

Form 10-Q REATA PHARMACEUTICALS For: Jun 30
 Tweet
Tweet Share
Share
1
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM
(Mark One)
For the quarterly period ended
OR
For the transition period from to
Commission File Number:
(Exact Name of Registrant as Specified in its Charter)
|
||
(State or other jurisdiction of incorporation or organization) |
|
(I.R.S. Employer |
|
|
|
|
||
(Address of principal executive offices) |
|
(Zip Code) |
(
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Securities Exchange Act of 1934:
Title of each class |
|
Trading Symbol(s) |
|
Name of each exchange on which registered |
|
|
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, an emerging growth company, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
|
☒ |
|
Accelerated filer |
|
☐ |
|
Non-accelerated filer |
|
☐ |
|
Smaller reporting company |
|
|
Emerging growth company |
|
|
|
|
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No
As of August 2, 2022, the registrant had
TABLE OF CONTENTS
- |
|
|
|
|
Page |
1 |
||
3 |
||
PART I. |
|
|
Item 1. |
4 |
|
|
4 |
|
|
5 |
|
|
6 |
|
|
7 |
|
|
8 |
|
Item 2. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
17 |
Item 3. |
44 |
|
Item 4. |
44 |
|
PART II. |
|
|
Item 1. |
45 |
|
Item 1A. |
45 |
|
Item 2. |
45 |
|
Item 3. |
45 |
|
Item 4. |
45 |
|
Item 5. |
45 |
|
Item 6. |
46 |
|
47 |
||
i
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Quarterly Report on Form 10-Q contains forward-looking statements that involve substantial risks and uncertainties. We make such forward-looking statements pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995 and other federal securities laws. In this Quarterly Report on Form 10-Q, all statements, other than statements of historical or present facts, including statements regarding our future financial condition, future revenues, projected costs, prospects, business strategy, and plans and objectives of management for future operations, are forward-looking statements. In some cases, you can identify forward-looking statements by terminology such as “believe,” “will,” “may,” “might,” “estimate,” “continue,” “anticipate,” “intend,” “target,” “project,” “model,” “should,” “would,” “plan,” “expect,” “predict,” “could,” “seek,” “goals,” “potential,” and similar terms or expressions that concern our expectations, strategy, plans, or intentions. These forward-looking statements include, but are not limited to, statements about:
1
Any forward-looking statements in this Quarterly Report on Form 10-Q reflect our current views with respect to future events or to our future financial performance and involve known and unknown risks, uncertainties, and other factors that may cause our actual results, performance, or achievements to be materially different from any future results, performance, or achievements expressed or implied by these forward-looking statements. Given these uncertainties, you should not place undue reliance on these forward-looking statements.
You should read this Quarterly Report on Form 10-Q and the documents that we have filed as exhibits to this Quarterly Report on Form 10-Q completely and with the understanding that our actual future results may be materially different from what we expect. Except as required by law, we assume no obligation to update or revise these forward-looking statements for any reason, even if new information becomes available in the future.
2
DEFINED TERMS
Unless the context requires otherwise, references to “Reata,” “the Company,” “we,” “us,” or “our” in this Quarterly Report on Form 10-Q refer to Reata Pharmaceuticals, Inc. and its subsidiaries. We also have used several other terms in this Quarterly Report on Form 10-Q, most of which are explained or defined below.
Abbreviated Term |
|
Defined Term |
AbbVie |
|
AbbVie Inc. |
ADPKD |
|
Autosomal dominant polycystic kidney disease |
ADL |
|
Activities of Daily Living |
AE |
|
Adverse event |
ALS |
|
Amyotrophic lateral sclerosis |
ATP |
|
Adenosine triphosphate |
bardoxolone |
|
Bardoxolone methyl |
BXLS |
|
Blackstone Life Sciences, LLC |
CKD |
|
Chronic kidney disease |
CMC |
|
Chemistry manufacturing controls |
COVID-19 |
|
Coronavirus disease |
CRL |
|
Complete Response Letter |
CRO |
|
Contract research organization |
DPNP |
|
Diabetic peripheral neuropathic pain |
eGFR |
|
Estimated glomerular filtration rate |
EMA |
|
European Medicines Agency |
ESKD |
|
End stage kidney disease |
Exchange Act |
|
Securities Exchange Act of 1934 |
FA-COMS |
|
Clinical Outcome Measures in Friedreich’s ataxia |
FDA |
|
United States Food and Drug Administration |
FXN |
|
Frataxin |
GFR |
|
Glomerular filtration rate |
GGT |
|
Gamma-glutamyl transferase |
Kyowa Kirin |
|
Kyowa Kirin Co., Ltd. |
LTIP Plan |
|
Second Amended and Restated Long Term Incentive Plan |
MAA |
|
Marketing Authorization Application |
mFARS |
|
Modified Friedreich’s Ataxia Rating Scale |
MMRM |
|
Mixed Model Repeated Measures |
NDA |
|
New Drug Application |
PGIC |
|
Patient global impression of change |
PK |
|
Pharmacokinetic |
registrational trial |
|
An adequate and well-controlled trial designed to be sufficient to apply for regulatory approval of a drug candidate, although notwithstanding the Company’s design a regulatory agency may determine that further clinical studies or data are required |
RSU |
|
Restricted Stock Unit |
SAE |
|
Serious adverse event |
SEC |
|
U.S. Securities and Exchange Commission |
U.S. GAAP |
|
Accounting principles generally accepted in the United States |
3
PART I - FINANCIAL INFORMATION
Item 1. Financial Statements.
Reata Pharmaceuticals, Inc.
Consolidated Balance Sheets
(in thousands, except share data)
|
|
June 30, 2022 |
|
|
December 31, 2021 |
|
||
|
|
(unaudited) |
|
|
|
|
||
Assets |
|
|
|
|
|
|
||
Cash and cash equivalents |
|
$ |
|
|
$ |
|
||
Marketable debt securities |
|
|
|
|
|
|
||
Prepaid expenses and other current assets |
|
|
|
|
|
|
||
Total current assets |
|
|
|
|
|
|
||
Property and equipment, net |
|
|
|
|
|
|
||
Operating lease right-of-use-assets |
|
|
|
|
|
|
||
Other assets |
|
|
|
|
|
|
||
Total assets |
|
$ |
|
|
$ |
|
||
Liabilities and stockholders’ equity |
|
|
|
|
|
|
||
Accounts payable |
|
$ |
|
|
$ |
|
||
Accrued direct research liabilities |
|
|
|
|
|
|
||
Other current liabilities |
|
|
|
|
|
|
||
Operating lease liabilities, current |
|
|
|
|
|
|
||
Deferred revenue |
|
|
|
|
|
|
||
Total current liabilities |
|
|
|
|
|
|
||
Other long-term liabilities |
|
|
|
|
|
|
||
Operating lease liabilities, noncurrent |
|
|
|
|
|
|
||
Liability related to sale of future royalties, net |
|
|
|
|
|
|
||
Total noncurrent liabilities |
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
Stockholders’ equity: |
|
|
|
|
|
|
||
Common stock A, $ |
|
|
|
|
|
|
||
Common stock B, $ |
|
|
|
|
|
|
||
Additional paid-in capital |
|
|
|
|
|
|
||
Accumulated deficit |
|
|
( |
) |
|
|
( |
) |
Total stockholders’ equity |
|
|
|
|
|
|
||
Total liabilities and stockholders’ equity |
|
$ |
|
|
$ |
|
||
See accompanying notes.
4
Reata Pharmaceuticals, Inc.
Unaudited Consolidated Statements of Operations
(in thousands, except share and per share data)
|
|
Three Months Ended |
|
|
Six Months Ended |
|
||||||||||
|
|
June 30 |
|
|
June 30 |
|
||||||||||
|
|
2022 |
|
|
2021 |
|
|
2022 |
|
|
2021 |
|
||||
Collaboration revenue |
|
|
|
|
|
|
|
|
|
|
|
|
||||
License and milestone |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
Other revenue |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Total collaboration revenue |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Expenses |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Research and development |
|
|
|
|
|
|
|
|
|
|
|
|
||||
General and administrative |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Depreciation |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Total expenses |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Other income (expense), net |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Loss before taxes on income |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Benefit from (provision for) taxes on income |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
Net loss |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
Net loss per share—basic and diluted |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
Weighted-average number of common shares used in |
|
|
|
|
|
|
|
|
|
|
|
|
||||
See accompanying notes.
5
Reata Pharmaceuticals, Inc.
Unaudited Consolidated Statements of Stockholders’ Equity
(in thousands, except share and per share data)
|
|
Three Months Ended June 30, 2022 |
|
|||||||||||||||||||||||||
|
|
Common Stock A |
|
|
Common Stock B |
|
|
Additional |
|
|
Total |
|
|
Total |
|
|||||||||||||
|
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Deficit |
|
|
Equity |
|
|||||||
Balance at March 31, 2022 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||||
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Compensation expense |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Exercise of options |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||||
Issuance of common stock |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
Conversion of common |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Balance at June 30, 2022 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||||
|
|
Six Months Ended June 30, 2022 |
|
|||||||||||||||||||||||||
|
|
Common Stock A |
|
|
Common Stock B |
|
|
Additional |
|
|
Total |
|
|
Total |
|
|||||||||||||
|
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Deficit |
|
|
Equity |
|
|||||||
Balance at December 31, 2021 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||||
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Compensation expense |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Exercise of options |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||||
Issuance of common stock |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
Conversion of common |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Balance at June 30, 2022 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||||
|
|
Three Months Ended June 30, 2021 |
|
|||||||||||||||||||||||||
|
|
Common Stock A |
|
|
Common Stock B |
|
|
Additional |
|
|
Total |
|
|
Total |
|
|||||||||||||
|
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Deficit |
|
|
Equity |
|
|||||||
Balance at March 31, 2021 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||||
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Compensation expense |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Exercise of options |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
Issuance of common stock |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|||
Conversion of common |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Balance at June 30, 2021 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||||
|
|
Six Months Ended June 30, 2021 |
|
|||||||||||||||||||||||||
|
|
Common Stock A |
|
|
Common Stock B |
|
|
Additional |
|
|
Total |
|
|
Total |
|
|||||||||||||
|
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Deficit |
|
|
Equity |
|
|||||||
Balance at December 31, 2020 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||||
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Compensation expense |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Exercise of options |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
Conversion of common |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Issuance of Common Stock |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|||
Balance at June 30, 2021 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||||
See accompanying notes.
6
Reata Pharmaceuticals, Inc.
Unaudited Consolidated Statements of Cash Flows
(in thousands)
|
|
Six Months Ended |
|
|||||
|
|
June 30 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Operating activities |
|
|
|
|
|
|
||
Net loss |
|
$ |
( |
) |
|
$ |
( |
) |
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
||
Depreciation |
|
|
|
|
|
|
||
Amortization of debt issuance costs and imputed interest |
|
|
|
|
|
|
||
Non-cash interest expense on liability related to sale of future royalty |
|
|
|
|
|
|
||
Stock-based compensation expense |
|
|
|
|
|
|
||
Amortization of discount (premium) on marketable debt securities |
|
|
( |
) |
|
|
|
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
||
Income tax receivable and payable |
|
|
|
|
|
|
||
Prepaid expenses, other current assets and other assets |
|
|
( |
) |
|
|
( |
) |
Accounts payable |
|
|
( |
) |
|
|
|
|
Accrued direct research, other current and long-term liabilities |
|
|
( |
) |
|
|
( |
) |
Operating lease obligations |
|
|
|
|
|
|
||
Deferred revenue |
|
|
( |
) |
|
|
( |
) |
Net cash used in operating activities |
|
|
( |
) |
|
|
( |
) |
Investing activities |
|
|
|
|
|
|
||
Purchases of property and equipment |
|
|
( |
) |
|
|
( |
) |
Purchases of marketable securities |
|
|
( |
) |
|
|
|
|
Net cash used in investing activities |
|
|
( |
) |
|
|
( |
) |
Financing activities |
|
|
|
|
|
|
||
Exercise of options |
|
|
|
|
|
|
||
Net cash provided by financing activities |
|
|
|
|
|
|
||
Net decrease in cash and cash equivalents |
|
|
( |
) |
|
|
( |
) |
Cash and cash equivalents at beginning of year |
|
|
|
|
|
|
||
Cash and cash equivalents at end of period |
|
$ |
|
|
$ |
|
||
Non-cash activity: |
|
|
|
|
|
|
||
Right-of-use assets obtained in exchange for lease obligations |
|
$ |
|
|
$ |
|
||
Purchases of equipment in accounts payable, accrued direct research, other current, and long-term liabilities |
|
$ |
|
|
$ |
|
||
Acquisition of property and equipment through tenant improvement allowance |
|
$ |
|
|
$ |
|
||
See accompanying notes.
7
Reata Pharmaceuticals, Inc.
Notes to Unaudited Consolidated Financial Statements
1. Description of Business
Reata Pharmaceuticals, Inc.’s (Reata, the Company, we, us, or our) mission is to identify, develop, and commercialize innovative therapies that change patients’ lives for the better. The Company focuses on small-molecule therapeutics with novel mechanisms of action for the treatment of severe, life-threatening diseases with few or no approved therapies. The Company’s lead programs are omaveloxolone in a rare neurological disease called Friedreich’s ataxia (FA) and bardoxolone methyl (bardoxolone) in rare forms of chronic kidney disease (CKD). Both of the Company’s lead product candidates activate the transcription factor Nrf2 to normalize mitochondrial function, restore redox balance, and resolve inflammation. Because mitochondrial dysfunction, oxidative stress, and inflammation are features of many diseases, the Company believes omaveloxolone, bardoxolone, and our next-generation Nrf2 activators have many potential clinical applications. Reata possesses exclusive, worldwide rights to develop, manufacture, and commercialize omaveloxolone, bardoxolone, and our next-generation Nrf2 activators, excluding certain Asian markets for bardoxolone in certain indications, which are licensed to Kyowa Kirin Co., Ltd. (Kyowa Kirin). In addition, we are developing RTA 901, the lead product candidate from our Hsp90 modulator program, in neurological indications. We are the exclusive licensee of RTA 901 and have worldwide commercial rights.
The Company’s consolidated financial statements include the accounts of all majority-owned subsidiaries. Accordingly, the Company’s share of net earnings and losses from these subsidiaries is included in the consolidated statements of operations. Intercompany profits, transactions, and balances have been eliminated in consolidation.
Prior period reclassifications
Certain prior period amounts in the consolidated financial statements have been reclassified to conform to the current period presentation. Specifically, Operating lease obligations have been reclassed out of Accrued direct research, other current and long-term liabilities in prior periods to conform with the current period presentation on the consolidated statements of cash flows.
2. Summary of Significant Accounting Policies
Basis of Presentation
The accompanying unaudited consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States (U.S. GAAP) for interim financial information and with the instructions to Form 10-Q and Article 10 of Regulation S-X. Accordingly, they do not include all of the information and notes required by U.S. GAAP for complete financial statements. In the opinion of management, all adjustments (consisting of normal recurring adjustments) considered necessary for a fair presentation have been included. Operating results for the six months ended June 30, 2022 are not necessarily indicative of the results that may be expected for the year ending December 31, 2022. The consolidated balance sheet at December 31, 2021, has been derived from the audited consolidated financial statements at that date but does not include all of the information and footnotes required by U.S. GAAP for complete financial statements. For further information, refer to the annual consolidated financial statements and footnotes thereto of the Company.
Summary of Significant Accounting Policies
The significant accounting policies used in the preparation of these condensed consolidated financial statements for the six months ended June 30, 2022 are consistent with those discussed in Note 2 to the consolidated financial statements in the Company’s Annual Report on Form 10-K for the year ended December 31, 2021.
8
Investments in Marketable Securities and Cash Equivalents
The Company invests excess cash balances in marketable debt securities and classifies its investments as held-to-maturity on facts and circumstances present at the time the Company purchased the securities. At each balance sheet date presented, the Company classified all of its investments in debt securities as held-to maturity and as current assets as they represent the investment of funds available for current operations.
The Company’s marketable debt securities are classified as cash equivalents if the original maturity, from the date of purchase, is 90 days or less, and as marketable debt securities if the original maturity, from the date of purchase, is in excess of 90 days. The carrying amount of cash equivalents approximate fair value. As of June 30, 2022 and December 31, 2021, cash and cash equivalents comprise funds in cash, money market accounts, and treasury securities. For the marketable debt securities, the Company performs its own review of prices received from the independent pricing services by comparing these prices to other sources and for our marketable debt securities, the Company confirms those securities are trading in active markets.
The Company considers all available evidence to evaluate if an impairment loss exists, and if so, marks the investment to market through a charge to the Company’s consolidated statements of operations and comprehensive loss. The Company did not record any impairment charges related to our marketable debt securities during the six months ended June 30, 2022.
3. Collaboration Agreements
Subsequent to the 2019 reacquisition of certain rights originally licensed to AbbVie Inc. (AbbVie) (see “AbbVie,” below), the Company’s collaboration revenue and deferred revenue have been generated primarily from licensing fees and reimbursements for expenses received under our exclusive license with Kyowa Kirin (the Kyowa Kirin Agreement).
Kyowa Kirin
In December 2009, the Company entered into an exclusive license with Kyowa Kirin to develop and commercialize bardoxolone in the licensed territory. The terms of the agreement include payment to the Company of a nonrefundable, up-front license fee of $
The up-front payment and regulatory milestones are accounted for as a single unit of accounting. The Company regularly evaluates its remaining performance obligation under the Kyowa Kirin Agreement. Accordingly, revenue may fluctuate from period to period due to changes to its estimated performance obligation period and variable considerations. The Company began recognizing revenue related to the up-front payment upon execution of the Kyowa Kirin Agreement.
In March 2021, the Company’s performance obligation period under the Kyowa Kirin Agreement was extended to June 2022, which decreased quarterly revenue recognition by approximately $
On July 27, 2021, Kyowa Kirin submitted a New Drug Application (NDA) in Japan to the Ministry of Health, Labour and Welfare for bardoxolone for improvement of renal function in patients with Alport syndrome. Based on this submission, the Company earned a $
9
AbbVie
In September 2010, the Company entered into a license agreement with AbbVie (the AbbVie License Agreement) for an exclusive license to develop and commercialize bardoxolone in the Licensee Territory (as defined in the AbbVie License Agreement).
In December 2011, the Company entered into a collaboration agreement with AbbVie (the Collaboration Agreement) to jointly research, develop, and commercialize the Company’s portfolio of second and later generation oral Nrf2 activators.
In October 2019, the Company and AbbVie entered into an Amended and Restated License Agreement (the Reacquisition Agreement) pursuant to which the Company reacquired the development, manufacturing, and commercialization rights concerning its proprietary Nrf2 activator product platform originally licensed to AbbVie in the AbbVie License Agreement and the Collaboration Agreement. In exchange for such rights, the Company agreed to pay AbbVie $
The Company recognized interest expense related to the Reacquisition Agreement of approximately $
4. Liability Related to Sale of Future Royalties
On June 24, 2020, the Company closed on the Development and Commercialization Funding Agreement with an affiliate of Blackstone Life Sciences, LLC (BXLS), which provides funding for the development and commercialization of bardoxolone for the treatment of CKD caused by Alport syndrome, autosomal dominant polycystic kidney disease (ADPKD), and certain other rare CKD indications in return for future royalties (the Development Agreement). The Development Agreement includes a $
In addition, concurrent with the Development Agreement, the Company entered into a common stock purchase agreement (the Purchase Agreement) with affiliates of BXLS to sell an aggregate of
The Company concluded that there were
10
The following table shows the activity within the liability related to sale of future royalties for the six months ended June 30, 2022:
|
Liability Related to Sale of Future Royalties |
|
|
|
(in thousands) |
|
|
Balance at December 31, 2021 |
$ |
|
|
Non-cash interest expense recognized |
|
|
|
Balance at June 30, 2022 |
|
|
|
Less: Unamortized transaction cost |
|
( |
) |
Carrying value at June 30, 2022 |
$ |
|
|
5. Marketable Debt Securities
During the quarter ended June 30, 2022, the Company invested its excess cash balances in marketable debt securities and, at each balance sheet date presented, the Company classified all of its investments in debt securities as held to maturity and as current assets as they mature within 12 months and represent the investment of funds available for current operations.
The following tables summarize our marketable debt securities (in thousands), as of June 30, 2022:
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
Amortized |
|
|
Gross |
|
|
Gross |
|
|
Fair Value (1) |
|
||||
Marketable debt securities: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
U.S. treasury securities |
|
|
|
|
|
|
|
( |
) |
|
|
|
||||
Total |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
(1)
6. Other Income (Expense), Net
|
|
Three Months Ended |
|
|
Six Months Ended |
|
||||||||||
|
|
June 30 |
|
|
June 30 |
|
||||||||||
|
|
2022 |
|
|
2021 |
|
|
2022 |
|
|
2021 |
|
||||
|
|
(in thousands) |
|
|||||||||||||
Other income (expense), net |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Investment income |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
Interest expense |
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
Non-cash interest expense on liability |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Other income (expense) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Total other income (expense), net |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
Investment Income
Interest income consists primarily of interest generated from our cash and cash equivalents and marketable debt securities.
11
Interest Expense
Interest expense consists primarily of the imputed interest from the amount due to AbbVie under the Reacquisition Agreement.
Non-Cash Interest Expense on Liability Related to Sale of Future Royalties
Non-cash interest expense consists of recognition of interest expense based on the Company’s current estimate of future royalties expensed to be paid over the estimated term of the Development Agreement.
Other Income (Expense)
Other income (expense) consists primarily of gains and losses on foreign currency exchange.
7. Leases
The Company headquarters is located in Plano, Texas, where it leases approximately
On February 4, 2022, the Company
On March 8, 2022, the Company
The Company has an additional lease of a single-tenant, build-to-suit building of approximately
For the six months ended June 30, 2022, the Company paid $
Supplemental balance sheet and other information related to the Company’s operating leases is as follows:
|
|
|
|
As of June 30, |
|
|||||
|
|
|
|
2022 |
|
|
2021 |
|
||
Weighted-average remaining lease term (in years) |
|
|
|
|
|
|
||||
Weighted-average discount rate |
|
|
|
|
% |
|
|
% |
||
12
Maturities of lease liabilities by fiscal year for the Company’s operating leases:
|
|
As of June 30, 2022 |
|
|
|
|
(in thousands) |
|
|
2022 (remaining six months) |
|
$ |
|
|
2023 (1) |
|
|
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
Thereafter |
|
|
|
|
Total lease payments (1) |
|
|
|
|
Less: Imputed interest |
|
|
( |
) |
Present value of lease liabilities |
|
$ |
|
|
(1)
8. Income Taxes
The following table summarizes income tax benefit expense and effective income tax rate:
|
|
Three Months Ended |
|
|
Six Months Ended |
|
||||||||||
|
|
June 30 |
|
|
June 30 |
|
||||||||||
|
|
2022 |
|
|
2021 |
|
|
2022 |
|
|
2021 |
|
||||
|
|
(in thousands, except for percentage data) |
|
|||||||||||||
Benefit from (provision for) taxes on income |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
Effective income tax rate |
|
|
% |
|
|
% |
|
|
% |
|
|
% |
||||
The Company’s effective tax rate for the three months and six months ended June 30, 2022, varies with the statutory rate primarily due to changes in the valuation allowance related to certain deferred tax assets generated or utilized in the applicable period.
Deferred tax assets are regularly reviewed for recoverability by jurisdiction and valuation allowances are established based on historical and projected future taxable losses and the expected timing of the reversals of existing temporary differences. The Company has recorded valuation allowances against the majority of its deferred tax assets as of June 30, 2022, and the Company expects to maintain these valuation allowances until there is sufficient evidence that future earnings can be achieved, which is uncertain at this time.
9. Stock-Based Compensation
The following table summarizes time-based and performance-based stock compensation expense reflected in the consolidated statements of operations:
|
|
Three Months Ended |
|
|
Six Months Ended |
|
||||||||||
|
|
June 30 |
|
|
June 30 |
|
||||||||||
|
|
2022 |
|
|
2021 |
|
|
2022 |
|
|
2021 |
|
||||
|
|
(in thousands) |
|
|||||||||||||
Research and development |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
General and administrative |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Total stock compensation expense |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
13
Restricted Stock Units (RSUs)
The following table summarizes RSU activity as of June 30, 2022, under the Second Amended and Restated Long Term Incentive Plan (LTIP Plan):
|
|
Number of |
|
|
Weighted-Average |
|
||
Outstanding at January 1, 2022 |
|
|
|
|
$ |
|
||
Granted |
|
|
|
|
|
|
||
Vested |
|
|
( |
) |
|
|
|
|
Forfeited |
|
|
( |
) |
|
|
|
|
Outstanding at June 30, 2022 |
|
|
|
|
$ |
|
||
As of June 30, 2022, total unrecognized compensation expense related to RSU and performance-based RSU awards that were deemed probable of vesting was approximately $
Stock Options
The following table summarizes stock option activity as of June 30, 2022, under the LTIP Plan and standalone option agreements:
|
|
Number of |
|
|
Weighted- |
|
||
Outstanding at January 1, 2022 |
|
|
|
|
$ |
|
||
Granted |
|
|
|
|
|
|
||
Exercised |
|
|
( |
) |
|
|
|
|
Forfeited |
|
|
( |
) |
|
|
|
|
Expired |
|
|
( |
) |
|
|
|
|
Outstanding at June 30, 2022 |
|
|
|
|
$ |
|
||
Exercisable at June 30, 2022 |
|
|
|
|
$ |
|
||
As of June 30, 2022, total unrecognized compensation expense related to stock options was approximately $
The total intrinsic value of all outstanding options and exercisable options as of June 30, 2022 was $
The number of weighted average options that were not included in the diluted earnings per share calculation because the effect would have been anti-dilutive represented
14
10. Employee Benefit Plans
In 2010,
11. Commitments and Contingencies
Litigation
From time to time, the Company is a party to legal proceedings in the course of its business, including the matters described below. The outcome of any such legal proceedings, regardless of the merits, is inherently uncertain. In addition, litigation and related matters are costly and may divert the attention of our management and other resources that would otherwise be engaged in other activities. If the Company were unable to prevail in any such legal proceedings, its business, results of operations, liquidity and financial condition could be adversely affected. The Company recognizes accruals for litigations to the extent that it can conclude that a loss is both probable and reasonably estimable and recognizes legal expenses as incurred.
Bardoxolone Securities Litigation
In late 2021 and early 2022, certain putative stockholders of the Company filed complaints in the United States District Court for the Eastern District of Texas alleging violations of the federal securities laws against the Company and certain of its executives, including its Chief Executive Officer; its Chief Operating Officer, Chief Financial Officer, and President; and its Chief Innovation Officer (in one of the suits). On April 22, 2022, the suits were consolidated and a lead plaintiff was appointed. On June 21, 2022, the lead plaintiff filed a complaint against the Company, the aforementioned executives, certain current and former member of the Company’s Board of Directors, and underwriters in connection with secondary offerings of Company stock in 2019 and 2020. The complaint alleges, among other things, that the Company made false and misleading statements regarding the sufficiency of the Phase 2 and Phase 3 CARDINAL studies to support an NDA for bardoxolone in the treatment of CKD caused by Alport syndrome, and the Company’s interactions with the FDA concerning potential approval for bardoxolone. The complaint asserts claims under the Securities Act of 1933 and the Securities Exchange Act of 1934 (Exchange Act). The plaintiffs seek, among other things, a class action designation, an award of damages, and costs and expenses, including attorney fees and expert fees. The Company believes that the allegations contained in the complaint are without merit and intends to defend the case. The Company cannot predict at this point the length of time that this action will be ongoing or the liability, if any, which may arise therefrom.
Derivative Lawsuit
An alleged stockholder of the Company filed a derivative action in the Court of Chancery of the State of Delaware against certain current and former directors of the Company and naming the Company as a nominal defendant. The plaintiff asserts claims in the complaint of breach of fiduciary duty and unjust enrichment concerning the alleged payment of excessive compensation to the non-employee directors of the Company between fiscal years 2019 and 2021. The plaintiff seeks, among other things, an order awarding damages and costs and expenses, including attorneys and expert fees, and directing the Board of Directors to reform and improve its corporate governance and internal procedures relating to the award of non-employee director compensation.
The defendants believe that the allegations contained in the complaint are without merit and intend to defend the case. The Company cannot predict at this point the length of time that this action will be ongoing or the liability, if any, which may arise therefrom.
15
Indemnifications
Accounting Standards Codification 460, Guarantees, requires that, upon issuance of a guarantee, the guarantor must recognize a liability for the fair value of the obligations it assumes under that guarantee.
As permitted under Delaware law and in accordance with the Company’s bylaws, officers and directors are indemnified for certain events or occurrences, subject to certain limits, while the officer or director is or was serving in such capacity. The maximum amount of potential future indemnification is unlimited; however, the Company has obtained director and officer insurance that limits its exposure and may enable recoverability of a portion of any future amounts paid. The Company believes the fair value for these indemnification obligations is minimal. Accordingly, the Company has not recognized any liabilities relating to these obligations as of June 30, 2022.
The Company has certain agreements with licensors, licensees, collaborators, and vendors that contain indemnification provisions. In such provisions, the Company typically agrees to indemnify the licensor, licensee, collaborator, or vendor against certain types of third-party claims. The Company accrues for known indemnification issues when a loss is probable and can be reasonably estimated. There were
16
Item 2. Management’s Discussion and Analysis of Financial Condition and Results of Operations
You should read the following discussion and analysis of our financial condition and results of operations together with our consolidated financial statements and related notes and other financial information appearing in this Quarterly Report on Form 10-Q. Some of the information contained in this discussion and analysis or set forth elsewhere in this Quarterly Report on Form 10-Q, including information with respect to our plans and strategy for our business, operations, and product candidates, includes forward-looking statements that involve risks and uncertainties. Factors that may cause actual results to differ materially from current expectations include, among other things, those described under the headings “Risk Factors” and “Cautionary Note Regarding Forward-Looking Statements” and discussed elsewhere in this Quarterly Report on Form 10-Q.
Overview
We are a clinical-stage biopharmaceutical company focused on identifying, developing, and commercializing innovative therapies that change patients’ lives for the better. We concentrate on small-molecule therapeutics with novel mechanisms of action for the treatment of severe, life-threatening diseases with few or no approved therapies. Our lead programs are omaveloxolone in Friedreich’s ataxia and bardoxolone in rare forms of CKD. Both of our lead product candidates activate the transcription factor Nrf2 to normalize mitochondrial function, restore redox balance, and resolve inflammation. Because mitochondrial dysfunction, oxidative stress, and inflammation are features of many diseases, we believe omaveloxolone, bardoxolone, and our next-generation Nrf2 activators have many potential clinical applications. We possess exclusive, worldwide rights to develop, manufacture, and commercialize omaveloxolone, bardoxolone, and our next-generation Nrf2 activators, excluding certain Asian markets for bardoxolone in certain indications, which are licensed to Kyowa Kirin. In addition, we are developing RTA 901, the lead product candidate from our Hsp90 modulator program, in neurological indications. We are the exclusive licensee of RTA 901 and have worldwide commercial rights.
Recent Key Developments
Omaveloxolone for Friedreich’s Ataxia
The U.S. Food and Drug Administration (FDA) has granted Fast Track Designation, Orphan Drug Designation, and Rare Pediatric Disease Designation to omaveloxolone for the treatment of Friedreich’s ataxia. In March 2022, we completed rolling submission of an NDA to the FDA for omaveloxolone for the treatment of patients with Friedreich’s ataxia. In May 2022, the FDA accepted our NDA for filing and granted Priority Review. We recently completed a mid-cycle communication meeting with the FDA and submitted additional data and analyses to the FDA. See “Programs in Neurological Diseases – Omaveloxolone in Patients with Friedreich’s Ataxia – Regulatory Guidance and Regulatory Interactions in the U.S.” below.
The FDA advised us that it is planning to hold an advisory committee meeting to discuss the application. The Prescription Drug User Fee Act (PDUFA) date, the FDA action date for the application, is scheduled for November 30, 2022.
We are continuing to complete the regulatory procedures and submissions required prior to filing a Marketing Authorization Application (MAA) in Europe for approval of omaveloxolone for the treatment of patients with Friedreich’s ataxia. We have received a positive opinion from the Pediatric Committee on our Pediatric Investigation Plan with a commitment to seek scientific advice for additional input on the protocol design, and we also received European Medicines Agency (EMA) Follow-Up Protocol Assistance feedback regarding our nonclinical and chemistry manufacturing controls (CMC) programs. The EMA feedback indicated that there were no identified impediments to our planned MAA submission and included agreement that certain nonclinical studies, including 2-year carcinogenicity study data, may be submitted after approval. We plan to submit an MAA to the EMA for omaveloxolone in the fourth quarter of 2022.
RTA 901 for Neurological Indications, Including Diabetic Peripheral Neuropathic Pain
In the first quarter of 2022, we initiated additional Phase 1 clinical pharmacology studies of RTA 901, including a drug-drug interaction study which has been fully enrolled. Following completion of these Phase 1 studies, we plan to initiate a randomized, double-blind placebo-controlled Phase 2 trial of RTA 901 in diabetic patients with peripheral neuropathic pain in the fourth quarter of 2022.
17
Bardoxolone in Patients with CKD Caused by Alport Syndrome
We received a Complete Response Letter (CRL) from the FDA in February 2022 with respect to its review of our NDA for bardoxolone in the treatment of patients with CKD caused by Alport syndrome. The CRL indicated the FDA cannot approve the NDA in its present form. We have recently requested a Type C meeting to discuss the program and continue to work with the FDA to confirm our next steps on our Alport syndrome program.
In October 2021, we submitted an MAA to the EMA for bardoxolone in the treatment of patients with CKD caused by Alport syndrome. In the first quarter of 2022, we received the 120-day list of questions from the EMA. We are in the process of preparing our responses. We previously requested a 90-day extension for our responses which was granted by the EMA. We plan to submit additional long term extension data from the ongoing EAGLE trial to address the comments and questions raised by the EMA and have been granted a further 30-day extension to complete our responses.
Bardoxolone in Patients with Autosomal Dominant Polycystic Kidney Disease (ADPKD)
We are currently enrolling patients in FALCON, a Phase 3, international, multi-center, randomized, double-blind, placebo-controlled trial studying the safety and efficacy of bardoxolone in patients with ADPKD, randomized one-to-one to active drug or placebo. FALCON is enrolling 850 patients in a broad range of ages with an estimated glomerular filtration rate (eGFR) between 30 and 90 mL/min/1.73 m2. The primary endpoint is eGFR change from baseline at Week 108 (8 weeks after planned drug discontinuation at Week 100). More than 580 patients are currently enrolled in the trial.
Background: Our Programs
The following chart outlines each of our programs by indication and phase of development: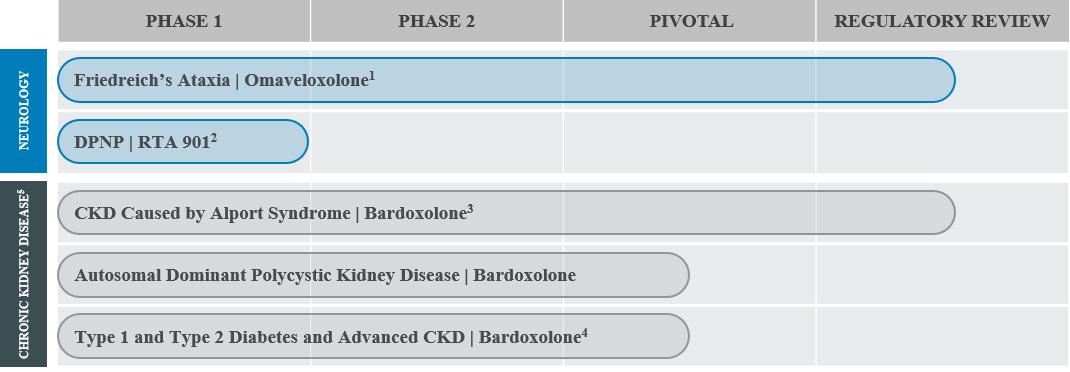
1NDA accepted for filing in May 2022, granted Priority Review, and assigned a PDUFA date of November 30, 2022.
2DPNP: Diabetic peripheral neuropathic pain.
3On February 25, 2022, we received a CRL from the FDA. We have recently requested a Type C meeting to discuss the program and continue to work with the FDA to confirm our next steps on our Alport syndrome program. MAA in the European Union is under review.
4AYAME trial conducted in Japan by our strategic collaborator in CKD, Kyowa Kirin. Kyowa Kirin expects the last patient visit to occur in the second half of 2022.
5Based on the outcome of AYAME and FALCON trials, and our discussions with the FDA regarding the bardoxolone program, we will decide future development plans for bardoxolone in additional forms of CKD.
Programs in Neurological Diseases
We are developing omaveloxolone for the treatment of patients with Friedreich’s ataxia, an inherited, debilitating, and degenerative neuromuscular disorder that is usually diagnosed during adolescence and can ultimately lead to premature death. In May 2022, the FDA accepted for filing our NDA for omaveloxolone for the treatment of patients with Friedreich’s ataxia and granted Priority Review.
Because mitochondrial dysfunction is a key feature of many neuromuscular diseases, we believe omaveloxolone may be broadly applicable to treat neurological diseases by activating Nrf2 to normalize and improve mitochondrial
18
function and adenosine triphosphate (ATP) production. We plan to pursue the development of omaveloxolone and our other Nrf2 activators for one or more additional neurological diseases.
We are also developing RTA 901 for the treatment of neurological diseases. RTA 901 is a highly potent and selective C-terminal modulator of Hsp90, which has a critical role in mitochondrial function, protein folding, and inflammation. RTA 901 has demonstrated profound efficacy in a wide range of animal models of neurological disease, including diabetic neuropathy, neuroinflammation, and neuropathic pain. We plan to initiate a randomized, placebo-controlled Phase 2 trial in DPNP in the fourth quarter of 2022.
Omaveloxolone in Patients with Friedreich’s Ataxia
Patients with Friedreich’s ataxia experience progressive loss of coordination, muscle weakness, and fatigue, which commonly progress to motor incapacitation and wheelchair reliance. Based on literature and proprietary research, we believe Friedreich’s ataxia affects approximately 5,000 children and adults in the United States and 22,000 individuals globally. According to data provided by IQVIA in 2020, there are approximately 4,000 projected patients diagnosed with Friedreich’s ataxia in the United States. The FDA has granted Orphan Drug Designation, Fast Track Designation, and Rare Pediatric Disease Designation to omaveloxolone for the treatment of Friedreich’s ataxia. The European Commission has granted Orphan Drug Designation in Europe to omaveloxolone for the treatment of Friedreich’s ataxia.
Diagnosis of Friedreich’s ataxia typically occurs by genetic testing, and most people in the United States with Friedreich’s ataxia are diagnosed in their teens and early twenties. Patients with Friedreich’s ataxia experience progressive loss of coordination, muscle weakness, and fatigue that commonly results in motor incapacitation, with patients requiring a wheelchair in their twenties. The mean age of death for patients with Friedreich’s ataxia is in the mid-thirties. Childhood-onset Friedreich’s ataxia can occur as early as age five, is more common than later-onset Friedreich’s ataxia, and normally involves more rapid disease progression. Currently, there are no approved therapies for the treatment of Friedreich’s ataxia.
MOXIe Part 2 Trial Results
Part 2 of our Phase 2 trial, called MOXIe (MOXIe Part 2), was an international, multi-center, double-blind, placebo-controlled, randomized, registrational trial that enrolled 103 patients with Friedreich’s ataxia at 11 trial sites in the United States, Europe, and Australia. MOXIe Part 2 was one of the largest global, interventional trials ever completed in Friedreich’s ataxia. Patients were randomized one-to-one to omaveloxolone or placebo. MOXIe Part 2 was completed in October 2019. The primary analysis population excluded patients with severe pes cavus (n=82), a musculoskeletal foot deformity that may interfere with the patient’s ability to perform some components of the neurological exam used to score the primary endpoint of the trial. Safety analyses were evaluated in the all-randomized population (n=103).
The primary endpoint for the trial was the change in the Modified Friedreich’s Ataxia Rating Scale (mFARS) score for omaveloxolone relative to placebo after 48 weeks of treatment. Omaveloxolone treatment demonstrated statistically significant evidence of efficacy for the primary endpoint of the trial, producing a placebo-corrected -2.40 point mean improvement in mFARS (n=82; p=0.014). Patients treated with omaveloxolone experienced a mean improvement in mFARS of -1.55 points from baseline, while patients treated with placebo experienced a mean worsening in mFARS of +0.85 points from baseline. Further, the observed placebo-corrected improvements in mFARS were time-dependent, increasing over the course of treatment with the largest improvement observed after 48 weeks of treatment.
19
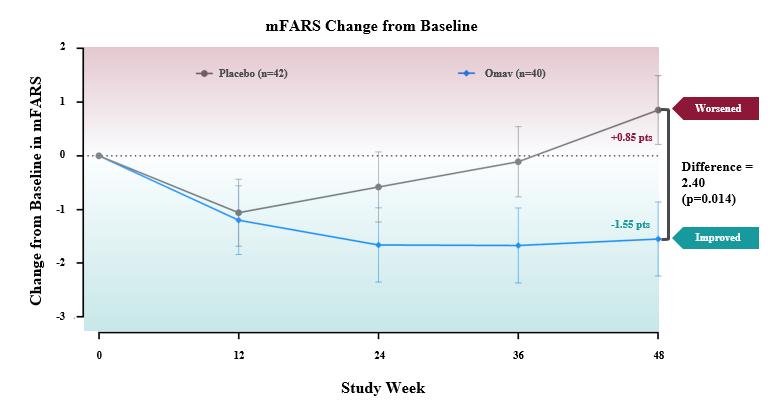
Additionally, all secondary endpoints either favored the omaveloxolone arm or were neutral. Patients on omaveloxolone experienced a nominal improvement in the Activities of Daily Living (ADL) questionnaire, with all nine questions favoring the omaveloxolone arm. On average, ADL scores for patients on omaveloxolone did not change from baseline, while placebo-treated patients worsened. Both patient global impression of change (PGIC) and clinical global impression of change numerically favored omaveloxolone, and improvement in PGIC correlated with the observed improvement in mFARS.
Omaveloxolone was reported to be generally well-tolerated. Four (8%) omaveloxolone patients and two (4%) placebo patients discontinued trial drug due to an adverse event (AE). The reported AEs were generally mild to moderate in intensity, and the most common AEs (i.e., reported in > 10% of omaveloxolone-treated patients) observed more frequently (>5% difference) in omaveloxolone compared to placebo were headache, nausea, increased aminotransferases, fatigue, abdominal pain, diarrhea, oropharyngeal pain, muscle spasms, back pain, and decreased appetite. Increases in aminotransferases are a pharmacological effect of omaveloxolone. In preclinical studies, omaveloxolone has been shown to increase production of aminotransferases in vitro, which we believe are related to restoration of mitochondrial function. In MOXIe Part 2, the aminotransferase increases were associated with improvements (reductions) in total bilirubin and were not associated with any evidence of liver injury.
In MOXIe Part 2, the overall rate of serious adverse events (SAEs) was low, with five patients in the omaveloxolone group and three patients in the placebo group reporting SAEs. No new safety signals were identified, and the reported SAEs were sporadic and generally expected in Friedreich’s ataxia patients. In the patients who reported SAEs while receiving omaveloxolone, none led to discontinuation.
MOXIe Extension Trial
The open-label MOXIe Extension trial is ongoing and enrolled a total of 149 patients (57 patients from MOXIe Part 1 and 92 patients from MOXIe Part 2). A total of 73 out of 75 (97%) patients without severe pes cavus who completed MOXIe Part 2 were enrolled in the MOXIe Extension, including 39 patients previously randomized to placebo (the placebo-to-omaveloxolone group) and 34 patients previously randomized to omaveloxolone (the omaveloxolone-to-omaveloxolone group). Due to the COVID-19 pandemic, not all patients had mFARS assessments performed at each time point. The ongoing safety review of the MOXIe Extension trial has not identified any new safety signals, including cardiovascular safety.
Delayed-Start Analysis Results from August 2021 Data Cut-Off (Used in Clinical Modules of NDA submission)
The intent of the post-hoc Delayed-Start Analysis is to evaluate whether omaveloxolone has a persistent effect on Friedreich’s ataxia disease course. Conceptually, this analysis evaluates whether the treatment effect that was
20
observed in the placebo-controlled MOXIe Part 2 trial is maintained in the MOXIe Extension trial when all patients are receiving omaveloxolone. If the treatment effect is maintained between those originally randomized to placebo (the placebo-to-omaveloxolone group) versus those originally randomized to omaveloxolone (the omaveloxolone-to-omaveloxolone group), then it demonstrates evidence of a persistent effect on the course of the disease. If the treatment effect is not maintained, and the patients originally randomized to placebo are able to achieve the same absolute response and “catch up” to the patients initially randomized to omaveloxolone, the results are consistent with a symptomatic treatment that does not affect the underlying course of the disease.
Two timepoints were compared in the analysis. The first timepoint was at Week 48, the final week of treatment in the placebo-controlled MOXIe Part 2 trial. The second timepoint was at Week 72 of the open-label MOXIe Extension in which all patients received omaveloxolone. A non-inferiority test was used to evaluate if the difference in mFARS between groups observed at the first timepoint was maintained or non-inferior at the second timepoint. The analysis methods, including the specified non-inferiority margin, were based on literature (Liu-Seifert, 2015a, 2015b). When comparing treatment groups using this methodology, maintaining a negative difference between treatment groups in mFARS is evidence of a persistent treatment effect.
The Delayed-Start Analysis used in clinical modules in our initial NDA rolling submission for omaveloxolone was based on data from an August 2021 data cut-off. In this analysis 58 of 73 patients from MOXIe Part 2 without severe pes cavus who enrolled into MOXIe Extension had at least 72 weeks of exposure in MOXIe Extension, and 28 of these patients had at least 120 weeks of exposure in the MOXIe Extension.
Results of this analysis demonstrated that the between-group difference in mFARS observed at the end of the placebo-controlled MOXIe Part 2 period (least squares mean difference = -2.25 ± 1.07) was preserved at MOXIe Extension Week 72 in the delayed-start period (LS mean difference = -3.51 ± 1.45). Consistent with a persistent treatment effect on disease, the upper limit of the 90% confidence interval (CI) for the difference estimate was less than zero (-0.615), meeting the threshold for demonstrating significant evidence of non-inferiority.
Delayed-Start Analysis Primary Endpoint (Non-Inferiority Test)1
|
Placebo-Controlled Week 48 (Δ1) |
Delayed-Start Week Ex. 72 (Δ2) |
|
||
Difference (LS Mean ± SE) |
-2.25 ± 1.07 p=0.037 |
-3.51 ± 1.45 p=0.016 |
|
||
Estimate = Δ2 – 0.5 × Δ1 |
-2.39 ± 1.38 |
|
|
||
Upper Limit of 1-sided 90% CI for Estimate |
-0.615 |
|
|
||
1Non-Inferiority test performed using a MMRM analysis with a Toeplitz covariance structure. |
|
||||
The graphical representation of changes from baseline in mFARS for omaveloxolone and placebo groups shows the separation at the end of the placebo-controlled period is maintained in the open-label period at Extension Week 72 and beyond.
21
Change from Baseline in mFARS (Patients without Severe Pes Cavus)
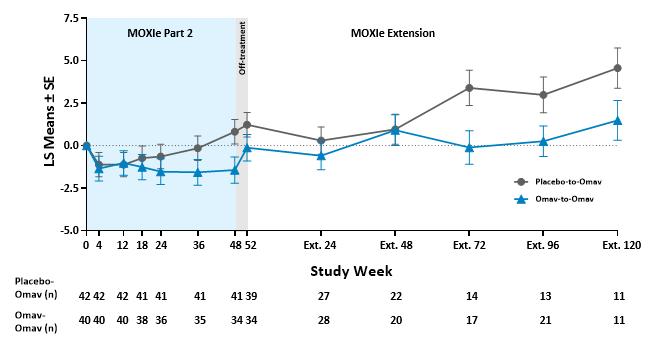
Many of the visits at Week 48 and Week 72 of the MOXIe Extension were scheduled during the initial peak of COVID-19 cases during Spring to Fall 2020. The mFARS assessment must be conducted in the clinic, and many in-clinic visits did not occur due to COVID-19 related travel restrictions and site closures during this period. Apart from the data at MOXIe Extension Week 48, parallel trajectories were seen in LS Mean mFARS change from baseline between the placebo-to-omaveloxolone group and the omaveloxolone-to-omaveloxolone group in the MOXIe Extension.
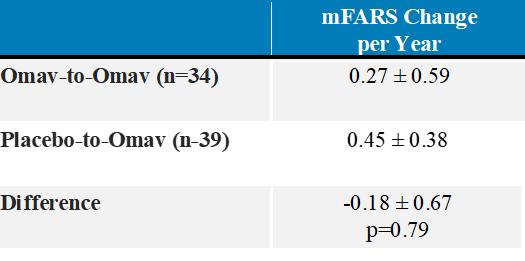
A longitudinal analysis was also performed to calculate annualized slopes incorporating all available data from the MOXIe Extension, which showed similar mean slopes in mFARS for the placebo-to-omaveloxolone group (0.45 ± 0.38 points per year) when compared to the omaveloxolone-to-omaveloxolone group (0.27 ± 0.59 points per year) with no significant difference between slopes (difference = -0.18 ± 0.67; p=0.79).
MOXIe Extension Annualized mFARS Change per Year (± SE)
Results from the Delayed-Start Analysis indicate a persistent omaveloxolone treatment effect on the disease course of Friedreich’s ataxia. Patients who received omaveloxolone during the double-blind MOXIe Part 2 had a benefit that could not be recovered by patients initially randomized to placebo who began omaveloxolone one year later in the MOXIe Extension. Notably, patients previously randomized to omaveloxolone in MOXIe Part 2 continued to show mean mFARS values that were similar to their original baseline after over three years of treatment.
Regulatory Guidance and Regulatory Interactions in the U.S.
Regulatory Guidance in the U.S. We are relying on section 115 of the Food and Drug Administration Modernization Act (FDAMA 115) and the December 2019 draft guidance thereunder from FDA on “Substantial Evidence of Effectiveness” as the basis for seeking approval of omaveloxolone in the U.S. The guidance provides that, if a sponsor has not conducted two adequate well-controlled studies, there are two alternative pathways to demonstrate substantial evidence of efficacy for drug approval; either:
22
Confirmatory evidence could include, for example, adequate and well-controlled clinical investigations in a related disease area, certain types of real world evidence such as extensive data on outcomes that provide further support for the lack of effect seen in the control group in the randomized trial, compelling mechanistic evidence in the setting of well-understood disease pathophysiology (e.g., pharmacodynamic data or compelling data from nonclinical testing), or well-documented natural history of the disease.
FDA may consider a number of factors when determining whether reliance on a single adequate and well-controlled clinical investigation plus confirmatory evidence is appropriate. These factors may include the persuasiveness of the single trial; the robustness of the confirmatory evidence; the seriousness of the disease, particularly where there is an unmet medical need; the size of the patient population; and whether it is ethical and practicable to conduct more than one adequate and well-controlled clinical investigation.
Regulatory Interactions Prior to NDA Acceptance. Following the announcement of the positive data from the MOXIe Part 2 study in October 2019, we met with the FDA in a Type C meeting in which the FDA provided us with guidance that it did not have any concerns with the reliability of the mFARS primary endpoint results in the MOXIe Part 2 study. Nevertheless, the FDA was not convinced that the MOXIe Part 2 results could support a single study approval without additional evidence that lends persuasiveness to the results. We believe their communication to us reflects their interpretation of the p-value, which while statistically significant, may not have met their threshold for a single study approval. This level of significance is challenging to generate in rare disease settings, such as Friedreich’s ataxia, since limited numbers of patients are available to enroll in clinical trials and they progress at a relatively slow rate over decades.
The FDA acknowledged the unmet need of patients with Friedreich’s ataxia, reiterated its commitment to facilitate the development of omaveloxolone within the constraints of the regulatory standards, and emphasized its willingness to consider all available options to meet the regulatory standards. In order to support a regulatory pathway of a single adequate and well-controlled clinical study plus confirmatory evidence for approval, we engaged with the FDA to determine what additional evidence they would consider to support an NDA submission and approval. The FDA subsequently requested a Delayed-Start Analysis.
During the first quarter of 2021, we submitted results from the Delayed-Start Analysis from a February 2021 data cut-off to the FDA as additional supporting evidence of effectiveness, and in May 2021 we received a communication from the FDA suggesting that, after a preliminary review of briefing materials for the Type C meeting, including the Delayed-Start Analysis, a pre-NDA meeting would be the most appropriate format for a discussion of the development program for omaveloxolone in Friedreich’s ataxia.
In the third quarter of 2021, we completed our pre-NDA meeting with the FDA. The purpose of the pre-NDA meeting was to discuss the content of Reata’s planned NDA submission including the nonclinical data and CMC packages, data standard plan, and the overall content plan. In the meeting, we stated that we believed that the MOXIe data, along with the Delayed-Start Analysis, would provide sufficient clinical data to support a full approval. The FDA stated that the proposed primary and supportive efficacy data appear reasonable, though the Delayed-Start Analysis was viewed as exploratory. The FDA noted that the ability of the data to support full approval, and the adequacy of the data and the determination of which data may be supportive of efficacy, would be a matter of review.
In November 2021, the FDA granted omaveloxolone Fast Track Designation for the treatment of Friedreich’s ataxia, providing eligibility for FDA programs such as Priority Review and rolling submission of the NDA, if relevant criteria are met. The FDA granted our request for a rolling submission, and, in March 2022, we completed submission of the NDA. In May 2022, the FDA accepted our NDA for filing and granted Priority Review Designation. The FDA advised us that it is planning to hold an advisory committee meeting to discuss the application. The PDUFA date is scheduled for November 30, 2022.
Recent Mid-Cycle Meeting Interactions. We recently completed a mid-cycle communication meeting with the FDA. The purpose of the mid-cycle communication meeting is for the FDA to provide the sponsor with an update of the status of the NDA review, including any issues identified. As is customary with the review of all NDAs, the FDA may identify other issues, and it may request additional information as it continues to review the NDA.
While we have not received formal minutes from the FDA, in the preliminary agenda for, and during, the mid-cycle communication meeting, the FDA stated that it has not identified any new significant issues but it continues to
23
have concerns regarding the strength of the efficacy evidence. The FDA noted specific points of discussion for the meeting including:
With respect to a potential label, the FDA requested additional justification or literature to support the relevance of proposed biomarkers (ferritin, gamma-glutamyl transferase (GGT), alanine aminotransferase (ALT), and aspartate aminotransferase (AST)) to Nrf2 activation and how that would correlate with treatment benefit in Friedreich’s ataxia patients.
The FDA did not identify any significant clinical safety issues. The FDA stated that the safety review is ongoing, and they are continuing to evaluate the cardiac safety of omaveloxolone in patients with Friedreich’s ataxia. They have not identified any other major safety concerns at this stage of their review.
During the mid-cycle meeting, we proposed to address their concerns in three ways. First, regarding the interpretability of the Delayed-Start Analysis, we noted that the reduction in number of patients was primarily due to missed visits due to the COVID-19 pandemic and not due to study discontinuations. We then presented updated results from the Delayed-Start Analysis using a March 2022 data cut-off, which contain new, later time points and increased numbers of patients at later time points than the prior analysis. The FDA acknowledged these data and agreed that we could submit the updated data in a subsequent submission. See “Results from March 2022 Data Cut-Off of Delayed-Start Analysis of the MOXIe Extension” below.
Second, we proposed to submit a new propensity-matched matched analysis of MOXIe Extension data using the largest, most robust Friedreich’s ataxia natural history study to provide additional clinical data that could be considered confirmatory evidence. We noted that in the FDA’s draft guidance on Substantial Evidence of Effectiveness that confirmatory evidence could include comparison to reliable, systematically collected and well documented natural history of patients with the disease, especially when the natural history is well defined, the external control population is similar to the treated population, and standard of care and concomitant treatments are not substantially different. We described how Friedreich’s ataxia meets all of these criteria. The FDA acknowledged and agreed to allow us to submit a propensity score-matched comparison of the progression in mFARS of subjects in the Clinical Outcome Measures in Friedreich’s ataxia (FA-COMS) natural history study to the patients in our MOXIe Extension study (the Propensity-Matched Analysis). See “Post Hoc Propensity-Matched Analysis of MOXIe Extension” below.
Third, we discussed an additional NDA amendment containing compelling mechanistic evidence in the setting of Friedreich’s ataxia’s well-understood disease pathophysiology, which could also serve as confirmatory evidence. Friedreich’s ataxia is caused by genetic mutations in the frataxin gene that result in insufficient amounts of functional frataxin protein. At the cellular level, this deficiency is associated with impaired mitochondrial energy production, which correlates with impaired neurologic function as measured by mFARS scores in FA patients. Substantial evidence demonstrates that Nrf2 levels and activity are suppressed in FA, and suppressed Nrf2 activity directly contributes to the pathophysiology of FA. Omaveloxolone induces Nrf2, which coordinates the expression of several enzymes and the production of key cofactors that increase mitochondrial energy production. Data from MOXIe Part 2 showed an association between omaveloxolone-induced Nrf2 activity and measures of neurological function, with larger increases in Nrf2 target levels associated with larger improvements in mFARS scores. The FDA also acknowledged this information and agreed that we could submit additional mechanistic evidence that could serve as confirmatory evidence.
In this context, we also addressed the mid-cycle meeting comment about the lack of a dose-response relationship in MOXIe Part 1. We noted that, based on the small size and short duration of MOXIe Part 1, it was not intended to demonstrate definitive efficacy. However, the study did demonstrate dose-dependent increases in Nrf2 target proteins.
Post-Mid-Cycle Meeting Submissions. Since the mid-cycle communication meeting, we have subsequently submitted the following additional data and analyses to the FDA as NDA updates and amendments.
24
A detailed description of each of these submissions is included below.
Recently, we identified a specific data entry error in certain mFARS assessments at one of the MOXIe trial sites. We have determined that these errors had a negligible impact on the results of MOXIe Part 2, the Delayed-Start Analysis, and the Propensity-Matched Analysis. We have submitted a report to the FDA explaining the issue and the negligible impact on the studies.
The FDA may elect not to review data submitted in the new submissions or to delay the NDA review process and PDUFA date in response to any of these new submissions or any other issues the FDA identifies.
Results from March 2022 Data Cut-Off of Delayed-Start Analysis of the MOXIe Extension (Recently Submitted to FDA)
We recently submitted an update to the Delayed-Start Analysis to the FDA using a data cut-off in March 2022. In this updated analysis 63 of 73 patients from MOXIe Part 2 without severe pes cavus who enrolled into MOXIe Extension had at least 120 weeks of exposure in MOXIe Extension, and 40 of these patients had at least 144 weeks of exposure in the MOXIe Extension.
Results of this analysis demonstrated that the between-group difference in mFARS observed at the end of the placebo-controlled MOXIe Part 2 period (LS mean difference = -2.17 ± 1.09) was preserved at MOXIe Extension Week 72 in the delayed-start period (LS mean difference = -2.91 ± 1.44). Consistent with a persistent treatment effect on disease, the upper limit of the 90% CI for the difference estimate was less than zero (-0.09), meeting the threshold for demonstrating significant evidence of non-inferiority. Additionally, the between group difference in mFARS was maintained at Extension Week 96, 120, and 144 (LS mean difference = -2.19 ± 1.38, -2.74 ± 1.26, and -2.58 ± 1.47 respectively) and the threshold for non-inferiority was met at Week 120 with an upper limit of the 90% CI of -0.106.
Delayed-Start Analysis (Non-Inferiority Test)1
|
Placebo-Controlled Week 48 (Δ1) |
Delayed-Start Week Ex. 72 (Δ2) |
Delayed-Start Week Ex. 96 (Δ2) |
Delayed-Start Week Ex. 120 (Δ2) |
Delayed-Start Week Ex. 144 (Δ2) |
Difference (LS Mean ± SE) |
-2.17 ± 1.089 p=0.0471 |
-2.91 ± 1.437 p=0.0433 |
-2.19 ± 1.375 p=0.1128 |
-2.74 ± 1.264 p=0.037 |
-2.58 ± 1.468 p=0.0796 |
Estimate = Δ2– 0.5 × Δ1 |
- |
-1.826 ± 1.3535 |
-1.10 ± 1.2991 |
-1.657 ± 1.2086 |
-1.496 ± 1.4630 |
Upper Limit of 1-sided 90% CI for Estimate |
- |
-0.090* |
0.567 |
-0.106* |
0.382 |
1Non-Inferiority test performed using a MMRM analysis with a Toeplitz covariance structure. *Threshold for non-inferiority was met.
|
|||||
The graphical representation of changes from baseline in mFARS for omaveloxolone and placebo groups shows the separation at the end of the placebo-controlled period is maintained in the open-label period at Extension Week 72 through Extension Week 144.
25
Change from Baseline in mFARS (Patients without Severe Pes Cavus)
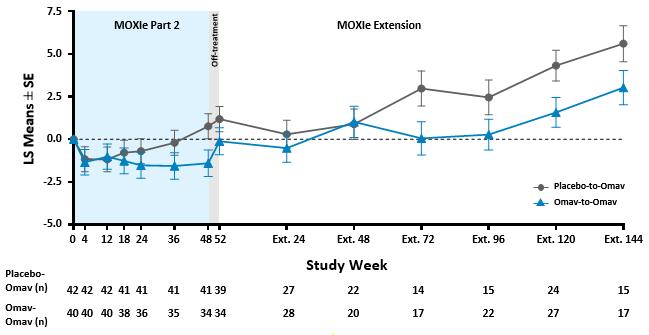
A longitudinal analysis was also performed on the updated data to calculate annualized slopes incorporating all available data from the MOXIe Extension through Week 144. The results showed no significant difference in slope between the placebo-to-omaveloxolone group and the omaveloxolone-to-omaveloxolone group (p=0.66).
MOXIe Extension Annualized mFARS Slope Through Extension Week 144 (± SE)
|
mFARS Change per Year |
Omaveloxolone-to-Omaveloxolone (n=34) |
0.446 ± 0.6340 |
Placebo-to-Omaveloxolone (n=39) |
0.760 ± 0.2773 |
Difference |
-0.314 ± 0.7118 p=0.6602 |
The resulting parallel trajectories between both treatment groups are consistent with the hypothesis that earlier omaveloxolone treatment altered the disease course. Patients who received omaveloxolone during the double-blind MOXIe Part 2 had a benefit that could not be recovered by patients initially randomized to placebo who began omaveloxolone one year later in MOXIe Extension.
Post Hoc Propensity-Matched Analysis of MOXIe Extension (Recently Submitted to FDA)
The intent of the Post-Hoc Propensity-Matched Analysis of MOXIe Extension is to facilitate interpretation of the open-label MOXIe Extension study using an external control, thus providing confidence in the durability of the treatment response. Accruing data in MOXIe Extension provides longer term follow-up for disease progression in patients receiving omaveloxolone; however, there is no long-term placebo arm for comparison.
MOXIe Extension data were compared to natural history external controls using propensity matching to provide longer term efficacy data in support of the statistically significant benefit demonstrated by pivotal MOXIe Part 2. Conceptually, the analysis compares the mFARS progression of omaveloxolone-treated patients in the open-label MOXIe Extension trial to the observed progression of propensity score-matched untreated patients in an external control group. Propensity matching is a statistical methodology utilized to identify and match patients in the MOXIe Extension study with patients in the external control group who are expected to have a similar disease progression based on the prognostic factors used to calculate the propensity scores.
The largest natural history study of Friedreich’s ataxia, FA-COMS, is a global, multi-center, longitudinal, prospective observational study that has enrolled more than 1,250 patients, with follow-up as long as 19 years in some patients. Clinical outcome measures, including mFARS, are assessed annually. The mFARS is a clinician-observed/performance-based outcome, all FA-COMS sites are tertiary care centers specializing in Friedreich’s ataxia, and all mFARS assessments are conducted in a standardized manner by trained neurologists with experience in Friedreich’s ataxia and other ataxias. The score for each component of mFARS is based on measurements of a patient’s functional ability using the same standardized set of instructions in both studies. Therefore, the significant
26
overlap in sites, which provides a similar testing environment to patients across studies, and the use of the same standardized instructions for the FARS assessment make the methodology for mFARS assessments across the 2 studies highly comparable. The FA-COMS database constitutes a well-established, reliable, and well documented source of natural history data for Friedreich’s ataxia.
Patients from FA-COMS were matched to MOXIe Extension patients using propensity scores based on multiple covariates: sex, baseline age, age of Friedreich’s ataxia onset, baseline mFARS score, and baseline gait score. Selection of these covariates was based on clinical relevance (i.e., factors considered prognostic for Friedreich’s ataxia progression) and availability in both studies. The change from baseline in mFARS at Year 3 for MOXIe Extension patients compared to the propensity score-matched FA-COMS patients was analyzed as the primary efficacy endpoint using mixed model repeated measures (MMRM) analysis. Change from baseline in mFARS at Year 1 and Year 2 were secondary endpoints. Data from MOXIe Part 1 and MOXIe Part 2 were not included in this propensity-matched analysis. Only data from MOXIe Extension were compared to natural history data. The matching was carried out as optimal 1:1 matching without replacement which resulted in equal sized groups for analysis.
For inclusion in each of the study populations patients must have had (1) baseline mFARS, (2) at least one post-baseline mFARS within 3 years after baseline, and (3) values for all propensity score model covariates (i.e., sex, baseline mFARS score, age at baseline, age of Friedreich’s ataxia onset, and baseline gait score). The MOXIe Extension study population included 136 patients. Of these, 95 patients were essentially treatment naïve at the time of entry into MOXIe Extension (i.e., includes those patients who received placebo in MOXIe Part 2 or had participated in MOXIe Part 1 and been off treatment for a long time), and the other 41 patients were continuing treatment (i.e., includes those patients who received omaveloxolone in MOXIe Part 2). The primary natural history (i.e., FA-COMS) study population included 598 patients eligible for matching with the MOXIe Extension population. There are three primary analysis populations, each based on a propensity score match with the FA-COMS study population: (1) Primary Pooled (n=272; 136 in each group), (2) Placebo-to-Omaveloxolone (n=190; 95 in each group), and (3) Omaveloxolone-to-Omaveloxolone (n=82; 41 in each group). Demographics and baseline characteristics were highly comparable between MOXIe Extension patients and the matched FA-COMS external control groups.
Propensity-Matched Analysis: Demographics and Baseline Characteristics Used for Propensity Score Calculation (Primary Pooled Population)
Characteristic |
Statistic |
Matched FA-COMS (n=136) |
MOXIe Extension (n=136) |
Age (years) |
Mean (SD) |
26.2 (13.72) |
26.6 (7.26) |
Age at Friedreich’s ataxia Onset |
Mean (SD) |
15.2 (10.48) |
15.5 (5.30) |
Sex, Female |
N (%) |
70 (51.5%) |
70 (51.5%) |
mFARS |
Mean (SD) |
41.0 (16.10) |
42.2 (12.60) |
Gait |
Mean (SD) |
2.7 (1.69) |
2.8 (1.36) |
In the Primary Pooled Population (n=272; 136 patients in each treatment group), by Year 3, patients in the FA-COMS matched set progressed 6.61 mFARS points whereas patients treated with omaveloxolone in MOXIe Extension progressed 3.00 points for a difference of -3.61 mFARS points (nominal p=0.0001). In this analysis, progression in mFARS was 55% slower in MOXIe Extension patients treated with omaveloxolone compared to matched untreated patients in the FA-COMS study.
27
Propensity-Matched Analysis: LS Mean Change from Baseline in mFARS Scores (Primary Pooled Population)
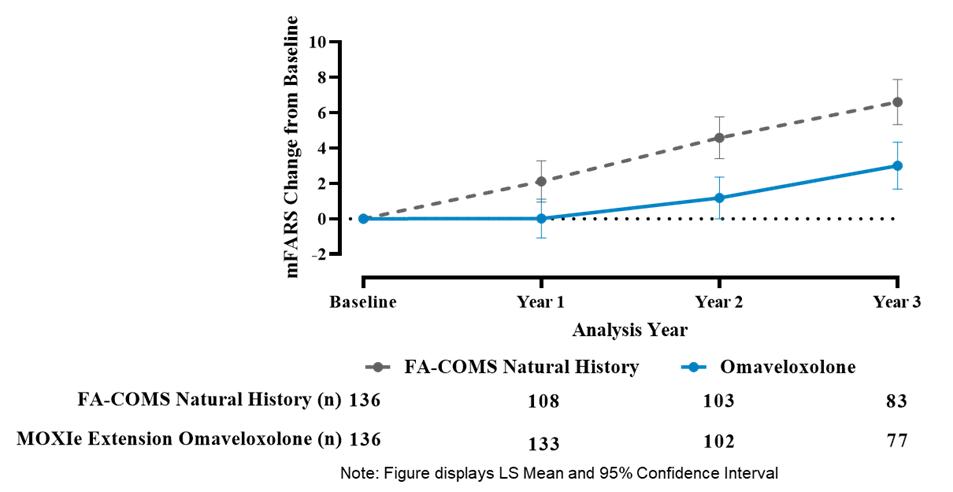
|
Baseline |
Year 1 |
Year 2 |
Year 3 |
||||
|
N |
Mean (SD) |
N |
LS Mean (SE) |
N |
LS Mean (SE) |
N |
LS Mean (SE) |
MOXIe Extension |
136 |
42.22 (12.60) |
133 |
0.015 (0.56) |
102 |
1.18 (0.59) |
77 |
3.00 (0.66) |
Matched FA-COMS |
136 |
41.03 (16.10) |
108 |
2.11 (0.59) |
103 |
4.58 (0.59) |
83 |
6.61 (0.65) |
Difference |
- |
- |
- |
-2.10 (0.81) p=0.0101 |
- |
-3.41 (0.84) p< 0.0001 |
- |
-3.61 (0.93) p=0.0001 |
In the pivotal MOXIe Part 2, omaveloxolone treated patients had experienced a reduction (i.e., an improvement) in mFARS score relative to the placebo group. The MOXIe Extension patients from the Primary Placebo-to-Omaveloxolone Population were essentially treatment-naïve at the time of entry into MOXIe Extension. After initiation of omaveloxolone treatment, at Year 1 this group experienced an improvement of neurologic function (as assessed by mFARS) relative to baseline. At Year 1, the treatment difference (-2.75 mFARS points; nominal p=0.0035) was similar in magnitude to the pivotal MOXIe Part 2 treatment difference in the primary analysis population (i.e., the Full Analysis Set) at Week 48 (-2.40 mFARS points).
28
Propensity-Matched Analysis: LS Mean Change in mFARS Scores (Primary Placebo-to-Omaveloxolone Population)
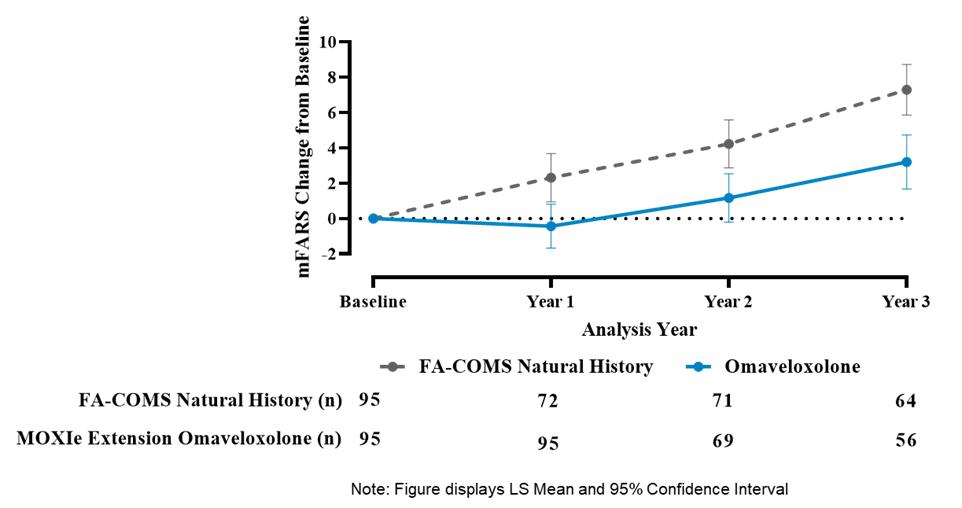
|
Baseline |
Year 1 |
Year 2 |
Year 3 |
||||
|
N |
Mean (SD) |
N |
LS Mean (SE) |
N |
LS Mean (SE) |
N |
LS Mean (SE) |
MOXIe Extension |
95 |
42.81 (12.79) |
95 |
-0.43 (0.63) |
69 |
1.18 (0.69) |
56 |
3.21 (0.76) |
Match FA-COMS |
95 |
44.54 (18.04) |
72 |
2.32 (0.69) |
71 |
4.23 (0.68) |
64 |
7.29 (0.72) |
Difference |
- |
- |
- |
-2.75 (0.94) p=0.0035 |
- |
-3.06 (0.97) p=0.0017 |
- |
-4.09 (1.05) p=0.0001 |
In the Primary Omaveloxolone-to- Omaveloxolone Population, the treatment effect favored omaveloxolone treated patients relative to FA-COMS external controls at Year 1 and Year 2. A smaller difference between treatment groups was observed at Year 1 in the Omaveloxolone-to- Omaveloxolone Population than in the Placebo-to-Omaveloxolone Population. The MOXIe Extension patients in the Omaveloxolone-to- Omaveloxolone Population did not experience an improvement from baseline likely because they were in their second year of treatment with active drug. Of note, this group did experience persistence of benefit relative to the matched FA-COMS external control. The Omaveloxolone-to- Omaveloxolone Population represents the patients treated for the longest total duration, having previously received omaveloxolone in MOXIe Part 2. The treatment effect favored MOXIe Extension at each visit and consistently increased over time in this population in comparison to the FA-COMS external control group.
Propensity-Matched Analysis: LS Mean Change in mFARS Scores (Primary Omaveloxolone-to-Omaveloxolone Population)
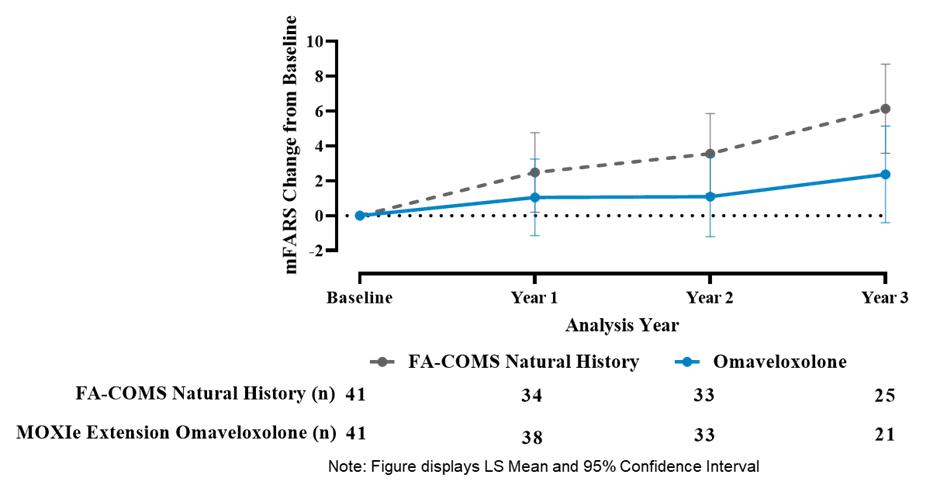
29
|
Baseline |
Year 1 |
Year 2 |
Year 3 |
||||
|
N |
Mean (SD) |
N |
LS Mean (SE) |
N |
LS Mean (SE) |
N |
LS Mean (SE) |
MOXIe Extension |
41 |
40.85 (12.19) |
38 |
1.05 (1.09) |
33 |
1.10 (1.13) |
21 |
2.38 (1.33) |
Match FA-COMS |
41 |
39.64 (16.80) |
34 |
2.48 (1.12) |
33 |
3.57 (1.13) |
25 |
6.14 (1.24) |
Difference |
- |
- |
- |
-1.43 (1.56) p=0.3641 |
- |
-2.47 (1.60) p=0.1265 |
- |
-3.76 (1.82) p=0.0400 |
The FA-COMS study constitutes a well-established, reliable, and well documented source of natural history data for Friedreich’s ataxia. We acknowledge the limitations of a post-hoc, cross-study comparison. However, in the situation of a rare disease such as Friedreich’s ataxia, we propose these data comparing the long-term efficacy outcomes for omaveloxolone to propensity-matched natural history data provide confirmatory evidence of effectiveness for omaveloxolone for the treatment of Friedreich’s ataxia.
Mechanistic Validation of Nrf2 Target Biomarkers in Friedreich’s ataxia
During the mid-cycle communication meeting discussion, the division requested to see the pharmacodynamic data contextualized with a discussion of the supporting independent literature regarding proposed biomarkers for label purposes. We have responded to the FDA’s request and, as briefly summarized below, we provided additional information including an integrated and detailed presentation of the disease pathophysiology of Friedreich’s ataxia, a review of the available pharmacodynamic data, justification of the relevance of these data in Friedreich’s ataxia, and an explanation of the relationship between the mechanistic data and the observed biomarker and clinical treatment effects in patients treated with omaveloxolone. As stated in the meeting, we believe these data do not just address their discussion topics regarding labeling but could also provide confirmatory evidence.
Friedreich’s ataxia is caused by genetic mutations in the frataxin (FXN) gene that result in insufficient amounts of FXN. At the cellular level, FXN deficiency is associated with impaired mitochondrial function, redox imbalance, and iron dysregulation. Additionally, lower mitochondrial function correlates with impaired neurologic function as measured by mFARS score in patients with Friedreich’s ataxia. Substantial evidence demonstrates that Nrf2 levels and activity are suppressed in cells from patients with Friedreich’s ataxia and in preclinical animal models of the disease. Although the molecular mechanism by which frataxin deficiency suppresses Nrf2 has not been fully characterized, dysregulated Nrf2 signaling is a common early upstream event that contributes to impaired mitochondrial energy production and redox imbalance in patients with Friedreich’s ataxia.
Nrf2 is a transcription factor that binds to specific DNA sequences called antioxidant response elements (AREs) that are located in the promoter regions of Nrf2 target genes. Binding to AREs allows Nrf2 to increase the transcription of its target genes, which play key roles in cellular metabolism and bioenergetic processes. Nrf2 target genes include γ-glutamyl transferase (GGT1), which plays a key role in redox balance, and ferritin heavy chain 1 (FTH1) and ferritin light chain (FTL), which combine to form ferritin, a protein involved in iron handling. By coordinating the expression of several enzymes and the production of key cofactors, Nrf2 activation restores redox balance, regenerates reducing equivalents, and modulates iron handling. Together, these effects increase the production of cellular energy (i.e., ATP) within the mitochondria in Friedreich’s ataxia patient fibroblasts and Friedreich’s ataxia disease models.
Treatment with omaveloxolone in MOXIe Part 1 resulted in dose-dependent increases in Nrf2 activity, as assessed by serum ferritin and GGT levels. Data from MOXIe Part 2 showed an association between omaveloxolone-induced Nrf2 activity and measures of neurological function, with larger increases in Nrf2 target levels associated with larger improvements in mFARS scores. Taken together, these data indicate that omaveloxolone rescues Nrf2 activity that is suppressed in patients with Friedreich’s ataxia and that the increase in Nrf2 activity is associated with a therapeutic benefit in these patients.
Regulatory Interactions in Europe
We are continuing to complete the regulatory procedures and submissions required prior to filing an MAA in Europe for approval of omaveloxolone for the treatment of patients with Friedreich’s ataxia. We have received a positive opinion from the Pediatric Committee on our Pediatric Investigation Plan with a commitment to seek scientific advice for additional input on the protocol design, and we also received EMA Follow-Up Protocol Assistance feedback regarding our nonclinical and CMC programs. The EMA feedback indicated that there were no identified impediments
30
to our planned MAA submission and included agreement that certain nonclinical studies, including 2-year carcinogenicity study data, may be submitted after approval. We plan to submit an MAA to the EMA for omaveloxolone in the fourth quarter of 2022.
Omaveloxolone in Other Neurological Indications
Omaveloxolone is a promising platform molecule. Because mitochondrial dysfunction is a key feature of many neurological and neuromuscular diseases, we believe omaveloxolone may be broadly applicable to treat such diseases by activating Nrf2 to normalize and improve mitochondrial function and ATP production.
Based on our understanding of the pathophysiology of neurological diseases characterized by mitochondrial dysfunction, inflammation, and oxidative stress, we believe omaveloxolone may be applicable to diseases such as progressive supranuclear palsy, Parkinson’s disease, frontotemporal dementia, Huntington’s disease, amyotrophic lateral sclerosis (ALS), Alzheimer’s disease, and epilepsy. Consistent with this, we have observed promising activity of omaveloxolone and our other Nrf2 activators in preclinical models of many of these diseases.
Our Nrf2 activators reduced seizure frequency in refractory, progressive epilepsy models and restored mitochondrial function in patient biopsy samples and preclinical models of Friedreich’s ataxia, ALS, familial and sporadic Parkinson’s disease, and frontotemporal dementia. In clinical trials, improvements in neuromuscular function have been observed in patients with Friedreich’s ataxia treated with omaveloxolone as assessed by mFARS, and improvements in mitochondrial function, as measured by reductions in blood lactate and heart rate, have been observed in patients with primary mitochondrial disease. Accordingly, we believe that omaveloxolone has the potential to treat a number of neurological and neuromuscular diseases that currently have few or no effective therapies, and we plan to pursue the development of omaveloxolone and our other Nrf2 activators for one or more of these diseases.
RTA 901 in Neurological Diseases
RTA 901 is the lead product candidate from our Hsp90 modulator program, which includes highly potent and selective C-terminal modulators of Hsp90. We have observed favorable activity of RTA 901 in a range of preclinical models of neurological disease, including models of diabetic neuropathy, neuroinflammation, and neuropathic pain.
Historically, other companies have explored N-terminal Hsp90 inhibitors for cancer therapeutics; however, this approach has been associated with multiple AEs including peripheral neuropathy and ocular toxicity. Binding at the C-terminus of Hsp90 leads to increased transcription of Hsp70, a cytoprotective and molecular chaperone gene, which facilitates cell survival in response to stress without the deleterious activities of N-terminal inhibition.
In preclinical rodent disease models, we observed that RTA 901 administered orally once-daily rescued existing nerve function, restored thermal and mechanical sensitivity, and improved nerve conductance velocity and mitochondrial function. These effects are dose-dependent, reversible, and Hsp70-dependent.
We completed a Phase 1 SAD/MAD trial of oral, once-daily RTA 901 in healthy adult volunteers to evaluate the safety, tolerability, and PK profile. The PK was approximately dose-proportional up to the highest doses evaluated with a half-life ranging from two to nine hours. Human exposures easily exceeded the exposures necessary for efficacy in multiple animal models. No safety or tolerability concerns were reported. In the first quarter of 2022, we initiated additional Phase 1 clinical pharmacology studies of RTA 901, including a drug-drug interaction study which has been fully enrolled. Following completion of these Phase 1 studies, we plan to initiate a randomized, double-blind placebo-controlled Phase 2 trial of RTA 901 in diabetic patients with peripheral neuropathic pain in the fourth quarter of 2022. We are the exclusive licensee of RTA 901 and have worldwide commercial rights.
Programs in Chronic Kidney Disease
We and Kyowa Kirin, our strategic collaborator in CKD in Japan, are developing bardoxolone for the treatment of CKD in multiple indications, including CKD caused by Alport syndrome, ADPKD, and type 1 and 2 diabetic CKD. CKD is characterized by a progressive worsening in the rate at which the kidney filters waste products from the blood, called the glomerular filtration rate (GFR). eGFR is an estimate of GFR that nephrologists use to track the decline in kidney function and progression of CKD. When GFR gets too low, patients develop end-stage kidney disease (ESKD) and require dialysis or a kidney transplant to survive.
Bardoxolone in Patients with CKD Caused by Alport Syndrome
31
Alport syndrome is a rare, genetic form of CKD caused by mutations in the genes encoding type IV collagen, which is a major structural component of the glomerular basement membrane in the kidney. The kidneys of patients with Alport syndrome progressively lose the capacity to filter waste products out of the blood, which can lead to ESKD and the need for chronic dialysis treatment or a kidney transplant. Alport syndrome affects both children and adults and can manifest as early as the first decade of life and causes average annual declines in eGFR of approximately four to five mL/min/1.73 m2. In patients with the most severe forms of the disease, approximately 50% progress to dialysis by age 25, 90% by age 40, and nearly 100% by age 60. There are currently no approved therapies to treat CKD caused by Alport syndrome.
The Alport Syndrome Foundation has estimated that Alport syndrome affects approximately 30,000 to 60,000 people in the United States. According to data provided by IQVIA in 2020, there are approximately 14,000 projected patients diagnosed with Alport syndrome in all stages of CKD in the United States. However, recent literature suggests that a large number of patients with Alport syndrome are either undiagnosed or misdiagnosed with other forms of CKD.
On February 25, 2022, we received a CRL from the FDA with respect to its review of our NDA for bardoxolone in the treatment of patients with CKD caused by Alport syndrome. The CRL indicated that the FDA cannot approve the NDA in its present form. Based on its review, the FDA concluded that it does not believe the submitted data demonstrates that bardoxolone is effective in slowing the loss of kidney function in patients with Alport syndrome and reducing the risk of progression to kidney failure and has requested additional data to support the efficacy and safety of bardoxolone. Their conclusion was based on efficacy and safety concerns primarily set forth in the FDA’s briefing book and discussed at the Cardiovascular and Renal Drugs Advisory Committee meeting held on December 8, 2021.
The FDA stated that the issues could be resolved by providing evidence of effectiveness that includes evidence from an adequate and well-controlled study showing a clinically relevant effect on the rate of loss of kidney function in patients with Alport syndrome or, alternatively, an effect on a clinical outcome (i.e., an endpoint that captures how patients with Alport syndrome feel, function, or survive). In addition, the FDA stated that we would need to address whether bardoxolone has a clinically relevant effect on the QT interval and show that the demonstrated clinical benefits of bardoxolone outweigh its risks. The FDA welcomed continued discussion on the details of a path forward. We have recently requested a Type C meeting to discuss the program and continue to work with the FDA to confirm our next steps on our Alport syndrome program.
In October 2021, we submitted an MAA to the EMA for bardoxolone in the treatment of patients with CKD caused by Alport syndrome. In the first quarter of 2022, we received the 120-day list of questions from the EMA. We are in the process of preparing our responses. We previously requested a 90-day extension for our responses which was granted by the EMA. We plan to submit additional long-term extension data from the ongoing EAGLE trial to address the comments and questions raised by the EMA and have been granted a further 30-day extension to complete our responses.
Bardoxolone in Patients with Autosomal Dominant Polycystic Kidney Disease
ADPKD is a rare and serious hereditary form of CKD caused by a genetic defect in PKD1 or PKD2 genes leading to the formation of fluid-filled cysts in the kidneys and other organs. Cyst growth can cause the kidneys to expand up to five to seven times their normal volume, leading to pain and progressive loss of kidney function. Inflammation appears to play a role in cyst growth and is associated with disease progression in ADPKD.
ADPKD affects both men and women of all racial and ethnic groups and is the leading inheritable cause of kidney failure with an estimated diagnosed population of 140,000 patients and an estimated prevalent population of 400,000 patients in the United States. Despite current standard-of-care treatment, an estimated 50% of ADPKD patients progress to ESKD and require dialysis or a kidney transplant by 60 years of age. The only therapy currently approved for ADPKD is JYNARQUE® (tolvaptan), developed by Otsuka Pharmaceuticals Co., Ltd., which was approved in the United States in 2018 to slow kidney function decline in adults at risk of rapidly progressing ADPKD.
We are currently enrolling patients in FALCON, an international, multi-center, randomized, double-blind, placebo-controlled trial studying the safety and efficacy of bardoxolone in patients with ADPKD randomized one-to-one to active drug or placebo. FALCON is enrolling 850 patients in a broad range of ages, with an eGFR between 30 and 90 mL/min/1.73 m2. The primary endpoint of the study is the off-treatment eGFR change from baseline at Week 108 (eight weeks after planned drug discontinuation at Week 100). All patients will be asked to return at Week 108
32
independent of the time of study drug discontinuation. The secondary endpoint is the eGFR change from baseline at Week 100. More than 580 patients are currently enrolled in the trial.
In the first quarter of 2022, we finalized a protocol amendment to FALCON, and initiated submission of the amendment to the FDA and other relevant health authorities. The major protocol amendment modifications include changing the primary endpoint of off-treatment eGFR change from baseline at Week 52 (or four weeks after drug discontinuation in Year 1) to eGFR change from baseline at Week 108 (eight weeks after planned drug discontinuation at Week 100). The major protocol amendment is under review in the applicable countries where FALCON is being conducted. We have received queries regarding the proposed changes from several countries and are actively working through the regulatory processes to secure approval to implement the amendment as agreed with the FDA. There is a possibility that approval for the amendment may either be delayed or denied in one or more countries. Failure to receive approval for the protocol amendment in multiple countries could materially adversely affect the trial results.
AYAME Trial in Diabetic CKD Conducted by Kyowa Kirin
Upon completion of Kyowa Kirin’s Phase 2 TSUBAKI trial of bardoxolone in patients with Stage 3 and 4 diabetic CKD in Japan, and after discussions with the Pharmaceuticals and Medical Devices Agency, Kyowa Kirin initiated a Phase 3 outcomes trial called AYAME in patients with Stage 3 or 4 diabetic CKD in Japan. The primary endpoint is time to onset of a ≥ 30% decrease in eGFR from baseline or ESKD. The secondary endpoints are time to onset of a ≥ 40% decrease in eGFR from baseline or ESKD, time to onset of a ≥ 53% decrease in eGFR from baseline or ESKD, time to onset of ESKD, and change in eGFR from baseline at each evaluation time point. Kyowa Kirin completed patient enrollment in AYAME in June 2019 and expects the last patient visit to occur in the second half of 2022.
United States Commercial Readiness
With the acceptance of our NDA for omaveloxolone and a Priority Review designation, we are advancing commercial launch preparations in the United States. We are in the process of building the commercial infrastructure necessary to effectively support the commercialization of omaveloxolone for the treatment of Friedreich’s ataxia, if and when we receive regulatory approval. Commercial launch preparation for omaveloxolone in Friedreich’s ataxia will continue to advance in step with the regulatory progress.
Our ability to launch omaveloxolone is dependent on the successful defense of an NDA and approval by the FDA. We have hired commercial leadership and intend to build the teams, infrastructure, systems, and processes necessary for the launch of omaveloxolone. This will include sales, marketing, market access, patient support, and distribution. Additionally, we are expanding quality and compliance functions to support commercialization. A trade name for omaveloxolone has been selected.
One challenge unique to rare-disease commercialization is patient identification due to the very small and sometimes heterogeneous disease populations. Our management team is experienced in maximizing patient identification for both clinical development and commercialization purposes in rare diseases. Commercial infrastructure for orphan products typically consists of a targeted, specialty sales force that calls on a limited and focused group of physicians and personnel involved in sales management, internal sales support, marketing, patient access, and distribution.
Ex-United States Commercial Readiness
Our ability to launch omaveloxolone is dependent on the successful filing and defense of an MAA and approval by EMA or other regulatory agencies. Outside of the United States, where appropriate and depending on the terms of our contractual arrangements, we plan, either alone, or with new collaboration partners, to commercialize our products. Our strategic collaborator Kyowa Kirin has all rights to commercialize bardoxolone in its territories. We are refining our strategy and market assessments with respect to potential launches in the EU, and we continue to evaluate market opportunities for our products in other global markets. Commercial launch preparation for omaveloxolone in Friedreich’s ataxia outside of the United States will advance in step with the regulatory progress.
Update on Adjustments to Operations Due to COVID-19
When COVID-19 emerged as a global pandemic in the first quarter of 2020, we were quick to respond and were an early adopter of a work-from-home policy, with the exception of the laboratory, which continued to operate throughout under strict safety protocols. For all remote employees, we provided appropriate workstation equipment
33
as well as training and resources to support employees’ mental and emotional wellbeing. We continue to operate under a hybrid work schedule encouraging office-based employees to work with their respective functional leaders to agree to flexible work schedules that meet both business and personal needs. For field-based medical affairs personnel, we are working with health care professionals to schedule in-person meetings/attend conferences where practical and in accordance with policy and regulatory guidance. We continue to update our Pandemic Management Plan as circumstances dictate in order to minimize risk and protect the wellbeing of employees.
Corporate Overview
To date, we have focused most of our efforts and resources on developing our product candidates and conducting preclinical studies and clinical trials. We have historically financed our operations primarily through revenue generated from our collaborations with AbbVie and Kyowa Kirin, from sales of our securities, secured loans, and a strategic financing from BXLS. We have not received any payments or revenue from collaborations other than nonrefundable upfront, milestone, and cost sharing payments from our collaborations with AbbVie and Kyowa Kirin, from the Development Agreement with BXLS, and from reimbursements of expenses under the terms of our agreement with Kyowa Kirin. We have incurred losses in each year since our inception, other than in 2014. As of June 30, 2022, we had $147.2 million of cash and cash equivalents and an accumulated deficit of $1,403.0 million. We continue to incur significant research and development and other expenses related to our ongoing operations. Despite contractual product development commitments and the potential to receive future payments from Kyowa Kirin, we anticipate that we will continue to incur losses for the foreseeable future, and we anticipate that our losses will increase as we continue our development of, seek regulatory approval for, and potentially commercialize our product candidates. If we do not successfully develop and obtain regulatory approval of our existing product candidates or any future product candidates and effectively manufacture, market, and sell any products that are approved, we may never generate revenue from product sales. Furthermore, even if we do generate revenue from product sales, we may never again achieve or sustain profitability on a quarterly or annual basis. Our prior losses, combined with expected future losses, have had and will continue to have an adverse effect on our stockholders’ equity and working capital. Our failure to become and remain profitable could depress the market price of our Class A common stock and could impair our ability to raise capital, expand our business, diversify our product offerings, or continue our operations.
The probability of success for each of our product candidates and clinical programs and our ability to generate product revenue and become profitable depend upon a variety of factors, including the quality of the product candidate, clinical results, investment in the program, competition, manufacturing capability, commercial viability, and our collaborators’ ability to successfully execute our development and commercialization plans. We will also require additional capital through equity, debt, or royalty financings or collaboration arrangements in order to fund our operations and execute on our business plans, and there is no assurance that such financing or arrangements will be available to us on commercially reasonable terms or at all. For a description of the numerous risks and uncertainties associated with product development and raising additional capital, see “Risk Factors” included in our Annual Report on Form 10-K for the year ended December 31, 2021.
Financial Operations Overview
Revenue
Our revenue to date has been generated primarily from licensing fees received under our collaborative license agreements and reimbursements for expenses. We currently have no approved products and have not generated any revenue from the sale of products to date. In the future, we may generate revenue from product sales, royalties on product sales, reimbursements for collaboration services under our current collaboration agreements, or license fees, milestones, or upfront payments if we enter into any new collaborations or license agreements. We expect that our future revenue will fluctuate from quarter to quarter for many reasons, including the uncertain timing and amount of any such payments and sales.
Our license and milestone revenue has been generated primarily from the Kyowa Kirin Agreement, the AbbVie License Agreement, and the Collaboration Agreement and consists of upfront payments and milestone payments. License revenue recorded with respect to the Kyowa Kirin Agreement, the AbbVie License Agreement, and the Collaboration Agreement consists solely of the recognition of deferred revenue. Under our revenue recognition policy, collaboration revenue associated with upfront, non-refundable license payments received under our license and collaboration agreements are deferred and recognized ratably over the expected term of the performance obligations under each agreement. Under the Reacquisition Agreement, we no longer have performance obligations under the
34
AbbVie License Agreement and the Collaboration Agreement. Under the Kyowa Kirin Agreement, we will not recognize any deferred revenue subsequent to June 30, 2022.
Research and Development Expenses
The largest component of our total operating expenses has historically been our investment in research and development activities, including the clinical development of our product candidates. From our inception through June 30, 2022, we have incurred a total of $1,169.1 million in research and development expense, a majority of which relates to the development of bardoxolone and omaveloxolone. We expect our research and development expense to continue to increase in the future as we advance our product candidates through clinical trials and expand our product candidate portfolio. The process of conducting the necessary clinical research to obtain regulatory approval is costly and time-consuming, and we consider the active management and development of our clinical pipeline to be crucial to our long-term success. The actual probability of success for each product candidate and preclinical program may be affected by a variety of factors, including the safety and efficacy data for product candidates, investment in the program, competition, manufacturing capability, and commercial viability.
Research and development expenses include:
Research and development costs are expensed as incurred. Costs for certain development activities such as clinical trials are highly judgmental and are recognized based on an evaluation of the progress to completion of specific tasks using information and data provided to us by our vendors and our clinical sites.
We base our expense accruals related to clinical trials on our estimates of the services received and efforts expended pursuant to contracts with multiple research institutions and CROs that conduct and manage clinical trials and preclinical studies on our behalf. The financial terms of these agreements vary from contract to contract and may result in uneven payment flows. Payments under some of these contracts depend on factors such as the successful enrollment of patients and the completion of clinical trial milestones. In accruing costs, we estimate the time period over which services will be performed and the level of effort to be expended in each period.
To date, we have not experienced material changes in our estimates of accrued research and development expenses after a reporting period. However, due to the nature of estimates, we cannot assure you that we will not make changes to our estimates in the future as we become aware of additional information about the status or conduct of our clinical trials and other research activities.
Currently, Kyowa Kirin has allowed us to conduct clinical studies of bardoxolone in certain rare forms of kidney diseases in Japan and has reimbursed us the majority of the costs for our CARDINAL study in Japan. Kyowa Kirin is the in-country caretaker in our FALCON study in Japan and we are reimbursing Kyowa Kirin for the costs of a certain number of patients in the study.
35
The following table summarizes our research and development expenses incurred during the three and six months ended June 30:
|
|
Three Months Ended |
|
|
Six Months Ended |
|
||||||||||
|
|
June 30 |
|
|
June 30 |
|
||||||||||
|
|
2022 |
|
|
2021 |
|
|
2022 |
|
|
2021 |
|
||||
|
|
(in thousands) |
|
|||||||||||||
Bardoxolone |
|
$ |
7,956 |
|
|
$ |
14,979 |
|
|
$ |
14,418 |
|
|
$ |
26,365 |
|
Omaveloxolone |
|
|
7,284 |
|
|
|
482 |
|
|
|
13,881 |
|
|
|
2,954 |
|
RTA 901 |
|
|
2,226 |
|
|
|
3,121 |
|
|
|
3,633 |
|
|
|
3,827 |
|
Other research and development expenses |
|
|
21,865 |
|
|
|
21,484 |
|
|
|
47,204 |
|
|
|
41,800 |
|
Total research and development expenses |
|
$ |
39,331 |
|
|
$ |
40,066 |
|
|
$ |
79,136 |
|
|
$ |
74,946 |
|
The program-specific expenses summarized in the table above include costs that we directly allocate to our product candidates. Our other research and development expenses include salaries, benefits, stock-based compensation and preclinical, research, and discovery costs, which we do not allocate on a program-specific basis.
General and Administrative Expenses
General and administrative expenses consist primarily of employee-related expenses for executive, operational, finance, legal, compliance, and human resource functions. Other general and administrative expenses include personnel expense, facility-related costs, professional fees, accounting and legal services, depreciation expense, other external services, and expenses associated with obtaining and maintaining our intellectual property rights.
We anticipate that our general and administrative expenses will increase in the future as we increase our headcount to support our continued research and development and potential commercialization of our product candidates. We have also incurred, and anticipate incurring in the future, increased expenses associated with being a public company, including exchange listing and SEC requirements, director and officer insurance premiums, legal, audit and tax fees, compliance with the Sarbanes-Oxley Act, regulatory compliance programs, and investor relations costs. Additionally, if and when we believe the first regulatory approval of one of our product candidates appears likely, we anticipate an increase in payroll and related expenses as a result of our preparation for commercial operations, especially for the sales and marketing of our product candidates.
Other Income (Expense), Net
Other income (expense) includes interest and gains earned on our cash and cash equivalents, and marketable debt securities, amortization of debt issuance costs, imputed interest on long term payables, foreign currency exchange gains and losses, and non-cash interest expense on liability related to the sale of future royalties.
Benefit from (Provision for) Taxes on Income
Provision for taxes on income consists of net loss, taxed at federal tax rates and adjusted for certain permanent differences. Realization of deferred tax assets is generally dependent upon future earnings by jurisdiction, of which the timing and amount are uncertain for the majority of our deferred tax assets, and valuation allowances are maintained against them. Changes in valuation allowances also affect the tax provision.
36
Results of Operations
Comparison of the Three Months Ended June 30, 2022 and 2021 (unaudited)
The following table sets forth our results of operations for the three months ended June 30:
|
|
2022 |
|
|
2021 |
|
|
Change $ |
|
|
Change % |
|
||||
|
|
(in thousands, except for percentage data) |
|
|||||||||||||
Collaboration revenue |
|
|
|
|
|
|
|
|
|
|
|
|
||||
License and milestone |
|
$ |
754 |
|
|
$ |
803 |
|
|
$ |
(49 |
) |
|
|
(6 |
) |
Other revenue |
|
|
8 |
|
|
|
1,418 |
|
|
|
(1,410 |
) |
|
|
(99 |
) |
Total collaboration revenue |
|
|
762 |
|
|
|
2,221 |
|
|
|
(1,459 |
) |
|
|
(66 |
) |
Expenses |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Research and development |
|
|
39,331 |
|
|
|
40,066 |
|
|
|
(735 |
) |
|
|
(2 |
) |
General and administrative |
|
|
25,143 |
|
|
|
21,998 |
|
|
|
3,145 |
|
|
|
14 |
|
Depreciation |
|
|
273 |
|
|
|
287 |
|
|
|
(14 |
) |
|
|
(5 |
) |
Total expenses |
|
|
64,747 |
|
|
|
62,351 |
|
|
|
2,396 |
|
|
|
4 |
|
Other income (expense), net |
|
|
(9,571 |
) |
|
|
(13,223 |
) |
|
|
3,652 |
|
|
|
28 |
|
Loss before taxes on income |
|
|
(73,556 |
) |
|
|
(73,353 |
) |
|
|
(203 |
) |
|
|
(0 |
) |
Benefit from (provision for) taxes on income |
|
|
1 |
|
|
|
653 |
|
|
|
(652 |
) |
|
** |
|
|
Net loss |
|
$ |
(73,555 |
) |
|
$ |
(72,700 |
) |
|
$ |
(855 |
) |
|
|
(1 |
) |
** Percentage not meaningful
Revenue
License and milestone revenue represented approximately 99% and 36% of total revenue for the three months ended June 30, 2022 and 2021, respectively, and consisted of the recognition of Kyowa Kirin deferred revenue. License and milestone revenue decreased by 6%, primarily due to the Company reaching the end of the performance obligation of the Kyowa Kirin Agreement during the current period.
Other revenue decreased in the three months ended June 30, 2022, compared to the three months ended June 30, 2021, primarily due to a decrease in reimbursements of expenses from Kyowa Kirin for manufacturing and non-clinical study expenses incurred.
Expenses
The following table summarizes our expenses, including as a percentage of total expenses, for the three months ended June 30:
|
|
2022 |
|
|
% of Total |
|
|
2021 |
|
|
% of Total |
|
||||
|
|
(in thousands, except for percentage data) |
|
|||||||||||||
Research and development |
|
$ |
39,331 |
|
|
|
60 |
% |
|
$ |
40,066 |
|
|
|
64 |
% |
General and administrative |
|
|
25,143 |
|
|
|
39 |
% |
|
|
21,998 |
|
|
|
35 |
% |
Depreciation |
|
|
273 |
|
|
|
1 |
% |
|
|
287 |
|
|
|
1 |
% |
Total expenses |
|
$ |
64,747 |
|
|
|
|
|
$ |
62,351 |
|
|
|
|
||
37
Research and Development Expenses
Research and development expenses decreased by $0.7 million, or 2%, for the three months ended June 30, 2022, remained consistent when compared to the three months ended June 30, 2021.
Research and development expenses, as a percentage of total expenses, was 60% and 64% for the three months ended June 30, 2022 and 2021, respectively. The decrease of 4% was due to the proportionately larger increase in general and administrative expenses, compared to research and development expenses.
General and Administrative Expenses
General and administrative expenses increased by $3.1 million, or 14%, for the three months ended June 30, 2022, compared to the three months ended June 30, 2021. The increase was primarily due to rent expense related to the new headquarters building lease that commenced in December 2021.
General and administrative expenses, as a percentage of total expenses, was 39% and 35%, for the three months ended June 30, 2022 and 2021, respectively. The increase of 4% was due to the proportionately larger increase in general and administrative expenses, compared to research and development expenses.
Other Income (Expense), Net
Other income (expense), net decreased by $3.7 million for the three months ended June 30, 2022, compared to the three months ended June 30, 2021. The decrease was primarily due to a decrease in effective interest rate in non-cash interest expense on liability related to the sale of future royalties and a decrease of interest expense attributable to the payable to AbbVie under to the Reacquisition Agreement, which was fully satisfied in 2021.
Benefit from (Provision for) Taxes on Income
Benefit from taxes on income decreased by $0.6 million for the three months ended June 30, 2022, compared to the three months ended June 30, 2021, primarily due to interest earned on tax benefit in 2021 resulting from CARES Act.
Results of Operations
Comparison of the Six Months Ended June 30, 2022 and 2021 (unaudited)
The following table sets forth our results of operations for the six months ended June 30:
|
|
2022 |
|
|
2021 |
|
|
Change $ |
|
|
Change % |
|
||||
|
|
(in thousands, except for percentage data) |
|
|||||||||||||
Collaboration revenue |
|
|
|
|
|
|
|
|
|
|
|
|
||||
License and milestone |
|
$ |
1,648 |
|
|
$ |
1,598 |
|
|
$ |
50 |
|
|
|
3 |
|
Other revenue |
|
|
29 |
|
|
|
1,568 |
|
|
|
(1,539 |
) |
|
|
(98 |
) |
Total collaboration revenue |
|
|
1,677 |
|
|
|
3,166 |
|
|
|
(1,489 |
) |
|
|
(47 |
) |
Expenses |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Research and development |
|
|
79,136 |
|
|
|
74,946 |
|
|
|
4,190 |
|
|
|
6 |
|
General and administrative |
|
|
49,984 |
|
|
|
42,703 |
|
|
|
7,281 |
|
|
|
17 |
|
Depreciation |
|
|
581 |
|
|
|
561 |
|
|
|
20 |
|
|
|
4 |
|
Total expenses |
|
|
129,701 |
|
|
|
118,210 |
|
|
|
11,491 |
|
|
|
10 |
|
Other income (expense), net |
|
|
(19,343 |
) |
|
|
(25,780 |
) |
|
|
6,437 |
|
|
|
25 |
|
Loss before taxes on income |
|
|
(147,367 |
) |
|
|
(140,824 |
) |
|
|
(6,543 |
) |
|
|
(5 |
) |
Benefit from (provision for) taxes on income |
|
|
(30 |
) |
|
|
669 |
|
|
|
(699 |
) |
|
** |
|
|
Net loss |
|
$ |
(147,397 |
) |
|
$ |
(140,155 |
) |
|
$ |
(7,242 |
) |
|
|
(5 |
) |
** Percentage not meaningful
Revenue
License and milestone revenue represented approximately 98% and 50% of total revenue for the six months ended June 30, 2022 and 2021, respectively, and consisted of the recognition of Kyowa Kirin deferred revenue.
38
License and milestone revenue increased by 3% during the six months ended June 30, 2022, compared to the six months ended June 30, 2021, primarily due to the achievement of a regulatory milestone in July 2021, variable consideration previously considered constrained, under the Kyowa Kirin Agreement.
Other revenue decreased in the six months ended June 30, 2022, compared to the six months ended June 30, 2021, primarily due to a decrease in reimbursements of expenses from Kyowa Kirin for manufacturing and non-clinical study expenses incurred.
Expenses
The following table summarizes our expenses, including as a percentage of total expenses, for the six months ended June 30:
|
|
2022 |
|
|
% of Total |
|
|
2021 |
|
|
% of Total |
|
||||
|
|
(in thousands, except for percentage data) |
|
|||||||||||||
Research and development |
|
$ |
79,136 |
|
|
|
60 |
% |
|
$ |
74,946 |
|
|
|
63 |
% |
General and administrative |
|
|
49,984 |
|
|
|
39 |
% |
|
|
42,703 |
|
|
|
36 |
% |
Depreciation |
|
|
581 |
|
|
|
1 |
% |
|
|
561 |
|
|
|
1 |
% |
Total expenses |
|
$ |
129,701 |
|
|
|
|
|
$ |
118,210 |
|
|
|
|
||
Research and Development Expenses
Research and development expenses increased by $4.2 million, or 6% for the six months ended June 30, 2022, compared to the six months ended June 30, 2021. The increase was primarily due to increased personnel and personnel-related costs to support the product development activities.
Research and development expenses, as a percentage of total expenses, was 60% and 63% for the six months ended June 30, 2022 and 2021, respectively. The decrease of 3% was due to the proportionately larger increase in general and administrative expenses, compared to research and development expenses.
General and Administrative Expenses
General and administrative expenses increased by $7.3 million, or 17%, for the six months ended June 30, 2022, compared to the six months ended June 30, 2021. The increase was primarily due to rent expense related to the new headquarters building lease that commenced in December 2021.
General and administrative expenses, as a percentage of total expenses, was 39% and 36%, for the six months ended June 30, 2022 and 2021, respectively. The increase of 3% was due to the proportionately larger increase in general and administrative expenses, compared to research and development expenses.
Other Income (Expense), Net
Other income (expense), net decreased by $6.4 million for the six months ended June 30, 2022,compared to the six months ended June 30, 2021. The decrease was primarily due to a decrease in effective interest rate in non-cash interest expense on liability related to the sale of future royalties and a decrease of interest expense attributable to the payable to AbbVie under to the Reacquisition Agreement, which was fully satisfied in 2021.
We periodically reassess the expected royalty payments under the Development Agreement, and to the extent such payment is greater or less than the initial estimate, we adjust the amortization. Based on our review in the first quarter of 2022, we lowered our previous estimate of future sales for which royalties will be paid. Accordingly, we have prospectively adjusted and recognized lower non-cash interest expense for the six months ended June 30, 2022.
Benefit from (Provision for) Taxes on Income
For the six months ended June 30, 2022 compared to the six months ended June 30, 2021, taxes increased by $0.7 million, primarily related to benefit recognized in 2021 from interest earned on tax refunds related to CARES Act.
39
Liquidity and Capital Resources
Since our inception, we have funded our operations primarily through collaboration and license agreements, the sale of preferred and common stock, the sale of royalty interests, and secured loans. Through June 30, 2022, we have raised gross cash proceeds of $476.6 million through the sale of convertible preferred stock and $785.0 million from payments under license and collaboration agreements. We also obtained $1,222.1 million in net proceeds from our initial public offering, follow-on offerings, and the sale of our Class A common stock under the Purchase Agreement, and $299.0 million in net proceeds from the sale of future royalties under the Development Agreement. We have not generated any revenue from the sale of any products. As of June 30, 2022, we had available cash and cash equivalents of approximately $147.2 million. Our cash and cash equivalents are invested in accordance with our investment policy, primarily with a view to liquidity and capital preservation.
Cash Flows
The following table sets forth the primary sources and uses of cash for each of the six months ended June 30:
|
|
2022 |
|
|
2021 |
|
||
|
|
(in thousands) |
|
|||||
|
|
|
|
|||||
Net cash (used in) provided by: |
|
|
|
|
|
|
||
Operating activities |
|
$ |
(107,307 |
) |
|
$ |
(70,611 |
) |
Investing activities |
|
|
(336,532 |
) |
|
|
(462 |
) |
Financing activities |
|
|
735 |
|
|
|
8,630 |
|
Net change in cash and cash equivalents |
|
$ |
(443,104 |
) |
|
$ |
(62,443 |
) |
Operating Activities
Net cash used in operating activities was $107.3 million for the six months ended June 30, 2022, consisting primarily of a net loss of $147.4 million adjusted for non-cash items including stock-based compensation expense of $29.3 million, non-cash interest expense on liability related to sale of future royalty of $20.1 million, and a net decrease in operating assets and liabilities of $9.6 million. The significant items in the change in operating assets that impacted our use of cash in operations include a decrease of $5.7 million in accounts payable due to timing of payments, and a decrease of $5.1 million in direct research and other current and long-term liabilities, primarily due to annual bonus payments.
Net cash used in operating activities was $70.6 million for the six months ended June 30, 2021, consisting primarily of a net loss of $140.2 million adjusted for non-cash items including stock-based compensation expense of $27.9 million, non-cash interest expense on liability related to sale of future royalty of $22.4 million, depreciation, amortization of issuance costs, and imputed interest expense of $4.0 million, and a net increase in operating assets and liabilities of $15.2 million. The significant items in the change in operating assets that impacted our use of cash in operations include a decrease of $22.2 million in income tax receivable due to the receipt of a CARES Act refund and an increase of $3.6 million in accounts payable due to timing of payments, offset by an increase of $5.0 million in prepaid expense, other current assets, and other assets due to insurance premium paid and a decrease of $4.0 million in accrued direct research and other current and long-term liabilities primarily due to the change in timing of bonus payments from December to March of the following year, which began with the December 2020 payments being delayed to March 2021.
Investing Activities
Net cash used in investing activities was $336.5 million and $0.5 million for the six months ended June 30, 2022 and 2021 , respectively. The increase is due to the purchase of $334.0 million in marketable securities during the period.
Financing Activities
Net cash provided by financing activities was $0.7 million and $8.6 million for the six months ended June 30, 2022 and 2021, respectively, primarily consisting of stock options exercises.
40
Operating Capital Requirements
To date, we have not generated any revenue from product sales. We do not know when or whether we will generate any revenue from product sales. We do not expect to generate significant revenue from product sales unless and until we obtain regulatory approval of and commercialize one or more of our current or future product candidates. We anticipate that we will continue to generate losses for the foreseeable future, and we expect the losses to increase as we continue the development of, and seek regulatory approvals for, our product candidates, and begin to commercialize any approved products. We are subject to all the risks related to the development and commercialization of novel therapeutics, including those described under the heading “Risk Factors” included in our most recent Annual Report on Form 10-K, and we may encounter unforeseen expenses, difficulties, complications, delays, and other unknown factors that may adversely affect our business. We continue to incur additional costs associated with operating as a public company. We anticipate that we will need substantial additional funding in connection with our continuing operations.
In October 2019, we entered into the 2019 Lease Agreement, relating to a new headquarter building lease of approximately 327,400 square feet of office and laboratory space located in Plano, Texas.
In July 2021, Kyowa Kirin announced the submission of an NDA in Japan for bardoxolone for improvement of renal function in patients with Alport syndrome. We earned a $5.0 million milestone related to this event that was received and began to be recognized in the third quarter of 2021.
In December 2020, we closed a follow-on underwritten public offering of 2,000,000 shares of our Class A common stock for gross proceeds of $281.7 million. Net proceeds to us from the offering were approximately $277.5 million, after deducting underwriting discounts and commissions and offering expenses.
In June 2020, we closed on the Development Agreement and Purchase Agreement, each dated June 10, 2020, under which certain BXLS entities paid us an aggregate of $350.0 million in exchange for future royalties on bardoxolone and an aggregate of 340,793 shares of our Class A common stock at $146.72 per share.
Our longer term liquidity requirements will require us to raise additional capital, such as through additional equity, debt, or royalty financings or collaboration arrangements. Our future capital requirements will depend on many factors, including the receipt of milestones under our Kyowa Kirin Agreement and the timing of our expenditures related to clinical trials. We believe our existing cash and cash equivalents will be sufficient to enable us to fund our operations through the fourth quarter of 2024. However, we anticipate opportunistically raising additional capital before that time through equity offerings, collaboration or license agreements, additional debt financings, or royalty financings in order to maintain adequate capital reserves. In addition, we may choose to raise additional capital at any time for the further development of our existing product candidates and may also need to raise additional funds sooner to pursue other development activities related to additional product candidates. Decisions about the timing or nature of any financing will be based on, among other things, our perception of our liquidity and of the market opportunity to raise equity, debt, or royalty financing. Additional securities may include common stock, preferred stock, or debt securities. We may explore strategic collaborations or license arrangements for any of our product candidates. If we do explore any arrangements, there can be no assurance that any agreement will be reached, and we may determine to cease exploring a potential transaction for any or all of the assets at any time. If an agreement is reached, there can be no assurance that any such transaction would provide us with a material amount of additional capital resources.
41
Until we can generate a sufficient amount of revenue from our product candidates, if ever, we expect to finance future cash needs through public or private equity or debt offerings, loans, royalty financings, and collaboration or license transactions. Recent and continued volatility in global financial markets may reduce our ability to access capital, which could negatively affect our liquidity. Additional capital may not be available on reasonable terms, if at all. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we may have to significantly delay, scale back, or discontinue the development or commercialization of one or more of our product candidates. If we raise additional funds through the issuance of additional equity or debt securities, it could result in dilution to our existing stockholders or increased fixed payment obligations, and any such securities may have rights senior to those of our common stock. If we incur indebtedness or obtain royalty financing, we could become subject to covenants that would restrict our operations and potentially impair our competitiveness, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell, or license intellectual property rights, and other operating restrictions that could adversely affect our ability to conduct our business, and any such debt or royalty financing could be secured by some or all of our assets. Any of these events could significantly harm our business, financial condition, and prospects. For a description of the numerous risks and uncertainties associated with product development and raising additional capital, see “Risk Factors” included in our Annual Report on Form 10-K for the year ended December 31, 2021.
Our forecast of the period through which our financial resources will be adequate to support our operations is a forward-looking statement and involves risks and uncertainties, and actual results could vary as a result of a number of factors. We have based this estimate on assumptions that may prove to be wrong, and we could utilize our available capital resources sooner than we currently expect. Our future funding requirements, both near- and long-term, will depend on many factors, including, but not limited to:
If we cannot expand our operations or otherwise capitalize on our business opportunities because we lack sufficient capital, our business, financial condition, and results of operations could be materially adversely affected.
42
Contractual Obligations and Commitments
We have various contractual obligations and other commitments that require payments at certain specified periods. The following table summarizes our contractual obligations and commitments as of June 30, 2022 (unaudited):
|
|
Payments due by period |
|
|||||||||||||||||
|
|
Less than |
|
|
1 to 3 |
|
|
4 to 5 |
|
|
6 years and beyond |
|
|
Total |
|
|||||
|
|
(unaudited, in thousands) |
|
|||||||||||||||||
Operating lease obligations(1) |
|
$ |
15,607 |
|
|
$ |
16,522 |
|
|
$ |
27,965 |
|
|
$ |
176,152 |
|
|
$ |
236,246 |
|
Total contractual obligations |
|
$ |
15,607 |
|
|
$ |
16,522 |
|
|
$ |
27,965 |
|
|
$ |
176,152 |
|
|
$ |
236,246 |
|
(1) Above table assumes one year rent abatement is applied beginning in June 2023 following FDA approval of omaveloxolone.
The terms of the Development Agreement require us to pay potential future royalty payments based on product development success. The above table excludes such obligations as the amount and timing of such obligations are unknown or uncertain, which are further described in Note 4, Liability Related to Sale of Future Royalties, to Consolidated Financial Statements contained in this Quarterly Report on Form 10-Q.
Clinical Trials
As of June 30, 2022, we have several on-going clinical trials in various stages. Under agreements with various CROs and clinical trial sites, we incur expenses related to clinical trials of our product candidates and potential other clinical candidates. The timing and amounts of these disbursements are contingent upon the achievement of certain milestones, patient enrollment, and services rendered or as expenses are incurred by the CROs or clinical trial sites. Therefore, we cannot estimate the potential timing and amount of these payments, and they have been excluded from the table above.
Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of our financial condition and results of operations are based on our consolidated financial statements, which have been prepared in accordance with U.S. GAAP. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, and expenses and the disclosure of contingent assets and liabilities in our financial statements. On an ongoing basis, we evaluate our estimates and judgments, including those related to revenue recognition, accrued research and development expenses, income taxes, and stock-based compensation. We base our estimates on historical experience, known trends and events, and various other factors that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
Our significant accounting policies are described in Note 2 of Part I, Item 1 of this Quarterly Report on Form 10-Q and in Part I, Item 7, “Critical Accounting Policies and Significant Judgments and Estimates” in our Annual Report on Form 10-K. There have been no changes to our critical accounting policies and estimates since our Annual Report on Form 10-K for the year ended December 31, 2021.
Off-Balance Sheet Arrangements
Since our inception, we have not had any relationships with unconsolidated organizations or financial partnerships, such as structured finance or special purpose entities, that would have been established for the purpose of facilitating off-balance sheet arrangements, and we have not engaged in any other off-balance sheet arrangements, as defined in the rules and regulations of the SEC.
Recent Accounting Pronouncements
For a discussion of recent accounting pronouncements, please see Note 2, Summary of Significant Accounting Policies, of Notes to Consolidated Financial Statements contained in this Quarterly Report on Form 10-Q.
43
Item 3. Quantitative and Qualitative Disclosures About Market Risk.
We are exposed to market risks in the ordinary course of our business. These market risks are principally limited to interest rate fluctuations. We had cash and cash equivalents of $147.2 million and marketable debt securities of $334.3 million at June 30, 2022, primarily invested in U.S. government treasuries. Our primary exposure to market risk is interest rate sensitivity, which is affected by changes in the general U.S. interest rates, particularly if our investments are in short-term securities. The primary objective of our investment activities is to preserve principal and liquidity while maximizing income without significantly increasing risk. We do not enter into investments for trading or speculative purposes. Due to the short-term nature of our investment portfolio, we do not believe an immediate and hypothetical increase of 100 basis points in interest rates would have a material effect on the fair market value of our portfolio, and accordingly we do not expect a sudden change in market interest rates to affect materially our operating results or cash flows.
We contract with research, development, and manufacturing organizations and investigational sites globally. Generally, these contracts are denominated in United States dollars. However, we may be subject to fluctuations in foreign currency rates in connection with agreements not denominated in United States dollars. We do not hedge our foreign currency exchange rate risk.
Item 4. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as of June 30, 2022. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized, and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company’s management, including its principal executive and principal financial officers, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives, and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of June 30, 2022, our Chief Executive Officer and Chief Financial Officer concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
Changes in Internal Control Over Financial Reporting
There have been no changes in our internal control over financial reporting, as such term is defined in Rules 13a-15(f) and 15d-15(f) promulgated under the Exchange Act, during the six months ended June 30, 2022, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
44
PART II — OTHER INFORMATION
Item 1. Legal Proceedings.
For a discussion of material pending legal proceedings, please read Note 10, Commitments and Contingencies – Litigation, to our condensed consolidated financial statements included in Part I, Item I, “Financial Statements (Unaudited),” of this Quarterly Report on Form 10-Q, which is incorporated into this item by reference.
Item 1A. Risk Factors.
In addition to other information set forth in this Quarterly Report on Form 10-Q, you should carefully consider the risk factors and other cautionary statements described under the heading “Risk Factors” included in our Annual Report on Form 10-K for the year ended December 31, 2021, which could materially affect our businesses, financial condition, or future results. Additional risks and uncertainties currently unknown to us, or that we currently deem to be immaterial, also may materially adversely affect our business, financial condition, or future results. There have been no material changes in our risk factors from those described in the Annual Report on Form 10-K for the year ended December 31, 2021.
Item 2. Unregistered Sales of Equity Securities and Use of Proceeds.
None.
Item 3. Defaults Upon Senior Securities.
None.
Item 4. Mine Safety Disclosures.
None.
Item 5. Other Information.
None.
45
Item 6. Exhibits.
Exhibit Number |
|
Description |
|
|
|
3.1 |
|
|
|
|
|
3.2 |
|
|
10.1# |
|
|
|
|
|
10.2*# |
|
|
10.3* |
|
|
|
|
|
31.1* |
|
|
|
|
|
31.2* |
|
|
|
|
|
32.1** |
|
|
|
|
|
32.2** |
|
|
|
|
|
101.INS* |
|
Inline XBRL Instance Document - The cover page interactive data file does not appear in the interactive data file because its XBRL tags are embedded within the inline XBRL document |
|
|
|
101.SCH* |
|
Inline XBRL Taxonomy Extension Schema Document |
|
|
|
101.CAL* |
|
Inline XBRL Taxonomy Extension Calculation Linkbase Document |
|
|
|
101.DEF* |
|
Inline XBRL Taxonomy Extension Definition Linkbase Document |
|
|
|
101.LAB* |
|
Inline XBRL Taxonomy Extension Label Linkbase Document |
|
|
|
101.PRE* |
|
Inline XBRL Taxonomy Extension Presentation Linkbase Document |
|
|
|
104 |
|
Cover Page Interactive Data File (embedded within the Inline XBRL document) |
|
|
|
* Filed herewith.
** Furnished herewith.
# Information in this exhibit identified by three asterisks [***] is confidential and has been omitted pursuant to
Item 601(b)(10)(iv) of Regulation S-K because it is not material and is the type of information that the Company customarily treats as private or confidential. An unredacted copy of this exhibit will be furnished to the Securities and Exchange Commission on a supplemental basis upon request.
46
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
Date: August 8, 2022 |
REATA PHARMACEUTICALS, INC. |
||
|
|
|
|
|
By: |
|
/s/ J. Warren Huff |
|
Name: |
|
J. Warren Huff |
|
Title: |
|
Chief Executive Officer |
|
By: |
|
/s/ Manmeet S. Soni |
|
Name: |
|
Manmeet S. Soni |
|
Title: |
|
Chief Operating Officer, Chief Financial Officer, and President |
47
Exhibit 10.2
SEVENTH SUPPLEMENT TO EXCLUSIVE LICENSE AND SUPPLY AGREEMENT
Regarding Phase 3 Clinical Study in Japan
This Seventh Supplement (herein so called), effective as of February 28, 2022 (the “Supplement Effective Date”), to the Exclusive License and Supply Agreement, effective as of December 24, 2009 (the “Original Agreement”), is by and between Reata Pharmaceuticals Holdings, LLC, a limited liability company organized and existing under the laws of Delaware, USA, with an address at 5320 Legacy Drive, Plano, Texas 75024 (“Reata”), as assignee of the Original Agreement from Reata Pharmaceuticals, Inc., a Delaware corporation and the original signatory to the Original Agreement, as assignor (“Reata Pharmaceuticals”), and Kyowa Kirin Co., Ltd., a company organized and existing under the laws of Japan, with an address at 1-9-2 Ohtemachi, Chiyoda-ku, Tokyo, 100-0004, Japan (“Kyowa Kirin”). Reata and Kyowa Kirin are sometimes hereinafter referred to each as a “Party” and collectively as the “Parties”.
WHEREAS, pursuant to the Original Agreement, Reata Pharmaceuticals granted to Kyowa Kirin an exclusive, royalty-bearing license under the Licensed Technology to research, develop, use, sell, offer for sale, import, and export Licensed Compound and Licensed Product in the Field in the Territory.
WHEREAS, pursuant to the Original Agreement, including the Sixth Supplement to Exclusive License and Supply Agreement (the “Sixth Supplement”), Kyowa Kirin is conducting portions of a Phase 3 clinical study of RTA 402 (which is included in the Licensed Compound) for the indication of autosomal dominant polycystic kidney disease (“ADPKD”) in Japan with a corresponding Protocol Number 402-C-1808 (“Study”) by participating in the global Phase 3 clinical study of RTA 402 for the indication of ADPKD conducted by Reata Pharmaceuticals, as a sponsor of the Study.
WHEREAS, Kyowa Kirin wishes to amend the cost allocation of the Study set out in the Sixth Supplement and perform the extended Study named as “AN EXTENDED ACCESS PROGRAM TO ASSESS LONG TERM SAFETY OF BARDOXOLONE METHYL IN PATIENTS WITH CHRONIC KIDNEY DISEASE” with a corresponding Protocol Number 402-C-1803 (“Extended Study”).
WHEREAS, in order to clarify and agree on obligation and responsibility of the Parties for the Study and Extended Study, the Parties wish to supplement the terms of the Original Agreement as set forth herein.
NOW, THEREFORE, in consideration of the foregoing premises and the mutual covenants herein contained, the Parties hereby agree as follows:
1. Obligation and Responsibility of the Parties
1.1 Notwithstanding the provisions of the Sixth Supplement, Kyowa Kirin shall bear the Study costs (the “Study Costs”) necessary for enrollment of up to [***] ([***]) patients, and Reata shall bear the Study Costs necessary for enrollment above [***] ([***]) patients up to [***] ([***]) patients according to the terms of the Sixth Supplement. Any increase in the Study Costs that are allocable to the excess enrollment above [***] ([***]) patients shall be equally borne by the Parties. Study Costs shall mean any external costs to be paid by Kyowa Kirin to any third parties for the Study. In addition to the Study Costs, Reata shall bear any internal costs of Kyowa Kirin for the Study necessary for enrollment above [***] ([***]) patients up to [***] ([***]) with a FTE rate of JPY [***] (the “Internal Costs”). Kyowa Kirin shall invoice Reata each quarter for the Study Costs and the Internal Costs borne by Reata, and Reata shall pay each invoice within thirty (30) days from the end of the calendar month in which Reata receives the invoice.
Certain identified information has been excluded from this exhibit because it is both not material and is of the type that the registrant treats as private or confidential. This redacted information has been marked in this exhibit with three asterisks[***].
1.2 The Parties agree that all patients who enroll in the Study and are eligible and willing to enroll in the Extended Study, may enroll in the Extended Study.
1.3 Kyowa Kirin shall be an in-country (Japan) caretaker of the Extended Study and solely responsible for the conduct thereof. For clarity, relevant provisions including but not limited to the Article IV (CLINICAL DEVELOPMENT) and the Article V (REGULATORY MATTERS) of the Original Agreement shall apply and Kyowa Kirin will conduct the Extended Study in accordance with those provisions. Notwithstanding the forgoing, Kyowa Kirin shall bear the Extended Study costs (the “Extended Study Costs”) necessary for enrollment of up to [***] ([***]) patients who were enrolled in the Study, and Reata shall bear the Extended Study Costs necessary for enrollment above [***] ([***]) patients up to [***] ([***]) patients who were enrolled in the Study. Any increase in the Extended Study Costs that are allocable to the excess enrollment above [***] ([***]) patients who were enrolled in the Study shall be equally borne by the Parties. Extended Study Costs shall mean any external costs to be paid by Kyowa Kirin to any third parties for the Extended Study. Kyowa Kirin shall invoice Reata each quarter for the Extended Study Costs borne by Reata, and Reata shall pay each invoice within thirty (30) days from the end of the calendar month in which Reata receives the invoice.
1.4 An agreement related to the Extended Study with a contract research organization (“CRO”) shall be entered into by and between Reata Pharmaceuticals, Kyowa Kirin and the CRO.
1.5 The number of enrollment of patients in the Study (“Enrollment Number”) shall be mutually agreed by the Parties in writing (including by email), provided that the Study Costs for the excess enrollment above the Enrollment Number shall be equally borne by the Parties if the Enrollment Number is above [***] ([***]) and informed consent from the patient(s) for such excess enrollment is obtained by the date Kyowa Kirin notifies Reata in writing (including by email).
2. Original Agreement
Except as supplemented and amended by this Seventh Supplement, the Original Agreement and all the Supplements to Exclusive License and Supply Agreement between the Parties, or between Kyowa Kirin and Reata Pharmaceuticals, shall remain in full force and effect pursuant to their terms
3. Other Provisions
3.1 This Seventh Supplement shall become effective as of the Supplement Effective Date and shall continue until the termination of the Original Agreement.
3.2 Except where specifically defined herein, capitalized terms used herein shall have the same meanings ascribed to them in the Original Agreement.
3.3 The headings to the several Articles hereof are not part of this Seventh Supplement, but are merely guides or labels to assist the locating and reading the several Articles hereof.
3.4 Except as stated herein, all terms and conditions of the Original Agreement and all the Supplements to the Original Agreement shall remain in full force and effect during the effective period of the Original Agreement.
3.5 This Seventh Supplement may be executed by electronic signature as agreed between the Parties, which shall be deemed to be an original.
IN WITNESS WHEREOF, the Parties have executed this Seventh Supplement to be effective as of the Supplement Effective Date.
KYOWA KIRIN CO., LTD.
By: /s/ Yasuo Fujii
Name: Yasuo Fujii
Title: Executive Officer, Director, Business Development Department
Date: February 28, 2022
REATA PHARMACEUTCALS HOLDINGS, LLC
By: /s/ Manmeet S. Soni
Name: Manmeet S. Soni
Title: Chief Operating Officer, Chief Financial Officer and President
Date: February 28, 2022
Exhibit 10.3
REATA PHARMACEUTICALS, INC.
INDEMNIFICATION AGREEMENT
This Agreement (“Agreement”) is made and entered into as of July 11, 2022, by and between Reata Pharmaceuticals, a Delaware corporation (the “Company”), and Steven W. Ryder, M.D. (“Indemnitee”).
RECITALS
A. Highly competent and experienced persons are reluctant to serve corporations as directors, executive officers or in other capacities unless they are provided with adequate protection through insurance and indemnification against claims and actions against them arising out of their service to and activities on behalf of the Company.
B. The Board of Directors of the Company (the “Board”) has determined that the inability to attract and retain such persons would be detrimental to the best interests of the Company and its stockholders and that the Company should act to assure such persons that there will be increased certainty of such protection in the future.
C. The Board has also determined that it is reasonable, prudent and necessary for the Company, in addition to purchasing and maintaining directors’ and officers’ liability insurance (or otherwise providing for adequate arrangements of self-insurance), contractually to obligate itself to indemnify such persons to the fullest extent permitted by applicable law so that they will serve or continue to serve the Company free from undue concern that they will not be adequately protected.
D. Indemnitee is willing to serve, continue to serve and to take on additional service for or on behalf of the Company, but only on the condition that Indemnitee be so indemnified to the fullest extent permitted by law, as permitted herein.
E. Article Thirteen of the Amended and Restated Certificate of Incorporation of the Company provides for indemnification of directors and officers to the fullest extent permitted by law.
In consideration of the foregoing and the mutual covenants herein contained, and other good and valuable consideration, the sufficiency and receipt of which are hereby acknowledged, the parties hereby agree as follows:
ARTICLE I
Certain Definitions
As used herein, the following words and terms shall have the following respective meanings (whether singular or plural):
“Acquiring Person” means any Person other than (i) the Company, (ii) any of the Company’s Subsidiaries, (iii) any employee benefit plan of the Company or of a Subsidiary of the Company or of a Company owned directly or indirectly by the stockholders of the Company in substantially the same proportions as their ownership of stock of the Company, or (iv) any trustee or other fiduciary holding securities under an employee benefit plan of the Company or of a Subsidiary of the Company or of a Company owned directly or indirectly by the stockholders of the Company in substantially the same proportions as their ownership of stock of the Company.
“Change in Control” means the occurrence of any of the following events:
(i) The acquisition, after the date of this Agreement, by any Person of beneficial ownership (within the meaning of Rule 13d-3 promulgated under the Exchange Act) of 40% or more of either (x) the then outstanding shares of Common Stock of the Company (the “Outstanding Company Common Stock”) or (y) the combined voting power of the then outstanding voting securities of the Company entitled to vote generally in the election of directors (the “Outstanding Company Voting Securities”); provided, however, that for purposes of this Subparagraph (i), the following acquisitions shall not constitute a Change of Control: (A) any acquisition directly from the Company, (B) any acquisition by the Company, (C) any acquisition by any employee benefit plan (or related trust) sponsored or maintained by the Company or any corporation controlled by the Company or (D) any acquisition by any corporation pursuant to a transaction which complies with clauses (A), (B) and (C) of paragraph (iii) below; or
(ii) Members of the Incumbent Board cease for any reason to constitute at least a majority of the Board; or
(iii) Consummation of a reorganization, merger or consolidation or sale or other disposition of all or substantially all of the assets of the Company or an acquisition of assets of another entity (a “Business Combination”), in each case, unless, following such Business Combination, (A) all or substantially all of the individuals and entities who were the beneficial owners, respectively, of the Outstanding Company Common Stock and Outstanding Company Voting Securities immediately prior to such Business Combination beneficially own, directly or indirectly, more than 50% of, respectively, the then outstanding shares of common equity and the combined voting power of the then outstanding voting securities entitled to vote generally in the election of directors or other similar governing body, as the case may be, of the entity resulting from such Business Combination (including, without limitation, an entity which as a result of such transaction owns the Company or all or substantially all of the Company’s assets either directly or through one or more subsidiaries) in substantially the same proportions as their ownership, immediately prior to such Business Combination of the Outstanding Company Common Stock and Outstanding Company Voting Securities, as the case may be, (B) no Person (excluding any employee benefit plan (or related trust) of the Company or the entity resulting from such Business Combination) beneficially owns, directly or indirectly, 40% or more of, respectively, the then outstanding shares of common equity of the entity resulting from such Business Combination or the combined voting power of the then outstanding voting securities of such entity except to the extent that such ownership results solely from ownership of the Company that existed prior to the Business Combination and (C) at least a majority of the members of the board of directors or other
2
similar governing body of the entity resulting from such Business Combination were members of the Incumbent Board at the time of the execution of the initial agreement, or of the action of the Board, providing for such Business Combination; or
(iv) Approval by the stockholders of the Company of a complete liquidation or dissolution of the Company.
“Claim” means an actual or threatened claim or request for relief which was, is or may be made by reason of anything done or not done by Indemnitee in, or by reason of any event or occurrence related to, Indemnitee’s Corporate Status, including any threatened, pending or completed action, suit, arbitration, investigation, inquiry, alternate dispute resolution mechanism, administrative or legislative hearing, or any other proceeding (including, without limitation, any securities laws action, suit, arbitration, alternative dispute resolution mechanism, hearing, or procedure) whether civil, criminal, administrative, arbitrative or investigative and whether or not based upon events occurring, or actions taken, before the date hereof, and any appeal in or related to any such action, suit, arbitration, investigation, hearing or procedure and any inquiry or investigation (including discovery), whether conducted by or in the right of the Company or any other Person, that Indemnitee in good faith believes could lead to any such action, suit, arbitration, alternative dispute resolution mechanism, hearing or other proceeding or appeal thereof.
“Corporate Status” means the status of a person who is, becomes or was a director, officer, employee, agent or fiduciary of the Company or is, becomes or was serving at the request of the Company as a director, officer, partner, member, venturer, proprietor, trustee, employee, agent, fiduciary or similar functionary of another foreign or domestic corporation, partnership, limited liability company, joint venture, sole proprietorship, trust, employee benefit plan or other enterprise. For purposes of this Agreement, the Company agrees that Indemnitee’s service on behalf of or with respect to any Subsidiary of the Company shall be deemed to be at the request of the Company.
“DGCL” means the Delaware General Corporation Law and any successor statute thereto, as either of them may from time to time be amended.
“Disinterested Director” with respect to any request by Indemnitee for indemnification hereunder, means a director of the Company who at the time of the vote is not a named defendant or respondent in the Claim in respect of which indemnification is sought by Indemnitee.
“Exchange Act” means the Securities Exchange Act of 1934.
“Expenses” means all attorneys’ fees and disbursements, retainers, accountant’s fees and disbursements, private investigator fees and disbursements, court costs, transcript costs, fees and expenses of experts, witness fees and expenses, costs and obligations under any bond posted in connection with any Claim, travel expenses, duplicating costs, printing and binding costs, telephone charges, postage, delivery service fees and all other disbursements, costs or expenses of the types customarily incurred in connection with prosecuting, defending (including affirmative defenses and counterclaims), preparing to prosecute or defend, investigating, being or preparing
3
to be a witness in, or otherwise participating in or preparing to participate in (including on appeal) a Claim and all interest or finance charges attributable to any thereof. Should any payments by the Company under this Agreement be determined to be subject to any federal, state or local income or excise tax, “Expenses” shall also include such amounts as are necessary to place Indemnitee in the same after-tax position (after giving effect to all applicable taxes) as Indemnitee would have been in had no such tax been determined to apply to such payments. Also, in this Agreement “witness” includes responding (or objecting) to a discovery request, whether in writing or in an oral deposition, in any Claim.
“Final Adjudication” means a final adjudication by a court from which there is no further right of appeal or a final adjudication of an arbitration pursuant to Section 5.1 if Indemnitee elects to seek such arbitration.
“Incumbent Board” means the individuals who, as of the date of this Agreement, constitute the Board and any other individual who becomes a director of the Company after that date and whose election or appointment by the Board or nomination for election by the Company’s stockholders was approved by a vote of at least a majority of the directors then comprising the Incumbent Board, but excluding, for this purpose, any such individual whose initial assumption of office occurs as a result of an actual or threatened election contest with respect to the election or removal of directors or other actual or threatened solicitation of proxies or consents by or on behalf of a Person other than the Incumbent Board.
“Independent Counsel” means a law firm, or a member of a law firm, that is experienced in matters of corporation law and neither contemporaneously is, nor in the five years theretofore has been, retained to represent: (a) the Company, any subsidiary of the Company, or Indemnitee in any matter material to either such Person (other than as Independent Counsel under this Agreement or similar agreements), (b) any other party to the Claim giving rise to a claim for indemnification hereunder or (c) the beneficial owner, directly or indirectly, of securities of the Company representing 5% or more of the combined voting power of the Company’s then outstanding voting securities, or Person controlled by such beneficial owner (other than, in each such case under clauses (a) through (c)), with respect to matters concerning the rights of Indemnitee under this Agreement, or of other indemnitees under similar indemnification agreements). Notwithstanding the foregoing, the term “Independent Counsel” shall not include any person who, under the applicable standards of professional conduct then prevailing, would have a conflict of interest in representing either the Company or Indemnitee in an action to determine Indemnitee’s rights under this Agreement.
“Independent Directors” means the directors on the Board that are independent directors as defined in NASDAQ Marketplace Rule 4200(a)(15) or successor provision, or, if the Company’s Common Stock is not then quoted on the NASDAQ Global Select Market, that qualify as independent, disinterested, or a similar term as defined in the rules of the principal securities exchange or inter-dealer quotation system on which the Company’s Common Stock is then listed or quoted.
4
“Person” means any individual, entity or group (within the meaning of Sections 13(d)(3) and 14(d)(2) of the Exchange Act).
“Potential Change in Control” shall be deemed to have occurred if (i) any Person shall have announced publicly an intention to effect a Change in Control, or commenced any action (such as the commencement of a tender offer for the Company’s Outstanding Company Common Stock or Outstanding Company Voting Securities or the solicitation of proxies for the election of any of the Company’s directors) that, if successful, could reasonably be expected to result in the occurrence of a Change in Control; (ii) the Company enters into an agreement, the consummation of which would constitute a Change in Control; or (iii) any other event occurs which the Board declares to be a Potential Change of Control.
“Subsidiary” means, with respect to any Person, any corporation or other entity of which a majority of the voting power of the voting equity securities or equity interest is owned, directly or indirectly, by that Person.
“Voting Securities” means any securities that vote generally in the election of directors, in the admission of general partners, or in the selection of any other similar governing body.
ARTICLE II
Services by Indemnitee
Indemnitee is serving as a director of the Company. Indemnitee may from time to time also agree to serve, as the Company may request from time to time, in another capacity for the Company (including an officer or director position) or as a director, officer, partner, member, venturer, proprietor, trustee, employee, agent, fiduciary or similar functionary of another foreign or domestic corporation, partnership, joint venture, limited liability company, sole proprietorship, trust, employee benefit plan or other enterprise. Indemnitee and the Company each acknowledge that they have entered into this Agreement as a means of inducing Indemnitee to serve, or continue to serve, the Company in such capacities. Indemnitee may at any time and for any reason resign from such position or positions (subject to any other contractual obligation or any obligation imposed by operation of law). The Company shall have no obligation under this Agreement to continue Indemnitee in any such position or positions.
ARTICLE III
Indemnification
Section 3.1 General. Subject to the provisions set forth in Article IV, the Company shall indemnify, and advance Expenses to, Indemnitee to the fullest extent permitted by applicable law in effect on the date hereof and to such greater extent as applicable law may hereafter from time to time permit. The other provisions set forth in this Agreement are provided in addition to and as a means of furtherance and implementation of, and not in limitation of, the obligations expressed in this Article III. No requirement, condition to or limitation of any right to indemnification or to advancement of Expenses under this Article III shall in any way limit the rights of Indemnitee under Article VII.
5
Section 3.2 Additional Indemnity of the Company. Indemnitee shall be entitled to indemnification pursuant to this Section 3.2 if, by reason of anything done or not done by Indemnitee in, or by reason of any event or occurrence related to, Indemnitee’s Corporate Status, Indemnitee is, was or becomes, or is threatened to be made, a party to, or witness or other participant in any Claim. Pursuant to this Section 3.2, Indemnitee shall be indemnified against any and all Expenses, judgments, penalties (including excise or similar taxes), fines and amounts paid in settlement (including all interest, assessments and other charges paid or payable in connection with or in respect of any such Expenses, judgments, penalties, fines and amounts paid in settlement) actually and reasonably incurred by Indemnitee or on Indemnitee’s behalf in connection with such Claim, issue or matter therein. Notwithstanding the foregoing, the obligations of the Company under this Section 3.2 shall be subject to the condition that no determination (which, in any case in which Independent Counsel is involved, shall be in a form of a written opinion) shall have been made pursuant to Article IV that Indemnitee would not be permitted to be indemnified under applicable law. Nothing in this Section 3.2 shall limit the benefits of Section 3.1, Section 3.3 or any other Section hereunder.
Section 3.3 Advancement of Expenses. The Company shall pay, on a current and as-incurred basis, all Expenses reasonably incurred by, or in the case of retainers to be incurred by, or on behalf of Indemnitee (or, if applicable, reimburse Indemnitee for any and all Expenses reasonably incurred by Indemnitee and previously paid by Indemnitee) in connection with any Claim, whether brought by the Company or otherwise, in advance of the later of (a) the final, non-appealable determination or resolution of all such Claims and (b) any determination respecting entitlement to indemnification pursuant to Article IV hereof (and shall continue to pay such Expenses after such determination and until it shall ultimately be determined (in a Final Adjudication) that Indemnitee is not entitled to be indemnified by the Company against such Expenses). Such payments and advances shall be made within 10 days after the receipt by the Company of a written request from Indemnitee requesting such payment or payments from time to time, whether prior to or after the final, non-appealable determination or resolution of such Claim. Any such payment by the Company is referred to in this Agreement as an “Expense Advance.” Any dispute as to the reasonableness of the incurrence of any Expense shall not delay an Expense Advance by the Company, and the Company agrees that any such dispute shall be resolved only upon the final, non-appealable determination or resolution of the respective underlying Claim involving Indemnitee. Indemnitee hereby undertakes and agrees that Indemnitee will reimburse and repay the Company without interest for any Expense Advances to the extent that it shall ultimately be determined (in a Final Adjudication) that Indemnitee is not entitled under the law to be indemnified by the Company against such Expenses. Indemnitee shall not be required to provide collateral or otherwise secure the undertaking and agreement described in the prior sentence. The Company shall make all Expense Advances pursuant to this Section 3.3 without regard to the financial ability of the Indemnitee to make repayment and without regard to whether or not the Indemnitee may ultimately be found to be entitled to indemnification under the provisions of this Agreement.
Section 3.4 Indemnification for Additional Expenses. The Company shall indemnify Indemnitee against any and all costs and expenses (of the types described in the definition of
6
Expenses in Article I) and, if requested by Indemnitee, shall (within two business days of that request) advance those costs and expenses to Indemnitee, that are incurred by Indemnitee in connection with any claim asserted against, or action brought by, Indemnitee for (i) indemnification or an Expense Advance by the Company under this Agreement or any other agreement or provision of the Company’s Certificate of Incorporation or Bylaws now or hereafter in effect relating to any Claim, (ii) recovery under any directors’ and officers’ liability insurance policies maintained by the Company, or (iii) enforcement of, or claims for breaches of, any provision of this Agreement, in each of the foregoing situations regardless of whether Indemnitee ultimately is determined to be entitled to that indemnification, Expense Advance payment, insurance recovery, enforcement, or damage claim, as the case may be, and regardless of whether the nature of the proceeding with respect to such matters is judicial, by arbitration, or otherwise.
Section 3.5 Partial Indemnity. If Indemnitee is entitled under any provision of this Agreement to indemnification by the Company for some or a portion of the Expenses, judgments, fines, penalties, and amounts paid in settlement of a Claim but not, however, for all of the total amount thereof, the Company shall nevertheless indemnify Indemnitee for the portion thereof to which Indemnitee is entitled. Moreover, notwithstanding any other provision of this Agreement, to the extent that Indemnitee has been successful on the merits or otherwise in defense of any or all Claims, or in defense of any issue or matter therein, including dismissal without prejudice, Indemnitee shall be indemnified against all Expenses incurred in connection therewith.
ARTICLE IV
Procedure for Determination of Entitlement to Indemnification
Section 4.1 Request by Indemnitee. To obtain indemnification under this Agreement, Indemnitee shall, at such time as determined by Indemnitee in Indemnitee’s sole discretion, submit to the Company a written request, including therein or therewith such documentation and information as is reasonably available to Indemnitee and is reasonably necessary to determine whether and to what extent Indemnitee is entitled to indemnification. The Secretary or an Assistant Secretary of the Company shall, promptly upon receipt of such a request for indemnification, advise the Board in writing that Indemnitee has requested indemnification. Nevertheless, any failure of Indemnitee to provide a request to the Company, or to provide such a requestwithin any time frame, shall not relieve the Company of any liability that it may have to Indemnitee hereunder.
Section 4.2 Determination of Request. Upon written request by Indemnitee for indemnification pursuant to the first sentence of Section 4.1 hereof, a determination, if required by applicable law, with respect to whether Indemnitee is permitted under applicable law to be indemnified shall be made in accordance with the terms of Section 4.5, in the specific case as set forth in this Section 4.2:
(a) If a Potential Change in Control or a Change in Control shall have occurred, by Independent Counsel (selected in accordance with Section 4.3) in a written opinion to the Board and Indemnitee, unless Indemnitee shall request that such determination be made by the Board, or a committee of the Board, in which case by the person or persons or in the manner provided for in clause (i) or (ii) of paragraph (b) below; or
7
(b) If a Potential Change in Control or a Change in Control shall not have occurred, then the determination shall be made by one of the following, in Indemnitee’s sole discretion, as the Indemnitee requests in writing: (i) by the Board by a majority vote of the Disinterested Directors even though less than a quorum of the Board, or (ii) by a majority vote of a committee solely of two or more Disinterested Directors designated to act in the matter by a majority vote of all Disinterested Directors even though less than a quorum of the Board, or (iii) by Independent Counsel selected by the Board or a committee of the Board by a vote as set forth in clauses (i) or (ii) of this paragraph (b), or if such vote is not obtainable or such a committee cannot be established, by a majority vote of all directors, or (iv) by the stockholders of the Company in a vote that excludes the shares held by directors who are not Disinterested Directors.
If it is so determined that Indemnitee is permitted to be indemnified under applicable law, payment to Indemnitee shall be made within 10 days after such determination. Nothing contained in this Agreement shall require that any determination be made under this Section 4.2 prior to the final, non-appealable determination or resolution of a Claim involving Indemnitee for which indemnification is sought hereunder; provided, that Expense Advances shall continue to be made by the Company pursuant to, and to the extent required by, the provisions of Article III. Indemnitee shall cooperate with the person or persons making such determination with respect to Indemnitee’s entitlement to indemnification, including providing to such person upon reasonable advance request any documentation or information that is not privileged or otherwise protected from disclosure and that is reasonably available to Indemnitee and reasonably necessary to such determination. Any costs or expenses (including attorneys’ fees and disbursements) incurred by Indemnitee in so cooperating with the person or persons making such determination shall be borne by the Company (irrespective of the determination as to Indemnitee’s entitlement to indemnification), and the Company shall indemnify and hold harmless Indemnitee therefrom.
Section 4.3 Independent Counsel. If the determination of entitlement to indemnification is to be made by Independent Counsel, the Independent Counsel shall be selected by Indemnitee, and Indemnitee shall give written notice to the Company, within 10 days after submission of Indemnitee’s request for indemnification, specifying the identity and address of the Independent Counsel so selected unless Indemnitee shall request that such selection be made by the Disinterested Directors or a committee of the Board, in which event the Company shall give written notice to Indemnitee within 10 days after receipt of Indemnitee’s request for the Board or a committee of the Disinterested Directors to make such selection, specifying the identity and address of the Independent Counsel so selected. In either event, (i) such notice to Indemnitee or the Company, as the case may be, shall be accompanied by a written confirmation by the Independent Counsel so selected that it satisfies the requirements of the definition of “Independent Counsel” in Article I and that it agrees to serve in such capacity and (ii) Indemnitee or the Company, as the case may be, may, within seven days after such written notice of selection shall have been given, deliver to the Company or to Indemnitee, as the case may be, a written objection to such selection. Any objection to the selection of Independent Counsel pursuant to this Section 4.3 may be asserted only on the ground that the Independent Counsel so selected does not meet the requirements of the definition of “Independent Counsel” in Article I, and the objection shall
8
set forth with particularity the factual basis of such assertion. If such written objection is timely made, the Independent Counsel so selected may not serve as Independent Counsel unless and until a court of competent jurisdiction (the “Court”) has determined that such objection is without merit or such objection is withdrawn. In the event of a timely written objection to a choice of Independent Counsel, the party originally selecting the Independent Counsel shall have seven days to make an alternate selection of Independent Counsel and to give written notice of such selection to the other party, after which time such other party shall have five days to make a written objection to such alternate selection. If, within 30 days after submission of Indemnitee’s request for indemnification pursuant to Section 4.1, no Independent Counsel shall have been selected and not objected to, either the Company or Indemnitee may petition the Court for resolution of any objection that shall have been made by the Company or Indemnitee to the other’s selection of Independent Counsel and/or for the appointment as Independent Counsel of a person selected by the Court or by such other person as the Court shall designate, and the person with respect to whom an objection is so resolved or the person so appointed shall act as Independent Counsel under Section 4.2. The Company shall pay any and all fees and expenses reasonably incurred by, such Independent Counsel in connection with acting pursuant to Section 4.2, and the Company shall pay all fees and expenses reasonably incurred incident to the procedures of this Section 4.3, regardless of the manner in which such Independent Counsel was selected or appointed. Upon the due commencement of any judicial proceeding or arbitration pursuant to Section 5.1, Independent Counsel shall be discharged and relieved of any further responsibility in such capacity (subject to the applicable standards of professional conduct then prevailing).
Section 4.4 Establishment of a Trust. In the event of a Potential Change in Control or a Change in Control, the Company shall, upon written request by Indemnitee, create a trust for the benefit of Indemnitee (the “Trust”) and from time to time upon written request of Indemnitee shall fund the Trust in an amount sufficient to satisfy any and all Expenses reasonably anticipated at the time of each such request to be incurred in connection with investigating, preparing for, and defending any Claim, and any and all judgments, fines, penalties, and settlement amounts of any and all Claims from time to time actually paid or claimed, reasonably anticipated, or proposed to be paid. The amount to be deposited in the Trust pursuant to the foregoing funding obligation shall be determined by the Independent Counsel (or other person(s) making the determination of whether Indemnitee is permitted to be indemnified by applicable law). The terms of the Trust shall provide that, upon a Change in Control, (i) the Trust shall not be revoked or the principal thereof invaded, without the written consent of Indemnitee; (ii) the trustee of the Trust shall advance to Indemnitee, within ten days of a request by Indemnitee, any and all Expenses reasonably incurred by, or in case of retainer to be incurred by, or on behalf of Indemnitee (or, if applicable, reimburse Indemnitee for any Expense reasonably incurred by Indemnitee and previously paid by Indemnitee), with any required determination concerning the reasonableness of the Expenses to be made by the Independent Counsel (and Indemnitee hereby agrees to reimburse the Trust under the circumstances in which Indemnitee would be required to reimburse the Company for Expense Advances under Section 3.3 of this Agreement); (iii) the Trust shall continue to be funded by the Company in accordance with the funding obligation set forth above; (iv) the trustee of the Trust shall promptly pay to Indemnitee all amounts for which Indemnitee shall be entitled to indemnification pursuant to this Agreement; and (v) all unexpended funds in the Trust shall revert to the Company upon a final determination by the Independent Counsel or a Final Adjudication,
9
as the case may be, that Indemnitee has been fully indemnified under the terms of this Agreement. The trustee of the Trust shall be chosen by Indemnitee and shall be an institution that is not affiliated with Indemnitee. Nothing in this Section 4.4 shall relieve the Company of any of its obligations under this Agreement.
Section 4.5 Presumptions and Effect of Certain Proceedings.
(a) Indemnitee shall be presumed to be entitled to indemnification under this Agreement upon submission of a request for indemnification under Section 4.1, and the Company shall have the burden of proof in overcoming that presumption in reaching a determination contrary to that presumption. Such presumption shall be used by Independent Counsel (or other person or persons determining entitlement to indemnification) as a basis for a determination of entitlement to indemnification unless the Company provides information sufficient to overcome such presumption by clear and convincing evidence or unless the investigation, review and analysis of Independent Counsel (or such other person or persons) convinces Independent Counsel by clear and convincing evidence that the presumption should not apply.
(b) If the person or persons empowered or selected under Article IV of this Agreement to determine whether Indemnitee is entitled to indemnification shall not have made a determination within 60 days after receipt by the Company of the request by Indemnitee therefor, the determination of entitlement to indemnification shall be deemed to have been made and Indemnitee shall be entitled to such indemnification; provided, however, that such 60-day period may be extended for a reasonable time, not to exceed an additional 30 days, if the person making the determination with respect to entitlement to indemnification in good faith requires such additional time for the obtaining or evaluating of documentation and/or information relating to such determination; and provided, further, that the 60-day limitation set forth in this Section 4.5(b) shall not apply and such period shall be extended as necessary (i) if within 30 days after receipt by the Company of the request for indemnification under Section 4.1 Indemnitee and the Company have agreed, and the Board has resolved, to submit such determination to the stockholders of the Company pursuant to Section 4.2(b) for their consideration at an annual meeting of stockholders to be held within 90 days after such agreement and such determination is made thereat, or a special meeting of stockholders is called within 30 days after such receipt for the purpose of making such determination, such meeting is held for such purpose within 60 days after having been so called and such determination is made thereat, or (ii) if the determination of entitlement to indemnification is to be made by Independent Counsel pursuant to Section 4.2(a) of this Agreement, in which case the applicable period shall be as set forth in Section 5.1(c).
(c) The termination of any Claim, issue or matter by judgment, order, settlement (whether with or without court approval) or conviction, or upon a plea of nolo contendere or its equivalent, shall not (except as otherwise expressly provided in this Agreement) by itself adversely affect the rights of Indemnitee to indemnification or create a presumption that Indemnitee failed to meet any particular standard of conduct, that Indemnitee had any
10
particular belief, or that a court has determined that indemnification is not permitted by applicable law. Indemnitee may be found to have failed to meet any particular standard of conduct in respect of any Claim, issue or matter only after Indemnitee shall have been so adjudged by the Court or arbitrator, as applicable, after exhaustion of all appeals therefrom.
(d) For purposes of the second sentence of Section 3.5, a settlement or other resolution of a Claim short of final judgment may be successful if it permits a party to avoid expense, delay, distraction, disruption and uncertainty. For purposes of the second sentence of Section 3.5, in the event that any Claim to which Indemnitee is a party is resolved in any manner other than by adverse judgment against Indemnitee (including settlement of such Claim with or without payment of money or other consideration), it shall be presumed that Indemnitee has been successful on the merits or otherwise in suchClaim. Anyone seeking to overcome this presumption shall have the burden of proof by clear and convincing evidence.
(e) The failure of the Company (including by its directors or Independent Counsel) to have made a determination before the commencement of any action pursuant to this Agreement that indemnification is proper because Indemnitee has met the applicable standard of conduct shall not be a defense to the action or create a presumption that Indemnitee has not met the standard of conduct.
ARTICLE V
Certain Remedies of Indemnitee
Section 5.1 Indemnitee Entitled to Adjudication in an Appropriate Court. If (a) a determination is made pursuant to Article IV that Indemnitee is not entitled to indemnification under this Agreement; (b) there has been any failure by the Company to make timely payment or advancement of any amounts due hereunder (including, without limitation, any Expense Advances); or (c) the determination of entitlement to indemnification is to be made by Independent Counsel pursuant to Section 4.2 and such determination shall not have been made and delivered in a written opinion within 60 days after the latest of (i) such Independent Counsel’s being appointed, (ii) the overruling by the Court of objections to such counsel’s selection, or (iii) expiration of all periods for the Company or Indemnitee to object to such counsel’s selection, Indemnitee shall be entitled to commence an action seeking an adjudication in the Court of Indemnitee’s entitlement to such indemnification or advancements due hereunder, including, without limitation, Expense Advances. Alternatively, Indemnitee, in Indemnitee’s sole discretion, may seek an award in arbitration to be conducted by a single arbitrator pursuant to the commercial arbitration rules of the American Arbitration Association. Indemnitee shall commence such action seeking an adjudication or an award in arbitration within 180 days following the date on which Indemnitee first has the right to commence such action pursuant to this Section 5.1, or such right shall expire. The Company agrees not to oppose Indemnitee’s right to seek any such adjudication or award in arbitration and it shall continue to pay Expense Advances pursuant to Section 3.3 until it shall ultimately be determined (in a Final Adjudication) that Indemnitee is not entitled to be indemnified by the Company against such Expenses.
11
Section 5.2 Adverse Determination Not to Affect any Judicial Proceeding. If a determination shall have been made pursuant to Article IV that Indemnitee is not entitled to indemnification under this Agreement, any judicial proceeding or arbitration commenced pursuant to this Article V shall be conducted in all respects as a de novo trial or arbitration on the merits, and Indemnitee shall not be prejudiced by reason of such initial adverse determination. In any judicial proceeding or arbitration commenced pursuant to this Article V, Indemnitee shall be presumed to be entitled to indemnification or advancement of Expenses, as the case may be, under this Agreement and the Company shall have the burden of proof in overcoming such presumption and to show by clear and convincing evidence that Indemnitee is not entitled to indemnification or advancement of Expenses, as the case may be.
Section 5.3 Company Bound by Determination Favorable to Indemnitee in any Judicial Proceeding or Arbitration. If a determination shall have been made or deemed to have been made pursuant to Article IV that Indemnitee is entitled to indemnification, the Company shall be irrevocably bound by such determination in any judicial proceeding or arbitration commenced pursuant to this Article V, and shall be precluded from asserting that such determination has not been made or that the procedure by which such determination was made is not valid, binding and enforceable.
Section 5.4 Company Bound by the Agreement. The Company shall be precluded from asserting in any judicial proceeding or arbitration commenced pursuant to this Article V that the procedures and presumptions of this Agreement are not valid, binding and enforceable and shall stipulate in any such court or before any such arbitrator that the Company is bound by all the provisions of this Agreement. Without limiting the generality of the preceding sentence, the Company shall not seek from a court, or agree to, a “bar order” that would have the effect of prohibiting or limiting Indemnitee’s rights to advancement of any Expenses under this Agreement.
ARTICLE VI
Contribution
Section 6.1 Contribution Payment.
(a) Whether or not the indemnification provided in Article III hereof is available, in respect of any threatened, pending or completed action, suit or Claim in which the Company is jointly liable with Indemnitee (or would be if joined in such action, or Claim), the Company shall pay, in the first instance, the entire amount of any judgment or settlement of such action, suit or Claim without requiring Indemnitee to contribute to such payment, and the Company hereby waives and relinquishes any right of contribution it may have against Indemnitee. The Company shall not enter into any settlement of any action, suit or Claim in which the Company is jointly liable with Indemnitee (or would be if joined in such action, suit or Claim) unless such settlement provides for a full and final release of all claims asserted against Indemnitee.
(b) Without diminishing or impairing the obligations of the Company set forth in the preceding subparagraph, if, for any reason, Indemnitee shall elect or be required to pay
12
all or any portion of any judgment or settlement in any threatened, pending or completed action, suit or Claim in which the Company is jointly liable with Indemnitee (or would be if joined in such action, suit or Claim), the Company shall contribute to the amount of Expenses, judgments, fines and amounts paid in settlement actually and reasonably incurred and paid or payable by Indemnitee in proportion to the relative benefits received by the Company and all officers, directors or employees of the Company, other than Indemnitee, who are jointly liable with Indemnitee (or would be if joined in such action, suit or Claim), on the one hand, and Indemnitee, on the other hand, from the transaction or events from which such action, suit or Claim arose; provided, however, that the proportion determined on the basis of relative benefit may, to the extent necessary to conform to law, be further adjusted by reference to the relative fault of the Company and all officers, directors or employees of the Company other than Indemnitee who are jointly liable with Indemnitee (or would be if joined in such action, suit or Claim), on the one hand, and Indemnitee, on the other hand, in connection with the transaction or events that resulted in such Expenses, judgments, fines or settlement amounts, as well as any other equitable considerations which applicable law may require to be considered.
(c) The Company hereby agrees, to the fullest extent permitted by applicable law, to fully indemnify and hold Indemnitee harmless from any claims of contribution which may be brought by officers, directors or employees of the Company, other than Indemnitee, who may be jointly liable with Indemnitee.
(d) To the fullest extent permissible under applicable law and without diminishing or impairing the obligations of the Company set forth in the preceding subparagraphs of this Section 6.1, if the indemnification provided for in this Agreement is unavailable to Indemnitee for any reason whatsoever, the Company, in lieu of indemnifying Indemnitee, shall contribute to the amount incurred by Indemnitee, whether for judgments, fines, penalties, excise taxes, amounts paid or to be paid in settlement and/or for Expenses, in connection with any claim relating to an indemnifiable event under this Agreement, in such proportion as is deemed fair and reasonable in light of all of the circumstances of such Claim in order to reflect (i) the relative benefits received by the Company and Indemnitee as a result of the event(s) and/or transaction(s) giving cause to suchClaim; and/or (ii) the relative fault of the Company (and its directors, officers, employees and agents) and Indemnitee in connection with such event(s) and/or transaction(s).
Section 6.2 Relative Fault. The relative fault of the Indemnitee, on the one hand, and of the Company and any and all other parties (including officers and directors of the Company other than Indemnitee) who may be at fault with respect to such matter shall be determined (i) by reference to the relative fault of Indemnitee as determined by the court or other governmental agency assessing the contribution amounts or (ii) to the extent such court or other governmental agency does not apportion relative fault, by the Independent Counsel (or such other party which makes a determination under Article IV) after giving effect to, among other things, the degree of which their actions were motivated by intent to gain personal profit or advantage, the degree to which their liability is primary or secondary, the degree to which their conduct is active or passive, the degree of the knowledge, access to information, and opportunity to prevent or correct the
13
subject matter of the Claims and other relevant equitable considerations of each party. The Company and Indemnitee agree that it would not be just and equitable if contribution pursuant to this Section 6.2 were determined by pro rata allocation or by any other method of allocation which does not take account of the equitable considerations referred to in this Section 6.2.
ARTICLE VII
Miscellaneous
Section 7.1 Non-Exclusivity. The rights of Indemnitee to receive indemnification and advancement of Expenses under this Agreement shall be in addition to, and shall not be deemed exclusive of, any other rights Indemnitee shall under the DGCL or other applicable law, the charter or bylaws of the Company, any other agreement, vote of stockholders or a resolution of directors, or otherwise. Every other right or remedy of Indemnitee shall be cumulative of the rights and remedies granted Indemnitee hereunder. No amendment or alteration of the charter or bylaws of the Company or any provision thereof shall adversely affect Indemnitee’s rights hereunder, and such rights shall be in addition to any rights Indemnitee may have under the charter, bylaws and the DGCL or other applicable law. To the extent that there is a change in the DGCL or other applicable law (whether by statute or judicial decision) that allows greater indemnification by agreement than would be afforded currently under the Company’s charter or bylaws and this Agreement, it is the intent of the parties hereto that Indemnitee shall enjoy by virtue of this Agreement the greater benefit so afforded by such change. Any amendment, alteration or repeal of the DGCL that adversely affects any right of Indemnitee shall be prospective only and shall not limit or eliminate any such right with respect to any Claim involving any occurrence or alleged occurrence of any action or omission to act that took place before the effective date of such amendment or repeal.
Section 7.2 Insurance and Subrogation.
(a) To the extent that the Company maintains an insurance policy or policies providing liability insurance for directors, officers, employees, agents or fiduciaries of the Company or for individuals serving at the request of the Company as directors, officers, partners, members, venturers, proprietors, trustees, employees, agents, fiduciaries or similar functionaries of another foreign or domestic corporation, partnership, limited liability company, joint venture, sole proprietorship, trust, employee benefit plan or other enterprise, Indemnitee shall be covered by such policy or policies in accordance with its or their terms to the maximum extent of the coverage available for any such director, officer, employee, agent or fiduciary under such policy or policies.
(b) In the event of any payment by the Company under this Agreement for which reimbursement is available under any insurance policy or policies obtained by the Company, the Company shall be subrogated to the extent of such payment to all of the rights of recovery of Indemnitee under such insurance policy or policies, who shall execute all papers required and take all action necessary to secure such rights, including execution of such documents as are necessary to enable the Company to bring suit to enforce such rights, provided that all Expenses relating to such action shall be borne by the Company.
14
(c) The Company shall not be liable under this Agreement to make any payment of amounts otherwise indemnifiable hereunder if and to the extent that Indemnitee has otherwise actually received such payment under the Company’s charter or bylaws or any insurance policy, contract, agreement or otherwise.
(d) If Indemnitee is a director of the Company, the Company will advise the Board of any proposed material reduction in the coverage for Indemnitee to be provided by the Company’s directors’ and officers’ liability insurance policy and will not effect such a reduction with respect to Indemnitee without the prior approval of at least 80% of the Independent Directors of the Company.
(e) If Indemnitee is a director of the Company during the term of this Agreement and if Indemnitee ceases to be a director of the Company for any reason, the Company shall procure a run-off directors’ and officers’ liability insurance policy with respect to claims arising from facts or events that occurred before the time Indemnitee ceased to be a director of the Company and covering Indemnitee, which policy, without any lapse in coverage, will provide coverage for a period of six years after the time Indemnitee ceased to be a director of the Company and will provide coverage (including amount and type of coverage and size of deductibles) that are substantially comparable to the Company’s directors’ and officers’ liability insurance policy that was most protective of Indemnitee in the 12 months preceding the time Indemnitee ceased to be a director of the Company and that is reasonably satisfactory to Indemnitee; provided, however, that:
(i) this obligation shall be suspended during the period immediately following the time Indemnitee ceases to be a director of the Company if and only so long as the Company has a directors’ and officers’ liability insurance policy in effect covering Indemnitee for such claims that, if it were a run-off policy, would meet or exceed the foregoing standards, but in any event this suspension period shall end when a Change in Control occurs; and
(ii) no later than the end of the suspension period provided in the preceding clause (i) (whether because of failure to have a policy meeting the foregoing standards or because a Change in Control occurs), the Company shall procure a run-off directors’ and officers’ liability insurance policy meeting the foregoing standards and lasting for the remainder of the six-year period.
(f) Notwithstanding the preceding clause (e) including the suspension provisions therein, if Indemnitee ceases to be an officer or a director of the Company in connection with a Change in Control or at or during the one-year period following the occurrence of a Change in Control, the Company shall procure a run-off directors’ and officers’ liability insurance policy covering Indemnitee that is reasonably satisfactory to Indemnity, meets the foregoing standards in clause (e), and lasts for a six-year period upon the Indemnitee’s ceasing to be an officer or a director of the Company in such circumstances.
15
(g) If at the time of the receipt of a notice of a Claim pursuant to the terms hereof, the Company has directors’ and officers’ liability insurance in effect, the Company shall give prompt notice of the commencement of such Claim to the insurers in accordance with the procedures set forth in the respective policies. The Company shall thereafter take all necessary or desirable action to cause such insurers to pay, on behalf of the Indemnitee, all amounts payable as a result of such Claim in accordance with the terms of such policies.
Section 7.3 Self Insurance of the Company; Other Arrangements. The parties hereto recognize that the Company may, but except as provided in Section 7.2(d), Section 7.2(e), and Section 7.2(f) is not required to, procure or maintain insurance or other similar arrangements, at its expense, to protect itself and any person, including Indemnitee, who is or was a director, officer, employee, agent or fiduciary of the Company or who is or was serving at the request of the Company as a director, officer, partner, member, venturer, proprietor, trustee, employee, agent, fiduciary or similar functionary of another foreign or domestic corporation, partnership, limited liability company, joint venture, sole proprietorship, trust, employee benefit plan or other enterprise against any expense, liability or loss asserted against or incurred by such person, in such a capacity or arising out of the person’s status as such a person, whether or not the Company would have the power to indemnify such person against such expense or liability or loss.
Except as provided in Section 7.2(d), Section 7.2(e) and Section 7.2(f), in considering the cost and availability of such insurance, the Company (through the exercise of the business judgment of its directors and officers) may, from time to time, purchase insurance which provides for certain (i) deductibles, (ii) limits on payments required to be made by the insurer, or (iii) coverage which may not be as comprehensive as that previously included in insurance purchased by the Company or its predecessors. The purchase of insurance with deductibles, limits on payments and coverage exclusions, even if in the best interest of the Company, may not be in the best interest of Indemnitee. As to the Company, purchasing insurance with deductibles, limits on payments and coverage exclusions is similar to the Company’s practice of self-insurance in other areas. In order to protect Indemnitee who would otherwise be more fully or entirely covered under such policies, the Company shall, to the maximum extent permitted by applicable law, indemnify and hold Indemnitee harmless to the extent (i) of such deductibles, (ii) of amounts exceeding payments required to be made by an insurer, or (iii) of amounts that prior policies of directors’ and officers’ liability insurance held by the Company or its predecessors have provided for payment to Indemnitee, if by reason of Indemnitee’s Corporate Status Indemnitee is or is threatened to be made a party to anyClaim. The obligation of the Company in the preceding sentence shall be without regard to whether the Company would otherwise be required to indemnify such officer or director under the other provisions of this Agreement, or under any law, agreement, vote of stockholders or directors or other arrangement. Without limiting the generality of any provision of this Agreement, the procedures in Article IV hereof shall, to the extent applicable, be used for determining entitlement to indemnification under this Section 7.3.
Section 7.4 Certain Settlement Provisions. The Company shall have no obligation to indemnify Indemnitee under this Agreement for amounts paid in settlement of a Claim without the Company’s prior written consent. The Company shall not settle any Claim in any manner that would impose any fine or other obligation on Indemnitee without Indemnitee’s prior written
16
consent. Neither the Company nor Indemnitee shall unreasonably withhold their consent to any proposed settlement.
Section 7.5 Duration of Agreement. This Agreement shall continue for so long as Indemnitee serves as a director, officer, employee, agent or fiduciary of the Company or, at the request of the Company, as a director, officer, partner, member, venturer, proprietor, trustee, employee, agent, fiduciary or similar functionary of another foreign or domestic corporation, partnership, limited liability company, joint venture, sole proprietorship, trust, employee benefit plan or other enterprise, and thereafter shall survive until and terminate upon the later to occur of: (a) the expiration of 20 years after the latest date that Indemnitee shall have ceased to serve in any such capacity; (b) the final non-appealable determination or resolution of all pending Claims in respect of which Indemnitee is granted rights of indemnification or advancement of Expenses hereunder and of any proceeding commenced by Indemnitee pursuant to Article IV relating thereto; or (c) the expiration of all statutes of limitation applicable to possible Claims arising out of Indemnitee’s Corporate Status.
Section 7.6 Notice by Each Party. Indemnitee shall promptly notify the Company in writing upon being served with any summons, citation, subpoena, complaint, indictment, information or other document or communication relating to any Claim for which Indemnitee may be entitled to indemnification or advancement of Expenses hereunder; provided, however, that any failure of Indemnitee to so notify the Company shall not adversely affect Indemnitee’s rights under this Agreement except to the extent the Company shall have been materially prejudiced as a direct result of such failure. The Company shall promptly notify Indemnitee in writing as to the pendency of any Claim that may involve a claim against Indemnitee for which Indemnitee may be entitled to indemnification or advancement of Expenses hereunder.
Section 7.7 Amendment. This Agreement may not be modified or amended except by a written instrument executed by or on behalf of each of the parties hereto.
Section 7.8 Waivers. The observance of any term of this Agreement may be waived (either generally or in a particular instance and either retroactively or prospectively) by the party entitled to enforce such term only by a writing signed by the party against which such waiver is to be asserted. Unless otherwise expressly provided herein, no delay on the part of any party hereto in exercising any right, power or privilege hereunder shall operate as a waiver thereof, nor shall any waiver on the part of any party hereto of any right, power or privilege hereunder operate as a waiver of any other right, power or privilege hereunder nor shall any single or partial exercise of any right, power or privilege hereunder preclude any other or further exercise thereof or the exercise of any other right, power or privilege hereunder.
Section 7.9 Entire Agreement. This Agreement and the documents expressly referred to herein constitute the entire agreement between the parties hereto with respect to the matters covered hereby, and any other prior or contemporaneous oral or written understandings or agreements with respect to the matters covered hereby, including without limitation any prior indemnification agreements, are expressly superseded by this Agreement.
17
Section 7.10 Severability. If any provision of this Agreement (including any provision within a single section, paragraph or sentence), or the application of such provision to any Person or circumstance, shall be judicially declared to be invalid, unenforceable or void, such decision will not have the effect of invalidating or voiding the remainder of this Agreement or affect the application of such provision to other Persons or circumstances, it being the intent and agreement of the parties that this Agreement shall be deemed amended by modifying such provision to the extent necessary to render it valid, legal and enforceable while preserving its intent, or if such modification is not possible, by substituting therefor another provision that is valid, legal and enforceable and that achieves the same objective. Any such finding of invalidity or unenforceability shall not prevent the enforcement of such provision in any other jurisdiction to the maximum extent permitted by applicable law.
Section 7.11 Notices. All notices and other communications hereunder shall be in writing and shall be deemed given upon (a) transmitter’s confirmation of a receipt of a facsimile transmission if during normal business hours of the recipient, otherwise on the next business day, (b) confirmed delivery of a standard overnight courier or when delivered by hand or (c) the expiration of five business days after the date mailed by certified or registered mail (return receipt requested), postage prepaid, to the parties at the following addresses (or at such other addresses for a party as shall be specified by like notice):
If to the Company, to it at:
Reata Pharmaceuticals, Inc.
5320 Legacy Dr.
Plano, Texas 75024
Attn: Corporate Secretary
Facsimile: 469-442-4740
If to Indemnitee, to Indemnitee at:
15 Vani Court
Westport, CT 06880
or to such other address or to such other individuals as any party shall have last designated by notice to the other parties. All notices and other communications given to any party in accordance with the provisions of this Agreement shall be deemed to have been given when delivered or sent to the intended recipient thereof in accordance with and as provided in the provisions of this Section 7.11.
Section 7.12 Governing Law. This Agreement and the legal relations among the parties shall, to the fullest extent permitted by law, be governed by, and construed and enforced in accordance with , the laws of the State of Delaware without regard to its conflict of laws rule.
18
Section 7.13 Submission to Jurisdiction. The Company and Indemnitee hereby irrevocably and unconditionally (a) agree that any action or proceeding arising out of or in connection with this Agreement (other than an arbitration provided for in Section 5.1) shall be brought only in the Court of Chancery of the State of Delaware (the “Delaware Court”), and not in any other state or federal court in the United States of America or any court in any other country, (b) consent to submit to the exclusive jurisdiction of the Delaware Court for the purposes of any action or proceeding arising out of or in connection with this Agreement, (c) waive any objection to the laying of venue of any such action or proceeding in the Delaware Court, and (d) waive, and agree not to plead or to make, any claim that any such action or proceeding brought in the Delaware Court has been brought in an improper or otherwise inconvenient forum.
Section 7.14 Certain Construction Rules.
(a) The article and section headings contained in this Agreement are for reference purposes only and shall not affect in any way the meaning or interpretation of this Agreement. As used in this Agreement, unless otherwise provided to the contrary, (1) all references to days shall be deemed references to calendar days and (2) any reference to a “Section” or “Article” shall be deemed to refer to a section or article of this Agreement. The words “hereof,” “herein” and “hereunder” and words of similar import referring to this Agreement refer to this Agreement as a whole and not to any particular provision of this Agreement. Whenever the words “include,” “includes” or “including” are used in this Agreement, they shall be deemed to be followed by the words “without limitation.” Unless otherwise specifically provided for herein, the term “or” shall not be deemed to be exclusive. Whenever the context may require, any pronoun used in this Agreement shall include the corresponding masculine, feminine or neuter forms and the singular form of nouns, pronouns and verbs shall include the plural and vice versa.
(b) For purposes of this Agreement, references to “other enterprises” shall include employee benefit plans; references to “fines” shall include any excise taxes assessed on a person with respect to any employee benefit plan; references to “serving at the request of the Company” shall include any service as a director, officer, employee or agent of the Company which imposes duties on, or involves services by, such director, nominee, officer, employee or agent with respect to an employee benefit plan, its participants or beneficiaries; and a person who acted in good faith and in a manner the person reasonably believed to be in the interests of the participants and beneficiaries of an employee benefit plan shall be deemed to have acted in a manner “not opposed to the best interest of the Company” for purposes of this Agreement and the DGCL.
(c) In the event of a merger, consolidation or amalgamation of the Company with or into any other entity, references to the “Company” shall include the entity surviving or resulting from the merger, consolidation or amalgamation as well as the Company, and Indemnitee shall stand in the same position under this Agreement with respect to the surviving or resulting entity as Indemnitee would stand with respect to the Company if its existence had continued upon and after the merger, consolidation or amalgamation.
19
Section 7.15 Counterparts. This Agreement may be executed in two or more counterparts, each of which shall be deemed to be an original and all of which together shall be deemed to be one and the same instrument, notwithstanding that both parties are not signatories to the original or same counterpart.
Section 7.16 Certain Persons Not Entitled to Indemnification. Notwithstanding any other provision of this Agreement (but subject to Section 7.1), Indemnitee shall not be entitled to indemnification or advancement of Expenses pursuant to the terms of this Agreement with respect to any Claim, issue or matter therein, brought or made by Indemnitee against the Company, except as specifically provided in Article III, Article IV or Section 7.3. In addition, the Company shall not be obligated pursuant to the terms of this Agreement:
(a) To indemnify Indemnitee if (and to the extent that) a final, non-appealable decision by a court or arbitration body having jurisdiction in the matter shall determine that such indemnification is not lawful; or
(b) To indemnify Indemnitee for the payment to the Company of profits pursuant to Section 16(b) of the Exchange Act, or Expenses incurred by Indemnitee for Claims in connection with such payment under Section 16(b) of the Exchange Act.
Section 7.17 Indemnification for Negligence, Gross Negligence, etc. Without limiting the generality of any other provision hereunder, it is the express intent of this Agreement that Indemnitee be indemnified and Expenses be advanced regardless of Indemnitee’s acts of negligence, gross negligence, intentional or willful misconduct to the extent that indemnification and advancement of Expenses is allowed pursuant to the terms of this Agreement and under applicable law.
Section 7.18 Mutual Acknowledgments. Both the Company and Indemnitee acknowledge that, in certain instances, applicable law (including applicable federal law that may preempt or override applicable state law) or public policy may prohibit the Company from indemnifying the directors, officers, employees, agents or fiduciaries of the Company under this Agreement or otherwise. For example, the Company and Indemnitee acknowledge that the U.S. Securities and Exchange Commission has taken the position that indemnification of directors, officers and controlling Persons of the Company for liabilities arising under federal securities laws is against public policy and, therefore, unenforceable. Indemnitee understands and acknowledges that the Company has undertaken or may be required in the future to undertake with the Securities and Exchange Commission to submit the question of indemnification to a court in certain circumstances for a determination of the Company’s right under public policy to indemnify Indemnitee. In addition, the Company and Indemnitee acknowledge that federal law prohibits indemnifications for certain violations of the Employee Retirement Income Security Act of 1974, as amended.
Section 7.19 Enforcement. The Company agrees that its execution of this Agreement shall constitute a stipulation by which it shall be irrevocably bound in any court or arbitration in which a proceeding by Indemnitee for enforcement of Indemnitee’s rights hereunder shall have been
20
commenced, continued or appealed, that its obligations set forth in this Agreement are unique and special, and that failure of the Company to comply with the provisions of this Agreement will cause irreparable and irremediable injury to Indemnitee, for which a remedy at law will be inadequate. As a result, in addition to any other right or remedy Indemnitee may have at law or in equity with respect to breach of this Agreement, Indemnitee shall be entitled to injunctive or mandatory relief directing specific performance by the Company of its obligations under this Agreement. The Company agrees not to seek, and agrees to waive any requirement for the securing or posting of, a bond in connection with Indemnitee’s seeking or obtaining such relief.
Section 7.20 Successors and Assigns. All of the terms and provisions of this Agreement shall be binding upon, shall inure to the benefit of and shall be enforceable by the parties hereto and their respective successors, assigns, heirs, executors, administrators, legal representatives.
Section 7.21 Period of Limitations. No legal action shall be brought and no cause of action shall be asserted by or on behalf of the Company or any affiliate of the Company against Indemnitee or Indemnitee’s spouse, heirs, executors, or personal or legal representatives after the expiration of one year from the date of accrual of that cause of action, and any claim or cause of action of the Company or its affiliate shall be extinguished and deemed released unless asserted by the timely filing of a legal action within that one-year period; provided, however, that for any claim based on Indemnitee’s breach of fiduciary duties to the Company or its stockholders, the period set forth in the preceding sentence shall be three years instead of one year; and provided, further, that, if any shorter period of limitations is otherwise applicable to any such cause of action, the shorter period shall govern.
[signatures on following page]
21
IN WITNESS WHEREOF, this Agreement has been duly executed and delivered to be effective as of the date first above written.
REATA PHARMACEUTICALS, INC.
By: /s/ J. Warren Huff
Name: J. Warren Huff
Title: Chief Executive Officer
INDEMNITEE:
By: /s/ Steven W. Ryder
Name: Steven W. Ryder, M.D.
22
Exhibit 31.1
CERTIFICATION PURSUANT TO
RULES 13a-14(a) AND 15d-14(a) UNDER THE SECURITIES EXCHANGE ACT OF 1934,
AS ADOPTED PURSUANT TO SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, J. Warren Huff, certify that:
Date: August 8, 2022 |
|
By: |
/s/ J. Warren Huff |
|
|
|
J. Warren Huff |
|
|
|
Chief Executive Officer |
|
|
|
(Principal Executive Officer) |
Exhibit 31.2
CERTIFICATION PURSUANT TO
RULES 13a-14(a) AND 15d-14(a) UNDER THE SECURITIES EXCHANGE ACT OF 1934,
AS ADOPTED PURSUANT TO SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, Manmeet S. Soni, certify that:
Date: August 8, 2022 |
|
By: |
/s/ Manmeet S. Soni |
|
|
|
Manmeet S. Soni |
|
|
|
Chief Operating Officer, Chief Financial Officer and President |
|
|
|
(Principal Financial Officer) |
Exhibit 32.1
CERTIFICATION PURSUANT TO
18 U.S.C. SECTION 1350, AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the Quarterly Report of Reata Pharmaceuticals, Inc. (the “Company”) on Form 10-Q for the quarter ended June 30, 2022 as filed with the Securities and Exchange Commission on the date hereof (the “Report”), I, J. Warren Huff, as Chief Executive Officer of the Company, certify, pursuant to 18 U.S.C. § 1350, as adopted pursuant to § 906 of the Sarbanes-Oxley Act of 2002, that:
Date: August 8, 2022 |
|
By: |
/s/ J. Warren Huff |
|
|
|
J. Warren Huff |
|
|
|
Chief Executive Officer |
|
|
|
(Principal Executive Officer) |
Exhibit 32.2
CERTIFICATION PURSUANT TO
18 U.S.C. SECTION 1350, AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the Quarterly Report of Reata Pharmaceuticals, Inc. (the “Company”) on Form 10-Q for the quarter ended June 30, 2022 as filed with the Securities and Exchange Commission on the date hereof (the “Report”), I, Manmeet S. Soni, as Chief Operating Officer, Chief Financial Officer and President of the Company, certify, pursuant to 18 U.S.C. § 1350, as adopted pursuant to § 906 of the Sarbanes-Oxley Act of 2002, that:
Date: August 8, 2022 |
|
By: |
/s/ Manmeet S. Soni |
|
|
|
Manmeet S. Soni |
|
|
|
Chief Operating Officer, Chief Financial Officer and President |
|
|
|
(Principal Financial Officer) |
Serious News for Serious Traders! Try StreetInsider.com Premium Free!
You May Also Be Interested In
- Irish Residential Properties Sub-Fund 1: Form 8.3 - Irish Residential Properties REIT PLC
- Proposed Increase in Size of Offer for Subscription and Re-Opening of Offer for Subscription to Further Applications
- Progress on ABN AMRO share buyback programme 12 April – 18 April 2024
Create E-mail Alert Related Categories
SEC FilingsSign up for StreetInsider Free!
Receive full access to all new and archived articles, unlimited portfolio tracking, e-mail alerts, custom newswires and RSS feeds - and more!
