stock split of the Series B Preferred Stock or any like event.
Dividends
The Series B Certificate of Designation will provide, among other things, that we shall not pay any dividends on shares of common stock (other than dividends in the form of common stock) unless and until such time as we pay dividends on each share of Series B Preferred Stock on an as-converted basis. Other than as set forth in the previous sentence, the Series B Certificate of Designation will provide that no other dividends shall be paid on shares of Series B Preferred Stock and that we shall pay no dividends (other than dividends in the form of common stock) on shares of common stock unless we simultaneously comply with the previous sentence.
Repurchase Restrictions
The Series B Certificate of Designation will not provide for any restriction on the repurchase of Series B Preferred Stock by us while there is any arrearage in the payment of dividends on the Series B Preferred Stock. There will be no sinking fund provisions applicable to the Series B Preferred Stock.
57
Redemption
We will not be obligated to redeem or repurchase any shares of Series B Preferred Stock. Shares of Series B Preferred Stock will not otherwise be entitled to any redemption rights or mandatory sinking fund or analogous fund provisions.
Exchange Listing
We do not intend to apply for listing of the Series B Preferred Stock on any securities exchange or other trading system.
Warrants
The material terms and provisions of the warrants being offered pursuant to this prospectus are summarized below. This summary of some provisions of the warrants is not complete. For the complete terms of the warrants, you should refer to the form of warrant filed as an exhibit to the registration statement of which this prospectus is a part. Pursuant to a warrant agency agreement between us and American Stock Transfer & Trust Company LLC, as warrant agent, the warrants will be issued in book-entry form and shall initially be represented only by one or more global warrants deposited with the warrant agent, as custodian on behalf of The Depository Trust Company, or "DTC", and registered in the name of Cede & Co., a nominee of DTC, or as otherwise directed by DTC.
Exercise of Warrants
Each Unit includes a Series 1 warrant and a Series 2 warrant, each to purchase one share of our common stock. The exercise price of the Series 1 warrants is equal to 125% of the assumed public offering price of a Class A Unit. The exercise price of the Series 2 warrants is equal to 125% of the assumed public offering price of a Class A Unit The Series 1 warrants will be immediately exercisable and expires on the earlier of (i) 12 months from the date of issuance and (ii) 30 calendar days following the public announcement of Positive Interim Results (as defined on page 63) related to the diarrhea results from the HALT-D Investigator Initiated Trial. The Series 2 warrants will be immediately exercisable and expire on the earlier of (i) 5 years from the date of issuance and (ii) 30 calendar days following the public announcement by the Company that a pivotal trial using crofelemer (Mytesi, or the same or similar product with a different name) for the treatment of cancer therapy related diarrhea has met its primary endpoint in accordance with the protocol.
The warrants issued in this offering will be governed by the terms of global warrants held in book-entry form. The holder of a warrant will not be deemed a holder of our underlying common stock until the warrant is exercised. Subject to limited exceptions, a holder of warrants will not have the right to exercise any portion of its warrants if the holder (together with such holder's affiliates, and any persons acting as a group together with such holder or any of such holder's affiliates) would beneficially own a number of shares of common stock in excess of 4.99% (or, at the election of the purchaser prior to the date of issuance, 9.99%) of the shares of our common stock then outstanding after giving effect to such exercise.
The exercise price and the number of shares issuable upon exercise of the warrants is subject to appropriate adjustment in the event of recapitalization events, stock dividends, stock splits, stock combinations, reclassifications, reorganizations or similar events affecting our common stock. The warrant holders must pay the exercise price in cash upon exercise of the warrants, unless such warrant holders are utilizing the cashless exercise provision of the warrants.
Upon the holder's exercise of a warrant, we will issue the shares of common stock issuable upon exercise of the warrant within two trading days following our receipt of a notice of exercise, provided that payment of the exercise price has been made (unless exercised to the extent permitted via the
58
"cashless" exercise provision). Prior to the exercise of any warrants to purchase common stock, holders of the warrants will not have any of the rights of holders of the common stock purchasable upon exercise, including the right to vote, except as set forth therein.
Warrant holders may exercise warrants only if the issuance of the shares of common stock upon exercise of the warrants is covered by an effective registration statement, or an exemption from registration is available under the Securities Act and the securities laws of the state in which the holder resides. We intend to use commercially reasonable efforts to have the registration statement, of which this prospectus forms a part, effective when the warrants are exercised. The warrant holders must pay the exercise price in cash upon exercise of the warrants unless there is not an effective registration statement or, if required, there is not an effective state law registration or exemption covering the issuance of the shares underlying the warrants (in which case, the warrants may only be exercised via a "cashless" exercise provision).
Fundamental Transaction
In the event we consummate a merger or consolidation with or into another person or other reorganization event in which our common stock are converted or exchanged for securities, cash or other property, or we sell, lease, license, assign, transfer, convey or otherwise dispose of all or substantially all of our assets or we or another person acquire 50% or more of our outstanding shares of common stock, then following such event, the holders of the warrants will be entitled to receive upon exercise of such warrants the same kind and amount of securities, cash or property which the holders would have received had they exercised their warrants immediately prior to such fundamental transaction. Any successor to us or surviving entity shall assume the obligations under the warrants.
Notwithstanding the foregoing, in the event of a fundamental transaction (other than certain fundamental transactions where the Company remains the surviving company) as described above, the holder may, subject to certain conditions, require the Company or a successor entity to purchase the warrant from the holder by paying to the holder an amount in cash equal to the Black-Scholes value of the remaining unexercised portion of the warrant on the effective date of such change of control; provided, however, that, if the change of control is not within the Company's control, including not approved by the Company's board of directors, the holder will only be entitled to receive from the Company or any successor entity, as of the date of consummation of such change of control, the same type or form of consideration (and in the same proportion), at the Black-Scholes value of the unexercised portion of the warrant, that is being offered and paid to the holders of our common stock in connection with the change of control, whether that consideration is in the form of cash, stock or any combination thereof, or whether the holders of common stock are given the choice to receive from among alternative forms of consideration in connection with the change of control.
Call Option for Series 2 Warrants
The Series 2 warrants are callable by us in certain circumstances. Subject to certain exceptions, in the event that the Series 2 warrants are outstanding, following the date that is 180 days after the closing date, (i) the volume weighted average price of our common stock for each of 30 consecutive trading days (the "Measurement Period"), which Measurement Period commences after the date that is 180 days after the closing date, exceeds 300% of the initial exercise price (subject to adjustment for forward and reverse stock splits, recapitalizations, stock dividends and similar transactions), (ii) the average daily trading volume for such Measurement Period exceeds $500,000 per trading day and (iii) the holder is not in possession of any information that constitutes or might constitute, material non-public information which was provided by the Company, and subject to the beneficial ownership limitation for the warrants described further above, then we may, within one trading day of the end of such Measurement Period, upon notice (a "Call Notice"), call for cancellation of all or any portion of the Series 2 warrants for which a notice of exercise has not yet been delivered (a "Call") for
59
consideration equal to $0.0001 per share. Any portion of a Series 2 warrant subject to such Call Notice for which a notice of exercise shall not have been received by the Call Date (as hereinafter defined) will be canceled at 6:30 p.m. (New York City time) on the tenth trading day after the date the Call Notice is received by the Holder (such date and time, the "Call Date"). Our right to call the Series 2 warrants shall be exercised ratably among the holders based on the outstanding Series 2 warrants.
Exchange Listing
We do not intend to apply for listing of the warrants on any securities exchange or other trading system.
60
We are a commercial stage pharmaceuticals company focused on developing and commercializing novel, sustainably derived gastrointestinal products on a global basis. Our wholly-owned subsidiary, Napo Pharmaceuticals, Inc. ("Napo"), focuses on developing and commercializing proprietary human gastrointestinal pharmaceuticals for the global marketplace from plants used traditionally in rainforest areas. Our Mytesi (crofelemer) product is approved by the U.S. Food and Drug Administration ("FDA") for the symptomatic relief of noninfectious diarrhea in adults with HIV/AIDS on antiretroviral therapy.
Jaguar was founded in San Francisco, California as a Delaware corporation on June 6, 2013. Napo formed Jaguar to develop and commercialize animal health products. Effective as of December 31, 2013, Jaguar was a wholly-owned subsidiary of Napo, and, until May 13, 2015, Jaguar was a majority-owned subsidiary of Napo. On July 31, 2017, the merger of Jaguar Animal Health, Inc. and Napo became effective, at which point Jaguar Animal Health's name changed to Jaguar Health, Inc. and Napo began operating as a wholly-owned subsidiary of Jaguar focused on human health and the ongoing commercialization of, and development of follow-on indications for Mytesi. Most of the activities of the Company are now focused on the commercialization of Mytesi and development of follow-on indications for crofelemer and a second-generation anti-secretory product, lechlemer. In the field of animal health, we have limited activities which are focused on developing and commercializing first-in-class gastrointestinal products for dogs, dairy calves, foals, and high value horses.
We believe Jaguar is poised to realize a number of synergistic, value adding benefits— an expanded pipeline of potential blockbuster human follow-on indications of crofelemer, and a second-generation anti-secretory agent—upon which to build global partnerships. As previously announced, Jaguar, through Napo, now holds extensive global rights for Mytesi, and crofelemer manufacturing may occur at two FDA-inspected and approved facilities, including a newly constructed, multimillion-dollar commercial manufacturing facility. Additionally, several of the drug product candidates in Jaguar's Mytesi pipeline are backed by strong Phase 2 and proof of concept evidence from completed human clinical trials.
Mytesi is a novel, first-in-class anti-secretory agent which has a basic normalizing effect locally on the gut, and this mechanism of action has the potential to benefit multiple disorders. Mytesi is in development for multiple possible follow-on indications, including cancer therapy-related diarrhea (CTD); orphan-drug indications for infants and children with congenital diarrheal disorders (CDDs) and short bowel syndrome (SBS); supportive care for inflammatory bowel disease (IBD); irritable bowel syndrome (IBS); and for idiopathic/functional diarrhea. In addition, a second-generation proprietary anti-secretory agent, lechlemer, is in development for cholera. Mytesi has received orphan-drug designation for SBS.
Napo currently has a direct sales force of 19 sales representatives, a national sales director and two regional sales directors covering U.S. geographies with the highest potential. With support provided by concomitant marketing, promotional activities, patient empowerment programs and medical education initiatives described below, we expect continued growth in the number of patients treated with Mytesi.
The goal of Napo's internal sales team is to deliver a frequent and consistent selling message to targeted, high-volume prescribers of antiretroviral therapies (ART) and to gastroenterologists who see large numbers of HIV patients. In December 2017 we released the results of a survey of 350 people living with HIV and AIDS regarding the topic of "Talking to Your Doctor About Symptoms". The survey results show that diarrhea remains prevalent in those living with HIV/AIDS, as 27% of respondents living with HIV/AIDS reported that they currently have diarrhea, while 56% reported that they have had diarrhea in the past. Additionally, the results of a recent Napo-sponsored survey of 271 U.S. board certified gastroenterologists indicate that the number one GI complaint for people
61
living with HIV/AIDS is diarrhea, and 93% of U.S. gastroenterologists see patients with HIV/AIDS in their practice.
Key to the success of our sales representatives in growing Mytesi sales is differentiating and targeting the right doctors—those HIV specialists who are high prescribers of ART medications and those gastrointestinal doctors who see large populations of people living with HIV/AIDS. The target list of prescribers for our sales reps includes a pool of approximately 3,100 high volume ART prescribing HIV specialists, and gastroenterologists who see the largest number of people living with HIV/AIDs, and we've strategically placed our sales force in the US geographies with the highest potential, including San Francisco, southern California, Arizona, Nevada, Miami/southern Florida, northern Florida, New York City/Long Island, Massachusetts, Rhode Island, New Hampshire, Connecticut, New Jersey/eastern Pennsylvania, northern Texas, southern Texas, Chicago, Alabama, Mississippi, Louisiana, Maryland/DC/eastern Virginia, North Carolina/South Carolina, Michigan, Indianapolis, Ohio and Atlanta.
In June 2018, Napo entered into an agreement with RedHill Biopharma, a specialty biopharmaceutical company primarily focused on late clinical-stage development and commercialization of proprietary drugs for gastrointestinal diseases and cancer, to establish a U.S. co-promotion program for Mytesi.
RedHill's specialized, GI-focused field sales force is promoting Mytesi to health care practitioners in 36 U.S. territories that contain significant numbers of HIV patients and health care practitioners that are not currently covered by Napo's field sales force. In these geographies, RedHill sales representatives target gastroenterologists who see large populations of people living with HIV, along with nurse practitioners and physician assistants. RedHill field representatives also target lower-level prescribers of anti-retroviral infectious disease specialists in regions currently covered by Napo's sales force. Four RedHill inside sales representatives actively target health care practitioners in other regions not covered by the Napo or RedHill field representatives. We believe this copromotion program will play an important role in extending the reach of our commercial efforts into the GI medical community in support of the treatment of people living with HIV (PLWH) with Mytesi. Under the terms of the Agreement, RedHill is compensated based on performance, and the program can be extended by agreement between the two companies, as it was in January 2019.
Medical education presentations led by health care practitioners (HCPs) participating in the Napo Speakers Bureau—a group that includes HIV/AIDS specialists, infectious disease specialists, gastroenterologists, colorectal surgeons, and nurse practitioners—focus on the prevalence and pathophysiology of gastrointestinal consequences of HIV infection and on the latest treatment options for HIV-related diarrhea. Presentations given by patient advocate members provide information to PLWH about the prevalence of diarrhea in PLWH and offer guidance about talking to HCPs regarding diarrhea-related concerns.
On July 24, 2018, we announced the results of an analysis conducted to examine whether the rate of HIV-associated diarrhea has changed over time. The analysis of data, sourced from the National Institutes of Health (NIH) clinicaltrials.gov database, revealed that 18% of HIV patients experience diarrhea and the rates have not declined significantly over time. The analysis includes data from 38 U.S. clinical trials from 2008-2016 in more than 21,000 patients. The results were reported at the International AIDS 2018 Conference (AIDS 2018) on Tuesday, July 24 in Amsterdam, Netherlands. The poster is available on the AIDS 2018 website at this link: https://programme.aids2018.org//PAGMaterial/eposters/4900.pdf.
With the introduction of newer antiretroviral (ARV) drug therapy, there has been a reduction in the severity of ARV-induced diarrhea. However, a significant portion of this patient population still suffers from diarrhea caused by HIV enteropathy, which is due to direct and indirect effects of HIV on the intestinal mucosa. Chronic diarrhea remains a significant complaint of people living with
62
HIV/AIDS, particularly those who are older and have lived with the virus in their gut for 10+ years. According to data from the U.S. Centers for Disease Control and Prevention, currently more than 50% of people living with HIV are over age 50; by 2020 this figure will increase to 70%.
Crofelemer (Mytesi) data from a supplemental analysis of the ADVENT trial was featured in a poster presentation at the 9th International Aids Society (IAS) Conference on HIV Science held in July 2017 in Paris, France. The presentation was titled Long-Term Crofelemer Use Gives Clinically Relevant Reductions in HIV-Related Diarrhea. IAS features the latest HIV science, including basic, clinical and prevention research, and brings together a broad cross section of HIV professionals from around the world with a focus on implementation—moving scientific advances into practice. The results indicate that at the end of the study period, more than 50% of the patients treated had complete resolution of their diarrhea; and 83% had at least a 50% reduction in diarrhea. Entry criteria required at least 7 watery stools in a week, and the average was 20 (with some patients having as high at 67 watery stools in a week).
Napo continues to pursue AIDS Drug Assistance Program (ADAP) formulary listing. ADAPs provide life-saving HIV treatments to low income, uninsured, and underinsured individuals living with HIV/AIDS in all 50 states and the territories. The ADAP program provides Mytesi free of charge to patients who qualify and copay support for some patients who have insurance coverage. In the third quarter of 2018, Mytesi was added to the ADAP formularies in New York, Tennessee, Mississippi and DC. As announced January 24, 2019, Mytesi has also been added to the formulary for Florida's ADAP, which is the third largest in the U.S. based on enrollment. As a result of this addition, based on data from healthcare research firm Decision Resource Group, approximately 86% of ADAP-eligible US lives now have access to Mytesi, which is now on the ADAP formularies for 30 states, including the five programs with the largest enrollment.
As we announced April 10, 2018, Napo has signed an agreement with the ADAP Crisis Task Force. The agreement establishes a reduced price provided by Napo ADAPs in all U.S. states and territories for purchases of Mytesi. Formed in 2002, the Task Force negotiates reduced drug prices for all ADAPs. Task Force membership is currently comprised of representatives from Arizona, California, Florida, Illinois, Massachusetts, New York, North Carolina, Tennessee, Texas, Virginia, and Washington state HIV/AIDS divisions. Per the terms of the agreement, all state ADAPs are guaranteed the same reduced price for the drug. ADAPs provide HIV-related services and approved medications to more than half a million people in the U.S. each year, and we expect this agreement to help further expand the number of patients able to benefit from the novel, first-in-class anti-secretory mechanism of action of Mytesi.
As announced June 26, 2019, we signed an agreement with Integrium, LLC, a clinical research organization, in support of a study to evaluate the effect of Mytesi on the gastrointestinal microbiome in people living with HIV. The study is being funded by an investment in Jaguar by California-based PoC Capital. We look forward to adding microbiome data to our overall understanding of the gut health of Mytesi patients, especially as Jaguar works towards expanding Mytesi access to new groups of patients who need symptomatic relief from diarrhea and diarrhea-related symptoms, such as bloating and abdominal discomfort.
Mytesi is currently reimbursed by Medicaid in all 50 states. It is also currently covered on 100% of the top 10 commercial insurance plan national formularies, representing more than 245 million U.S. lives. Additionally, Napo operates a co-pay coupon program, which helps ensure that the majority of participating patients do not have a Mytesi co-pay greater than $25. Information about the NapoCares Patient Assistance Program, which assists patients with benefit verification, prior authorization, and claims appeals, can be found at mytesi.com/mytesi-savings.html.
63
Pipeline within a product—crofelemer
According to the World Health Organization, there are nearly 1.7 billion cases of diarrheal disease globally every year, and the disease caused an estimated 1.5 million deaths in 2012. Although not all types of diarrhea are secretory in nature, we view the current, initial approval of Mytesi as the opening of the door to an important pipeline—underscored by the current approval by the FDA of the Chemistry, Manufacturing and Controls (CMC) for this natural product, as well as acknowledgement by the FDA of the safety of the product for chronic use for the approved indication.
Crofelemer is in development for the symptomatic relief of cancer therapy-related diarrhea (CTD). A significant proportion of patients undergoing cancer therapy experience diarrhea. Novel targeted cancer therapy agents, such as epidermal growth factor receptor antibodies and tyrosine kinase inhibitors, with or without cycle chemotherapy agents, may activate intestinal chloride secretory pathways leading to increased chloride secretion into the gut lumen, coupled with significant loss of water that would result in secretory diarrhea.
As part of the Company's near-term plan, Napo had a meeting with the FDA in March 2019 to review the protocol for Napo's planned Phase 3 clinical trial in cancer subjects to evaluate the effects of Mytesi in prevention and/or relief of CTD. Participants in the meeting, which was with the FDA's Division of Gastroenterology and Inborn Errors Products (DGIEP) and FDA's Division of Oncology Products, included Pravin Chaturvedi, Ph.D., Napo's/Jaguar's Chairman of the Scientific Advisory Board (SAB) and Acting Chief Scientific Officer, regulatory affairs, medical safety monitoring, and biostatistics specialists, and academic key opinion leaders (KOLs)/SAB members from leading oncology treatment institutions, one of whom will serve as the principal investigator for Napo's planned trial. Following the meeting with FDA, we reached agreement with the DGIEP in the following key areas related to our planned investigational new drug application (IND) submission for a supplemental new drug application (sNDA) for crofelemer (Mytesi) for the symptomatic relief of CTD:
- •
- Acceptance of the nonclinical safety package for crofelemer from NDA 202292 (the NDA for Mytesi's currently approved HIV indication) without
the need for any additional nonclinical or preclinical safety studies for our planned sNDA
- •
- Acceptance of the Chemistry, Manufacturing and Controls (CMC) submissions for use of 125-mg delayed release crofelemer tablets (Mytesi) from
NDA 202292, with the proviso of requiring additional details on the drug product specification assays and a summary of assay results for Mytesi lots that are planned to be used in the proposed single
pivotal clinical trial in CTD
- •
- No additional drug-drug interaction studies are required for crofelemer at this time
Napo's planned next steps are to continue interactions with the FDA and KOLs/SAB members from leading oncology treatment institutions to incorporate the input from our dialog with FDA into the pivotal Phase 3 protocol. Our goal is to ensure that the protocol addresses the need for a treatment for CTD, the practicalities of patient enrollment and trial design, and that expert statisticians from both Napo and FDA agree on endpoints relevant to crofelemer's mechanism of action. Our planned study for diarrhea related to CTD is analogous to the successful pivotal program we ran for Mytesi's currently approved HIV indication, and as part of risk mitigation we intend to use the same formulation and dosing as the current commercialized Mytesi.
In support of our focus on the potential CTD indication, an ongoing investigator initiated trial (IIT) utilizing Mytesi is underway. Enrollment is ongoing for the HALT-D study in breast cancer patients receiving regimens containing Herceptin and Perjeta ("HALT-D Investigator Initiated Trial"). Interim results from the study, which is sponsored by Georgetown University and funded by Genentech, a member of the Roche Group, are expected to be read out in mid-2019. The study's primary endpoint has an 81% power to detect a 40% difference in the percent and/or number of patients experiencing any grade of diarrhea for two consecutive days at a p value of 0.1. (The statistical power of a study,
64
sometimes referred to as a study's sensitivity, is a measure of how likely the study is to distinguish an actual effect from one of chance). For the sake of clarity, the estimates of the percent of patients experiencing such diarrhea is postulated to be 60% in the placebo patients and 20% in the study's crofelemer-treated arms. The interim analysis, which is being conducted to ensure that the study has a chance to ultimately achieve the primary endpoint, will determine whether or not the study has a power of at least 20% to detect such a difference when 23 patients have been randomized. The interim analysis will be deemed positive and the trial will continue if the power is 20% or greater (the "Positive Interim Results").
According to data appearing in "Treatment Guidelines for CID" (chemotherapy-induced diarrhea) in the April 2004 issue of Gastroenterology and Endoscopy News, diarrhea is the most common adverse event reported in chemotherapy patients. Approved third-party supportive care products for chemotherapy-induced nausea and vomiting (CINV) include Sustol, Aloxi, Akynzeo and Sancuso. According to Transparency Market Research, sales of therapeutics for the prevention of CINV approximated $620 million in 2013, and sales of such therapeutics are expected to reach $1 billion in 2020.
Diarrhea has been reported as the most common side effect of the recently approved CDK 4/6 inhibitor abemaciclib and the pan-HER TKI neratinib, with occurrence ranging from 86% to >95% and grade 3 over 40%, in published studies. Diarrhea in this patient population has the potential to cause dehydration, potential infections, and non-adherence to treatment. A novel anti-diarrheal like Mytesi may hold promise for treating secretory diarrhea—and therefore also support long-term cancer treatment adherence—in this population.
As we announced on January 22, 2018, Napo has accepted a request for support submitted by Dr. Mohamad Miqdady, Chief of Pediatric Gastroenterology, Hepatology and Nutrition at Sheikh Khalifa Medical City (SKMC) in Abu Dhabi, for an investigator-initiated trial of crofelemer, the active pharmaceutical ingredient in Mytesi, for congenital diarrheal disorders (CDDs) in children.
CDDs are a group of rare, chronic intestinal channel diseases, with onset in early infancy, that are characterized by severe, lifelong diarrhea and a lifelong need for nutritional intake either parenterally or with a feeding tube. CDDs are related to specific genetic defects inherited as autosomal recessive traits. The incidence of CDDs is prevalent in regions where consanguineous marriages (related by blood) is part of the culture. CDDs are directly associated with serious secondary conditions including dehydration, metabolic acidosis, and failure to thrive, prompting the need for immediate therapy to prevent death and limit lifelong disability.
SKMC is the Abu Dhabi public health system's flagship institution and the largest hospital in the United Arab Emirates (UAE), consisting of a 586-bed tertiary hospital, 14 outpatient specialty clinics, and the Abu Dhabi Blood Bank, all of which are accredited by Joint Commission International, the oldest and largest healthcare standards-setting and accrediting body in the United States. Dr. Miqdady is an American Board certified in Pediatric Gastroenterology, Hepatology and Nutrition, and he is a member of Napo's Scientific Advisory Board.
Napo intends to submit documentation in the second half of 2019 to the U.S. FDA for the formulation of crofelemer appropriate for feeding tube administration to support this investigation.
As announced on June 5, 2017, Napo has received orphan-drug designation from the FDA for pediatric short bowel syndrome (SBS). The Orphan Drug Act provides for granting special status to a drug or biological product to treat a rare disease or condition upon request of a sponsor. Orphan-drug designation qualifies the sponsor of the drug for various development incentives, including extended exclusivity, tax credits for qualified clinical testing, and relief of filing fees.
As announced June 11, 2019, Napo will receive preclinical services from the National Institute of Allergy and Infectious Diseases ("NIAID") to support the development of lechlemer for the proposed
65
cholera indication. Under NIAID's suite of preclinical services, NIAID-funded contractors will conduct toxicology testing for 7-day rat and dog studies. NIAID is part of the National Institutes of Health, which is an agency of the U.S. Department of Health and Human Services.
Jaguar's and Napo's portfolio development strategy involves meeting with Key Opinion Leaders (KOLs) to identify indications that are potentially high-value because they address important medical needs that are significantly or globally unmet, obtain input on protocol practicality and protocol generation, and then strategically sequencing indication development priorities, second-generation product pipeline development, and partnering goals on a global basis, as well as identifying possible opportunities for a Special Protocol Assessment (SPA) from the FDA. When granted, SPA provides that, upon request, FDA will evaluate within 45 days certain protocols to assess whether they are adequate to meet scientific and regulatory requirements identified by the sponsor. In 2007, under the SPA process, Napo obtained agreement with the FDA for the design of the pivotal study protocol for the currently approved indication of crofelemer (Mytesi) for the symptomatic relief of noninfectious diarrhea in adults with HIV/AIDS on antiretroviral therapy. The 2007 SPA agreement was an important milestone for Napo, allowing Napo to address and mitigate regulatory uncertainty prior to the completion of its final Phase 3 trial of crofelemer for its currently approved indication.
In October 2017, Napo established a scientific advisory board for each potential follow-on indication currently planned for Mytesi. Napo has developed relationships with physicians and patient advocates around the world who are recognized specialists and key opinion leaders (KOLs) in the planned Mytesi follow-on indications. The two charts below provide the names, credentials and affiliations of current Napo scientific advisory board members and KOL advisors to Napo.
We are confident that our scientific advisory boards will provide expert, actionable input regarding all aspects of development, including trial design, for Mytesi for our follow-on indications—each of which addresses a significant, global, unmet medical need.
Napo's HIV Scientific Advisory Board has focused primarily on physician education, and community and global awareness regarding the importance and availability of solutions for neglected comorbidities, such as the first-in-class anti-secretory mechanism of action of Mytesi for its currently approved indication.
Napo Scientific Advisory Board (SAB) Members
Pravin Chaturvedi, PhD |
Chair of Napo's Scientific Advisory Boards; 25+ years drug development experience in pharmaceutical/biotech field; Successfully developed crofelemer (Mytesi) (first pivotal adaptive design) |
HIV Physicians Scientific Advisory Board
David Asmuth, MD |
Infectious diseases specialist and Professor of Medicine, UC Davis Health | |
Gary Blick, MD, AAHIVS |
Founder of Health Care Advocates International and BEAT AIDS Project Zimbabwe |
|
Christine Wanke, MD |
Director of the Nutrition and Infection Unit; Associate Chair and Professor, Department of Public Health and Community Medicine; Professor, Department of Medicine, Tufts University School of Medicine; Professor, Sackler School of Biomedical Science; Professor, Friedman School of Nutrition Science and Policy |
66
Cancer Therapy-Related Diarrhea Scientific Advisory Board
Lee Schwartzberg, MD, FACP |
Executive Director of the West Cancer Center, a multispecialty oncology practice affiliated with the University of Tennessee; Chief, Division of Hematology/Oncology, the University of Tennessee Health Science Center | |
Eric Roeland, M.D. |
Attending Physician, Center for Palliative Care, Harvard Medical School |
|
Hope Rugo, MD |
Clinical Professor of Medicine, Director Breast Oncology and Clinical Trials Education, Division of Hematology and Oncology, University of California San Francisco |
IBD Scientific Advisory Board
Corey Siegel, MD, MS |
Associate Professor of Medicine; Associate Professor of The Dartmouth Institute; Director of the Inflammatory Bowel Disease Center at the Dartmouth-Hitchcock Medical Center |
Pediatric Indications (SBS and CDD) Scientific Advisory Board
Mohammed Miqdady, MD |
Chief of Pediatric Gastroenterology, Hepatology & Nutrition at Sheikh Khalifa Medical City in Abu Dhabi | |
Martin, MD |
Professor, Department of Pediatrics, David Geffen School of Medicine at UCLA |
|
Sue Rhee, MD |
Division Chief, Pediatric Gastroenterology, Hepatology and Nutrition Pediatric gastroenterologist and liver specialist, UCSF Benioff Children's Hospital |
Key Opinion Leader (KOL) Advisors to Napo (on an as-needed basis)
KOL Advisors: Cancer Therapy-Related Diarrhea
Herbert DuPont, MD |
Professor and Director, Center for Infectious Diseases, University of Texas Houston School of Public Health | |
Pablo C. Okhuysen, M.D. |
Department of Infectious Diseases, Infection Control, and Employee Health, Division of Internal Medicine, MD Anderson |
67
KOL Advisors: Diarrhea Related to IBD
David Rubin, MD |
Joseph B. Kirsner Professor of Medicine Section Chief, Gastroenterology, Hepatology and Nutrition Co-Director, Digestive Diseases Center, University of Chicago Medicine | |
Charles Bernstein, MD |
Distinguished Professor of Medicine and Bingham Chair in Gastroenterology Research, University of Manitoba |
|
William Sandborn, MD |
Director, Inflammatory Bowel Disease Center Chief, Division of Gastroenterology Professor of Medicine, US San Diego Health |
|
Scott Lee, MD |
Associate Professor of Medicine, Digestive Health Center, University of Washington Medical Center |
|
Edward Loftus, Jr., MD |
Consultant, Division of Gastroenterology and Hepatology, Department of Internal Medicine, Mayo Clinic |
|
Douglas Wolf, MD |
Medical Director of IBD Research at Atlanta Gastroenterology Associates. Clinical Assistant Professor of Medicine, Emory University School of Medicine |
|
Brooks D. Cash, MD, AGAF, FACG, FACP, FASGE |
Division Director, Gastroenterology, Hepatology, and Nutrition Visiting Professor of Medicine, The University of Texas McGovern Medical School |
KOL Advisors: Pediatric Indications (SBS and CDD)
Jay Thiagarajah, MD, PhD |
Attending Physician, Division of Gastroenterology, Hepatology and Nutrition, Boston Children's Hospital. Instructor of Pediatrics, Harvard Medical School | |
James Goldenring, M.D., Ph.D. |
Professor of Surgery, Vanderbilt University School of Medicine. Paul W. Sanger Chair in Experimental Surgery. Professor of Cell and Developmental Biology |
KOL Advisors: Diarrhea Related to HIV and other Infectious Diseases
Herbert DuPont, MD |
Professor and Director, Center for Infectious Diseases, University of Texas Houston School of Public Health | |
Pradip Bardhan, MBBS, MD |
Chief Physician at ICDDR,B, Bangladesh |
|
Patrick Clay, Pharm D |
Consultant |
|
Paulo Pacheco, MD |
Clinical Assistant Professor, Department of Medicine, New York University Langone Health |
|
Elie Schochet, MD, FACS |
Colorectal surgeon, Holy Cross Medical Group |
68
KOL Advisors: Diarrhea Related to IBS
Anthony Lembo, MD |
Director of the GI Motility and Functional Bowel Disorders Program at Beth Israel Deaconess Medical Center and Associate Professor of Medicine at Harvard Medical School | |
Doug Drossman, MD |
Co-Director Emeritus, UNC Center for Functional GI and Motility Disorders Adjunct Professor of Medicine and Psychiatry, University of North Carolina School of Medicine |
|
William Chey, MD |
Professor of Internal Medicine and Professor of Nutritional Sciences, University of Michigan School of Public Health |
According to a 2017 report from Research and Markets, the combined global market for prescription and OTC gastrointestinal agents is expected to reach $21 billion by 2025. Jaguar estimates that a first-in-class anti-secretory agent should be able to achieve a significant portion of the market share.
Our management team has significant experience in gastrointestinal product development for both humans and animals. Napo was founded 30 years ago to perform drug discovery and development by leveraging the knowledge of traditional healers working in rainforest areas. Ten members of the Jaguar and Napo team have been together for more than 15 years. Dr. Steven King, our executive vice president of sustainable supply, ethnobotanical research and intellectual property, and Lisa Conte, our founder, president and CEO, have worked together for more than 30 years. Together, these dedicated personnel successfully transformed crofelemer, which is extracted from trees growing in the rainforest, to Mytesi, which is a natural, sustainably harvested, FDA-approved drug.
There are significant barriers to entry for Mytesi (crofelemer). Through Napo, we hold an extensive global patent portfolio. At the present time we hold approximately 141 issued worldwide patents, with coverage in many cases that extends until 2031. These issued patents cover multiple indications including HIV-AIDS diarrhea, IBS, IBD, manufacturing, enteric protection from gastric juices, among others. We also have approximately 24 pending patent applications worldwide in the human health areas that are being prosecuted.
Mytesi is the first oral drug approved by the FDA under botanical guidance, which provides another barrier to entry from potential generic competition. The FDA requires that the manufacturer of crofelemer use a validated proprietary bioassay to release the drug substance and drug product of Mytesi. While most generic products are fashioned to meet chemical release specifications that are in the public domain, the specifics of this assay are not publicly available. There is no pathway by which a generic product can be developed for a drug approved under botanical guidance. In addition, Mytesi is not systemically absorbed, so the classic approach of creating a generic drug by matching pharmacokinetic blood levels is not possible. A generic player would have to conduct costly and risky clinical trials.
While Jaguar's commercial and development efforts have evolved to focus primarily on Mytesi and human pipeline indications since its merger with Napo, the Company is continuing limited initiatives related to Canalevia, its drug product candidate for treatment of chemotherapy-induced diarrhea ("CID") in dogs, and Equilevia, its non-prescription, personalized, premium product for total gut health in equine athletes. CID in dogs is typically caused by the same mechanism of action as in humans, and hence the work in dogs serves as a preclinical proof of concept for the diarrhea in humans that is related to targeted cancer therapy.
As announced June 19, 2019, the Target Animal Safety technical section of the Company's application for conditional approval of Canalevia for CID in dogs is expected to be submitted to the FDA's Center for Veterinary Medicine (CVM) in the third quarter of 2019. This technical section,
69
which is the last of the four technical sections Jaguar is required to file for Canalevia for the proposed CID indication, will contain data from a 2017 target animal safety study indicating that the NOAEL (no-observed-adverse-effect level) of Canalevia in dogs is approximately six times greater than previously demonstrated, and that Canalevia is also safe for use in puppies.
The safety of residues of veterinary drugs is most commonly addressed through the conduct of target animal safety studies that provide for the determination of the NOAEL.
The 2017 toxicology study is the first study to demonstrate the safety of Canalevia in puppies as young as 12 weeks of age. Prior crofelemer toxicology studies only involved adult dogs.
As previously announced, Jaguar has received MUMS (Minor Use and Minor Species) designation status from the FDA for Canalevia for the indication of CID in dogs. MUMS designation is modeled on the orphan-drug designation for human drug development and offers possible financial incentives to encourage MUMS drug development, such as the availability of grants to help with the cost of developing the MUMS drug. Additionally, as announced on March 8, 2018, the CVM has indicated that Jaguar's Reasonable Expectation of Effectiveness (RxE) technical section is complete towards conditional approval of Canalevia. As announced March 20, 2019, Jaguar has completed the filing with CVM of the Chemistry, Manufacturing, and Controls (CMC) technical section in support of the Company's application for conditional approval of Canalevia for treatment of CID in dogs. Completion of the pharmacokinetics analysis of the blood samples from the target animal safety study is the last step remaining in getting the Target Animal Safety technical section ready to submit to CVM. We anticipate filing the Target Animal Safety technical section with CVM in the third quarter of 2019. If Canalevia is approved for CID in dogs, we expect to conduct the commercial launch of Canalevia for this indication in 2020.
Crofelemer is extracted from the Croton lechleri tree, which we sustainably harvest and manage through programs that we have been developing over the past 29 years. This process has involved working with communities to plant trees, obtaining permits for export, and creating a supply network that is robust and reliable.
We continue to have working relationships with partners that began in the 1990s. Additionally, through the establishment of a nonprofit called the Healing Forest Conservancy (HFC), our team has created a long-term mechanism for benefit sharing that recognizes the intellectual contribution of indigenous populations. This program is intended to contribute to the continued strength and effectiveness of the valued and strategically important relationships we have carefully cultivated over the past 29 years.
Product Pipeline
In addition to our Mytesi (crofelemer) product that is approved by the U.S. FDA for the symptomatic relief of noninfectious diarrhea in adults with HIV/AIDS on antiretroviral therapy, we are also developing a pipeline of prescription drug product candidates to address unmet needs in gastrointestinal health through Napo. Mytesi (crofelemer) is a novel, first-in-class anti-secretory agent which has a basic normalizing effect locally on the gut, and this mechanism of action has the potential to benefit multiple disorders. Clinical trials demonstrated that nearly 80% of Mytesi users experienced an improvement in their diarrhea over a four-week period. At week 20 of the pivotal trial, over half the patients had no watery stools, or a 100% decrease, and 83% had at least a 50% decrease in watery stools. Our Mytesi pipeline currently includes prescription drug product candidates for four follow-on indications, several of which are backed by Phase 2 evidence from completed Phase 2 trials. In addition, a second-generation proprietary anti-secretory agent is in development for cholera.
70
Napo Prescription Drug Product Candidates
Product Candidates
|
Indication | Completed Milestones | Current Phase of Development |
Anticipated Near-Term Milestones* |
||||
|---|---|---|---|---|---|---|---|---|
Mytesi |
Cancer therapy-related diarrhea (CTD) | • Investigator-initiated (IIT) clinical trial funded by Genentech, a member of the Roche Group • Met with FDA in March 2019 to discuss the anticipated protocol for a planned pivotal trial |
Phase 3 | • Availability of interim data IIT for Genentech-Roche-funded trial in mid-2019 |
||||
|
||||||||
Mytesi |
Supportive care for IBD | • Safety • Multiple Phase 2 studies completed in various secretory diarrhea (not IBD) |
Phase 2 | • Protocol development with KOLs for discussions with FDA |
||||
|
||||||||
Formulation of crofelemer |
Rare disease indications (SBS & CDD) | • Phase 1 study • Orphan-drug designation for SBS |
Phase 2 | • Formulation/IIT, Abu Dhabi, Protocol design |
||||
|
||||||||
Mytesi |
Irritable bowel syndrome—diarrhea predominant (IBS-D) | • Phase 1 study • Two Phase 2 studies completed |
Phase 2 | • Publication of supplemental analysis of Phase 2 data |
||||
|
||||||||
Mytesi |
Idiopathic/functional diarrhea | • Safety • Multiple Phase 2 studies completed in various secretory diarrhea • IIT request accepted |
Phase 2 | • Initiation of IIT |
||||
|
||||||||
SB-300 (lechlemer) |
Second-generation anti-secretory agent for multiple indications including cholera | • Animal and human studies in secretory diarrhea; successful cholera trial design for anti-secretory mechanism of action with API |
Pre IND | • Pre-clinical toxicology funded by NIAID • Formulation / POC |
- *
- Clinical trials are funding dependent
Estimated Size of Mytesi Target Markets
We believe the medical need for Mytesi is significant, compelling, and unmet, and that doctors are looking for a drug product with a mechanism of action that is distinct from the options currently available to resolve diarrhea. A growing percentage of HIV patients have lived with the virus in their gut for 10+ years, often causing gut enteropathy and chronic or chronic-episodic diarrhea. According to
71
data from the U.S. Centers for Disease Control and Prevention, by 2020 more than 70% of Americans with HIV are expected to be 50 and older.(1)
Market
|
Number of Competitors for Mytesi's Approved/ Anticipated Labelled Indication |
Market Size/Potential | ||
|---|---|---|---|---|
HIV-D |
0 | We estimate the U.S. market revenue potential for Mytesi to be approximately $100 million in gross annual sales | ||
CTD |
0 |
An estimated 650,000 U.S. cancer patients receive chemotherapy in an outpatient oncology clinic.(2) Comparable supportive care (i.e. CINV) product sales of ~$620 million in 2013, which is projected to reach $1.0 billion by 2020(3) |
||
IBD |
0 |
Estimated 1,171,000 Americans have IBD(4) |
||
IBS-D |
3 |
Most IBS products have estimated revenue potential of greater than $1.0 billion(5) |
||
CDD/SBS |
0 |
Financial benefits of Orphan-drug Designation |
||
Cholera (hydration maintenance) PRV (SB-300) |
0 |
In recent transactions by other companies, priority review vouchers have sold for $67 million to $350 million(6) |
- (1)
- HIV
Among People Aged 50 and Older (https://www.cdc.gov/hiv/group/age/olderamericans/index.html)
- (2)
- Centers
for Disease Control and Prevention. Preventing Infections in Cancer Patients: Information for Health Care Providers
(cdc.gov/cancer/preventinfections/providers.htm)
- (3)
- Heron
Therapeutics, Inc. Form 10-K for the fiscal year ended December 31, 2016
- (4)
- Kappelman,
M. et al. Recent Trends in the Prevalence of Crohn's Disease and Ulcerative Colitis in a Commercially Insured US Population. Dig Dis Sci.
2013 Feb; 58(2): 519-525
- (5)
- Merrill
Lynch forecasts peak US sales of roughly $1.5 bn for Ironwood's Linzess
(http://247wallst.com/healthcare-business/2015/04/27/key-analyst-sees-nearly-30-upside-in-ironwood); Rodman & Renshaw estimate peak annual sales of Synergy Pharmaceuticals' Trulance at $2.3 bn
in 2021 (Source: https://www.benzinga.com/analyst-ratings/analyst-color/17/03/9224181/analyst-synergy-pharma-could-achieve-
sustainable-profita) - (6)
- In
Aug. 2015, AbbVie Inc. bought a priority review voucher from United Therapeutics Corp for $350 million
(http://www.reuters.com/article/us-abbvie-priorityreview/abbvie-buys-special-review-
voucher-for-350-million-idUSKCN0QO1LQ20150819). In July 2014, BioMarin announced that it had sold a priority review voucher to Sanofi and Regeneron for $67.5 million. (https://investors.biomarin.com/2014-07-30-BioMarin-Sells-Priority-Review-Voucher-for-67-5-Million).
72
The following diagram illustrates the mechanism of action of our human and animal gastrointestinal drug products and drug product candidates, which normalize chloride and water flow and transit time of fluids within the intestinal lumen.
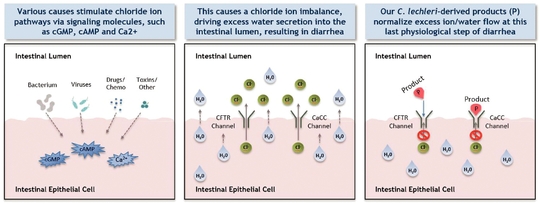
Business Strategy
Our goal is to become a leading pharmaceutical company with first-in-class, sustainably derived products that address significant unmet gastrointestinal medical needs globally. To accomplish this goal, we plan to:
Expand Mytesi by leveraging our significant gastrointestinal knowledge, experience and intellectual property portfolio
Mytesi is a novel, first-in-class anti-secretory agent which has a basic normalizing effect locally on the gut, and this mechanism of action has the potential to benefit multiple gastrointestinal disorders. Our Mytesi (crofelemer) product is approved by the U.S. FDA for the symptomatic relief of noninfectious diarrhea in adults with HIV/AIDS on antiretroviral therapy. Jaguar, through Napo, holds extensive global rights for Mytesi. Mytesi is in development for multiple possible follow-on indications, including diarrhea related to targeted cancer therapy; orphan-drug indications for infants and children with congenital diarrheal disorders and short bowel syndrome; supportive care for inflammatory bowel disease; irritable bowel syndrome; and for idiopathic/functional diarrhea. In addition, a second-generation proprietary anti-secretory agent is in development for cholera.
Our management team collectively has more than 100 years of experience in the development of gastrointestinal prescription drug and non-prescription products. This experience covers all aspects of product development, including discovery, preclinical and clinical development, GMP manufacturing, and regulatory strategy. Key members of this team successfully developed Mytesi.
Establish and expand commercial capabilities in Mytesi sales and marketing efforts
As announced on August 7, 2017, we appointed Pete Riojas, a 31-year pharmaceutical industry veteran, to lead Napo's direct sales organization, which is comprised of Mytesi field sales representatives strategically positioned to cover U.S. geographies with the highest potential. With support provided by concomitant marketing, promotional activities, patient empowerment programs, including an integrated social digital campaign, and medical education initiatives described below, we expect a proportional response in the number of patients treated with Mytesi.
In June 2018, as stated above, Napo entered into an agreement with RedHill Biopharma, a specialty biopharmaceutical company primarily focused on late clinical-stage development and commercialization of proprietary drugs for gastrointestinal diseases and cancer, to establish a U.S. co-promotion program for Mytesi. RedHill's specialized, GI-focused field sales force promotes Mytesi
73
to health care practitioners in 36 U.S. territories that contain significant numbers of HIV patients and health care practitioners that are not currently covered by Napo's field sales force. In these regions, RedHill sales representatives target gastroenterologists who see large populations of people living with HIV, along with nurse practitioners and physician assistants. RedHill field representatives also target lower-level ART prescribing infectious disease specialists in regions currently covered by Napo's sales force. Four RedHill inside sales representatives actively target health care practitioners in other regions not covered by the Napo or RedHill field representatives. We believe this co-promotion program will play a significant role in extending the reach of our commercial efforts into the GI medical community in support of the treatment of people living with HIV (PLWH) with Mytesi. Under the terms of the Agreement, RedHill is compensated based on performance, and the program can be extended by agreement between the two companies, as it was in January 2019.
Leverage our relationships with key opinion leaders regarding development of follow-on indications
Approximately 25 key opinion leaders (KOLs) who are recognized specialists in HIV patient care, CTD, IBD, IBS, cholera, SBS, CDD and equine gut health, are currently participating in our scientific advisory board or KOL advisory program in some manner.
Establish partnerships to support moving pipeline indications to pivotal clinical trials
Jaguar is actively pursuing development of a robust pipeline of potential follow-on indications for crofelemer, and the Company's goal is to establish partnerships to support moving pipeline indications to pivotal clinical trials.
Strategically sequence the development of follow-on indications of Mytesi and seek geographically-focused licensing opportunities
As announced September 24, 2018, Jaguar and Knight Therapeutics Inc. ("Knight") entered into a Distribution, License and Supply Agreement that grants Knight the exclusive right to commercialize Mytesi and related products in Canada and Israel.
Although it is possible that we may enter into additional corporate partnering relationships related to Mytesi, our intention would be to retain all or co-commercialization and promotional rights in the U.S., so that we do not become primarily a royalty-collecting organization, and we are opposed to entering into any Mytesi partnering relationship that would require splitting indications. We are seeking to put limited geographically-focused partnerships in place in the near term, while also considering possibilities for a worldwide partnership with a leading global entity (excluding the US exclusive commercial rights) in the field of gastrointestinal care and cancer in the long term.
Reduce risks relating to product development
Risk reduction is a key focus of our product development planning. Mytesi is approved for chronic indication, providing us the ability to leverage this corresponding safety data when seeking approval for planned follow-on indications that are also chronic or chronic episodic indications. Crofelemer manufacturing is being conducted at two FDA-inspected and approved locations, including a newly constructed, multimillion-dollar commercial manufacturing facility. In an effort to reduce risk further, we have implemented the following approach: First, we meet with key opinion leaders, typically at medical conferences—as we did in 2017 at Digestive Disease Week for IBS and IBD, the American Society of Clinical Oncology annual meeting, and the Multinational Association of Supportive Care and Congress. Next, we confirm unmet medical needs with these key opinion leaders and discuss the practicality of patient enrollment and trial implementation. We then generate protocols to discuss with the FDA, seeking, when possible, special protocol assessments. Our goal, by the time we start devoting significant funds to a clinical trial, is to have de-risked the program as much as we believe we possibly can, in particular the regulatory pathway. We believe this approach will lead to better long-term outcomes for our products in development.
74
We believe that Jaguar is poised to realize a number of synergistic, value adding benefits—and an expanded pipeline of important human follow-on indications and a second-generation anti secretory agent—upon which to build global partnerships.
In May 2016, the New Drug Application ("NDA") and commercial rights for human applications of crofelemer (Mytesi) previously licensed to Salix Pharmaceuticals, Inc. ("Salix") were transferred to Napo. The active pharmaceutical ingredient ("API") in Mytesi is crofelemer, our proprietary gastrointestinal anti-secretory agent sustainably harvested from the rainforest.
Diarrhea is a common adverse event seen with chemotherapy agents typically used in breast and colon cancers, and in particular in the more recently introduced therapeutic classes of epidermal growth factor receptor ("EGFR") monoclonal antibodies and tyrosine kinase inhibitors ("TKI") often used for chronic adjuvant care management of cancer. The increased need for and use of these agents has made diarrhea one of the most disabling issues for cancer patients.
We will seek partnerships outside the United States for the above indications, while focusing on development, and commercial access in the United States directly. We are also focused on investigating (lechlemer) for various gastrointestinal indications. Lechlemer is a proprietary Jaguar pharmaceutical product, a standardized botanical extract distinct from crofelemer, also sustainably derived from the Croton lechleri tree.
We believe lechlemer, which has the same mechanism of action as crofelemer and is significantly less costly to produce, may support efforts to receive a priority review voucher from the U.S. FDA for a cholera indication. Priority review vouchers are granted by the FDA to drug developers as an incentive to develop treatments for neglected diseases and rare pediatric diseases. Additionally, we believe lechlemer represents a long-term pipeline opportunity as a second-generation anti-secretory agent, on a global basis, for multiple gastrointestinal diseases—especially in resource-constrained countries where cost of goods is a factor, in part, because requirements often exist in such regions for drug prices to decrease annually.
The Company has presented Phase 2 data on crofelemer for the treatment of devastating dehydration in cholera patients from the renowned International Centre for Diarrhoeal Disease Research (icddr,b) in Bangladesh, and Napo plans to follow the same study design for a trial conducted in association with icddr,b in support of development of lechlemer for the potential cholera-related indication.
As announced June 11, 2019, Napo, will receive preclinical services from the National Institute of Allergy and Infectious Diseases ("NIAID") to support the development of lechlemer for the proposed cholera indication. Under NIAID's suite of preclinical services, NIAID-funded contractors will conduct toxicology testing for 7-day rat and dog studies. NIAID is part of the National Institutes of Health, which is an agency of the U.S. Department of Health and Human Services.
Our portfolio development strategy is based on identifying indications that are potentially high-value because they address important medical needs that are significantly or globally unmet, and then strategically sequencing indication development priorities, second-generation product pipeline development, and partnering goals on a global basis.
Our technology for proprietary gastrointestinal disease products is central to the product pipelines of both veterinary and human indications. Crofelemer is also the API in Canalevia, our lead prescription drug product candidate, intended for the treatment of chemotherapy-induced diarrhea in dogs. We expect our first veterinary prescription product launch will be Canalevia for chemotherapy-induced diarrhea, an interesting commercial synergy with the pursuit of follow-on indications for Mytesi.
75
Mytesi Clinical Data
Mytesi has been clinically demonstrated to have:
- •
- Minimal absorption, with plasma concentrations below the level of detection
- •
- No clinically relevant drug-drug interactions
- •
- No effect on viral load or CD4 counts
- •
- Adverse events comparable to those with placebo
The efficacy of Mytesi 125-mg delayed-release tablets twice daily was evaluated in a randomized, double-blind, 24-week, multicenter study (the ADVENT trial) comprised of a placebo-controlled (1 month) treatment period and a placebo-free (5 month) treatment period. The study enrolled HIV-positive patients on stable ART with a history of diarrhea for 1 month or more. In the Mytesi 125mg bid group, more than twice as many patients (18% vs. 8% on placebo, p<0.01) achieved the highly rigorous endpoint defined as reduction to £2 watery stools per week for 2 out of the 4 weeks in the placebo-controlled period (the average baseline in the ADVENT population was 20 watery stools per week).
In a supplemental analysis of the ADVENT study population, 78% of patients in the Mytesi 125mg BID group experienced a decrease in watery stools at week 4. Among these patients that experienced a decrease, 61% had at least a 50% decrease in watery stools. At week 20, 89% of patients in the Mytesi BID group experienced a decrease in watery stools. Among these patients that experienced a decrease, 83% had at least a 50% decrease in watery stools, and over half of patients had no watery stools at all (100% decrease).
Products in Development
Cancer Therapy-Related Diarrhea (CTD)
CTD is a common problem with a relevant mechanism for crofelemer
| National Cancer Institute Criteria for Grading Severity of Diarrhea | ||||||||
|---|---|---|---|---|---|---|---|---|
| |
Grade 1 | Grade 2 | Grade 3 | Grade 4 | ||||
Patients without a colostomy |
Increase of <4 stools per day over pretreatment | Increase of 4 to 6 stools per day or nocturnal stools | Increase of ³7 stools per day or incontinence; need for parenteral support for hydration | Physiologic consequences requiring intensive care; hemodynamic collapse | ||||
Diarrhea is a common adverse event seen with chemotherapy agents in the therapeutic classes of epidermal growth factor receptor ("EGFR") tyrosine kinase inhibitors ("TKI's") and EGFR monoclonal antibodies (for breast, lung, and other malignancies). The increased need for and use of these agents has made diarrhea one of the most disabling issues for cancer patients. Crofelemer offers the potential for an appropriate mechanism of action against this likely secretory diarrhea and has prompted interest among physicians concerned about this diarrheal symptom, stimulating the aforementioned investigator-initiated trials. Diarrhea is also a common adverse event seen with chemotherapy agents used in colorectal and gastric cancers, and chronic maintenance chemotherapy. There are currently no anti-diarrhea agents approved generally for chemotherapy-induced diarrhea.
76
Clinical Study
A study titled HALT-D: DiarrHeA Prevention and ProphyLaxis with Crofelemer in HER2 Positive Breast Cancer Patients Receiving Trastuzumab, Pertuzumab, and Docetaxel or Paclitaxel with or without Carboplatin is currently underway in conjunction with Georgetown University. The primary objective of the study is to characterize the incidence and severity of diarrhea in patients receiving investigational therapy in the setting of prophylactic anti-diarrheal management.
As announced June 18, 2019, a nonclinical pilot study to evaluate the severity of diarrhea caused by a specified dose of a specific TKI in healthy dogs has been completed. A primary nonclinical study, expected to begin in July 2019, will evaluate the effects of crofelemer on diarrhea induced in healthy dogs by the same TKI. Both studies, which are being funded by a third-party cancer agent manufacturer, are intended to provide additional scientific rationale and support for the use of crofelemer in providing symptomatic relief of noninfectious diarrhea in human patients receiving TKI-and/or-other targeted cancer therapy containing regimens in future human clinical investigations.
The nonclinical pilot study involved healthy Beagle dogs, which were given a once-daily administration of the specific TKI for 14 days. The pilot study allowed determination about whether the TKI dose needs to be modified in the primary study to achieve moderate-to-severe diarrhea but not very severe diarrhea that would mandate multiple dose interruptions or drug holidays. The primary study will compare three treatment groups of dogs randomized to receive crofelemer with the same TKI over a 28-day treatment period.
Irritable Bowel Syndrome—Diarrhea Predominant (IBS-D)
Diarrhea is a common symptom of irritable bowel syndrome (IBS), a frustrating, underdiagnosed and undertreated condition. IBS-D is a subtype characterized mainly by loose or watery stools at least 25 percent of the time. According to the U.S. FDA, studies estimate that IBS affects 10 to 15 percent of adults in the United States.
Abdominal pain is the key symptom of IBS, and the pain, which is associated with a change in stool frequency or consistency, can be severe. To improve the diagnosis and outcomes for IBS patients and to update clinicians on the latest research, Dr. William Chey, a gastroenterologist and professor of medicine and nutrition sciences at the University of Michigan, along with an international team of collaborators, compiled Rome IV, an updated compendium of diagnostic criteria on functional GI disorders such IBS. Rome IV contains a chapter titled Centrally Mediated Disorders of Gastrointestinal Pain.
Although new agents for IBS-D have come on the market, there is an unmet medical need for long-term, safe management of the abdominal pain associated with IBS-D. We recognize that patients suffering from IBS-D may require a poly-pharmacy approach to lifetime management of their disease. Mytesi, which represents a novel mechanistic approach with the benefit of a long-term safety profile, could possibly be an important addition to the treatment of IBS-D, if approved for this indication.
Mytesi has been demonstrated to be safe for chronic use, and two studies provide statistically significant results of crofelemer use for abdominal pain in women.
The largest group of IBS sufferers are those with the subtype referred to as IBS-M (mixed diarrhea and constipation). IBS-M is also referred to as IBS-A, because the condition often involves frequent alternating between IBS-D and IBS-C (constipation predominant). IBS-M is distressing for patients as well as difficult to diagnose and manage, and is often associated with pain and urgency as well as significant abdominal distension and bloating. No approved drugs currently exist for IBS-M. Leading gastroenterologists have stated that IBS-C drugs may cause diarrhea in an IBS-M patient, and an IBS-D drug may cause significant constipation. Since Mytesi has not caused constipation in clinical trials or real-world experience, we therefore believe an opportunity exists for an IBS-M indication for
77
Mytesi. Resultingly, and due to the demonstrated safety of Mytesi for chronic use and its demonstrated benefit for abdominal pain in women, Napo is considering expanding development efforts to evaluate the IBS-M indication.
Clinical Study
Crofelemer has been tested in safety studies and two significant Phase 2 studies for d-IBS (diarrhea-predominate Irritable Bowel Syndrome) as detailed below.
Completed Studies—IBS-D
Phase 2a—a randomized double-blind placebo-controlled, dose-ranging (placebo, 125 mg, 250 mg, and 500 mg bid) study over a 12-week treatment period in 246 patients with d-IBS (Rome II criteria), including both males and females, whose average age was 50 years old.
n=245
subjects
61 placebo
62 125 mg crofelemer BID
59 250 mg crofelemer BID
62 500 mg crofelemer BID
IBS symptoms (pain, urgency, stool frequency and consistency, and adequate relief) were self-reported by the patients via an interactive voice response system. Patients needed to exhibit active disease during the two-week baseline period as defined by a mean daily stool frequency greater than or equal to 2/day, pain score greater than or equal to 1 and stool consistency greater than or equal to 3 (5-point Lickert scale for pain and consistency) to be enrolled. Patients received treatment for 12 weeks followed by a two-week treatment free period.
The protocol-specified primary efficacy measure was daily stool consistency. Statistical analysis of the primary endpoint found no significant differences between placebo and any of the crofelemer dose groups (p ³ 0.1434) and no significant dose relationship was seen with regard to change from Baseline to Month 3 in stool consistency scores (p = 0.1165) in the ITT population.
A supplementary analysis of Rome Foundation-defined stool consistency and abdominal pain showed positive results. Responders were subjects who had stool consistency score of ³ 4 for < 25% of days in a given week and ³ 30% improvement in abdominal pain scores a given week (i.e., Rome Foundation-defined stool consistency and abdominal pain responders).
When we look at a supplemental analysis at a reduction in a composite abdominal pain/stool consistency endpoint, the regulatory endpoint in accordance with FDA guidance, we see at the 125 mg dose bid a significant 15% difference with just women patients compared to placebo; and a significant 11% when we include both men and women. The current IBS-d products on the market have a 7-8% reduction (Viberzi and Xifaxan).
In this analysis, Rome Foundation-defined stool consistency and abdominal pain responders were significantly more likely during the entire 3 months in the 125 mg BID group when compared with placebo (24.2% versus 13.1%, p = 0.0399) and there was a statistical trend in favor of crofelemer 125 mg BID during Months 1 through 2 (27.4% versus 16.4%, p = 0.0640). Similar positive effects of crofelemer 125 mg BID were observed in female subjects (n = 183). When the supplementary analysis was applied to the female patients, crofelemer at a dose of 125 mg BID was superior to placebo at Month 3 (26.1% vs 10.9%, p=0.0337).
78
- •
- Results: The 125mg bid of crofelemer exhibited a consistent response during each month among most efficacy endpoints in women with d-IBS
reaching statistical significance (p<0.05) for pain.
- •
- Crofelemer had little effect on the stool consistency score, though there was a trend toward reduced stool frequency.
- •
- Treatment benefits were not apparent in men, although relatively few men enrolled in the trial (13-16/group).
- •
- As with previous trials of crofelemer, no drug-related serious adverse events were reported. Adverse event rates were similar across all dose groups, although in the two highest doses (250 and 500 mg bid) there were a higher percentage of dropouts. There were no drug-related or dose-related differences in constipation. During the two-week treatment-free follow-up period symptoms approached baseline levels.
Safety: Crofelemer at doses of 125, 250 and 500 mg had a safety profile that was generally similar to placebo among men and women with IBS-D.
Phase 2—A Randomized, double-blind, placebo-controlled study to assess the safety and efficacy of crofelemer for the symptomatic treatment of diarrhea predominant irritable bowel syndrome (d-IBS) in 240 female subjects 18 years or older with active d-IBS according to the Rome II criteria for the diagnosis of d-IBS.
The study consisted of a 2-week screening period and a 12-week blinded treatment period followed by a 4-week treatment-free follow-up period. During the 12-week treatment period 240 subjects were given 125 mg of crofelemer BID or placebo BID and recorded daily assessments of their IBS symptoms in the interactive voice response system.
The primary endpoint was the change from baseline for overall percentage of abdominal pain/discomfort free days (PFDs). On a daily basis, respondents recorded the intensity of their abdominal pain/discomfort for that day using the 5-point Likert scale: 0=none, 1=mild, 2=moderate, 3=intense, 4=severe. Any day that a score of zero (0) was recorded was considered a PFD.
Stool consistency and abdominal pain endpoints were analyzed using definitions of symptom improvement from a recent FDA guidance on IBS endpoints (March 2010) and recommendations of the Rome Foundation (letter dated 28 June 2010) concerning the IBS endpoints described in this guidance.
Results: The overall increase in pain-free days (protocol-specified primary endpoint) for subjects in the crofelemer group was not statistically significant when compared with subjects in the placebo group (p = 0.5107)
A supplementary analysis of abdominal pain showed positive results. Responders were subjects who had ³ 30% improvement in abdominal pain scores a given week (i.e., FDA-defined abdominal pain responders; this definition of abdominal pain responders was presented in the March 2010 guidance on IBS endpoints).
In this analysis, abdominal pain responders were significantly more likely during Months 1 through 2 (58.3% versus 45.0%, p = 0.0303) and during the entire 3 months (54.2% versus 42.5%, p = 0.0371) in the crofelemer group when compared to placebo.
Safety: The overall safety profile for crofelemer 125 mg BID for 12 weeks was comparable to that observed with placebo and was consistent with the IBS population under study.
79
Rare Pediatric Disease Indications: Congenital Diarrheal Disorders and Short Bowel Syndrome (SBS)
Congenital diarrheal disorders (CDD) are a group of rare, chronic intestinal channel diseases, occurring in early infancy, that are characterized by severe, lifelong diarrhea and a lifelong need for nutritional intake either parenterally or with a feeding tube. CDDs are related to specific genetic defects inherited as autosomal recessive traits, and the incidence of CDDs is much more prevalent in regions where consanguineous marriage is part of the culture. CDDs are directly associated with serious secondary conditions including dehydration, metabolic acidosis, and failure to thrive, prompting the need for immediate therapy to prevent death and limit lifelong disability.
Potential Orphan-Drug: Congenital Diarrheal Disorders (CDD) & Short Bowel Syndrome (SBS)
Clinical Study—CDD
We have completed safety studies of crofelemer in children as young as 3 months of age, and Napo has accepted a request for support submitted by Dr. Mohamad Miqdady, Chief of Pediatric Gastroenterology, Hepatology and Nutrition at Sheikh Khalifa Medical City (SKMC) in Abu Dhabi, for an investigator-initiated trial of crofelemer, the active pharmaceutical ingredient in Mytesi, for CDD in children.
A pre-clinical study in mice, conducted by an independent third-party investigator, is underway to support possible orphan-drug designation for crofelemer for Congenital Diarrheal Disorders (CDD). This animal model study is examining the effects of crofelemer on diarrhea caused by microvillous inclusion disease (MVID), a very rare autosomal recessive disorder which belongs to the CDD category.
SBS is a complex condition characterized by malabsorption of fluids and nutrients due to congenital deficiencies or surgical resection of small bowel segments. Consequently, patients suffer from symptoms such as debilitating diarrhea, malnutrition, dehydration and imbalances of fluids and salts. This could be due to either a genetic disorder or premature birth. In countries such as the United Arab Emirates and Saudi Arabia, SBS occurs with much higher incidence. Napo recently visited with medical centers in this region.
We have received orphan-drug status for Mytesi (crofelemer) for the SBS pediatric indication and are pursuing orphan-drug status for CDD. The mission of the FDA Office of Orphan Products Development is to advance the evaluation and development of products (drugs, biologics, devices, or medical foods) that demonstrate promise for the diagnosis and/or treatment of rare diseases or conditions.
IBD—Supportive Care:
Key opinion leaders ("KOLs") identified an unmet need to treat diarrhea in IBD patients, particularly in specific subsets of patients. KOLs felt all IBD patients who undergo ileal pouch-anal anastomosis (IPAA) surgery suffer severe, chronic diarrhea following the procedure. Because this is a highly-motivated patient population with a low placebo-responder risk, we believe a relatively small proof-of-concept trial is the appropriate next step from a development standpoint.
KOLs felt crofelemer's novel mechanism of action may also prove to be an effective treatment for diarrhea that results from bile acid malabsorption, which has been shown to occur in approximately 30% of patients with IBD.
Additionally, KOLs felt crofelemer's novel mechanism of action may prove to be an effective treatment for diarrhea experienced by patients receiving IV infusions of Entyvio, a Takeda Pharmaceuticals prescription medicine used in adults with moderate to severe ulcerative colitis or Crohn's disease. Secretory diarrhea occurs when the intestine does not complete absorption of
80
electrolytes and water from luminal contents. This can happen when a nonabsorbable, osmotically active substance is ingested ("osmotic diarrhea") or when electrolyte absorption is impaired ("secretory diarrhea").
Secretory diarrhea can result from bacterial toxins, luminal secretagogues (such as bile acids or laxatives), reduced absorptive surface area caused by disease or resection, circulating secretagogues (such as various hormones, drugs, and poisons), and medical problems that compromise regulation of intestinal function. These studies in acute diarrhea support the normalizing aspect of the mechanism of action, regardless of the cause of the diarrhea, and are supportive of the supportive care indication under development in IBD patients.
Clinical Study
Mytesi has safety studies that support chronic use for the current approved indication, and has demonstrated statistically significant results in multiple supportive care settings, though not specifically in IBD patients. Next steps would include a Phase 2 proof of concept study for supportive care in patients with IBD.
Completed Study—Travelers' Diarrhea (supportive care)
Phase 2—A study of crofelemer in 184 persons in a double-blind, placebo-controlled study for the symptomatic treatment of acute diarrhea among travelers to Jamaica and Mexico.
The study was designed to evaluate the effectiveness of crofelemer in the treatment of travelers' diarrhea.
A total of 184 persons from the United States who acquired diarrhea in Jamaica or Mexico were enrolled in a double-blind, placebo-controlled study examining the effectiveness of three doses of crofelemer in reducing illness. Subjects were treated with 125 mg, 250 mg, or 500 mg crofelemer or a matching placebo four times a day for 2 days. Subjects kept daily diaries of symptoms and were seen each day for 3 days. Of the subjects, 169 (92%) were included in the efficacy analysis.
The most common etiological agent identified was enterotoxigenic Escherichia coli, found in 19% of subjects. The mean time interval from taking the first dose of medication until passage of the last unformed stool during 48-hour therapy (TLUS48) was 38.7 hours for the placebo group.
TLUS48 was shortened by crofelemer:
30.6 h for the 125-mg dose group (p = 0.005);
30.3 h for the 250-mg group; and
32.6 h for the 500-mg group (p = 0.01).
Treatment failures were seen in 29.3% in the placebo group compared with 7.3% (p = 0.01), 4.3 (p = 0.002), and 9.8 (p = 0.026) in the three treatment groups. Crofelemer was well tolerated at all doses.
The study provided statistically significant results of crofelemer use for shortening the duration of travelers' diarrhea. This antisecretory approach works directly against the pathophysiology of travelers' diarrhea and is not likely to potentiate invasive forms of diarrhea or to produce posttreatment constipation.
Cholera/General Watery Diarrhea
According to the Centers for Disease Control and Prevention of the U.S. Department of Health & Human Services, Cholera is an acute, diarrheal illness caused by infection of the intestine with the
81
bacterium Vibrio cholerae. An estimated 3-5 million cases and over 100,000 deaths occur each year around the world. The infection is often mild or without symptoms, but can sometimes be severe. Approximately one in 10 (5-10%) of infected persons will have severe disease characterized by profuse watery diarrhea, vomiting, and leg cramps. In these people, rapid loss of body fluids leads to dehydration and shock. Without treatment, death can occur within hours. At this time, for example, the largest cholera outbreak in recorded history is occurring in Yemen.
We are investigating lechlemer for the indication of cholera/general watery diarrhea. Lechlemer is a distinct and proprietary Napo pharmaceutical formulation of a standardized botanical extract, also sustainably derived from the Croton lechleri tree. We believe lechlemer represents a long-term pipeline opportunity as a second-generation anti-secretory agent, on a global basis, for multiple gastrointestinal diseases. Additionally, we believe lechlemer, which has the same mechanism of action as crofelemer and is significantly less costly to produce, may support efforts to receive a priority review voucher from the U.S. FDA for a cholera indication. Priority review vouchers are granted by the FDA to drug developers as an incentive to develop treatments for neglected diseases and rare pediatric diseases. If approved for this indication, lechlemer could serve as long-term pipeline anti-secretory agent for cholera/general watery diarrhea in geographies where cost of goods is a critical factor, for example, in resource-constrained regions and countries in which a requirement exists for drug prices to decrease annually.
Clinical Study
We have initiated CMC and have multiple animal and human studies in secretory diarrheas. We have also completed a successful trial design for cholera with an anti-secretory mechanism of action, published studies with crofelemer in patients with cholera and other acute severe watery diarrhea disease.
Completed Studies—Cholera and Severe Acute Dehydrating Watery Diarrhea
Phase 2 study of crofelemer in the treatment acute, severely dehydrating watery diarrhea with confirmed cholera with the use of an antibiotic (azithromycin) and oral rehydration therapy in 100 adult patients between 18 and 55 in Bangladesh.
A total of 100 adult patients, from Bangladesh, between the ages of 18 and 55, with acute, severely dehydrating watery diarrhea with confirmed cholera were treated with crofelemer on a background of an antibiotic (azithromycin) and oral rehydration therapy. After a four-hour period of rapid rehydration therapy, patients were randomized 1:2:2 to placebo or 125 mg or 250 mg oral dose of crofelemer. Crofelemer or placebo doses were administered about one hour after the oral administration of azithromycin (1 gm dose). The primary objective was to evaluate the safety and effects of crofelemer on reducing the watery stool output normalized to body weight (mL/kg) in the first 24 hours on the background of azithromycin and rehydration therapy. Crofelemer was well tolerated and there were no drug related adverse events in this study. Both doses of crofelemer produced approximately 25-30% reduction in median watery stool volumes in the 0-6 and 0-12 hour period following initiation of therapy. Crofelemer showed a strong trend in the reduction of watery stool output in the 0-6 hour and 0-12 hour intervals (p=0.07). Upon exclusion of three outlier patients, the crofelemer dose of 125 mg produced a statistically significant reduction in the normalized stool output (p=0.028) and the dose of 250 mg crofelemer showed a strong trend for reduction of watery stool output (p=0.07).
In another study, the effects of crofelemer were evaluated in patients with acute dehydrating watery diarrhea caused by various bacterial pathogens, such as enterotoxic strains of Escherichia coli (ETEC) and Vibrio cholerae infection. Crofelemer was evaluated in adult Indian patients with acute watery diarrhea of less than 24-hour duration with suspected bacterial infections, without the use of antibiotics. In this study, patients received oral doses of placebo or crofelemer at a dose of 250 mg
82
every 6 hours for 2 days on the background of oral rehydration therapy only. The use of antibiotics was prohibited in this study. A total of 98 patients were randomized into this study (47 in placebo group and 51 in the crofelemer group). Primary endpoints for this study were changes in stool weight, frequency, consistency, duration of diarrhea. Secondary endpoints included the assessment of clinical symptoms scored as total of 7-item GI index. Clinical success was defined as no diarrhea within 48 hours from study start date and treatment failure was defined as no improvement/worsening of symptoms after 24 hours, fever, bloody stools or dehydration.
Results: 98 patients (51 crofelemer, 47 placebo) were enrolled in the study. 16 patients (4 in the crofelemer group and 12 in the placebo group) used antibiotics and were considered as treatment failures and were excluded from the "per protocol efficacy analysis". Groups were similar in age, weight, vital signs, stool frequency, consistency, dehydration and GI index.
The crofelemer group had improvement over baseline and compared to placebo at day 3. More specifically, crofelemer showed superior effects in reducing stool weight (61% vs 11%), stool frequency (65% vs 21%), reversion to soft stool (92% vs 49%) and improved the 7-item GI index (70% C vs 33% P), (all p<0.05).
Crofelemer was well tolerated with no related serious adverse events or concerning changes in lab values. Progression to dehydration and report of fecal incontinence was more common in placebo group (p<0.05).
Conclusions: Clinical success (cessation of diarrhea within 48 hours of 1st dose) was achieved in 79% of crofelemer patients compared to 28% placebo patients (p<0.05).
Other Product Potential Future Indications
Institutional Diarrhea
Patients in medical institutions such as hospitals often experience diarrhea following infection with Clostridium difficile, an anaerobic bacillus shed in feces. According to the Centers for Disease Control and Prevention of the U.S. Department of Health & Human Services, any surface, device, or material (e.g., commodes, bathing tubs, and electronic rectal thermometers) that becomes contaminated with feces may serve as a reservoir for the C. difficile spores, which are transferred to patients mainly via the hands of healthcare personnel who have touched a contaminated surface or item. We believe development of an approved formulation of crofelemer for use in C. difficile has the potential to help patients infected with C. difficile leave the hospital sooner, help keep patients infected with C. difficile out of the hospital, and aid in controlling C. difficile contagion in institutional settings, which would also represent a significant economic benefit.
Competition
There are several significantly larger pharmaceutical companies competing with us in the gastrointestinal segment. These companies include Valeant Pharmaceuticals International, Merck & Co., Inc., and Allergan plc as well as smaller pharmaceutical companies.
Diarrhea in adult patients living with HIV/AIDs. We are not aware of any other FDA-approved drugs for the symptomatic relief of diarrhea in HIV/AIDs patients. HIV/AIDs patients also use loperamide and over the counter anti-diarrheal remedies such as Mylanta or Kaopectate to treat their diarrhea, but these medicines affect motility and can result in rebound diarrhea.
Diarrhea predominant irritable bowel syndrome. Two drugs were approved in 2015 for the treatment of diarrhea predominant irritable bowel syndrome, Allergan plc's Virbezi and Xifaxan which is marketed by Valeant Pharmaceuticals International. Also, Lotronex was approved by the FDA in 2000 but was withdrawn from the market and later reintroduced in 2002 under a Risk Management
83
Program. With the exception of Lotronex, the sponsors of Verbezi and Xifaxan employ extensive media and print promotion for the commercialization of these products. We are seeking a partner to further the clinical development and commercialization of crofelemer for d-IBS. There are currently numerous trials on going for d-IBS.
Pediatric diarrhea. Acute diarrhea in children is commonly treated by a change in diet, oral rehydration therapy and/or antibiotics, assuming the cause of the diarrhea is bacterial in nature. Children aged 12 and younger are advised not to use anti-motility drugs (loperamide for example) unless directed to do so by a physician. There are recent clinical trials for probiotics and zinc sulfate. Other recent anti-diarrheal studies in children include a safety and tolerability study of Fidaxomicin for C difficile associated diarrhea.
Cancer therapy-related diarrhea. We are not aware of any FDA-approved drugs specifically indicated for cancer therapy-related diarrhea, including chemotherapy-related diarrhea. A recent Phase IIb trial of elsiglutide for the treatment of chemotherapy induced diarrhea in colorectal cancer patients did not meet statistical significance. Opioids and over the counter drugs are commonly used to treat chemotherapy induced diarrhea, but these drugs affect motility. Certain tyrosine-kinase inhibitor chemotherapy agents have diarrhea as a significant side effect. For example, FDA guidance suggests diarrhea prophylaxis prior to initiating adjuvant therapy with neratinib.
Congenital Diarrheal Disorders and Short Bowel Syndrome. We are not aware of any FDA-approved drugs specifically indicated for Congenital Diarrheal Disorders and Short Bowel Syndrome.
Cholera. We are not aware of any FDA-approved drugs specifically indicated as an anti-secretory agent for use to address the devastating dehydration in cholera patients.
Irritable Bowel Syndrome (IBS). If we receive regulatory approval for Mytesi for IBS, we expect to compete with major pharmaceutical and biotechnology companies that operate in the gastrointestinal space, such as Sucampo AG, Takeda Pharmaceuticals, Allergan, Inc., Ironwood Pharmaceuticals, Inc., Synergy Pharmaceuticals Inc., Sebela Pharmaceuticals, Inc. and Salix Pharmaceuticals. Because Mytesi is approved with chronic safety and several of the other agents have safety concerns, there is likely to be an opportunity for a polypharmacetuical approach to long-term management of these patients, removing a direct competitive scenario from Mytesi's potential entry to the marketplace and disease indication.
To our knowledge, there are currently no FDA-approved anti-secretory products, in particular which act locally in the gut with the chronic safety profile of crofelemer, in development or on the market. Crofelemer represents a new tool in gastrointestinal disease management.
Distribution and Marketing Agreements
Effective January 16, 2019, Napo Pharmaceuticals, Inc. engaged Cardinal Health as its exclusive third party logistics distribution agent for commercial sales for the Company's Mytesi product and to perform certain other services which include, without limitation, storage, distribution, returns, customer support, financial support, EDI and system access support (Exclusive Distribution Agreement).
In addition to the terms and conditions of the Agreement, Cardinal Health's purchase of products, and assumption of title therein, is set forth in the Title Model Addendum. The Title Model Addendum states that upon receipt of product at the 3PL Facility (Cardinal Health in La Vergne, Tennessee) from the Company, title and risk of loss for the Mytesi product purchased by Cardinal Health (excluding consigned inventory) shall pass to Cardinal Health, and title and risk of loss for consigned inventory shall remain with Client until purchased by Cardinal Health in accordance with this Addendum. Napo Pharmaceuticals, Inc. considers Cardinal Health the Company's exclusive customer for Mytesi products per the Cardinal Health Exclusive Distribution agreement.
84
Manufacturing
The plant material used to manufacture is crude plant latex ("CPL") extracted and purified from Croton lechleri, a widespread and naturally regenerating tree in the rainforest that is managed as part of sustainable harvesting programs. The tree is found in several South American countries and has been the focus of long-term sustainable harvesting research and development work. Napo's collaborating suppliers obtain CPL and arrange for the shipment of CPL to Napo's third-party contract manufacturer.
Napo's third-party contract manufacturer, India-based Glenmark Pharmaceuticals Ltd. (Glenmark), a research-driven, global, integrated pharmaceutical company, is Napo's primary manufacturer of crofelemer, the active pharmaceutical ingredient in Mytesi. Glenmark processes CPL into crofelemer utilizing a proprietary manufacturing process. The processing occurs at two FDA-approved Glenmark facilities. Additionally, Napo plans to establish a third processing site, which will be operated by Indena S.p.A., a Milan, Italy-based contract manufacturer dedicated to the identification, development and production of high-quality active principles derived from plants, for use in the pharmaceutical, health food and personal care industries. Indena has completed the required technology transfer and pilot manufacturing and has the equipment in place for the initiation of scale up and validation activities to ultimately support commercial scale manufacturing.
Canalevia will be manufactured by the same process used to manufacture the API that was used in the animal safety studies and the human studies in support of the approval of Mytesi. Napo has also licensed this intellectual property to third parties in connection with its agreements related to the manufacture of crofelemer.
In May 2014 and June 2014, and as amended in February 2015, we entered into binding memorandums of understanding with Indena S.p.A. to negotiate a definitive commercial supply agreement for the manufacture of crofelemer and the botanical extract, SB-300.
We have contracts in place with all the manufacturers and third party testing labs required to manufacture Mytesi and lechlemer. We are finalizing a master service agreement with Glenmark for the manufacture of crofelemer which addresses cost of goods reductions at increasing scale. We are in the process of evaluating alternative and secondary third parties to reduce costs associated with finished product manufacture and the assays necessary to the release specifications of Mytesi.
Proprietary Library of Medicinal Plants
We possess a proprietary library of more than 2,300 medicinal plants.
Intellectual Property
Trademarks
We plan to market all of our products under a trademark or trademarks we select and we will own all rights, title and interest, including all goodwill, associated with such trademarks. Mytesi is a registered trademark owned by Napo.
License Agreements
Termination, Asset Transfer and Transition Agreement
On September 19, 2017 (the "Transfer Date"), Napo entered into the Termination, Asset Transfer and Transition Agreement (the "Glenmark Transition Agreement") with Glenmark. The Glenmark Transition Agreement supersedes the Glenmark Collaboration Agreement and returns to Napo certain rights which Napo licensed to Glenmark pursuant to the Glenmark Collaboration Agreement related to
85
the development and commercialization of crofelemer for certain specified human indications in India and 140 other countries largely in developing regions (the "Transferred Assets").
As a result of the execution of the Glenmark Transition Agreement, we, through Napo, now hold extensive global rights for Mytesi, and also hold commercial rights to the existing regulatory approvals for crofelemer in Brazil, Ecuador, Zimbabwe and Botswana.
In consideration for Glenmark's assignment and transfer of the Transferred Assets to Napo, Napo agreed to pay Glenmark in cash, within 45 days after receipt by Napo, 25% of any payment that Napo receives from a third party to whom Napo grants a license or sublicense or with whom Napo partners in respect of, or sells or otherwise transfers the certain specified human indications in India and 140 other countries largely in developing regions any of the Transferred Assets, subject to certain limitations, until Glenmark has received a total of $7 million. As additional consideration for the assignment and transfer of the Transferred Assets, Napo agreed (i) to enter into a manufacturing and supply agreement with Glenmark for crofelemer, which will be manufactured at either or both of Glenmark's facilities in India (this master service agreement is in draft form, though not yet fully executed) and (ii) to transfer and assign to Glenmark all right, title and interest in and to certain required dedicated equipment used to manufacture crofelemer located at Glenmark's Ankleshwar facility, subject to certain limitations.
Patent Portfolio
Napo
Napo owns a portfolio of patents and patent applications covering formulations of and methods of treatment with proanthocyanidin polymers isolated from Croton spp. or Calophyllum spp., including Mytesi (crofelemer). The patent family associated with International Patent publication WO1998/16111 relates to enteric protected formulations of proanthocyanidin polymers isolated from Croton spp. or Calophyllum spp., including crofelemer, and methods of treating watery diarrhea using these enteric protected formulations. There is one U.S. patent in force in this family, US 7,341,744, which has a term until at least June 23, 2019, which term has been extended under 35 U.S.C. 156 by 1,075 days. Based upon the June 23, 2019 expiration date, the expiration date for crofelemer is June 2, 2022, to account for the regulatory delay in obtaining human marketing approval for crofelemer.
Napo additionally owns a family of patents arising from International Patent Application Publication WO2012/058664 that cover methods of treating HIV associated diarrhea and HAART associated diarrhea with proanthocyanidin polymers isolated from Croton spp. or Calophyllum spp., including crofelemer. In the U.S., there are two issued patents, US 8,962,680 and US 9,585,868, both of which expire October 31, 2031. Outside the U.S., patent protection for methods of treating HIV associated diarrhea has been obtained in Australia, Europe, Hong Kong, Japan, Kenya, Mexico, Russia, Ukraine, South Africa, and Zimbabwe, with expiration dates of October 31, 2031, and applications are pending in Brazil, Hong Kong, Canada, China, and Malaysia. Napo also has patent families related to methods of treating diarrhea predominant irritable bowel syndrome, methods of treating constipation predominant irritable bowel syndrome, and methods of treating inflammatory bowel disease, familial adenomatous polyposis and colon cancer, with proanthocyanidin polymers isolated from Croton spp. or Calophyllum spp., including crofelemer. In particular, for diarrhea predominant irritable bowel syndrome, Napo has two issued U.S. patents, US 8,846,113 and US 9,980,938, which expire on February 9, 2027, as well as issued patents in Australia, Canada, Europe, Gulf States, Hong Kong, Japan, South Korea, Mexico, New Zealand, Singapore, and Taiwan and pending applications in Bangladesh, Bolivia, Chile, Mexico, Panama, Peru, Paraguay, Thailand, and Venezuela, all of which are estimated to expire April 30, 2027; for constipation predominant irritable bowel syndrome, Napo has three issued U.S. patents, with terms to at least April 30, 2027, patents in Australia, Canada, Europe, Hong Kong, Mexico, New Zealand, and Singapore, all of which are estimated to expire April 30, 2027;
86
and for inflammatory bowel disease, familial adenomatous polyposis and/or colon cancer, Napo has two issued U.S. patents, US 8,852,649 and US 9,987,250 with terms until at least January 4, 2028, as well as issued patents in Australia, Hong Kong, and Europe and Canada, which have estimated expiration dates of April 30, 2027. Napo has a pending U.S. non provisional application for the treatment of chemotherapy induced diarrhea (CID) with crofelemer filed on March 9, 2018 and two International Patent Applications on other human indications including for the treatments of short bowel syndrome and congenital diarrhea disorder filed on May 31, 2018.
For methods of manufacturing proanthocyanidin polymers isolated from Croton spp. or Calophyllum spp., including crofelemer, Napo owns issued patents in India, South Africa, and Eurasia with terms at least until August 26, 2029. Napo also owns issued patents in India, Russia, and South Africa and pending applications in Argentina, Brazil, and Venezuela that also cover methods of manufacturing proanthocyanidin polymers isolated from Croton spp. or Calophyllum spp., including crofelemer, with terms at least until January 17, 2032. Lastly, Napo owns two U.S. patents covering a formulation of NP 500 (nordihydroguiaretic acid (NDGA)) and its use in treating a metabolic disorder that have terms until April 23, 2031.
Government Regulation
The FDA and comparable regulatory authorities in state and local jurisdictions and in other countries impose substantial and burdensome requirements upon companies involved in the clinical development, manufacture, marketing and distribution of prescription drugs such as those Napo is developing. These agencies and other federal, state and local entities regulate, among other things, the research and development, testing, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping, approval, advertising and promotion, distribution, post-approval monitoring and reporting, sampling and export and import of Napo's drug product candidates. To comply with the regulatory requirements in each of the jurisdictions in which Napo is seeking to market and subsequently sell its prescription products, Napo is establishing processes and resources to provide oversight of the development, approval processes and launch of its products and to position those products in order to gain market share.
U.S. Government Regulation
In the United States, the FDA approves and regulates drugs under the Federal Food, Drug, and Cosmetic Act ("FDCA"), and its implementing regulations.
The process of obtaining regulatory approvals and the subsequent compliance with applicable federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to a variety of administrative or judicial sanctions, such as the FDA's refusal to approve pending NDAs, withdrawal of an approval, imposition of a clinical hold, issuance of warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties.
The process required by the FDA before a drug may be marketed in the United States generally involves the following:
- •
- completion of pre-clinical laboratory tests, animal studies and formulation studies in compliance with the FDA's good laboratory practice, or
GLP, regulations;
- •
- submission to the FDA of an investigational new drug application, or IND, which must become approved before human clinical trials may begin;
87
- •
- approval by an institutional review board, or IRB, of the study protocol and informed consent forms for the clinical site before each trial may
be initiated. Multiple sites may necessitate the involvement of multiple IRBs and submissions;
- •
- performance of adequate and well-controlled human clinical trials in accordance with good clinical practice, or GCP, requirements to establish
the safety and efficacy of the proposed drug product for each indication;
- •
- submission to the FDA of an NDA which would include the study reports of the clinical trials, chemistry and manufacturing of the active
pharmaceutical ingredient and the final dosage form as well as other required sections to be included in the NDA;
- •
- satisfactory completion of an FDA advisory committee review, if applicable;
- •
- satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance
with current good manufacturing practice, or cGMP, requirements and to assure that the facilities, methods and controls are adequate to preserve the drug's identity, strength, quality and purity; and
- •
- FDA review and approval of the NDA.
Pre-clinical Studies
Pre-clinical studies include laboratory evaluation of the drug product's chemistry, toxicity and formulation, as well as animal studies to assess potential safety and efficacy. An IND sponsor must submit the results of the pre-clinical tests, together with manufacturing information, analytical data and any available clinical data or literature, among other things, to the FDA as part of an IND. Some pre-clinical testing may continue even after the IND is submitted. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to one or more proposed clinical trials and places the clinical trial on a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND may not result in the FDA allowing clinical trials to commence.
Clinical Trials
Clinical trials involve the administration of the investigational new drug to human subjects pursuant to a clinical protocol, under the supervision of qualified investigators in accordance with GCP requirements, which include the requirement that all research subjects provide their informed consent in writing for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives or endpoints of the trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA under the IND. In addition, an IRB at each institution participating in the clinical trial must review and approve the plan for any clinical trial before it commences at that institution. Information about certain clinical trials must be submitted within specific timeframes to the National Institutes of Health, or NIH, for public dissemination on their www.clinicaltrials.govwebsite.
Human clinical trials are typically conducted in three sequential phases, which may overlap or be combined:
- •
- Phase 1: The drug is initially introduced into healthy human subjects or patients with the target disease or condition and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an early indication of its effectiveness depending on the endpoints set forth in the protocol.
88
- •
- Phase 2: The drug is administered to a limited patient population to identify possible adverse effects and safety risks, to
preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage.
- •
- Phase 3: The drug is administered to an expanded patient population, generally at geographically dispersed clinical trial sites, in well-controlled clinical trials to generate enough data to statistically evaluate the efficacy and safety of the product for approval, to establish the overall risk-benefit profile of the product, and to provide adequate information for the labeling of the product.
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if serious adverse events occur. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, or at all. Furthermore, the FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB's requirements or if the drug has been associated with unexpected serious harm to patients.
Special Protocol Assessment
The special protocol assessment, or SPA, process is designed to facilitate the FDA's review and approval of drugs by allowing the FDA to evaluate issues related to the adequacy of certain clinical trials, including Phase 3 clinical trials that can form the primary basis for a drug product's efficacy claim in an NDA. Upon specific request by a clinical trial sponsor, the FDA will evaluate the protocol and respond to a sponsor's questions regarding, among other things, primary efficacy endpoints, trial conduct and data analysis, within 45 days of receipt of the request.
The FDA ultimately assesses whether the protocol design and planned analysis of the trial are acceptable to support regulatory approval of the product candidate with respect to effectiveness of the indication studied. All agreements and disagreements between the FDA and the sponsor regarding a SPA must be clearly documented in a SPA letter or the minutes of a meeting between the sponsor and the FDA.
Even if the FDA agrees to the design, execution and analyses proposed in protocols reviewed under the SPA process, the FDA may revoke or alter its agreement under the following circumstances:
- •
- public health concerns emerge that were unrecognized at the time of the protocol assessment;
- •
- the director of the review division determines that a substantial scientific issue essential to determining safety or efficacy has been
identified after testing has begun;
- •
- a sponsor fails to follow a protocol that was agreed upon with the FDA; or
- •
- the relevant data, assumptions, or information provided by the sponsor in a request for SPA change, are found to be false statements or misstatements, or are found to omit relevant facts.
A documented SPA may be modified, and such modification will be deemed binding on the FDA review division, except under the circumstances described above, if FDA and the sponsor agree in writing to modify the protocol and such modification is intended to improve the study.
Marketing Approval
Assuming successful completion of the required clinical testing, the results of the pre-clinical studies and clinical trials, together with detailed information relating to the product's chemistry, manufacture, controls and proposed labeling, among other things, are submitted to the FDA as part of
89
an NDA requesting approval to market the product for one or more indications. In most cases, the submission of an NDA is subject to a substantial application user fee. Under the Prescription Drug User Fee Act ("PDUFA"), guidelines that are currently in effect, the FDA has a goal of ten months from the date of "filing" of a standard NDA for a new molecular entity to review and act on the submission. This review typically takes twelve months from the date the NDA is submitted to FDA because the FDA has approximately two months to make a "filing" decision.
In addition, under the Pediatric Research Equity Act of 2003 ("PREA"), as amended and reauthorized, certain NDAs or supplements to an NDA must contain data that are adequate to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations; this would include information which supports dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults or full or partial waivers from the pediatric data requirements.
The FDA may also require submission of a risk evaluation and mitigation strategy, or REMS, plan to ensure that the benefits of the drug outweigh its risks. The REMS plan could include medication guides, physician communication plans, assessment plans, and/or elements to assure safe use, such as restricted distribution methods, patient registries, or other risk minimization tools.
The FDA may also require other information as part of the NDA filing such as an environmental impact statement. The FDA can waive some or delay compliance with some of these requirements.
The FDA conducts a preliminary review of all NDAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews an NDA to determine, among other things, whether the drug is safe and effective and whether the facility in which it is manufactured, processed, packaged or held meets standards designed to assure the product's continued safety, quality and purity.
The FDA may refer an application for a novel drug to an advisory committee. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA may inspect one or more clinical trial sites to assure compliance with GCP requirements.
After evaluating the NDA and all related information, including the advisory committee recommendation, if any, and inspection reports regarding the manufacturing facilities and clinical trial sites, the FDA may issue an approval letter, or, in some cases, a complete response letter. A complete response letter must contain a statement of specific items that prevent the FDA from approving the application and will also contain conditions that must be met in order to secure final approval of the NDA and may require additional clinical or pre-clinical testing in order for FDA to reconsider the application. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If and when those conditions have
90
been met to the FDA's satisfaction, the FDA will typically issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications.
Even if the FDA approves a product, it may limit the approved indications for use of the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies, Phase 4 clinical trials, be conducted to further assess a drug's safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution and use restrictions or other risk management mechanisms under a REMS, which can materially affect the potential market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-marketing studies or surveillance programs. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes, and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Post-Approval Requirements
Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims are subject to prior FDA review and approval. New secondary indications must be supported by clinical trials or data. There also are continuing, annual user fee requirements for any marketed products and the establishments at which such products are manufactured, as well as new application fees for supplemental applications with clinical data.
The FDA may impose a number of post-approval requirements as a condition of approval of an NDA. For example, the FDA may require post-marketing testing, including Phase 4 clinical trials, and surveillance to further assess and monitor the product's safety and effectiveness after commercialization.
In addition, drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies, and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with cGMP requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP requirements and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in mandatory revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, include, but are not limited to:
- •
- restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls;
91
- •
- fines, warning letters or holds on post-approval clinical trials;
- •
- refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product approvals;
- •
- product seizure or detention, or refusal to permit the import or export of products; or
- •
- injunctions or the imposition of civil or criminal penalties.
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability. In addition, the FDA regulates the purity and or consistency of the approved product. Products, if deemed adulterated can lead to serious consequences as set forth above as well as civil and criminal penalties.
Foreign Government Regulation
To the extent that any of Napo's product candidates, once approved, are sold in a foreign country, Napo may be subject to similar foreign laws and regulations, which may include, for instance, applicable post-marketing requirements, including safety surveillance, anti-fraud and abuse laws and implementation of corporate compliance programs and reporting of payments or other transfers of value to healthcare professionals.
In order to market Napo's future products in the EEA (which is comprised of the 28 Member States of the EU plus Norway, Iceland and Liechtenstein) and many other foreign jurisdictions, a sponsor must obtain separate regulatory approvals. More concretely, in the EEA, medicinal products can only be commercialized after obtaining a Marketing Authorization, or MA. There are two types of marketing authorizations:
- •
- the Community MA, which is issued by the European Commission through the Centralized Procedure, based on the opinion of the Committee for
Medicinal Products for Human Use of the European Medicines Agency, or EMA, and which is valid throughout the entire territory of the EEA. The Centralized Procedure is mandatory for certain types of
products, such as biotechnology medicinal products, orphan medicinal products and medicinal products indicated for the treatment of AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and
viral diseases. The Centralized Procedure is optional for products containing a new active substance not yet authorized in the EEA, or for products that constitute a significant therapeutic,
scientific or technical innovation or which are in the interest of public health in the EU; and
- •
- National MAs, which are issued by the competent authorities of the Member States of the EEA and only cover their respective territory, are available for products not falling within the mandatory scope of the Centralized Procedure. Where a product has already been authorized for marketing in a Member State of the EEA, this National MA can be recognized in another Member State through the Mutual Recognition Procedure. If the product has not received a National MA in any Member State at the time of application, it can be approved simultaneously in various Member States through the Decentralized Procedure.
Under the above described procedures, before granting the MA, the EMA or the competent authorities of the Member States of the EEA make an assessment of the risk-benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy.
In the EEA, new products authorized for marketing, or reference products, qualify for eight years of data exclusivity and an additional two years of market exclusivity upon marketing authorization. The data exclusivity period prevents generic or biosimilar applicants from relying on the pre-clinical and
92
clinical trial data contained in the dossier of the reference product when applying for a generic or biosimilar marketing authorization in the EU during a period of eight years from the date on which the reference product was first authorized in the EU. The market exclusivity period prevents a successful generic or biosimilar applicant from commercializing its product in the EU until 10 years have elapsed from the initial authorization of the reference product in the EU. The 10-year market exclusivity period can be extended to a maximum of eleven years if, during the first eight years of those 10 years, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies.
In the EEA, marketing authorization applications for new medicinal products not authorized have to include the results of studies conducted in the pediatric population, in compliance with a pediatric investigation plan, or PIP, agreed with the EMA's Pediatric Committee ("PDCO"). The PIP sets out the timing and measures proposed to generate data to support a pediatric indication of the drug for which marketing authorization is being sought. The PDCO can grant a deferral of the obligation to implement some or all of the measures of the PIP until there are sufficient data to demonstrate the efficacy and safety of the product in adults. Further, the obligation to provide pediatric clinical trial data can be waived by the PDCO when these data is not needed or appropriate because the product is likely to be ineffective or unsafe in children, the disease or condition for which the product is intended occurs only in adult populations, or when the product does not represent a significant therapeutic benefit over existing treatments for pediatric patients. Once the marketing authorization is obtained in all Member States of the EU and study results are included in the product information, even when negative, the product is eligible for six months' supplementary protection certificate extension. For orphan-drug designated medicinal products, the 10-year period of market exclusivity is extended to 12 years.
Other U.S. Healthcare Laws
In addition to FDA restrictions on marketing of pharmaceutical and biological products, other U.S. federal and state healthcare regulatory laws restrict business practices in the pharmaceutical industry, which include, but are not limited to, state and federal anti-kickback, false claims, data privacy and security and physician payment and drug pricing transparency laws.
The U.S. federal Anti-Kickback Statute prohibits, among other things, any person or entity from knowingly and willfully offering, paying, soliciting, receiving or providing any remuneration, directly or indirectly, overtly or covertly, to induce or in return for purchasing, leasing, ordering, or arranging for or recommending the purchase, lease, or order of any good, facility, item or service reimbursable, in whole or in part, under Medicare, Medicaid or other federal healthcare programs. The term "remuneration" has been broadly interpreted to include anything of value. The Anti-Kickback Statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers, formulary managers and beneficiaries on the other. Although there are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, the exceptions and safe harbors are drawn narrowly. Practices that involve remuneration that may be alleged to be intended to induce prescribing, purchases, or recommendations may be subject to scrutiny if they do not meet the requirements of a statutory or regulatory exception or safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the U.S. federal Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all its facts and circumstances. Several courts have interpreted the statute's intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the statute has been violated.
93
Additionally, the intent standard under the U.S. federal Anti-Kickback Statute was amended by the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively, the ACA, to a stricter standard such that a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. In addition, the ACA codified case law that a claim including items or services resulting from a violation of the U.S. federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. The majority of states also have anti-kickback laws, which establish similar prohibitions and in some cases may apply to items or services reimbursed by any third-party payor, including commercial insurers.
The federal false claims and civil monetary penalties laws, including the civil False Claims Act, prohibit any person or entity from, among other things, knowingly presenting, or causing to be presented, a false, fictitious or fraudulent claim for payment to, or approval by, the federal government, knowingly making, using, or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government, or from knowingly making a false statement to avoid, decrease or conceal an obligation to pay money to the U.S. federal government. A claim includes "any request or demand" for money or property presented to the U.S. government. Actions under the civil False Claims Act may be brought by the Attorney General or as a qui tam action by a private individual in the name of the government. Violations of the civil False Claims Act can result in very significant monetary penalties and treble damages. Several pharmaceutical and other healthcare companies have been prosecuted under these laws for, among other things, allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. Other companies have been prosecuted for causing false claims to be submitted because of the companies' marketing of products for unapproved, or off-label, uses. Companies also have been prosecuted for allegedly violating the Anti-Kickback Statute and False Claims Act as a result of impermissible arrangements between companies and healthcare practitioners or as a result of the provision of remuneration by the companies to the healthcare practitioners. In addition, the civil monetary penalties statute imposes penalties against any person who is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent. Many states also have similar fraud and abuse statutes or regulations that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor.
Violations of fraud and abuse laws, including federal and state anti-kickback and false claims laws, may be punishable by criminal and civil sanctions, including fines and civil monetary penalties, the possibility of exclusion from federal healthcare programs (including Medicare and Medicaid), disgorgement and corporate integrity agreements, which impose, among other things, rigorous operational and monitoring requirements on companies. Similar sanctions and penalties, as well as imprisonment, also can be imposed upon executive officers and employees of such companies. Given the significant size of actual and potential settlements, it is expected that the government authorities will continue to devote substantial resources to investigating healthcare providers' and manufacturers' compliance with applicable fraud and abuse laws.
The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, created additional federal criminal statutes that prohibit, among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the U.S. federal Anti-Kickback Statute, the ACA broadened the reach of certain criminal healthcare fraud statutes created under HIPAA by amending the intent requirement such that
94
a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
In addition, there has been a recent trend of increased federal and state regulation of payments made to physicians and certain other healthcare providers. The ACA imposed, among other things, new annual reporting requirements through the Physician Payments Sunshine Act for covered manufacturers for certain payments and "transfers of value" provided to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Failure to submit timely, accurately and completely the required information for all payments, transfers of value and ownership or investment interests may result in civil monetary penalties of up to an aggregate of $150,000 per year and up to an aggregate of $1 million per year for "knowing failures." Covered manufacturers must submit reports by the 90th day of each subsequent calendar year. In addition, certain states require implementation of compliance programs and compliance with the pharmaceutical industry's voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, impose restrictions on marketing practices and/or tracking and reporting of gifts, compensation and other remuneration or items of value provided to physicians and other healthcare professionals and entities.
Napo may also be subject to data privacy and security regulation by both the federal government and the states in which Napo conducts its business. HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and their respective implementing regulations, including the Final HIPAA Omnibus Rule published on January 25, 2013, impose specified requirements relating to the privacy, security and transmission of individually identifiable health information held by covered entities and their business associates. Among other things, HITECH made HIPAA's security standards directly applicable to "business associates," defined as independent contractors or agents of covered entities that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney's fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same requirements, thus complicating compliance efforts.
Coverage and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any pharmaceutical products for which Napo obtains regulatory approval. In the United States and markets in other countries, patients who are prescribed treatments for their conditions and providers performing the prescribed services generally rely on third-party payors to reimburse all or part of the associated healthcare costs. Patients are unlikely to use Napo's products unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of Napo's products. Sales of any products for which Napo receives regulatory approval for commercial sale will therefore depend, in part, on the availability of coverage and adequate reimbursement from third-party payors. Third-party payors include government authorities, managed care plans, private health insurers and other organizations. In the United States, the process for determining whether a third-party payor will provide coverage for a pharmaceutical or biological product typically is separate from the process for setting the price of such product or for establishing the reimbursement rate that the payor will pay for the product once coverage is approved. Third-party payors may limit coverage to specific products on an approved list, also known as a formulary, which might not include all of the FDA-approved products for a particular indication. A decision by a third-party payor not to cover Napo's product candidates could reduce physician utilization of Napo's products once approved and have a material adverse effect
95
on Napo's sales, results of operations and financial condition. Moreover, a third-party payor's decision to provide coverage for a pharmaceutical or biological product does not imply that an adequate reimbursement rate will be approved. Adequate third-party reimbursement may not be available to enable Napo to maintain price levels sufficient to realize an appropriate return on Napo's investment in product development. Additionally, coverage and reimbursement for products can differ significantly from payor to payor. One third-party payor's decision to cover a particular medical product or service does not ensure that other payors will also provide coverage for the medical product or service, or will provide coverage at an adequate reimbursement rate. As a result, the coverage determination process will require Napo to provide scientific and clinical support for the use of Napo's products to each payor separately and will be a time-consuming process.
In the EEA, governments influence the price of products through their pricing and reimbursement rules and control of national health care systems that fund a large part of the cost of those products to consumers. Some jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed to by the government. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare the cost effectiveness of a particular product candidate to currently available therapies. Other member states allow companies to fix their own prices for medicines, but monitor and control company profits. The downward pressure on health care costs in general, particularly prescription products, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in some countries, cross border imports from low-priced markets exert a commercial pressure on pricing within a country.
The containment of healthcare costs has become a priority of federal, state and foreign governments, and the prices of pharmaceutical or biological products have been a focus in this effort. Third-party payors are increasingly challenging the prices charged for medical products and services, examining the medical necessity and reviewing the cost-effectiveness of pharmaceutical or biological products, medical devices and medical services, in addition to questioning safety and efficacy. If these third-party payors do not consider Napo's products to be cost-effective compared to other available therapies, they may not cover Napo's products after FDA approval or, if they do, the level of payment may not be sufficient to allow Napo to sell its products at a profit.
Healthcare Reform
A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Government authorities and other third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medical products. For example, in March 2010, the ACA was enacted, which, among other things, increased the minimum Medicaid rebates owed by most manufacturers under the Medicaid Drug Rebate Program; introduced a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected; extended the Medicaid Drug Rebate Program to utilization of prescriptions of individuals enrolled in Medicaid managed care plans; imposed mandatory discounts for certain Medicare Part D beneficiaries as a condition for manufacturers' outpatient drugs coverage under Medicare Part D; subjected drug manufacturers to new annual fees based on pharmaceutical companies' share of sales to federal healthcare programs; created a new Patient Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; creation of the Independent Payment Advisory Board, once empaneled, will have authority to recommend certain changes to the Medicare program that could result in reduced payments for prescription drugs; and establishment of a Center for Medicare Innovation at the CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending. Since its enactment, the U.S. federal government has delayed or suspended implementation of certain provisions of the ACA. In addition, there have been judicial
96
and Congressional challenges to certain aspects of the ACA, and Napo expects there will be additional challenges and amendments to the ACA in the future. For example, in January 2017, the U.S. House of Representatives and Senate passed legislation, which, if signed into law, would repeal certain aspects of the ACA. In addition, Congress could consider subsequent legislation to replace those elements of the ACA if so repealed.
Napo expects that the ACA, as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and lower reimbursement, and additional downward pressure on the price that Napo receives for any approved product. Any reduction in reimbursement from Medicare or other government-funded programs may result in a similar reduction in payments from private payors. Moreover, recently there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products. The implementation of cost containment measures or other healthcare reforms may prevent Napo from being able to generate revenue, attain profitability or commercialize Napo's drugs.
Additionally, on August 2, 2011, the Budget Control Act of 2011 created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation's automatic reduction to several government programs. This included aggregate reductions of Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013 and, due to subsequent legislative amendments to the statute, will stay in effect through 2025 unless additional action is taken by Congress. On January 2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several types of providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. More recently, there has been heightened governmental scrutiny recently over the manner in which manufacturers set prices for their marketed products, which have resulted in several recent Congressional inquiries and proposed bills designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for pharmaceutical products.
Napo expects that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for Napo's products once approved or additional pricing pressures.
Animal Health Business
The development, approval and sale of animal health products are governed by the laws and regulations of each country in which we intend to seek approval, where necessary, to market and subsequently sell our prescription drug and non-drug products. To comply with these regulatory requirements, we are establishing processes and resources to provide oversight of the development, approval processes and launch of its products and to position those products in order to gain market share in each respective market.
Certain U.S. federal regulatory agencies are charged with oversight and regulatory authority of animal health products in the United States. These agencies, depending on the product and its intended use may include the FDA, the USDA and the Environmental Protection Agency. The approval of prescription drugs intended for animal use is regulated by the FDA's Center for Veterinary Medicine ("CVM"). In addition, the Drug Enforcement Administration regulates animal therapeutics that are classified as controlled substances. In addition, the Federal Trade Commission may in the case of non-drug products, regulate the marketing and advertising claims being made.
97
Marketing Exclusivity
We are currently planning on seeking MUMS designation for some of our prescription drug products and if we receive such a designation, we will be entitled to a seven-year marketing exclusivity, which means that we will face no competition from another sponsor marketing the same drug in the same dosage form for the same intended use. If we were to lose such designation or not receive such designation but our application as a new animal drug is found to be a new chemical entity that meets the criteria described by the FDA, we would be entitled to a five-year marketing exclusivity. In order to receive this five-year exclusivity, the FDA would have to find in its approval of our application that our NADA contains an API not previously approved in another application, that the application itself is an original application, not a supplemental application, and that our application included the following studies: one or more investigations to demonstrate substantial evidence of effectiveness of the drug for which we are seeking approval; animal safety studies and human food safety studies (where applicable). If the NADA is seeking approval of a drug for which we have received conditional approval, we, upon approval would still be entitled to a five-year marketing exclusivity provided we meet the criteria as set forth above. If, however, our NADA is for a drug for which the FDA has determined that the drug contains an API that has previously been approved, regardless of whether the original approval was for use in humans or not, we may only be entitled to a three-year marketing exclusivity provided that the NADA is an original, not supplemental, application and contains both safety and efficacy studies demonstrating the safety and efficacy of the drug which is the subject of the application. We have received MUMS designation for Canalevia for the indication of chemotherapy-induced diarrhea, or CID, in dogs. Additionally, the FDA has indicated that the use of Canalevia for the treatment of EID in dogs qualifies as a "minor use", which means that Canalevia is eligible for conditional approval for the indication of EID in dogs.
Labeling
The FDA plays a significant role in regulating the labeling, advertising and promotion of animal drugs. This is also true of regulatory agencies in the EU and other territories. In addition, advertising and promotion of animal health products is controlled by regulations in many countries. These rules generally restrict advertising and promotion to those claims and uses that have been reviewed and approved by the applicable agency. We will conduct a review of advertising and promotional material for compliance with the local and regional requirements in the markets where we eventually may sell its product candidates.
Other Regulatory Considerations
We believe regulatory rules relating to human food safety, food additives, or drug residues in food will not apply to the products we currently are developing because our animal prescription drug product candidates are not intended for use in production animals, with the exception of horses, which qualify as food animals in Europe and Canada; and our non-prescription products are not regulated by section 201(g) of the Federal Food, Drug, and Cosmetic Act, which the FDA is authorized to administer.
Our animal prescription drug product candidates currently in development, if approved, may eventually face generic competition in the United States and in the EU after the period of exclusivity has expired. In the United States, a generic animal drug may be approved pursuant to an abbreviated new animal drug application (ANADA). With an ANADA, a generic applicant is not subject to the submission of new clinical and safety data but instead must only show that the proposed generic product is a copy of the novel drug product, and bioequivalent to the approved novel product. However, if our product candidates are the first approved by the FDA or the EMA as applicable for use in animals, they will be eligible for a five-year marketing exclusivity in the United States and
98
10 years in the EU thereby prohibiting generic entry into the market. If the product has MUMS designation it has a seven-year marketing exclusivity.
We do not believe that our animal non-prescription products are currently subject to regulation in the United States. The CVM only regulates those animal supplements that fall within the FDA's definition of an animal drug, food or feed additive. The Federal Food Drug and Cosmetic Act defines food as "articles used for food or drink for man or other animals and articles used as components of any such article." Animal foods are not subject to pre-market approval and are designed to provide a nutritive purpose to the animals that receive them. Feed additives are defined as those articles that are added to an animal's feed or water as illustrated by the guidance documents. Our non-prescription products are not added to food, are not ingredients in food nor are they added to any animal's drinking water. Therefore, our non-prescription products do not fall within the definition of a food or feed additive. The FDA seeks to regulate such supplements as food or food additives depending on the intended use of the product. The intended use is demonstrated by how the article is included in a food, or added to the animals' intake (i.e., through its drinking water). If the intended use of the product does not fall within the proscribed use making the product a food, it cannot be regulated as a food. There is no intent to make our non-prescription products a component of an animal food, either directly or indirectly. A feed additive is a product that is added to a feed for any reason including the top dressing of an already prepared feed. Some additives, such as certain forage, are deemed to be Generally Recognized as Safe ("GRAS"), and therefore, not subject to a feed Additive Petition approval prior to use. However, the substances deemed GRAS are generally those that are recognized as providing nutrients as a food does. We do not believe that our non-prescription products fit within this framework either. Finally, a new animal drug refers to drugs intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in animals. Our non-prescription products are not intended to diagnose, cure, mitigate, treat or prevent disease and therefore, do not fit within the definition of an animal drug. Our non-prescription products are intended to support a healthy gut, support fluid retention, and normalize stool formation in animals suffering from scours. Additionally, because a previously marketed human formulation of the botanical extract in our non-prescription products was considered a dietary supplement subject to the Dietary Supplement Health and Education Act of 1994 (and not regulated as a drug by the FDA), we do not believe that the FDA would regulate the animal formulation used in our non-prescription products in a different manner. We do not believe that our non-prescription products fit the definition of an animal drug, food or food additive and therefore are not regulated by the FDA at this time.
In addition to the foregoing, we may be subject to state, federal and foreign healthcare and/or veterinary medicine laws, including but not limited to anti-kickback laws, as we may from time to time enter consulting and other financial arrangements with veterinarians, who may prescribe or recommend our products. If our financial relationships with veterinarians are found to be in violation of such laws that apply to us, we may be subject to penalties.
Legal Proceedings
From time to time, we may become involved in litigation relating to claims arising from the ordinary course of business. Other than as set forth below, there are currently no claims or actions pending against us, the ultimate disposition of which could have a material adverse effect on our results of operations, financial condition or cash flows.
March 2018 Demand Letter relating to 2018 Special Meeting of Stockholders
While not a legal proceeding, on March 27, 2018, we received a demand letter from a law firm representing a purported stockholder, relating to certain approvals obtained at a special meeting of stockholders on March 12, 2018 (the "2018 Special Meeting"). The demand letter alleges that we miscalculated the votes with respect to (i) the proposal to amend our Third Amended and Restated
99
Certificate of Incorporation as filed with Secretary of State of the State of Delaware on March 15, 2018 (the "COI"), which increased the authorized shares of Common Stock from 250,000,000 to 500,000,000 (the "Share Increase Proposal") and (ii) the proposal to amend the COI to effect a reverse stock split at a ratio of not less than 1-for-1.2 and not greater than 1-for-10 (the "Former Reverse Stock Split Proposal").We did not implement the Former Reverse Stock Split Proposal. In addition, at the 2018 annual meeting of stockholders held on May 18, 2018, stockholders approved amendments to the COI to (i) effect a reverse stock split at a ratio of not less than 1-for-11 and not greater than 1-for-15 and (ii) decrease the number of authorized shares of Common Stock to 150,000,000.
On September 5, 2018, we responded to the law firm, indicating that the Board unanimously rejected the demands set forth in the demand letter (the "Demand Letter Claims"). While no proceedings with respect to the demand letter were ever initiated, we believe that the allegations set forth in the demand letter were without merit and we would have vigorously defended against any such proceeding. The Demand Letter Claims were settled with a release of all such claims in March 2019 without any material financial settlement costs incurred by us.
July 2017 Complaint Relating to the Merger
On July 20, 2017, a putative class action complaint was filed in the United States District Court, Northern District of California, Civil Action No. 3:17-cv-04102, by Tony Plant (the "Plaintiff") on behalf of shareholders of the Company who held shares on April 12, 2017 and were entitled to vote at the 2017 Special Shareholders Meeting, against the Company and certain individuals who were directors as of the date of the vote (collectively, the "Defendants"), in a matter captioned Tony Plant v. Jaguar Animal Health, Inc., et al., making claims arising under Section 14(a) and Section 20(a) of the Exchange Act and Rule 14a-9, 17 C.F.R. § 240.14a-9, promulgated thereunder by the SEC. The claims alleged false and misleading information provided to investors in the Joint Proxy Statement/Prospectus on Form S-4 (File No. 333-217364) declared effective by the Commission on July 6, 2017 related to the solicitation of votes from shareholders to approve the merger and certain transactions related thereto. The Company accepted service of the complaint and summons on behalf of itself and the United States-based director Defendants on November 1, 2017. The Company has not accepted service on behalf of, and Plaintiff has not yet served, the non-U.S.-based director Defendants.
On October 3, 2017, Plaintiff filed a motion seeking appointment as lead plaintiff and appointment of Monteverde & Associates PC as lead counsel. That motion was granted. Plaintiff filed an amended complaint against the Company and the United States-based director Defendants on January 10, 2018. The Defendants filed a motion to dismiss on March 12, 2018, for which oral arguments were held on June 14, 2018. The court dismissed the amended complaint on September 20, 2018. Plaintiff was entitled to amend that complaint within 20 days from the date of dismissal. On October 10, 2018, Plaintiff amended the amended complaint to focus on the Company's commercial strategy in support of Equilevia and the related disclosure statements in the Form S-4 described above. On November 6, 2018, the Defendants moved to dismiss the second amended complaint. The Defendants argue in their motion that the second amended complaint fails to state a claim upon which relief can be granted because the omissions and misrepresentations alleged in the complaint are immaterial as a matter of law. The court denied the Defendants' motion to dismiss on June 28, 2019. Unless the due date is extended by agreement of the parties, the Defendants' answer to the second amended complaint is due on July 12, 2019. If the Plaintiff were able to prove his allegations in this matter and to establish the damages he asserts, then an adverse ruling could have a material impact on the Company. The Company believes that it is not probable that an asset has been impaired or a liability has been incurred as of the date of the financial statements and the amount of any potential loss is not reasonably estimable.
100
Corporate Information
We were incorporated in the State of Delaware on June 6, 2013. Our principal executive offices are located at 201 Mission Street, Suite 2375, San Francisco, CA 94015 and our telephone number is (415) 371-8300. Our website address is www.jaguar.health. The information contained on, or that can be accessed through, our website is not part of this prospectus. Our voting common stock is listed on the NASDAQ Capital Market and trades under the symbol "JAGX." On July 31, 2017, we completed the acquisition of Napo pursuant to the Agreement and Plan of Merger, dated March 31, 2017, by and among the Company, Napo, Napo Acquisition Corporation, and Napo's representative.
Employees
As of March 31, 2019, we had 41 employees. Six employees hold D.V.M. or Ph.D. degrees. 13 of our employees are engaged in research and development activities and 20 employees are engaged in sales and marketing. None of our employees are represented by labor unions or covered by collective bargaining agreements.
Description of Properties
Our corporate headquarters are located in San Francisco, California, where we lease 6,311 rentable square feet of office space from CA-Mission Street Limited Partnership. Our lease agreement expires on September 30, 2020. We believe that our existing facilities are adequate for our near term needs.
101
DIRECTORS AND CORPORATE GOVERNANCE
The following table lists our directors and their respective ages and positions as of June 27, 2019:
Name
|
Age | Position | ||
|---|---|---|---|---|
James J. Bochnowski(1)(2)(3) |
75 | Chairman of the Board (Class I) | ||
Lisa A. Conte |
60 | Chief Executive Officer, President and Director (Class I) | ||
Jeffery C. Johnson(2)(3) |
47 | Director (Class III) | ||
Greg J. Divis |
52 | Director (Class III) | ||
John Micek III(1)(3) |
66 | Director (Class II) | ||
Jiahao Qui |
33 | Director (Class II) | ||
Jonathan B. Siegel(1)(2) |
46 | Director (Class I) | ||
Murray David MacNaughtan |
52 | Director (Class III) |
- (1)
- Member
of the audit committee.
- (2)
- Member
of the compensation committee.
- (3)
- Member of the nominating committee.
Executive Officers
Lisa A. Conte. Ms. Conte has served as our President, Chief Executive Officer and a member of our board of directors since she founded the company in June 2013. From 2001 to 2014, Ms. Conte served as the Chief Executive Officer of Napo Pharmaceuticals, Inc., a biopharmaceutical company she founded in November 2001. In 1989, Ms. Conte founded Shaman Pharmaceuticals, Inc., a natural product pharmaceutical company. Ms. Conte is also currently a member of the board of directors of Healing Forest Conservatory, a California not-for-profit public benefit corporation. Ms. Conte holds an M.S. in Physiology and Pharmacology from the University of California, San Diego, and an M.B.A. and A.B. in Biochemistry from Dartmouth College.
We believe Ms. Conte is qualified to serve on our board of directors due to her extensive knowledge of our company and experience with our product and product candidates, as well as her experience managing and raising capital for public and private companies.
Steven R. King, Ph.D. Dr. King has served as our Executive Vice President of Sustainable Supply, Ethnobotanical Research and Intellectual Property since March 2014 and as our Secretary since September 2014. From 2002 to 2014, Dr. King served as the Senior Vice President of Sustainable Supply, Ethnobotanical Research and Intellectual Property at Napo Pharmaceuticals, Inc. Prior to that, Dr. King served as the Vice President of Ethnobotany and Conservation at Shaman Pharmaceuticals, Inc. Dr. King has been recognized by the International Natural Products and Conservation Community for the creation and dissemination of research on the long-term sustainable harvest and management of Croton lechleri, the widespread source of crofelemer. Dr. King is currently a member of the board of directors of Healing Forest Conservatory, a California not-for-profit public benefit corporation. Dr. King holds a Ph.D. in Biology from the Institute of Economic Botany of the New York Botanical Garden and an M.S. in Biology from the City University of New York.
Karen S. Wright. Ms. Wright has served as our Chief Financial Officer since December 15, 2015. Prior to joining us, Ms. Wright served as head of finance for Clene Nanomedicine, Inc., beginning in August 2014. From June 2011 to May 2014, Ms. Wright served as vice president of finance and corporate controller at Veracyte, Inc., and from 2006 to 2011, she served as vice president of finance, corporate controller and principal accounting officer of VIA Pharmaceuticals, Inc. Ms. Wright holds a B.S. in Accounting and Marketing from the University of California Walter A. Haas School of Business.
102
Non-Employee Directors
James J. Bochnowski. Mr. Bochnowski has served as a member of our board of directors since February 2014 and as Chairman of our board since June 2014. Since 1988, Mr. Bochnowski has served as the founder and Managing Member of Delphi Ventures, a venture capital firm. In 1980, Mr. Bochnowski co-founded Technology Venture Investors. Mr. Bochnowski holds an M.B.A. from Harvard University Graduate School of Business and a B.S. in Aeronautics and Astronautics from Massachusetts Institute of Technology.
We believe Mr. Bochnowski is qualified to serve on our board of directors due to his significant experience with venture capital backed healthcare companies and experience as both an executive officer and member of the board of directors of numerous companies.
Jeffery C. Johnson. Mr. Johnson has served as a member of our board of directors since March 2018. Mr. Johnson is a partner at Sagard Holdings, ULC and is an investment manager at Sagard Capital Partners Management Corp. Mr. Johnson has been employed by entities affiliated with Sagard Holdings, ULC since 2012. From 2006 to 2011, he served as portfolio manager and senior analyst at Evercore Asset Management. Before that, Mr. Johnson was a portfolio manager and analyst at Citigroup Asset Management where he was responsible for consumer and healthcare investments. He began his career at strategy consulting firm Braxton Associates. He also serves on the board of directors of Peak Achievement Athletics and previously served on the board of directors of Vein Clinics of America. Mr. Johnson received his M.B.A. in Finance and Accounting from the Kellogg School of Management in 1999. Mr. Johnson was elected to our board of directors pursuant to the terms of the Series A Preferred Stock Purchase Agreement, dated as of March 23, 2018, by and between the Company and Sagard Capital Partners, L.P., and the Series A Certificate of Designation, which gives the Preferred Stock holders the right to elect two Class III directors so long as they are entitled to vote in the aggregate 5% or more of all of the votes entitled to be cast by holders of all voting securities of the Company at any meeting of stockholders.
We believe Mr. Johnson is qualified to serve on our board of directors due to his experience evaluating, investing in and managing companies in the health care sector for Sagard Holdings, ULC, and for other investment firms he was previously employed by.
Jiahao Qiu. Mr. Qiu has served as a member of our board of directors since February 2014. Mr. Qiu has been employed at BioVeda Management, Ltd., a life science investment firm, as associate (2010-2012), senior associate (2012-2014) and Principal since April 2014. From 2009 to 2010, he served as an interpreter for the Delegation of the European Union to China. Mr. Qiu holds a B.S. in Biotechnology from the Jiao Tong University in Shanghai, China.
We believe Mr. Qiu is qualified to serve on our board of directors due to his experience with evaluating, managing and investing in life science portfolio companies for BioVeda Management, Ltd.
Greg J. Divis. Mr. Divis has served as a member of our board of directors since June 2018. Mr. Divis currently serves as the Chief Operating Officer of Avadel Pharmaceuticals, an emerging branded specialty pharmaceutical company he joined in 2017. Prior to Avadel he served as an Executive-in-Residence and Operating Partner for Linden Capital, a healthcare-focused middle market private equity firm. Previous roles also include President and Chief Executive Officer of Lumara Health, a specialty-branded pharmaceutical company focused on women's health, where Mr. Divis led the successful turnaround and transformation of the business resulting in a series of transactions culminating in the successful sale to AMAG Pharmaceuticals. Mr. Divis has also held such notable roles as Vice President, Business Development & Lifecycle Management at Sanofi-Aventis, and Vice-President and General Manager, UK and Ireland, for Schering-Plough Corporation. He currently serves on the Board of Directors of Mobius Therapeutics and previously served on the Board of Tolero Pharmaceuticals. Mr. Divis is a graduate of the University of Iowa.
103
We believe Mr. Divis is qualified to serve on our board of directors due to his extensive experience in the pharmaceutical industry and experience as both an executive officer and member of the board of directors of other companies.
Murray David MacNaughtan. Mr. MacNaughtan has served as a member of our board of directors since June 2018. Mr. MacNaughtan is a Partner at Sagard Holdings, and Head of the Sagard Healthcare Royalty Partners team based in Toronto. Prior to this role, he was a Senior Principal with Canada Pension Plan Investment Board, leading their intellectual property investment strategy under the direction of Adam Vigna. Mr. MacNaughtan has over 25 years of experience in the biopharmaceutical industry, with roles in operations, business development, and finance, including over 15 years of investment experience with royalty-based opportunities.Mr. MacNaughtan holds a B.Sc. and M.Sc. in Applied Science from Queen's University and an MBA from the University of Toronto, Rotman School of Business.
Mr. MacNaughtan has known Mr. Alagheband for over 15 years and worked with him at DRI Capital for 7 years. He has known Mr. Manchanda for over a decade, working with him at DRI Capital for two years. Mr. MacNaughtan earned a B.Sc. and M.Sc. in Applied Science from Queen's University in Ontario, and an M.B.A. from the University of Toronto.
We believe Mr. MacNaughtan is qualified to serve on our board of directors due to his extensive experience in specialty finance and the pharmaceutical industry.
John Micek III. Mr. Micek has served as a member of our board of directors since April 2016. From 2000 to 2010, Mr. Micek was managing director of Silicon Prairie Partners, LP, a Palo Alto, California based family-owned venture fund. Since 2010, Mr. Micek has been managing partner of Verdant Ventures, a merchant bank dedicated to sourcing and funding university and corporate laboratory spinouts in areas including pharmaceuticals and cleantech. Mr. Micek serves on the board of directors of Armanino Foods of Distinction, Innovare Corporation and JAL/Universal Assurors. He is also a board member and the Chief Executive Officer and Chief Financial Officer of Enovo Systems and from March 2014 to August 2015 he served as interim Chief Financial Officer for Smith Electric Vehicles, Inc. Mr. Micek is a cum laude graduate of Santa Clara University and the University of San Francisco School of Law, and is a practicing California attorney specializing in financial services.
We believe Mr. Micek is qualified to serve on our board of directors due to his many years of executive experience in management and on boards of director.
Jonathan B. Siegel. Mr. Siegel has served as a member of our board of director since March 2018. Mr. Siegel is founder of JBS Healthcare Ventures, which pursues investments in public and private healthcare entities. In 2017 he left Kingdon Capital ("Kingdon"), where he was principal of the firm, a member of the executive committee and the sector head for healthcare. He joined Kingdon in 2011 and has more than 18 years of investment experience. Prior to joining Kingdon, Mr. Siegel was with SAC Capital Advisors from 2005 to 2011, serving as a portfolio manager for healthcare starting in 2007. Before joining SAC, he was an associate director of pharmaceutical and specialty pharmaceutical research with Bear, Stearns & Co., a research associate with Dresdner Kleinwort Wasserstein, specializing in pharmaceuticals, a consultant to the Life Sciences Division of Computer Sciences Corporation, a research associate at the Novartis Center for Immunobiology, Harvard Medical School, Beth Israel Deaconess Medical Center, and a research assistant at Tufts University School of Medicine. Additionally, he previously served on the board of KV Pharmaceutical Company. Mr. Siegel received a B.S. in Psychology from Tufts University in 1995 and an M.B.A. from Columbia Business School in 1999.
We believe Mr. Siegel is qualified to serve on our board of directors due to his extensive experience in the pharmaceutical investment sector.
104
Family Relationships
There are no family relationships among any of our directors or executive officers.
Board Composition and Risk Oversight
Our business and affairs are managed under the direction of our board of directors, which consists of eight members. Certain members of our board of directors were elected pursuant to the provisions of a voting agreement among certain of our major stockholders, as amended. See "Certain Relationships and Related Persons Transactions—Voting Agreement" for more information regarding the voting agreement.
Six of the eight directors that comprise our board are independent within the meaning of the independent director rules of the NASDAQ Stock Market, LLC, or NASDAQ.
Our board of directors are divided into three classes of directors. At each annual meeting of stockholders, a class of directors will be elected for a three-year term to succeed the class whose term is then expiring. The terms of the directors will expire upon the election and qualification of successor directors at the annual meeting of stockholders to be held during the years 2020 for the Class II directors, 2021 for the Class III directors, and 2022 for the Class I directors.
The Class I directors are Ms. Conte, Mr. Bochnowski, and Mr. Siegel.
The Class II directors are Mr. Qiu and Mr. Micek.
The Class III directors are Mr. Johnson, Mr. Divis and Mr. MacNaughtan.
We expect that any additional directorships resulting from an increase in the number of directors will be distributed among the three classes so that, as nearly as possible, each class will consist of one-third of the directors. The division of our board of directors into three classes with staggered three-year terms may delay or prevent a change of our management or a change in control.
Our board of directors has an active role, as a whole and at the committee level, in overseeing the management of our risks. Our board of directors is responsible for general oversight of risks and regular review of information regarding our risks, including credit risks, liquidity risks and operational risks. Our compensation and nominating committees are responsible for overseeing the management of risks relating to our executive compensation plans and arrangements and the risks associated with the independence of our board of directors and potential conflicts of interest. Our audit committee is responsible for overseeing the management of our risks relating to accounting matters and financial reporting. While each committee is responsible for evaluating certain risks and overseeing the management of such risks, our entire board of directors is regularly informed through discussions from committee members about such risks. Our board of directors believes its administration of risk oversight function has not affected our board of directors' leadership structure.
Director Independence
Our common stock is listed on The Nasdaq Capital Market. Under Nasdaq rules, independent directors must comprise a majority of a listed company's board of directors. In addition, Nasdaq rules require that, subject to specified exceptions, each member of a listed company's Audit, Compensation and Nominating Committee must be independent. Audit Committee members must also satisfy the independence criteria set forth in Rule 10A-3 under the Exchange Act. Under Nasdaq rules, a director will only qualify as an "independent director" if, in the opinion of the company's board of directors, such person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director.
105
To be considered independent for purposes of Rule 10A-3, a member of an audit committee of a listed company may not, other than in his or her capacity as a member of the audit committee, our board of directors, or any other board committee (1) accept, directly or indirectly, any consulting, advisory, or other compensatory fee from the listed company or any of its subsidiaries or (2) be an affiliated person of the listed company or any of its subsidiaries.
Our board of directors periodically undertakes a review of its composition, the composition of its committees and the independence of our directors and considered whether any director has a material relationship with us that could compromise his or her ability to exercise independent judgment in carrying out his or her responsibilities. Based upon information requested from and provided by each director concerning his or her background, employment and affiliations, including family relationships, our board of directors has determined that six of our eight directors (i.e., Mr. Bochnowski, Mr. Siegel, Mr. Micek, Mr. Qiu, Mr. Divis and Mr. MacNaughtan) do not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and that each of these directors is "independent" as that term is defined under the Nasdaq rules. Our board of directors also determined that Mr. Micek (chairperson), Mr. Siegel and Mr. Bochnowski, who comprise our Audit Committee, Mr. Bochnowski (chairperson), Mr. Siegel and Mr. Johnson, who comprise our Compensation Committee, and Mr. Johnson (chairperson), Mr. Bochnowski and Mr. Micek, who comprise our Nominating Committee, satisfy the independence standards for those committees established by applicable SEC rules and the Nasdaq rules and listing standards.
In making this determination, our board of directors considered the relationships that each non-employee director has with us and all other facts and circumstances our board of directors deemed relevant in determining independence, including the beneficial ownership of our capital stock by each non-employee director.
Board Committees
Audit Committee
The members of our Audit Committee are Mr. Micek, Mr. Siegel and Mr. Bochnowski. Mr. Micek is the chairperson of the Audit Committee. Our Audit Committee's responsibilities include:
- •
- appointing, approving the compensation of, and assessing the independence of our registered public accounting firm;
- •
- overseeing the work of our independent registered public accounting firm, including through the receipt and consideration of reports from that
firm;
- •
- reviewing and discussing with management and our independent registered public accounting firm our annual and quarterly financial statements
and related disclosures;
- •
- monitoring our internal control over financial reporting, disclosure controls and procedures and code of conduct;
- •
- discussing our risk management policies;
- •
- establishing policies regarding hiring employees from our independent registered public accounting firm and procedures for the receipt and
retention of accounting related complaints and concerns;
- •
- reviewing and approving or ratifying any related person transactions; and
- •
- preparing the Audit Committee report required by SEC rules.
106
All audit and non-audit services, other than de minimis non-audit services, to be provided to us by our independent registered public accounting firm must be approved in advance by our Audit Committee.
Our board of directors has determined that each of Mr. Micek, Mr. Siegel and Mr. Bochnowski is an independent director under Nasdaq rules and under Rule 10A-3. All members of our Audit Committee meet the requirements for financial literacy under the applicable rules and regulations of the SEC and Nasdaq. Our board of directors has determined that Mr. Micek is an "audit committee financial expert," as defined by applicable SEC rules, and has the requisite financial sophistication as defined under the applicable Nasdaq rules and regulations.
The Audit Committee held four meetings in 2018. The Audit Committee has adopted a written charter approved by the board of directors, which is available on our website at https://jaguarhealth.gcs-web.com/corporate-governance.
Compensation Committee
The members of our Compensation Committee are Mr. Bochnowski, Mr. Siegel and Mr. Johnson. Mr. Bochnowski is the chairperson of the Compensation Committee. Our Compensation Committee's responsibilities include:
- •
- determining, or making recommendations to our board of directors with respect to, the compensation of our Chief Executive Officer;
- •
- determining, or making recommendations to our board of directors with respect to, the compensation of our other executive officers;
- •
- overseeing and administering our cash and equity incentive plans;
- •
- reviewing and making recommendations to our board of directors with respect to director compensation;
- •
- reviewing and discussing at least annually with management our "Compensation Discussion and Analysis" disclosure if and to the extent then
required by SEC rules; and
- •
- preparing the Compensation Committee report and necessary disclosure in our annual proxy statement in accordance with applicable SEC rules.
Our board has determined that each of Mr. Bochnowski, Mr. Siegel and Mr. Johnson is independent under the applicable Nasdaq rules and regulations, is a "non-employee director" as defined in Rule 16b-3 promulgated under the Exchange Act, and is an "outside director" as that term is defined in Section 162(m) of the Internal Revenue Code of 1986, as amended.
The Compensation Committee held three meeting in 2018. All compensation-related matters were approved at the board of directors' level. The Compensation Committee has adopted a written charter approved by the board of directors, which is available on our website at https://jaguarhealth.gcs-web.com/corporate-governance.
Nominating Committee
The members of our Nominating Committee are Mr. Johnson, Mr. Bochnowski and Mr. Micek. Mr. Johnson is the chairperson of the Nominating Committee. Our Nominating Committee's responsibilities include:
- •
- identifying individuals qualified to become members of our board of directors;
- •
- evaluating qualifications of directors;
107
- •
- recommending to our board of directors the persons to be nominated for election as directors and to each of the committees of our board of
directors; and
- •
- overseeing an annual evaluation of our board of directors.
The Nominating Committee did not hold any meetings in 2018. All nomination-related matters were approved at the board of directors' level. The Nominating Committee has adopted a written charter approved by the board of directors, which is available on our website at https://jaguarhealth.gcs-web.com/corporate-governance.
Code of Ethics and Conduct
We have adopted a Code of Business Conduct and Ethics that applies to our directors, officers and employees, including our President and Chief Executive Officer, our Chief Financial Officer and other employees who perform financial or accounting functions. The Code of Business Conduct and Ethics sets forth the basic principles that guide the business conduct of our employees. A current copy of the code is available on our website at https://jaguarhealth.gcs-web.com/corporate-governance. We intend to disclose future amendments to certain provisions of our code of business conduct and ethics, or waivers of such provisions on our website to the extent required by applicable rules and exchange requirements. The inclusion of our website address in this prospectus does not incorporate by reference the information on or accessible through our website into this prospectus.
Compensation Committee Interlocks and Insider Participation
None of the members of our Compensation Committee has ever been an officer or employee of our company. None of our executive officers currently serves, or in the past year has served, as a member of the board of directors or Compensation Committee or other board committee performing equivalent functions of any entity that has one or more of its executive officers serving on our board of directors or Compensation Committee.
Limitation of Liability and Indemnification
Our third amended and restated certificate of incorporation, as amended, and amended and restated bylaws contain provisions that limit the personal liability of our directors for monetary damages to the fullest extent permitted by Delaware law. Delaware law provides that directors of a corporation will not be personally liable to us or our stockholders for monetary damages for any breach of fiduciary duties as directors, except liability for:
- •
- any breach of the director's duty of loyalty to us or our stockholders;
- •
- any act or omission not in good faith or that involves intentional misconduct or a knowing violation of law;
- •
- unlawful payments of dividends or unlawful stock repurchases or redemptions as provided in Section 174 of the DGCL; or
- •
- any transaction from which the director derived an improper personal benefit.
Such limitation of liability does not apply to liabilities arising under federal securities laws and does not affect the availability of equitable remedies, such as injunctive relief or rescission.
Our third amended and restated certificate of incorporation provides that we indemnify our directors to the fullest extent permitted by Delaware law. In addition, our amended and restated bylaws provide that we indemnify our directors and officers to the fullest extent permitted by Delaware law. Our amended and restated bylaws also provide that we shall advance expenses incurred by a director or officer in advance of the final disposition of any action or proceeding, and permit us to secure
108
insurance on behalf of any officer, director, employee or other agent for any liability arising out of his or her actions in that capacity, regardless of whether we would otherwise be permitted to indemnify him or her under the provisions of Delaware law. We have entered and expect to continue to enter into agreements to indemnify our directors, executive officers and other employees as determined by our board of directors. With certain exceptions, these agreements provide for indemnification for related expenses including, among others, attorneys' fees, judgments, fines and settlement amounts incurred by any of these individuals in any action or proceeding. We believe that these bylaw provisions and indemnification agreements are necessary to attract and retain qualified persons as directors and officers. We also maintain directors' and officers' liability insurance.
The limitation of liability and indemnification provisions in our third amended and restated certificate of incorporation and amended and restated bylaws and our indemnification agreements, may discourage stockholders from bringing a lawsuit against our directors for breach of their fiduciary duty of care. They may also reduce the likelihood of derivative litigation against our directors and officers, even though an action, if successful, might benefit us and other stockholders. Furthermore, a stockholder's investment may be adversely affected to the extent that we pay the costs of settlement and damage awards against directors and officers. There is no pending litigation or proceeding involving any of our directors, officers or employees for which indemnification is sought, and we are not aware of any threatened litigation that may result in claims for indemnification.
Board Leadership Structure
Our Bylaws and corporate governance guidelines provide our board of directors with flexibility in its discretion to combine or separate the positions of Chairperson of the board of directors and chief executive officer. As a general policy, our board of directors believes that separation of the positions of Chairperson and chief executive officer reinforces the independence of the board of directors from management, creates an environment that encourages objective oversight of management's performance and enhances the effectiveness of the board of directors as a whole. We expect and intend the positions of Chairperson of the board and chief executive officer to be held by two individuals in the future.
109
COMPENSATION OF DIRECTORS AND EXECUTIVE OFFICERS
Summary Compensation Table (2018 and 2017)
The total compensation paid to the Company's Principal Executive Officer and its two highest compensated executive officers other than the Principal Executive Officer, respectively, for services rendered in 2018 and 2017, as applicable, is summarized as follows:
| |
Year | Salary ($) |
Bonus ($) |
Option awards ($)(1) |
All other compensation ($)(2) |
Total ($) |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Lisa A. Conte |
2018 | 480,000 | $ | 30,000 | 490,046 | 19,014 | 1,019,060 | ||||||||||||
President and Chief Executive Officer |
2017 | 440,000 | — | 184,990 | 17,599 | 642,589 | |||||||||||||
Steven R. King, Ph.D. |
2018 | 287,045 | — | 154,206 | 36,316 | 783,362 | |||||||||||||
Executive Vice President, Sustainable |
2017 | 280,500 | — | 59,202 | 32,032 | 393,457 | |||||||||||||
Supply, Ethnobotanical Research and Intellectual Property |
|||||||||||||||||||
Karen S. Wright |
2018 | 280,667 | 30,000 | 120,509 | — | 431,175 | |||||||||||||
Chief Financial Officer and |
2017 | 240,000 | 5,000 | 554,108 | — | 300,108 | |||||||||||||
Treasurer(3) |
|||||||||||||||||||
Footnotes to Summary Compensation Table
- (1)
- Represents
the dollar amounts recognized for financial statement reporting purposes with respect to the fiscal year (for stock option awards) determined under FASB
ASC Topic 718. The following are the options held by each executive officer as of December 31, 2018:
- a.
- Ms. Conte—an
aggregate of 10,491 shares were granted as follows: 152 shares granted April 1, 2014, 81 shares granted July 2, 2015,
106 shares granted July 7, 2015, 66 shares granted April 1, 2016 which became effective at the annual stockholders' meeting of June 14, 2016, 302 shares granted
September 22, 2016, 16 shares granted December 19, 2016, 272 shares granted December 21, 2017, 3,092 shares granted on March 12, 2018 and 6,399 shares granted on
June 1, 2018. The weighted average exercise price of all of Ms. Conte option grants is $475.92.
- b.
- King—an
aggregate of 3,672 shares were granted as follows: 88 shares granted April 1, 2014, 47 shares granted July 2, 2015 which became
effective at the annual stockholders' meeting of June 14, 2016, 25 shares granted April 1, 2016 which became effective at the annual stockholders' meeting of June 14, 2016, 21
shares granted September 22, 2016, 4 shares granted December 19, 2016, 90 shares granted December 21, 2017, 1,264 shares granted on March 12, 2018 and 2,133 shares granted
on June 1, 2018. The weighted average exercise price of all of Dr. King's option grants is $468.15.
- c.
- Ms. Wright—an
aggregate of 2,350 shares were granted as follows: 19 shares granted November 23, 2015, 3 shares granted April 1, 2016
which became effective at the annual stockholders' meeting of June 14, 2016, 98 shares granted September 22, 2016, 2 shares granted December 19, 2016, 51 shares granted
December 21, 2017, 951 shares granted on March 12, 2018 and 1,226 shares granted on June 1, 2018. The weighted average exercise price of all of Ms. Wright's option grants
is $415.15.
- d.
- All of the April 1, 2014 option grants vested 25% on January 1, 2015 (nine months from grant date), with the remainder vesting equally over the following 27 months such that the options are vested in full on April 1, 2017. Ms. Wright's November 23, 2015 option vested 25% on September 9, 2016, with the remainder vesting equally over the following 27 months such that the option is vested in full on November 9, 2018. All of the July 2, 2015 options were granted
110
contingent upon approval of the Company's stockholders at the June 14, 2016 annual stockholders' meeting and vest 1/36th per month beginning one month after grant date, with the remainder vesting equally over the following 35 months such that the option is vested in full on July 2, 2018. Ms. Conte's July 7, 2015 option was likewise granted contingent upon approval of the Company's stockholders at the June 14, 2016 annual stockholders' meeting and vests 1/36th per month beginning one month after grant date, with the remainder vesting equally over the following 35 months such that the option is vested in full on July 7, 2018. All of the options granted on April 1, 2016 which became effective at the annual stockholders' meeting of June 14, 2016, September 22, 2016, December 19, 2016 vest 1/36th per month beginning one month after grant, with the remainder vesting equally over the following 35 months such that the option is vested in full on December 19, 2019. All of the December 21, 2017 options grants vested in full as of March 31, 2018 if the option holder was an employee on that date. All of the March 12, 2018 options grants vest 1/36th per month beginning one month after grant, with the remainder vesting equally over the following 35 months such that the option is vested in full on March 12, 2021. All of the June 1, 2018 options grants vest 1/36th per month beginning one month after grant, with the remainder vesting equally over the following 35 months such that the option is vested in full on June 1, 2021.
- (2)
- Amounts
shown in this column reflect incremental health insurance premiums paid for such executive's family members.
- (3)
- Ms. Wright has served as Chief Financial Officer and Treasurer since December 15, 2015.
Narrative to Summary Compensation Table
Understanding our history is key to the understanding of our compensation structure for 2017 and 2018. After our initial public offering closed on May 18, 2015, the executive officers of privately-held Jaguar Health, Inc. (f/k/a Jaguar Animal Health, Inc.) became our named executive officers.
Base Salary
On July 2, 2015, the Compensation Committee increased Ms. Conte's annual base salary from $400,000 to $440,000 and Dr. King's annual base salary from $255,000 to $280,500. The pay increases were effective June 15, 2015. On December 15, 2015, the Company's board of directors appointed Karen S. Wright as the Company's new Chief Financial Officer. Ms. Wright's annual base salary is $240,000. On April 12, 2018, the Compensation Committee increased Ms. Conte's annual base salary from $440,000 to $500,000, Dr. King's annual base salary from $280,500 to $290,317, and Ms. Wright's annual base salary from $240,000 to $301,000, all effective May 31, 2018.
Bonuses
We paid a performance based bonus to Ms. Wright of $5,000 in 2017 and a one-time cash bonus of $30,000 to both Ms. Conte and Ms. Wright in 2018.
Equity Compensation
Ms. Conte and Dr. King received stock option grants at the time they were hired by privately-held Jaguar Animal Health, Inc. Such options generally vest over time, with 25% of the options vesting after nine months of employment and monthly vesting thereafter with full vesting after three years. Ms. Wright received stock option grants with a similar vesting schedule at the time they were hired by us. The board of directors periodically grants additional options to the current named executive officers that typically vest ratably over a three-year period.
111
All stock options and RSUs issued to our current named executive officers vest and become exercisable upon a change in control.
Outstanding Equity Awards at 2018 Fiscal Year End
The following table provides information regarding outstanding equity awards held by our named executive officers as of December 31, 2018.
| |
|
Number of Securities Underlying Unexercised Options |
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
|
|
Stock Option expiration date |
||||||||||
| |
Options Vesting Commencement Date |
Option exercise price |
|||||||||||
| |
Exercisable | Unexercisable | |||||||||||
Lisa A. Conte |
4/1/2014 | 152 | — | (1) | $ | 2,656.50 | 4/1/2024 | ||||||
|
7/2/2015 | 81 | — | (2) | $ | 5,344.23 | 7/2/2025 | ||||||
|
7/7/2015 | 106 | — | (3) | $ | 5,291.75 | 7/7/2025 | ||||||
|
4/1/2016 | 58 | 8 | (5) | $ | 1,658.92 | 4/1/2026 | ||||||
|
9/22/2016 | 227 | 75 | (6) | $ | 1,312.44 | 9/22/2026 | ||||||
|
12/19/2016 | 10 | 6 | (7) | $ | 776.96 | 12/19/2026 | ||||||
|
12/21/2017 | 272 | — | (8) | $ | 129.56 | 12/21/2027 | ||||||
|
3/12/2018 | 1,374 | 1,718 | (9) | $ | 587.97 | 3/12/2028 | ||||||
|
6/1/2018 | 1,422 | 4,977 | (10) | $ | 190.86 | 6/1/2028 | ||||||
Steven R. King, Ph.D. |
4/1/2014 | 88 | — | (1) | $ | 2,656.50 | 4/1/2024 | ||||||
|
7/2/2015 | 47 | — | (2) | $ | 5,344.23 | 7/2/2025 | ||||||
|
4/1/2016 | 23 | 2 | (5) | $ | 1,658.92 | 4/1/2026 | ||||||
|
9/22/2016 | 16 | 5 | (6) | $ | 1,312.44 | 9/22/2026 | ||||||
|
12/19/2016 | 2 | 2 | (7) | $ | 776.96 | 12/19/2026 | ||||||
|
12/21/2017 | 90 | — | (8) | $ | 129.56 | 12/21/2027 | ||||||
|
3/12/2018 | 562 | 702 | (9) | $ | 587.97 | 3/12/2028 | ||||||
|
6/1/2018 | 474 | 1,659 | (10) | $ | 190.86 | 6/1/2028 | ||||||
Karen S. Wright |
11/9/2015 | 19 | — | (4) | $ | 2,141.90 | 11/23/2025 | ||||||
|
4/1/2016 | 3 | — | (5) | $ | 1,658.92 | 4/1/2026 | ||||||
|
9/22/2016 | 74 | 24 | (6) | $ | 1,312.44 | 9/22/2026 | ||||||
|
12/19/2016 | 1 | 1 | (7) | $ | 776.96 | 12/19/2026 | ||||||
|
12/21/2017 | 51 | — | (8) | $ | 129.56 | 12/21/2027 | ||||||
|
3/12/2018 | 422 | 529 | (9) | $ | 587.97 | 3/12/2028 | ||||||
|
6/1/2018 | 272 | 954 | (10) | $ | 190.86 | 6/1/2028 | ||||||
- (1)
- On
January 1, 2015, 25% of each of such named executive officer's shares vested and became exercisable. The remainder of the shares were vested in
approximately equal monthly installments through April 1, 2017, subject to continued service with us through each relevant vesting date.
- (2)
- The
shares were granted on July 2, 2015 contingent upon the approval of the stockholders at the June 14, 2016 annual stockholders' meeting and vest
1/36th per month beginning one month after grant date, with the remainder vested equally over the following 35 months such that the option was fully vested on July 2, 2018,
subject to continued service with us through each relevant vesting date.
- (3)
- The shares were granted on July 7, 2015 contingent upon the approval of the stockholders at the June 14, 2016 annual stockholders' meeting and vested 1/36th per month beginning one month after grant date, with the remainder vested equally over the following 35 months such that the option was fully vested on July 7, 2018, subject to continued service with us through each relevant vesting date.
112
- (4)
- The
shares were granted on November 23, 2015. On August 9, 2016, 25% of such named executive officer's shares vested and became exercisable. The
remainder of the shares vested in approximately equal monthly installments through November 9, 2018.
- (5)
- The
options were granted on April 1, 2016, which became effective at the annual stockholders' meeting of June 14, 2016, and vest 1/36th per
month beginning one month after grant, with the remainder vesting equally over the following 35 months such that the option is vested in full on April 1, 2019, subject to continued
service with us through each relevant vesting date.
- (6)
- The
options were granted on September 22, 2016 and vest 1/36th per month beginning one month after grant, with the remainder vesting equally over the
following 35 months such that the option is vested in full on September 22, 2019, subject to continued service with us through each relevant vesting date.
- (7)
- The
options were granted on December 19, 2016 and vest 1/36th per month beginning one month after grant, with the remainder vesting equally over the
following 35 months such that the option is vested in full on December 19, 2019, subject to continued service with us through each relevant vesting date.
- (8)
- The
options were granted on December 21, 2017 and vested 100% on March 31, 2018.
- (9)
- The
options were granted on March 12, 2018 and vest 1/36th per month over thirty-six months such that the option is vested in full on March 12,
2021, subject to continued service with us through each relevant vesting date.
- (10)
- The options were granted on June 1, 2018 and vest 1/36th per month over thirty-six months such that the option is vested in full on June 12, 2021, subject to continued service with us through each relevant vesting date.
Offering-Related Stock Option Grants to Executive Officers, Directors and Certain Employees
Our Compensation Committee has recommended that our board of directors grant stock options pursuant to the 2014 Stock Incentive Plan to our executive officers and certain employees upon consummation of this offering ("2019 Grant"). Our Compensation Committee has also determined to consider at a later date subsequent to the offering whether to recommend to the full Board options grants, if any, to Board members for their Board services to the Company. The purpose of this proposed grant is to improve our ability to motivate and retain the services of our executive officers, directors and key employees in light of the fact that all outstanding stock options held by such individuals are currently significantly out of the money, and following the stock option to RSU exchange approved by our shareholders on February 28, 2019 with a projected offer date in Fall 2019, the aggregate options and RSUs held by our executive officers and key employees will equal approximately 0.1% (or 0%, if all outstanding options with an exercise price in excess of $10 / share are excluded) of the issued and outstanding shares of Common Stock on a fully diluted basis (and assuming conversion of all outstanding convertible securities, including but not limited to conversion of our Series A Preferred Stock into shares of Common Stock in accordance with our Certificate of Incorporation as amended from time to time without any regulatory limitations, all issued and outstanding warrants, notes, RSUs and stock options (whether issued under or outside the 2014 Stock Incentive Plan and the like)) as of the closing of this offering. If granted, 2019 Grant options are expected to cover up to an aggregate of 12.0% of the issued and outstanding shares of Common Stock on a fully diluted basis (and assuming conversion of all outstanding convertible securities, including but not limited to conversion of our Series A Preferred Stock into shares of Common Stock in accordance with our Certificate of Incorporation as amended from time to time without any regulatory limitations, all issued and outstanding warrants, notes, RSUs and stock options (whether issued under or outside the 2014 Stock Incentive Plan and the like)) as of the closing of this offering, of which approximately
113
4.9% are recommended to be allocated to our executive officers, comprised of Lisa A. Conte, Steven R. King and Karen S. Wright.
If granted, the exercise price of the stock options will be the closing price of the Common Stock on the date immediately prior to the grant. These stock options are expected to vest monthly in equal proportions over a period of three years beginning on the date of grant, provided that a portion of the options (ranging from one to five months' worth of options, based on the number of years of employment) would vest immediately upon their grant.
Executive Employment Agreements
Lisa A. Conte
In March 2014, we entered into an offer letter with Ms. Conte to serve as our Chief Executive Officer, effective March 1, 2014, in an at-will capacity. Under this offer letter, Ms. Conte's annual base salary is $400,000, she is eligible for an annual target bonus of 30% of her base salary. Effective June 15, 2015, our board of directors has reviewed the terms of Ms. Conte's employment arrangement in connection with its annual compensation review, and has adjusted Ms. Conte's base salary to $440,000. Ms. Conte is entitled to participate in all employee benefit plans, including group health care plans and all fringe benefit plans. Effective May 1, 2018, the Compensation Committee adjusted Ms. Conte's base salary to $500,000.
In April 2014, Ms. Conte was granted a stock option to purchase 160,383 shares of Common Stock at an exercise price of $2.54 per share. The option has a 10-year term and vests as follows: 25% vested on January 1, 2015, 9 months after the grant date, with the remainder vesting equally over the next 27 months such that the option was vested in full on April 1, 2017. On June 2, 2014, Ms. Conte was granted 17,820 RSUs, or RSUs. Fifty percent of the shares of Common Stock underlying the RSUs vested and were issued on January 1, 2016, and the remaining 50% will vest and be issuable on July 1, 2017 pursuant to the terms of the RSU agreement. In the event of a change in control, as defined in the Jaguar Health, Inc. 2013 Equity Incentive Plan (the "2013 Plan"), the vesting of all outstanding awards granted to Ms. Conte under the 2013 Plan will accelerate if Ms. Conte's service with us is terminated without cause within twelve months of the change in control.
Steven R. King, Ph.D.
In February 2014, we entered into an offer letter with Dr. King to serve as our Executive Vice President, Sustainable Supply, Ethnobotanical Research and Intellectual Property, effective March 1, 2014, in an at-will capacity. Under the offer letter, Dr. King's annual base salary of $255,000, he is eligible for an annual target bonus of 30% of his base salary, and he is eligible to participate in the employee benefit plans we offer to our other employees. Effective June 15, 2015, our board of directors has reviewed the terms of Dr. King's employment arrangement in connection with its annual compensation review, and has adjusted Dr. King's base salary to $280,500. Dr. King is entitled to participate in all employee benefit plans, including group health care plans and all fringe benefit plans. Effective May 1, 2018, the Compensation Committee adjusted Dr. King's base salary to $290,317.
In April 2014, Dr. King was granted a stock option to purchase 93,556 shares of Common Stock at an exercise price of $2.54 per share. The option has a 10-year term and vests as follows: 25% vested on January 1, 2015, 9 months after the grant date, with the remainder vesting equally over the next 27 months such that the option was vested in full on April 1, 2017. In June 2014, Dr. King was granted 10,395 RSUs. Fifty percent of the shares of Common Stock underlying the RSUs vested and were issued on January 1, 2016, and the remaining 50% will vest and be issuable on July 1, 2017 pursuant to the terms of the RSU agreement. In the event of a change in control, as defined in the 2013 Plan, the vesting of all outstanding awards granted to Dr. King under the 2013 Plan will accelerate if Dr. King's service with us is terminated without cause within twelve months of the change in control.
114
Karen S. Wright
In October 2015, we entered into an offer letter with Ms. Wright to serve as our Executive Vice President, Finance, effective November 9, 2015, in an at-will capacity. On December 15, 2015 the board of directors approved Ms. Wright's appointment to serve as our Chief Finance Officer. Under the offer letter, Ms. Wright's annual base salary is $240,000, she is eligible for an annual target bonus of 25% of her base salary, and she is eligible to participate in the employee benefit plans we offer to our other employees. Effective May 1, 2018, the Compensation Committee adjusted Ms. Wright's base salary to $301,000.
In November 2015, Ms. Wright was granted a stock option to purchase 20,000 shares of Common Stock at an exercise price of $2.04 per share. The option has a 10-year term and vests as follows: 25% vested on August 9, 2016, 9 months after the hire date, with the remainder vesting equally over the next 27 months such that the option is vested in full on November 9, 2018.
The following table summarizes the total compensation earned in 2017 and 2018 for the Company's non-management directors. Ms. Conte receives no additional compensation for her service as a director. Messrs. Johnson and Siegel did not join the board of directors until March 2018, and Messrs. Divis and MacNaughtan did not join the board of directors until June 2018, and therefore, did not receive any compensation for 2018.
| |
Year | Fees Earned or Paid in Cash ($) |
Option awards ($)(4) |
Total ($) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
James J. Bochnowski |
2018 | — | 160,622 | 114,976 | |||||||||
|
2017 | — | 88,941 | 88,941 | |||||||||
Folkert W. Kamphuis(1) |
2018 | — | 109,931 | 109,931 | |||||||||
|
2017 | — | 88,410 | 88,410 | |||||||||
Jiahao Qui |
2018 | — | 18,065 | 18,065 | |||||||||
|
2017 | — | 16,435 | 16,435 | |||||||||
Zhi Yang(2) |
2018 | — | 18,065 | 18,065 | |||||||||
|
2017 | — | 16,435 | 16,435 | |||||||||
John Micek III |
2018 | — | 108,760 | 108,760 | |||||||||
|
2017 | — | 42,861 | 42,861 | |||||||||
Ari Azhir(3) |
2018 | — | 30,327 | 30,327 | |||||||||
|
2017 | — | 20,380 | 20,380 | |||||||||
Jeffery C. Johnson |
2018 | — | 29,286 | 29,286 | |||||||||
|
2017 | — | — | — | |||||||||
Greg J. Divis |
2018 | — | 15,223 | 15,223 | |||||||||
|
2017 | — | — | — | |||||||||
Jonathan B. Siegel |
2018 | — | 29,286 | 29,286 | |||||||||
|
2017 | — | — | — | |||||||||
Murray David MacNaughtan |
2018 | — | 15,223 | 15,223 | |||||||||
|
2017 | — | — | — | |||||||||
Footnote to Compensation of Directors Table
- (1)
- Mr. Kamphuis's
3-year term on the board of directors ended effective May 18, 2018.
- (2)
- Dr. Yang's
3-year term on the board of directors ended effective May 18, 2018.
- (3)
- Dr. Azhir resigned from the board of directors effective March 29, 2018.
115
- (4)
- Represents the dollar amounts recognized for financial statement reporting purposes with respect to the fiscal year (for stock option awards) determined under FASB ASC Topic 718. The aggregate number of options held by each non-management director officer as of December 31, 2018 was as follows: Mr. Bochnowski holds an aggregate of 4,771 options (37 options granted in fiscal year 2014, 19 options granted in fiscal year 2015, 96 options granted in fiscal year 2016 and 4,619 options granted in fiscal year 2018); Mr. Kamphuis holds an aggregate of 887 options (47 shares granted in fiscal year 2015; 81 shares granted in fiscal year 2016 and 759 shares granted in fiscal year 2018); Mr. Qiu holds an aggregate of 195 options (9 shares granted in fiscal year 2015; 1 shares granted in fiscal year 2016 and 185 shares granted in fiscal year 2015); Dr. Yang holds an aggregate of 195 options (9 shares granted in fiscal year 2015, 1 share in fiscal year 2016, 185 shares in fiscal year 2017; Mr. Micek III holds an aggregate of 2,751 options (102 shares granted fiscal year 2016 and 2,649 shares granted fiscal year 2018); Dr. Azhir holds an aggregate of 654 options (93 shares granted in fiscal year 2016 and 561 shares granted in fiscal year 2017); Mr. Johnson holds an aggregate of 1,498 options (1,498 shares granted fiscal year 2018); Mr. Divis holds an aggregate of 1,498 options (1,498 shares granted fiscal year 2018); Mr. Siegel holds an aggregate of 1,498 options (1,498 shares granted fiscal year 2018); and Mr. MacNaughtan holds an aggregate of 1,498 options (1,498 shares granted fiscal year 2018). The option awards included in the table above do not include the options that the Company intends to grant to our directors, executive officers and certain other employees in connection with this offering, as further described in "Compensation of Directors and Executive Officers—Offering-Related Stock Option Grants to Executive Officers, Directors and Certain Employees."
116
CERTAIN RELATIONSHIPS AND RELATED PERSON TRANSACTIONS
Participation in this Offering
Certain investors, including certain of our officers, directors and existing investors and their affiliated entities, have agreed to purchase an aggregate of approximately $3.55 million of securities in this offering at the public offering price and on the same terms as the other purchasers in this offering. The underwriters will receive the same underwriting discounts and commissions on any securities purchased by these entities as they will on any other securities sold to the public in this offering.
The following includes a summary of transactions since January 1, 2018, to which we have been a party in which the amount involved exceeded or will exceed the lesser of (i) $120,000 and (ii) one percent (1%) of the average of our total assets at year-end for the prior two fiscal years, and in which any of our directors, executive officers or beneficial owners of more than 5% of our capital stock or any member of the immediate family of any of the foregoing persons had or will have a direct or indirect material interest. Compensation arrangements for our directors and executive officers are described under "Compensation of Directors and Executive Officers".
Transactions with Sagard
Sagard Consent and Waiver to Refinancing of Existing Notes
On May 28, 2019, in consideration for the consent and waiver by Sagard to the refinancing of approximately $10.5 million outstanding aggregate amount of convertible promissory notes issued by Napo pursuant to the Amended and Restated Note Purchase Agreement, dated March 31, 2017, by and between Napo, Kingdon Associates, M. Kingdon Offshore Master Fund L.P., Kingdon Family Partnership, L.P., and Kingdon Credit Master Fund L.P. (collectively, the "Kingdon Purchasers"), the Company agreed to pay Sagard a consent fee in an amount equal to (i) $250,000, if paid in cash, or (ii) $400,000, if paid in shares of Common Stock, where the number of shares issuable will be equal to $400,000 divided by the closing price of the Common Stock on the trading day immediately preceding payment. The consent fee is payable five days after the closing of an underwritten public offering of Common Stock pursuant to an effective registration statement on Form S-1, provided that if such offering does not occur before July 31, 2019, the fee will be paid no later than July 31, 2019. The Company also agreed to use commercially reasonable efforts to ensure that the amount owed pursuant to the Exchange Notes does not exceed $10,535,900 in the aggregate.
Sagard Participation in Notes Financing
On May 30, 2019, the Company entered into a securities purchase agreement with Sagard, pursuant to which the Company issued a promissory note in the principal amount of $500,000 (the "Sagard Note") and a 5-year warrant (the "Sagard Warrant") to purchase $375,000 in shares of Common Stock (the "Sagard Warrant Shares"). The Sagard Note bears interest at a rate of 12% per annum and matures on July 31, 2019 (the "Maturity Date"). In connection with this transaction, the Company also entered into a registration rights agreement with Sagard, pursuant to which the Company agreed to register the Sagard Warrant Shares within 60 calendar days following the Maturity Date and to use reasonable best efforts to cause such registration statement to be declared effective within 180 days following the Maturity Date. The exercise price for the Sagard Warrant is the price per share at which the Company issues Common Stock in the Public Offering, provided that if the Company has not consummated a Public Offering by the Maturity Date, then the Sagard Warrant exercise price will be equal to the closing sales price of the Common Stock on the Maturity Date. The Company will use the proceeds for working capital and other general corporate purposes.
117
Preferred Stock Offering
Preferred Stock Purchase Agreement
On March 23, 2018, we entered into the Preferred Stock Purchase Agreement with Sagard, pursuant to which we, in a private placement, agreed to issue and sell to Sagard 5,524,926 shares of Preferred Stock (the "Preferred Shares"), for an aggregate purchase price of $9,199,001. The Preferred Stock Purchase Agreement also provides for customary representations, warranties and covenants among the parties. Among other things, the Preferred Stock Purchase Agreement requires that we (i) file prior to the initial closing the Series A Certificate of Designation and (ii) enter into a registration rights agreement with Sagard providing for the registration of shares of our Common Stock, issuable upon conversion of the Preferred Shares (the "Conversion Shares"). In addition, so long as Sagard or its affiliates own, in the aggregate, no less than 50% or more of the cumulative amount of the Preferred Shares and Conversion Shares issued in the Preferred Stock Offering, Sagard and its affiliates have the right to purchase (x) 100% of the first $10 million of any new securities issued by us and thereafter (y) a pro rata portion of any new securities that we may issue from time to time, subject to certain exceptions specified in the Preferred Stock Purchase Agreement. The Preferred Shares are subject to a 12-month lock-up period, which period may be shortened in limited circumstances specified in the Preferred Stock Purchase Agreement.
The Preferred Stock Purchase Agreement also provides that Sagard has the right to designate at least one non-voting observer (subject to increase to two if at any time two designees of the Preferred Shares and the Conversion Shares are not represented on the board of directors) to attend meetings of the Board, the board of directors of any of our subsidiaries and each committee of any of the foregoing (a "Board Observer"). In addition, at such time as no shares of Preferred Stock are outstanding, and so long as Sagard holds (i) at least 35% of the total number of the Conversion Shares that have been issued upon conversion of all shares of Preferred Stock issued in the Preferred Stock Offering, Sagard shall be entitled thereunder to nominate two directors of the Company (each, a "Series A Director") and (ii) less than 35% but at least 20% of the total number of the Conversion Shares that have been issued upon conversion of all shares of Preferred Stock issued in the Preferred Stock Offering, Sagard shall be entitled thereunder to nominate one director of the Company.
Notwithstanding the foregoing, the number of Series A Directors shall be reduced to the extent necessary to comply with our obligations, if any, under the rules or regulations of the Nasdaq Stock Market (including Nasdaq Listing Rule 5640). The Preferred Stock Purchase Agreement provides that, if one Series A Director may not be appointed due to compliance with Nasdaq Listing Rule 5640, then Sagard shall be entitled to designate one additional Board Observer to attend meetings of the board of directors, the board of directors of any of our subsidiaries and each committee of any of the foregoing as an observer.
Terms of Series A Preferred Stock
The Series A Certificate of Designation authorizes 5,524,926 shares of Preferred Stock and provides for the rights, preferences and privileges of such Preferred Stock. Any reference to share prices in the below description of the Preferred Stock, including but not limited to the conversion price for the Preferred Shares and the amount of the liquidation preference per share, is subject to adjustment in the event of any stock dividend, stock split, reverse stock split, combination or other similar recapitalization, as further described in the Series A Certificate of Designation.
Dividends
Holders of shares of Preferred Stock are entitled to participate equally and ratably with the holders of shares of Common Stock in all dividends paid and distributions made to the holders of Common Stock on the shares of Common Stock on an as converted basis.
118
Election of Directors and Voting Rights
The holders of a majority of the outstanding shares of Preferred Stock are entitled to elect two (2) members of the Company's Board of Directors. Notwithstanding the foregoing, the number of Series A Directors shall be reduced to the extent necessary to comply with the Company's obligations, if any, under the rules or regulations of the Nasdaq Stock Market (including Nasdaq Listing Rule 5640).
The holders of shares of Preferred Stock have the right to vote with holders of shares of the Common Stock, voting together as one class on all other matters, with each share of Preferred Stock entitling the holder thereof to cast that number of votes per share as is equal to the aggregate number of shares of Common Stock into which it is then convertible, using the market value of the Common Stock on the date of the Preferred Stock Agreement (i.e., $0.1935) as the conversion price; provided that, at any time prior to the time the Company obtains stockholder approval, as required pursuant to Nasdaq Rule 5635(b), no holder (together with such holder's attribution parties) is permitted to have a number of votes in excess of such aggregate number of votes granted to the holders of 19.99% of the shares of Common Stock then outstanding (including any votes with respect to any shares of Common Stock and Preferred Stock beneficially owned by the holder or such holder's attribution parties).
Voluntary Conversion
Each share of Preferred Stock is convertible into 0.08571429 shares of Common Stock at an effective conversion price of $19.425 per share (based on an original price per Preferred Share of $1.665), provided that, at any time prior to the time the Company obtains stockholder approval, as required pursuant to Nasdaq Rule 5635(b) any conversion of Preferred Stock by a holder into shares of the Common Stock would be prohibited if, as a result of such conversion, the holder, together with such holder's attribution parties, would beneficially own more than 19.99% of the total number of shares of the Common Stock issued and outstanding after giving effect to such conversion. Subject to certain limited exceptions, the shares of Preferred Stock cannot be offered, pledged or sold by Sagard for one year from the date of issuance. The conversion price is subject to certain adjustments in the event of any stock dividend, stock split, reverse stock split, combination or other similar recapitalization.
Mandatory Conversion
The shares of Preferred Stock will be mandatorily converted upon the date and time, or the occurrence of an event, specified by vote or written consent of the holders of a majority of the then outstanding shares of Preferred Stock at a conversion price of $19.425 per share. In each case, the number of shares of Common Stock issuable upon such conversion will be limited to the extent necessary to satisfy limitations on beneficial ownership as described under "Voluntary Conversion" above.
Optional Redemption
At any time after the first anniversary of the issuance of the Preferred Shares, so long as certain call conditions specified in the Series A Certificate of Designation have been satisfied, the Company shall have the right to offer to redeem shares of Preferred Stock at a share price equal to two times the original share issue price of the Preferred Shares. The Company is only permitted to exercise this right to redeem two times, the first of which must be for an aggregate redemption price of $9,199,001 and the second of which must be for all remaining shares of Preferred Stock remaining. If a holder of Preferred Shares fails to accept the Company's redemption offer, such holder's shares of Preferred Stock shall be automatically converted into shares of Common Stock pursuant to the terms of "Mandatory Conversion" as described above.
119
Mandatory Redemption
If (i) the Company's consolidated net revenues attributable to the Mytesi products ("Mytesi Revenues") for the six-month period ended March 31, 2021 are less than $22 million, (ii) the average volume-weighted average price of the Common Stock for the thirty days immediately prior to the Measurement Date (as defined below) is less than $105.00 or (iii) the Company fails to file with the SEC on or before June 30, 2021 its quarterly report on Form 10-Q for the three months ended March 31, 2021, then the holders of at least a majority of shares of Preferred Stock then outstanding may require the Company to redeem all shares of Preferred Stock then outstanding at a per share purchase price equal to $2.3057. For purposes of the foregoing sentence, "Measurement Date" means the later of (x) April 30, 2021 and (y) the date on which the Company files its quarterly report on Form 10-Q for the three months ended March 31, 2021 (but in no event later than June 30, 2021).
The mandatory redemption right described above shall terminate if, prior to the Measurement Date, both (i) the Mytesi Revenues for any six-month period ending at the end of a calendar quarter are equal to or exceed $22 million and (ii) the average volume-weighted average price of the Common Stock for the thirty days immediately preceding the end of such calendar quarter is equal to or greater than $105.00.
Fundamental Change
The Series A Certificate of Designation provides the holders of Preferred Stock with a right to require the Company to repurchase shares of Preferred Stock at a price to be calculated pursuant to the terms of the Series A Certificate of Designation upon the occurrence of any of the following events (each a "Fundamental Change"):
(X) (a) any person or "group" (within the meaning of Rules 13d-3 and 13d-5 under the Exchange Act), other than certain "Permitted Holders" (and/or any direct transferee of shares of Preferred Stock from any Permitted Holder) (as defined in the Series A Certificate of Designation), (i) shall have acquired beneficial ownership of more than fifty percent (50%) or more on a fully diluted basis of the voting and/or economic interest in the capital stock of the Company (or surviving entity in a merger or consolidation, if applicable)) or (ii) shall have obtained the power (whether or not exercised) to elect a majority of the members of the Company's Board of Directors (or similar governing body); or (b) the occurrence of any "change of control" or similar event under any agreements relating to any indebtedness of the Company or its subsidiaries; or
(Y) except in the case of a Deemed Liquidation Event (as defined in the Series A Certificate of Designation and described below) in which holders of Preferred Stock receive, concurrently with the consummation of such Deemed Liquidation Event, a cash payment pursuant to Sections 2.1 and 2.4 of the Series A Certificate of Designation in full, in an amount equal to the "Fundamental Change Price" as defined in the Series A Certificate of Designation that would otherwise be payable, the Company or any of its subsidiaries enters into any transaction of merger or consolidation (except that a person may be merged with or into the Company or another wholly-owned subsidiary thereof so long as the Company or another wholly-owned subsidiary is the continuing or surviving person), or conveys, sells, leases, subleases (as lessor or sublessor), exchanges, transfers or otherwise disposes of, in one transaction or a series of transactions, all or substantially all of the consolidated business, assets or property of the Company and its subsidiaries.
The "Fundamental Change Price" for each share of Preferred Stock, as of any date, shall be calculated as the sum of (i) the amount payable in respect of such share under Section 2.1 of the Series A Certificate of Designation in the event of a "Liquidation Event" as of such date, plus (ii) any and all accrued and unpaid dividends upon the Preferred Stock, whether or not declared, as of the date
120
of the Fundamental Change, plus (iii) the "Participation Amount" as defined in the Series A Certificate of Designation.
Merger or Liquidation
Subject to the Fundamental Change provision described above, under the terms of the Series A Certificate of Designation, upon merger or consolidation resulting in a change of control, sale, lease, transfer, exclusive license or other disposition, in a single transaction or series of related transactions, by the Company or any subsidiary of the Company, of substantially all of the assets of the Company and its subsidiaries taken as a whole, or the sale or disposition (whether by merger, consolidation or otherwise) of Napo (as defined below) (or any successor in interest) or one or more other subsidiaries of the Company if substantially all of the assets of the Company and its subsidiaries taken as a whole are held by such subsidiary or subsidiaries (collectively, a "Deemed Liquidation"), liquidation, dissolution or winding up of the Company as determined under the Certificate (collectively, a "Liquidation Event"), each share of Preferred Stock will be entitled to a preference of $1.665 per share (or the equivalent of $19.425 per share on an as-converted to Common Stock basis) plus a participation right described below. Thereafter, the holders of Common Stock then outstanding shall be entitled to receive an amount per share of Common Stock (in stock or cash as determined under the Series A Certificate of Designation) equal to $19.425 (as adjusted for stock splits, reverse splits, stock dividends, reclassifications, recapitalizations and/or other similar events). Thereafter, all of the remaining assets of the Company and/or proceeds from a Deemed Liquidation or Liquidation Event, as applicable, will in general be divided pro rata among the holders of the shares of Preferred Stock and the shares of Common Stock, on an as converted basis (all as more fully specified and calculated under the Series A Certificate of Designation).
Covenants
Pursuant to the terms of the Preferred Stock as provided in the Series A Certificate of Designation, so long as any shares of Preferred Stock are outstanding, the Company may not agree, among other things, without the prior written consent or vote of the holders of at least a majority of the then outstanding shares of Preferred Stock, to: (a) amend, alter, repeal or waive any provision of the Series A Certificate of Designation, (b) amend, alter or repeal any provision of the Company's charter documents in a manner that would adversely affect the powers, privileges, preferences or rights of the Preferred Stock, (c) create additional classes or series of capital stock having rights, preferences or privileges senior to or pari passu with the Preferred Stock or (d) increase or decrease the authorized number of shares of Preferred Stock.
So long as Sagard or its affiliates own at least 35% of the shares of Preferred Stock that were originally issued, the Series A Certificate of Designation provides that the Company may not, among other things, without the prior written consent or vote of the holders of at least a majority of the then outstanding shares of Preferred Stock, (a) authorize or issue any capital stock of any subsidiary which is not wholly-owned by the Company, (b) declare or pay dividends on the Company's equity securities or redeem any of the Company's equity securities, (c) incur, guarantee or assume any indebtedness, subject to certain limited exceptions, (d) grant or incur any lien, subject to certain limited exceptions, (e) enter into any transaction for the acquisition of all or substantially of the equity interests or assets of another person, subject to certain limited exceptions (f) make any investments, subject to certain limited exceptions or (g) enter into any transactions with the Company's affiliates, subject to certain limited exceptions.
Services Agreement
On March 23, 2018, the Company entered into a management services agreement with Sagard Capital Partners Management Corp. ("SCPM"), an affiliate of Sagard, pursuant to which SCPM will
121
provide certain consulting and management advisory services to the Company over a three-year period (the "Initial Term") for an annual fee of $450,000, which fees will be paid in equal installments over the Initial Term beginning in the second year of the Initial Term (the "Services Agreement"). The Services Agreement may be terminated earlier than the initial three-year term (i) upon mutual consent of the parties, (ii) by either party following a breach of the Services Agreement by the other party that remains uncured following 30 days' written notice thereof, (iii) in SCPM's sole discretion with 10 day's prior written notice, or (iv) upon the consummation of a Deemed Liquidation (so long as all accrued and unpaid fees payable thereunder as of such termination have been paid in full) or a Fundamental Change in which all of the Company's shares of Preferred Stock are repurchased by the Company.
As described above, Jeffery C. Johnson, a member of the Company's board of directors, is an investment manager at SCPM, and David MacNaughtan, a member of the Company's board of directors, is a partner at Sagard Holdings, an affiliate of SCPM.
Transactions with CVP
On February 26, 2018, the Company entered into a securities purchase agreement with Chicago Venture Partners L.P. ("CVP"), pursuant to which the Company issued to CVP a promissory note in the aggregate principal amount of $2,240,909 for an aggregate purchase price of $1,560,000 (the "Feb 2018 Note"). The Feb 2018 Note carries an original issue discount of $655,909, and the initial principal balance also includes $25,000 to cover CVP's transaction expenses. The Feb 2018 Note bears interest at the rate of 8% per annum and matures on August 26, 2019.
On March 21, 2018, the Company entered into a securities purchase agreement with CVP, pursuant to which the Company issued to CVP a promissory note in the aggregate principal amount of $1,090,341 for an aggregate purchase price of $750,000 (the "March 2018 Note"). The March 2018 Note carries an original issue discount of $315,341, and the initial principal balance also includes $25,000 to cover CVP's transaction expenses. The March 2018 Note bears interest at the rate of 8% per annum and matures on September 21, 2019.
Effective August 13, 2018, the Company entered into an acknowledgement agreement with CVP extending the maturity date of the $2,155,000 secured convertible promissory note dated July 29, 2017 (the "June 2017 Note") from August 2, 2018 to August 26, 2019 and also extending the maturity date of the $1,587,500 secured promissory note dated December 8, 2017 (the "Dec 2017 Note" and, together with the June 2017 Note, the Feb 2018 Note and the March 2018 Note, the "CVP Notes") from September 8, 2018 to August 26, 2019.
On May 28, 2019, Jaguar entered into a guaranty and suretyship agreement, pursuant to which it guaranteed payment and performance of all obligations of Napo, arising under and in connection with approximately $10.5 million outstanding aggregate amount of convertible promissory notes issued by Napo pursuant to the Amended and Restated Note Purchase Agreement, dated March 31, 2017, by and between Napo, Kingdon Associates, M. Kingdon Offshore Master Fund L.P., Kingdon Family Partnership, L.P., and Kingdon Credit Mater Fund L.P. (collectively, the "Existing Notes"). The Existing Notes bear interest at a rate of 10% per annum and mature on December 31, 2019.
On May 28, 2019, Jaguar and Napo (collectively, the "Borrower") entered into an exchange agreement with Chicago Venture Partners, L.P. ("CVP"), the holder of the Existing Notes, pursuant to which CVP exchanged the Existing Notes for a secured promissory note in the original principal amount of $10,535,900.42 the ("Exchange Note 1") and a secured promissory note in the original principal amount of $2,296,926.16 ("Exchange Note 2" and together with Exchange Note 1, the "Exchange Notes"). The Exchange Notes bear interest at the rate of 10% per annum and mature on December 31, 2020. The outstanding balance of Exchange Note 2 is equal to the exchange fee that Jaguar agreed to pay CVP in consideration of certain accommodations granted to Jaguar and Napo, including but not limited to the extension of the maturity dates of the Existing Notes and the legal and
122
other fees incurred by CVP in connection with the effectuation of the transactions contemplated under the Exchange Agreement.
CVP also entered into security agreements with Jaguar (the "Jaguar Security Agreement") and Napo (the "Napo Security Agreement", and together with the Jaguar Security Agreement, the "Security Agreements"), pursuant to which CVP will receive (i) a security interest in substantially all of the Company's assets as security for the Company's obligations under Exchange Note 2 and (ii) a security interest in substantially all of Napo's assets as security for Napo's obligations under Exchange Note 1 and Exchange Note 2. Notwithstanding the foregoing, (a) the amount owing under Exchange Note 2 will not be considered part of the obligations secured by the Napo Security Agreement until such time as Jaguar receives permission from a third party and (b) the security interest granted under the Jaguar Security Agreement will be automatically terminated and released upon Jaguar's receipt of a waiver from such third party.
In January through June 2019, the Company entered into exchange agreements with CVP, pursuant to which the Company issued 1,150,793 of Common Stock in the aggregate to CVP in exchange for a reduction of approximately $11.2 million in the principal amount of the CVP Notes and Exchange Notes. The shares of Common Stock that were exchanged for portions of the secured promissory notes were issued in reliance on the exemption from registration provided under Section 3(a)(9) of the Securities Act.
Transactions with Oasis Capital
On January 7, 2019, the Company entered into a common stock purchase agreement (the "January CSPA") with Oasis Capital, relating to an offering (the "Original Equity Line Offering") of an aggregate of up to 80,476 shares (the "Original Shares") of Common Stock, of which 76,190 of such Original Shares are being offered in a primary offering consisting of an equity line of credit. Under the terms of the January CSPA, the Company has the right to "put," or sell, up to 76,190 shares of Common Stock (the "January Purchase Shares") to Oasis Capital for an amount equal to the product of (i) the number of January Purchase Shares set forth on the applicable put notice (minus the deposit and clearing fees associated with such purchase) and (ii) a fixed price of $52.50 per share or such other price agreed upon between the Company and Oasis Capital. The Company had the option to increase the equity line of credit by an additional 114,285 shares of Common Stock by notifying Oasis Capital at any time after the effective date of the January CSPA (the "January Upsize Option"). On March 18, 2019, the Company delivered a notice to Oasis Capital of its decision to exercise the January Upsize Option. The Company has sold the Original Shares and all 114,285 shares of Common Stock under the January Upsize Option to Oasis Capital.
On March 24, 2019, the Company entered into a securities purchase agreement (the "Purchase Agreement") with Oasis Capital, pursuant to which the Company agreed to issue and sell, in a registered public offering by the Company directly to Oasis Capital (the "RDO"), an aggregate of 19,019 shares of Common Stock (the "RDO Shares") at an offering price of $14.00 per share for gross proceeds of approximately $266,266 before deducting the placement agent fee and related offering expenses.
On April 1, 2019, the Company entered into another common stock purchase agreement (the "April CSPA") with Oasis Capital relating to an offering (the "April Equity Line Offering") of an aggregate of up to 285,714 shares (the "April Purchase Shares") of the Company's common stock, all of which are being offered in a primary offering consisting of an equity line of credit. Under the terms of the April CSPA, the Company has the right to "put," or sell, the April Purchase Shares to Oasis Capital for an amount equal to the product of (i) the number of April Purchase Shares set forth in the applicable put notice (minus the deposit and clearing fees associated with such purchase) and (ii) a fixed price of $19.60 per share or such other price agreed upon between the Company and Oasis
123
Capital. The Company had the option to increase the equity line of credit by an additional 285,714 shares of Common Stock by notifying Oasis Capital at any time after the effective date of the April CSPA.
Effective June 14, 2019, we halted all future offers and sales of our voting common stock, par value $0.0001 per share under the April CSPA and terminated the April CSPA. Between April 1, 2019, the date of the April CSPA, and June 14, 2019, we sold an aggregate of 4,824 shares of Common Stock pursuant to the CSPA for aggregate gross proceeds of approximately $100,000.
Transactions with Lisa A. Conte
Lisa A. Conte has served as our President, Chief Executive Officer and a member of our board of directors since she founded the company in June 2013. On July 18, 2018, Ms. Conte purchased 21 shares of common stock for an aggregate purchase price of $1,668.60. Ms. Conte, Mr. Bochnowski, and Mr. Siegel purchased promissory notes in the aggregate of $525,000 pursuant to the Securities Purchase Agreements entered into between the Company and certain investors beginning on March 18, 2019.
Transactions with Jonathan B. Siegel
On March 29, 2018, our board of directors appointed Mr. Jonathan B. Siegel to fill the vacancy created by Dr. Azhir's resignation and serve as Class I director of the Company until the 2019 annual meeting of stockholders or until his successor is elected and qualified.
Mr. Siegel was formerly a principal and member of the executive committee of Kingdon Capital ("Kingdon") and the head for Kingdon's healthcare sector. As described further above, on March 31, 2017, Napo entered into the Kingdon NPA with the Kingdon Purchasers, which are affiliates of Kingdon, pursuant to which Napo issued approximately $10 million in aggregate principal amount of secured promissory notes. Napo's obligations under the Kingdon Notes were secured by a security interest in substantially all of Napo's assets, including Napo intellectual property. CVP acquired these notes from Kingdon on May 28, 2019.
On July 16, 17, and 18, 2018, JBS Healthcare Ventures LLC purchased an aggregate of 214 shares of common stock in the open market for an aggregate purchase price of $18,653. Mr. Siegel is the sole member of JBS Healthcare Ventures LLC. Ms. Conte, Mr. Bochnowski, and Mr. Siegel purchased Bridge Notes in the aggregate of $525,000 pursuant to the Securities Purchase Agreements entered into between the Company and certain investors beginning on March 18, 2019.
Transactions with James J. Bochnowski
Mr. Bochnowski has served as a member of our board of directors since February 2014 and as Chairman of our board since June 2014. Ms. Conte, Mr. Bochnowski, and Mr. Siegel purchased promissory notes in the aggregate of $525,000 pursuant to the Securities Purchase Agreements entered into between the Company and certain investors beginning on March 18, 2019.
Transactions with Charles C. Conte
Charles C. Conte is the brother of Lisa A. Conte, who Conte has served as our President, Chief Executive Officer and a member of our board of directors since she founded the company in June 2013.
On September 11, 2018, Charles C. Conte purchased a convertible promissory note in the aggregate principal amount of $111,250 convertible at a price of $59.50 per share of common stock and a warrant exercisable for 484 shares of common stock with an exercise price of $86.10 per share.
124
Indemnification Agreements
We have entered into indemnification agreements with each of our directors and officers. These agreements, among other things, require us or will require us to indemnify each director to the fullest extent permitted by Delaware law, including indemnification of expenses such as expenses, judgments, penalties, fines and amounts paid in settlement to the extent legally permitted incurred by the director or officer in any action or proceeding, including any action or proceeding by or in right of us, arising out of the person's services as a director or officer.
125
The following table sets forth certain information with respect to the beneficial ownership of our common stock as of June 7, 2019 by:
- •
- each person, or group of affiliated persons, who is known by us to be the beneficial owner of more than 5% of our outstanding common stock;
- •
- each of our directors;
- •
- each of our executive officers; and
- •
- all of our executive officers and directors as a group.
Beneficial ownership is determined in accordance with the rules and regulations of the SEC and includes voting or investment power with respect to our common stock. Shares of our common stock subject to options, warrants or RSUs that are currently exercisable or vested, or exercisable or subject to vesting within 60 days of the date of this prospectus are considered outstanding and beneficially owned by the person holding the options, warrants, or RSUs for the purpose of calculating the percentage ownership of that person but not for the purpose of calculating the percentage ownership of any other person. Except as otherwise noted, the persons and entities in this table have sole voting and investing power with respect to all of the shares of our common stock beneficially owned by them, subject to community property laws, where applicable. The information is not necessarily indicative of beneficial ownership for any other purpose, including for purposes of Section 13(d) and Section 13(g) of the Securities Act.
The column titled "Percentage of Shares Beneficially Owned—Before Offering" is based on a total of 1,006,743 shares of common stock outstanding as of June 7, 2019. The total shares of common stock outstanding may be adjusted for the purpose of calculating the percentage ownership of a person that has options, warrants or RSUs that are currently exercisable or vested, or exercisable or subject to vesting within 60 days of the date of this prospectus but not for the purpose of recalculating the percentage ownership of any other person. The column titled "Percentage of Shares Beneficially Owned—After Offering" is based on 9,400,787 shares of common stock to be outstanding after this offering, and includes the 2,521,008 Class A Units or 12,000 Class B Units, or some combination thereof, assuming conversion of all Series B Preferred Shares included in the Class B Units, which we are selling in this offering.
126
Except as otherwise set forth below, the address of each beneficial owner listed in the table below is c/o Jaguar Health, Inc., 201 Mission Street, Suite 2375, San Francisco, California 94105.
| |
|
Percentage of Shares Beneficially Owned |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Name and Address of Beneficial Owner
|
Number of Shares Beneficially Owned |
Before Offering(16) |
After Offering |
|||||||
5% Stockholders |
||||||||||
Sagard Capital Partners, L.P.(1) |
473,565 | 32.55 | % | — | * | |||||
Oasis Capital, LLC(2) |
214,338 | 20.09 | % | — | * | |||||
Jonathan Glaser(3) |
88,605 | 7.90 | % | — | * | |||||
Executive Officers and Directors |
||||||||||
Lisa A. Conte(4) |
13,923 | 1.30 | % | — | * | |||||
Steven R. King, Ph.D.(5) |
1,995 | — | * | — | * | |||||
Karen S. Wright(6) |
1,294 | — | * | — | * | |||||
James J. Bochnowski(7) |
51,331 | 4.60 | % | — | * | |||||
Jeffery C. Johnson(8) |
631 | — | * | — | * | |||||
John Micek III(9) |
1,277 | — | * | — | * | |||||
Jiahao Qiu(10) |
121 | — | * | — | * | |||||
Jonathan B. Siegel(11) |
8,757 | — | * | — | * | |||||
Greg Divis(12) |
506 | — | * | — | * | |||||
Murray David MacNaughtan(13) |
506 | — | * | — | * | |||||
All executive officers and directors as a group (10 persons)(14) |
80,431 | 6.82 | % | — | * | |||||
- *
- Less
than 1%.
- (1)
- As
reported on Schedule 13D/A filed on June 3, 2019. The address for the reporting person is 280 Park Avenue, 3rd Floor
West, New York, NY 10017.
- (2)
- As
reported on Form 13G/A filed on March 29, 2019, in addition to subsequent transactions. The address for the reporting person is 208 Ponce de
Leon Ave., Ste. 1600, San Juan, Puerto Rico, 00918.
- (3)
- As
reported on Form 13G/A filed on February 14, 2019, in addition to subsequent transactions. The address for the reporting person is 11601 Wilshire
Boulevard, Suite 2180, Los Angeles, CA 90025.
- (4)
- Represents
(i) 32 shares of Common Stock, (ii) 5,649 shares of Common Stock issuable to Ms. Conte under stock options that are exercisable or
will become exercisable in the 60 days subsequent to June 7, 2019, and (iii) 8,242 shares of Common Stock issuable to Ms. Conte under warrants that are exercisable in the
60 days subsequent to June 7, 2019. The weighted average exercise price of the 5,649 stock options is $477.28.
- (5)
- Represents
(i) 6 shares of Common Stock and (ii) 1,988 shares of Common Stock issuable to Dr. King under stock options that are exercisable or
will become exercisable in the 60 days subsequent to June 7, 2019. The weighted average exercise price of the 1,988 stock options is $470.33.
- (6)
- Represents
1,294 shares of Common Stock issuable to Ms. Wright under stock options that are exercisable or will become exercisable in the 60 days
subsequent to June 7, 2019. The weighted average exercise price of the 1,294 stock options is $415.80.
- (7)
- Includes (i) 1,036 shares of Common Stock and (ii) 2,218 shares of Common Stock issuable to Mr. Bochnowski under stock options that are exercisable or will become exercisable in the 60 days subsequent to June 7, 2019, and (iii) 48,077 shares of Common Stock issuable to Mr. Bochnowski
127
under warrants that are exercisable in the 60 days subsequent to June 7, 2019. All securities other than stock options are held by the Bochnowski Family Trust. Mr. Bochnowski is a co-trustee and beneficiary of such trust and shares voting and investment control over such shares with his spouse. The weighted average exercise price of the 2,218 stock options is $345.73.
- (8)
- Represents
631 shares of Common Stock issuable to Mr. Johnson under stock options that are exercisable or will become exercisable in the 60 days
subsequent to June 7, 2019. Mr. Johnson is one of Sagard Capital Partners, L.P.'s ("Sagard") two director designees in accordance with the terms of the Series A Certificate
of Designation of the Preferred Stock and is part of the Sagard executive management team. The weighted average exercise price of the 631 stock options is $166.94.
- (9)
- Represents
1,277 shares of Common Stock issuable to Mr. Micek under stock options that are exercisable or will become exercisable in the 60 days
subsequent to June 7, 2019. The weighted average exercise price of the 1,277 stock options is $303.69.
- (10)
- Represents
121 shares of Common Stock issuable to Mr. Qui under stock options that are exercisable or will become exercisable in the 60 days
subsequent to June 7, 2019. The weighted average exercise price of the 121 stock options is $598.28.
- (11)
- Represents
(i) 571 shares of Common Stock, (ii) 631 shares of Common Stock issuable to Mr. Siegel under stock options that are exercisable or
will become exercisable in the 60 days subsequent to June 7, 2019, and (iii) 7,555 shares of Common Stock issuable to Mr. Siegel under warrants that are exercisable in the
60 days subsequent to June 7, 2019. The weighted average exercise price of the 631 stock options is $166.94.
- (12)
- Represents
506 shares of Common Stock issuable to Mr. Divis under stock options that are exercisable or will become exercisable in the 60 days
subsequent to June 7, 2019. The weighted average exercise price of the 506 stock options is $95.20.
- (13)
- Represents
506 shares of Common Stock issuable to Mr. MacNaughtan under stock options that are exercisable or will become exercisable in the 60 days
subsequent to June 7, 2019. Mr. MacNaughtan is one of Sagard's two director designees in accordance with the terms of the Series A Certificate of Designation of the Preferred
Stock. The weighted average exercise price of the 506 stock options is $95.20.
- (14)
- See notes (4) - (13)
128
MATERIAL U.S. FEDERAL INCOME TAX CONSIDERATIONS
The following is a general discussion of material U.S. federal income considerations relating to the purchase, ownership and disposition of our common stock, Series B Preferred Stock or warrants. This discussion is based on current provisions of the Internal Revenue Code of 1986, as amended, (the "Internal Revenue Code"), existing and proposed U.S. Treasury Regulations promulgated or proposed thereunder and current administrative and judicial interpretations thereof, all as in effect as of the date of this prospectus and all of which are subject to change or to differing interpretation, possibly with retroactive effect. We have not sought and will not seek any rulings from the Internal Revenue Service (the "IRS") regarding the matters discussed below. There can be no assurance that the IRS or a court will not take a contrary position.
This discussion is limited to U.S. holders and non-U.S. holders who hold our common stock, Series B Preferred Stock or warrants as capital assets within the meaning of Section 1221 of the Internal Revenue Code (generally, as property held for investment). This discussion does not address all aspects of U.S. federal income taxation, such as the U.S. alternative minimum income tax and the additional tax on net investment income, nor does it address any aspect of state, local or non-U.S. taxes, or U.S. federal taxes other than income taxes, such as federal estate taxes. This discussion does not consider any specific facts or circumstances that may apply to a holder and does not address the special tax considerations that may be applicable to particular holders, such as:
- •
- insurance companies;
- •
- tax-exempt organizations;
- •
- financial institutions;
- •
- brokers or dealers in securities;
- •
- regulated investment companies;
- •
- pension plans;
- •
- controlled foreign corporations;
- •
- passive foreign investment companies;
- •
- corporations that accumulate earnings to avoid U.S. federal income tax;
- •
- certain U.S. expatriates;
- •
- U.S. persons that have a "functional currency" other than the U.S. dollar;
- •
- persons that acquire our common stock as compensation for services;
- •
- owners that hold our common stock, Series B Preferred Stock or warrants as part of a straddle, hedge, conversion transaction, synthetic
security or other integrated investment;
- •
- entities that are treated as disregarded entities for U.S. federal income tax purposes (regardless of their places of organization or
formation) and their investors; and
- •
- partnerships or other entities treated as partnerships for U.S. federal income tax purposes and their investors.
If any entity taxable as a partnership for U.S. federal income tax purposes holds our common stock, Series B Preferred Stock or warrants, the U.S. federal income tax treatment of a partner in the partnership generally will depend on the status of the partner, the activities of the partnership and certain determinations made at the partner level. An investor in a partnership or entity treated as disregarded for U.S. federal income tax purposes should consult his, her or its own tax advisor
129
regarding the applicable tax consequences relating to the purchase, ownership and disposition of our common stock, Series B Preferred Stock or warrants.
For purposes of this discussion, the term "U.S. holder" means a beneficial owner of our common stock, Series B Preferred Stock or warrants that is, for U.S. federal income tax purposes:
- •
- an individual who is a citizen or resident of the United States;
- •
- a corporation created or organized in or under the laws of the United States or of any political subdivision of the United States;
- •
- an estate the income of which is subject to U.S. federal income taxation regardless of its source; or
- •
- a trust, if (i) a U.S. court is able to exercise primary supervision over the administration of the trust and one or more U.S. persons have authority to control all substantial decisions of the trust or (ii) the trust has a valid election to be treated as a U.S. person under applicable U.S. Treasury Regulations.
A "non-U.S. holder" is a beneficial owner of our common stock, Series B Preferred Stock or warrants that is, for U.S. federal income tax purposes, an individual, corporation, estate or trust that is not a U.S. holder.
Prospective investors should consult their own tax advisors regarding the U.S. federal, state, local and non-U.S. income and other tax considerations of purchasing, holding and disposing of our common stock, Series B Preferred Stock or warrants.
U.S. Holders
Purchase of Units
For U.S. federal income tax purposes, the purchase of a Class A Unit will be treated as the purchase of three components: a component consisting of one share of our common stock, a component consisting of one Series 1 warrant to purchase one share of our common stock and a component consisting of one Series 2 warrant to purchase one share of our common stock. The purchase of a Class B Unit will be treated as the purchase of three components: a component consisting of one share of our Series B Preferred Stock, a component consisting of one Series 1 warrant to purchase one share of our common stock and a component consisting of one Series 2 warrant to purchase one share of our common stock. The purchase price for each Unit will be allocated between its components in proportion to the relative fair market value of each at the time the Unit is purchased by the holder. This allocation of the purchase price for each Unit will establish a holder's initial tax basis for U.S. federal income tax purposes in the shares and warrants that compose each Unit.
Exercise of Warrants
A U.S. holder generally will not recognize gain or loss on the exercise of a warrant and related receipt of shares of our common stock (unless cash is received in lieu of the issuance of a fractional share of our common stock). A U.S. holder's initial tax basis in the shares of our common stock received upon the exercise of a warrant will be equal to the sum of (a) such U.S. holder's tax basis in such warrant plus (b) the exercise price paid by such U.S. holder on the exercise of such warrant. A U.S. holder's holding period for the shares of our common stock received upon the exercise of a warrant will begin on the day after the date that the warrant is exercised.
In certain limited circumstances, a U.S. holder may be permitted to undertake a cashless exercise of warrants into shares of our common stock. The U.S. federal income tax treatment of a cashless exercise of warrants into shares of common stock is unclear, and the tax consequences of a cashless
130
exercise could differ from the consequences upon the exercise of a warrant described in the preceding paragraph. U.S. holders should consult their own tax advisors regarding the U.S. federal income tax consequences of a cashless exercise of warrants.
Certain Adjustments to the Warrants or Series B Preferred Stock
An adjustment to the number of shares of our common stock that will be issued upon the exercise of a warrant or conversion of a share of Series B Preferred Stock, or an adjustment to the exercise price of a warrant, may be treated as a constructive distribution to a U.S. holder of the warrant or share depending on the circumstances of such adjustment (for example, if such adjustment is to compensate for a distribution of cash or other property to our shareholders). Adjustments to the exercise price of warrants or conversion price of Series B Preferred Stock made pursuant to a bona fide reasonable adjustment formula that has the effect of preventing dilution of the interest of the holders thereof generally should not be considered to result in a constructive distribution. Any such constructive distribution would be taxable whether or not there is an actual distribution of cash or other property. See the more detailed discussion of the rules applicable to distributions made by us under the heading "Distributions on Common Stock or Series B Preferred Stock" below.
Expiration of the Warrants without Exercise
Upon the lapse or expiration of a warrant, a U.S. holder will recognize a loss in an amount equal to such U.S. holder's tax basis in the warrant. Any such loss generally will be a capital loss and will be long-term capital loss if the warrant is held for more than one year. Deductions for capital losses are subject to certain limitations.
Conversion of Series B Preferred Stock
A U.S. holder generally will not recognize gain or loss upon the conversion of a share of Series B Preferred Stock into common stock. A U.S. holder's initial tax basis in the shares of our common stock received upon the conversion of a share of Series B Preferred Stock will be equal to such U.S. holder's tax basis in the share of Series B Preferred Stock. A U.S. holder's holding period for the shares of our common stock received upon the conversion of a share of Series B Preferred Stock will include the U.S. holder's holding period in such share of Series B Preferred Stock.
Distributions on Common Stock or Series B Preferred Stock
If we pay distributions of cash or property with respect to our common stock or Series B Preferred Stock (including constructive distributions as described above under the heading "Certain Adjustments to the Warrants or Series B Preferred Stock"), those distributions generally will constitute dividends for U.S. federal income tax purposes to the extent paid from our current or accumulated earnings and profits, as determined under U.S. federal income tax principles. If a distribution exceeds our current and accumulated earnings and profits, the excess will be treated as a tax-free return of the U.S. holder's investment, up to such holder's tax basis in its shares of our common stock or Series B Preferred Stock. Any remaining excess will be treated as capital gain, subject to the tax treatment described below under the heading "Gain on Sale, Exchange or Other Taxable Disposition."
Gain on Sale, Exchange or Other Taxable Disposition
Upon the sale or other taxable disposition of common shares, Series B Preferred Stock or warrants, a U.S. holder generally will recognize capital gain or loss in an amount equal to the difference between (a) the amount of cash plus the fair market value of any property received and (b) such U.S. holder's tax basis in such common shares, Series B Preferred Stock or warrants sold or otherwise disposed of. Such gain or loss generally will be long-term capital gain or loss if, at the time
131
of the sale or other disposition, the common shares, Series B Preferred Stock or warrants have been held by the U.S. holder for more than one year. Preferential tax rates may apply to long-term capital gain of a U.S. holder that is an individual, estate, or trust. Deductions for capital losses are subject to certain limitations.
Non-U.S. Holders
Distributions on Common Stock or Series B Preferred Stock
If we pay distributions of cash or property with respect to our common stock or Series B Preferred Stock, those distributions generally will constitute dividends for U.S. federal income tax purposes to the extent paid from our current or accumulated earnings and profits, as determined under U.S. federal income tax principles. If a distribution exceeds our current and accumulated earnings and profits, the excess will be treated as a tax-free return of the non-U.S. holder's investment, up to such holder's tax basis in its shares of our common stock or Series B Preferred Stock. Any remaining excess will be treated as capital gain, subject to the tax treatment described below under the heading "Gain on Sale, Exchange or Other Taxable Disposition." Dividends paid to a non-U.S. holder generally will be subject to withholding of U.S. federal income tax at a 30% rate or such lower rate as may be specified by an applicable income tax treaty between the United States and such holder's country of residence. In the case of any constructive distribution, it is possible that this tax would be withheld from any amount owed to the non-U.S. holder, including, but not limited to, distributions of cash, common stock or sales proceeds subsequently paid or credited to that holder. If we are unable to determine, at the time of payment of a distribution, whether the distribution will constitute a dividend, we may nonetheless choose to withhold any U.S. federal income tax on the distribution as permitted by U.S. Treasury Regulations.
Distributions that are treated as effectively connected with a trade or business conducted by a non-U.S. holder within the United States are generally not subject to the 30% withholding tax if the non-U.S. holder provides a properly executed IRS Form W- 8ECI stating that the distributions are not subject to withholding because they are effectively connected with the non-U.S. holder's conduct of a trade or business in the United States. If a non-U.S. holder is engaged in a trade or business in the United States and the distribution is effectively connected with the conduct of that trade or business, the distribution will generally have the consequences described above for a U.S. holder (subject to any modification provided under an applicable income tax treaty). Any U.S. effectively connected income received by a non-U.S. holder that is treated as a corporation for U.S. federal income tax purposes may also, under certain circumstances, be subject to an additional "branch profits tax" at a 30% rate (or such lower rate as may be specified by an applicable income tax treaty).
A non-U.S. holder who claims the benefit of an applicable income tax treaty between the United States and such holder's country of residence generally will be required to provide a properly executed IRS Form W-8BEN or W-8BEN-E, as applicable, and satisfy applicable certification and other requirements. A non-U.S. holder that is eligible for a reduced rate of U.S. withholding tax under an income tax treaty generally may obtain a refund or credit of any excess amounts withheld by timely filing an appropriate claim with the IRS. Non-U.S. holders should consult their own tax advisors regarding their entitlement to benefits under a relevant income tax treaty.
Gain on Sale, Exchange or Other Taxable Disposition
Subject to the discussion below in "Information Reporting and Backup Withholding" and "Foreign Account Tax Compliance Act," a non-U.S. holder generally will not be subject to U.S. federal income
132
tax on gain recognized on a sale, exchange or other taxable disposition of our common stock, Series B Preferred Stock or warrants unless:
- •
- the gain is effectively connected with the non-U.S. holder's conduct of a trade or business in the United States and, if an applicable income
tax treaty so provides, the gain is attributable to a permanent establishment maintained by the non-U.S. holder in the United States; in these cases, the non-U.S. holder will be taxed on a net income
basis at the regular graduated rates and in the manner applicable to a U.S. holder, and, if the non-U.S. holder is a corporation, an additional branch profits tax at a rate of 30%, or a lower rate as
may be specified by an applicable income tax treaty, may also apply;
- •
- the non-U.S. holder is an individual present in the United States for 183 days or more in the taxable year of the disposition and certain other conditions are met, in which case the non-U.S. holder will be subject to a 30% tax (or such lower rate as may be specified by an applicable income tax treaty) on the amount by which such non-U.S. holder's capital gains allocable to U.S. sources exceed capital losses allocable to U.S. sources during the taxable year of the disposition; or
we are or were a "U.S. real property holding corporation" during the shorter of the five-year period ending on the date of the disposition or the period that the non-U.S. holder held our common stock. Generally, a corporation is a "U.S. real property holding corporation" if the fair market value of its "U.S. real property interests" (within the meaning of the Internal Revenue Code) equals or exceeds 50% of the sum of the fair market value of its worldwide real property interests plus its other assets used or held for use in a trade or business. We believe that we are not currently, and we do not anticipate becoming, a "U.S. real property holding corporation" for U.S. federal income tax purposes.
Information Reporting and Backup Withholding
Distributions on, and the payment of the proceeds of a disposition of, our common stock, Series B Preferred Stock or warrants generally will be subject to information reporting if made within the United States or through certain U.S.-related financial intermediaries. Information returns are required to be filed with the IRS and copies of information returns may be made available to the tax authorities of the country in which a holder resides or is incorporated under the provisions of a specific treaty or agreement.
Backup withholding may also apply if the holder fails to provide certification of exempt status or a correct U.S. taxpayer identification number and otherwise comply with the applicable backup withholding requirements. Generally, a holder will not be subject to backup withholding if it provides a properly completed and executed IRS Form W-9 or appropriate IRS Form W-8, as applicable. Backup withholding is not an additional tax. Amounts withheld under the backup withholding rules may be refunded or credited against the holder's U.S. federal income tax liability, if any, provided certain information is timely filed with the IRS.
Foreign Account Tax Compliance Act
Legislation commonly referred to as the Foreign Account Tax Compliance Act, or FATCA, generally imposes a U.S. federal withholding tax of 30% on certain payments to certain non-U.S. entities (including certain intermediaries) unless such persons comply with FATCA's information reporting and withholding regime. This regime and its requirements are different from, and in addition to, the certification requirements described elsewhere in this discussion. The FATCA withholding rules apply to dividend payments.
The United States has entered into, and continues to negotiate, intergovernmental agreements (each, an "IGA") with a number of other jurisdictions to facilitate the implementation of FATCA. An IGA may significantly alter the application of FATCA and its information reporting and withholding requirements with respect to any particular investor. FATCA is particularly complex and its application remains uncertain. Prospective investors should consult their own tax advisors regarding how these rules may apply in their particular circumstances.
133
We have entered into an underwriting agreement dated July , 2019 with Ladenburg Thalmann & Co. Inc., as the representative of the underwriters (the "representative") named below and the sole book-running manager of this offering. Subject to the terms and conditions of the underwriting agreement, the underwriters have agreed to purchase the number of our securities set forth opposite its name below.
Underwriters
|
Class A Units |
Class B Units |
Warrants | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Ladenburg Thalmann & Co. Inc. |
[ ] | [ ] | [ ] | |||||||
| | | | | | | | | | | |
Total |
[ ] | [ ] | [ ] | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
A copy of the underwriting agreement will be filed as an exhibit to the registration statement of which this prospectus is a part.
We have been advised by the underwriters that they propose to offer the securities directly to the public at the public offering price set forth on the cover page of this prospectus. The underwriters may sell shares of common stock, Series B Preferred Stock or warrants separately to purchasers or may sell a combination of such securities to purchasers in any proportion. Any securities sold by the underwriters to securities dealers will be sold at the public offering price less a selling concession not in excess of $[ ] per share and $[ ] per warrant.
The underwriting agreement provides that subject to the satisfaction or waiver by the representative of the conditions contained in the underwriting agreement, the underwriters are obligated to purchase and pay for all of the securities offered by this prospectus.
No action has been taken by us or the underwriters that would permit a public offering of the shares of common stock, shares of Series B Preferred Stock, shares of common stock underlying the Series B Preferred Stock and warrants to purchase common stock, in any jurisdiction outside the United States where action for that purpose is required. None of our securities included in this offering may be offered or sold, directly or indirectly, nor may this prospectus or any other offering material or advertisements in connection with the offer and sales of any of the securities offered hereby be distributed or published in any jurisdiction except under circumstances that will result in compliance with the applicable rules and regulations of that jurisdiction. Persons who receive this prospectus are advised to inform themselves about and to observe any restrictions relating to this offering of securities and the distribution of this prospectus. This prospectus is neither an offer to sell nor a solicitation of any offer to buy the securities in any jurisdiction where that would not be permitted or legal.
The underwriters have advised us that they do not intend to confirm sales to any account over which they exercise discretionary authority.
134
Underwriting Discount and Expenses
The following table summarizes the underwriting discount and commission to be paid to the underwriter assuming no exercise of the over-allotment option and the full exercise of the over-allotment option.
| |
Per Class A Unit(1) |
Per Class B Unit(1) |
Total | Total With No Exercise of the Over- Allotment Option |
Total With Full Exercise of the Over- Allotment Option |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Public offering price |
$ | [ ] | $ | [ ] | $ | 0.01 | $ | [ ] | $ | [ ] | ||||||
Underwriting discount to be paid to the underwriters by us(2)(3) |
$ | [ ] | $ | [ ] | $ | [ ] | $ | [ ] | $ | [ ] | ||||||
Proceeds to us (before expenses) |
$ | [ ] | $ | [ ] | $ | [ ] | $ | [ ] | $ | [ ] | ||||||
- (1)
- The
public offering price and underwriting discount corresponds to (x) in respect of the Class A Units (i) a public offering price per share of
common stock of $4.76, (ii) a public offering price per warrant of $4.76, and (y) in respect of the Class B Units (i) a public offering price per share of Series B
Preferred Stock of $1,000 and (ii) a public offering price per warrant of $4.76.
- (2)
- We
have also agreed to pay to the underwriters a 1% management fee equal to 1% of the total gross proceeds from the offering and have agreed to reimburse the
accountable expenses of the underwriter, including legal fees, in this offering, up to a maximum of $110,000.
- (3)
- We have granted a 45-day option to the underwriter to purchase 378,151 additional shares of common stock and/or warrants to purchase shares of common stock (up to 15% of the number of shares of common stock (including the number of shares of common stock issuable upon conversion of shares of Series B Preferred Stock) and the number of shares of common stock underlying the warrants sold in the primary offering) at the public offering price per share of common stock and the public offering price per warrant set forth above less the underwriting discounts and commissions, solely to cover over- allotments, if any.
We estimate the total expenses payable by us for this offering to be approximately $1.8 million, which amount includes (i) the underwriting discount of $0.96 million ($0.99 million if the underwriters' over-allotment option is exercised in full), (ii) the management fee of 1% of the gross proceeds of the offering, (iii) the reimbursement of the accountable expenses of the representative equal to $110,000 including the legal fees of the representative being paid by us, (iv) a potential tail fee of up to $[ ] million and a warrant to purchase [ ] shares of Common Stock (as described further below), and (v) other estimated company expenses of approximately $0.62 million, which includes legal, accounting, printing costs and various fees associated with the registration and listing of our shares. We have agreed to pay a previous underwriter of our underwritten public offering of securities that closed on October 2, 2018 (the "October 2018 Offering"), a tail fee equal to 8% cash compensation for the gross proceeds raised, and 8% warrant coverage of the number of shares of Common Stock placed, in any public or private offering consummated within twelve months of the expiration or termination of our engagement with the previous underwriter by any investor contacted by the previous underwriter in the October 2018 Offering.
The securities we are offering are being offered by the underwriters subject to certain conditions specified in the underwriting agreement.
135
Over-allotment Option
We have granted to the underwriters an option exercisable not later than 45 days after the date of this prospectus to purchase up to a number of additional shares of common stock and/or Series 1 and/or Series 2 warrants to purchase shares of common stock not to exceed 15% of the number of shares of common stock sold in the primary offering (including the number of shares of common stock issuable upon conversion of shares of Series B Preferred Stock, but excluding shares of common stock underlying the warrants issued in this offering and any shares of common stock issued upon any exercise of the underwriter's over-allotment option) and/or 15% of the Series 1 and/or Series 2 warrants sold in the primary offering at the public offering price per share of common stock and the public offering price per warrant set forth on the cover page hereto less the underwriting discounts and commissions. The underwriters may exercise the option solely to cover overallotments, if any, made in connection with this offering. If any additional shares of common stock and/or warrants are purchased pursuant to the over-allotment option, the underwriters will offer these shares of common stock and/or warrants on the same terms as those on which the other securities are being offered.
Determination of Offering Price
Our common stock is currently traded on The NASDAQ Capital Market under the symbol "JAGX." On June 27, 2019, the closing price of our common stock was $4.76 per share. We do not intend to apply for listing of the Series B Preferred Stock or the warrants on any securities exchange or other trading system.
The public offering price of the securities offered by this prospectus was determined by negotiation between us and the underwriters. Among the factors that were considered in determining the public offering price of the shares:
- •
- our history and our prospects;
- •
- the industry in which we operate;
- •
- our past and present operating results;
- •
- the previous experience of our executive officers; and
- •
- the general condition of the securities markets at the time of this offering.
The offering price stated on the cover page of this prospectus should not be considered an indication of the actual value of the shares of common stock or shares of preferred stock sold in this offering. That price is subject to change as a result of market conditions and other factors and we cannot assure you that the shares of common stock sold in this offering can be resold at or above the public offering price.
Lock-up Agreements
Our officers and directors, and certain 5% or greater shareholders, including Sagard, have agreed with the representative to be subject to a lock-up period of 90 days following the date of this prospectus. This means that, during the applicable lock-up period, such persons may not offer for sale, contract to sell, sell, distribute, grant any option, right or warrant to purchase, pledge, hypothecate or otherwise dispose of, directly or indirectly, any shares of our common stock or any securities convertible into, or exercisable or exchangeable for, shares of our common stock. Certain limited transfers are permitted during the lock-up period if the transferee agrees to these lock-up restrictions. We have also agreed, in the underwriting agreement, to similar lock-up restrictions on the issuance and sale of our securities for 90 days following the effectiveness of the underwriting agreement, although we will be permitted to issue stock options or stock awards to directors, officers and employees under our existing plans. The lock-up period is subject to an additional extension to accommodate for our reports
136
of financial results or material news releases. The representative may, in its sole discretion and without notice, waive the terms of any of these lock-up agreements.
Other Relationships
Subject to the satisfaction of certain conditions, we have granted the representative a right of first refusal to act as lead or sole bookrunner or exclusive placement agent in connection with any subsequent public or private offering of equity securities or other capital markets financing by us. This right of first refusal extends for 12 months from the closing date of this offering. The terms of any such engagement of the representative will be determined by separate agreement.
Transfer Agent and Registrar
The transfer agent of our common stock is AST. There address is 6201 15th Avenue, Brooklyn, NY 11219.
Stabilization, Short Positions and Penalty Bids
The underwriters may engage in syndicate covering transactions stabilizing transactions and penalty bids or purchases for the purpose of pegging, fixing or maintaining the price of our common stock:
- •
- Syndicate covering transactions involve purchases of securities in the open market after the distribution has been completed in order to cover
syndicate short positions. Such a naked short position would be closed out by buying securities in the open market. A naked short position is more likely to be created if the underwriters are
concerned that there could be downward pressure on the price of the securities in the open market after pricing that could adversely affect investors who purchase in the offering.
- •
- Stabilizing transactions permit bids to purchase the underlying security so long as the stabilizing bids do not exceed a specific maximum and
are engaged in for the purpose of preventing or retarding a decline in the market price of the shares of common stock while this offering is in progress.
- •
- Penalty bids permit the underwriters to reclaim a selling concession from a syndicate member when the securities originally sold by the syndicate member are purchased in a stabilizing or syndicate covering transaction to cover syndicate short positions.
These syndicate covering transactions, stabilizing transactions, and penalty bids may have the effect of raising or maintaining the market prices of our securities or preventing or retarding a decline in the market prices of our securities. As a result the price of our common stock may be higher than the price that might otherwise exist in the open market. Neither we nor the underwriters make any representation or prediction as to the effect that the transactions described above may have on the price of our common stock. These transactions may be effected on The NASDAQ Capital Market, in the over-the-counter market or on any other trading market and, if commenced, may be discontinued at any time.
In connection with this offering, the underwriters also may engage in passive market making transactions in our common stock in accordance with Regulation M during a period before the commencement of offers or sales of shares of our common stock in this offering and extending through the completion of the distribution. In general, a passive market maker must display its bid at a price not in excess of the highest independent bid for that security. However, if all independent bids are lowered below the passive market maker's bid that bid must then be lowered when specific purchase limits are exceeded. Passive market making may stabilize the market price of the securities at a level above that which might otherwise prevail in the open market and, if commenced, may be discontinued at any time.
137
Neither we, nor the underwriters make any representation or prediction as to the direction or magnitude of any effect that the transactions described above may have on the prices of our securities. In addition, neither we nor the underwriters make any representation that the underwriters will engage in these transactions or that any transactions, once commenced will not be discontinued without notice.
Indemnification
We have agreed to indemnify the underwriters against certain liabilities, including certain liabilities arising under the Securities Act or to contribute to payments that the underwriters may be required to make for these liabilities.
138
The validity of the securities offered hereby will be passed upon by our counsel, Reed Smith LLP, Palo Alto, California. Sheppard, Mullin, Richter & Hampton LLP, New York, New York, is acting as counsel for the underwriters in connection with certain legal matters in connection with this offering.
The financial statements of the Company as of December 31, 2018 and 2017 and for each of the two years in the period ended December 31, 2018 incorporated by reference in this prospectus have been so incorporated in reliance on the reports of BDO USA, LLP, an independent registered public accounting firm (the reports on the financial statements contains an explanatory paragraph regarding the Company's ability to continue as a going concern), incorporated herein by reference, given on the authority of said firm as experts in auditing and accounting.
WHERE YOU CAN FIND MORE INFORMATION
We are subject to the reporting requirements of the Securities Exchange Act of 1934, as amended, and file annual, quarterly and current reports, proxy statements and other information with the SEC. You may read and copy these reports, proxy statements and other information at the SEC's public reference facilities at 100 F Street, N.E., Room 1580, Washington, D.C. 20549. You can request copies of these documents by writing to the SEC and paying a fee for the copying cost. Please call the SEC at 1-800-SEC-0330 for more information about the operation of the public reference facilities. SEC filings are also available at the SEC's web site at http://www.sec.gov.
This prospectus, which constitutes a part of the registration statement on Form S-1 that we have filed with the SEC under the Securities Act, does not contain all of the information in the registration statement and its exhibits. For further information with respect to us and the common stock offered by this prospectus, you should refer to the registration statement and the exhibits filed as part of that document. Statements contained in this prospectus as to the contents of any contract or any other document referred to are not necessarily complete, and in each instance, we refer you to the copy of the contract or other document filed as an exhibit to the registration statement. Each of these statements is qualified in all respects by this reference.
We also maintain a website at www.jaguar.health, through which you can access our SEC filings. The information set forth on, or accessible from, our website is not part of this prospectus or the accompanying prospectus.
INCORPORATION OF INFORMATION BY REFERENCE
The SEC allows us to "incorporate by reference" information that we file with them. Incorporation by reference allows us to disclose important information to you by referring you to those other documents. The information incorporated by reference is an important part of this prospectus, and information that we file later with the SEC will automatically update and supersede this information. This prospectus omits certain information contained in the registration statement, as permitted by the SEC. You should refer to the registration statement, including the exhibits, for further information about us and the securities we may offer pursuant to this prospectus. Statements in this prospectus or the accompanying prospectus regarding the provisions of certain documents filed with, or incorporated by reference in, the registration statement are not necessarily complete and each statement is qualified in all respects by that reference. Copies of all or any part of the registration statement, including the documents incorporated by reference or the exhibits, may be obtained upon payment of the prescribed
139
rates at the offices of the SEC listed above in "Where You Can Find More Information." The documents we are incorporating by reference are:
- •
- our Annual Report on
Form 10-K for the fiscal year ended December 31, 2018 filed on April 10, 2019;
- •
- our definitive proxy statement and
definitive additional materials, on Schedule 14A, relating to our Annual Meeting of Stockholders held on May 24, 2019, filed April 29, 2019;
- •
- our Quarterly Report on
Form 10-Q for the fiscal quarter ended March 31, 2019 filed on May 21, 2019;
- •
- Our Current Reports on Form 8-K filed on
January 8, 2019,
February 26, 2019,
March 5, 2019,
March 15, 2019,
March 19, 2019,
March 22, 2019,
March 25, 2019 (as subsequently amended on Form 8-K/A on
March 26, 2019),
April 1, 2019,
April 4, 2019,
April 8, 2019,
April 11, 2019,
April 19, 2019,
May 2, 2019,
May 17, 2019,
May 24, 2019
(two filings),
May 29, 2019,
June 3, 2019,
June 6, 2019,
June 14, 2019 (as subsequently amended on Form 8-K/A on
June 28, 2019),
June 25, 2019 and July 5, 2019;
- •
- the description of our common
stock contained in our registration statement on Form 8-A filed on October 30, 2014 (Registration No. 001-36714) with the SEC, including any amendment or report filed for the
purpose of updating such description; and
- •
- all reports and other documents subsequently filed by us pursuant to Sections 13(a), 13(c), 14 and 15(d) of the Exchange Act after the date of this prospectus and prior to the termination or completion of the offering of securities under this prospectus shall be deemed to be incorporated by reference in this prospectus and to be a part hereof from the date of filing such reports and other documents.
Unless otherwise noted, the SEC file number for each of the documents listed above is 001-36714.
In addition, all reports and other documents filed by us pursuant to the Exchange Act after the date of this prospectus shall be deemed to be incorporated by reference into this prospectus.
Any statement contained in this prospectus, the accompanying prospectus, or in a document incorporated or deemed to be incorporated by reference into this prospectus or the accompanying prospectus will be deemed to be modified or superseded for purposes of this prospectus to the extent that a statement contained in this prospectus, the accompanying prospectus, or any other subsequently filed document that is deemed to be incorporated by reference into this prospectus modifies or supersedes the statement. Any statement so modified or superseded will not be deemed, except as so modified or superseded, to constitute a part of this prospectus or accompanying prospectus.
You may request, orally or in writing, a copy of any or all of the documents incorporated herein by reference. These documents will be provided to you at no cost, by contacting: Investor Relations, Jaguar Health, Inc., 201 Mission Street, Suite 2375, San Francisco, CA, 94105 or call (415) 371-8300.
You should rely only on information contained in, or incorporated by reference into, this prospectus and the accompanying prospectus. We have not authorized anyone to provide you with information different from that contained in this prospectus and the accompanying prospectus. We are not making offers to sell the securities in any jurisdiction in which such an offer or solicitation is not authorized or in which the person making such offer or solicitation is not qualified to do so or to anyone to whom it is unlawful to make such offer or solicitation.
140
2,521,008 Class A Units, consisting of Common Stock and Warrants or 12,000 Class B Units consisting of Series B Convertible Preferred Stock
and Warrants
(and 2,521,008 shares of common stock underlying shares of Series B Convertible Preferred Stock and 5,042,016 shares of Common Stock underlying Warrants)

PROSPECTUS
Sole Book-Running Manager
Ladenburg Thalmann
, 2019
Part II—INFORMATION NOT REQUIRED IN PROSPECTUS
ITEM 13. OTHER EXPENSES OF ISSUANCE AND DISTRIBUTION.
The following table sets forth an itemized statement of the expenses (excluding underwriting discounts) that are payable by us in connection with the registration, offer and sale of the common stock described in this registration statement. With the exception of the SEC registration fee, the FINRA filing fee and The NASDAQ Capital Market listing fee, the amounts set forth below are estimates.
| |
Amount to be Paid |
|||
|---|---|---|---|---|
SEC registration fee |
$ | 10,181 | ||
FINRA filing fee |
4,363 | |||
Accounting fees and expenses |
90,000 | |||
Legal fees and expenses |
460,000 | |||
Printing and related expenses |
50,000 | |||
Transfer agent and registrar fees |
25,000 | |||
Miscellaneous |
40,000 | |||
| | | | | |
Total |
$ | 675,181 | ||
| | | | | |
| | | | | |
| | | | | |
ITEM 14. INDEMNIFICATION OF DIRECTORS AND OFFICERS.
Section 102(b)(7) of the DGCL authorizes a corporation in its certificate of incorporation to eliminate or limit personal liability of directors of the corporation for violations of the directors' fiduciary duty of care. However, directors remain liable for breaches of duties of loyalty, failing to act in good faith, engaging in intentional misconduct, knowingly violating a law, paying a dividend or approving a stock repurchase which was illegal under DGCL Section 174 or obtaining an improper personal benefit. In addition, equitable remedies for breach of fiduciary duty of care, such as injunction or recession, are available.
Our current certificate of incorporation eliminates the personal liability of the members of our board of directors to the fullest extent permitted by the DGCL. Any repeal or modification of that provision by the stockholders of the corporation will not adversely affect any right or protection of a director of the corporation existing at the time of such repeal or modification.
Section 145 of the DGCL provides that a corporation has the power to indemnify a director, officer, employee or agent of the corporation, or a person serving at the request of the corporation for another corporation, partnership, joint venture, trust or other enterprise in related capacities against expenses (including attorneys' fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by the person in connection with an action, suit or proceeding to which he was or is a party or is threatened to be made a party to any threatened, ending or completed action, suit or proceeding by reason of such position, if such person acted in good faith and in a manner he reasonably believed to be in or not opposed to the best interests of the corporation, and, in any criminal action or proceeding, had no reasonable cause to believe his conduct was unlawful, except that, in the case of actions brought by or in the right of the corporation, no indemnification shall be made with respect to any claim, issue or matter as to which such person shall have been adjudged to be liable to the corporation unless and only to the extent that the Court of Chancery or other adjudicating court determines that, despite the adjudication of liability but in view of all of the circumstances of the case, such person is fairly and reasonably entitled to indemnity for such expenses which the Court of Chancery or such other court shall deem proper.
II-1
Our current bylaws provide for indemnification of our officers and directors to the fullest extent permitted by the DGCL.
We have entered into indemnification agreements with each of our directors, and intend to enter into such agreements with each of our officers prior to this offering, pursuant to which we agreed, to the maximum extent permitted by applicable law and subject to the specified terms and conditions set forth in each agreement, to indemnify a director or officer who acts on our behalf and is made or threatened to be made a party to any action or proceeding against expenses, judgments, fines and amounts paid in settlement that are incurred by such officer or director in connection with the action or proceeding. The indemnification provisions apply whether the action was instituted by a third party or by us.
We have purchased and maintain insurance on behalf of our officers and directors that provides coverage for expenses and liabilities incurred by them in their capacities as officers and directors.
In any underwriting agreement we enter into in connection with the sale of common stock being registered hereby, the underwriters will agree to indemnify, under certain conditions, us, our directors, our officers and persons who control us within the meaning of the Securities Act against certain liabilities.
ITEM 15. RECENT SALES OF UNREGISTERED SECURITIES.
Common Stock Purchase Agreement
Shares of our common stock were sold pursuant to the common stock purchase agreement with Aspire Capital Fund, LLC, as disclosed on our Form 8-K filed with the SEC on June 9, 2016.
2016 Securities Purchase Agreement
On November 22, 2016, we entered into a Securities Purchase Agreement with certain institutional investors, pursuant to which we sold securities to such investors in a private placement transaction (the "2016 Private Placement"). In the 2016 Private Placement, we sold an aggregate of 23,809 shares of our common stock at a price of $42.00 per share for gross proceeds of approximately $1.0 million, as disclosed on our Form 8-K filed with the SEC on November 29, 2016.
Invesco Asset Management Limited
On February 21, 2017, Invesco Asset Management Limited ("Invesco") delivered a signed commitment letter to us, pursuant to which Invesco agreed, subject to the terms and conditions of such letter, to purchase, simultaneously with the consummation of the merger, $3.0 million of our common stock at a price equal to $64.75 per share, as disclosed on our Form 10-Q filed with the SEC on May 15, 2017.
Settlement of Outstanding Napo Debt
Pursuant to the Agreement and Plan of Merger, dated March 31, 2017 (the "Merger Agreement"), by and among the Company, Napo, Napo Acquisition Corporation ("Merger Sub"), and Napo's representative (the "Merger"), Napo entered into a Settlement and Discounted Payoff Agreement with Nantucket Investments Limited ("Nantucket") and the lenders named therein (the "Settlement Agreement") pursuant to which Napo agreed, simultaneously with the consummation of the merger, (a) to make a cash payment to Nantucket of no less than $8 million, which will reduce the outstanding principal obligations under the Litigation Financing Agreement, dated October 10, 2014, by and between Napo and Nantucket (the "Financing Agreement"), and (b) in satisfaction as a compromise for the outstanding obligations under the Financing Agreement and the release of any lien or security interest in respect of such outstanding obligations, (x) to transfer to Nantucket 38,905 shares of Jaguar
II-2
common stock owned by Napo and (y) pursuant to the merger agreement, to cause Jaguar to issue to Nantucket 27,719 newly issued shares of Jaguar voting common stock and 548,286 newly issued shares of Jaguar non-voting common stock, as disclosed on our Form 8-K filed with the SEC on March 30, 2017.
Sales Pursuant to Share Purchase Agreement
On June 28, 2017, pursuant to a share purchase agreement, we issued 1,428 shares of our common stock to an existing investor for gross proceeds of $50,000, as disclosed on our Form 10-Q filed with the SEC on August 9, 2017.
Completion of Acquisition or Disposition of Assets
On July 31, 2017, the Company completed its merger with Napo pursuant to the Merger Agreement, as disclosed on our Form 8-K filed with the SEC on August 1, 2018. In connection with the Merger, (i) each issued and outstanding share of Napo common stock (other than dissenting shares and shares held by the Company or Napo) was converted into a contingent right to receive (x) up to a whole number of shares of Company common stock comprising in the aggregate up to approximately 20.2% of the fully diluted shares of Company common stock immediately following the consummation of the merger, which contingent right will vest only if the resale of certain shares of Company common stock (the "Tranche A Shares") issued by the Company to Nantucket pursuant to the Napo debt settlement provides Nantucket with specified cash returns over a specified period of time (the "Hurdle Amounts"), and (y) if the applicable Hurdle Amount is achieved before all of the Tranche A Shares are sold, additional shares of Company common stock (equal to 50% of the unsold Tranche A Shares), which will be distributed pro rata among holders of contingent rights and holders of Napo restricted stock units, (ii) existing creditors of Napo (inclusive of Nantucket) were issued in the aggregate approximately 612,900 shares of Company non-voting common stock and 32,606 shares of Company voting common stock in full satisfaction of all existing indebtedness then owed by Napo to such creditors, and (iii) Invesco, an existing Napo stockholder, was issued an aggregate of approximately 46,332 shares of Company common stock in return for $3 million of new funds invested into the Company by such investor, which were immediately loaned to Napo to partially facilitate the extinguishment of the debt that Napo owed to Nantucket. The minimum Hurdle Amount needed for the vesting of the contingent rights will vary depending on a number of factors (including, among other things, the time period over which Nantucket receives specified cash returns in connection with the resale of the Tranche A Shares), and Napo stockholders may not receive any shares of Jaguar common stock in certain circumstances (including if the minimum Hurdle Amount is not satisfied).
Exercise of Warrants
On July 31, 2017, the Company entered into Warrant Exercise Agreements (the "Exercise Agreements") with certain investors who own the right to purchase up to an aggregate of 23,809 shares of common stock at an exercise price of $70.00 per share (the "Series C Warrants"). Such holders (the "Existing Holders") own, in the aggregate, Series C Warrants exercisable for 12.976 shares of Common Stock. Pursuant to the Exercise Agreements, the Exercising Holders and the Company agreed that the Exercising Holders would exercise their Series C Warrants with respect to 12.976 shares of Common Stock underlying such Series C Warrants for a reduced exercise price equal to $28.00 per share, as disclosed on our Form 8-K filed with the SEC on July 31, 2017.
KCSA Strategic Communications
On July 31, 2017, we issued 926 shares of our voting common stock to KCSA Strategic Communications ("KCSA") pursuant to the Merger Agreement and an agreement between Napo and
II-3
KCSA, as a complete settlement and satisfaction of Napo's outstanding obligations to KCSA, as disclosed on our Form 10-Q filed with the SEC on November 20, 2017.
On October 30, 2017, pursuant to a debt settlement agreement dated October 30, 2017, we issued 3,359 shares of common stock to KCSA, which together with the 926 shares of common stock previously issued on July 31, 2017, fully satisfied $60,000 in debt incurred by us for services rendered by KCSA, as disclosed on our Form 10-K filed with the SEC on April 9, 2018.
L2 Capital, LLC
Shares of our common stock were sold pursuant to the common stock purchase agreement with L2 Capital, LLC, as disclosed on our Form 8-K filed with the SEC on November 24, 2017.
Chicago Venture Partners, L.P.
In November and December 2017, through a series of partial redemptions pursuant to the terms of the Secured Convertible Promissory Note issued to Chicago Venture Partners, L.P. (the "CVP Note") as disclosed in our Form 8-K filed with the SEC on July 3, 2017, we issued 57,142 shares of common stock to redeem $601,311.80 of the CVP Note, as disclosed on our Form 10-K filed with the SEC on April 9, 2018.
Sales Pursuant to Share Purchase Agreement
In December 2017, pursuant to a share purchase agreement dated December 27, 2017, we issued 57,285 shares of our common stock to certain investors for gross proceeds of $401,000, as disclosed on our Form 10-K filed with the SEC on April 9, 2018.
First Amendment to Note Purchase Agreement and Notes
On December 29, 2017, Napo, our wholly-owned subsidiary, entered into an amendment (the "First Amendment") to the Note Purchase Agreement and Notes with each of the purchasers (the "Purchasers") party to the Note Purchase Agreement, dated March 1, 2017, by and among the Napo and the Purchasers (as amended, the "Note Purchase Agreement"). In connection with the First Amendment, Napo amended the original issue discount exchangeable promissory notes previously issued to the Purchasers on March 1, 2017 (the "First Tranche Notes") and April 27, 2017 (the "Second Tranche Notes" and together with the First Tranche Notes, the "Notes") pursuant to the Note Purchase Agreement to, among other things, (a) increase the principal amount outstanding under the First Tranche Notes and the Second Tranche Notes by twelve percent (12%), (b) lower the price at which the Notes are exchangeable for shares (the "Exchange Shares") of the Company's common stock (the "Common Stock") from $39.20 per share to $14.00 per share, and (c) extend the maturity date of the First Tranche Notes from December 1, 2017 to February 15, 2018 and the Second Tranche Notes from January 27, 2018 to April 1, 2018.
In connection with the First Amendment, the Company also issued 35,601 shares of common stock to the Purchasers as repayment of $299,050.08 principal amount of the First Tranche Notes, as disclosed on our Form 8-K filed with the SEC on January 2, 2018. Following such repayment and the 12% increase to the outstanding balance of the Notes described above, $435,949.92 and $735,000.00 principal amount remain outstanding under the First Tranche Notes and Second Tranche Notes, respectively.
II-4
Investor Awareness, Inc.
In January 2018, pursuant to a consulting agreement dated August 14, 2017, we issued 714 shares of our common stock to Investor Awareness, Inc. as partial consideration for financial public relations services rendered, as disclosed on our Form 10-Q filed with the SEC on May 5, 2018.
Issuance Pursuant to Share Purchase Agreements
In January 2018, pursuant to a share purchase agreement dated January 18, 2018, we issued 131,655 shares of our common stock to certain investors for gross proceeds of approximately $954,000, as disclosed on our Form 10-Q filed with the SEC on May 5, 2018.
Second Amendment to Note Purchase Agreement and Notes
On February 16, 2018, Napo, our wholly-owned subsidiary, entered into the Second Amendment to Note Purchase Agreement and Notes and Payoff Agreement (the "Second Amendment") with each of the Purchasers party to the Note Purchase Agreement, to extend the maturity date of the Second Tranche Notes from April 1, 2018 to May 1, 2018.
In connection with the Second Amendment, the Company also issued 54,049 shares of common stock of the Company to the Purchasers as repayment of the remaining $435,949.92 aggregate principal amount of the original issue discount exchangeable promissory notes previously issued by Napo to the Purchasers on March 1, 2017 pursuant to the Note Purchase Agreement and $18,063.24 in accrued and unpaid interest thereon, ad disclosed on our Form 8-K filed with the SEC on February 16, 2018.
Chicago Venture Partners, L.P.
In January through March 2018, through a series of partial redemptions pursuant to the terms of the CVP Note, as disclosed in our Form 8-K filed with the SEC on July 3, 2017, we issued 122,037 shares of common stock to redeem $950,000 of the CVP Note, including accrued and unpaid interest thereon, as disclosed on our Form 10-Q filed with the SEC on May 5, 2018.
Sagard Capital Partners, L.P.
On March 23, 2018, pursuant to a stock purchase agreement, we issued 78,927 shares of our Series A Convertible Participating Preferred Stock, $0.0001 par value per share, to Sagard Capital Partners, L.P. for gross proceeds of $9,199,001, as disclosed on our Form 10-Q filed with the SEC on May 5, 2018.
Sales Pursuant to Share Purchase Agreements
On March 23, 2018, pursuant to share purchase agreements, we issued 420,168 shares of our common stock to certain investors for gross proceeds of approximately $5 million. We used net proceeds from the offering to repay certain aged payables relating to our acquisition of Napo in July 2017, as disclosed on our Form 10-Q filed with the SEC on May 5, 2018.
Letter of Credit and Warrant
To satisfy the letter of credit requirement in the Office Lease Agreement with CA-Mission Street Limited Partnership ("Landlord"), effective as of August 30, 2018, Pacific Capital Management, LLC (the "LC Facilitator"), one of the Company's existing shareholders, caused its financial institution to issue a letter of credit in the amount of $475,000 (the "Letter of Credit") on behalf of the Company in favor of Landlord pursuant to the terms of the Landlord Letter of Credit & Warrant Issuance Agreement, dated August 28, 2018, by and between the Company and the LC Facilitator. In consideration of the LC Facilitator causing a Letter of Credit from LC Facilitator's financial institution
II-5
to be issued to the Landlord, the Company issued to the LC Facilitator a five-year warrant (the "Facilitator Warrant") to purchase 9,579 shares of common stock, subject to adjustment for reclassification or change of the common stock, stock splits, dividends, distributions or changes to the exercise price of the Facilitator Warrant in accordance with the terms of the Facilitator Warrant. The Facilitator Warrant is exercisable commencing after the 7-month anniversary date of issuance, and the exercise price of the Facilitator Warrant is the lower of (i) $59.50 per share and (ii) the average of the closing sales price of the common stock for the 30 consecutive trading days commencing on September 4, 2018.
Charles C. Conte
On September 11, 2018, Charles C. Conte purchased a convertible promissory note in the aggregate principal amount of $111,250 with an exercise price of $59.50 per share of common stock and a warrant exercisable for 484 shares of common stock with an exercise price of $86.10 per share.
L2 Capital, LLC
On September 11, 2018, L2 Capital, LLC purchased a convertible promissory note in the aggregate principal amount of $445,000 with an exercise price of $59.50 per share of common stock and a warrant exercisable for 2,648 shares of common stock with an exercise price of $63.00 per share.
Securities Purchase Agreements
On March 18, 2019, Jaguar began entering into securities purchase agreements (each, a "Securities Purchase Agreement") with selected accredited investors (the "Investors" and each, an "Investor") pursuant to which Jaguar will sell promissory notes ("Notes") to such Investors. As an inducement for entering into the Securities Purchase Agreement, each Investor also received warrants exercisable for shares of Common Stock (the "Investor Warrants"). The initial offering closed on March 18, 2019, and as of June 27, 2019, $5,050,000 aggregate principal amount of Notes were issued in offerings and the proceeds from such offerings were paid to the Company.
CVP
In January through June 2019, Jaguar entered into exchange agreements with Chicago Venture Partners L.P. ("CVP"), pursuant to which the Company issued 1,150,793 shares of Common Stock in the aggregate to CVP in exchange for a reduction of approximately $11.2 million in the outstanding amount of the CVP Notes and Exchange Notes. The shares of Common Stock that were exchanged for portions of the secured promissory notes were issued in reliance on the exemption from registration provided under Section 3(a)(9) of the Securities Act.
The offers, sales, and issuances of the securities described above were deemed to be exempt from registration under the Securities Act in reliance on Section 4(a)(2) of the Securities Act, Regulation D or Regulation S promulgated thereunder as transactions by an issuer not involving a public offering. The recipients of securities in each of these transactions acquired the securities for investment only and not with a view to or for sale in connection with any distribution thereof and appropriate legends were affixed to the securities issued in these transactions. Each of the recipients of securities in these transactions was an accredited or sophisticated person and had adequate access, through employment, business or other relationships, to information about us.
ITEM 16. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES.
- (a)
- Exhibits. The exhibits are incorporated by reference from the Exhibit Index attached hereto.
II-6
The undersigned registrant hereby undertakes to provide to the underwriters at the closing specified in the underwriting agreement certificates in such denominations and registered in such names as required by the underwriters to permit prompt delivery to each purchaser.
The undersigned registrant hereby undertakes that:
(a) For purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective.
(b) For the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer, or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question of whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
II-7
II-8
II-9
II-10
II-11
II-12
II-13
II-14
II-15
II-16
II-17
II-18
- *
- Filed
herewith.
- **
- To
be filed by amendment.
- ***
- Previously
filed.
- †
- Confidential
treatment granted as to portions of the exhibit. Confidential materials omitted and filed separately with the Securities and Exchange
Commission.
- ‡
- Management contract or compensatory plan or arrangement.
II-19
Pursuant to the requirements of the Securities Act of 1933, the registrant has duly caused this Registration Statement to be signed on its behalf by the undersigned, thereunto duly authorized in the City of San Francisco, State of California, on July 5, 2019.
| JAGUAR HEALTH, INC. | ||||||
By: |
/s/ LISA A. CONTE |
|||||
| Name: | Lisa A. Conte | |||||
| Title: | Chief Executive Officer and President | |||||
Pursuant to the requirements of the Securities Act of 1933, this Registration Statement has been signed by the following persons in the capacities and on the date indicated.
Signature
|
Title
|
Date
|
||||
|---|---|---|---|---|---|---|
| /s/ LISA A. CONTE Lisa A. Conte |
Chief Executive Officer, President and Director (Principal Executive Officer) | July 5, 2019 | ||||
/s/ KAREN S. WRIGHT Karen S. Wright |
Chief Financial Officer and Treasurer (Principal Financial and Accounting Officer) |
July 5, 2019 |
||||
* James J. Bochnowski |
Chairman of the Board |
July 5, 2019 |
||||
Jiahao Qiu |
Director |
|||||
* Greg J. Divis |
Director |
July 5, 2019 |
||||
* Jeffery C. Johnson |
Director |
July 5, 2019 |
||||
* Jonathan B. Siegel |
Director |
July 5, 2019 |
||||
II-20
Signature
|
Title
|
Date
|
||||
|---|---|---|---|---|---|---|
| * Murray David MacNaughtan |
Director | July 5, 2019 | ||||
By: |
/s/ LISA A. CONTE Lisa A. Conte, Attorney-in-Fact |
|||||
II-21
QuickLinks -- Click here to rapidly navigate through this document
![]()
Consent of Independent Registered Public Accounting Firm
Jaguar Health, Inc.
San Francisco, California
We hereby consent to the incorporation by reference in the Prospectus constituting a part of this Registration Statement of our report dated April 10, 2019, relating to the consolidated financial statements of Jaguar Health, Inc., which is incorporated by reference in that Prospectus. Our report contains an explanatory paragraph regarding the Company's ability to continue as a going concern.
We also consent to the reference to us under the caption "Experts" in the Prospectus.
![]()
San Jose, California
July 5, 2019
Consent of Independent Registered Public Accounting Firm
Serious News for Serious Traders! Try StreetInsider.com Premium Free!
You May Also Be Interested In
- Jaguar Health (JAGX) Appoints Miller Collis as SVP of Growth Strategy
- BuyBackTech Enabling Consumers to Cash In Old, Unused, and Broken Phones and Tablets
- Neurocrine Biosciences (NBIX) and Sentia Medical Sciences Extend Research Collaboration to Discover Novel CRF Peptides
Create E-mail Alert Related Categories
SEC FilingsRelated Entities
S1Sign up for StreetInsider Free!
Receive full access to all new and archived articles, unlimited portfolio tracking, e-mail alerts, custom newswires and RSS feeds - and more!
