

Form 8-K INTERCEPT PHARMACEUTICAL For: Jul 07
 Tweet
Tweet Share
ShareExhibit 99.1

Intercept Announces Positive Data in Fibrosis due to NASH from a New Analysis of its Phase 3 REGENERATE Study of Obeticholic Acid (OCA)
OCA 25 mg met the agreed primary endpoint of improvement in liver fibrosis without worsening of NASH at 18 months (p<0.0001), consistent with the original REGENERATE analysis
OCA 25 mg demonstrated double the response rate in reduction of liver fibrosis without worsening of NASH vs. placebo
Includes larger and more robust safety database of 2,477 patients
with nearly 1,000 on study drug for 4 years
Intercept to resubmit new drug application (NDA) in liver fibrosis due to NASH; pre-submission meeting with FDA scheduled later this month
Company to host conference call at 8:30 a.m. ET today
MORRISTOWN, NJ, July 7, 2022 -- Intercept Pharmaceuticals, Inc. (Nasdaq: ICPT), a biopharmaceutical company focused on the development and commercialization of novel therapeutics to treat progressive non-viral liver diseases, today announced positive topline results from a new interim analysis of its ongoing pivotal Phase 3 REGENERATE trial of OCA in patients with liver fibrosis due to nonalcoholic steatohepatitis (NASH). This is the second analysis in which OCA has met the primary endpoint for the intent-to-treat (ITT) population in REGENERATE and based on these results, Intercept will be re-submitting its NDA for OCA in liver fibrosis due to NASH.
In this new interim analysis of the ITT population from REGENERATE (n=931), 22.4% of subjects randomized to once-daily oral OCA 25 mg met the primary endpoint of achieving at least one stage of fibrosis improvement with no worsening of NASH at month 18 on liver biopsy compared with 9.6% of subjects on placebo (p<0.0001). These results are consistent with the original positive analysis announced in February 2019, which also showed that OCA 25 mg had a statistically significant effect on fibrosis improvement (p=0.0002). The population in this new interim analysis mirrored the original analysis population from 2019 but used a consensus panel approach to histology reads, in line with recent U.S. Food and Drug Administration (FDA) guidance, while the original analysis relied on results from individual central readers. A numerically greater proportion of individuals in the OCA 25 mg treatment group compared to placebo achieved the endpoint of resolution of NASH with no worsening of liver fibrosis but, consistent with the original analysis, this did not reach statistical significance. For the primary objective to be met, the study was required to achieve only one of the two primary endpoints.
Safety was evaluated in 2,477 subjects who took at least one dose of study drug (placebo, OCA 10 mg, or OCA 25 mg). Compared to the original analysis, the safety population in this new interim analysis had significantly longer exposure to study drug (median 42 months vs. 15 months), yielding more than 8,000 total patient-years and 3.4 times more exposure. Nearly 1,000 subjects had been on study drug for four years.
Treatment-emergent adverse events (TEAEs), treatment-emergent serious adverse events (TESAEs), and deaths were generally balanced across the OCA and placebo treatment groups. The most common TEAE was pruritus (24% in placebo, 33% in OCA 10 mg and 55% in OCA 25 mg) and pruritus was the most common cause for treatment discontinuation. Serious gallbladder-related events occurred in <3% of subjects in any treatment group and, consistent with its known mechanism of action, OCA 25 mg had higher rates of biliary events including gallstones.
Independent groups of experts reviewed certain categories of safety events to provide a blinded adjudication as specifically requested by FDA. These included events pertaining to hepatic safety (excluding clinical outcomes), cardiovascular and renal. Topline analysis through four years of treatment showed a numerically higher number of adjudicated hepatic safety events for OCA 25 mg, the vast majority of which were mild in severity. For adjudicated core major adverse cardiovascular events and adjudicated acute kidney injury events, frequency of events was low and balanced across treatment groups.
Consistent with its mechanism of action as an FXR agonist, OCA treatment was associated with an increase in LDL at Month 1 which returned to near baseline values by Month 12. Changes in other blood chemistries including ALP (alkaline phosphatase), GGT (gamma-glutamyl transferase), AST (aspartate aminotransferase), and ALT (alanine aminotransferase) also showed evidence of FXR mediated activity, supporting the dose-response of OCA.
“Achieving a statistically significant histology endpoint has proven to be an extremely high bar in clinical trials for NASH. We are thrilled that OCA has demonstrated consistent improvement in fibrosis based on a second methodology and we now have two positive, statistically significant results for this primary endpoint from our pivotal REGENERATE trial. These results demonstrate the antifibrotic effect of OCA and are consistent with our belief that OCA can be an important treatment for people living with fibrosis due to NASH,” said Jerry Durso, President and Chief Executive Officer of Intercept. “The weight of evidence in both safety and efficacy has notably increased and provides a more robust benefit:risk profile of OCA. We look forward to our meeting with FDA later this month to discuss the resubmission of our NDA for OCA in fibrosis due to NASH.”
Dr. Arun Sanyal, Chair, Division of Gastroenterology/Hepatology/Nutrition & Director, Stravitz-Sanyal Liver Institute, VCU Health, Department of Internal Medicine, said, “Today marks a significant milestone for patients with fibrosis due to NASH, a disease that currently has no approved therapy. These data demonstrate that once daily oral OCA has the potential to improve fibrosis. Further, the safety and tolerability profile of OCA based on a sizeable dataset should give clinicians confidence that we are a step closer to a treatment option.”
Analysis Methodology and Efficacy Results
In REGENERATE, patients with biopsy-proven stage 2 or 3 liver fibrosis due to NASH were randomized 1:1:1 to receive placebo (n=311), OCA 10 mg (n=312), or OCA 25 mg (n=308) once daily. A second biopsy was conducted in these 931 subjects at Month 18 for the planned interim analysis.
The new interim analysis was based on a reassessment of the baseline and Month 18 liver biopsies using a consensus reading methodology. Consensus panels were comprised of three board-certified pathologists who demonstrated accuracy and reproducibility in their assessments of liver biopsies during proficiency testing. The consensus panels in this new interim analysis reviewed digitized whole slide images of the same glass slides of liver biopsy tissue that were evaluated in the original analysis using individual central readers.
The results of the new interim analysis from REGENERATE are shown in the following table:
|
Placebo n=311 |
OCA 10 mg n=312 |
OCA 25 mg n=308 | |||||
| At least one stage of fibrosis improvement with no worsening of NASH* | 9.6% |
14.1% p=NS |
22.4% p<0.0001 |
||||
| Resolution of NASH‡ with no worsening of liver fibrosis |
3.5%
|
6.1% p=NS |
6.5% p=NS |
||||
|
*Defined as no worsening of hepatocellular ballooning, no worsening of lobular inflammation and no worsening of steatosis
‡Defined as the overall histopathologic interpretation of (i) no fatty liver disease or (ii) fatty liver disease (simple or isolated steatosis) without steatohepatitis AND a nonalcoholic fatty liver disease (NAFLD) activity score of 0 for ballooning and 0-1 for inflammation | |||||||
Intercept plans to share these data in an upcoming scientific forum.
OCA has not been approved for the treatment of NASH by any regulatory authority in any geography and is considered an investigational treatment for this indication.
About the REGENERATE Study
REGENERATE (Randomized Global Phase 3 Study to Evaluate the Impact on NASH with Fibrosis of Obeticholic Acid Treatment) is an ongoing Phase 3, randomized, double-blind, placebo-controlled, multicenter, international study assessing the safety and efficacy of obeticholic acid (OCA) on clinical outcomes in patients with liver fibrosis due to NASH. A pre-specified 18-month interim analysis was conducted on 931 subjects who had scheduled biopsy at Month 18 to assess the effect of OCA on liver histology comparing Month 18 biopsies with baseline biopsies. REGENERATE is fully enrolled with 2,480 randomized participants and is expected to continue through clinical outcomes for verification and description of clinical benefit. The end-of-study analysis will evaluate the effect of OCA on all-cause mortality and liver-related clinical outcomes, as well as long-term safety.
About Intercept
Intercept is a biopharmaceutical company focused on the development and commercialization of novel therapeutics to treat progressive non-viral liver diseases, including primary biliary cholangitis (PBC) and nonalcoholic steatohepatitis (NASH). For more information, please visit www.interceptpharma.com or connect with the company on Twitter and LinkedIn.
About Liver Fibrosis due to NASH
Nonalcoholic steatohepatitis (NASH) is a serious progressive liver disease caused by excessive fat accumulation in the liver that induces chronic inflammation, resulting in progressive fibrosis (scarring) that can lead to cirrhosis, eventual liver failure, cancer and death. Advanced fibrosis is associated with a substantially higher risk of liver-related morbidity and mortality in patients with NASH. There are currently no medications approved for the treatment of NASH.
Conference Call on July 7, 2022 at 8:30 a.m. ET
Intercept will hold a conference call to discuss the new data analysis from its Phase 3 REGENERATE study in patients with liver fibrosis due to NASH today at 8:30 a.m. ET. The conference call will be available via a listen-only webcast on the investor page of our website at http://ir.interceptpharma.com. To join the webcast, click here. Participants who wish to ask a question may register here to receive dial-in numbers and a unique PIN to join the call. While not required, it is recommended you join 10 minutes prior to the event start. A replay of the call will be available on our website shortly following the completion of the call and will be available for one year.
Cautionary Note Regarding Forward-Looking Statements
This press release contains forward-looking statements, including, but not limited to, statements regarding:
| · | The progress, timing, and results of our REGENERATE clinical trial, including the safety and efficacy of obeticholic acid (“OCA”) for the treatment of liver fibrosis due to nonalcoholic steatohepatitis (“NASH”), and the use of a consensus panel biopsy-reading methodology. |
| · | Our planned New Drug Application (“NDA”) pre-submission meeting with the FDA regarding OCA for the treatment of liver fibrosis due to NASH. |
| · | Our planned resubmission of an NDA to the FDA for OCA for the treatment of liver fibrosis due to NASH, and the potential timing, review, acceptance, and approval of the NDA. |
| · | The potential commercial success of OCA for NASH, as well as our strategy, future operations, future financial position, future revenue, projected costs, prospects, plans, and objectives. |
These statements constitute forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. The words “anticipate,” “believe,” “estimate,” “expect,” “intend,” “may,” “plan,” “predict,” “project,” “target,” “potential,” “will,” “would,” “could,” “should,” “possible,” “continue” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. Readers are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date of this release, and we undertake no obligation to update any forward-looking statement except as required by law. These forward-looking statements are based on estimates and assumptions by our management that, although believed to be reasonable, are inherently uncertain and subject to a number of risks. The following represent some, but not necessarily all, of the factors that could cause actual results to differ materially from historical results or those anticipated or predicted by our forward-looking statements:
| · | The success of our existing business and operations, including Ocaliva for PBC. |
| · | Our ability to successfully commercialize Ocaliva for PBC and, if approved, OCA for NASH. |
| · | Our ability to maintain our regulatory approval of Ocaliva for PBC. |
| · | Our ability to timely and cost-effectively file for and obtain regulatory approval of our product candidates on an accelerated basis or at all, including OCA for NASH. |
| · | Our ability to address the issues raised in the complete response letter (“CRL”) received in June 2020 with respect to OCA for NASH. |
| · | Any advisory committee recommendation or dispute resolution determination that OCA for NASH should not be approved or approved only under certain conditions. |
| · | Any future determination that the regulatory applications and subsequent information we submit for our product candidates, including OCA for NASH, do not contain adequate clinical or other data or meet applicable regulatory requirements for approval. |
| · | Conditions that may be imposed by regulatory authorities on our marketing approvals for our products and product candidates, including OCA for NASH, such as the need for clinical outcomes data (and not just results based on achievement of a surrogate endpoint), any risk mitigation programs such as a REMS, and any related restrictions, limitations and/or warnings contained in the label of any of our products or product candidates. |
| · | Any potential side effects associated with Ocaliva for PBC, OCA for NASH, or our other product candidates that could delay or prevent approval, require that an approved product be taken off the market, require the inclusion of safety warnings or precautions, or otherwise limit the sale of such product or product candidate, including in connection with our update to Ocaliva prescribing information in May 2021 contraindicating Ocaliva for patients with PBC and decompensated cirrhosis, a prior decompensation event, or compensated cirrhosis with evidence of portal hypertension. |
| · | The initiation, timing, cost, conduct, progress, and results of our research and development activities, preclinical studies and clinical trials, including any issues, delays or failures in identifying patients, enrolling patients, treating patients, retaining patients, meeting specific endpoints, or completing and timely reporting the results of our NASH or PBC clinical trials. |
| · | The outcomes of interactions with regulators, including the FDA, regarding our clinical trials. |
| · | Our ability to establish and maintain relationships with, and the performance of, third-party manufacturers, contract research organizations and other vendors upon whom we are substantially dependent for, among other things, the manufacture and supply of our products, including Ocaliva for PBC and, if approved, OCA for NASH, and our clinical trial activities. |
| · | Our ability to identify, develop, and successfully commercialize our products and product candidates, including our ability to successfully launch OCA for NASH, if approved. |
| · | Our ability to obtain and maintain intellectual property protection for our products and product candidates, including our ability to cost-effectively file, prosecute, defend, and enforce any patent claims or other intellectual property rights. |
| · | The size and growth of the markets for our products and product candidates and our ability to serve those markets. |
| · | The degree of market acceptance of Ocaliva for PBC and, if approved, OCA for NASH or our other product candidates among physicians, patients, and healthcare payors. |
| · | The availability of adequate coverage and reimbursement from governmental and private healthcare payors for our products, including Ocaliva for PBC and, if approved, OCA for NASH, and our ability to obtain adequate pricing for such products. |
| · | Our ability to establish and maintain effective sales, marketing, and distribution capabilities, either directly or through collaborations with third parties. |
| · | Competition from existing drugs or new drugs that become available. |
| · | Our ability to attract and retain key personnel to manage our business effectively. |
| · | Our ability to prevent or defend against system failures or security or data breaches due to cyber-attacks, or cyber intrusions, including ransomware, phishing attacks and other malicious intrusions. |
| · | Our ability to comply with data protection laws; costs and outcomes relating to any disputes, governmental inquiries or investigations, regulatory proceedings, legal proceedings or litigation, including any securities, intellectual property, employment, product liability or other litigation. |
| · | Our collaborators’ election to pursue research, development and commercialization activities. |
| · | Our ability to establish and maintain relationships with collaborators with development, regulatory and commercialization expertise. |
| · | Our need for and ability to generate or obtain additional financing. |
| · | Our estimates regarding future expenses, revenues and capital requirements and the accuracy thereof. |
| · | Our use of cash, cash equivalents and short-term investments. |
| · | Our ability to acquire, license and invest in businesses, technologies, product candidates and products. |
| · | Our ability to manage the growth of our operations, infrastructure, personnel, systems and controls. |
| · | Our ability to obtain and maintain adequate insurance coverage. |
| · | Continuing threats from COVID-19, including additional waves of infections, and their impacts including quarantines and other government actions, delays relating to our regulatory applications, disruptions relating to our ongoing clinical trials or involving our contract research organizations, study sites or other clinical partners, disruptions relating to our supply chain or involving our third-party manufacturers, distributors or other distribution partners, and facility closures or other restrictions, and impact of the foregoing on our results of operations and financial position. |
| · | The impact of general economic, industry, market, regulatory, or political conditions. |
| · | The other risks and uncertainties identified in our periodic filings filed with the U.S. Securities and Exchange Commission, including our latest Annual Report on Form 10-K and/or Quarterly Report on Form 10-Q. |
CONTACT
For more information about Intercept, please contact:
For investors:
Nareg Sagherian, Executive Director, Global Investor Relations
For media:
Karen Preble, Executive Director, Global Corporate Communications
Exhibit 99.2

REGENERATE Topline Data 07 July 2022 Click to add text Click to add text

Cautionary Note Regarding Forward - Looking Statements This presentation contains forward - looking statements, including, but not limited to, statements regarding: The progress, timing , and results of our REGENERATE clinical trial, including the safety and efficacy of obeticholic acid (“OCA”) for the treatment of liver fibrosis due to nonalcoholic steatohepatitis (“NASH”), and the use of a consensus pan el biopsy - reading methodology; our planned New Drug Application (“NDA”) pre - submission meeting with the FDA regarding OCA for the treatment of liver fibrosis due to NASH; our planned resubmission of an ND A to the FDA for OCA for the treatment of liver fibrosis due to NASH, and the potential timing, review, acceptance, and approval of the NDA; and the potential commercial success of OCA for NASH, as well as our str ate gy, future operations, future financial position, future revenue, projected costs, prospects, plans, and objectives. These statements constitute forward - looking statements within the meaning of Section 27A of the Securities Act of 1933, as amend ed, and Section 21E of the Securities Exchange Act of 1934, as amended. The words “anticipate,” “believe,” “estimate,” “expect,” “intend,” “may,” “plan,” “predict,” “project,” “target,” “potential,” “will,” “wo uld,” “could,” “should,” “possible,” “continue” and similar expressions are intended to identify forward - looking statements, although not all forward - looking statements contain these identifying words. Readers are cautioned not to pl ace undue reliance on these forward - looking statements, which speak only as of the date of this presentation, and we undertake no obligation to update any forward - looking statement except as required by law. These forwa rd - looking statements are based on estimates and assumptions by our management that, although believed to be reasonable, are inherently uncertain and subject to a number of risks. The following represent some, but not necessarily all, of the factors that could cause actual results to differ materially from historical results or those anticipated or predicted by our forward - looking statements: The success of our existing business and operations, including Ocaliva for PBC; our ability to successfully commercialize Ocaliva for PBC and, if approved, OCA for NASH; our ability to maintain our regulatory approval of Ocaliva for PBC; our ability to timely and cost - effectively file for and obtain regulatory approval of our product candidates on an accelerated basis or at all, including OCA for NASH; our ability to address the issues raised in the complete response letter (“ CRL”) received in June 2020 with respect to OCA for NASH; any advisory committee recommendation or dispute resolution determination that OCA for NASH should not be approved or approved only under certain co ndi tions; any future determination that the regulatory applications and subsequent information we submit for our product candidates, including OCA for NASH, do not contain adequate clinical or other data or m eet applicable regulatory requirements for approval; conditions that may be imposed by regulatory authorities on our marketing approvals for our products and product candidates, including OCA for NASH, such as th e n eed for clinical outcomes data (and not just results based on achievement of a surrogate endpoint), any risk mitigation programs such as a REMS, and any related restrictions, limitations and/or warnings contained i n t he label of any of our products or product candidates; any potential side effects associated with Ocaliva for PBC, OCA for NASH, or our other product candidates that could delay or prevent approval, require that an approved product b e taken off the market, require the inclusion of safety warnings or precautions, or otherwise limit the sale of such product or product candidate, including in connection with our update to Ocaliva prescribing information in May 2021 contraindicating Ocaliva for patients with PBC and decompensated cirrhosis, a prior decompensation event, or compensated cirrhosis with evidence of portal hypertension; the ini tia tion, timing, cost, conduct, progress, and results of our research and development activities, preclinical studies and clinical trials, including any issues, delays or failures in identifying patients, enroll ing patients, treating patients, retaining patients, meeting specific endpoints, or completing and timely reporting the results of our NASH or PBC clinical trials; the outcomes of interactions with regulators, including the FDA, re gar ding our clinical trials; our ability to establish and maintain relationships with, and the performance of, third - party manufacturers, contract research organizations and other vendors upon whom we are substantially depe ndent for, among other things, the manufacture and supply of our products, including Ocaliva for PBC and, if approved, OCA for NASH, and our clinical trial activities; our ability to identify, develop, and successfully c ommercialize our products and product candidates, including our ability to successfully launch OCA for NASH, if approved; our ability to obtain and maintain intellectual property protection for our products and pr odu ct candidates, including our ability to cost - effectively file, prosecute, defend, and enforce any patent claims or other intellectual property rights; the size and growth of the markets for our products and product candidat es and our ability to serve those markets; the degree of market acceptance of Ocaliva for PBC and, if approved, OCA for NASH or our other product candidates among physicians, patients, and healthcare payors; the availab ili ty of adequate coverage and reimbursement from governmental and private healthcare payors for our products, including Ocaliva for PBC and, if approved, OCA for NASH, and our ability to obtain adequate pricing for such products; our ability to establis h and maintain effective sales, marketing, and distribution capabilities, either directly or through collaborations with third parties; competition from existing drugs or n ew drugs that become available; our ability to attract and retain key personnel to manage our business effectively; our ability to prevent or defend against system failures or security or data breaches due to cyber - attacks, or cybe r intrusions, including ransomware, phishing attacks and other malicious intrusions; our ability to comply with data protection laws; costs and outcomes relating to any disputes, governmental inquiries or investigations, regu lat ory proceedings, legal proceedings or litigation, including any securities, intellectual property, employment, product liability or other litigation; our collaborators’ election to pursue research, development and com mercialization activities; our ability to establish and maintain relationships with collaborators with development, regulatory and commercialization expertise; our need for and ability to generate or obtain additional finan cin g; our estimates regarding future expenses, revenues and capital requirements and the accuracy thereof; our use of cash, cash equivalents and short - term investments; our ability to acquire, license and invest in bu sinesses, technologies, product candidates and products; our ability to manage the growth of our operations, infrastructure, personnel, systems and controls; our ability to obtain and maintain adequate insurance covera ge; continuing threats from COVID - 19, including additional waves of infections, and their impacts including quarantines and other government actions, delays relating to our regulatory applications, disruptions relat ing to our ongoing clinical trials or involving our contract research organizations, study sites or other clinical partners, disruptions relating to our supply chain or involving our third - party manufacturers, distributors or ot her distribution partners, and facility closures or other restrictions, and impact of the foregoing on our results of operations and financial position; the impact of general economic, industry, market, regulatory, or political con ditions; and the other risks and uncertainties identified in our periodic filings filed with the U.S. Securities and Exchange Commission, including our latest Annual Report on Form 10 - K and/or Quarterly Report on Form 10 - Q. 2

Topline Results Overview • The planned Interim Analysis of the REGENERATE F2 and F3 month 18 (M18) liver biopsies was conducted under a Consensus Methodology. • The same population of subjects, and the same biopsies that were read in the Original Analysis have been re - read under this new methodology, in line with the FDA's guidance for “consensus” histology reads. • The new Consensus Methodology results have shown that OCA 25 mg has again demonstrated a statistically significant and clinically meaningful doubling of the antifibrotic effect versus placebo responders. • Since the prior analysis and submission, a larger population with longer duration on study has created a more robust safety database. • Treatment emergent adverse events (TEAEs), treatment emergent serious adverse events (TESAEs), and deaths were generally balanced across the OCA and placebo treatment groups. • The most common TEAE was pruritus (24% in placebo, 33% in OCA 10 mg, and 55% in OCA 25 mg) which was also the most common cause for treatment discontinuation. • Independent groups of experts have reviewed certain categories of safety events to provide a blinded adjudication as specifically requested by FDA during the prior review. • Overall safety and tolerability results enable a robust assessment for chronic dosing of OCA. 3

Demographics and Baseline Disease Characteristics (N=2477) ▪ Characteristics were balanced acros s treatment groups ▪ Mean Age ~55 years ▪ ~60% female, 90% white ▪ Mean BMI ~34 kg/m 2 ▪ ~60 % with diabetes at BL ▪ 1 in 4 patients have mild renal insufficiency ▪ Biomarkers consistent with advanced disease ▪ Histology showed 53% F3, 36% F2 4

OCA 25 mg Antifibrotic Effect at M18 is Double the Effect of Placebo Statistical Significance and Clinical Relevance Now Demonstrated Twice EFFICACY: At least one stage improvement in fibrosis without worsening of NASH • Results from Consensus Methodology were consistent with the Original Analysis conducted by central pathologists and confirmed the antifibrotic treatment effect of OCA 25 mg at M18 – The consensus panel analysis showed a doubling of the proportion of responders versus the placebo group: pbo 9.6% vs OCA 25 mg 22.4% , p<0.0001 (2019: pbo 11.9% vs OCA 25 mg 23.1%, p=0.0002) – The antifibrotic treatment effect of OCA 10 mg 14.1% (NS) observed at M18 was consistent with the 2019 analysis ( 17.6%, NS ) • A notable dose - response was observed for fibrosis improvement in both planned interim analyses with two different reading methodologies EFFICACY: Resolution of NASH with no worsening of fibrosis • NASH Resolution with no worsening of fibrosis showed a numerically higher response in both OCA treatment groups compared with placebo, but the proportion of responders for OCA 10 mg and OCA 25 mg did not reach statistical significance
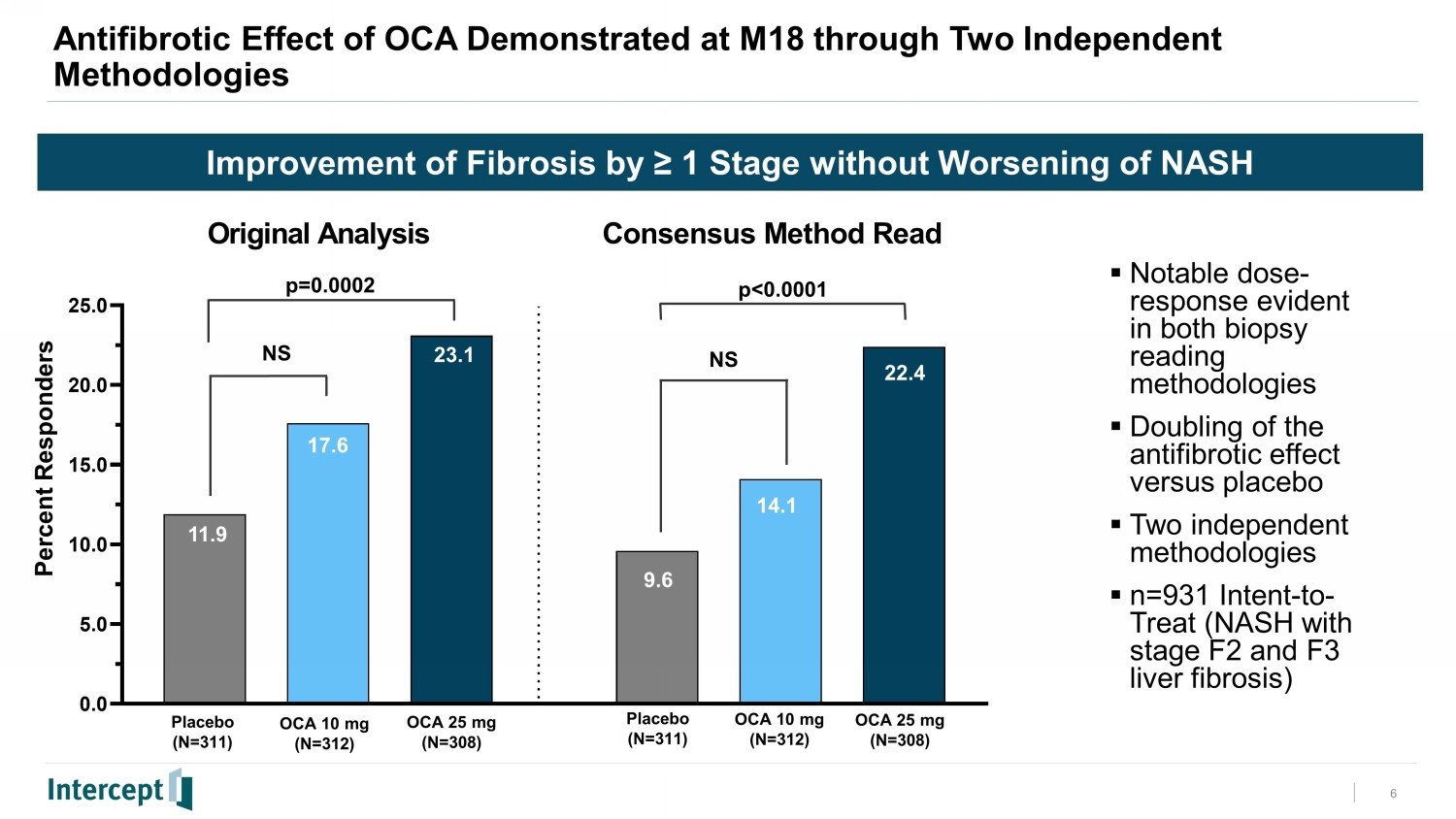
6 Improvement of Fibrosis by ≥ 1 Stage without Worsening of NASH 0.0 5.0 10.0 15.0 20.0 25.0 Original Analysis Consensus Method Read 11.9 23.1 17.6 22.4 14.1 22.4 Percent Responders Antifibrotic Effect of OCA Demonstrated at M18 through Two Independent Methodologies p=0.0002 p<0.0001 Placebo (N=311) OCA 10 mg (N=312) OCA 25 mg (N=308) Placebo (N=311) OCA 10 mg (N=312) OCA 25 mg (N=308) 9.6 ▪ Notable dose - response evident in both biopsy reading methodologies ▪ Doubling of the antifibrotic effect versus placebo ▪ Two independent methodologies ▪ n=931 Intent - to - Treat (NASH with stage F2 and F3 liver fibrosis) NS NS

7 Resolution of NASH with No Worsening of Fibrosis 0 5 10 15 20 25 11.7 6.5 11.2 6.1 8.0 3.5 Original Analysis Consensus Method Read 11.7% 11.2 3.5% 6.5% NS NS Percent Responders Resolution of NASH for OCA 10 mg and OCA 25 mg at M18 Shows Consistent Effect Compared to Placebo Cohort 6.1 6.5 Placebo (N=311) OCA 10 mg (N=312) OCA 25 mg (N=308) Placebo (N=311) OCA 10 mg (N=312) OCA 25 mg (N=308) ▪ Consistent trend for both doses of OCA shows numerically more responders than placebo ▪ Two independent methodologies ▪ n=931 Intent - to - Treat (NASH with stage F2 and F3 liver fibrosis)
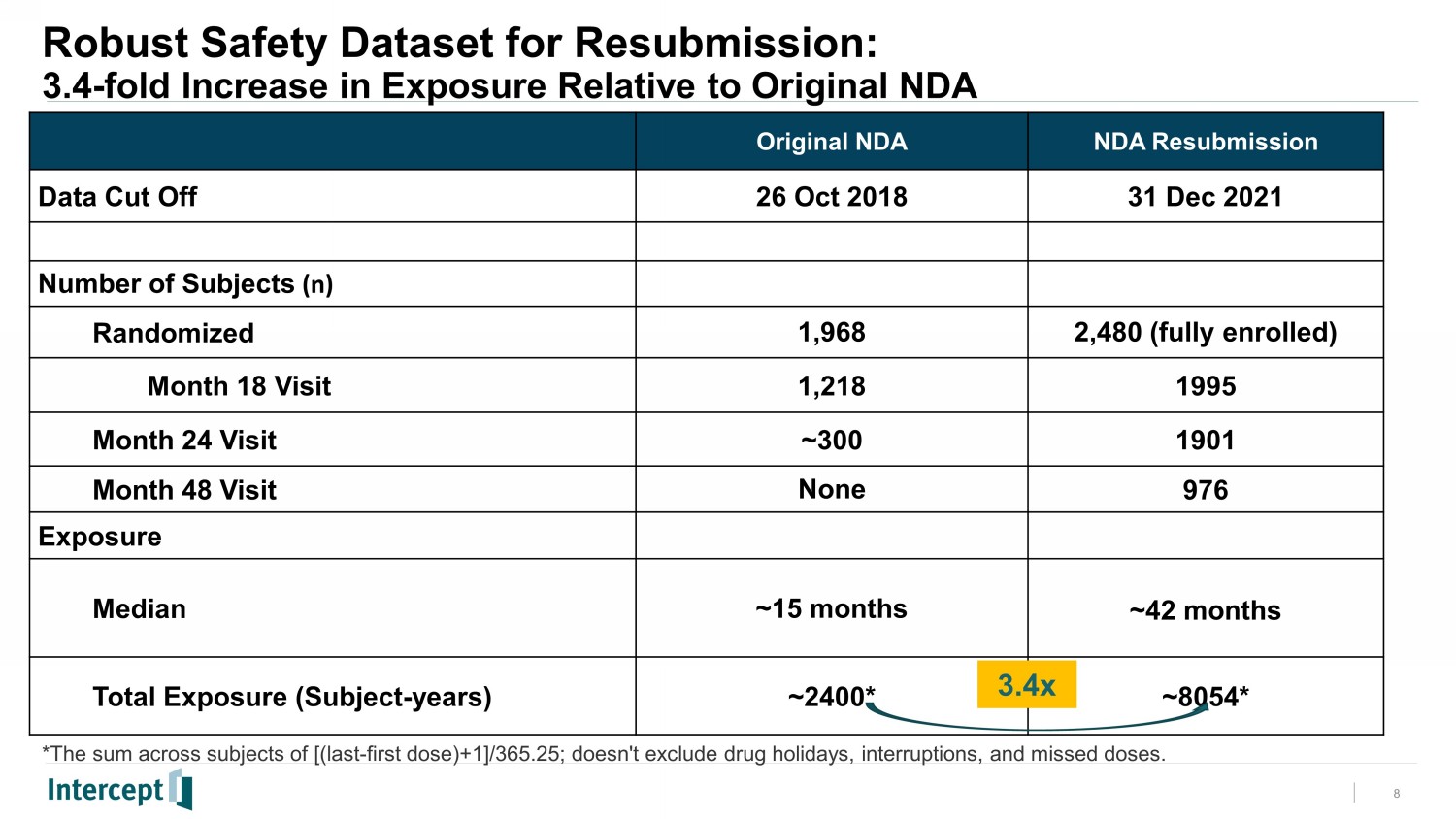
Robust Safety Dataset for Resubmission: 3.4 - fold Increase in Exposure Relative to Original NDA 8 Original NDA NDA Resubmission Data Cut Off 26 Oct 2018 31 Dec 2021 Number of Subjects (n) Randomized 1,968 2,480 (fully enrolled) Month 18 Visit 1,218 1995 Month 24 Visit ~300 1901 Month 48 Visit None 976 Exposure Median ~15 months ~42 months Total Exposure (Subject - years) ~2400* ~8054* *The sum across subjects of [(last - first dose)+1]/365.25; doesn't exclude drug holidays, interruptions, and missed doses. 3.4x

Summary of Safety & Tolerability • Treatment emergent adverse events (TEAEs), treatment emergent serious adverse events (TESAEs) and deaths were generally balanced across the OCA and placebo treatment groups. • The most common TEAE was pruritus (24% in placebo, 33% in OCA 10 mg and 55% in OCA 25 mg) which was also the most common cause for treatment discontinuation. • Serious gallbladder related events occurred in <3% of subjects; consistent with the known MOA, OCA 25 mg had higher rates of biliary events including gallstones. • Adjudicated hepatic safety events were numerically higher for OCA 25 mg and generally mild in severity. • Frequency for adjudicated Core MACE (Major Adverse Cardiovascular Events [cardiovascular death, non - fatal MI and non - fatal stroke]) was low and balanced among treatment groups. Consistent with its MOA, OCA treatment was associated with an increase in LDL at Month 1 and returned to near baseline values by Month 12. • Frequency of adjudicated acute kidney injury events was low (<1%) and balanced among the treatment groups. • Overall safety and tolerability results enable a robust assessment for chronic dosing of OCA. 9
Serious News for Serious Traders! Try StreetInsider.com Premium Free!
You May Also Be Interested In
- New Spirits and Seltzers Brand, Pink Sand Spirits, Co., Announces a Groundbreaking Partnership with the Global Food Distributor, Sysco, in The Bahamas
- 5 Things to Know About First-Ever Starbucks Promises Day
- TuminEwap to Collaborate with Japanese Financial Giant, Accelerating Expansion in Asian Markets
Create E-mail Alert Related Categories
SEC FilingsSign up for StreetInsider Free!
Receive full access to all new and archived articles, unlimited portfolio tracking, e-mail alerts, custom newswires and RSS feeds - and more!
