

Form 10-K ARROWHEAD PHARMACEUTICAL For: Sep 30
 Tweet
Tweet Share
ShareUNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
__________________________________________________________________
FORM 10-K
__________________________________________________________________
(Mark One)
| ANNUAL REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |||||
For the fiscal year ended September 30 , 2022
| TRANSITION REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |||||
For the transition period from to
Commission file number 001-38042
__________________________________________________________________
(Exact name of registrant as specified in its charter)
__________________________________________________________________
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | ||||
(626 ) 304-3400
(Address and telephone number of principal executive offices)
__________________________________________________________________
Securities registered pursuant to Section 12(b) of the Exchange Act:
Title of each class | Trading Symbol(s) | Name of each exchange on which registered | ||||||
Securities registered pursuant to Section 12(g) of the Exchange Act: None
Indicate by a check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes x No o
Indicate by a check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes x No o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| x | Accelerated filer | o | Non-accelerated filer | o | o | |||||||||||||||||||||||||||
| o | ||||||||||||||||||||||||||||||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. o
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. x
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No x
The aggregate market value of issuer’s voting and non-voting outstanding common stock held by non-affiliates was approximately $4.1 billion based upon the closing stock price of issuer’s common stock on March 31, 2022. Shares of common stock held by each officer and director and by each person who is known to own 10% or more of the outstanding common stock have been excluded in that such persons may be deemed to be affiliates of the Company. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
As of November 16, 2022, 106,005,722 shares of the issuer’s Common Stock were issued and outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
F-1 | ||||||||
FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains certain forward-looking statements within the meaning of Section 27A of the Securities Act of 1933 and Section 21E of the Securities Exchange Act of 1934, and we intend that such forward-looking statements be subject to the safe harbors created thereby. For this purpose, any statements contained in this Annual Report on Form 10-K except for historical information may be deemed to be forward-looking statements. Without limiting the generality of the foregoing, words such as “may,” “will,” “expect,” “believe,” “anticipate,” “intend,” “plan,” “project,” “could,” “estimate,” “target,” “forecast,” or “continue” or the negative of these words or other variations thereof or comparable terminology are intended to identify forward-looking statements. In addition, any statements that refer to projections of our future financial performance, trends in our business, or other characterizations of future events or circumstances are forward-looking statements.
The forward-looking statements included herein are based on current expectations of our management based on available information and involve a number of risks and uncertainties, all of which are difficult or impossible to predict accurately, and many of which are beyond our control. As such, our actual results may differ materially from those expressed in any forward-looking statements. Factors that may cause or contribute to such differences include, but are not limited to, those discussed in more detail in “Item 1. Business” and “Item 1A. Risk Factors” of Part I and “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” of Part II of this Annual Report on Form 10-K. Readers should carefully review these risks, as well as the additional risks described in other documents we file from time to time with the Securities and Exchange Commission (the “SEC”). In light of the significant risks and uncertainties inherent in the forward-looking information included herein, the inclusion of such information should not be regarded as a representation by us or any other person that such results will be achieved, and readers are cautioned not to place undue reliance on such forward-looking information. Except as may be required by law, we disclaim any intent to revise the forward-looking statements contained herein to reflect events or circumstances after the date hereof or to reflect the occurrence of unanticipated events.
PART I
Unless otherwise noted, (1) the term “Arrowhead” refers to Arrowhead Pharmaceuticals, Inc., a Delaware corporation and its Subsidiaries, (2) the terms “Company,” “we,” “us,” and “our,” refer to the ongoing business operations of Arrowhead and its Subsidiaries, whether conducted through Arrowhead or a subsidiary of Arrowhead, (3) the term “Subsidiaries” refers to Arrowhead Madison Inc. (“Arrowhead Madison”), Arrowhead Australia Pty Ltd (“Arrowhead Australia”), and Visirna Therapeutics Inc. (“Visirna”) (4) the term “common stock” refers to Arrowhead’s common stock, (5) the term “preferred stock” refers to Arrowhead’s preferred stock and (6) the term “stockholder(s)” refers to the holders of Arrowhead common stock.
ITEM 1.BUSINESS
Overview
Arrowhead develops medicines that treat intractable diseases by silencing the genes that cause them. Arrowhead's pipeline of 12 development programs ranges from pre-clinical discovery to late-stage clinical development, and it is focused on developing its most advanced therapies for late-stage clinical trials. Using a broad portfolio of RNA chemistries and efficient modes of delivery, Arrowhead therapies trigger the RNA interference mechanism to induce rapid, deep and durable knockdown of target genes.

RNA Interference and the Benefits of RNAi Therapeutics
RNA interference (“RNAi”) is a mechanism present in living cells that inhibits the expression of a specific gene, thereby affecting the production of a specific protein. RNAi-based therapeutics may leverage this natural pathway of gene silencing to target and shut down specific disease-causing genes.
Small molecule and antibody drugs have proven effective at inhibiting certain cell surface, intracellular, and extracellular targets. However, other drug targets have proven difficult to inhibit with traditional drug-based and biologic therapeutics. Developing effective drugs for these targets would have the potential to address large underserved markets for the treatment of many diseases. Using the ability to specifically silence any gene, RNAi therapeutics may be able to address previously “undruggable” targets, unlocking the market potential of such targets.
1
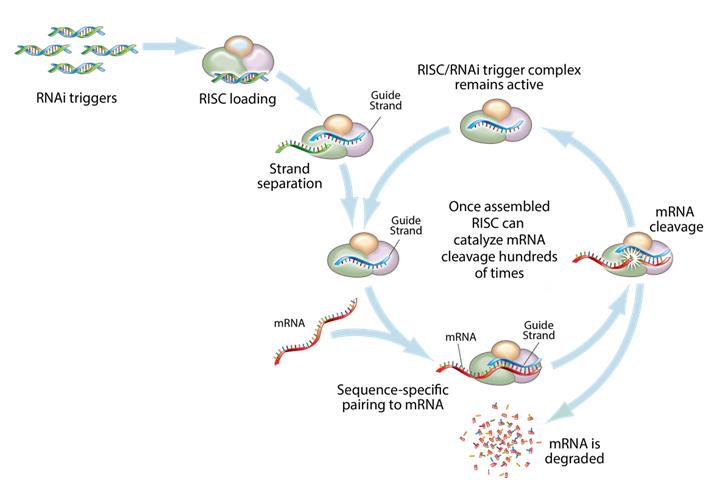
This figure depicts the mechanism by which gene silencing occurs. Double stranded RNAi triggers are introduced into a cell and are loaded into the RNA-induced silencing complex, (“RISC”). The strands are then separated, leaving an active RISC/RNAi trigger complex. This complex can then pair with and degrade the complementary messenger RNAs (“mRNA”) and stop the production of the target proteins. RNAi is a catalytic process, so each RNAi trigger can degrade mRNA hundreds of times, which results in a relatively long duration of effect for RNAi therapeutics.
Key Benefits of RNAi as a Therapeutic Modality
•Silences the expression of disease associated genes;
•Potential to address any target in the transcriptome including previously “undruggable” targets;
•Rapid lead identification;
•High specificity;
•Opportunity to use multiple RNA sequences in one drug product for synergistic silencing of related targets; and
•RNAi therapeutics are uniquely suited for personalized medicine through target and cell specific delivery and gene knockdown.
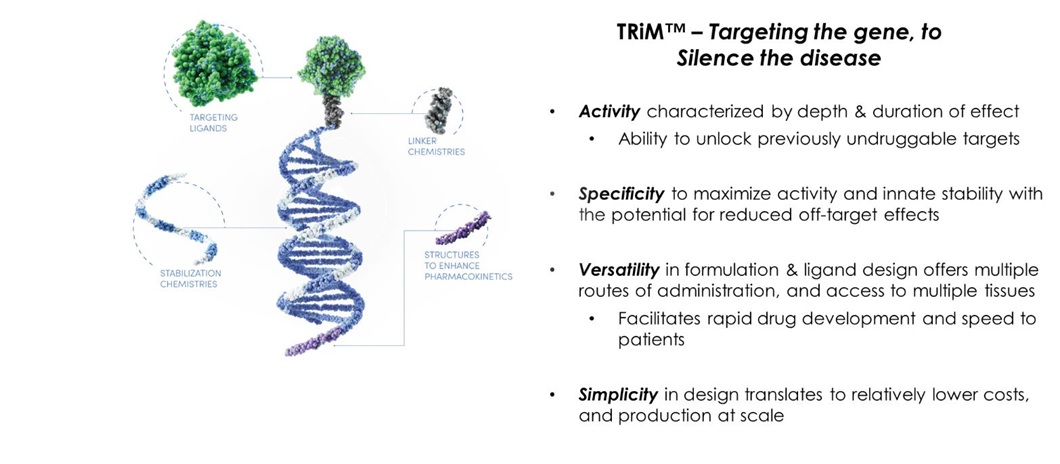
Targeted RNAi Molecule (TRiMTM) Platform
Arrowhead’s Targeted RNAi Molecule (TRiMTM) platform utilizes ligand-mediated delivery and is designed to enable tissue-specific targeting while being structurally simple. Targeting has been core to Arrowhead’s development philosophy and the TRiMTM platform builds on more than a decade of work on actively targeted drug delivery vehicles. Arrowhead scientists have discovered ways to progressively “TRiM” away extraneous features and chemistries and retain optimal pharmacologic activity.
The TRiMTM platform is comprised of a highly potent RNA trigger identified using Arrowhead’s proprietary trigger selection rules and algorithms with the following components optimized, as needed, for each drug candidate: a high affinity targeting ligand; various linker and chemistries; structures that enhance pharmacokinetics; and highly potent RNAi triggers with sequence specific stabilization chemistries.
Therapeutics developed with the TRiMTM platform offer several advantages: simplified manufacturing and reduced costs; multiple routes of administration; and potential for improved safety because there are less metabolites from smaller molecules, thereby reducing the risk of intracellular buildup. Arrowhead also believes that for RNAi to reach its true potential, it must target organs outside the liver. Arrowhead is leading this expansion with the TRiMTM platform, which has shown the potential to reach multiple tissues, including liver, lung, tumor and muscle, and Arrowhead believes that it has the potential to reach other tissues as well.
2
RNA Chemistries
The structure and chemistries of the oligonucleotide molecules used to trigger the RNAi mechanism can be tailored for optimal activity. Arrowhead’s broad portfolio of RNA trigger structures and chemistries, including some proprietary structures, enable the Company to optimize each drug candidate on a target-by-target basis and utilize the combination of structure and chemical modifications that yield the most potent RNAi trigger.
As a component of the TRiMTM platform, Arrowhead’s design philosophy for RNA chemical modifications is to start with a structurally simple molecule and add only selective modification and stabilization chemistries as necessary to achieve the desired level of target knockdown and duration of effect. The conceptual framework for the stabilization strategy starts with a more sophisticated RNAi trigger screening and selection process that identifies potent sequences rapidly in locations that others may miss.
Pipeline
Arrowhead is focused on developing innovative drugs for diseases with a genetic basis, typically characterized by the overproduction of one or more proteins that are involved with disease. The depth and versatility of Arrowhead's RNAi technologies enables Arrowhead to potentially address conditions in virtually any therapeutic area and pursue disease targets that are not otherwise addressable by small molecules and biologic. Arrowhead is focused on bringing the promise of RNAi to address diseases outside of the liver, and its pipeline now includes disease targets in the liver, lung, muscle and other undisclosed tissue types.
3
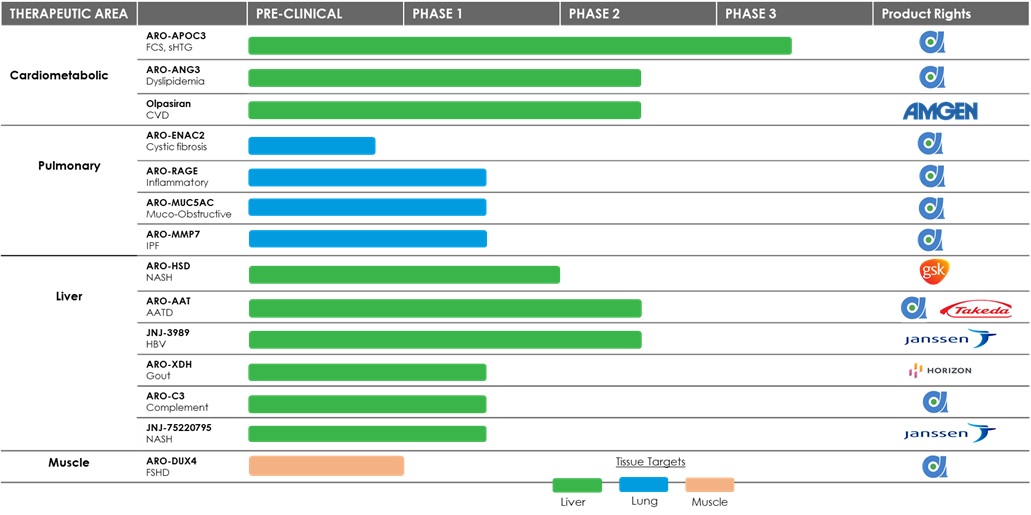
ARO-APOC3
ARO-APOC3 is designed to reduce production of Apolipoprotein C-III (apoC-III), a component of triglyceride rich lipoproteins (TRLs) including Very Low Density Lipoprotein (VLDL) and chylomicrons and a key regulator of triglyceride metabolism. Arrowhead believes that knocking down the hepatic production of apoC-III may result in reduced VLDL synthesis and assembly, enhanced breakdown of TRLs, and better clearance of VLDL and chylomicron remnants. Arrowhead is currently investigating ARO-APOC3 in two Phase 2b clinical trials and one Phase 3 clinical trial.
Hypertriglyceridemia: Elevated triglyceride levels are an independent risk factor for cardiovascular disease. Severely elevated triglycerides (often over 2,000 mg/dL) in patients with familial chylomicronemia syndrome (FCS), a rare genetic disorder, can result in potentially fatal acute pancreatitis.
Study Name: Study to Evaluate ARO-APOC3 in Adults With Severe Hypertriglyceridemia (SHASTA-2)
A Double-Blind, Placebo-Controlled Phase 2b Study to Evaluate the Efficacy and Safety of ARO-APOC3 in Adults With Severe Hypertriglyceridemia
ClinicalTrials.gov Identifier: NCT04720534
Study Name: Study of ARO-APOC3 in Adults With Mixed Dyslipidemia (MUIR)
A Double-Blind, Placebo-Controlled Phase 2b Study to Evaluate the Efficacy and Safety of ARO-APOC3 in Adults With Mixed Dyslipidemia
ClinicalTrials.gov Identifier: NCT04998201
Study Name: Study of ARO-APOC3 in Adults With FCS (PALISADE)
A Phase 3 Study to Evaluate the Efficacy and Safety of ARO-APOC3 in Adults With Familial Chylomicronemia Syndrome
ClinicalTrials.gov Identifier: NCT05089084
ARO-ANG3
ARO-ANG3 is designed to reduce production of angiopoietin-like protein 3 (ANGPTL3), a liver synthesized inhibitor of lipoprotein lipase and endothelial lipase. ANGPTL3 inhibition has been shown to lower serum LDL, serum and liver triglyceride and has genetic validation as a novel target for cardiovascular disease. Arrowhead is currently investigating ARO-ANG3 in two Phase 2b clinical trials.
Dyslipidemia and Hypertriglyceridemia: Dyslipidemia and hypertriglyceridemia are risk factors for atherosclerotic coronary heart disease and cardiovascular events.
Study Name: Study of ARO-ANG3 in Adults With Mixed Dyslipidemia (ARCHES-2)
A Double-blind, Placebo-controlled Phase 2b Study to Evaluate the Efficacy and Safety of ARO-ANG3 in Adults With Mixed Dyslipidemia
ClinicalTrials.gov Identifier: NCT04832971
4
Study Name: Study of ARO-ANG3 in Participants With Homozygous Familial Hypercholesterolemia (HoFH) (GATEWAY)
Phase 2 Study to Evaluate the Safety and Efficacy of ARO-ANG3 in Subjects with Homozygous Familial Hypercholesterolemia (HoFH)
ClinicalTrials.gov Identifier: NCT05217667
ARO-ENaC2
ARO-ENaC2 is designed to reduce production of the epithelial sodium channel alpha subunit (αENaC) in the airways of the lung. In cystic fibrosis patients, increased ENaC activity contributes to airway dehydration and reduced mucociliary transport.
Cystic Fibrosis: Cystic fibrosis (CF) is a rare disease caused by a genetic mutation that leads to mucus buildup in the lungs and pancreas. In CF lung disease, patients can have difficulty breathing and experience frequent and persistent lung infections.
ARO-RAGE
ARO-RAGE is designed to reduce production of the Receptor for Advanced Glycation End products (RAGE) as a potential treatment for various inflammatory pulmonary diseases. Arrowhead is currently investigating ARO-RAGE in a Phase 1/2 clinical trial.
Study Name: Study of ARO-RAGE in Healthy Subjects and Patients With Asthma
A Phase 1/2a Study Evaluating the Effects of ARO-RAGE in Healthy Subjects and Patients With Asthma ClinicalTrials.gov Identifier: NCT05276570
ARO-MUC5AC
ARO-MUC5AC is designed to reduce production of mucin 5AC (MUC5AC) as a potential treatment for various muco-obstructive pulmonary diseases. Arrowhead is currently investigating ARO-MUC5AC in a phase 1/2 clinical trial.
Study Name: Study of ARO-MUC5AC in Healthy Subjects and Patients With Asthma
A Phase 1/2a Study to Evaluate the Effects of ARO-MUC5AC in Healthy Subjects and Patients with Asthma
ClinicalTrials.gov Identifier: NCT05292950
ARO-MMP7
ARO-MMP7 is designed to reduce expression of matrix metalloproteinase 7 (MMP7) as a potential treatment for idiopathic Pulmonary Fibrosis (IPF). Arrowhead is currently investigating ARO-MMP7 in a Phase 1/2 clinical trial.
Study Name: Study of ARO-MMP7 Inhalation Solution in Healthy Subjects and Patients With Idiopathic Pulmonary Fibrosis
A Phase 1/2a Study Evaluating the Effects of ARO-MMP7 Inhalation Solution in Healthy Subjects and Patients With Idiopathic Pulmonary Fibrosis
ClinicalTrials.gov Identifier: NCT05537025
ARO-C3
ARO-C3 is designed to reduce production of complement component 3 (C3) as a potential therapy for patients with various complement mediated or complement associated renal and hematological diseases. Arrowhead is currently investigating ARO-C3 in a Phase 1/2 clinical trial.
Paroxysmal Nocturnal Hematuria: Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, acquired, clonal disorder involving hematopoietic stem cells and is characterized by destruction of red blood cells, blood clots, and impaired bone marrow function. Silencing hepatic C3 expression may be a viable therapeutic approach for systemic C3 inhibition in PNH.
Complement-Mediated Renal Disease: A number of rare renal diseases result from uncontrolled activation of the alternative pathway of complement, leading to progressive glomerular damage, proteinuria, hematuria, and impaired kidney function, and often resulting in end-stage renal disease (ESRD). In addition, dysregulation of the alternative complement pathway has been shown to play a role in the pathogenesis and progression of disease in some of the more common glomerulopathies. Silencing C3 may be a therapeutic approach for treatment of these conditions.
Study Name: Study of ARO-C3 in Adult Healthy Volunteers and Patients With Paroxysmal Nocturnal Hemoglobinuria and Complement-Mediated Renal Disease
5
A Phase 1/2a Dose-Escalating Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and/or Pharmacodynamics of ARO-C3 in Adult Healthy Volunteers and in Adult Patients With Paroxysmal Nocturnal Hemoglobinuria and Adult Patients With Complement-Mediated Renal Disease
ClinicalTrials.gov Identifier: NCT05083364
ARO-DUX4
ARO-DUX4 is designed to target the gene that encodes human double homeobox 4 (DUX4) protein as a potential treatment for patients with facioscapulohumeral muscular dystrophy.
Facioscapulohumeral Muscular Dystrophy: Facioscapulohumeral muscular dystrophy (FSHD) is an autosomal dominant disease associated with the failure to maintain complete epigenetic suppression of DUX4 expression in differentiated skeletal muscle, leading to overexpression of DUX4, which is myotoxic and can lead to muscle degeneration. As DUX4 expression is recognized as the cause of muscle pathology in FSHD patients, Arrowhead believes that the selective targeting and knockdown of DUX4 using RNAi may prevent or reverse downstream myotoxicity and lead to muscle repair and improvement in muscle function in patients. There are currently no effective treatments specifically for FSHD.
Collaboration and License Agreements
Glaxosmithkline Intellectual Property (No. 3) Limited (“GSK”)
On November 22, 2021, GSK and Arrowhead entered into an Exclusive License Agreement (the “GSK License Agreement”). Under the GSK License Agreement, GSK has received an exclusive license for ARO-HSD. The exclusive license is worldwide with the exception of greater China, for which Arrowhead retained rights to develop and commercialize ARO-HSD. Arrowhead has completed its Phase 1/2 study of ARO-HSD, and GSK is now wholly responsible for all clinical development and commercialization of ARO-HSD in its territory. Under the terms of the agreement, Arrowhead has received an upfront payment of $120 million and is eligible for additional payments of $30 million at the start of Phase 2 and $100 million upon achieving a successful Phase 2 trial readout and the first patient dosed in a Phase 3 trial. Furthermore, should the Phase 3 trial read out positively, and the potential new medicine receives regulatory approval in major markets, the deal provides for commercial milestone payments to Arrowhead of up to $190 million at first commercial sale, and up to $590 million in sales-related milestone payments. Arrowhead is further eligible to receive tiered royalties on net product sales in a range of mid-teens to twenty percent.
ARO-HSD
ARO-HSD is designed to reduce production of HSD17B13, a hydroxysteroid dehydrogenase involved in the metabolism of hormones, fatty acids and bile acids. Published human genetic data indicate that a loss of function mutation in HSD17B13 provides strong protection against nonalcoholic steatohepatitis (NASH) cirrhosis and alcoholic hepatitis and cirrhosis. Arrowhead completed a Phase 1/2 clinical trial and GSK is preparing to begin a Phase 2b clinical trial.
Nonalcoholic Steatohepatitis: NASH is liver inflammation and damage caused by a buildup of fat in the liver. This can cause scarring of the liver and in advanced cases can lead to cirrhosis.
Study Name: Study of ARO-HSD in Healthy Volunteers and Patients With Non-Alcoholic Steatohepatitis (NASH) or Suspected NASH
A Phase 1/2a Single and Multiple Dose-Escalating Study to Evaluate the Safety, Tolerability, Pharmacokinetics and Pharmacodynamic Effects of ARO-HSD in Normal Healthy Volunteers as Well as in Patients With NASH or Suspected NASH
ClinicalTrials.gov Identifier: NCT04202354
Horizon Therapeutics Ireland DAC (“Horizon”)
On June 18, 2021, Horizon and Arrowhead entered into a collaboration and license agreement (the “Horizon License Agreement”). Under the terms of the Horizon License Agreement, Horizon received a worldwide exclusive license for ARO-XDH, a previously undisclosed discovery-stage investigational RNAi therapeutic being developed by Arrowhead as a potential treatment for people with uncontrolled gout. Arrowhead conducted all activities through the preclinical stages of development of ARO-XDH, and Horizon is now wholly responsible for clinical development and commercialization of ARO-XDH. In July 2021, Arrowhead received $40 million as an upfront payment and is eligible to receive up to $660 million in potential development, regulatory and sales milestones. Arrowhead is also eligible to receive royalties in the low- to mid-teens range on net product sales.
ARO-XDH
6
ARO-XDH is designed to reduce production of xanthine dehydrogenase (XDH) as a potential treatment for people with uncontrolled gout. Gout is a serious and painful form of arthritis that is caused by excess uric acid in the blood. In the United States, there are more than nine million gout patients and approximately one-third of those patients are treated with oral urate-lowering therapies. However, a meaningful portion of treated patients do not respond sufficiently to treatment and therefore continue to experience painful and debilitating gout symptoms. XDH represents a clinically validated target that is the primary source of serum uric acid (sUA). High levels of sUA, if left untreated or undertreated, can potentially lead to serious long-term or even permanent damage to the bones, joints and organs.
Takeda Pharmaceutical Company Limited (“Takeda”)
On October 7, 2020, Takeda and Arrowhead entered into an Exclusive License and Co-Funding Agreement (the “Takeda License Agreement”). Under the Takeda License Agreement, Takeda and Arrowhead will co-develop Arrowhead’s ARO-AAT program, Arrowhead’s second-generation subcutaneously administered RNAi therapeutic candidate being developed as a treatment for liver disease associated with alpha-1 antitrypsin deficiency. Within the United States, ARO-AAT, if approved, will be co-commercialized under a 50/50 profit sharing structure. Outside the United States, Takeda will lead the global commercialization strategy and will receive an exclusive license to commercialize ARO-AAT, while Arrowhead will be eligible to receive tiered royalties of 20% to 25% on net sales. In January 2021, Arrowhead received $300.0 million as an upfront payment and is eligible to receive potential development, regulatory and commercial milestone payments of up to $595.0 million.
Fazirsiran (formerly TAK-999 and ARO-AAT)
Fazirsiran is a subcutaneously administered RNAi therapeutic being developed as a treatment for liver disease associated with alpha-1 antitrypsin deficiency (AATD), which is a rare genetic disorder that severely damages the liver and lungs of affected individuals. Fazirsiran is designed to reduce production of the mutant Z-AAT protein by silencing the AAT gene in order to prevent accumulation of Z-AAT in the liver, allow clearance of the accumulated Z-AAT protein, prevent repeated cycles of cellular damage, and possibly prevent or even reverse the progression of liver fibrosis.
Goal of Fazirsiran Treatment: The goal of fazirsiran treatment is prevention and potential reversal of Z-AAT accumulation-related liver injury and fibrosis. Reduction of inflammatory Z-AAT protein, which has been clearly defined as the cause of progressive liver disease in AATD patients, is important as it is expected to halt the progression of liver disease and allow fibrotic tissue repair.
Alpha-1 Antitrypsin Deficiency (AATD): AATD is a genetic disorder associated with liver disease in children and adults, and pulmonary disease in adults. AAT is a circulating glycoprotein protease inhibitor that is primarily synthesized and secreted by liver hepatocytes. Its physiologic function is the inhibition of neutrophil protease to protect healthy lung tissues during inflammation and prevent tissue damage. The most common disease variant, the Z mutant, has a single amino acid substitution that results in improper folding of the protein. The mutant protein cannot be effectively secreted and accumulates in globules in the hepatocytes. This triggers continuous hepatocyte injury, leading to fibrosis, cirrhosis, and increased risk of hepatocellular carcinoma.
Current Treatments: Individuals with the homozygous PiZZ genotype have severe deficiency of functional AAT leading to pulmonary disease and hepatocyte injury and liver disease. Lung disease in this patient population is frequently treated with AAT augmentation therapy. However, augmentation therapy does nothing to treat liver disease, and there is no specific therapy for hepatic manifestations. There is a significant unmet need as liver transplant, with its attendant morbidity and mortality, is currently the only available cure.
Clinical Trials:
Study Name: Safety, Tolerability and Effect on Liver Histologic Parameters of ARO-AAT (SEQUOIA)
A Placebo-Controlled, Multi-dose, Phase 2/3 Study to Determine the Safety, Tolerability and Effect on Liver Histologic Parameters in Response to ARO-AAT in Patients with Alpha-1 Antitrypsin Deficiency (AATD)
ClinicalTrials.gov Identifier: NCT03945292
Janssen Pharmaceuticals, Inc. (“Janssen”)
On October 3, 2018, Janssen, part of the Janssen Pharmaceutical Companies of Johnson & Johnson, and Arrowhead entered into a License Agreement (the “Janssen License Agreement”) and a Research Collaboration and Option Agreement (the “Janssen Collaboration Agreement”). Arrowhead also entered into a stock purchase agreement with JJDC, Inc. (“JJDC”), Johnson & Johnson's venture capital arm (“JJDC Stock Purchase Agreement”). Under the Janssen License Agreement, Janssen has received a worldwide, exclusive license to Arrowhead’s JNJ-3989 (ARO-HBV) program, Arrowhead’s third-generation subcutaneously administered RNAi therapeutic candidate being developed as a potential
7
therapy for patients with chronic hepatitis B virus infection. Beyond Arrowhead’s Phase 1/2 study of JNJ-3989 (ARO-HBV), which Arrowhead was responsible for completing, Janssen is wholly responsible for clinical development and commercialization of JNJ-3989 (ARO-HBV). Under the Janssen Collaboration Agreement, Janssen was able to select three new targets against which Arrowhead would develop clinical candidates. These candidates were subject to certain restrictions and did not include candidates that already were in Arrowhead’s pipeline. Arrowhead was obligated to perform discovery, optimization and preclinical research and development, entirely funded by Janssen, which on its own or in combination with Janssen development work, would have been sufficient to allow the filing of a U.S. Investigational New Drug Application (“IND”) or equivalent, at which time Janssen would have the option to take an exclusive license. If the option was exercised, Janssen would have been wholly responsible for clinical development and commercialization of each optioned candidate. Under the terms of the agreements taken together, Arrowhead has received $175.0 million as an upfront payment, $75.0 million in the form of an equity investment by JJDC in Arrowhead’s common stock under the JJDC Stock Purchase Agreement, and milestone and option payments totaling $73.0 million, and Arrowhead may receive up to $1.6 billion in development and sales milestone payments for the Janssen License Agreement, and up to $0.6 billion in development and sales milestone payments for the remaining target covered under the Janssen Collaboration Agreement. Arrowhead is further eligible to receive tiered royalties on product sales up to mid-teens under the Janssen License Agreement and up to low-teens under the Janssen Collaboration Agreement. During 2022, Janssen’s option period expired unexercised for two of the three candidates (ARO-JNJ2 and ARO-JNJ3) under the Janssen Collaboration Agreement.
JNJ-3989 (also referred to as JNJ-73763989 and formerly referred to as ARO-HBV)
JNJ-3989 is being developed in collaboration with Janssen as a potential therapy for patients with chronic hepatitis B infection, when used in combination with other therapeutic modalities. JNJ-3989 is a subcutaneous RNAi therapy candidate which is designed to silence all HBV gene products and intervenes upstream of the reverse transcription process where current standard-of-care nucleotide and nucleoside analogues act. Arrowhead believes this, especially the elimination of hepatitis B surface antigen (HBsAg), may allow the body’s natural immune defenses to clear the virus and potentially lead to a functional cure. JNJ-3989 is currently being investigated in multiple Phase 2 clinical trials being conducted by Janssen. The Phase 1/2a study and its preceding studies were conducted by Arrowhead.
Clinical Trials:
Study Name: A Study of Different Combination Regimens Including JNJ-73763989 and/or JNJ-56136379 for the Treatment of Chronic Hepatitis B Virus Infection (REEF-1)
A Phase 2b, Multicenter, Double-blind, Active-controlled, Randomized Study to Investigate the Efficacy and Safety of Different Combination Regimens Including JNJ-73763989 and/or JNJ-56136379 for the Treatment of Chronic Hepatitis B Virus Infection
ClinicalTrials.gov Identifier: NCT03982186
Study Name: A Study of JNJ 73763989+JNJ 56136379+Nucleos(t)Ide Analog (NA) Regimen Compared to NA Alone in e Antigen Negative Virologically Suppressed Participants With Chronic Hepatitis B Virus Infection
A Randomized, Double Blind, Placebo-controlled Phase 2b Study to Evaluate Efficacy, Pharmacokinetics, and Safety of 48-week Study Intervention With JNJ 73763989+JNJ 56136379+Nucleos(t)Ide Analog (NA) Regimen Compared to NA Alone in e Antigen Negative Virologically Suppressed Participants With Chronic Hepatitis B Virus Infection
ClinicalTrials.gov Identifier: NCT04129554
Study Name: A Study of JNJ-73763989 in Healthy Chinese Adult Participants
A Randomized, Open-Label, Parallel, Single Dose Study to Investigate Pharmacokinetics, Safety, and Tolerability of JNJ-73763989 in Healthy Chinese Adult Participants
ClinicalTrials.gov Identifier: NCT04586439
Study Name: A Study to Evaluate the Effect of Hepatic Impairment on JNJ-73763989
A Phase 1, Single-Dose, Open-Label, Parallel-Group Study to Evaluate the Effect of Hepatic Impairment on the Pharmacokinetics of JNJ-73763989
ClinicalTrials.gov Identifier: NCT04208386
Study Name: A Study of JNJ-73763989 + Nucleos(t)Ide Analog in Participants Co-Infected With Hepatitis B and Hepatitis D Virus (REEF-D)
A Phase 2, Multicenter, Randomized, Double-blind, Placebo-Controlled Study With Deferred Active Treatment to Investigate the Efficacy, Safety, and Pharmacokinetics of JNJ-73763989 + Nucleos(t)Ide Analog in Participants Co-Infected With Hepatitis B and Hepatitis D Virus
ClinicalTrials.gov Identifier: NCT04535544
8
Study Name: A Study of JNJ-73763989 + JNJ-56136379 + Nucleos(t)Ide Analog (NA) Regimen With or Without Pegylated Interferon Alpha-2a (PegIFN-α2a) in Treatment-Naive Participants With Hepatitis B e Antigen (HBeAg) Positive Chronic Hepatitis B Virus (HBV) Infection and Normal Alanine Aminotransferase (ALT)
A Phase 2, Randomized, Open-label, Multicenter Study to Evaluate Efficacy, Pharmacokinetics, Safety, and Tolerability of Response-guided Treatment With JNJ-73763989 + JNJ-56136379 + Nucleos(t)Ide Analog Regimen With or Without Pegylated Interferon Alpha-2a in Treatment-naive Patients With HBeAg Positive Chronic Hepatitis B Virus Infection and Normal ALT
ClinicalTrials.gov Identifier: NCT04439539
Study Name: A Study to Assess Intrahepatic and Peripheral Changes of Immunologic and Virologic Markers in Chronic Hepatitis B Virus Infection (INSIGHT)
A Phase 2 Randomized, Open-label, Parallel-group, Multicenter Study to Assess Intrahepatic and Peripheral Changes of Immunologic and Virologic Markers in Response to Combination Regimens Containing JNJ-73763989 and Nucleos(t)Ide Analog With or Without JNJ-56136379 in Patients With Chronic Hepatitis B Virus Infection
ClinicalTrials.gov Identifier: NCT04585789
Study Name: A Study of JNJ-73763989 in Adult Participants With Renal Impairment
An Open-label, Single-dose, Parallel-group Study to Evaluate the Effect of Renal Impairment on the Pharmacokinetics of JNJ-73763989 in Adult Participants
ClinicalTrials.gov Identifier: NCT04963738
Study Name: A Study of JNJ-73763989, Pegylated Interferon Alpha-2a and Nucleos(t)Ide Analogs in Participants With Chronic Hepatitis B Virus Infection (PENGUIN-2)
A Phase 2, Open-label, Multicenter Study to Assess Efficacy, Safety, Tolerability, and Pharmacokinetics of Treatment With JNJ-73763989, Nucleos(t)Ide Analogs, and Pegylated Interferon Alpha-2a in Patients With Chronic Hepatitis B Virus Infection
ClinicalTrials.gov Identifier: NCT05005507
Study Name: A Study of JNJ-73763989, JNJ-56136379, Nucleos(t)Ide Analogs, and Pegylated Interferon Alpha-2a in Virologically Suppressed Participants With Chronic Hepatitis B Virus Infection (PENGUIN)
A Phase 2, Open-label, Single-arm, Multicenter Study to Assess Efficacy, Safety, Tolerability, and Pharmacokinetics of Treatment With JNJ-73763989, JNJ-56136379, Nucleos(t)Ide Analogs, and Pegylated Interferon Alpha-2a in Virologically Suppressed Patients With Chronic Hepatitis B Virus Infection
ClinicalTrials.gov Identifier: NCT04667104
JNJ-75220795 (formerly referred to as ARO-JNJ1)
JNJ-75220795 (ARO-JNJ1) is an investigational therapeutic being developed by Janssen. It utilizes Arrowhead’s proprietary TRIMTM platform and is designed to reduce expression in the liver of patatin like phospholipase domain containing 3 (PNPLA3) as a potential treatment for patients with non-alcoholic steatohepatitis (NASH).
Clinical Trials:
A Single and Multiple Ascending Dose Study of Subcutaneously Administered JNJ-75220795
A Double-Blind, Placebo-Controlled, Randomized, Multipart, Single and Multiple Ascending Dose Study to Investigate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Subcutaneously Administered JNJ-75220795 ClinicalTrials.gov Identifier: NCT04844450
A Study of JNJ-75220795 in Japanese Participants
A Double-Blind, Placebo-Controlled, Randomized, Single Ascending Dose Study to Investigate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Subcutaneously Administered JNJ-75220795 in Japanese Participants ClinicalTrials.gov Identifier: NCT05039710
Amgen Inc. (“Amgen”)
On September 28, 2016, Amgen and Arrowhead entered into two collaboration and license agreements and a common stock purchase agreement. Under the Second Collaboration and License Agreement (the “Olpasiran Agreement”), Amgen has received a worldwide, exclusive license to Arrowhead’s novel RNAi Olpasiran (previously referred to as AMG 890 or ARO-LPA) program. These RNAi molecules are designed to reduce elevated lipoprotein(a), which is a genetically validated, independent risk factor for atherosclerotic cardiovascular disease. Under the prior collaboration and license agreement (the “First Collaboration and License Agreement” or the “ARO-AMG1 Agreement”), Amgen received an option to a worldwide, exclusive license for ARO-AMG1, an RNAi therapy for an undisclosed genetically validated cardiovascular target. Under both agreements, Amgen is wholly responsible for clinical development and
9
commercialization. Under the terms of the agreements taken together, Arrowhead has received $35.0 million in upfront payments, $21.5 million in the form of an equity investment by Amgen in Arrowhead’s common stock, and $30.0 million in milestone payments, and may receive up to an additional $400.0 million in remaining development, regulatory and sales milestone payments. Arrowhead is further eligible to receive up to low double-digit royalties for sales of products under the Olpasiran Agreement. In July 2019, Amgen informed Arrowhead that it would not be exercising its option for an exclusive license for ARO-AMG1, and as such, there will be no further milestone or royalty payments under the ARO-AMG1 Agreement. See Note 13 — Subsequent Events of Notes to Consolidated Financial Statements of Part IV, “Item 15. Exhibits and Financial Statement Schedules.”
Olpasiran (formerly AMG 890 and ARO-LPA)
Olpasiran is designed to reduce production of apolipoprotein A, a key component of lipoprotein(a), which has been genetically linked with increased risk of cardiovascular diseases, independent of cholesterol and LDL levels. Amgen completed a Phase 2 clinical study evaluating the efficacy, safety, and tolerability of Olpasiran in subjects with elevated levels of lipoprotein(a). Amgen reported Phase 2 clinical results at the American Heart Association (AHA) Scientific Sessions in November 2022 and simultaneously published in the New England Journal of Medicine. Amgen intends to evaluate Olpasiran in a Phase 3 study to assess the impact of Olpasiran on major cardiovascular events in participants with atherosclerotic cardiovascular disease and elevated lipoprotein(a), in a double-blind, randomized, placebo-controlled, multicenter study. Amgen plans to begin enrolling subjects in December 2022, which will trigger a $25.0 million milestone payment to Arrowhead.
Joint Venture and License Agreement with Visirna Therapeutics, Inc. (“Visirna”)
On April 25, 2022, Visirna and Arrowhead entered into a License Agreement (the “Visirna License Agreement”), pursuant to which Visirna received an exclusive license to develop, manufacture and commercialize four of Arrowhead’s RNAi-based investigational cardiometabolic medicines in Greater China (including the People’s Republic of China, Hong Kong, Macau and Taiwan). Pursuant to a Share Purchase Agreement entered into simultaneously with the Visirna License Agreement (the “Visirna SPA”), Arrowhead acquired a majority stake in Visirna (after accounting for shares reserved for Visirna’s employee stock ownership plan) as partial consideration for the Visirna License Agreement. Under the Visirna SPA, entities affiliated with Vivo Capital also acquired a minority stake in Visirna in exchange for $60 million in upfront capital to support the operations of Visirna. As further consideration under the Visirna License Agreement, Arrowhead is also eligible to receive potential royalties on commercial sales.
Intellectual Property and Other Key Agreements
Arrowhead controls approximately 523 issued patents (including 331 directed to RNAi trigger molecules; 76 directed to targeting groups or targeting compounds; and 4 for hydrodynamic gene delivery), including European validations, and approximately 726 currently pending patent applications worldwide from 80 different patent families. Arrowhead’s patent applications have been filed throughout the world, including, in the United States, Argentina, ARIPO (Africa Regional Intellectual Property Organization), Australia, Brazil, Canada, Chile, China, Eurasian Patent Organization, Europe, GCC (Gulf Cooperation Council), Hong Kong, Israel, India, Indonesia, Iraq, Jordan, Japan, Lebanon, Mexico, New Zealand, OAPI (African Intellectual Property Organization), Peru, Philippines, Russian Federation, Saudi Arabia, Singapore, South Korea, Thailand, Taiwan, Uruguay, Venezuela, Vietnam, and South Africa.
RNAi Triggers: Arrowhead owns issued patents or has filed patent applications directed to RNAi trigger molecules, which serve as the foundation of Arrowhead’s TRiMTM platform, and are targeted to reduce expression of various gene targets, including the following:
10
| Patent Group | Estimated Year(s) of Expiration* | ||||
| HBV | 2032, 2036, 2037 | ||||
| AAT | 2035, 2038 | ||||
| LPA | 2036 | ||||
| α-ENaC | 2028, 2038 | ||||
| APOC3 | 2035, 2038 | ||||
| ANGPTL3 | 2038 | ||||
| HIF2α | 2034, 2036, 2040 | ||||
| HSD17B13 | 2039 | ||||
| PNPLA3 | 2041 | ||||
| DUX4 | 2041 | ||||
| Factor 12 | 2036, 2038 | ||||
| RRM2 | 2031 | ||||
| β-ENaC | 2031, 2040 | ||||
| β-Catenin | 2033 | ||||
| Cx43 | 2029 | ||||
| HCV | 2025 | ||||
| HIF1A | 2026 | ||||
| HRH1 | 2027 | ||||
| HSF1 | 2030, 2032 | ||||
| FRP-1 | 2026 | ||||
| KRAS | 2033 | ||||
| P2X3 | 2027 | ||||
| Mob-5 | 2027 | ||||
| PDtype4 | 2026 | ||||
| PI4Kinase | 2028 | ||||
| SYK | 2027 | ||||
| TNF-α | 2027, 2028 | ||||
*Assuming issuance of any pending patent applications, and excluding any patent term adjustments or patent term extensions.
Delivery Technologies: The delivery technology-related patents and patent applications, which include components used in Arrowhead’s TRiMTM platform, have been filed and/or issued in various jurisdictions worldwide including the United States, Argentina, Australia, Brazil, Canada, China, Eurasian Patent Organization, Europe (including validations in France, Germany, Italy, Spain, Switzerland, United Kingdom), GCC (Gulf Cooperation Council), Israel, India, Japan, Lebanon, Mexico, New Zealand, Philippines, Russia, South Africa, South Korea, Singapore, Taiwan, and Uruguay. Arrowhead also controls a patent directed to hydrodynamic nucleic acid delivery that issued in the United States. The approximate year of expiration for each of these various groups of patents are set forth below:
11
| Patent Group | Estimated Year(s) of Expiration* | ||||
| Targeting ligands and other RNAi delivery and platform technologies | |||||
| Targeting groups (Galactose derivative trimer-PK) | 2031 | ||||
| Targeting groups (αvβ3/αvβ5 integrin) | 2034, 2038, 2039 | ||||
| Targeting groups (αvβ6 integrin) | 2037, 2038, 2041 | ||||
| Targeting groups (Galactose derivative ligands) | 2037, 2037 | ||||
| RNAi agent design (5′-phosphate mimic) | 2037 | ||||
| Physiologically labile linkers | 2036 | ||||
| Biologically cleavable linkers | 2036 | ||||
| Trialkyne linkers | 2039 | ||||
| Muscle delivery platform | 2041, 2041 | ||||
| PK/PD lipid modifiers | 2041 | ||||
| Transferrin targeting | 2028 | ||||
| LDLR targeting | 2028 | ||||
| Peptide targeting (CPP-Arg) | 2028 | ||||
| Peptide targeting (YM3-10H) | 2032 | ||||
| Hydrodynamic delivery | |||||
| Third iteration | 2024 | ||||
*Assuming issuance of any pending patent applications, and excluding any patent term adjustments or patent term extensions.
The RNAi and drug delivery patent landscapes are complex and rapidly evolving. As such, Arrowhead may need to obtain additional patent licenses prior to commercialization of its candidates. Please see “Risk Factors” in Part I, Item 1A of this Annual Report on Form 10-K.
Acquisition of Assets from Novartis
On March 3, 2015, Novartis and Arrowhead entered into an Asset Purchase and Exclusive License Agreement (the “RNAi Purchase Agreement”) pursuant to which Arrowhead acquired Novartis’s RNAi assets and rights thereunder. Pursuant to the RNAi Purchase Agreement, Arrowhead acquired or was granted a license to certain patents and patent applications owned or controlled by Novartis related to RNAi therapeutics, was assigned Novartis’s rights under a license from Alnylam Pharmaceuticals, Inc. (“Alnylam”) (the “Alnylam-Novartis License”) and acquired a license to certain additional Novartis assets (the “Licensed Novartis Assets”). The patents acquired from Novartis include multiple patent families covering delivery technologies and RNAi-trigger design rules and modifications. The Licensed Novartis Assets include an exclusive, worldwide right and license, solely in the RNAi field, with the right to grant sublicenses through multiple tiers under or with respect to certain patent rights and know how relating to delivery technologies and RNAi-trigger design rules and modifications. Under the assigned Alnylam-Novartis License, Arrowhead acquired a worldwide, royalty-bearing, exclusive license with limited sublicensing rights to existing and future Alnylam intellectual property (including intellectual property that came under Alnylam’s control on or before March 31, 2016), excluding intellectual property concerning delivery technology, to research, develop and commercialize 30 undisclosed gene targets.
2012 License to Alnylam
In consideration for licenses obtained from Alnylam to certain RNAi intellectual property, in January 2012, Arrowhead granted Alnylam a worldwide, non-exclusive, sublicensable royalty-bearing license under its broad and target-specific DPC intellectual property rights to research, develop and commercialize RNAi-based products against a single undisclosed target in combination with DPC technology. Under the license to Alnylam, Alnylam may be obligated to pay Arrowhead development and sales milestone payments of up to the low double-digit millions of dollars for each licensed product that progresses through clinical trials, receives marketing approval and is the subject of a first commercial sale. Additionally, Alnylam may be obligated to pay Arrowhead low single-digit percentage royalties on sales of such products.
12
Non-Exclusively Licensed Patent Rights from Roche
On October 21, 2011, Arrowhead acquired the RNAi therapeutics business of Hoffmann-La Roche, Inc. and F. Hoffmann-La Roche Ltd. (collectively, “Roche”). The acquisition provided us with two primary sources of value:
•Broad freedom to operate with respect to key patents directed to the primary RNAi-trigger formats: canonical, UNA, meroduplex, and dicer substrate structures; and
•A large team of scientists experienced in RNAi and oligonucleotide delivery.
Pursuant to this acquisition, Roche assigned to Arrowhead its entire rights under certain licenses including: the License and Collaboration Agreement between Roche and Alnylam dated July 8, 2007 (the “Alnylam License”); the Non-Exclusive Patent License Agreement between Roche and MDRNA, Inc. dated February 12, 2009 (“MDRNA License”); and the Non-Exclusive License Agreement between Roche and City of Hope dated September 19, 2011 (the “COH License”) (collectively the “RNAi Licenses”).
The RNAi Licenses include licenses to patents related to modifications of double-stranded oligonucleotides, including modifications to the base, sugar, or internucleoside linkage, nucleotide mimetics, and end modifications, which do not abolish the RNAi activity of the double-stranded oligonucleotides. Also included are patents relating to modified double-stranded oligonucleotides, such as meroduplexes described in U.S. Patent No. 9,074,205 assigned to Marina Biotech (f/k/a MDRNA, Inc.), as well as U.S. Patent Nos. 8,314,227, 9,051,570, and 9,303,260 related to unlocked nucleotide analogs (“UNA”). The UNA patents were assigned by Marina Biotech to Arcturus Therapeutics, Inc., but remain part of the MDRNA License. The RNAi Licenses further include patents related to dicer substrates and uses of the double-stranded oligonucleotides that function through the mechanism of RNA interference, such as described in City of Hope’s U.S. Patent Nos. 8,084,599, 8,658,356, 8,691,786, 8,796,444, 8,809,515, and 9,518,262.
Government Regulation
Government authorities in the United States, at the federal, state, and local levels, and in other countries and jurisdictions, including the European Union (“EU”), extensively regulate, among other things, the research, development, testing, product approval, manufacture, quality control, manufacturing changes, packaging, storage, recordkeeping, labeling, promotion, advertising, sales, distribution, marketing, and import and export of drugs and biologic products. All of Arrowhead’s foreseeable product candidates are expected to be regulated as drugs. The processes for obtaining regulatory approval in the United States and in foreign countries and jurisdictions, along with compliance with applicable statutes and regulations and other regulatory authorities both pre- and post-commercialization, are a significant factor in the production and marketing of Arrowhead’s products and its R&D activities and require the expenditure of substantial time and financial resources.
Review and Approval of Drugs in the United States
The United States Food and Drug Administration (the “FDA”) and other government entities regulate drugs under the Federal Food, Drug, and Cosmetic Act (the “FDCA”), the Public Health Service Act, and the regulations promulgated under those statutes, as well as other federal and state statutes and regulations. Failure to comply with applicable legal and regulatory requirements in the United States at any time during the product development process, approval process, or after approval, may subject us to a variety of administrative or judicial sanctions, such as a delay in approving or refusal by the FDA to approve pending applications, withdrawal of approvals, delay or suspension of clinical trials, issuance of warning letters and other types of regulatory letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil monetary penalties, refusals of or debarment from government contracts, exclusion from the federal healthcare programs, restitution, disgorgement of profits, civil or criminal investigations by the FDA, U.S. Department of Justice, State Attorneys General, and/or other agencies, False Claims Act suits and/or other litigation, and/or criminal prosecutions.
An applicant seeking approval to market and distribute a new drug in the United States must typically undertake the following:
(1) completion of preclinical laboratory tests, animal studies, and formulation studies in compliance with the FDA’s good laboratory practice (“GLP”) regulations;
(2) submission to the FDA of an IND for human clinical testing, which must become effective without FDA objection before human clinical trials may begin;
(3) approval by an independent institutional review board (“IRB”), representing each clinical site before each clinical trial may be initiated;
13
(4) performance of adequate and well-controlled human clinical trials in accordance with the FDA’s current good clinical practice (“cGCP”) regulations, to establish the safety and effectiveness of the proposed drug product for each indication for which approval is sought;
(5) preparation and submission to the FDA of a New Drug Application (“NDA”);
(6) satisfactory review of the NDA by an FDA advisory committee, where appropriate or if applicable;
(7) satisfactory completion of one or more FDA inspections of the manufacturing facility or facilities at which the drug product, and the active pharmaceutical ingredient or ingredients thereof, are produced to assess compliance with current good manufacturing practice (“cGMP”) regulations and to assure that the facilities, methods, and controls are adequate to ensure the product’s identity, strength, quality, and purity;
(8) payment of user fees, as applicable, and securing FDA approval of the NDA; and
(9) compliance with any post-approval requirements, such as any Risk Evaluation and Mitigation Strategies (“REMS”) or post-approval studies required by the FDA.
Preclinical Studies and an IND
Preclinical studies can include in vitro and animal studies to assess the potential for adverse events and, in some cases, to establish a rationale for therapeutic use. The conduct of preclinical studies is subject to federal regulations and requirements, including GLP regulations. Other studies include laboratory evaluation of the purity, stability and physical form of the manufactured drug substance or active pharmaceutical ingredient and the physical properties, stability and reproducibility of the formulated drug or drug product. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and plans for clinical studies, among other things, to the FDA as part of an IND. Some preclinical testing, such as longer-term toxicity testing, animal tests of reproductive adverse events and carcinogenicity, may continue after the IND is submitted. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to a proposed clinical trial and places the trial on clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND may not result in the FDA allowing clinical trials to commence.
Following commencement of a clinical trial under an IND, the FDA may place a clinical hold on that trial. A clinical hold is an order issued by the FDA to the sponsor to delay a proposed clinical investigation or to suspend an ongoing investigation. A partial clinical hold is a delay or suspension of only part of the clinical work requested under the IND. For example, a specific protocol or part of a protocol is not allowed to proceed, while other protocols may do so. No more than 30 days after imposition of a clinical hold or partial clinical hold, the FDA will provide the sponsor a written explanation of the basis for the hold. Following issuance of a clinical hold or partial clinical hold, an investigation may only resume after the FDA has notified the sponsor that the investigation may proceed. The FDA will base that determination on information provided by the sponsor correcting the deficiencies previously cited or otherwise satisfying the FDA that the investigation can proceed.
Human Clinical Studies in Support of an NDA
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators in accordance with cGCP requirements, which include, among other things, the requirement that all research subjects provide their informed consent in writing before their participation in any clinical trial. Clinical trials are conducted under written study protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. In addition, an IRB representing each institution participating in the clinical trial must review and approve the plan for any clinical trial before it commences at that institution, and the IRB must conduct continuing review and reapprove the study at least annually. The IRB must review and approve, among other things, the study protocol and informed consent information to be provided to study subjects. An IRB must operate in compliance with FDA regulations. Information about certain clinical trials must be submitted within specific timeframes to the NIH for public dissemination on its ClinicalTrials.gov website.
Human clinical trials are typically conducted in three sequential phases, which may overlap or be combined:
Phase 1: The product candidate is initially introduced into healthy human subjects or patients with the target disease or condition and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an early indication of its effectiveness.
14
Phase 2: The product candidate is administered to a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage.
Phase 3: The product candidate is administered to an expanded patient population, generally at geographically dispersed clinical trial sites, in well-controlled clinical trials to generate enough data to statistically evaluate the efficacy and safety of the product for approval, to establish the overall risk-benefit profile of the product, and to provide adequate information for the labeling of the product.
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if serious adverse events occur. Phase 1, Phase 2, and Phase 3 clinical trials may not be completed successfully within any specified period, or at all. Furthermore, the FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution, or an institution it represents, if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients. The FDA will typically inspect one or more clinical sites in late-stage clinical trials to assure compliance with cGCP and the integrity of the clinical data submitted.
Submission of an NDA to the FDA
Assuming successful completion of required clinical testing and other requirements, the results of the preclinical and clinical studies, together with detailed information relating to the product’s chemistry, manufacture, controls and proposed labeling, among other things, are submitted to the FDA as part of an NDA requesting approval to market the drug product for one or more indications. Under federal law, the submission of most NDAs is additionally subject to an application user fee, currently $3.2 million for fiscal year 2023, for applications requiring clinical data, and the sponsor of an approved NDA is also subject to an annual program fee, currently $393,933 for fiscal year 2023. These fees are adjusted annually.
Under certain circumstances, the FDA will waive the application fee for the first human drug application that a small business, defined as a company with less than 500 employees, including employees of affiliates, submits for review. An affiliate is defined as a business entity that has a relationship with a second business entity if one business entity controls, or has the power to control, the other business entity, or a third-party controls, or has the power to control, both entities. In addition, an application to market a prescription drug product that has received orphan designation is not subject to a prescription drug user fee unless the application includes an indication for other than the rare disease or condition for which the drug was designated.
The FDA conducts a preliminary review of an NDA within 60 days of its receipt and informs the sponsor by the 74th day after the FDA’s receipt of the submission to determine whether the application is sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA has agreed to specified performance goals in the review process of NDAs. Most such applications are meant to be reviewed within ten months from the date of filing, and most applications for “priority review” products are meant to be reviewed within six months of filing. The review process may be extended by the FDA for three additional months to consider new information or clarification provided by the applicant to address an outstanding deficiency identified by the FDA following the original submission.
Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA will typically inspect one or more clinical sites to assure compliance with cGCP.
The FDA also may require submission of a REMS plan to mitigate any identified or suspected serious risks. The REMS plan could include medication guides, physician communication plans, assessment plans, and elements to assure safe use, such as restricted distribution methods, patient registries, or other risk minimization tools.
The FDA is required to refer an application for a novel drug to an advisory committee or explain why such referral was not made. Typically, an advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
15
The FDA’s Decision on an NDA
On the basis of the FDA’s evaluation of the NDA and accompanying information, including the results of the inspection of the manufacturing facilities, the FDA may issue an approval letter or a complete response letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application. If and when those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the NDA, the FDA will issue an approval letter. The FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
If the FDA approves a product, it may limit the approved indications for use for the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies be conducted to further assess the drug’s safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution restrictions or other risk management mechanisms, including REMS, which can materially affect the potential market and profitability of the product. After approval, the FDA may seek to prevent or limit further marketing of a product based on the results of post-market studies or surveillance programs. Some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval.
The product may also be subject to official lot release, meaning that the manufacturer is required to perform certain tests on each lot of the product before it is released for distribution. If the product is subject to official lot release, the manufacturer must submit samples of each lot, together with a release protocol showing a summary of the history of manufacture of the lot and the results of all of the manufacturer’s tests performed on the lot, to the FDA. The FDA may in addition perform certain confirmatory tests on lots of some products before releasing the lots for distribution. Finally, the FDA will conduct laboratory research related to the safety and effectiveness of drug products.
Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making available in the United States a drug for this type of disease or condition will be recovered from sales in the United States for that drug. Orphan drug designation entitles the applicant to incentives such as grant funding towards clinical study costs, tax advantages, and waivers of FDA user fees. Orphan drug designation must be requested before submitting an NDA, and both the drug and the disease or condition must meet certain criteria specified in the Orphan Drug Act and FDA’s implementing regulations at 21 C.F.R. Part 316. The granting of an orphan drug designation does not alter the standard regulatory requirements and process for obtaining marketing approval. Safety and effectiveness of a drug must be established through adequate and well-controlled studies.
After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. If a product that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other application to market the same drug for the same indication, except in very limited circumstances, for seven years. Orphan drug exclusivity does not prevent the FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition.
The FDA’s interpretation of the scope of orphan drug exclusivity may change. The FDA’s longstanding interpretation of the Orphan Drug Act is that exclusivity is specific to the orphan indication for which the drug was actually approved. As a result, the scope of exclusivity has been narrow and protected only against competition from the same “use or indication” rather than the broader “disease or condition.” In the September 2021 case Catalyst Pharmaceuticals, Inc. v. FDA, a federal circuit court set aside the FDA’s narrow interpretation and ruled that orphan drug exclusivity covers the full scope of the orphan-designated disease or condition regardless of whether the drug obtains approval only for a narrower use. The decision concerned amifampridine, a drug used to treat Lambert-Eaton myasthenic syndrome (LEMS). Depending on how the FDA applies the decision beyond this case, it may limit the drugs that can receive exclusivity.
Expedited Review and Accelerated Approval Programs
A sponsor may seek approval of its product candidate under programs designed to accelerate the FDA’s review and approval of NDAs. For example, Fast Track Designation may be granted to a drug intended for treatment of a serious or life-threatening disease or condition and data demonstrate its potential to address unmet medical needs for the disease or condition. The key benefits of Fast Track Designation are the eligibility for priority review, rolling review (submission of portions of an application before the complete marketing application is submitted), and accelerated approval, if relevant
16
criteria are met. The FDA may grant the NDA a priority review designation, which sets the target date for FDA action on the application at six months after the FDA accepts the application for filing. Priority review is granted where there is evidence that the proposed product would be a significant improvement in the safety or effectiveness of the treatment, diagnosis, or prevention of a serious condition. Priority review designation does not change the scientific/medical standard for approval or the quality of evidence necessary to support approval.
The FDA may approve an NDA under the accelerated approval program if the drug treats a serious condition, provides a meaningful advantage over available therapies, and demonstrates an effect on either (1) a surrogate endpoint that is reasonably likely to predict clinical benefit, or (2) on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. Post-marketing studies or completion of ongoing studies after marketing approval are generally required to verify the drug’s clinical benefit in relationship to the surrogate endpoint or ultimate outcome in relationship to the clinical benefit.
In addition, the Food and Drug Administration Safety and Innovation Act of 2012 (“FDASIA”) established the Breakthrough Therapy designation. A sponsor may seek FDA designation of its product candidate as a breakthrough therapy if the drug is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. If a drug is designated as breakthrough therapy, FDA will provide more intensive guidance on the drug development program and expedite its review.
Post-Approval Requirements
Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims, are subject to prior FDA review and approval. There also are continuing, annual user fee requirements for any marketed products and the establishments at which such products are manufactured, as well as new application fees for supplemental applications with clinical data.
In addition, drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with cGMP requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events or problems with manufacturing processes of unanticipated severity or frequency, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
•restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls;
•fines, warning letters or holds on post-approval clinical trials;
•refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product license approvals;
•product seizure or detention, or refusal to permit the import or export of products; or
•injunctions or the imposition of civil or criminal penalties.
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability.
17
In addition, the distribution of prescription pharmaceutical products is subject to the Prescription Drug Marketing Act (“PDMA”), which regulates the distribution of drugs and drug samples at the federal level and sets minimum standards for the registration and regulation of drug distributors by the states. Both the PDMA and state laws limit the distribution of prescription pharmaceutical product samples and impose requirements to ensure accountability in distribution.
Abbreviated New Drug Applications for Generic Drugs
In 1984, with passage of the Drug Price Competition and Patent Term Restoration Act of 1984 (commonly referred to as the “Hatch-Waxman Amendments”) amending the FDCA, Congress authorized the FDA to approve generic drugs that are the same as drugs previously approved by the FDA under the NDA provisions of the statute. To obtain approval of a generic drug, an applicant must submit an abbreviated new drug application (“ANDA”) to the agency. In support of such applications, a generic manufacturer may rely on the preclinical and clinical testing previously conducted for a drug product previously approved under an NDA, known as the reference listed drug (“RLD”). To reference that information, however, the ANDA applicant must demonstrate, and the FDA must conclude, that the generic drug does, in fact, perform in the same way as the RLD it purports to copy. Specifically, in order for an ANDA to be approved, the FDA must find that the generic version is identical to the RLD with respect to the active ingredients, the route of administration, the dosage form, and the strength of the drug.
At the same time, the FDA must also determine that the generic drug is “bioequivalent” to the innovator drug. Under the statute, a generic drug is bioequivalent to a RLD if “the rate and extent of absorption of the generic drug do not show a significant difference from the rate and extent of absorption of the RLD.” Upon approval of an ANDA, the FDA indicates that the generic product is “therapeutically equivalent” to the RLD and it assigns a therapeutic equivalence rating to the approved generic drug in its publication “Approved Drug Products with Therapeutic Equivalence Evaluations,” also referred to as the “Orange Book.” Physicians and pharmacists consider the therapeutic equivalence rating to mean that a generic drug is fully substitutable for the RLD. In addition, by operation of certain state laws and numerous health insurance programs, the FDA’s designation of a therapeutic equivalence rating often results in substitution of the generic drug without the knowledge or consent of either the prescribing physician or patient.
Under the Hatch-Waxman Amendments, the FDA may not approve an ANDA until any applicable period of nonpatent exclusivity for the RLD has expired. The FDCA provides a period of five years of data exclusivity for new drug containing a new chemical entity. In cases where such exclusivity has been granted, an ANDA may not be filed with the FDA until the expiration of five years unless the submission is accompanied by a Paragraph IV certification, in which case the applicant may submit its application four years following the original product approval. The FDCA also provides for a period of three years of exclusivity if the NDA includes reports of one or more new clinical investigations, other than bioavailability or bioequivalence studies, that were conducted by or for the applicant and are essential to the approval of the application. This three-year exclusivity period often protects changes to a previously approved drug product, such as a new dosage form, route of administration, combination or indication.
Hatch-Waxman Patent Certification and the 30 Month Stay
Upon approval of an NDA or a supplement thereto, NDA sponsors are required to list with the FDA each patent with claims that cover the applicant’s product or a method of using the product. Each of the patents listed by the NDA sponsor is published in the Orange Book. When an ANDA applicant files its application with the FDA, the applicant is required to certify to the FDA concerning any patents listed for the reference product in the Orange Book, except for patents covering methods of use for which the ANDA applicant is not seeking approval.
Specifically, the applicant must certify with respect to each patent that:
•the required patent information has not been filed;
•the listed patent has expired;
•the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or
•the listed patent is invalid, unenforceable or will not be infringed by the new product.
A certification that the new product will not infringe the already approved product’s listed patents or that such patents are invalid or unenforceable is called a Paragraph IV certification. If the applicant does not challenge the listed patents or indicate that it is not seeking approval of a patented method of use, the ANDA application will not be approved until all the listed patents claiming the referenced product have expired. If the ANDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit
18
within 45 days after the receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit or a decision in the infringement case that is favorable to the ANDA applicant.
To the extent that a Section 505(b)(2) applicant is relying on studies conducted for an already approved product, the applicant is required to certify to the FDA concerning any patents listed for the approved product in the Orange Book to the same extent that an ANDA applicant would. As a result, approval of a 505(b)(2) NDA can be stalled until all the listed patents claiming the referenced product have expired, until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired, and, in the case of a Paragraph IV certification and subsequent patent infringement suit, until the earlier of 30 months, settlement of the lawsuit or a decision in the infringement case that is favorable to the Section 505(b)(2) applicant.
Pediatric Studies and Exclusivity
Under the Pediatric Research Equity Act of 2003, an NDA or supplement thereto must contain data that are adequate to assess the safety and effectiveness of the drug product for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. With the enactment of FDASIA, sponsors must also submit pediatric study plans prior to the assessment data. Those plans must contain an outline of the proposed pediatric study or studies the applicant plans to conduct, including study objectives and design, any deferral or waiver requests, and other information required by regulation. The applicant, the FDA, and the FDA’s internal review committee must then review the information submitted, consult with each other, and agree upon a final plan. The FDA or the applicant may request an amendment to the plan at any time.
The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements. Additional requirements and procedures relating to deferral requests and requests for extension of deferrals are contained in FDASIA. Unless otherwise required by regulation, the pediatric data requirements do not apply to products with orphan designation.
Pediatric exclusivity is another type of non-patent marketing exclusivity in the United States and, if granted, provides for the attachment of an additional six months of marketing protection to the term of any existing regulatory exclusivity, including the non-patent and orphan exclusivity. This six-month exclusivity may be granted if an NDA sponsor submits pediatric data that fairly respond to a written request from the FDA for such data. The data do not need to show the product to be effective in the pediatric population studied; rather, if the clinical trial is deemed to fairly respond to the FDA’s request, the additional protection is granted. If reports of requested pediatric studies are submitted to and accepted by the FDA within the statutory time limits, whatever statutory or regulatory periods of exclusivity or patent protection cover the product are extended by six months. This is not a patent term extension, but it effectively extends the regulatory period during which the FDA cannot accept or approve another application.
Patent Term Restoration and Extension
A patent claiming a new drug product may be eligible for a limited patent term extension under the Hatch-Waxman Amendments. Those Amendments permit a patent restoration of up to five years for patent term lost during product development and the FDA regulatory review. The restoration period granted is typically one-half the time between the effective date of an IND and the submission date of a NDA, plus the time between the submission date of a NDA and ultimate approval. Patent term restoration cannot be used to extend the remaining term of a patent past a total of 14 years from the product’s approval date. Only one patent applicable to an approved drug product is eligible for the extension, and the application for the extension must be submitted prior to the expiration of the patent in question. The U.S. Patent and Trademark Office reviews and approves the application for any patent term extension or restoration in consultation with the FDA.
Legislative Developments
The 21st Century Cures Act (the “Cures Act”), which was signed into law in December 2016, includes provisions to accelerate the development and delivery of new treatments. For example, the Cures Act requires the FDA to establish a program to evaluate the potential use of real world evidence to help to support the approval of a new indication for an approved drug and to help to support or satisfy post-approval study requirements, to issue guidance on adaptive and novel clinical trial designs for new drugs, and to establish a process for qualifying drug development tools used to support FDA approval for marketing or investigational use of a drug. The Cures Act also permits the FDA to rely on qualified data summaries to support the approval of a supplemental application for an already approved drug. The FDA has continued to issue guidance focused on implementing the Cures Act requirements.
19
Review and Approval of Drug Products in the European Union and United Kingdom
In order to market any pharmaceutical product outside of the United States, a company must also comply with numerous and varying regulatory requirements of other countries and jurisdictions governing, among other things, research and development, testing, manufacturing, quality control, safety, efficacy, labeling, clinical trials, marketing authorization, packaging, storage, record keeping, reporting, export and import, advertising, marketing and other promotional practices involving pharmaceutical products, as well as commercial sales, distribution, authorization, approval and post-approval monitoring and reporting of its products. Whether or not a company obtains FDA approval for a pharmaceutical product, the company would need to obtain the necessary approvals by the comparable foreign regulatory authorities before it can commence clinical trials or marketing of the pharmaceutical product in those countries or jurisdictions. The approval process ultimately varies between countries and jurisdictions and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries and jurisdictions might differ from and be longer than that required to obtain FDA approval. Regulatory approval in one country or jurisdiction does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country or jurisdiction may negatively impact the regulatory process in others.
The United Kingdom (“UK”) formally left the EU on January 31, 2020 and the transition period, during which EU laws continued to apply to the UK, expired on December 31, 2020. This means EU laws now only apply to the UK in respect of Northern Ireland as laid out in the Protocol on Ireland and Northern Ireland. Following the transition period, the EU and the UK have concluded a trade and cooperation agreement (“TCA”), which was applied provisionally from January 1, 2021 and entered into force on May 1, 2021.
The TCA includes provisions affecting the life sciences sector (including on customs and tariffs), but areas for further discussion between the EU and the UK remain. In addition, there are some specific provisions concerning pharmaceuticals. These include the mutual recognition of Good Manufacturing Practice (“GMP”) and issued GMP documents. The TCA does not, however, contain wholesale mutual recognition of UK and EU pharmaceutical regulations and product standards.
Since January 1, 2021, the EU laws which have been transposed into UK law through secondary legislation continue to be applicable in the UK as “retained EU law.” As there is no general power to amend these regulations, the UK government has enacted the Medicines and Medical Devices Act 2021. The purpose of the act is to enable the existing regulatory frameworks in relation to human medicines and clinical trials of human medicines, among others, to be updated. The powers under the act may only be exercised in relation to specified matters and must safeguard public health.
Specified provisions of the Medicines and Medical Devices Act 2021 entered into force on February 11, 2021. The remaining provision came into effect within two months of February 11, 2021 or will otherwise come into effect as stipulated in subsequent statutory instruments. The Medicines and Medical Devices Act 2021 supplements the UK Medical Devices Regulations 2002 (“UK Regulations”), which are based on the EU Medical Devices Directive as amended to reflect the UK’s post-Brexit regulatory regime. Notably, the UK Regulations do not include any of the revisions that have been made by the EU Medical Devices Regulation (EU) 2017/745, which, since May 26, 2021, applies in all EU member states.
The UK’s Medicines and Healthcare products Regulatory Agency (“MHRA”) conducted a comprehensive consultation between September and November 2021 on proposals to develop a new UK regime for medical devices in the UK. The proposals include more closely aligning definitions for medical devices and in vitro medical devices with internationally recognized definitions and changing the classification of medical devices according to levels or risk. The proposals are intended to improve patient and public safety and increase the appeal of the UK market. The new regime is planned to come into force on July 1, 2023, which will align with the date from which the UK is due to stop accepting CE marked medical devices and require UKCA (“UK Conformity Assessed”) marking. It is contemplated that, in Northern Ireland, the amended regime could run in parallel with any existing or future EU rules in accordance with the Protocol on Ireland and Northern Ireland.
Drug and Biologic Development Process
The conduct of clinical trials in the EU is governed by the EU Clinical Trials Regulation (EU) No. 536/2014 (“CTR”) which entered into force on January 31, 2022. The CTR replaced the Clinical Trials Directive 2001/20/EC, (Clinical Trials Directive) and introduced a complete overhaul of the existing regulation of clinical trials for medicinal products in the EU.
Under the former regime, which will expire after a transition period of one or three years, respectively, as outlined below in more detail, before a clinical trial can be initiated, it must be approved in each EU member state where there is a site at which the clinical trial is to be conducted. The approval must be obtained from two separate entities: the National Competent Authority (“NCA”), and one or more Ethics Committees. The NCA of the EU member states in which the
20
clinical trial will be conducted must authorize the conduct of the trial, and the independent Ethics Committee must grant a positive opinion in relation to the conduct of the clinical trial in the relevant EU member state before the commencement of the trial. Any substantial changes to the trial protocol or other information submitted with the clinical trial applications must be submitted to or approved by the relevant NCA and Ethics Committees. Under the current regime all suspected unexpected serious adverse reactions to the investigated drug that occur during the clinical trial must be reported to the NCA and to the Ethics Committees of the EU member state where they occur.
A more unified procedure will apply under the new CTR. A sponsor will be able to submit a single application for approval of a clinical trial through the a centralized EU clinical trials portal. One national regulatory authority (the reporting EU member state proposed by the applicant) will take the lead in validating and evaluating the application, and consult and coordinate with the other concerned EU member states. If an application is rejected, it may be amended and resubmitted through the EU clinical trials portal. If an approval is issued, the sponsor may start the clinical trial in all concerned EU member states. However, a concerned EU member state may in limited circumstances declare an “opt-out” from an approval and prevent the clinical trial from being conducted in such EU member state. The CTR also aims to streamline and simplify the rules on safety reporting and introduces enhanced transparency requirements such as mandatory submission of a summary of the clinical trial results to the EU Database (“CTIS”). The CTR includes a three-year transition period. Member states will work in CTIS immediately after the system has gone live. For one year, until January 31, 2023, clinical trial sponsors can still choose whether to submit an initial clinical trial application in line with the former system (Clinical Trials Directive) or via CTIS. From January 31, 2023 on, submission of initial clinical trial applications via CTIS becomes mandatory, and by January 31, 2025, all ongoing trials approved under the former Clinical Trials Directive will be governed by the new Regulation and have to be transitioned to CTIS.
Under both the former regime and the new CTR, national laws, regulations, and the applicable Good Clinical Practice and Good Laboratory Practice standards must also be respected during the conduct of the trials, including the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (“ICH”) guidelines on Good Clinical Practice (“GCP”), and the ethical principles that have their origin in the Declaration of Helsinki.
During the development of a medicinal product, the European Medical Agency (“EMA”) and national regulators within the EU provide the opportunity for dialogue and guidance on the development program. At the EMA level, this is usually done in the form of scientific advice, which is given by the Committee for Medicinal Products for Human Use (“CHMP”) on the recommendation of the Scientific Advice Working Party (“SAWP”). A fee is incurred with each scientific advice procedure, but is significantly reduced for designated orphan medicines. Advice from the EMA is typically provided based on questions concerning, for example, quality (chemistry, manufacturing and controls testing), nonclinical testing and clinical studies, and pharmacovigilance plans and risk-management programs. Advice is not legally binding with regard to any future Marketing Authorization Application (“MAA”) of the product concerned.
Marketing Authorization Procedures
In the EU and in Iceland, Norway and Liechtenstein (together the European Economic Area or “EEA”), after completion of all required clinical testing, pharmaceutical products may only be placed on the market after obtaining a Marketing Authorization (“MA”). To obtain an MA of a drug under EU regulatory systems, an applicant can submit a MAA through, amongst others, a centralized or decentralized procedure.
The centralized procedure provides for the grant of a single MA by the European Commission (“EC”) that is valid for all EU member states and, after respective national implementing decisions, in the three additional member states of the EEA. The centralized procedure is compulsory for specific pharmaceutical products, including for medicines developed by means of certain biotechnological processes, products designated as orphan pharmaceutical products, advanced therapy pharmaceutical products and pharmaceutical products with a new active substance indicated for the treatment of certain diseases (AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and viral diseases). For pharmaceutical products containing a new active substance not yet authorized in the European Economic Area before May 20, 2004 and indicated for the treatment of other diseases, pharmaceutical products that constitute significant therapeutic, scientific or technical innovations or for which the grant of a MA through the centralized procedure would be in the interest of public health at EU level, an applicant may voluntarily submit an application for a marketing authorization through the centralized procedure.
Under the centralized procedure, the CHMP established at the EMA is responsible for conducting the initial assessment of a drug. The CHMP is also responsible for several post-authorization and maintenance activities, such as the assessment of modifications or extensions to an existing marketing authorization. Under the centralized procedure, the timeframe for the evaluation of an MAA by the EMA’s CHMP is, in principle, 210 days from receipt of a valid MAA. However, this timeline excludes clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the CHMP, so the overall process typically takes a year or more, unless the application is
21
eligible for an accelerated assessment. Accelerated assessment might be granted by the CHMP in exceptional cases when a pharmaceutical product is of major interest from the point of view of public health and in particular from the viewpoint of therapeutic innovation. On request, the CHMP can reduce the time frame to 150 days if the applicant provides sufficient justification for an accelerated assessment. The CHMP will provide a positive opinion regarding the application only if it meets certain quality, safety and efficacy requirements. However, the EC has final authority for granting the MA within 67 days after receipt of the CHMP opinion.
The decentralized procedure permits companies to file identical MA applications for a pharmaceutical product to the competent authorities in various EU member states simultaneously if such pharmaceutical product has not received marketing approval in any EU member state before. This procedure is available for pharmaceutical products not falling within the mandatory scope of the centralized procedure. The competent authority of a single EU member state, known as the reference EU member state, is appointed to review the application and provide an assessment report. Under this procedure, an applicant submits an application based on identical dossiers and related materials, including a draft summary of product characteristics, and draft labeling and package leaflet, to the reference EU member state and concerned EU member states. The reference EU member state prepares a draft assessment report and drafts of the related materials within 120 days after receipt of a valid application. Subsequently, each concerned EU member state must decide whether to approve the assessment report and related materials.
If an EU member state cannot approve the assessment report and related materials on the grounds of potential serious risk to public health, the disputed points are subject to a dispute resolution mechanism and may eventually be referred to the EC, whose decision is binding for all EU member states.
All new MAAs must include a Risk Management Plan (“RMP”), describing the risk management system that the company will put in place and documenting measures to prevent or minimize the risks associated with the product. The regulatory authorities may also impose specific obligations as a condition of the MA. RMPs and Periodic Safety Update Reports (“PSURs”) are routinely available to third parties requesting access, subject to limited redactions.
Marketing Authorizations have an initial duration of five years. After these five years, the authorization may subsequently be renewed on the basis of a reevaluation of the risk-benefit balance. Once renewed, the MA is valid for an unlimited period unless the EC or the national competent authority decides, on justified grounds relating to pharmacovigilance, to proceed with only one additional five-year renewal. Applications for renewal must be made to the EMA at least nine months before the five-year period expires.
Data and Market Exclusivity in the European Union
As in the United States, it may be possible to obtain a period of market and / or data exclusivity in the EU that would have the effect of postponing the entry into the marketplace of a competitor’s generic, hybrid or biosimilar product (even if the pharmaceutical product has already received an MA) and prohibiting another applicant from relying on the MA holder’s pharmacological, toxicological and clinical data in support of another MA for the purposes of submitting an application, obtaining MA or placing the product on the market. New Chemical Entities (“NCE”) approved in the EU qualify for eight years of data exclusivity and ten years of marketing exclusivity. The overall ten-year period can be extended to a maximum of eleven years if, during the first eight years of those ten years, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are deemed to bring a significant clinical benefit in comparison with existing therapies.
The data exclusivity period begins on the date of the product’s first MA in the EU. After eight years, a generic product application may be submitted and generic companies may rely on the MA holder’s data. However, a generic product cannot launch until two years later (or a total of 10 years after the first MA in the EU of the innovator product), or three years later (or a total of 11 years after the first MA in the EU of the innovator product) if the MA holder obtains MA for a new indication with significant clinical benefit within the eight-year data exclusivity period. Additionally, another noncumulative one-year period of data exclusivity can be added to the eight years of data exclusivity where an application is made for a new indication for a well-established substance, provided that significant preclinical or clinical studies were carried out in relation to the new indication. Another year of data exclusivity may be added to the eight years, where a change of classification of a pharmaceutical product has been authorized on the basis of significant pre-trial tests or clinical trials (when examining an application by another applicant for or holder of market authorization for a change of classification of the same substance the competent authority will not refer to the results of those tests or trials for one year after the initial change was authorized).
Products may not be granted data exclusivity since there is no guarantee that a product will be considered by the EU’s regulatory authorities to include a NCE. Even if a compound is considered to be a NCE and the MA applicant is able to gain the prescribed period of data exclusivity, another company nevertheless could also market another version of the
22
pharmaceutical product if such company can complete a full MAA with their own complete database of pharmaceutical tests, preclinical studies and clinical trials and obtain MA of its pharmaceutical product.
Orphan Designation and Exclusivity
The criteria for designating an orphan medicinal product in the EU are similar in principle to those in the United States. The EMA grants orphan drug designation if the medicinal product is intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition affecting no more than five in 10,000 persons in the EU (prevalence criterion). In addition, Orphan Drug Designation can be granted if, for economic reasons, the medicinal product would be unlikely to be developed without incentives and if there is no other satisfactory method approved in the EU of diagnosing, preventing, or treating the condition, or if such a method exists, the proposed medicinal product is a significant benefit to patients affected by the condition. An application for orphan drug designation (which is not a marketing authorization, as not all orphan-designated medicines reach the authorization application stage) must be submitted first before an application for marketing authorization of the medicinal product is submitted. The applicant will receive a fee reduction for the marketing authorization application if the orphan drug designation has been granted, but not if the designation is still pending at the time the marketing authorization is submitted, and sponsors must submit an annual report to EMA summarizing the status of development of the medicine. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. Designated orphan medicines are eligible for conditional marketing authorization.
The EMA’s Committee for Orphan Medicinal Products reassesses the orphan drug designation of a product in parallel with the review for a marketing authorization; for a product to benefit from market exclusivity it must maintain its orphan drug designation at the time of marketing authorization review by the EMA and approval by the EC. Additionally, any marketing authorization granted for an orphan medicinal product must only cover the therapeutic indication(s) that are covered by the orphan drug designation. Upon the grant of a marketing authorization, orphan drug designation provides up to ten years of market exclusivity in the orphan indication.
During the 10-year period of market exclusivity, with a limited number of exceptions, the regulatory authorities of the EU member states and the EMA may not accept applications for marketing authorization, accept an application to extend an existing marketing authorization or grant marketing authorization for other similar medicinal products for the same therapeutic indication. A similar medicinal product is defined as a medicinal product containing a similar active substance or substances as contained in a currently authorized orphan medicinal product, and which is intended for the same therapeutic indication. An orphan medicinal product can also obtain an additional two years of market exclusivity for an orphan-designated condition when the results of specific studies are reflected in the Summary of Product Characteristics (“SmPC”), addressing the pediatric population and completed in accordance with a fully compliant Pediatric Investigation Plan (“PIP”). No extension to any supplementary protection certificate can be granted on the basis of pediatric studies for orphan indications.
The 10-year market exclusivity may be reduced to six years if, at the end of the fifth year, it is established that the product no longer meets the criteria for orphan designation, i.e. the condition prevalence or financial returns criteria under Article 3 of Regulation (EC) No. 141/2000 on orphan medicinal products. When the period of orphan market exclusivity for an indication ends, the orphan drug designation for that indication expires as well. Orphan exclusivity runs in parallel with normal rules on data exclusivity and market protection. Additionally, a marketing authorization may be granted to a similar medicinal product (orphan or not) for the same or overlapping indication subject to certain requirements.
Pediatric Development
In the EU, companies developing a new pharmaceutical product are obligated to study their product in children and must therefore submit a PIP together with a request for agreement to the EMA. The EMA issues a decision on the PIP based on an opinion of the EMA’s Pediatric Committee (“PDCO”). Companies must conduct pediatric clinical trials in accordance with the PIP approved by the EMA, unless a deferral (e.g. until enough information to demonstrate its effectiveness and safety in adults is available) or waiver (e.g. because the relevant disease or condition occurs only in adults) has been granted by the EMA. The marketing authorization application for the pharmaceutical product must include the results of all pediatric clinical trials performed and details of all information collected in compliance with the approved PIP, unless a waiver or a deferral has been granted, in which case the pediatric clinical trials may be completed at a later date. Pharmaceutical products that are granted a marketing authorization on the basis of the pediatric clinical trials conducted in accordance with the approved PIP are eligible for a six month extension of the protection under a supplementary protection certificate (if any is in effect at the time of approval) or, in the case of orphan pharmaceutical products, a two year extension of the orphan market exclusivity. This pediatric reward is subject to specific conditions and is not automatically available when data in compliance with the approved PIP are developed and submitted. An approved PIP is also required when a marketing authorization holder wants to add a new indication, pharmaceutical form or route of administration for a medicine that is already authorized and covered by intellectual property rights.
23
Post-Approval Regulation
Similar to the United States, both MA holders and manufacturers of pharmaceutical products are subject to comprehensive regulatory oversight by the EMA, the EC and/or the competent regulatory authorities of the EU member states. This oversight applies both before and after grant of manufacturing licenses and marketing authorizations. It includes control of compliance with EU good manufacturing practices rules, manufacturing authorizations, pharmacovigilance rules and requirements governing advertising, promotion, sale, and distribution, recordkeeping, importing and exporting of pharmaceutical products.
Failure by us or by any of our third-party partners, including suppliers, manufacturers and distributors to comply with EU laws and the related national laws of individual EU member states governing the conduct of clinical trials, manufacturing approval, MA of pharmaceutical products and marketing of such products, both before and after grant of MA, manufacturing of pharmaceutical products, statutory health insurance, bribery and anti-corruption or other applicable regulatory requirements may result in administrative, civil or criminal penalties. These penalties could include delays or refusal to authorize the conduct of clinical trials or to grant MA, product withdrawals and recalls, product seizures, suspension, withdrawal or variation of the MA, total or partial suspension of production, distribution, manufacturing or clinical trials, operating restrictions, injunctions, suspension of licenses, fines and criminal penalties.
The holder of an EU MA for a pharmaceutical product must also comply with EU pharmacovigilance legislation and its related regulations and guidelines, which entail many requirements for conducting pharmacovigilance, or the assessment and monitoring of the safety of pharmaceutical products.
These pharmacovigilance rules can impose on holders of MAs the obligation to conduct a labor intensive collection of data regarding the risks and benefits of marketed pharmaceutical products and to engage in ongoing assessments of those risks and benefits, including the possible requirement to conduct additional clinical studies or post-authorization safety studies to obtain further information on a medicine’s safety, or to measure the effectiveness of risk-management measures, which may be time consuming and expensive and could impact our profitability. MA holders must establish and maintain a pharmacovigilance system and appoint an individual qualified person for pharmacovigilance who is responsible for oversight of that system. Key obligations include expedited reporting of suspected serious adverse reactions and submission of Periodic Safety Update Reports (“PSURs”) in relation to pharmaceutical products for which they hold MAs. The EMA reviews PSURs for pharmaceutical products authorized through the centralized procedure. If the EMA has concerns that the risk-benefit profile of a product has varied, it can adopt an opinion advising that the existing MA for the product be suspended, withdrawn or varied. The agency can advise that the MA holder be obliged to conduct post-authorization Phase 4 safety studies. If the EC agrees with the opinion, it can adopt a decision varying the existing MA. Failure by the MA holder to fulfill the obligations for which the European Commission’s decision provides can undermine the on-going validity of the MA.
More generally, non-compliance with pharmacovigilance obligations can lead to the variation, suspension or withdrawal of the marketing authorization for the pharmaceutical product or imposition of financial penalties or other enforcement measures.
The manufacturing process for pharmaceutical products in the EU is highly regulated and regulators may shut down manufacturing facilities that they believe do not comply with regulations. Manufacturing requires a manufacturing authorization, and the manufacturing authorization holder must comply with various requirements set out in the applicable EU laws, regulations and guidance, including Directive 2001/83/EC, Directive 2003/94/EC, Regulation (EC) No 726/2004 and the European Commission Guidelines for Good Manufacturing Practice (“GMP”). These requirements include compliance with EU GMP standards when manufacturing pharmaceutical products and active pharmaceutical ingredients, including the manufacture of active pharmaceutical ingredients outside of the EU with the intention to import the active pharmaceutical ingredients into the EU.
Similarly, the distribution of pharmaceutical products into and within the EU is subject to compliance with the applicable EU laws, regulations and guidelines, including the requirement to hold appropriate authorizations for distribution granted by the competent authorities of the EU member states. The manufacturer or importer must have a qualified person who is responsible for certifying that each batch of product has been manufactured in accordance with GMP, before releasing the product for commercial distribution in the EU or for use in a clinical trial. Manufacturing facilities are subject to periodic inspections by the competent authorities for compliance with GMP.
Advertising and Promotion
The advertising and promotion of our products is also subject to EU laws concerning promotion of pharmaceutical products, interactions with physicians, misleading and comparative advertising and unfair commercial practices. In addition, other national legislation of individual EU member states may apply to the advertising and promotion of pharmaceutical products and may differ from one country to another. These laws require that promotional materials and
24
advertising in relation to pharmaceutical products comply with the product’s SmPC as approved by the competent regulatory authorities. The SmPC is the document that provides information to physicians concerning the safe and effective use of the pharmaceutical product. It forms an intrinsic and integral part of the marketing authorization granted for the pharmaceutical product. Promotion of a pharmaceutical product that does not comply with the SmPC is considered to constitute off-label promotion. All advertising and promotional activities for the product must be consistent with the approved SmPC and therefore all off-label promotion of pharmaceutical products is prohibited in the EU. Direct-to-consumer advertising of prescription-only pharmaceutical products is prohibited in the EU. Violations of the rules governing the promotion of pharmaceutical products in the EU could be penalized by administrative measures, fines and imprisonment. These laws may further limit or restrict the advertising and promotion of our products to the general public and may also impose limitations on its promotional activities with healthcare professionals.
Pricing and Reimbursement Environment
Even if a pharmaceutical product obtains a marketing authorization in the EU, there can be no assurance that reimbursement for such product will be secured on a timely basis or at all. The EU member states are free to restrict the range of pharmaceutical products for which their national health insurance systems provide reimbursement, and to control the prices and reimbursement levels of pharmaceutical products for human use. An EU member state may approve a specific price or level of reimbursement for the pharmaceutical product, or alternatively adopt a system of direct or indirect controls on the profitability of the company responsible for placing the pharmaceutical product on the market, including volume-based arrangements, caps and reference pricing mechanisms.
Reference pricing used by various EU member states and parallel distribution, or arbitrage between low-priced and high-priced member states, can further reduce prices. In some countries, we may be required to conduct a clinical study or other studies that compare the cost-effectiveness of our product candidates, if any, to other available therapies in order to obtain or maintain reimbursement or pricing approval. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products. Historically, pharmaceutical products launched in the EU do not follow price structures of the United States and generally published and actual prices tend to be significantly lower. Publication of discounts by third-party payers or authorities and public tenders may lead to further pressure on the prices or reimbursement levels within the country of publication and other countries.
The so-called health technology assessment (“HTA”) of pharmaceutical products is becoming an increasingly common part of the pricing and reimbursement procedures in some EU member states, including France, Germany, Ireland, Italy and Sweden. The HTA process, which is governed by the national laws of these countries, is the procedure according to which the assessment of the public health impact, therapeutic impact, and the economic and societal impact of use of a given pharmaceutical product in the national healthcare systems of the individual country is conducted. HTA generally focuses on the clinical efficacy and effectiveness, safety, cost, and cost-effectiveness of individual pharmaceutical products as well as their potential implications for the healthcare system. Those elements of pharmaceutical products are compared with other treatment options available on the market. The outcome of HTA regarding specific pharmaceutical products will often influence the pricing and reimbursement status granted to pharmaceutical products by the regulatory authorities of individual EU member states. A negative HTA of one of our products by a leading and recognized HTA body could not only undermine our ability to obtain reimbursement for such product in the EU member state in which such negative assessment was issued, but also in other EU member states. For example, EU member states that have not yet developed HTA mechanisms could rely to some extent on the HTA performed in other countries with a developed HTA framework, when adopting decisions concerning the pricing and reimbursement of a specific pharmaceutical product.
On January 31, 2018, the European Commission adopted a proposal for a regulation on health technology assessment. This legislative proposal is intended to boost EU level cooperation among EU member states in assessing health technologies, including new pharmaceutical products, and providing the basis for cooperation at the EU level for joint clinical assessments in these areas. The proposal provides that EU member states will be able to use common HTA tools, methodologies and procedures across the EU, working together in four main areas, including joint clinical assessment of the innovative health technologies with the most potential impact for patients, joint scientific consultations whereby developers can seek advice from HTA authorities, identification of emerging health technologies to identify promising technologies early, and continuing voluntary cooperation in other areas. Individual EU member states will continue to be responsible for assessing non-clinical (e.g., economic, social, ethical) aspects of health technology, and making decisions on pricing and reimbursement. While EU member states could choose to delay participation in the joint work until three years after the rules enter into force, it will become mandatory after six years. The European Commission has stated that the role of the HTA regulation is not to influence pricing and reimbursement decisions in the individual EU member states, but there can be no assurance that the HTA regulation will not have effects on pricing and reimbursement
25
decisions. The HTA entered into force on January 11, 2022 and applies as of January 2025 followed by a further three-year transitional period during which EU member states must fully adapt to the new system.
To obtain reimbursement or pricing approval in some countries, including the EU member states, we may be required to conduct studies that compare the cost-effectiveness of our product candidates to other therapies that are considered the local standard of care. There can be no assurance that any country will allow favorable pricing, reimbursement and market access conditions for any of our products, or that we will be feasible to conduct additional cost-effectiveness studies, if required.
In certain of the EU member states, pharmaceutical products that are designated as orphan pharmaceutical products may be exempted or waived from having to provide certain clinical, cost-effectiveness and other economic data in connection with their filings for pricing/reimbursement approval.
European Data Laws
The collection and use of personal health data and other personal data in the EU is governed by the provisions of the European General Data Protection Regulation (EU) 2016/679 (“GDPR”), which came into force in May 2018 and related implementing laws in individual EU member states. The GDPR has a number of significant practical consequences, in particular for international data transfers, competent supervisory authorities and enforcement of the GDPR. The GDPR increased responsibility and liability in relation to personal data that we process.
The GDPR imposes a number of strict obligations and restrictions on the ability to process (processing includes collection, analysis and transfer of) personal data of individuals within the EU and in the EEA, including health data from clinical trials and adverse event reporting. The GDPR also includes requirements relating to the consent of the individuals to whom the personal data relates, the information provided to the individuals prior to processing their personal data or personal health data, notification obligations to the national data protection authorities and the security and confidentiality of the personal data. EU member states may also impose additional requirements in relation to health, genetic and biometric data through their national implementing legislation.
The GDPR also imposes specific restrictions on the transfer of personal data to countries outside of the EU/EEA that are not considered by the European Commission to provide an adequate level of data protection (including the United States). Appropriate safeguards are required to enable such transfers. Among the appropriate safeguards that can be used, the data exporter may use the standard contractual clauses (“SCCs”). In this respect, recent legal developments in Europe have created complexity and compliance uncertainty regarding certain transfers of personal data from the EU/EEA. For example, following the Schrems II decision of the Court of Justice of the EU on July 16, 2020, in which the Court invalidated the Privacy Shield under which personal data could be transferred from the EU/EEA to U.S. entities who had self-certified under the Privacy Shield scheme, there is uncertainty as to the general permissibility of international data transfers under the GDPR. The Court did not invalidate the then current SCCs, but ruled that data exporters relying on these SCCs are required to verify, on a case-by-case basis, if the law of the third country ensures an adequate level of data protection that is essentially equivalent to that guaranteed in the EU/EEA. In light of the implications of this decision, we may face difficulties regarding the transfer of personal data from the EU/EEA to third countries. On June 4, 2021, the EU Commission has issued a new set of SCCs which replace the old sets of SCCs that were adopted under the previous European Data Protection Directive 95/46. Since September 27, 2021, it is no longer possible to conclude contracts incorporating these previous versions of the SCCs. In addition, for contracts concluded before September 27, 2021, it is still possible to rely on the previous SCCs until the end of an additional 15 months transitional period (until December 27, 2022), provided that the processing operations which are the subject matter of the contract remain unchanged and reliance on previous SCCs ensures that the transfer is subject to appropriate safeguards. In addition, when relying on SCCs, the data exporters are required to conduct a transfer risk assessment to verify if anything in the law and/or practices of the third country may impinge on the effectiveness of the SCCs in the context of the transfer at stake and, if so, to identify and adopt supplementary measures that are necessary to bring the level of protection of the data transferred to the EU standard of essential equivalence. Where no supplementary measure is suitable, the data exporter should avoid, suspend or terminate the transfer. On June 18, 2021, the European Data Protection Board has adopted recommendations to assist data exporters with such assessment and their duty to identify and implement supplementary measures where they are needed to ensure compliance with the EU level of protection to the personal data they transfer to third countries. On March 25, 2022, the EU Commission and the U.S. announced that they have agreed in principle on a new Trans-Atlantic Data Privacy Framework. Following this statement, President Biden signed an Executive Order on ‘Enhancing Safeguards for United States Signals Intelligence Activities’ on October 7, 2022. Along with the Regulations issued by the Attorney General, the Executive Order implements into US law the agreement in principle announced in March 2022. On that basis, the European Commission will now prepare a draft adequacy decision and then launch its adoption procedure. While this new EU-US privacy framework is expected to entry into force in 2023, there is still some uncertainty around that new framework.
26
Failure to comply with the requirements of the GDPR and the related national data protection laws of the EU member states may result in significant monetary fines for noncompliance of up to €20 million or 4% of the annual global revenues of the noncompliant company, whichever is greater, other administrative penalties and a number of criminal offenses (punishable by uncapped fines) for organizations and in certain cases their directors and officers as well as civil liability claims from individuals whose personal data was processed. Data protection authorities from the different EU member states may still implement certain variations, enforce the GDPR and national data protection laws differently, and introduce additional national regulations and guidelines, which adds to the complexity of processing personal data in the EU. Guidance developed at both EU level and at the national level in individual EU member states concerning implementation and compliance practices are often updated or otherwise revised.
There is, moreover, a growing trend towards required public disclosure of clinical trial data in the EU which adds to the complexity of obligations relating to processing health data from clinical trials. Such public disclosure obligations are provided in the new EU Clinical Trials Regulation, EMA disclosure initiatives and voluntary commitments by industry. Failing to comply with these obligations could lead to government enforcement actions and significant penalties against us, harm to our reputation, and adversely impact our business and operating results. The uncertainty regarding the interplay between different regulatory frameworks, such as the Clinical Trials Regulation and the GDPR, further adds to the complexity that we face with regard to data protection regulation.
With regard to the transfer of data from the EU to the UK, the TCA provided for a transition period of up to six months as of January 1, 2021 to enable the European Commission to complete its adequacy assessment of the UK’s data protection laws. On June 28, 2021 the European Commission adopted two adequacy decisions for the UK: one under the GDPR and the other for the Law Enforcement Directive. Personal data may now freely flow from the EU to the UK since the UK is deemed to have an adequate data protection level. However, the adequacy of decisions are subject to a ‘sunset clause’ which entails that the decisions will automatically expire four years after their entry into force. Additionally, following the UK’s withdrawal from the EU and the EEA, companies have to comply also with the UK’s data protection laws (including the GDPR as incorporated into UK national law), the latter regime having the ability to separately fine up to the greater of £17.5 million or 4% of global turnover. Furthermore, transfers from the UK to other countries, including to the EEA, are subject to specific transfer rules under the UK regime. These UK transfer rules broadly mirror the EU GDPR rules. On February 2, 2022, the UK Secretary of State laid before the UK Parliament the international data transfer agreement (IDTA), the international data transfer addendum to the European Commission’s standard contractual clauses for international data transfers (Addendum) and a document setting out transitional provisions. The IDTA and Addendum came into force on March 25, 2022 and replaced the old EU SCCs. However, the transitional provisions, adopted with the IDTA and the Addendum, allow the continued use, until March 21, 2024, of any EU SCCs, valid as at December 31, 2020, as long as the contract was entered into before September 21, 2022.
Promotional Activities
In the EU, interactions between pharmaceutical companies and physicians are also governed by strict laws, regulations, industry self-regulation codes of conduct and physicians’ codes of professional conduct both at EU level and in the individual EU member states. The provision of benefits or advantages to physicians to induce or encourage the prescription, recommendation, endorsement, purchase, supply, order or use of pharmaceutical products is prohibited in the EU. The provision of benefits or advantages to physicians is also governed by the national anti-bribery laws of the EU member states. Violation of these laws could result in substantial fines and imprisonment.
Payments made to physicians in certain EU member states must be publicly disclosed. Moreover, agreements with physicians must often be the subject of prior notification and approval by the physician’s employer, his/her regulatory professional organization, and/or the competent authorities of the individual EU member states. These requirements are provided in the national laws, industry codes, or professional codes of conduct, applicable in the individual EU member states. Failure to comply with these requirements could result in reputational risk, public reprimands, administrative penalties, fines or imprisonment.
While the UK has left the EU, as mentioned above, it should be noted that the UK still has the strictest anti-bribery regime in Europe, the UK Bribery Act 2010. The Act is applicable English law and continues to apply to any company incorporated in or “carrying on business” in the UK, irrespective of where in the world the alleged bribery activity occurs.
Other Legislation Regarding Marketing, Authorization and Pricing of Pharmaceutical Products in the European Union
Other core legislation relating to the marketing, authorization and pricing of pharmaceutical products in the EU exists as regulations and directives, while the implementing acts and guidelines based on these may vary in each EU
27
member state. In addition, the respective national provisions of the member states, as well as self-committed codes of the pharmaceutical industry, must be observed. Such regulations and directives include the following:
•Directive 2001/83/EC, establishing the requirements and procedures governing the marketing authorization for medicinal products for human use, as well as the rules for the constant supervision of products following authorization. This Directive has been amended several times, most recently by Directive 2012/26/EU regarding pharmacovigilance, and the Falsified Medicines Directive 2011/62/EU.
•Regulation (EC) 726/2004, as amended, establishing procedures for the authorization, supervision and pharmacovigilance of medicinal products for human and veterinary use and establishing the EMA.
•Regulation (EC) 469/2009, establishing the requirements necessary to obtain a Supplementary Protection Certificate, which extends the period of patent protection applicable to medicinal products at the EU-level.
•Directive 89/105/EEC, ensuring the transparency of measures taken by the EU member states to set the prices and reimbursements of medicinal products. Specifically, while each member state has competence over the pricing and reimbursement of medicines for human use, they must also comply with this Directive, which establishes procedures to ensure that member state decisions and policies do not obstruct trade in medicinal products. The European Commission proposed to repeal and replace Directive 89/105/EEC, but this proposal was withdrawn in 2015.
•Directive 2003/94/EC, laying down the principles of good manufacturing practice in respect of medicinal products and investigational medicinal products for human use (the “GMP Directive”).
•Directive 2005/28/EC of April 8, 2005, laying down principles and detailed guidelines for good clinical practice as regards investigational medicinal products for human use, as well as the requirements for authorization of the manufacturing or importation of such products (the “GCP Directive”).
•Directives 2004/9/EC and 2004/10/EC laying down principles of good laboratory practices (“GLP”) including on the organizational process under which non-clinical health and safety studies are performed.
•Directive 2010/84/EU and Regulation (EU) 1235/2010 on pharmacovigilance laying down procedures for the authorization and supervision of medicinal products for human and veterinary use.
•Directive 2006/114/EC concerning misleading and comparative advertising.
•Directive 2005/29/EC regulating unfair business-to-consumer commercial practices that occur before, during and after a business-to-consumer transaction.
•Regulation (EC) 1223/2009 on Cosmetic Products, setting mandatory requirements for cosmetics which are available on the market within the EU.
•Regulation (EC) 1901/2006 on Pediatric Use, laying down rules to ensure that medicines for use in children are researched, developed and authorized appropriately.
•Directive (2004/109/EC) on Transparency laying down rules to improves the harmonization of information duties of issuers, whose securities are listed at a regulated market at a stock exchange within the EU.
Pharmaceutical Coverage, Pricing and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of products approved by the FDA and other government authorities. Sales of products will depend, in part, on the extent to which the costs of the products will be covered by third-party payors, including government health programs such as, in the United States, Medicare and Medicaid, commercial health insurers and managed care organizations. The process for determining whether a payor will provide coverage for a product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the product once coverage is approved. Third-party payors may limit coverage to specific products on an approved list, or formulary, which might not include all of the approved products for a particular indication.
In order to secure coverage and reimbursement for any product that might be approved for sale, a company may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of the product, in addition to the costs required to obtain FDA or other comparable regulatory approvals. A payor’s decision to provide coverage for a drug product does not necessarily imply that an adequate reimbursement rate will be approved. Third-party reimbursement may not be sufficient to maintain price levels high enough to realize an appropriate return on our investment in product development.
28
The containment of healthcare costs has become a priority of federal, state and foreign governments, and the prices of drugs have been a focus in this effort. Third-party payors are increasingly challenging the prices charged for medical products and services and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. If these third-party payors do not consider a product to be cost effective compared to other available therapies, they may not cover the product after approval as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow a company to sell its products at a profit. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost containment programs to limit the growth of government-paid health care costs, including price controls, risk sharing, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs. Adoption of such controls and measures and tightening of restrictive policies in jurisdictions with existing controls and measures, could limit payments for pharmaceuticals. As a result, the marketability of any product which receives regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement.
In addition, an increasing emphasis on managed care in the United States has increased and will continue to increase the pressure on drug pricing. Coverage policies, third-party reimbursement rates and drug pricing regulation may change at any time. In particular, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, contains provisions that may reduce the profitability of drug products, including, for example, increased rebates for drugs sold to Medicaid programs, extension of Medicaid rebates to Medicaid managed care plans, mandatory discounts for certain Medicare Part D beneficiaries and annual fees based on pharmaceutical companies’ share of sales to federal health care programs. Even if favorable coverage and reimbursement status is attained for one or more products that receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
In the EU, pricing and reimbursement schemes vary widely between member states. Some countries provide that drug products may be marketed only after a reimbursement price has been agreed. Some member states may require the completion of additional studies that compare the cost-effectiveness of a particular product candidate to currently available therapies. For example, the EU provides options for its member states to restrict the range of drug products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. EU member states may approve a specific price for a drug product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the drug product on the market. Other member states allow companies to fix their own prices for drug products but monitor and control company profits. The downward pressure on health care costs in general, particularly prescription drugs, has become intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in some countries, cross-border imports from low-priced markets exert competitive pressure that may reduce pricing within a country. Any country that has price controls or reimbursement limitations for drug products may not allow favorable reimbursement and pricing arrangements for any of our products.
Healthcare Laws and Regulations
Healthcare providers, physicians and third-party payors play important roles in the recommendation and prescription of drug products that are granted marketing approval. Arrangements with healthcare providers, physicians, third-party payors and customers are subject to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute products for which we obtain marketing approval. Restrictions under applicable federal and state healthcare laws and regulations, include the following:
•the federal Anti-Kickback Statute prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing any remuneration (in cash or in kind), directly or indirectly, to induce or reward either the referral of an individual for, or the purchase, lease, order or recommendation of, any item, facility or service for which payment may be made in whole or in part under a federal healthcare program such as Medicare and Medicaid;
•the federal Foreign Corrupt Practices Act prohibits, among other things, U.S. corporations and persons acting on their behalf from offering, promising, authorizing or making payments to any foreign government official (including certain healthcare professionals in many countries), political party, or political candidate in an attempt to obtain or retain business or otherwise seek preferential treatment abroad;
•the federal False Claims Act, which may be enforced by the U.S. Department of Justice or private whistleblowers who bring civil actions (qui tam actions) on behalf of the federal government, imposes civil penalties, as well as liability for treble damages and for attorneys’ fees and costs, on individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false
29
or fraudulent, making a false statement material to a false or fraudulent claim, or improperly avoiding, decreasing, or concealing an obligation to pay money to the federal government;
•the U.S. Department of Health and Human Services’ Civil Monetary Penalty authorities, which imposes administrative sanctions for, among other things, presenting or causing to be presented false claims for government payment and providing remuneration to government health program beneficiaries to influence them to order or receive healthcare items or services;
•the federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) imposes criminal and civil liability for, among other conduct, executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters;
•HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act and its implementing regulations, also imposes criminal and civil liability and penalties on those who violate requirements, including mandatory contractual terms, intended to safeguard the privacy, security, transmission and use of individually identifiable health information;
•the federal false statements statute relating to healthcare matters imposes criminal liability for knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services;
•the federal Physician Payment Sunshine Act requires manufacturers of drugs (among other products) to report to the Centers for Medicare and Medicaid Services within the U.S. Department of Health and Human Services information related to payments and other transfers of value to physicians and teaching hospitals, as well as physician ownership and investment interests in the reporting manufacturers;
•similar state and foreign laws and regulations, such as state anti-kickback and false claims laws, may apply (e.g., in the EU, where the implementation of EU-wide regulations as well as independent national legislation may vary for each EU member state) to sales or marketing arrangements and claims involving healthcare items or services reimbursed by nongovernmental third-party payors, including private insurers; and
•certain state laws require pharmaceutical companies to comply with voluntary compliance guidelines promulgated by a pharmaceutical industry association and relevant compliance guidance issues by HHS Office of Inspector General; bar drug manufacturers from offering or providing certain types of payments or gifts to physicians and other health care providers; and/or require disclosure of gifts or payments to physicians and other healthcare providers.
Various state and foreign laws also govern the privacy and security of health information in some circumstances; many of these laws differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
Research and Development Facilities
Arrowhead operates lab facilities in Madison, Wisconsin and San Diego, California where its research and development activities, including the development of RNAi therapeutics, take place. Arrowhead’s principal executive offices are located in Pasadena, California. A summary of research and development resources is provided below:
•329 R&D personnel as of September 30, 2022;
•State-of-the-art laboratories consisting of 132,000 total square feet;
•Cell culture laboratories;
•Complete animal facilities;
•Primate colony housed at the Wisconsin National Primate Research Center, an affiliate of the University of Wisconsin, and at other contract research organizations;
•In-house histopathology capabilities;
•Animal efficacy models for cardio metabolic, viral, lung and oncologic diseases;
•Animal safety screening and assessment;
•In-house drug substance manufacturing capabilities to produce and release GMP material (API);
•Polymer, peptide, oligonucleotide and small molecule synthesis, analytics capabilities (HPLC, NMR, LCMS, etc.);
30
•Drug metabolism and pharmacokinetics (DMPK), biodistribution, and clearance assessment and methodology capabilities;
•Conventional and confocal microscopy, flow cytometry, Luminex platform, qRT-PCR and clinical chemistry analytics.
Human Capital Management
As of September 30, 2022, Arrowhead employed 397 full time employees based at three facilities in the United States, including Pasadena and San Diego, California, and Madison, Wisconsin. The following table presents total number of employees as of September 30 by location.
| Site | 2022 | 2021 | |||||||||
| Pasadena, CA | 115 | 93 | |||||||||
| Madison, WI | 225 | 198 | |||||||||
| San Diego, CA | 57 | 38 | |||||||||
| Total | 397 | 329 | |||||||||
Arrowhead continued to add additional employees in fiscal year 2022 with a focus on expanding its in-house manufacturing capacity, as well as increasing expertise and bandwidth in clinical and preclinical research and development. Arrowhead continually evaluates the business need and opportunity and balances in-house expertise and capacity with outsourced expertise and capacity. Currently, Arrowhead outsources substantial clinical trial work to clinical research organizations and certain drug manufacturing to contract manufacturers.
Drug development is a complex endeavor which requires deep expertise and experience across a broad array of disciplines. Pharmaceutical companies both large and small compete for a limited number of qualified applicants to fill specialized positions. To attract qualified applicants to the Company, Arrowhead offers a total compensation package consisting of base salary and cash target bonus targeting the 50th to 75th percentile of market, and offers a comprehensive benefit package and equity compensation to every employee. Bonus opportunity and equity compensation increase as a percentage of total compensation based on level of responsibility. Actual bonus payout is based on performance.
A significant portion of Arrowhead’s employees have obtained advanced degrees in their professions. Arrowhead supports its employees’ further development with individualized development plans, mentoring, coaching, group training, conference attendance and financial support including tuition reimbursement.
Diversity and Inclusion
Arrowhead is committed maintaining a welcome, healthy and equitable environment where all employees can excel and contribute to its mission of bringing safe and effective medicine to patients in need. Arrowhead continues the formal training and processes initiated in 2021 to promote awareness of inclusion and diversity issues for management and employees, including anti-bias training and employee outreach and engagement. In fiscal year 2022, Arrowhead formed a Diversity, Equity, and Inclusion (DEI) committee comprised of a diverse group of employees across each of its worksites. Arrowhead’s DEI committee meets regularly, provides well-attended education and outreach opportunities to its employee base, and offers advice to its senior management concerning Arrowhead's efforts to build a more diverse, equitable, and inclusive workplace.
Investor Information
Arrowhead’s website address is http://www.arrowheadpharma.com. Arrowhead’s website address is not intended to function as a hyperlink and the information contained on its website is not, and should not be considered part of, and is not incorporated by reference into, this Annual Report on Form 10-K. Arrowhead’s reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), including its Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, Proxy Statements, and amendments to such periodic reports and Proxy Statements, are accessible through its website, free of charge, as soon as reasonably practicable after these reports are filed electronically with, or otherwise furnished to, the SEC. These SEC reports can be accessed through the “Investors” section of Arrowhead's website.
The SEC maintains an Internet website that contains reports, proxy and information statements, and other information regarding Arrowhead and other issuers that file electronically with the SEC. The SEC’s Internet website address is http://www.sec.gov.
31
ITEM 1A.RISK FACTORS
The Company’s business involves various risks and uncertainties in addition to the normal risks of business, some of which are discussed in this section. It should be noted that the Company’s business may be adversely affected by general economic conditions and other forces beyond the Company’s control. In addition, other risks and uncertainties not presently known or that the Company currently believes to be immaterial may also adversely affect the Company’s business. Any such risks or uncertainties, or any of the following risks or uncertainties, that develop into actual events could result in a material and adverse effect on the Company’s business, financial condition, results of operations, or liquidity.
The information discussed below should be considered carefully with the other information contained in this Annual Report on Form 10-K and the other documents and materials filed by the Company with the SEC, as well as news releases and other information publicly disseminated by the Company from time to time.
Risk Factors Summary
Risks Related to Our Discovery, Development, and Commercialization of Medicines
•Our prospects substantially depend on the success of our clinical-stage product candidates. If we and our licensees are unable to obtain approval for and commercialize these product candidates, our business could be materially harmed.
•There are substantial risks inherent in attempting to commercialize our new drugs, and, as a result, we may not be able to successfully develop products for commercial use.
•Our product candidates are in clinical development, which is a lengthy and expensive process with uncertain outcomes and the potential for substantial delays. There can be no assurance that our product candidates will obtain regulatory approval, which is necessary before they can be commercialized.
•Our clinical trials may not yield successful results for the product candidates that we may identify and pursue for their intended uses, which would prevent, delay or limit the scope of regulatory approval and commercialization.
•Our clinical trials may reveal significant adverse events, toxicities or other side effects and may result in a safety profile that could inhibit regulatory approval or market acceptance of any of our product candidates.
•Clinical trials of our product candidates may not uncover all possible adverse events that patients may experience.
•Topline data may not accurately reflect the complete results of a particular study or trial.
•Results of earlier studies or clinical trials may not be predictive of future clinical trial results, and initial studies or clinical trials may not establish an adequate safety or efficacy profile for our product candidates to justify proceeding to advanced clinical trials or an application for regulatory approval.
•It may take us longer than we project to complete clinical trials, and we may not be able to complete them at all.
•We face potential product liability exposure, and if successful claims are brought against us, we may incur substantial liability for a product candidate and may have to limit its commercialization.
•The successful commercialization of our product candidates, if approved, will depend in part on the extent to which government authorities and health insurers establish adequate reimbursement levels and pricing policies.
•We may not enjoy the market exclusivity benefits of our orphan drug designations.
•Our success depends on the attraction and retention of senior management and scientists with relevant expertise.
Risks Related to Regulatory Review and Approval of Our Candidates
•Breakthrough Therapy designation for ARO-AAT may not lead to a faster development or review process.
•We and our licensees conduct clinical trials for product candidates outside the United States, and the FDA and comparable foreign regulatory authorities may not accept data from such trials.
•Even if we obtain FDA approval for products in the United States, we may never obtain approval to commercialize any product candidates outside of the United States, which would limit our ability to realize their full market potential.
•Even if our product candidates are approved for commercialization, failure to comply with regulatory requirements or unanticipated problems with our products may result in various adverse actions such as the suspension or withdrawal of one or more of our products, closure of a facility or enforcement of substantial penalties or fines.
Risks Related to Our Intellectual Property
•Our ability to protect our patents and other proprietary rights is uncertain, exposing us to the possible loss of competitive advantage.
32
•We are party to technology license agreements with third parties that require us to satisfy obligations to keep them effective and, if these agreements are terminated, our technology and our business could be seriously and adversely affected.
•We may be subject to patent infringement claims, which could result in substantial costs and liability and prevent us from commercializing our potential products.
•We license patent rights from third-party owners and we rely on such owners to obtain, maintain and enforce the patents underlying such licenses.
•Our technology licensed from various third parties may be subject to retained rights.
•Confidentiality agreements with employees and others may not adequately prevent disclosure of trade secrets and other proprietary information.
•We may not be able to effectively secure first-tier technologies when competing against other companies or investors.
Risks Related to Our Business Model
•Our business model assumes we will generate revenue by, among other activities, marketing or out-licensing the products we develop. Our drug candidates are in various stages of development and we have no approved products based on RNA interference and our delivery technologies. Accordingly, there is a limited amount of information about us upon which you can evaluate our business and prospects.
•We may need to establish additional relationships with strategic and development partners to fully develop our drug candidates and market any approved products.
•Our ability to generate milestone and royalty payments under our current and potential future licensing and collaboration agreements is substantially controlled by our partners, and as such, we will likely need other sources of financing to continue to develop our internal drug candidates.
•We may lose a considerable amount of control over our intellectual property and may not receive anticipated revenues in strategic transactions, particularly where the consideration is contingent on the achievement of development or sales milestones.
•We will need to achieve commercial acceptance of our drug candidates to generate revenues and achieve profitability.
•We rely on outside sources for various components and processes for our products.
•We have limited manufacturing capability and must rely on third-party manufacturers to manufacture our clinical supplies and commercial products, if and when approved, and if they fail to meet their obligations, the development and commercialization of our products could be adversely affected.
•We rely on third parties to conduct our clinical trials, and if they fail to fulfill their obligations, the development of our products may be adversely affected.
•We face competition from various entities including large pharmaceutical companies, small biotech companies, private companies, and research institutions.
•We may have difficulty expanding our operations successfully as we evolve our pipeline and move toward commercializing drugs.
•Our business and operations could suffer in the event of information technology system failures.
•Because we use biological materials, hazardous materials, chemicals and radioactive compounds, if we do not comply with laws regulating the protection of the environment and health and human safety, our business could be adversely affected.
•Litigation claims may result in financial losses or harm our reputation and may divert management resources.
•Our operations, including our relationships with healthcare providers, physicians and third-party payers, are subject to applicable anti-kickback, fraud and abuse, and other healthcare laws and regulations, which, in the event of a violation, exposes us to liability for criminal sanctions, civil penalties, and contractual damages, and reputational harm and diminished profits and future earnings.
Risks Related to Our Financial Condition
•We have a history of net losses, and we expect to continue to incur net losses and may not achieve or maintain profitability.
•We will require substantial additional funds to complete our research and development activities.
•If the estimates we make, or the assumptions on which we rely, in preparing our consolidated financial statements prove inaccurate, our actual results may vary from those reflected in our accruals.
•Our operating results may fluctuate significantly, which makes our future operating results difficult to predict and could cause our operating results to fall below expectations or our guidance.
•The investment of our cash, cash equivalents and fixed income marketable securities is subject to risks which may cause losses and affect the liquidity of these investments.
33
•Our ability to utilize net operating loss carryforwards and other tax benefits may be limited.
•Our business is subject to changing regulations for corporate governance and public disclosure that has increased both our costs and the risk of noncompliance.
•We could be subject to additional tax liabilities.
Risks Related to Investment and Securities
•Our Board of Directors has the authority to issue shares of “blank check” preferred stock, which may make an acquisition of the Company by another company more difficult.
•We do not intend to declare cash dividends on our common stock.
•If securities or industry analysts do not publish research reports about our business or if they make adverse recommendations regarding an investment in our stock, our stock price and trading volume may decline.
•The market for purchases and sales of our common stock may be limited, and the sale of a limited number of shares could cause the price to fall sharply.
•Our common stock price has fluctuated significantly over the last several years and may continue to do so in the future, without regard to our results of operations and prospects.
•Stockholder equity interest may be substantially diluted in any additional equity issuances.
Economic and Industry Risks
•Unfavorable global economic conditions, whether brought about by material global crises, health epidemics, military conflicts and war, geopolitical and trade disputes or other factors, may adversely affect our business and financial results.
•Drug development is time consuming, expensive and risky.
•The healthcare system is under significant financial pressure to reduce costs, which could reduce payment and reimbursement rates for drugs.
•Regulatory standards are subject to change over time, making it difficult to accurately predict the likelihood of marketing approval even when clinical trials meet their endpoints.
Risks Related to Our Discovery, Development, and Commercialization of Medicines
Our prospects substantially depend the success of our clinical-stage product candidates. If we and our licensees are unable to obtain approval for and commercialize these product candidates, our business could be materially harmed.
Our future success is substantially dependent on the ability of our company and our licensees to timely complete clinical trials and obtain marketing approval for, and then successfully commercialize our clinical-stage product candidates. We are not permitted to market or promote our product candidates before we receive marketing approval from the FDA and comparable foreign regulatory authorities, and we may never receive such marketing approvals.
The success of developing and commercializing our product candidates will depend on several factors, including the following:
•obtaining positive data that supports demonstration of efficacy, safety and tolerability profiles and durability of effect for our product candidates that are satisfactory to the FDA or any comparable foreign regulatory authority for marketing approval;
•successful and timely enrollment of appropriate patients for the indications included in our current and future clinical trials;
•potential variability of patient outcomes;
•our ability to address any potential delays resulting from factors related to the COVID-19 pandemic;
•the extent of any required post-marketing approval commitments to applicable regulatory authorities;
•the maintenance of existing or the establishment of new supply arrangements with third-party drug product suppliers and manufacturers for clinical development and, if approved, commercialization of our product candidates;
•the maintenance of existing or the establishment of new scaled production arrangements with third-party manufacturers to obtain finished products that are appropriately packaged for sale;
•obtain and maintaining patent protection, trade secret protection and regulatory exclusivity, both in the United States and internationally;
•protection of our rights in our intellectual property portfolio, including our licensed intellectual property;
•establishing sales, marketing and distribution capabilities and the successful launch of commercial sales of our product candidates if and when approved for marketing, whether alone or in collaboration with others;
•a continued acceptable safety profile following any marketing approval;
•commercial acceptance by patients, the medical community and third-party payors; and
34
•our ability to compete with other therapies.
We do not have complete control over many of these factors, including certain aspects of clinical development and the regulatory submission process, potential threats to our intellectual property rights and the manufacturing, marketing, distribution and sales efforts of any future collaborator. For development programs that are licensed to third parties, we generally do not have control over the design or conduct of clinical trials and will not have discretion over marketing decisions. If we are not successful with respect to one or more of these factors in a timely manner or at all, we could experience significant delays or an inability to successfully commercialize any product candidates from our lead programs, which would materially harm our business. If we do not receive marketing approvals for such product candidates, we may not be able to continue our operations.
There are substantial risks inherent in attempting to commercialize our new drugs, and, as a result, we may not be able to successfully develop products for commercial use.
Scientific research and development requires significant amounts of capital and takes a long time to reach commercial viability if it can be achieved at all. To date, our research and development projects have not produced commercially viable drugs and may never do so. During the research and development process, we may experience technological barriers that we may be unable to overcome. Because we use platform technology to develop drug candidates, toxicology signals that may emerge in the course of testing of one particular candidate may apply broadly across our drug candidate platform. Further, certain underlying premises in our development programs are not proven and many of the drug targets that we are pursuing have not yet been validated clinically. For instance, the reduction of the production of mutant alpha-1 antitrypsin in the liver may not lead to a reduction of globules in the liver, and even if it leads to a reduction in such globules, this may not lead to other beneficial hepatic changes. It is also unknown at this time what changes in the liver may be required to gain regulatory approval and/or favorable reimbursement for a drug that reduces the production of mutant alpha-1 antitrypsin in the liver. Similar uncertainties and risks exist that are specific to each of our development programs. Because of these and similar uncertainties, it is possible that no commercial products will be successfully developed. If we are unable to successfully develop commercial products, we will be unable to generate revenue or build a sustainable or profitable business.
Our product candidates are in clinical development, which is a lengthy and expensive process with uncertain outcomes and the potential for substantial delays. There can be no assurance that our product candidates will obtain regulatory approval, which is necessary before they can be commercialized.
The sale of human therapeutic products in the United States and foreign jurisdictions is subject to extensive and time-consuming regulatory approval which requires, among other things:
•controlled research and human clinical testing;
•establishment of the safety and efficacy of the product;
•government review and approval of a submission containing manufacturing, preclinical and clinical data; and
•adherence to cGMP regulations during production and storage.
Since 2011, we have focused substantially all of our efforts and financial resources on identifying, acquiring and developing our product candidates, including conducting lead optimization, nonclinical studies, preclinical studies and clinical trials, and providing general administrative support for these operations. And, the clinical-stage product candidates we currently have under development will require significant development, preclinical and clinical testing and investment of significant funds to gain regulatory approval before they can be approved for commercialization. The results of our research and human clinical testing of our products may not meet regulatory requirements. Some of our product candidates, if approved, may require the completion of post-market studies. There can be no assurance that any of our products will be further developed and approved. The process of completing clinical testing and obtaining required approvals will take several years and require the use of substantial resources. Further, there can be no assurance that product candidates employing a new technology will be shown to be safe and effective in clinical trials or receive applicable regulatory approvals. If we fail to obtain regulatory approvals for any or all of our products, we will not be able to market such product and our operations may be adversely affected.
Our clinical trials may not yield successful results for the product candidates that we may identify and pursue for their intended uses, which would prevent, delay or limit the scope of regulatory approval and commercialization.
We must demonstrate our product candidates’ safety and efficacy in humans for each target indication through extensive clinical testing. We may experience numerous unforeseen events during, or as a result of, the testing process that could delay or prevent commercialization of any products, including the following:
•the results of preclinical studies may be inconclusive, or they may not be indicative of results that will be obtained in human clinical trials;
35
•safety and efficacy results attained in early human clinical trials may not be indicative of results that are obtained in later clinical trials;
•after reviewing test results, we may abandon projects that we might previously have believed to be promising;
•we or our regulators may suspend or terminate clinical trials because the participating subjects or patients are being exposed to unacceptable health risks; and
•our product candidates may not have the desired effects or may include undesirable side effects or other characteristics that preclude regulatory approval or limit their commercial use if approved.
We cannot be certain that current clinical trials or any future clinical trials, whether conducted by us or our licensees, will be successful. Additionally, any safety concerns observed in any one of our clinical trials in our targeted indications could limit the prospects for regulatory approval of our product candidates in those and other indications, which could have a material adverse effect on our business, financial condition and results of operation. Success in clinical trials in a particular indication does not ensure that a product candidate will be successful in other indications. Similarly, approval of a product candidate in a particular indication does not ensure that the product candidate will be successful in other indications. Moreover, results acceptable to support approval in one jurisdiction may be deemed inadequate by another regulatory authority to support regulatory approval in that other jurisdiction. To the extent that the results of the trials are not satisfactory to the FDA or comparable foreign regulatory authorities for support of a marketing application, we may be required to expend significant resources, which may not be available to us, to conduct additional trials in support of potential approval of our product candidates. Even if regulatory approval is secured for a product candidate, the terms of such approval may limit the scope and use of the specific product candidate, which may also limit its commercial potential.
Our clinical trials may reveal significant adverse events, toxicities or other side effects and may result in a safety profile that could inhibit regulatory approval or market acceptance of any of our product candidates.
In order to obtain marketing approval for any of our product candidates, we must demonstrate the safety and efficacy of the product candidate for the relevant clinical indication or indications through preclinical studies and clinical trials as well as additional supporting data. As is the case with pharmaceuticals generally, it is likely that there may be side effects and adverse events (“AEs”) associated with the use of our products or product candidates. If our product candidates are associated with undesirable side effects in preclinical studies or clinical trials, or have unexpected characteristics, we may need to interrupt, delay or abandon their development or limit development to more narrow uses or subpopulations in which the undesirable side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective.
If further significant adverse events or other side effects are observed in any of our current or future clinical trials, we may have difficulty recruiting patients to the clinical trials, patients may drop out of our trials, or we may be required to abandon the trials or our development efforts of that product candidate altogether. We, the FDA, the EMA, other applicable regulatory authorities or an institutional review board may suspend clinical trials of a product candidate at any time for various reasons, including a belief that subjects in such trials are being exposed to unacceptable health risks or adverse side effects. Some potential therapeutics developed in the biotechnology industry that initially showed therapeutic promise in early-stage studies have later been found to cause side effects that prevented their further development. Even if the side effects do not preclude the drug from obtaining or maintaining marketing approval, undesirable side effects may inhibit market acceptance of the approved product due to its tolerability relative to other therapies. Any of these developments could materially harm our business, financial condition and prospects.
Clinical trials of our product candidates may not uncover all possible adverse events that patients may experience.
Clinical trials are conducted in representative samples of the potential patient population, which may have significant variability. By design, clinical trials are based on a limited number of subjects and are of limited duration of exposure to the product, to determine whether the product candidate demonstrates the substantial evidence of efficacy and safety necessary to obtain regulatory approval. As with the results of any statistical sampling, we cannot be sure that all side effects of our product candidates may be uncovered. It may be the case that only with a significantly larger number of patients exposed to the product candidate for a longer duration may a more complete safety profile be identified. Further, even larger clinical trials may not identify rare significant AEs, and the duration of such studies may not be sufficient to identify when those events may occur. Other products have been approved by the regulatory authorities for which safety concerns have been uncovered following approval. Such safety concerns have led to labeling changes, restrictions on distribution through use of a REMS, or withdrawal of products from the market, and any of our product candidates may be subject to similar risks.
Although to date our current drug candidates have generally evidenced an acceptable safety profile in clinical trials, patients treated with our products, if approved, may experience previously unreported adverse reactions or minor incidences of adverse reactions may manifest with greater frequency in subsequent larger trials, and it is possible that the FDA or other regulatory authorities may ask for additional safety data as a condition of, or in connection with, our efforts
36
to obtain approval of our product candidates. If toxicities, adverse events or any other safety problems occur or are identified after our products, if any, reach the market, we may make the decision or be required by regulatory authorities to conduct additional clinical safety trials, amend the labeling of our products or add additional warnings to the labeling, recall our products, or even withdraw approval for our products.
Topline data may not accurately reflect the complete results of a particular study or trial.
We may publicly disclose topline or interim data from time to time, which is based on a preliminary analysis of then-available efficacy and safety data which are based on preliminary analysis of key efficacy and safety data, and the results and related findings and conclusions are subject to change following a more comprehensive review of the data related to the particular study or trial. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the topline results that we report may differ from future results of the same studies, or different conclusions or considerations may qualify such results, once additional data have been received and fully evaluated. Topline data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, topline data should be viewed with caution until the final data are available. Further, others, including regulatory agencies, may not accept or agree with our assumptions, estimations, calculations, conclusions or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization of the particular drug candidate or drug and our company in general. In addition, the information we may publicly disclose regarding a particular study or clinical trial is based on what is typically extensive information, and you or others may not agree with what we determine is the material or otherwise appropriate information to include in our disclosure, and any information we determine not to disclose may ultimately be deemed significant with respect to future decisions, conclusions, views, activities or otherwise regarding a particular drug, drug candidate or our business. If the topline data that we report differ from a future analysis of results, or if others, including regulatory authorities, disagree with the conclusions reached, our ability to obtain approval for and commercialize our product candidates, our business, operating results, prospects or financial condition may be harmed.
Results of earlier studies or clinical trials may not be predictive of future clinical trial results, and initial studies or clinical trials may not establish an adequate safety or efficacy profile for our product candidates to justify proceeding to advanced clinical trials or an application for regulatory approval.
The results of nonclinical and preclinical studies and clinical trials may not be predictive of the results of later-stage clinical trials, and interim results of clinical trials do not necessarily predict final results. The results of preclinical studies and clinical trials in one set of patients or disease indications, or from preclinical studies or clinical trials that we did not lead, may not be predictive of those obtained in another. In some instances, there can be significant variability in safety or efficacy results between different clinical trials of the same product candidate due to numerous factors, including changes in trial procedures set forth in protocols, differences in the size and type of the patient populations, changes in and adherence to the dosing regimen and other clinical trial protocols and the rate of dropout among clinical trial participants. In addition, preclinical and clinical data are often susceptible to various interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain marketing approval. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy profile despite having progressed through nonclinical studies and initial clinical trials. Many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials after achieving positive results in early-stage development, or after achieving positive results in pivotal trials, and we cannot be certain that we will not face similar setbacks. Even if early-stage clinical trials are successful, we may need to conduct additional clinical trials of our product candidates in additional patient populations or under different treatment conditions before we are able to seek approvals from the FDA and regulatory authorities outside the United States to market and sell these product candidates. Our failure to obtain marketing approval for our product candidates for commercially viable indications, or at all, would substantially harm our business, prospects, financial condition and results of operations.
It may take us longer than we project to complete clinical trials, and we may not be able to complete them at all.
Although for planning purposes we project the commencement, continuation and completion of our clinical trials, a number of factors, including scheduling conflicts with participating clinicians and clinical institutions, and difficulties in identifying or enrolling patients who meet trial eligibility criteria, may cause significant delays. Enrollment of clinical trials may be particularly difficult in orphan diseases or limited-sized patient populations. The FDA or other regulatory bodies may require additional, longer or broader clinical trials to establish safety and effectiveness, notwithstanding guidance the Company may have received from those bodies during clinical trial planning and execution. Further, the cost for conducting clinical trials is significant and if our cash resources become limited we may not be able to commence, continue
37
and/or complete our clinical trials. We may not commence or complete clinical trials involving any of our product candidates as projected or may not conduct them successfully.
We face potential product liability exposure, and if successful claims are brought against us, we may incur substantial liability for a product candidate and may have to limit its commercialization.
The use of our product candidates in clinical trials and the sale of any products for which we obtain marketing approval expose us to the risk of product liability claims. Product liability claims might be brought against us by clinical trial participants, consumers, healthcare providers, pharmaceutical companies, or others selling our products. If we cannot successfully defend ourselves against these claims, we may incur substantial liabilities. Regardless of merit or eventual outcomes of such claims, product liability claims may result in:
•decreased demand for our product candidates;
•impairment of our business reputation;
•withdrawal of clinical trial participants;
•costs of litigation;
•substantial monetary awards to patients or other claimants; and
•loss of revenues.
Our insurance coverage may not be sufficient to reimburse us for all expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive and, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses.
The successful commercialization of our product candidates, if approved, will depend in part on the extent to which government authorities and health insurers establish adequate reimbursement levels and pricing policies.
Sales of any approved drug candidate will depend in part on the availability of coverage and reimbursement from third-party payers such as government insurance programs, including Medicare and Medicaid, private health insurers, health maintenance organizations and other health care related organizations, who are increasingly challenging the price of medical products and services. Accordingly, coverage and reimbursement may be uncertain. Adoption of any drug by the medical community may be limited if third-party payers will not offer adequate coverage. Additionally, significant uncertainty exists as to the reimbursement status of newly-approved drugs. Cost control initiatives may decrease coverage and payment levels for any drug and, in turn, the price that we will be able to charge and/or the volume of our sales. We are unable to predict all changes to the coverage or reimbursement methodologies that will be applied by private or government payers. Any denial of private or government payer coverage or inadequate reimbursement could harm our business and reduce our revenue. With respect to our partnered product candidates, we will be reliant on that partner to obtain reimbursement from government and private payors for the drug, if approved, and any failure of that partner to establish adequate reimbursement could have a negative impact on our revenues and profitability.
In addition, both the federal and state governments in the United States and foreign governments continue to propose and pass new legislation, regulations, and policies affecting coverage and reimbursement rates, which are designed to contain or reduce the cost of health care. Further federal and state proposals and healthcare reforms are likely, which could limit the prices that can be charged for the product candidates that we develop and may further limit our commercial opportunity. For example, the Inflation Reduction Act of 2022 (“IRA”) includes several measures intended to lower the cost of prescription drugs and related healthcare reforms, including limits on price increases and subjecting an escalating number of drugs to annual price negotiations with CMS (The Centers for Medicare & Medicaid Services). We cannot be sure whether additional legislation or rulemaking related to the IRA will be issued or enacted, or what impact, if any, such changes will have on the profitability of any of our drug candidates, if approved for commercial use, in the future. There also may be future changes unrelated to the IRA that result in reductions in potential coverage and reimbursement levels for our product candidates, if approved and commercialized, and we cannot predict the scope of any future changes or the impact that those changes would have on our operations.
If future reimbursement for approved product candidates, if any, is substantially less than we project, or rebate obligations associated with them are substantially greater than we expect, our future net revenue and profitability could be materially diminished.
We may not enjoy the market exclusivity benefits of our orphan drug designations.
Although we may obtain orphan designations in the treatment of certain diseases our products are intended to treat, the designation may not be applicable to any particular product we might get approved and that product may not be the first product to receive approval for that indication. Under the Orphan Drug Act, the first product with an orphan designation receives market exclusivity, which prohibits the FDA from approving the “same” drug for the same indication. The FDA has stated that drugs can be the “same” even when they are not identical but has not provided guidance with respect to how
38
it will determine “sameness” for RNAi drugs. It is possible that another RNAi drug could be approved for the treatment of a disease that one of our orphan products is intended to treat before our product is approved, which means that we may not obtain orphan drug exclusivity and could also potentially be blocked from approval until the first product’s orphan drug exclusivity period expires or we demonstrate, if we can, that our product is superior. Further, even if we obtain orphan drug exclusivity for a product, that exclusivity may not effectively protect the product from competition because different drugs can be approved for the same condition. Even after an orphan drug is approved and granted orphan drug exclusivity, the FDA can subsequently approve the same drug for the same condition if the FDA concludes that the later drug is safer, more effective or makes a major contribution to patient care. Further, orphan drug exclusivity can be lost if the FDA later determines that the request for designation was materially defective or if the applicant is unable to assure the availability of sufficient quantities of the drug to meet the needs of patients with the disease or condition for which the drug was designated.
Our success depends on the attraction and retention of senior management and scientists with relevant expertise.
Our future success depends to a significant extent on the continued services of our key employees, including our senior scientific, technical and managerial personnel. We do not maintain key person life insurance for any of our executives and we do not maintain employment agreements with many senior employees. Competition for qualified employees in the pharmaceutical industry is high, and our ability to execute our strategy will depend in part on our ability to continue to attract and retain qualified scientists, management and other employees. This will depend in part on our ability to create and maintain a desirable workplace culture, which may be impacted by employee preferences for remote working. In addition, the market for qualified employees in the pharmaceutical industry is experiencing labor shortages and inflationary pressures are causing salaries and wages to increase, all of which exacerbates these competitive dynamics. If we are unable to find, hire and retain qualified individuals, we will have difficulty implementing our business plan in a timely manner, or at all.
Risks Related to Regulatory Review and Approval of Our Product Candidates
Breakthrough Therapy designation for ARO-AAT may not lead to a faster development or review process.
We have been granted a Breakthrough Therapy designation for ARO-AAT in the United States for the treatment of liver disease associated with AATD. Breakthrough Therapy designation is intended to facilitate the development and expedite the review of new therapies to treat serious conditions with unmet medical needs by providing sponsors with the opportunity for frequent interactions and additional drug development guidance with the FDA and its senior managers. Breakthrough Therapy designation applies to the combination of the drug candidate and the specific indication for which it is being studied. Product candidates that receive Breakthrough Therapy designation may receive more frequent interactions with the FDA regarding the product candidate’s development plan and clinical trials and may be eligible for the FDA’s Rolling Review.
Despite receiving Breakthrough Therapy designation, ARO-AAT may not actually benefit from faster clinical development or regulatory review or approval any sooner than other product candidates that do not have such designation, or at all. Furthermore, such a designation does not increase the likelihood that ARO-AAT will receive marketing approval in the United States. The FDA may also rescind Breakthrough Therapy designation if it determines that ARO-AAT no longer meets the relevant criteria.
We and our licensees conduct clinical trials for product candidates outside the United States, and the FDA and comparable foreign regulatory authorities may not accept data from such trials.
We and our licensees currently conduct clinical trials outside the United States. The acceptance by the FDA or comparable foreign regulatory authority of study data from clinical trials conducted outside the United States or another jurisdiction may be subject to certain conditions or may not be accepted at all. In cases where data from foreign clinical trials are intended to serve as the basis for marketing approval in the United States, the FDA will generally not approve the application on the basis of foreign data alone unless (i) the data are applicable to the U.S. population and U.S. medical practice; (ii) the trials were performed by clinical investigators of recognized competence and pursuant to GCP regulations; and (iii) the data may be considered valid without the need for an on-site inspection by the FDA or, if the FDA considers such as inspection to be necessary, the FDA is able to validate the data through an on-site inspection or other appropriate means. Additionally, the FDA’s clinical trial requirements, including sufficient size of patient populations and statistical powering, must be met. Many foreign regulatory authorities have similar approval requirements. In addition, such foreign trials would be subject to the applicable local laws of the foreign jurisdictions where the trials are conducted. There can be no assurance that the FDA or any comparable foreign regulatory authority will accept data from trials conducted outside of the United States or the applicable jurisdiction. If the FDA or any comparable foreign regulatory authority does not accept such data, it would result in the need for additional trials, which would be costly and time-consuming and delay aspects of
39
our business plan, and which may result in product candidates that we may develop not receiving approval or clearance for commercialization in the applicable jurisdiction.
Even if we obtain FDA approval for products in the United States, we may never obtain approval to commercialize any product candidates outside of the United States, which would limit our ability to realize their full market potential.
In order to market any products outside of the United States, we must establish and comply with numerous and varying regulatory requirements of other countries regarding safety and effectiveness. Clinical trials conducted in one country may not be accepted by regulatory authorities in other countries, and regulatory approval in one country does not mean that regulatory approval will be obtained in any other country. Approval processes vary among countries and can involve additional product testing and validation and additional or different administrative review periods from those in the United States, including additional preclinical studies or clinical trials. In many jurisdictions outside the United States, a product candidate must be approved for reimbursement before it can be approved for sale in that jurisdiction. In some cases, the price that we intend to charge for our products is also subject to approval before a product can be marketed in that jurisdiction, even after establishing safety and efficacy in a clinical setting.
Seeking foreign regulatory approval could result in difficulties and costs and require additional nonclinical studies or clinical trials which could be costly and time-consuming. Regulatory requirements can vary widely from country to country and could delay or prevent the introduction of our product candidates in those countries. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval. We do not have any product candidates approved for sale in international markets, and we do not have experience in obtaining regulatory approval in international markets. If we fail to comply with regulatory requirements in international markets or to obtain and maintain required approvals, or if regulatory approval in international markets is delayed, our target market will be reduced and our ability to realize the full market potential of our products will be harmed.
Even if our product candidates are approved for commercialization, failure to comply with regulatory requirements or unanticipated problems with our products may result in various adverse actions such as the suspension or withdrawal of one or more of our products, closure of a facility or enforcement of substantial penalties or fines.
If regulatory approval to sell any of our product candidates is received, regulatory agencies will subject any marketed product(s) and the facilities where they are manufactured to continual review and periodic inspection. If previously unknown problems with a product, manufacturing and laboratory facilities or regulatory requirements are discovered, such as adverse events of unanticipated severity or frequency, problems with a manufacturing process or laboratory facility, or failure to comply with applicable regulatory approval requirements, a regulatory agency may impose restrictions or penalties on that product or on us. Such restrictions or penalties may include, among other things:
•restrictions on the marketing or manufacturing of the product, the withdrawal of the product from the market or product recalls;
•warning letters or holds on clinical trials;
•refusal by the FDA to approve pending applications or supplements to approved applications filed by us or suspension or revocation of approvals;
•product seizure or detention, or refusal to permit the import or export of our product candidates; and
•closure of the facility, enforcement of substantial fines, injunctions, or the imposition of civil or criminal penalties.
Risks Related to Our Intellectual Property
Our ability to protect our patents and other proprietary rights is uncertain, exposing us to the possible loss of competitive advantage.
We have licensed rights to pending patents and have filed and expect to continue to file patent applications. Researchers sponsored by us may also file patent applications that we may need to license. Such patent applications may not be available for licensing or may not be economically feasible to license. Certain of our patents may not be granted or may not contain claims of the necessary breadth because, for example, prior patents exist. If a particular patent is not granted, the value of the invention described in the patent would be diminished. Further, even if these patents are granted, they may be difficult to enforce. Even if ultimately successful, efforts to enforce our patent rights could be expensive, distracting for management, cause our patents to be invalidated or held unenforceable, and thus frustrate commercialization of products. Even if patents are issued and are enforceable, others may develop similar, superior or parallel technologies to any technology developed by us and not infringe on our patents. Our technology may prove to infringe upon patents or rights owned by others. Patent prosecution and maintenance is expensive, and we may be forced to curtail prosecution or maintenance if our cash resources are limited. Thus, the patents held by or licensed to us may not afford us any meaningful competitive advantage. In addition, the laws of some foreign countries in which we do business, including through our joint ventures, do not protect intellectual property rights to the same extent or in the same manner as the laws of the United
40
States. As a result, we may encounter significant problems in protecting and defending our intellectual property both in the United States and abroad. If we are unable to adequately protect our owned intellectual property or derive sufficient value from our licensed or owned intellectual property, the value of your investment may decline.
We are party to technology license agreements with third parties that require us to satisfy obligations to keep them effective and, if these agreements are terminated, our technology and our business could be seriously and adversely affected.
We are party to license agreements to incorporate third-party proprietary technologies into our drug products under development. These license agreements require us to pay royalties and satisfy other conditions. If we fail to satisfy our obligations under these agreements, the terms of the licenses may be materially modified, such as by rendering currently exclusive licenses non-exclusive, or may give our licensors the right to terminate their respective agreement with us, which could limit our ability to implement our current business plan and harm our business and financial condition.
We may be subject to patent infringement claims, which could result in substantial costs and liability and prevent us from commercializing our potential products.
Because the intellectual property landscape in the fields in which we participate is rapidly evolving and interdisciplinary, it is difficult to conclusively assess our freedom to operate without infringing on third-party rights. However, if granted marketing approval, we are currently aware of certain patent rights held by third parties that, if found to be valid and enforceable, could be alleged to render one or more of our drug candidates infringing. If a claim should be brought and is successful, we may be required to pay substantial damages, be forced to abandon any affected drug candidates and/or seek a license from the patent holder. In addition, any patent infringement claims brought against us, whether or not successful, may cause us to incur significant expenses and divert the attention of our management and key personnel from other business concerns. These could negatively affect our results of operations and prospects. We cannot be certain that patents owned or licensed by us will not be challenged, potentially successfully, by others.
In addition, if our product candidates are found to infringe the intellectual property rights of third parties, these third parties may assert infringement claims against our customers, licensees and other parties with whom we have business relationships, and we may be required to indemnify those parties for any damages they suffer as a result of these claims. The claims may require us to initiate or defend protracted and costly litigation on behalf of customers, licensees, and other parties regardless of the merits of these claims. If any of these claims succeed, we may be forced to pay damages on behalf of those parties or may be required to obtain licenses for the products they use. If we cannot obtain all necessary licenses on commercially reasonable terms, we may be unable to continue selling such products.
We license patent rights from third-party owners and we rely on such owners to obtain, maintain and enforce the patents underlying such licenses.
We are a party to a number of licenses that give us rights to third-party intellectual property that is necessary or useful for our business. We also expect to enter into additional licenses to third-party intellectual property in the future.
Our success will depend in part on the ability of our licensors to obtain, maintain and enforce patent protection for our licensed intellectual property, in particular, those patents to which we have secured exclusive rights. Our licensors may not successfully prosecute the patent applications to which we are licensed. Even if patents are issued in respect of these patent applications, our licensors may fail to maintain these patents, may determine not to pursue litigation against other companies that are infringing these patents, or may pursue such litigation less aggressively than we would. Without protection for the intellectual property we license, other companies might be able to offer substantially identical products for sale, which could adversely affect our competitive business position and harm our business prospects.
Our technology licensed from various third parties may be subject to retained rights.
Our licensors often retain certain rights under their agreements with us, including the right to use the underlying technology for noncommercial academic and research use, to publish general scientific findings from research related to the technology, and to make customary scientific and scholarly disclosures of information relating to the technology. It is difficult to monitor whether our licensors limit their use of the technology to these uses, and we could incur substantial expenses to enforce our rights to our licensed technology in the event of misuse.
Confidentiality agreements with employees and others may not adequately prevent disclosure of trade secrets and other proprietary information.
In order to protect our proprietary technology and processes, we rely in part on confidentiality agreements with our collaborators, employees, consultants, outside scientific collaborators and sponsored researchers, and other advisors. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. As our organization grows, so does the risk of
41
unauthorized disclosure of confidential information. In addition, while we undertake efforts to protect our trade secrets and other confidential information from disclosure, others may independently discover trade secrets and proprietary information, and in such cases, we may not be able to assert any trade secret rights against such party. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our competitive business position.
We may not be able to effectively secure first-tier technologies when competing against other companies or investors.
Our future success may require that we acquire patent rights and know-how to new or complimentary technologies. However, we compete with a substantial number of other companies that may also compete for technologies we desire. In addition, many venture capital firms and other institutional investors, as well as other pharmaceutical and biotech companies, invest in companies seeking to commercialize various types of emerging technologies. Many of these companies have greater financial, scientific and commercial resources than us. Therefore, we may not be able to secure the technologies we desire. Furthermore, should any commercial undertaking by us prove to be successful, there can be no assurance competitors with greater financial resources will not offer competitive products and/or technologies.
Risks Related to Our Business Model
Our business model assumes we will generate revenue by, among other activities, marketing or out-licensing the products we develop. Our drug candidates are in various stages of development and we have no approved products based on RNA interference and our delivery technologies. Accordingly, there is a limited amount of information about us upon which you can evaluate our business and prospects.
We have no approved drugs and thus have not begun to market or generate revenues from the commercialization of any product candidates. Because no drug candidates generated with our product platform have been approved, we have not yet demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the biopharmaceutical area. For example, to execute our business plan, we will need to successfully:
•Execute product development activities using technologies that have not yet generated an approved product;
•Build, maintain, and protect a strong intellectual property portfolio;
•Demonstrate safety and efficacy of our drug candidates in multiple human clinical studies;
•Receive FDA approval and approval from similar foreign regulatory bodies;
•Gain market acceptance for the development and commercialization of any drugs we develop;
•Ensure our products are reimbursed by commercial and/or government payors at a rate that permits commercial viability;
•Develop and maintain successful strategic relationships with suppliers, distributors, and commercial licensing partners;
•Manage our spending and cash requirements as our expenses will increase in the near term if we add programs and additional preclinical and clinical trials; and
•Effectively market any products for which we obtain marketing approval.
If we are unsuccessful in accomplishing these objectives, we may not be able to develop products, raise capital, expand our business or continue our operations.
We may need to establish additional relationships with strategic and development partners to fully develop our drug candidates and market any approved products.
Over the past several years we have entered into license and collaboration agreements with Takeda, Janssen, Amgen, Horizon, GSK and Visirna. Our business strategy includes obtaining additional collaborations with other pharmaceutical and biotech companies to support the development of our RNAi therapeutics and other drug candidates. We do not possess all of the financial and development resources necessary to develop and commercialize all of the products that may result from our technologies. Unless we expand our product development capacity and enhance our internal marketing capability, we may need to make appropriate arrangements with strategic partners to develop and commercialize any drug candidates that may be approved. We may not be able to attract such partners, and even if we are able to enter into such partnerships, the terms may be less favorable than anticipated. Further, entering into partnership agreements may limit our commercialization options and/or require us to share revenues and profits with our partners. If we do not find appropriate partners, or if our existing arrangements or future agreements are not successful, our ability to develop and commercialize products could be adversely affected. Even if we are able to find collaborative partners, the overall success of the development and commercialization of product candidates in those programs will depend largely on the efforts of other parties and will be beyond our control, particularly as partnered programs progress and our licensees may elect to
42
assume greater control over these programs. In addition, in the event we pursue our commercialization strategy through collaboration or licenses to third parties, there are a variety of technical, business and legal risks, including:
•We may not be able to control the amount and timing of resources that our collaborators may be willing or able to devote to the development or commercialization of our drug candidates or to their marketing and distribution; and
•Disputes may arise between us and our collaborators that result in the delay or termination of the research, development or commercialization of our drug candidates or that result in costly litigation or arbitration that diverts our management’s resources.
The occurrence of any of the above events or other related events could impair our ability to generate revenues and harm our business and financial condition.
Our ability to generate milestone and royalty payments under our current and potential future licensing and collaboration agreements is substantially controlled by our partners, and as such, we will likely need other sources of financing to continue to develop our internal drug candidates.
Under our licensing and collaboration agreements with Amgen, Janssen, Takeda, Horizon, GSK and Visirna, our partners substantially control clinical development and commercialization for all of the candidates covered under those agreements in their relevant territories. To the extent that (i) our partners’ interests in advancing these candidates or targets changes, (ii) unforeseen scientific issues with the candidates arise, or (iii) the pace at which our partners move the candidates through clinical trials toward commercialization slows, our ability to collect milestones and royalties may be significantly diminished. This would further cause us to rely upon other sources of financing to continue to develop our other internal drug candidates.
We may lose a considerable amount of control over our intellectual property and may not receive anticipated revenues in strategic transactions, particularly where the consideration is contingent on the achievement of development or sales milestones.
Our business model has been to develop new technologies and to utilize the intellectual property created through the research and development process to develop commercially successful products. If the acquirers of our technologies fail to achieve performance milestones, we may not receive a significant portion of the total value of any sale, license or other strategic transaction.
We will need to achieve commercial acceptance of our drug candidates to generate revenues and achieve profitability.
Even if our research and development efforts yield technologically feasible applications, we may not successfully develop commercial products. Drug development takes years of study in human clinical trials prior to regulatory approval, and, even if we are successful, it may not be on a timely basis. During our drug development period, superior competitive technologies may be introduced which could diminish or extinguish the potential commercial uses for our drug candidates. Additionally, the degree to which the medical community and consumers will adopt any product we develop is uncertain. The rate and degree of market acceptance of our products will depend on a number of factors, including the establishment and demonstration in the medical community of the clinical efficacy and safety of our products, their potential advantage over alternative treatments, and the costs to patients and third-party payors, including insurance companies and Medicare. Recent efforts in the United States and abroad to reduce overall healthcare spending has put significant pressure on the price of prescription drugs and certain companies have been publicly criticized for the relatively high cost of their therapies. These pressures may force us to sell any approved drugs at a lower price than we or analysts may anticipate or may result in lower levels of reimbursement and coverage from third parties.
We cannot predict whether significant commercial market acceptance for our products, if approved, will ever develop, and we cannot reliably estimate the projected size of any such potential market. Our revenue growth and achievement of consistent profitability will depend substantially on our ability to introduce products that will be accepted by the medical community. If we are unable to cost-effectively achieve acceptance of our technology among the medical establishment and patients, or if the associated products do not achieve wide market acceptance, our business will be materially and adversely affected.
We rely on outside sources for various components and processes for our products.
We rely on third parties for various components and processes for our product candidates. We may not be able to achieve multiple sourcing because there may be no acceptable second source, other companies may choose not to work with us, or the component or process sought may be so new that a second source does not exist or does not exist on acceptable terms. There may be a disruption or delay in the performance of our third-party contractors, suppliers or collaborators which is beyond our control. If such third parties are unable to satisfy their commitments to us, the development of our products would be adversely affected. Therefore, it is possible that our development plans will have to be slowed down or stopped completely at times due to our inability to obtain required raw materials, components, and
43
outsourced processes at an acceptable cost, if at all, or to get a timely response from vendors, particularly as a result of recent labor market and global supply chain constraints.
We have limited manufacturing capability and must rely on third-party manufacturers to manufacture our clinical supplies and commercial products, if and when approved, and if they fail to meet their obligations, the development and commercialization of our products could be adversely affected.
We have limited manufacturing capabilities and experience and we rely, and expect to continue to rely, on third-party manufacturers for the production of some of our product candidates for clinical trials and potential future commercialization. If we were to experience an unexpected loss or interruption of supply for any of our product candidates, whether as a result of manufacturing, supply or storage issues or otherwise, we could experience delays, disruptions, suspensions or terminations of, or be required to restart or repeat, any pending or ongoing clinical trials. Further, our drug candidates are composed of multiple components and require specialized formulations for which scale-up and manufacturing could be difficult. We have limited experience in such scale-up and manufacturing requiring us to depend on a limited number of third parties, who may not be able to deliver in a timely manner, or at all. In order to develop products, apply for regulatory approvals, and commercialize our products, we will need to develop, contract for, or otherwise arrange for the necessary manufacturing capabilities. Our internal GMP manufacturing capabilities are limited to small-scale production of material. There are a limited number of manufacturers that supply synthetic oligonucleotides. There are risks inherent in pharmaceutical manufacturing that could affect the ability of our contract manufacturers to meet our delivery time requirements or provide adequate amounts of material to meet our needs. Included in these risks are synthesis and purification failures and contamination during the manufacturing process, which could result in unusable product and cause delays in our development process, as well as additional expense to us.
Additionally, our product candidates have not yet been manufactured for commercial use. If any of our product candidates become approved for commercial sale, we will need to establish either internal or third-party manufacturing capacity. Manufacturing partner requirements may require us to fund capital improvements, perhaps on behalf of third parties, to support the scale-up of manufacturing and related activities. We may not be able to establish scaled manufacturing capacity for an approved product in a timely or economic manner, if at all. If we or our third-party manufacturers are unable to provide commercial quantities of such an approved product, we will have to successfully transfer manufacturing technology to a different manufacturer. Engaging a new manufacturer for such an approved product could require us to conduct comparative studies or utilize other means to determine bioequivalence of the new and prior manufacturers’ products, which could delay or prevent our ability to commercialize such an approved product. If we or any of these manufacturers is unable or unwilling to increase its manufacturing capacity or if we are unable to establish alternative arrangements on a timely basis or on acceptable terms, the development and commercialization of such an approved product may be delayed or there may be a shortage in supply. Any inability to manufacture our product candidates or future approved drugs in sufficient quantities when needed would seriously harm our business. For example, recent global supply chain constraints have led to intermittent lab supply shortages, which are critical for our preclinical programs. While we are exploring alternative suppliers for certain critical materials, there can be no assurance that our efforts will be successful.
Manufacturers of our approved products, if any, must comply with cGMP requirements relating to methods, facilities and controls used in the manufacturing, processing and packaging of the product, which are intended to ensure that drug products are safe and that they consistently meet applicable requirements and specifications. These requirements include quality control, quality assurance, and the maintenance of records and documentation. Manufacturers of our approved products, if any, may be unable to comply with these cGMP requirements and with other FDA, state and foreign regulatory requirements. These requirements are enforced by the FDA and other health authorities through periodic announced and unannounced inspections of manufacturing facilities. A failure to comply with these requirements or to provide adequate and timely corrective actions in response to deficiencies identified in an inspection may result in enforcement action, including warning letters, fines and civil penalties, suspension of production, suspension or delay in product approval, product seizure or recall, plant shutdown, or the delay, withholding, or withdrawal of product approval. If the safety of any quantities supplied is compromised due to a manufacturer’s failure to adhere to applicable laws or for other reasons, we may not be able to obtain regulatory approval for or successfully commercialize our products, which would seriously harm our business.
We rely on third parties to conduct our clinical trials, and if they fail to fulfill their obligations, the development of our products may be adversely affected.
We rely on independent clinical investigators, contract research organizations and other third-party service providers to assist us in managing, monitoring and otherwise carrying out our clinical trials. We contract with certain third-parties to provide certain services, including site selection, enrollment, monitoring and data management services. We rely on these parties to carry out our clinical trials in compliance with GCP and other relevant requirements. Although we depend
44
heavily on these parties, we do not control them and therefore we cannot be assured that these third parties will adequately perform all of their contractual obligations to us. If our third-party service providers cannot adequately and timely fulfill their obligations to us, or if the quality and accuracy of our clinical trial data is compromised due to failure by such third parties to adhere to our protocols, GCP, or other regulatory requirements or if such third parties otherwise fail to meet deadlines or quality requirements, our development plans may be delayed or terminated. Further, if clinical study results are compromised, then we may need to repeat the affected studies, which could result in significant additional costs and delays to us.
We face competition from various entities including large pharmaceutical companies, small biotech companies, private companies, and research institutions.
Many of our competitors have greater financial resources and may have more experience in research and development, manufacturing, managing clinical trials and/or regulatory compliance than we do. Our competitors may compete with us for lead clinical trial investigators, clinical trial site locations and patient enrollment. These competitors may also compete with us on recruiting scientific and management personnel. Because our products are in various stages of preclinical and clinical development, along with many of the competing products, and given unpredictability inherent in drug development, it is difficult to predict which third parties may provide the most competition, and on what specific basis that competition may be based.
We may have difficulty expanding our operations successfully as we evolve our pipeline and move toward commercializing drugs.
Our future financial performance and our ability to commercialize products and compete effectively will depend, in part, on our ability to effectively manage future growth. We expect that as we increase the number of product candidates we are developing we will also need to expand our operations. This expected growth may place a strain on our administrative and operational infrastructure and information technology systems. As product candidates we develop enter and advance through clinical trials, we will need to expand our development, regulatory, manufacturing, marketing, and sales capabilities or contract with other organizations to provide these capabilities for us. We are currently planning to establish a sales and marketing infrastructure, although we have no institutional experience in the sale, marketing, or distribution of pharmaceutical products. To achieve commercial success for any approved product for which we retain sales and marketing rights, we must continue to develop a sales and marketing organization or outsource these functions to third parties. If we or our collaborators are unable to establish sales, marketing and distribution capabilities or enter into or maintain agreements with third parties to market and sell our product candidates, we may not be successful in commercializing our product candidates if and when they are approved. Further, as our operations expand due to our development progress, we expect that we will need to manage additional relationships with various collaborators, suppliers, and other organizations. Our ability to manage our operations and future growth will require us to continue to improve our operational, financial, information technology and management controls, reporting systems and procedures. We may not be able to effectively manage the expansion of our operations or implement improvements to our management information and control systems in an efficient or timely manner and may discover deficiencies in existing systems and controls.
Our business and operations could suffer in the event of information technology system failures.
Our internal computer systems and those of our contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, ransomware and other cyber-attacks, human error, natural disasters, terrorism, war, and telecommunication and electrical failures. Such events could cause interruption of our operations and loss of intellectual property. For example, the loss of preclinical trial data or data from completed or ongoing clinical trials for our product candidates could result in delays in our regulatory filings and development efforts and significantly increase our costs. Further, cybersecurity breaches may allow hackers access to our preclinical compounds, strategies, discoveries, trade secrets, and/or other confidential information. To the extent that any disruption or security breach were to result in a loss of or damage to our data, or inappropriate disclosure of confidential, proprietary or private information, we could incur liability or regulatory penalties, including under laws and regulations governing the protection of health and other personally identifiable information, we could lose valuable trade secret rights, the development of our product candidates could be delayed, and we could suffer reputational damage and damage to key business relationships. The risk of a cyber-security breach or other informational technology disruption, particularly through cyber-attacks, has generally increased as the number, intensity and sophistication of attempted attacks and intrusions from around the world have increased. We have experienced cyber-security attacks in the past, which to date have not had a material impact on our operations or development programs; however; there is no assurance that such impacts will not be material in the future.
Because we use biological materials, hazardous materials, chemicals and radioactive compounds, if we do not comply with laws regulating the protection of the environment and health and human safety, our business could be adversely affected.
45
Our research, development and manufacturing activities involve the use of potentially harmful biological materials as well as materials, chemicals and various radioactive compounds that could be hazardous to human health and safety or the environment. We store most of these materials and various wastes resulting from their use at our facilities in Madison, Wisconsin and San Diego, California pending ultimate use and disposal. We cannot completely eliminate the risk of contamination, which could cause interruption to our research and development and manufacturing efforts, injury to our employees and others, environmental damage, and liabilities under federal, state and local law. In such an event, we may be held liable for any resulting damages, and any liability could exceed our resources. Although we carry insurance in amounts and types that we consider commercially reasonable, we do not have insurance coverage for losses relating to an interruption of our research, development or manufacturing efforts caused by contamination, and the coverage or coverage limits of our insurance policies may not be adequate. If our losses exceed our insurance coverage, our financial condition would be affected.
Litigation claims may result in financial losses or harm our reputation and may divert management resources.
When the market price of a stock is volatile, holders of that stock have often initiated securities class action litigation against the company that issued the stock. We cannot predict with certainty the eventual outcome of such litigation, arbitration or third-party inquiry. We may not be successful in defending ourselves or asserting our rights in current or future lawsuits, investigations, or claims that have been or may be brought against us and, as a result, our business could be materially harmed. These lawsuits, arbitrations, investigations or claims may result in large judgments or settlements against us, any of which could have a negative effect on our financial performance and business. Additionally, lawsuits, arbitrations and investigations can be expensive to defend, whether or not the lawsuit, arbitration or investigation has merit, and the defense of these actions may divert the attention of our management and other resources that would otherwise be engaged in running our business.
Our operations, including our relationships with healthcare providers, physicians and third-party payers, are subject to applicable anti-kickback, fraud and abuse, and other healthcare laws and regulations, which, in the event of a violation, exposes us to liability for criminal sanctions, civil penalties, and contractual damages, and reputational harm and diminished profits and future earnings.
Our operations, including any arrangements that we enter into with healthcare providers, physicians, and third-party payers, are subject to broadly applicable fraud and abuse and other healthcare laws and regulations. Such laws and regulations, including applicable U.S. federal and state healthcare laws and regulations, as well as foreign laws, such as the federal Anti-Kickback Statute, the False Claims Act, the Health Insurance Portability and Accountability Act of 1996, or the Foreign Corrupt Practices Act, may constrain our operation and the business or financial arrangements through which we can market, sell and distribute any drug candidates for which we obtain marketing approval.
Efforts to confirm that our business arrangements with third parties comply with applicable healthcare laws and regulations involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we become subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion of products from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If any of the physicians or other healthcare providers or entities with whom we expect to do business are found to be not in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
Risks Related to Our Financial Condition
We have a history of net losses, and we expect to continue to incur net losses and may not achieve or maintain profitability.
We have incurred net losses since our inception and we expect that our operating losses will continue for the foreseeable future as we continue our drug development efforts and prepare for the potential commercialization of our product candidates. To achieve profitability, we must, either directly or through licensing and/or partnering relationships, meet certain milestones, successfully develop and obtain regulatory approval for one or more drug candidates and effectively manufacture, market and sell any drugs we successfully develop. Even if we successfully commercialize drug candidates that receive regulatory approval, we may not be able to realize revenues at a level that would allow us to achieve or sustain profitability. Accordingly, we may never generate significant revenue and, even if we do generate significant revenue, we may never achieve consistent profitability.
We will require substantial additional funds to complete our research and development activities.
46
Our business currently does not generate the cash that is necessary to finance our operations. Subject to the success of the research and development programs of our Company and our partners, and potential licensing or partnering transactions, we may need to raise additional capital to:
•Fund research and development infrastructure and activities relating to the development of our drug candidates, including preclinical and clinical trials and manufacturing to support these efforts;
•Fund a commercialization infrastructure and activities related to the sale, marketing, customer support, and distribution of our drug products if and when they become approved;
•Fund our general and administrative infrastructure and activities;
•Pursue business development opportunities for our technologies;
•Add to and protect our intellectual property; and
•Retain our management and technical staff.
Our future capital needs depend on many factors, including:
•The scope, duration, and expenditures associated with our research and development, including the progression of our clinical trials, with late-stage trials generally requiring greater capital than early-stage trials;
•Regulatory requirements for our clinical trials;
•The extent to which our research and development and clinical efforts are successful;
•Expenditures to build out or contract for sales, marketing and distribution capabilities as we prepare for the potential commercialization of our product candidates, if any;
•The outcome of potential partnering or licensing transactions, if any, and the extent to which our business development efforts result in the acquisition of new programs or technologies;
•Competing technological developments;
•Our intellectual property positions, if any, in our products; and
•The regulatory approval process and regulatory standards for our drug candidates.
We will need to raise additional funds through public or private equity offerings, debt financings or additional strategic alliances and licensing arrangements in the future to continue our operations. We may not be able to obtain additional financing on terms favorable to us, if at all. General market conditions may make it very difficult for us to seek financing from the capital markets, and the terms of any financing may adversely affect the holdings or the rights of our stockholders. For example, if we raise additional funds by issuing equity securities, further dilution to our stockholders will result, which may substantially dilute the value of investment. In addition, as a condition to providing additional funds to us, future investors may demand, and may be granted, rights superior to those of existing stockholders. Debt financing, if available, may involve restrictive covenants that could limit our flexibility in conducting future business activities and, in the event of insolvency, would be paid before holders of equity securities received any distribution of corporate assets. In order to raise additional funds through alliance, joint venture or licensing arrangements, we may be required to relinquish rights to our technologies or drug candidates or grant licenses on terms that are not favorable to us. If adequate funds are not available, we may have to further delay, reduce or eliminate one or more of our planned activities. These actions would likely reduce the market price of our common stock.
If the estimates we make, or the assumptions on which we rely, in preparing our consolidated financial statements prove inaccurate, our actual results may vary from those reflected in our accruals.
Our consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America. The preparation of these consolidated financial statements requires us to make estimates and judgments that affect the reported amounts of our assets, liabilities, revenues and expenses, the amounts of charges accrued by us and related disclosure of contingent assets and liabilities. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. We cannot assure you, however, that our estimates, or the assumptions underlying them, will be correct.
Our operating results may fluctuate significantly, which makes our future operating results difficult to predict and could cause our operating results to fall below expectations or our guidance.
Our quarterly and annual operating results have fluctuated and may continue to fluctuate significantly in the future, which makes it difficult for us to predict our future operating results. From time to time, we may enter into license or collaboration agreements or strategic partnerships with other companies that include development funding and significant upfront and milestone payments and/or royalties. These upfront and milestone payments may vary significantly from period to period and any such variance could cause a significant fluctuation in our operating results from one period to the next.
In addition, we measure compensation cost for stock-based awards made to employees at the grant date of the award, based on the fair value of the award, and recognize the cost as an expense over the employee’s requisite service
47
period. As the variables that we use as a basis for valuing these awards change over time, including our underlying stock price and stock price volatility, the magnitude of the expense that we must recognize may vary significantly.
Furthermore, our operating results may fluctuate due to a variety of other factors, may of which are outside of our control and may be difficult to predict, including the following:
•the timing and cost of, and level of investment in, research and development activities relating to our current and any future product candidates, which will change from time to time;
•our ability to enroll patients in clinical trials and the timing of enrollment;
•the cost of manufacturing our current and any future product candidates, which may vary depending on FDA guidelines and requirements, the quantity of production and the terms of our agreements with manufacturers;
•expenditures that we will or may incur to acquire or develop additional product candidates and technologies;
•the timing and outcomes of clinical trials for product candidates;
•the need to conduct unanticipated clinical trials or trials that are larger or more complex than anticipated;
•competition from existing and potential future products that compete with any of our product candidates, and changes in the competitive landscape of our industry, including consolidation among our competitors or partners;
•any delays in regulatory review or approval of any of our product candidates;
•the level of demand for any of our product candidates, if approved, which may fluctuate significantly and be difficult to predict;
•the risk/benefit profile, cost and reimbursement policies with respect to our product candidates, if approved, and existing and potential future products that compete with our product candidates;
•our ability to commercialize any of our product candidates, if approved, inside and outside of the United States, either independently or working with third parties;
•our ability to establish and maintain collaborations, licensing or other arrangements;
•our ability to adequately support future growth;
•potential unforeseen business disruptions that increase our costs or expenses;
•future accounting pronouncements or changes in our accounting policies; and
•the changing and volatile global economic environment.
The cumulative effect of these factors could result in large fluctuations and unpredictability in our quarterly and annual operating results. As a result, comparing our operating results on a period-to-period basis may not be meaningful. Investors should not rely on our past results as an indication of our future performance. This variability and unpredictability could also result in our failing to meet the expectations of industry or financial analysts or investors for any period. If our revenue or operating results fall below the expectations of analysts or investors or below any forecasts we may provide to the market, or if the forecasts we provide to the market are below the expectations of analysts or investors, the price of our common stock could decline substantially. Such a stock price decline could occur even when we have met any previously publicly stated guidance we may provide.
The investment of our cash, cash equivalents and fixed income securities is subject to risks which may cause losses and affect the liquidity of these investments.
At September 30, 2022, we had $482.3 million in cash, cash equivalents, fixed income securities and a certificate of deposit and we received an additional $250.0 million in proceeds from a royalty sale in November 2022. Our investments may also include commercial paper, securities issued by the U.S. government obligations, and money market funds meeting the criteria of our investment policy, which is focused on the preservation of our capital. These investments are subject to general credit, liquidity, and market and interest rate risks, particularly in the current economic environment. We may realize losses in the fair value of these investments or a complete loss of these investments, which would have a negative effect on our consolidated financial statements. In addition, should our investments cease paying or reduce the amount of interest paid to us, our interest income would suffer. The market risks associated with our investment portfolio may have an adverse effect on our results of operations, liquidity and financial condition.
Our ability to utilize net operating loss carryforwards and other tax benefits may be limited.
We have historically incurred net losses. Under the Internal Revenue Code of 1986, as amended (the “Code”), a corporation is generally allowed a deduction for net operating losses (NOLs) carried forward from a prior taxable year. Under that provision, we can carryforward our NOLs to offset our future taxable income, if any, until such NOLs are used or expire. As of September 30, 2022, we had federal and state NOL carryforwards of approximately $504.8 million and $626.5 million, respectively. As a result of the Coronavirus Aid, Relief, and Economic Security Act of 2020 (“CARES Act”) and legislation commonly referred to as the Tax Cuts and Jobs Act of 2017 (“2017 Tax Act”), NOLs arising before January 1, 2018, and NOLs arising after January 1, 2018, are subject to different rules. Under the CARES Act and 2017 Tax Act, federal NOLs incurred in 2018, 2019 and 2020 can generally be carried back five years, carried forward indefinitely and can offset 100% of future taxable income for tax years before January 1, 2021 and up to 80% of future
48
taxable income for tax years after December 31, 2020. Any NOLs arising on or after January 1, 2021, cannot be carried back, can generally be carried forward indefinitely and can offset up to 80% of future taxable income. It is uncertain if and to what extent various states will conform to the newly enacted federal tax law. These NOL carryforwards could expire unused before offsetting potential future income tax liabilities.
In addition, under Section 382 of the Code and corresponding provisions of state law, if a corporation undergoes an “ownership change,” which is generally defined as a greater than 50 percent change, by value, in its equity ownership over a three-year period, the corporation’s ability to use its pre-change NOL carryforwards and other pre-change tax attributes to offset its post-change income or taxes may be limited. It is possible that we have experienced an ownership change limitation. We may experience ownership changes in the future as a result of subsequent shifts in our stock ownership, some of which may be outside of our control.
If an ownership change occurs and our ability to use our NOL carryforwards is materially limited, it would harm our future operating results by effectively increasing our future tax obligations.
Our business is subject to changing regulations for corporate governance and public disclosure that has increased both our costs and the risk of noncompliance.
Each year we are required to evaluate our internal controls systems in order to allow management to report on and our Independent Registered Public Accounting Firm to attest to, our internal controls as required by Section 404 of the Sarbanes-Oxley Act. As a result, we continue to incur additional expenses and divert our management's time to comply with these regulations. In addition, if we cannot continue to comply with the requirements of Section 404 in a timely manner, we might be subject to sanctions or investigation by regulatory authorities, such as the SEC, the Public Company Accounting Oversight Board or The Nasdaq Global Select Market. Any such action could adversely affect our financial results and the market price of our common stock.
We could be subject to additional tax liabilities.
We are subject to U.S. federal, state, and local taxes in the United States and other countries. Significant judgment is required in evaluating our tax positions. During the ordinary course of business, there are many activities and transactions for which the ultimate tax determination is uncertain. In addition, our tax obligations and effective tax rates could be adversely affected by changes in the relevant tax, accounting and other laws, regulations, principles and interpretations, including those relating to income tax nexus, by recognizing tax losses or lower than anticipated earnings in jurisdictions where we have lower statutory rates and higher than anticipated earnings in jurisdictions where we have higher statutory rates, by changes in foreign currency exchange rates, or by changes in the valuation of our deferred tax assets and liabilities. We may be audited in various jurisdictions, and such jurisdictions may assess additional taxes, sales taxes and value-added taxes against us. Although we believe our tax estimates are reasonable, the final determination of any tax audits or litigation could be materially different from our historical tax provisions and accruals, which could have a material adverse effect on our operating results or cash flows in the period for which a determination is made.
Risks Related to Investment and Securities
Our Board of Directors has the authority to issue shares of “blank check” preferred stock, which may make an acquisition of the Company by another company more difficult.
We have adopted and may in the future adopt certain measures that may have the effect of delaying, deferring or preventing a takeover or other change in control of the Company that a holder of our common stock might consider in its best interest. For example, our Board of Directors, without further action by our stockholders, currently has the authority to issue up to 5,000,000 shares of preferred stock and to fix the rights (including voting rights), preferences and privileges of these shares (“blank check” preferred). Such preferred stock may have rights, including economic rights, senior to our common stock. These factors could also reduce the price that certain investors might be willing to pay for shares of our common stock and result in the market price being lower than it would be without these provisions.
We do not intend to declare cash dividends on our common stock.
We will not distribute cash to our stockholders unless and until we can develop sufficient funds from operations to meet our ongoing needs and implement our business plan. The time frame for that is unpredictable and investors should not expect dividends in the near future, if at all.
If securities or industry analysts do not publish research reports about our business or if they make adverse recommendations regarding an investment in our stock, our stock price and trading volume may decline.
The trading market for our common stock can be influenced by the research and reports that industry or securities analysts publish about our business. Currently, coverage of our Company by industry and securities analysts is limited.
49
Investors have many investment opportunities and may limit their investments to companies that receive greater coverage from analysts. If additional industry or securities analysts do not commence coverage of the Company, the trading price of our stock could be negatively impacted. If one or more of the analysts downgrade our stock or comment negatively on our prospects, our stock price may decline. If one or more of these analysts cease to cover our industry or us or fail to publish reports about the Company regularly, our common stock could lose visibility in the financial markets, which could also cause our stock price or trading volume to decline. Further, incorrect judgments, estimates or assumptions made by research analysts may adversely affect our stock price, particularly if subsequent performance falls below the levels that were projected by the research analyst(s), even if we did not set or endorse such expectations. Any of these events could cause further volatility in our stock price and could result in substantial declines in the value of our stock.
The market for purchases and sales of our common stock may be limited, and the sale of a limited number of shares could cause the price to fall sharply.
Although our common stock is listed for trading on the Nasdaq Global Select Market, at various times our securities are relatively thinly traded. Investor trading patterns could serve to exacerbate the volatility of the price of our stock. For example, mandatory sales of our common stock by institutional holders could be triggered if an investment in our common stock no longer satisfies their investment standards and guidelines. It may be difficult to sell shares of our common stock quickly without significantly depressing the value of the stock. Unless we are successful in developing continued investor interest in our stock, sales of our stock could result in major fluctuations in the price of the stock.
Our common stock price has fluctuated significantly over the last several years and may continue to do so in the future, without regard to our results of operations and prospects.
Because we are still a clinical-stage pharmaceutical company and have not yet commercialized a drug, there are few objective metrics by which our progress may be measured. Consequently, we expect that the market price of our common stock will continue to fluctuate significantly. We may not continue to generate substantial revenue from the license or sale of our technology for several years, if at all. In the absence of product revenue as a measure of our operating performance, we anticipate that investors and market analysts will assess our performance by considering factors such as:
•Announcements of developments related to our business;
•Our ability to enter into or extend investigation phase, development phase, commercialization phase and other agreements with new and/or existing partners;
•Announcements regarding the status of any or all of our collaborations or products, including clinical trial results;
•Market perception and/or investor sentiment regarding our technology;
•Announcements of actions taken by regulatory authorities, such as the U.S. Food and Drug Administration;
•Announcements regarding developments in the RNA interference or biotechnology fields in general;
•Announcements regarding clinical trial results with our products or competitors’ products;
•Market perception and/or announcements regarding other companies developing products in the field of biotechnology generally or specifically RNA interference;
•The issuance of competitive patents or disallowance or loss of our patent rights;
•The addition or departure of key executives; and
•Variations in our operating results.
We will not have control over many of these factors but expect that they may influence our stock price. As a result, our stock price may be volatile and such volatility could result in the loss of all or part of your investment.
Stockholder equity interest may be substantially diluted in any additional equity issuances.
We expect that significant additional capital will be needed in the future to continue our planned operations. To the extent we raise additional capital by issuing equity securities, our stockholders may experience substantial dilution. We may sell common stock, convertible securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time. If we sell common stock, convertible securities or other equity securities in more than one transaction, investors may be materially diluted by subsequent sales. These sales may also result in material dilution to our existing stockholders, and new investors could gain rights superior to our existing stockholders.
Economic and Industry Risks
Unfavorable global economic conditions, whether brought about by material global crises, health epidemics, military conflicts or war, geopolitical and trade disputes or other factors, may adversely affect our business and financial results.
Our business is sensitive to global economic conditions, which can be adversely affected by public health crises (including the COVID-19 pandemic) and epidemics, political and military conflict, trade and other international disputes, significant natural disasters (including as a result of climate change) or other events that disrupt macroeconomic conditions.
50
Adverse macroeconomic conditions, including inflation, slower growth or recession, new or increased tariffs and other barriers to trade, changes to fiscal and monetary policy or government budget dynamics (particularly in the pharmaceutical and biotech areas), tighter credit, higher interest rates, volatility in financial markets, high unemployment, labor availability constraints, currency fluctuations and other challenges in the global economy have in the past adversely affected, and may in the future adversely affect, us and our business partners and suppliers.
For example, trade policies and geopolitical disputes (including as a result of China-Taiwan relations) and other international conflicts can result in tariffs, sanctions and other measures that restrict international trade, and can materially adversely affect our business, particularly if these measures occur in regions where we source our components or raw materials. For example, tensions between the United States and China have led to a series of tariffs being imposed by the United States on imports from China mainland, as well as other business restrictions. Tariffs increase the costs of the components and raw materials we source. Countries may also adopt other measures, such as controls on imports or exports of goods, technology or data, that could adversely impact the Company’s operations and supply chain. These geopolitical risks could also adversely affect our joint venture in China.
Further, military conflicts or wars (such as the ongoing conflict between Russia and Ukraine) can cause exacerbated volatility and disruptions to various aspects of the global economy. The uncertain nature, magnitude, and duration of hostilities stemming from such conflicts, including the potential effects of sanctions and counter-sanctions, or retaliatory cyber-attacks on the world economy and markets, have contributed to increased market volatility and uncertainty, which could have an adverse impact on macroeconomic factors that affect our business and operations, such as worldwide supply chain issues. Additionally, the ongoing conflict between Russia and Ukraine has impacted our business decisions with respect to potential clinical trial sites in Europe. For example, a number of our clinical trial sites we had previously planned to use in Russia, Ukraine and Belarus were shut down and we have sought alternatives in other geographies. We cannot be certain of the overall impact of the conflict between Russia and Ukraine on our ability to conduct and complete our clinical trials as planned, and any interruptions of our clinical trials can result in significant delays or termination of the research, development or commercialization of our drug candidates, which could impair our ability to generate revenues and harm our business and financial condition. It is not possible to predict the short and long-term implications of military conflicts or wars or geopolitical tensions which could include further sanctions, uncertainty about economic and political stability, increases in inflation rate and energy prices, cyber-attacks, supply chain challenges and adverse effects on currency exchange rates and financial markets.
Additionally, our operations and facilities, as well as operations of our suppliers and manufacturers, may be located in areas that are prone to earthquakes and other natural disasters. Such operations and facilities are also subject to the risk of interruption by fire, drought, power shortages, nuclear power plant accidents and other industrial accidents, terrorist attacks and other hostile acts, ransomware and other cybersecurity attacks, labor disputes, public health crises (including the COVID-19 pandemic), and other events beyond the Company’s control. Global climate change is resulting in certain types of natural disasters occurring more frequently or with more intense effects. Such events can create delays or interruptions to the Company’s development efforts and inefficiencies in the Company’s supply and manufacturing chain. Significant delays in our development efforts could materially impact our ability to obtain regulatory approval and to commercialize our products. Any insurance we maintain against damage to our property and the disruption of our business due to disaster may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, or at all. Further, because the Company relies on single or limited sources for the supply and manufacture of many critical components, a business interruption affecting such sources would exacerbate any negative consequences to the Company.
Any public health crises, including the COVID-19 pandemic, may affect our operations and those of third parties on which we rely, including our business partners and suppliers. In the past three years, the COVID-19 pandemic has had, and likely will continue to have, an adverse impact on the global economy, including as a result of impacts associated with protective health measures that we, other businesses and governments are taking or might have to take again in the future to manage the pandemic. The extent to which the COVID-19 pandemic and measures taken in response thereto impact our business, results of operations and financial condition will depend on future developments which are highly uncertain and difficult to predict. These developments include, but are not limited to, future resurgences of the virus and its variants, actions taken to contain the virus or address its impact, and the timing, distribution and efficacy of vaccines and other treatments.
Without limiting the foregoing, we have experienced and/or may in the future experience:
•delays in receiving authorization from regulatory authorities to initiate any planned clinical trials, inspections, reviews and approvals of products;
•delays or difficulties enrolling patients in our clinical trials;
•delays in or disruptions to the conduct of preclinical programs and clinical trials;
51
•constraints on the movement of products and supplies through the supply chain, which can disrupt our ability to conduct clinical trials and develop our products;
•price increases in raw materials and capital equipment, as well as increasing price competition in our markets;
•adverse impacts on our workforce and/or key employees; and
•increased risk that counterparties to our contractual arrangements will become insolvent or otherwise unable to fulfill their contractual obligations.
Drug development is time consuming, expensive and risky.
We are focused on technology related to new and improved pharmaceutical candidates. Product candidates that appear promising in the early phases of development, such as in animal and early human clinical trials, often fail to reach the market for a number of reasons, such as:
•Clinical trial results may be unacceptable, even though preclinical trial results were promising;
•Inefficacy and/or harmful side effects in humans or animals;
•The necessary regulatory bodies, such as the U.S. Food and Drug Administration, may not approve our potential product for the intended use, or at all; and/or
•Manufacturing and distribution may be uneconomical.
For example, any positive preclinical results in animals may not be replicated in human clinical studies. These programs may be also found to be unsafe in humans, particularly at higher doses needed to achieve the desired levels of efficacy. Also, the positive safety results from single dose human clinical studies may not be replicated in other human studies, including multiple dose studies. Clinical and preclinical study results are frequently susceptible to varying interpretations by scientists, medical personnel, regulatory personnel, statisticians and others, which often delays, limits, or prevents further clinical development or regulatory approvals of potential products. Clinical trials can take many years to complete, including the process of study design, clinical site selection and the recruitment of patients. As a result, we can experience significant delays in completing clinical studies, which can increase the cost of developing a drug candidate and shorten the time that an approved product may be protected by patents. If our drug candidates are not successful in human clinical trials, we may be forced to curtail or abandon certain development programs. If we experience significant delays in commencing or completing our clinical studies, we could suffer from significant cost overruns, which could negatively affect our capital resources and our ability to complete these studies.
The healthcare system is under significant financial pressure to reduce costs, which could reduce payment and reimbursement rates for drugs.
Throughout the world and particularly in the United States, the healthcare system is under significant financial pressure to reduce costs. The price of pharmaceuticals has been a topic of considerable public discussion that could lead to price controls or other price-limiting strategies by payors that have the effect of lowering payment and reimbursement rates for drugs or otherwise making the commercialization of pharmaceuticals less profitable. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. These effects could reduce or eliminate our ability to return value to our stockholders.
Regulatory standards are subject to change over time, making it difficult to accurately predict the likelihood of marketing approval even when clinical trials meet their endpoints.
Regulatory standards are promulgated by various government entities and are subject to change based on factors such as scientific developments, public perceptions of risk, and political forces. Because clinical trials often take years to complete, it is sometimes possible for standards that exist during the conception and initiation of a clinical trial to change before the clinical trial is completed or reviewed by government regulators. For example, we may initiate clinical trials that are designed to show benefits on relatively short-term endpoints, but ultimately be required to show benefits in longer-term outcome studies. While some government entities have safeguards intended to ensure standards agreed upon by sponsors and regulators at the outset of a clinical trial are applied during regulatory review processes, those safeguards generally permit regulators to apply more rigorous standards where regulators believe doing so is necessary. As such, there can be no assurance that regulatory standards that are appropriate at the outset of a clinical trial program will not become more rigorous during the regulatory approval process and could potentially result in a delayed approval or denial of marketing authorization.
ITEM 1B.UNRESOLVED STAFF COMMENTS
None.
52
ITEM 2.PROPERTIES
On December 20, 2021, the Company completed a purchase of 13 acres of land in the Verona Technology Park in Verona, Wisconsin, which is being developed into an approximately 160,000 square foot drug manufacturing facility and an approximately 140,000 square foot laboratory and office facility which will support the Company’s process development and analytical activities. Additionally, the Company entered into a lease agreement for a new 144,000 square foot laboratory and office facility in San Diego, California to support discovery activities, which it currently anticipates to be available starting around April 2023. The following table summarizes the Company’s leased facilities as of September 30, 2022.
| Office Space (sq.ft) | Monthly Expenses | Primary Use | Lease Expiration | Lease Term (year) | |||||||||||||||||||||||||
| Pasadena, California | 49,000 | $ | 157,490 | Corporate Headquarters | April 2027 | 7.5 | |||||||||||||||||||||||
| Madison, Wisconsin | 111,000 | $ | 168,371 | Research Facility | September 2031 | 15.0 | |||||||||||||||||||||||
| San Diego, California | 21,000 | $ | 61,821 | Research Facility | January 2023 | 2.3 | |||||||||||||||||||||||
| San Diego, California | 10,000 | $ | 33,379 | Office Space | March 2023 | 1.2 | |||||||||||||||||||||||
ITEM 3.LEGAL PROCEEDINGS
Legal Proceedings are set forth in the Company’s financial statement schedules in Part IV, Item 15 of this Annual Report and are incorporated herein by reference. See Note 7 — Commitments and Contingencies of Notes to Consolidated Financial Statements of Part IV, “Item 15. Exhibits and Financial Statement Schedules.”
ITEM 4.MINE SAFETY DISCLOSURES
Not applicable.
53
PART II
ITEM 5.MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Shares of the Company’s common stock are traded on The Nasdaq Global Select Market under the symbol “ARWR.” There were approximately 100 holders of record of the Company’s common stock as of November 16, 2022.
Dividends
The Company has never paid dividends on its common stock and does not anticipate that it will do so in the foreseeable future.
Recent Sales of Unregistered Securities
None.
Repurchases of Equity Securities
None.
Performance Graph
The following performance graph shall not be deemed “soliciting material” or to be “filed” with the SEC, nor shall such information be incorporated by reference into any future filing under the Securities Act of 1933 or Securities Exchange Act of 1934, each as amended, except to the extent that we specifically incorporate it by reference into such filing. The graph compares the cumulative 5-year total return to stockholders on the Company’s common stock relative to the cumulative total returns of the Nasdaq Composite Index and the Nasdaq Biotechnology Index. The Company selected the Nasdaq Biotechnology Index because it believes the index reflects the market conditions within the industry in which the Company primarily operates. The comparison of total return on investment, defined as the change in year-end stock price plus reinvested dividends, for each of the periods assumes that $100 was invested on September 30, 2017, in each of the Company’s common stock, the Nasdaq Composite Index and the Nasdaq Biotechnology Index, with investment weighted on the basis of market capitalization.
54
The comparisons in the following graph are based on historical data and are not intended to forecast the possible future performance of the Company’s common stock.
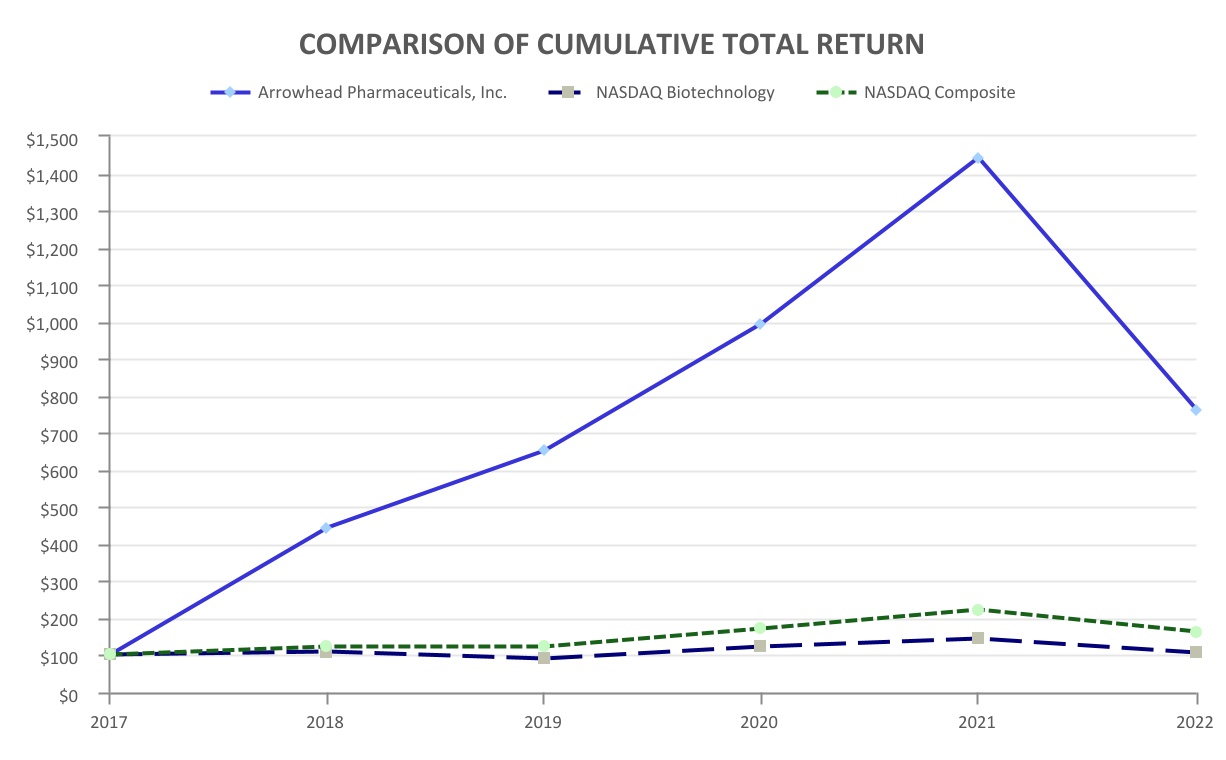
| $100 investment in stock or index | Ticker | 2017 | 2018 | 2019 | 2020 | 2021 | 2022 | |||||||||||||||||||||||||||||||||||||
| Arrowhead Pharmaceuticals, Inc. | ARWR | $ | 100.00 | $ | 442.73 | $ | 650.81 | $ | 994.46 | $ | 1,441.80 | $ | 763.28 | |||||||||||||||||||||||||||||||
| NASDAQ Biotechnology Index | ^NBI | $ | 100.00 | $ | 109.80 | $ | 89.54 | $ | 121.87 | $ | 145.58 | $ | 107.86 | |||||||||||||||||||||||||||||||
| NASDAQ Composite Index | ^IXIC | $ | 100.00 | $ | 123.87 | $ | 123.14 | $ | 171.91 | $ | 222.42 | $ | 162.80 | |||||||||||||||||||||||||||||||
ITEM 6. RESERVED
55
ITEM 7.MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
OVERVIEW
The Company develops medicines that treat intractable diseases by silencing the genes that cause them. Using a broad portfolio of RNA chemistries and efficient modes of delivery, the Company’s therapies trigger the RNAi mechanism to induce rapid, deep and durable knockdown of target genes. RNAi is a mechanism present in living cells that inhibits the expression of a specific gene, thereby affecting the production of a specific protein. RNAi-based therapeutics may leverage this natural pathway of gene silencing to target and shut down specific disease-causing genes.
The Company has focused its resources on therapeutics that exclusively utilize its high levels of pharmacologic activity in multiple animal models spanning several therapeutic areas. TRiMTM enabled therapeutics offer several potential advantages over prior generation and competing technologies, including: simplified manufacturing and reduced costs; multiple routes of administration including subcutaneous injection and inhaled administration; the ability to target multiple tissue types including liver, lung, muscle and others; and the potential for improved safety and reduced risk of intracellular buildup, because there are less metabolites from smaller, simpler molecules.
The Company’s pipeline includes:
•Hypertriglyceridemia - ARO-APOC3
•Dyslipidemia - ARO-ANG3
•Facioscapulohumeral muscular dystrophy - ARO-DUX4
•Complement mediated diseases - ARO-C3
•Muco-obstructive or inflammatory pulmonary conditions - ARO-RAGE and ARO-MUC5AC
•Idiopathic pulmonary fibrosis - ARO-MMP7
•Liver disease - ARO-HSD (out-licensed to GSK)
•Uncontrolled gout - ARO-XDH (out-licensed to Horizon)
•Non-alcoholic steatohepatitis (NASH) - NJ-75220795 (ARO-JNJ1, out-licensed to Janssen)
•Liver disease associated with alpha-1 antitrypsin deficiency (“AATD”) - Fazirsiran (ARO-AAT, a collaboration with Takeda)
•Chronic hepatitis B virus - JNJ-3989 (ARO-HBV, out-licensed to Janssen)
•Cardiovascular disease - Olpasiran (AMG 890 or ARO-LPA, out-licensed to Amgen)
The Company operates lab facilities in San Diego, California and Madison, Wisconsin, where its research and development activities, including the development of RNAi therapeutics, take place. The Company’s principal executive offices are located in Pasadena, California.
The Company continues to develop other clinical candidates for future clinical trials. Clinical candidates are tested internally and through GLP toxicology studies at outside laboratories. Drug materials for such studies and clinical trials are either manufactured internally or contracted to third-party manufacturers. The Company engages third-party contract research organizations (“CROs”) to manage clinical trials and works cooperatively with such organizations on all aspects of clinical trial management, including plan design, patient recruiting, and follow up. These outside costs, relating to the preparation for and administration of clinical trials, are referred to as “candidate costs.” If the clinical candidates progress through human testing, candidate costs will increase.
2022 Business Highlights
During fiscal year 2022, the Company continued to develop and advance its pipeline and partnered candidates and expanded its facilities to support its growing programs. The bullets below highlight some of those key developments; however, it is not all-inclusive and is meant to be read in conjunction with the entirety of management’s discussion and analysis, the Company’s Consolidated Financial Statements and notes thereto, and all other items contained within this Annual Report on Form 10-K.
•dosed the first patients in its PALISADE study, a Phase 3 clinical study to evaluate the safety and efficacy of ARO-APOC3 in adults with familial chylomicronemia syndrome (FCS);
•filed for regulatory clearance to begin a Phase 1/2a study of ARO-C3 and subsequently dosed the first subjects in AROC3-1001, a Phase 1/2 clinical study of ARO-C3, the Company’s investigational RNA interference (RNAi) therapeutic designed to reduce production of complement component 3 (C3) as a potential therapy for various complement mediated diseases;
•presented additional interim clinical data from AROHSD1001, AROAAT2002, and AROAPOC31001;
56
•completed enrollment in Phase 2b ARCHES-2 study of investigational ARO-ANG3 for patients with mixed dyslipidemia;
•filed for regulatory clearance to initiate Phase 1/2a study of ARO-RAGE and subsequently dosed first subjects for treatment of Asthma;
•filed for regulatory clearance to initiate Phase 1/2a study of ARO-MUC5AC and subsequently dosed first subjects for treatment of muco-obstructive lung disease;
•filed for regulatory clearance to initiate Phase 1/2a study of ARO-MMP7 for treatment of idiopathic pulmonary fibrosis (IPF);
•initiated and dosed the first patients in the Phase 2 GATEWAY clinical study of investigational ARO-ANG3 for the treatment of patients with homozygous familial hypercholersterolemia;
•entered into definitive agreements to form a joint venture, Visirna Therapeutics, Inc. with Vivo Capital through which the Company and Vivo Capital intend to expand the reach of innovative medicines in Greater China;
•hosted a pulmonary research & development (R&D) day to discuss the Company’s emerging pipeline of pulmonary targeted RNA interference (RNAi) therapeutic candidates that leverage its proprietary Targeted RNAi Molecule (TRiMTM) platform, including an announcement of its previously undisclosed candidate designed to reduce expression of matrix metalloproteinase 7 (MMP7) as a potential treatment for idiopathic pulmonary fibrosis (IPF);
•Entered into an exclusive license agreement with GSK for ARO-HSD;
•Janssen presented clinical data from REEF-1, a Phase 2b study of different combination regimens, including JNJ-73763989 (JNJ-3989), formerly called ARO-HBV, and/or JNJ-56136379 (JNJ-6379), and a nucleos(t)ide analog (NA) for the treatment of chronic hepatitis B virus infection (CHB);
•in conjunction with Takeda, announced results from a Phase 2 clinical study (AROAAT-2002) of investigational fazirsiran (TAK-999/ARO-AAT) for the treatment of liver disease associated with alpha-1 antitrypsin deficiency (AATD), and was recently published in the New England Journal of Medicine (NEJM) and presented in an oral presentation at The International Liver Congress™ 2022 - The Annual Meeting of the European Association for the Study of the Liver (EASL); and
•completed the purchase of 13 acres of land in the Verona Technology Park in Verona, Wisconsin and held a groundbreaking ceremony on the site. The site is being developed into an approximately 160,000 square foot drug manufacturing facility and an approximately 140,000 square foot laboratory and office facility which will support the Company’s process development and analytical activities. Additionally, the Company entered into a lease agreement for a new 144,000 square foot laboratory and office facility in San Diego, California to support discovery activities, which it currently anticipates to be available in April 2023.
2022 Financial Performance Summary
Net loss was $176.1 million for the year ended September 30, 2022 as compared to net loss of $140.8 million for the year ended September 30, 2021. Net loss per share – diluted was $1.67 for the year ended September 30, 2022 as compared to net loss per share – diluted of $1.36 for the year ended September 30, 2021. The increase in net losses for the year ended September 30, 2022 was due to an increase in research and development and general and administrative expenses as the Company’s pipeline of candidates has expanded and progressed through clinical trial phases, partially offset by an increase in revenue from its license and collaboration agreements, primarily from the License Agreements with GSK, Horizon and Takeda. For further information, see Part I, “Item 1. Business” and “Results of Operations - Revenue” below.
The Company had $108.0 million of cash and cash equivalents, $268.4 million in short-term investments, $105.9 million of long-term investments and $691.9 million of total assets as of September 30, 2022, as compared to $184.4 million of cash and cash equivalents, $126.7 million of marketable securities, $56.6 million in short-term investments, $245.6 million of long-term investments and $710.1 million of total assets as of September 30, 2021. Based upon the Company’s current cash and investment resources and operating plan, the Company expects to have sufficient liquidity to fund operations for at least the next twelve months.
Critical Accounting Estimates
Management makes certain judgments and uses certain estimates and assumptions when applying U.S. generally accepted accounting principles (“GAAP”) in the preparation of the Company’s Consolidated Financial Statements. On an
57
ongoing basis, the Company evaluates its estimates, judgments and assumptions. The Company bases its estimates on historical experience and on various other assumptions that it believes are reasonable, the results of which form the basis for making judgments about the carrying values of assets, liabilities and equity and the amount of revenue and expense. Actual results may vary from what the Company anticipates and different assumptions or estimates about the future could change its reported results. The Company believes the following accounting policies are the most critical to it, in that they require its most difficult, subjective or complex judgments in the preparation of the Company’s Consolidated Financial Statements. For further information, see Note 1, Organization and Significant Accounting Policies of the Notes to the Company’s Consolidated Financial Statements in Part IV, “Item 15. Exhibits and Financial Statement Schedules.”
Revenue Recognition—The Company adopted Financial Accounting Standards Board (“FASB”) Topic 606 – Revenue for Contracts from Customers which amended revenue recognition principles and provides a single, comprehensive set of criteria for revenue recognition within and across all industries. The Company has not yet achieved commercial sales of its drug candidates to date, however, this standard is applicable to its ongoing licensing and collaboration agreements. This is discussed further in Note 2, Collaboration and License Agreements of the Notes to the Company’s Consolidated Financial Statements in Part IV, “Item 15. Exhibits and Financial Statement Schedules.”
At contract inception, the Company assesses whether the goods or services promised within each contract are distinct and, therefore, represent a separate performance obligation, or whether they are not distinct and are combined with other goods and services until a distinct bundle is identified. The Company then determines the transaction price, which typically includes upfront payments and any variable consideration that it determines is probable to not cause a significant reversal in the amount of cumulative revenue recognized when the uncertainty associated with the variable consideration is resolved. The Company then allocates the transaction price to each performance obligation and recognizes the associated revenue when (or as) each performance obligation is satisfied.
The Company recognizes the transaction price allocated to upfront license payments as revenue upon delivery of the license to the customer and resulting ability of the customer to use and benefit from the license, if the license is determined to be distinct from the other performance obligations identified in the contract. These other performance obligations are typically to perform research and development services for the customer, often times relating to the candidate that the customer is licensing. If the license is not considered to be distinct from other performance obligations, the Company assesses the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied at a point in time or over time. If the performance obligation is satisfied over time, the Company then determines the appropriate method of measuring progress for purposes of recognizing revenue from license payments. The Company evaluates the measure of progress each reporting period and, if necessary, adjusts the related revenue recognition.
Typically, the Company’s collaboration agreements entitle it to additional payments upon the achievement of milestones or royalties on sales. The milestones are generally categorized into three types: development milestones, generally based on the initiation of toxicity studies or clinical trials; regulatory milestones, generally based on the submission, filing or approval of regulatory applications such as a Clinical Trial Application or a NDA in the United States; and sales-based milestones, generally based on meeting specific thresholds of sales in certain geographic areas. The Company evaluates whether it is probable that the consideration associated with each milestone or royalty will not be subject to a significant reversal in the cumulative amount of revenue recognized. Amounts that meet this threshold are included in the transaction price using the most likely amount method, whereas amounts that do not meet this threshold are excluded from the transaction price until they meet this threshold. At the end of each subsequent reporting period, the Company re-evaluates the probability of a significant reversal of the cumulative revenue recognized for its milestones and royalties, and, if necessary, adjusts its estimate of the overall transaction price. Any such adjustments are recorded on a cumulative catch-up basis, which would affect revenues and net income in the Company’s consolidated statements of operations and comprehensive loss. Typically, milestone payments and royalties are achieved after the Company’s performance obligations associated with the collaboration agreements have been completed and after the customer has assumed responsibility for the respective clinical or preclinical program. Milestones or royalties achieved after the Company’s performance obligations have been completed are recognized as revenue in the period the milestone or royalty was achieved. If a milestone payment is achieved during the performance period, the milestone payment would be recognized as revenue to the extent performance had been completed at that point, and the remaining balance would be recorded as deferred revenue.
The revenue standard requires the Company to assess whether a significant financing component exists in determining the transaction price. The Company performs this assessment at the onset of its licensing or collaboration agreements. Typically, a significant financing component does not exist because the customer is paying for a license or services in advance with an upfront payment. Additionally, future royalty payments are not substantially within the control of the Company or the customer.
58
The revenue standard requires the Company to allocate the arrangement consideration on a relative standalone selling price basis for each performance obligation after determining the transaction price of the contract and identifying the performance obligations to which that amount should be allocated. The relative standalone selling price is defined in the revenue standard as the price at which an entity would sell a promised good or service separately to a customer. If other observable transactions in which the Company has sold the same performance obligation separately are not available, the Company estimates the standalone selling price of each performance obligation. Key assumptions to determine the standalone selling price may include forecasted revenues, development timelines, reimbursement rates for personnel costs, discount rates and probabilities of technical and regulatory success.
Whenever we determine that goods or services promised in a contract should be accounted for as a combined performance obligation over time, the Company determines the period over which the performance obligations will be performed and revenue will be recognized. Revenue is recognized using either the proportional performance method or on a straight-line basis if efforts will be expended evenly over time. Labor hours, costs incurred or patient visits in clinical trials are typically used as the measure of performance. Significant management judgment is required in determining the level of effort required under an arrangement and the period over which the Company is expected to complete its performance obligations. If the Company determines that the performance obligation is satisfied over time, any upfront payment received is initially recorded as deferred revenue on the Company’s consolidated balance sheets.
Collaborative Arrangements—The Company analyzes its collaborative arrangements to assess whether such arrangements involve joint operating activities performed by parties that are both active participants in the activities and exposed to significant risks and rewards, and therefore an appropriate recognition method is determined and applied consistently, either by analogy to appropriate accounting literature or by applying a reasonable accounting policy election. For collaborative arrangements that are within the scope of FASB Topic 808—Collaborative Arrangements, the Company evaluates the income statement classification for presentation of amounts due to or owed from other participants associated with multiple units of account in a collaborative arrangement based on the nature of each activity. Payments or reimbursements that are the result of a collaborative relationship instead of a customer relationship, such as co-development and co-commercialization activities, are recorded as increases or decreases to research and development expense or general and administrative expense, as appropriate.
Contingent Consideration—The consideration for the Company’s acquisitions may include future payments that are contingent upon the occurrence of a particular event. The Company estimates the fair value of contingent consideration obligations through valuation models designed to estimate the probability of such contingent payments based on various assumptions and incorporating estimated success rates. These fair value measurements are based on significant inputs not observable in the market. Substantial judgment is employed in determining the appropriateness of these assumptions as of the acquisition date and for each subsequent period. Accordingly, changes in assumptions could have a material impact on the amount of contingent consideration expense the Company records in any given period. The Company determined the fair value of its contingent consideration obligation to be $0 at September 30, 2022 and 2021.
Leases—The Company classifies each of its leases as operating or financing considering factors such as the length of the lease term, the present value of the lease payments, the nature of the asset being leased, and the potential for ownership of the asset to transfer during the lease term. Leases with terms greater than one-year are recognized on the Company’s consolidated balance sheets as right-of-use assets that represent its right to use an underlying asset for the lease term, and lease liabilities that represent its obligation to make lease payments arising from the lease. Lease assets and liabilities are recognized at the lease commencement date based on the estimated present value of lease payments over the expected lease term minus the present value of any incentives, rebates or abatement expected to be received from the lessor. The Company did not include the extension option in the lease term.
The interest rate implicit in lease contracts is typically not readily determinable. As such, the Company utilizes the appropriate incremental borrowing rate, which is the rate incurred to borrow on a collateralized basis an amount equal to the lease payments over a similar term and in a similar economic environment. The Company records expense to recognize fixed lease payments on a straight-line basis over the expected lease term. Costs determined to be variable and not based on an index or rate are not included in the measurement of the lease liability and are expensed as incurred.
59
RESULTS OF OPERATIONS
The following data summarizes the Company’s results of operations for the following periods indicated:
| Year Ended September 30, | ||||||||||||||||||||
| 2022 | 2021 | 2020 | ||||||||||||||||||
| (in thousands, except per share amounts) | ||||||||||||||||||||
| Revenue | $ | 243,231 | $ | 138,287 | $ | 87,992 | ||||||||||||||
| Operating loss | $ | (178,507) | $ | (149,036) | $ | (93,159) | ||||||||||||||
| Net loss | $ | (176,063) | $ | (140,848) | $ | (84,553) | ||||||||||||||
| Net loss per share-diluted | $ | (1.67) | $ | (1.36) | $ | (0.84) | ||||||||||||||
Year Ended September 30, 2022 Compared to Year Ended September 30, 2021
Revenue
Total revenue for the year ended September 30, 2022 increased to $243.2 million, 75.9% from the same period of 2021. The increase was primarily driven by the revenue recognition associated with GSK, Horizon and Takeda license agreements, as discussed below. The Company has evaluated each agreement in accordance with FASB Topic 808–Collaborative Arrangements and Topic 606-Revenue for Contracts from Customers.
GSK
At the inception of the GSK License Agreement, the Company identified one distinct performance obligation. The Company determined that the key deliverables included the license and certain R&D services, including the Company’s responsibility to complete the Phase 1/2 study (the “GSK R&D Services”). Due to the specialized and unique nature of the GSK R&D Services and their direct relationship with the license, the Company determined that these deliverables represented one distinct bundle and, thus, one performance obligation. Beyond the GSK R&D Services, which are the responsibility of the Company, GSK will be responsible for managing future clinical development and commercialization in its territory.
The Company determined the initial transaction price totaled $120.0 million, including the upfront payment, which was collected in January 2022. The Company has excluded any future estimated milestones or royalties from this transaction price to date. The Company has allocated the total $120.0 million initial transaction price to its one distinct performance obligation for the ARO-HSD license and the associated GSK R&D Services. As the Company has completed its performance obligation related to this agreement, the upfront payment of $120.0 million was fully recognized as of September 30, 2022. There were $0 in contract assets recorded as accounts receivable and $0 in contract liabilities recorded as deferred revenue as of September 30, 2022.
The Company has also performed certain development and manufacturing activities, including drug substance and drug product manufacture under GMP conditions, for GSK pursuant to the GSK License Agreement, for which the Company has been reimbursed for its costs. The Company recognized $4.8 million in connection with these efforts for the year ended September 30, 2022. There were $4.8 million of contract assets recorded as accounts receivable and $0 of contract liabilities recorded as current deferred revenue as of September 30, 2022.
Horizon
At the inception of the Horizon License Agreement, the Company identified one distinct performance obligation. The Company determined that the key deliverables included the license and certain R&D services, including the Company’s responsibilities to conduct all activities through the preclinical stages of development of ARO-XDH (the “Horizon R&D Services”). Due to the specialized and unique nature of these Horizon R&D Services and their direct relationship with the license, the Company determined that these deliverables represented one distinct bundle and, thus, one performance obligation. Beyond the Horizon R&D Services, which are the responsibility of the Company, Horizon will be responsible for managing future clinical development and commercialization of ARO-XDH.
The Company determined the initial transaction price totaled $40.0 million, including the upfront payment. The Company has excluded any future estimated milestones or royalties from this transaction price to date. The Company allocates the total $40.0 million initial transaction price to its one distinct performance obligation for the ARO-XDH license and the associated Horizon R&D Services. Revenue is recognized on a straight-line basis over the estimated timeframe for completing the Horizon R&D Services. The Company determined that the straight-line basis was appropriate as its efforts will be expended evenly over the course of completing its performance obligation. Revenue for the years ended September 30, 2022 and 2021 were $26.7 million and $6.7 million, respectively. There were $0 in contract assets
60
recorded as accounts receivable and $6.7 million in contract liabilities recorded as deferred revenue as of September 30, 2022.
In addition, the Company has performed certain development and manufacturing activities, including drug substance and drug product manufacture under GMP conditions, for Horizon pursuant to the Horizon License Agreement. The Company recognized $2.5 million and $0 in connection with these efforts for the years ended September 30, 2022 and 2021, respectively. There were $1.3 million of contract assets recorded as accounts receivable and $0 of contract liabilities recorded as current deferred revenue as of September 30, 2022.
Takeda
At the inception of the Takeda License Agreement, the Company identified one distinct performance obligation. The Company determined that the key deliverables included the license and certain R&D services including the Company’s responsibilities to complete the initial portion of the SEQUOIA study, to complete the ongoing Phase 2 AROAAT2002 study and to ensure certain manufacturing of ARO-AAT drug product is completed and delivered to Takeda (the “Takeda R&D Services”). Due to the specialized and unique nature of these Takeda R&D Services and their direct relationship with the license, the Company determined that these deliverables represent one distinct bundle and, thus, one performance obligation. Beyond the Takeda R&D Services, which are the responsibility of the Company, Takeda will be responsible for managing future clinical development and commercialization outside the United States. Within the United States, the Company will also participate in co-development and co-commercialization efforts and will co-fund these efforts with Takeda as part of the 50/50 profit sharing structure within the United States. The Company considers the collaborative activities, including the co-development and co-commercialization, to be a separate unit of account within Topic 808, and as such, these co-funding amounts are recorded as research and development expenses or general and administrative expenses, as appropriate.
The Company determined the initial transaction price totaled $300.0 million, which includes the upfront payment. The Company has excluded any future milestones or royalties from this transaction price to date. The Company has allocated the total $300.0 million initial transaction price to its one distinct performance obligation for the ARO-AAT license and the associated Takeda R&D Services. Revenue is recognized using a proportional performance method (based on actual patient visits completed versus total estimated visits completed for the ongoing SEQUOIA and AROAAT2002 clinical studies). The Company recognized $85.8 million and $90.8 million in connection with these efforts for the years ended September 30, 2022 and 2021, respectively. There were $0 of contract assets recorded as accounts receivable and $123.4 of contract liabilities recorded as deferred revenue, of which $67.4 million was classified as current deferred revenue, as of September 30, 2022. The Company also recorded $8.6 million as accrued expenses as of September 30, 2022 that was primarily driven by co-development and co-commercialization activities.
Janssen
At the inception of Janssen License Agreement and Janssen Collaboration Agreement, the Company identified one distinct performance obligation. Regarding the Janssen License Agreement, the Company determined that the key deliverables included the license and certain R&D services including the Company’s responsibility to complete the Phase 1/2 study of JNJ-3989 (ARO-HBV) and the Company’s responsibility to ensure certain manufacturing of JNJ-3989 (ARO-HBV) drug product is completed and delivered to Janssen (the “Janssen R&D Services”). Due to the specialized and unique nature of these Janssen R&D Services and their direct relationship with the license, the Company determined that these deliverables represent one distinct bundle and, thus, one performance obligation. The Company also determined that Janssen’s option to require the Company to develop up to three new targets was not a material right and, thus, not a performance obligation at the onset of the agreement. The consideration for this option is accounted for separately.
The Company determined the transaction price totaled approximately $252.7 million, which includes the upfront payment, the premium paid by JJDC for its equity investment in the Company, two $25.0 million milestone payments related to JNJ-3989 (ARO-HBV), and estimated payments for reimbursable Janssen R&D Services to be performed. The Company has allocated the total $252.7 million initial transaction price to its one distinct performance obligation for the JNJ-3989 (ARO-HBV) license and the associated Janssen R&D Services. The Company has recognized this transaction price in its entirety as of September 30, 2021, as its performance obligations were substantially completed. Future milestones and royalties achieved will be recognized in their entirety when earned. There were no contract assets and liabilities recorded as of September 30, 2022.
The Company has conducted its discovery, optimization and preclinical research and development of JNJ-75220795 (ARO-JNJ1), ARO-JNJ2, and ARO-JNJ3 under the Janssen Collaboration Agreement. All costs and labor hours spent by the Company have been entirely funded by Janssen. Janssen’s option period expired unexercised for two of the three candidates (ARO-JNJ2 and ARO-JNJ3) under the Janssen Collaboration Agreement during 2022. In May 2021, Janssen exercised its option right for JNJ-75220795 (ARO-JNJ1), which resulted in a $10.0 million milestone payment to the Company. This $10.0 million milestone payment was recognized entirely as of September 30, 2021. The Company recognized $3.4 million and $0.5 million of revenue associated with these efforts during September 30, 2022 and 2021,
61
respectively. There were $0.1 million of contract assets recorded as accounts receivable and $0 of contract liabilities recorded as current deferred revenue as of September 30, 2022.
Amgen
Under the Olpasiran Agreement and the ARO-AMG1 Agreement, the Company has received $35.0 million in upfront payments, $21.5 million in the form of an equity investment by Amgen in the Company’s common stock, and $30.0 million in milestone payments, and may receive up to an additional $400.0 million in remaining development, regulatory and sales milestone payments. The Company is further eligible to receive up to low double-digit royalties for sales of products under the Olpasiran Agreement. The Company has substantially completed its performance obligations under the Olpasiran Agreement and the ARO-AMG1 Agreement. In July 2019, Amgen informed the Company that it would not be exercising its option for an exclusive license for ARO-AMG1, and as such, there will be no further milestone or royalty payments under the ARO-AMG1 Agreement. In July 2020, Amgen initiated a Phase 2 clinical study of Olpasiran, which resulted in a $20.0 million milestone payment to the Company. There was no revenue recorded associated with the Company’s agreement with Amgen for the years ended September 30, 2022 and 2021. There were no contract assets and liabilities recorded as of September 30, 2022.
62
Operating Expenses
The analysis below details the operating expenses and discusses the expenditures of the Company within the major expense categories. For purposes of comparison, the amounts for the years ended September 30, 2022 and 2021 are shown in the tables below.
Research and Development Expenses
R&D expenses are related to the Company’s research and development discovery efforts and related candidate costs, which are comprised primarily of outsourced costs related to the manufacturing of clinical supplies, toxicity/efficacy studies and clinical trial expenses. Internal costs primarily relate to discovery operations at the Company’s research facilities in San Diego, California and Madison, Wisconsin, including facility costs and laboratory-related expenses. The Company does not separately track R&D expenses by individual research and development projects, or by individual drug candidates. The Company operates in a cross-functional manner across projects and does not separately allocate facilities-related costs, candidate costs, discovery costs, compensation expenses, depreciation and amortization expenses, and other expenses related to research and development activities.
The following table provides details of research and development expenses:
| (in thousands) | Twelve Months Ended September 30, 2022 | % of Expense Category | Twelve Months Ended September 30, 2021 | % of Expense Category | Increase (Decrease) | ||||||||||||||||||||||||||||||
| $ | % | ||||||||||||||||||||||||||||||||||
| Candidate costs | $ | 136,904 | 46 | % | $ | 92,628 | 45 | % | $ | 44,276 | 48 | % | |||||||||||||||||||||||
| R&D discovery costs | 54,346 | 18 | % | 32,734 | 16 | % | 21,612 | 66 | % | ||||||||||||||||||||||||||
| Salaries | 51,931 | 18 | % | 40,179 | 19 | % | 11,752 | 29 | % | ||||||||||||||||||||||||||
| Facilities related | 12,948 | 4 | % | 7,694 | 4 | % | 5,254 | 68 | % | ||||||||||||||||||||||||||
| Total research and development expense, excluding non-cash expense | $ | 256,129 | 86 | % | $ | 173,235 | 84 | % | $ | 82,894 | 48 | % | |||||||||||||||||||||||
| Stock compensation | 32,371 | 11 | % | 25,742 | 12 | % | 6,629 | 26 | % | ||||||||||||||||||||||||||
| Depreciation and amortization | 8,807 | 3 | % | 7,365 | 4 | % | 1,442 | 20 | % | ||||||||||||||||||||||||||
| Total research and development expense | $ | 297,307 | 100 | % | $ | 206,342 | 100 | % | $ | 90,965 | 44 | % | |||||||||||||||||||||||
Candidate costs increased $44.3 million to $136.9 million for the year ended September 30, 2022 compared to $92.6 million for the year ended September 30, 2021. This increase was primarily due to the progression of the Company’s pipeline of candidates into and through clinical trials, which resulted in higher outsourced clinical trials, toxicity study and manufacturing costs. For example, the Company’s cardiometabolic candidates, ARO-ANG3 and ARO-APOC3, have advanced into Phase 2 and Phase 3 clinical trials.
R&D discovery costs increased $21.6 million to $54.3 million for the year ended September 30, 2022 compared to $32.7 million for the year ended September 30, 2021. This increase was due to the growth of the Company’s discovery efforts and continued advancement into novel therapeutic areas and tissue types.
Salaries and stock compensation expense consist of salary, bonuses, payroll taxes, related benefits and stock compensation for the Company’s R&D personnel. The increases in salaries and stock comp expenses were primarily due to an increase in R&D headcount that has occurred as the Company has expanded its pipeline of candidates, in addition to annual increases and performance bonuses. Stock compensation expense was based upon the valuation of stock options and restricted stock units granted to employees, directors and certain consultants.
Facilities-related expense included lease costs for the Company’s research and development facilities in San Diego, California and Madison, Wisconsin. Facilities-related costs increased $5.3 million to $12.9 million for the year ended September 30, 2022, compared to $7.7 million for the year ended September 30, 2021. This increase was mainly due to the additional lease expense as the Company expands discovery efforts to identify new drug candidates.
The increase of depreciation and amortization expense, a non-cash expense, relates to depreciation on lab equipment and leasehold improvements at the facilities.
The Company anticipates these R&D expenses to continue to increase as its pipeline of candidates grows and progresses to later phase clinical trials, in addition to inflationary pressure on goods/services and the labor market.
63
General & Administrative Expenses
The following table provides details of the Company’s general and administrative expenses:
| (in thousands) | Twelve Months Ended September 30, 2022 | % of Expense Category | Twelve Months Ended September 30, 2021 | % of Expense Category | Increase (Decrease) | ||||||||||||||||||||||||||||||
| $ | % | ||||||||||||||||||||||||||||||||||
| Salaries | $ | 16,646 | 14 | % | $ | 13,681 | 17 | % | $ | 2,965 | 22 | % | |||||||||||||||||||||||
| Professional, outside services, and others | 14,738 | 12 | % | 13,124 | 17 | % | 1,614 | 12 | % | ||||||||||||||||||||||||||
| Facilities related | 2,912 | 2 | % | 2,344 | 2 | % | 568 | 24 | % | ||||||||||||||||||||||||||
| Total general & administrative expense, excluding non-cash expense | $ | 34,296 | 28 | % | $ | 29,149 | 36 | % | $ | 5,147 | 18 | % | |||||||||||||||||||||||
| Stock compensation | 88,521 | 71 | % | 50,931 | 63 | % | 37,590 | 74 | % | ||||||||||||||||||||||||||
| Depreciation/amortization | 1,614 | 1 | % | 901 | 1 | % | 713 | 79 | % | ||||||||||||||||||||||||||
| Total general & administrative expense | $ | 124,431 | 100 | % | $ | 80,981 | 100 | % | $ | 43,450 | 54 | % | |||||||||||||||||||||||
Salaries expense increased $3.0 million to $16.6 million for the year ended September 30, 2022 compared to $13.7 million for the year ended September 30, 2021. The increase was driven by the combination of annual increases, performance bonuses and increased headcount needed as the Company has grown.
Professional, outside services, and others expense includes legal, consulting, patent expenses, business insurance expenses, other outside services, travel, and communication and technology expenses. This expense increased $1.6 million to $14.7 million for the year ended September 30, 2022 compared to $13.1 million for the year ended September 30, 2021. The increase was mainly due to software implementation and additional recruiting and administrative expenses in support of additional headcount.
Facilities related expense primarily includes rental costs for the Company’s corporate headquarters in Pasadena, California. Facilities related expense increased $0.6 million to $2.9 million for the year ended September 30, 2022 compared to $2.3 million for the year ended September 30, 2021. The increase was primarily due to additional rental expense.
Stock compensation expense, a non-cash expense, increased by $37.6 million to $88.5 million for the year ended September 30, 2022 compared to $50.9 million for the year ended September 30, 2021. The increase in the current period was due to a performance award that was achieved earlier than anticipated, as well as a modification of certain performance awards to include market conditions. The fair value of market condition-based awards was expensed ratably over the service period and was not adjusted for actual achievement.
The increase in depreciation and amortization expense, a noncash expense, was primarily related to amortization of leasehold improvements for the Company’s corporate headquarters.
The Company anticipates these general & administrative expenses to continue to increase as its pipeline of candidates grows and progresses to later phase clinical trials, in addition to inflationary pressure on goods/services and the labor market.
Other Income
Other income is primarily related to interest income and realized and unrealized gain/loss on investments. Other income decreased $2.4 million to $5.8 million for the year ended September 30, 2022 compared to $8.2 million for the year ended September 30, 2021. The decrease was due to lower yields on more recently purchased bonds and a realized loss on the sale of marketable securities, offset by various credits the Company received during 2022.
Year Ended September 30, 2021 Compared to Year Ended September 30, 2020
See “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” of the Company’s Form 10-K for the year ended September 30, 2021 for a discussion of changes in its results of operations from the year ended September 30, 2020 to the year ended September 30, 2021.
64
LIQUIDITY AND CAPITAL RESOURCES
The Company has historically financed its operations through the sale of its common stock and revenue from its licensing and collaboration agreements. Research and development activities have required significant capital investment since the Company’s inception and are expected to continue to require significant cash expenditure as the Company’s pipeline continues to expand and matures into later stage clinical trials. Additionally, the Company plans to expand its facilities with its purchase of land in Verona, Wisconsin, and its entry into a new lease in San Diego, California. Each of these expansions is designed to increase the Company’s internal manufacturing and discovery capabilities, and each will require significant capital investment.
The Company’s cash and cash equivalents decreased to $108.0 million at September 30, 2022 compared to $184.4 million at September 30, 2021. Cash invested in short-term fixed income securities was $268.4 million at September 30, 2022 compared to $56.6 million at September 30, 2021. Cash invested in long-term fixed income securities was $105.9 million at September 30, 2022, compared to $245.6 million at September 30, 2021. In April 2022, the Company sold all of its marketable securities for $122.3 million. In August 2020, the Company entered into an Open Market Sale Agreement (the “ATM” agreement), pursuant to which the Company may, from time to time, sell up to $250.0 million in shares of the Company’s common stock through Jefferies LLC. As of the year ended September 30, 2022, no shares have been issued under the ATM agreement. The Company believes its current financial resources are sufficient to fund its operations through at least the next twelve months.
The following table presents a summary of cash flows:
| Year Ended September 30, | |||||||||||||||||
| 2022 | 2021 | 2020 | |||||||||||||||
| (in thousands) | |||||||||||||||||
| Cash Flow from: | |||||||||||||||||
| Operating activities | $ | (136,131) | $ | 171,312 | $ | (95,801) | |||||||||||
| Investing activities | (5,417) | (141,678) | (240,778) | ||||||||||||||
| Financing activities | 65,186 | 11,305 | 257,948 | ||||||||||||||
| Net (decrease) increase in cash and cash equivalents | $ | (76,362) | $ | 40,939 | $ | (78,631) | |||||||||||
| Cash and cash equivalents at end of period | $ | 108,005 | $ | 184,434 | $ | 143,583 | |||||||||||
During the year ended September 30, 2022, cash flow used by operating activities was $136.1 million, which was primarily due to the ongoing expenses related to the Company’s research and development programs and general and administrative expenses, partially offset by the receipt of the $120.0 million upfront payment from GSK. Cash used in investing activities was $5.4 million, which was primarily related to the purchase of property and equipment of $52.8 million, offset by net sales of investments of $47.4 million. Cash provided by financing activities of $$65.2 million was primarily related to the formation of the joint venture, Visirna, as well as cash received from stock option exercises.
On November 9, 2022, the Company and Royalty Pharma Investments 2019 ICAV (“Royalty Pharma”) entered into a Royalty Purchase Agreement (the “Royalty Pharma Agreement”), pursuant to which Royalty Pharma agreed to pay up to $410.0 million in cash to the Company in consideration for the Company’s future royalty interest in Olpasiran, a small interfering RNA (siRNA) originally developed by the Company and licensed to Amgen in 2016 under the Olpasiran Agreement. Pursuant to the Royalty Pharma Agreement, Royalty Pharma paid $250.0 million upfront on November 9, 2022. See Note 13 — Subsequent Events of Notes to Consolidated Financial Statements of Part IV, “Item 15. Exhibits and Financial Statement Schedules.”
During the year ended September 30, 2021, the Company generated $171.3 million in cash from operating activities, which was primarily related to the Takeda license agreement’s $300.0 million upfront payment, partially offset by the ongoing expenses of the Company’s research and development programs and general and administrative expenses. Cash used in investing activities was $141.7 million, which was primarily related to the purchase of investments of $240.7 million and purchase of property and equipment of $23.6 million, partially offset by maturities of fixed-income securities of $122.6 million. Cash provided by financing activities of $11.3 million was due to cash received from stock option exercises.
On December 20, 2021, the Company completed a purchase of 13 acres of land in the Verona Technology Park in Verona, Wisconsin, which is being developed into an approximately 160,000 square foot drug manufacturing facility and an approximately 140,000 square foot laboratory and office facility which will support the Company’s process development and analytical activities. The Company intends to invest between $200.0 million and $260.0 million into the build out of the facilities with cash on hand. As part of this acquisition, the Company entered into a development agreement
65
with the City of Verona to construct certain infrastructure improvements within the tax incremental district and expects to be reimbursed up to $16.0 million by the City of Verona by future tax increment revenue generated from the developed property. The total amount of funding that City of Verona is expected to pay under the Tax Incremental Financing program is not guaranteed and will depend on future tax revenues generated from the developed property. The Company also expects receive up to $2.5 million of refundable Wisconsin state income tax credits from the Wisconsin Economic Development Corporation (WEDC) as incentives to invest in the local community and create new jobs.
See “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” of the Company’s Form 10-K for the year ended September 30, 2021 for a discussion of cash flows from the year ended Sep 30, 2020.
Contractual Obligations
For information related to the Company’s future commitments for its facility-related obligations and collaboration and licensing agreements, see Notes 8 and 2, respectively, of Notes to the Company’s Consolidated Financial Statements of Part IV, “Item 15. Exhibits and Financial Statement Schedules.” Commitments related to the Company’s clinical, manufacturing and business operation related agreements are $229.6 million as of the year ended September 30, 2022, but many of these agreements are cancellable.
ITEM 7A.QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
The Company is subject to market risk exposures primarily due to its investing activities. The primary market risk exposure is change in interest rates. Adverse changes to rates may occur due to changes in the liquidity of a market or to changes in market perceptions of creditworthiness and risk tolerance.
The Company does not hold any instruments for trading purposes and investment criteria are governed by its Investment Policy. As of September 30, 2022 and 2021, the Company had cash and cash equivalents of $108.0 million and $184.4 million, respectively, and short-term and long-term investments and marketable securities of $374.3 million and $429.0 million, respectively. At times, the Company has invested its cash reserves in corporate bonds typically with maturities of less than 3 years and historically classified these investments as held-to-maturity. The Company has also invested in mutual funds that invest in marketable debt securities such as U.S. government bonds, U.S. government agency bonds, corporate bonds, and other asset backed debt securities. Due to the relatively short-term nature of the investments that the Company holds, it does not believe that the results of operations or cash flows would be affected to any significant degree by a sudden change in market interest rates relative to its investment portfolio.
ITEM 8.FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The information required by this item is included in Item 15 of this Annual Report on Form 10-K.
ITEM 9.CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A.CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
The Company maintains disclosure controls and procedures designed to ensure that information required to be disclosed in its reports filed under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in the SEC rules and forms, and that such information is accumulated and communicated to its management, including its Chief Executive Officer and Chief Financial Officer, as appropriate, to allow for timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and management necessarily was required to apply its judgment in evaluating the cost benefit relationship of possible controls and procedures.
As required by Rule 13a-15(b) of the Exchange Act, the Company carried out an evaluation, under the supervision and with the participation of its management, including its Chief Executive Officer and Chief Financial Officer, of the
66
effectiveness of the design and operation of the Company’s disclosure controls and procedures as of the end of the period covered by this Annual Report on Form 10-K. Based on the foregoing, the Company’s Chief Executive Officer and Chief Financial Officer concluded that the Company’s disclosure controls and procedures were effective at the reasonable assurance level.
Management’s Annual Report on Internal Control over Financial Reporting
The Company’s management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) of the Exchange Act. The Company’s internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of its Consolidated Financial Statements for external purposes in accordance with GAAP.
This process includes those policies and procedures that:
(i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the Company’s assets;
(ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with GAAP, and that receipts and expenditures are being made only in accordance with authorizations of the Company’s management and directors; and
(iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the Company’s financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of the internal control over financial reporting to future periods are subject to risk that controls may become inadequate because either conditions change or the degree of compliance with policies or procedures may deteriorate.
Management has assessed the effectiveness of the Company’s internal control over financial reporting as of September 30, 2022. In making this assessment, the Company used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control-Integrated Framework (2013). Based on this assessment, management concluded that the Company’s internal control over financial reporting was effective as of September 30, 2022.
Rose, Snyder and Jacobs LLP, the independent registered public accounting firm that audited the Consolidated Financial Statements included in this 2022 Annual Report on Form 10-K, has issued an audit report on the effectiveness of the Company’s internal control over financial reporting as of September 30, 2022, which is included herein.
Changes in Internal Control Over Financial Reporting
There has been no change in the Company’s internal control over financial reporting during the Company’s most recent fiscal quarter that has materially affected, or is reasonably likely to materially affect, the Company’s internal control over financial reporting. The Company’s process for evaluating controls and procedures is continuous and encompasses constant improvement of the design and effectiveness of established controls and procedures and the remediation of any deficiencies which may be identified during this process.
ITEM 9B.OTHER INFORMATION
None.
ITEM 9C.DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
PART III
ITEM 10.DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE.
The information called for by this Item will be incorporated by reference from the Company’s Definitive Proxy Statement, under the headings Proposal One — Election of Directors, Equity Compensation Plan Information, Corporate Governance, Environmental and Social Commitment, and, if applicable, Delinquent Section 16(a) Reports, to be filed for
67
the Company’s 2023 Annual Meeting of Stockholders, which proxy statement will be filed no later than January 28, 2023 (the “Definitive Proxy Statement”).
ITEM 11.EXECUTIVE COMPENSATION
The information called for by this Item will be incorporated by reference from the Definitive Proxy Statement, under the heading Executive Compensation.
ITEM 12.SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS.
The information called for by this Item will be incorporated by reference from the Definitive Proxy Statement, under the heading Voting Securities of Principal Stockholders and Management.
ITEM 13.CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE.
The information called for by this Item will be incorporated by reference from the Definitive Proxy Statement, under the headings Review and Approval of Related-Party Transactions and Certain Relationships and Related Transactions, and Director Independence.
ITEM 14.PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information called for by this Item will be incorporated by reference from the Definitive Proxy Statement, under the heading Audit Fees.
PART IV
ITEM 15.EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
The following documents are filed as part of this Annual Report on Form 10-K:
(1)Financial Statements.
See Index to Financial Statements and Schedule on page F-1.
(2)Financial Statement Schedules.
See Index to Financial Statements and Schedule on page F-1. All other schedules are omitted as the required information is not present or is not present in amounts sufficient to require submission of the schedule, or because the information required is included in the Consolidated Financial Statements or notes thereto.
(3)Exhibits.
The following exhibits are filed (or incorporated by reference herein) as part of this Annual Report on Form 10-K:
| Incorporated by Reference Herein | ||||||||||||||||||||
| Exhibit Number | Description | Form | Date | |||||||||||||||||
| 1.1 | Quarterly Report on Form 10-Q, as Exhibit 1.1 | August 5, 2020 | ||||||||||||||||||
| 2.1† | Annual Report on Form 10-K as Exhibit 2.1 | December 20, 2011 | ||||||||||||||||||
| 2.2† | Quarterly Report on Form 10-Q, as Exhibit 2.1 | May 11, 2015 | ||||||||||||||||||
68
| Incorporated by Reference Herein | ||||||||||||||||||||
| Exhibit Number | Description | Form | Date | |||||||||||||||||
| 3.1 | Current Report on Form 8-K as Exhibit 3.3 | April 6, 2016 | ||||||||||||||||||
| 3.2 | Current Report on Form 8-K as Exhibit 3.4 | April 6, 2016 | ||||||||||||||||||
| 4.1 | Current Report on Form 8-K, as Exhibit 4.1 | April 6, 2016 | ||||||||||||||||||
| 4.2 | Registration Statement on Form S-3, as Exhibit 4.2 | December 2, 2019 | ||||||||||||||||||
| 4.3 | Current Report on Form 8-K, as Exhibit 4.1 | March 23, 2017 | ||||||||||||||||||
| 4.4 | Annual Report on Form 10-K, as Exhibit 4.4 | November 25, 2019 | ||||||||||||||||||
| 4.5 | Quarterly Report on Form 10-Q, as Exhibit 10.4 | February 7, 2019 | ||||||||||||||||||
| 10.1** | Schedule 14C, as Annex B | January 12, 2012 | ||||||||||||||||||
| 10.2** | Schedule 14C, as Annex A | December 20, 2013 | ||||||||||||||||||
| 10.3** | Current Report on Form 8-K, as Exhibit 10.1 | February 12, 2014 | ||||||||||||||||||
| 10.4** | Current Report on Form 8-K, as Exhibit 10.2 | February 12, 2014 | ||||||||||||||||||
| 10.5** | Schedule 14A, Exhibit A | January 28, 2021 | ||||||||||||||||||
| 10.6** | Registration Statement on Form S-8, Exhibit 99.1 | December 22, 2021 | ||||||||||||||||||
| 10.7** | Form of RSU Agreement for Employees (Arrowhead Pharmaceuticals, Inc. 2021 Incentive Plan- Inducement Award) | Registration Statement on Form S-8, Exhibit 99.2 | December 22, 2021 | |||||||||||||||||
| 10.8** | Form of Stock Option Grant (Arrowhead Pharmaceuticals, Inc. 2021 Incentive Plan- Inducement Award) | Registration Statement on Form S-8, Exhibit 99.3 | December 22, 2021 | |||||||||||||||||
| 10.9** | Annual Report on Form 10-K, as Exhibit 10.11 | December 14, 2006 | ||||||||||||||||||
| 10.10** | Current Report on Form 8-K, as Exhibit 10.1 | June 13, 2008 | ||||||||||||||||||
| 10.11** | Annual Report on Form 10-K, as Exhibit 10.8 | December 22, 2009 | ||||||||||||||||||
| 10.12† | Annual Report on Form 10-K, as Exhibit 10.36 | December 20, 2011 | ||||||||||||||||||
69
| Incorporated by Reference Herein | ||||||||||||||||||||
| Exhibit Number | Description | Form | Date | |||||||||||||||||
| 10.13† | Annual Report on Form 10-K, as Exhibit 10.33 | December 20, 2011 | ||||||||||||||||||
| 10.14† | Quarterly Report on Form 10-Q, as Exhibit 10.1 | August 12, 2014 | ||||||||||||||||||
| 10.15† | Annual Report on Form 10-K, as Exhibit 10.18 | December 14, 2016 | ||||||||||||||||||
| 10.16† | Annual Report on Form 10-K, as Exhibit 10.19 | December 14, 2016 | ||||||||||||||||||
| 10.17 | Amendment No. 1 to the Registration Statement on Form S-3, as Exhibit 10.1) | November 25, 2016 | ||||||||||||||||||
| 10.18† | Quarterly Report on Form 10-Q, as Exhibit 10.1 | February 7, 2019 | ||||||||||||||||||
| 10.19† | Annual Report on Form 10-Q, as Exhibit 10.19 | November 25, 2019 | ||||||||||||||||||
| 10.20† | Annual Report on Form 10-K, as Exhibit 10.20 | November 25, 2019 | ||||||||||||||||||
| 10.21† | Quarterly Report on Form 10-Q, as Exhibit 10.2 | February 7, 2019 | ||||||||||||||||||
| 10.22† | Annual Report on Form 10-K, as Exhibit 10.21 | November 25, 2019 | ||||||||||||||||||
| 10.23 | Quarterly Report on Form 10-Q, as Exhibit 10.3 | February 7, 2019 | ||||||||||||||||||
| 10.24† | Quarterly Report on Form 10-Q, as Exhibit 10.1 | February 4, 2021 | ||||||||||||||||||
| 10.25 | Quarterly Report on Form 10-Q, as Exhibit 10.1 | May 10, 2022 | ||||||||||||||||||
| 10.26† | Quarterly Report on Form 10-Q, as Exhibit 10.4 | August 5, 2021 | ||||||||||||||||||
| 10.27 | Quarterly Report on Form 10-Q, as Exhibit 10.1 | February 2, 2022 | ||||||||||||||||||
70
| Incorporated by Reference Herein | ||||||||||||||||||||
| Exhibit Number | Description | Form | Date | |||||||||||||||||
| 10.28 | Quarterly Report on Form 10-Q, as Exhibit 10.1 | February 9. 2016 | ||||||||||||||||||
| 10.29 | Annual Report on Form 10-K, as Exhibit 10.23 | November 23, 2020 | ||||||||||||||||||
| 10.30 | Annual Report on Form 10-K, as Exhibit 10.24 | November 23, 2020 | ||||||||||||||||||
| 10.31 | Annual Report on Form 10-K, as Exhibit 10.25 | November 23, 2020 | ||||||||||||||||||
| 10.32 | Annual report on Form 10-K, as Exhibit 10.26 | November 23, 2020 | ||||||||||||||||||
| 10.33 | Annual report on Form 10-K, as Exhibit 10.27 | November 23, 2020 | ||||||||||||||||||
| 10.34 | Quarterly Report on Form 10-Q, as Exhibit 10.3 | February 4, 2021 | ||||||||||||||||||
| 10.35 | Quarterly Report on Form 10-Q, as Exhibit 10.4 | February 4, 2021 | ||||||||||||||||||
| 10.36 | Quarterly Report on Form 10-Q, as Exhibit 10.1 | August 5, 2019 | ||||||||||||||||||
| 10.37 | First Amendment to Office Lease by and between Arrowhead Pharmaceuticals, Inc. and 177 Colorado Owner LLC., dated October 23, 2020 | Quarterly Report on Form 10-Q, as Exhibit 10.2 | February 4, 2021 | |||||||||||||||||
| 10.38† | Quarterly Report on Form 10-Q, as Exhibit 10.1 | May 7, 2020 | ||||||||||||||||||
| 10.39 | Quarterly Report on Form 10-Q, as Exhibit 10.1 | February 2, 2022 | ||||||||||||||||||
| 21.1* | ||||||||||||||||||||
| 23.1* | ||||||||||||||||||||
| 31.1* | ||||||||||||||||||||
| 31.2* | ||||||||||||||||||||
| 32.1*** | ||||||||||||||||||||
71
| Incorporated by Reference Herein | ||||||||||||||||||||
| Exhibit Number | Description | Form | Date | |||||||||||||||||
| 32.2*** | ||||||||||||||||||||
| 101.INS* | Inline XBRL Taxonomy Extension Instance Document | |||||||||||||||||||
| 101.SCH* | Inline XBRL Taxonomy Extension Schema Document | |||||||||||||||||||
| 101.CAL* | Inline XBRL Taxonomy Extension Calculation Linkbase Document | |||||||||||||||||||
| 101.LAB* | Inline XBRL Taxonomy Extension Label Linkbase Document | |||||||||||||||||||
| 101.PRE* | Inline XBRL Taxonomy Extension Presentation Linkbase Document | |||||||||||||||||||
| 101.DEF* | Inline XBRL Taxonomy Extension Definition Linkbase Document | |||||||||||||||||||
| 104* | The cover page from the Company’s Annual Report on Form 10-K for the year ended September 30, 2022, formatted in Inline XBRL (included as Exhibit 101) | |||||||||||||||||||
* Filed herewith
** Indicates compensation plan, contract or arrangement.
*** Furnished herewith
† Certain portions of this exhibit were redacted by means of marking such portions with asterisks because the identified portions are (i) not material and (ii) treated as private or confidential by the Company.
ITEM 16.FORM 10-K SUMMARY
None.
72
SIGNATURE
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report on Form 10-K to be signed on its behalf by the undersigned, thereunto duly authorized.
Dated: November 28, 2022
| ARROWHEAD PHARMACEUTICALS, INC. | ||||||||
| By: | /s/ Christopher Anzalone | |||||||
| Christopher Anzalone | ||||||||
| Chief Executive Officer | ||||||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report on Form 10-K has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated:
| Signature | Title | Date | ||||||||||||
| /s/ Christopher Anzalone | Chief Executive Officer, President and Director (Principal Executive Officer) | November 28, 2022 | ||||||||||||
| Christopher Anzalone | ||||||||||||||
| /s/ Kenneth A. Myszkowski | Chief Financial Officer (Principal Financial and Accounting Officer) | November 28, 2022 | ||||||||||||
| Kenneth A. Myszkowski | ||||||||||||||
| /s/ Douglass Given | Director, Chairman of the Board of Directors | November 28, 2022 | ||||||||||||
| Douglass Given | ||||||||||||||
| /s/ Mauro Ferrari | Director | November 28, 2022 | ||||||||||||
| Mauro Ferrari | ||||||||||||||
| /s/ Michael S. Perry | Director | November 28, 2022 | ||||||||||||
| Michael S. Perry | ||||||||||||||
| /s/ William Waddill | Director | November 28, 2022 | ||||||||||||
| William Waddill | ||||||||||||||
| /s/ Marianne De Backer | Director | November 28, 2022 | ||||||||||||
| Marianne De Backer | ||||||||||||||
| /s/ Adeoye Olukotun | Director | November 28, 2022 | ||||||||||||
| Adeoye Olukotun | ||||||||||||||
| /s/ Victoria Vakiener | Director | November 28, 2022 | ||||||||||||
| Victoria Vakiener | ||||||||||||||
73
INDEX TO FINANCIAL STATEMENTS AND SCHEDULE
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and
Stockholders of Arrowhead Pharmaceuticals, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Arrowhead Pharmaceuticals, Inc. and Subsidiaries (the Company) as of September 30, 2022 and 2021, and the related consolidated statements of operations and comprehensive income (loss), stockholders’ equity, and cash flows for each of the years in the three-year period ended September 30, 2022, and the related notes (collectively referred to as the financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of September 30, 2022 and 2021, and the results of its operations and its cash flows for each of the years in the three-year period ended September 30, 2022, in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company’s internal control over financial reporting as of September 30, 2022, based on criteria established in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO), and our report dated November 28, 2022, expressed an unqualified opinion.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
Revenue Recognition – Revenue Recognized Over Time
Description of the Matter
As discussed in Note 1 and Note 2 to the Consolidated Financial Statements, the Company earns its revenue through license and collaboration agreements. For performance obligations related to services that are required to be recognized over time, the Company measures its progress to completion using various measures, including an input measure of total labor costs incurred divided by total labor costs expected to be incurred, time elapsed, and an output measure of total patient visits divided by total patient visits expected. The selection of measurement criteria is based on the nature and phase of trials being conducted.
F-2
Auditing revenue recognition is complex and highly judgmental due to the variability and uncertainty associated with the Company’s assessment of measure of progress. Changes in these estimates would have a significant effect on the amount of revenue recognized.
How We Addressed the Matter in Our Audit
We obtained an understanding, evaluated the design, and tested the operating effectiveness of controls that address the risk of material misstatement of license and collaboration agreement revenue including those associated with cost to complete estimates. We tested controls over management’s process to collect, review, and approve the data used in assessing revenue recognized over time.
To test the measures of progress used for performance obligations related to services that are required to be recognized over time, our audit procedures included, among others, evaluating the appropriateness of the Company’s accounting policy for each type of arrangement, testing the identified measure of performance by reading contracts with customers, including all amendments, and reviewing the contract analyses prepared by management. We evaluated whether the selected measures of progress towards satisfaction of performance obligations were applied consistently. We also tested the completeness and accuracy of the underlying data used for the measure of progress by testing and or analyzing the underlying data and conducting interviews of project personnel.
We have served as the Company’s auditor since 2004.
November 28, 2022
F-3
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and
Stockholders of Arrowhead Pharmaceuticals, Inc.
Opinion on Internal Control over Financial Reporting
We have audited Arrowhead Pharmaceuticals, Inc. and its Subsidiaries (the Company’s) internal control over financial reporting as of September 30, 2022, based on criteria established in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of September 30, 2022, based on criteria established in Internal Control—Integrated Framework (2013) issued by COSO.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated balance sheets as of September 30, 2022 and 2021 and the related consolidated statements of operations and comprehensive income (loss), stockholders’ equity, and cash flows for each of the three years in the period ended September 30, 2022 and related notes, and our report dated November 28, 2022 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Rose, Snyder & Jacobs LLP
Encino, CA
November 28, 2022
F-4
Arrowhead Pharmaceuticals, Inc.
Consolidated Balance Sheets
(in thousands, except per share amounts)
| September 30, | |||||||||||
| 2022 | 2021 | ||||||||||
| ASSETS | |||||||||||
| Current assets: | |||||||||||
| Cash, cash equivalents and restricted cash | $ | $ | |||||||||
| Accounts receivable | |||||||||||
| Short-term investments | |||||||||||
| Marketable securities | |||||||||||
| Prepaid expenses | |||||||||||
| Other current assets | |||||||||||
| Total current assets | |||||||||||
| Property and equipment, net | |||||||||||
| Intangible assets, net | |||||||||||
| Long-term investments | |||||||||||
| Right-of-use assets | |||||||||||
| Other assets | |||||||||||
| Total Assets | $ | $ | |||||||||
| LIABILITIES, NONCONTROLLING INTEREST AND STOCKHOLDERS’ EQUITY | |||||||||||
| Current liabilities: | |||||||||||
| Accounts payable | $ | $ | |||||||||
| Accrued expenses | |||||||||||
| Accrued payroll and benefits | |||||||||||
| Lease liabilities | |||||||||||
| Deferred revenue | |||||||||||
| Total current liabilities | |||||||||||
| Long-term liabilities: | |||||||||||
| Lease liabilities, net of current portion | |||||||||||
| Deferred revenue, net of current portion | |||||||||||
| Total long-term liabilities | |||||||||||
| Commitments and contingencies (Note 7) | |||||||||||
| Noncontrolling interest and stockholders' equity: | |||||||||||
Common stock, $ Authorized | |||||||||||
| Additional paid-in capital | |||||||||||
| Accumulated other comprehensive loss | ( | ( | |||||||||
| Accumulated deficit | ( | ( | |||||||||
| Total Arrowhead Pharmaceuticals, Inc. stockholders' equity | |||||||||||
| Noncontrolling interest | |||||||||||
| Total noncontrolling interest and stockholders' equity | |||||||||||
| Total Liabilities, Noncontrolling Interest and Stockholders' Equity | $ | $ | |||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-5
Arrowhead Pharmaceuticals, Inc.
Consolidated Statements of Operations and Comprehensive Loss
(in thousands, except per share amounts)
| Year Ended September 30, | |||||||||||||||||
| 2022 | 2021 | 2020 | |||||||||||||||
| Revenue | $ | $ | $ | ||||||||||||||
| Operating expenses: | |||||||||||||||||
| Research and development | |||||||||||||||||
| General and administrative | |||||||||||||||||
| Total operating expenses | |||||||||||||||||
| Operating loss | ( | ( | ( | ||||||||||||||
| Other income (expense): | |||||||||||||||||
| Interest income, net | |||||||||||||||||
| Other income (expense), net | ( | ||||||||||||||||
| Total other income | |||||||||||||||||
| Loss before income tax expense and noncontrolling interest | ( | ( | ( | ||||||||||||||
| Income tax expense | |||||||||||||||||
| Net loss including noncontrolling interest | ( | ( | ( | ||||||||||||||
| Net loss attributable to noncontrolling interest, net of tax | ( | ||||||||||||||||
| Net loss attributable to Arrowhead Pharmaceuticals, Inc. | $ | ( | $ | ( | $ | ( | |||||||||||
| Net loss per share attriutable to Arrowhead Pharmaceuticals, Inc.: | |||||||||||||||||
| Basic | $ | ( | $ | ( | $ | ( | |||||||||||
| Diluted | $ | ( | $ | ( | $ | ( | |||||||||||
| Weighted-average shares used in calculating | |||||||||||||||||
| Basic | |||||||||||||||||
| Diluted | |||||||||||||||||
| Other comprehensive loss, net of tax: | |||||||||||||||||
| Foreign currency translation adjustments | ( | ( | |||||||||||||||
| Comprehensive loss | $ | ( | $ | ( | $ | ( | |||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-6
Arrowhead Pharmaceuticals, Inc.
Consolidated Statements of Stockholders’ Equity
(in thousands)
| Common Stock | Amount ($) | Additional Paid-In Capital | Accumulated Other Comprehensive Income (Loss) | Accumulated Deficit | Non- controlling Interest | Totals | |||||||||||||||||||||||||||||||||||
| Balance at September 30, 2019 | $ | $ | $ | ( | $ | ( | $ | ( | $ | ||||||||||||||||||||||||||||||||
| Stock-based compensation | — | — | — | — | — | ||||||||||||||||||||||||||||||||||||
| Exercise of stock options | — | — | — | ||||||||||||||||||||||||||||||||||||||
| Common stock - restricted stock units vesting | ( | — | — | — | |||||||||||||||||||||||||||||||||||||
| Common stock - issued for cash | — | — | — | ||||||||||||||||||||||||||||||||||||||
| Foreign currency translation adjustments | — | — | — | — | — | ||||||||||||||||||||||||||||||||||||
| Deconsolidation of Ablaris Therapeutics, Inc. | — | — | — | — | — | ||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | ( | — | ( | ||||||||||||||||||||||||||||||||||
| Balance at September 30, 2020 | $ | $ | $ | $ | ( | $ | $ | ||||||||||||||||||||||||||||||||||
| Common Stock | Amount ($) | Additional Paid-In Capital | Accumulated Other Comprehensive Income (Loss) | Accumulated Deficit | Non- controlling Interest | Totals | |||||||||||||||||||||||||||||||||||
| Balance at September 30, 2020 | $ | $ | $ | $ | ( | $ | $ | ||||||||||||||||||||||||||||||||||
| Stock-based compensation | — | — | — | — | — | ||||||||||||||||||||||||||||||||||||
| Exercise of stock options | — | — | — | ||||||||||||||||||||||||||||||||||||||
| Common stock - restricted stock units vesting | ( | — | — | — | |||||||||||||||||||||||||||||||||||||
| Foreign currency translation adjustments | — | — | — | ( | — | — | ( | ||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | ( | — | ( | ||||||||||||||||||||||||||||||||||
| Balance at September 30, 2021 | $ | $ | $ | ( | $ | ( | $ | $ | |||||||||||||||||||||||||||||||||
| Common Stock | Amount ($) | Additional Paid-In Capital | Accumulated Other Comprehensive Income (Loss) | Accumulated Deficit | Non- controlling Interest | Totals | |||||||||||||||||||||||||||||||||||
| Balance at September 30, 2021 | $ | $ | $ | ( | $ | ( | $ | $ | |||||||||||||||||||||||||||||||||
| Stock-based compensation | — | — | — | — | — | ||||||||||||||||||||||||||||||||||||
| Exercise of stock options | — | — | — | — | |||||||||||||||||||||||||||||||||||||
| Common stock - restricted stock units vesting | ( | — | — | — | |||||||||||||||||||||||||||||||||||||
| Foreign currency translation adjustments | — | — | — | ( | — | — | ( | ||||||||||||||||||||||||||||||||||
| Interest in joint venture | — | — | — | ||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | ( | ( | ( | ||||||||||||||||||||||||||||||||||
| Balance at September 30, 2022 | $ | $ | $ | ( | $ | ( | $ | $ | |||||||||||||||||||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-7
Arrowhead Pharmaceuticals, Inc.
Consolidated Statements of Cash Flows
(in thousands)
| Year Ended September 30, | |||||||||||||||||
| 2022 | 2021 | 2020 | |||||||||||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | |||||||||||||||||
| Net loss | $ | ( | $ | ( | $ | ( | |||||||||||
| Adjustments to reconcile net loss to net cash flow from operating activities: | |||||||||||||||||
| Stock-based compensation | |||||||||||||||||
| Net loss (gain) from investments | ( | ( | |||||||||||||||
| Depreciation and amortization | |||||||||||||||||
| Amortization of note premiums | |||||||||||||||||
| Changes in operating assets and liabilities: | |||||||||||||||||
| Accounts receivable | ( | ( | |||||||||||||||
| Prepaid expenses and other current assets | ( | ( | ( | ||||||||||||||
| Accounts payable | ( | ( | |||||||||||||||
| Accrued expenses | |||||||||||||||||
| Deferred revenue | ( | ( | |||||||||||||||
| Operating lease liabilities | |||||||||||||||||
| Other | ( | ||||||||||||||||
| Net cash (used in) provided by operating activities | ( | ( | |||||||||||||||
| CASH FLOWS FROM INVESTING ACTIVITIES: | |||||||||||||||||
| Purchases of property and equipment | ( | ( | ( | ||||||||||||||
| Purchases of investments | ( | ( | ( | ||||||||||||||
| Proceeds from sale of investments | |||||||||||||||||
| Net cash used in investing activities | ( | ( | ( | ||||||||||||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | |||||||||||||||||
| Proceeds from the exercises of stock options | |||||||||||||||||
| Proceeds from the issuance of common stock | |||||||||||||||||
| Proceeds from investment in joint venture | |||||||||||||||||
| Net cash provided by financing activities | |||||||||||||||||
| NET (DECREASE) INCREASE IN CASH AND CASH EQUIVALENTS | ( | ( | |||||||||||||||
| EFFECT OF EXCHANGE RATE ON CASH AND CASH EQUIVALENTS | ( | ( | |||||||||||||||
| CASH AND CASH EQUIVALENTS AT BEGINNING OF PERIOD | |||||||||||||||||
| CASH AND CASH EQUIVALENTS AT END OF PERIOD | $ | $ | $ | ||||||||||||||
| Supplementary disclosures: | |||||||||||||||||
| Interest paid | $ | $ | $ | ||||||||||||||
| Income Taxes (Paid) Refunded | $ | ( | $ | ( | $ | ||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-8
Arrowhead Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
NOTE 1. ORGANIZATION AND SIGNIFICANT ACCOUNTING POLICIES
General
Arrowhead Pharmaceuticals, Inc. and its subsidiaries (referred to herein collectively as the “Company”) are primarily engaged in developing medicines that treat intractable diseases by silencing the genes that cause them. Using a broad portfolio of RNA chemistries and efficient modes of delivery, the Company’s therapies trigger the RNA interference mechanism to induce rapid, deep and durable knockdown of target genes. RNA interference (“RNAi”) is a mechanism present in living cells that inhibits the expression of a specific gene, thereby affecting the production of a specific protein. The Company’s RNAi-based therapeutics may leverage this natural pathway of gene silencing to target and shut down specific disease-causing genes.
The following table presents the Company’s current pipeline:
| Therapeutic Area | Name | Stage | Product Rights | ||||||||
| Cardiometabolic | ARO-APOC3 | Two Phase 2b and one Phase 3 | Arrowhead | ||||||||
| ARO-ANG3 | Two Phase 2b | Arrowhead | |||||||||
| Olpasiran | Phase 3 | Amgen | |||||||||
| Pulmonary | ARO-ENAC2 | Pre-Clinical | Arrowhead | ||||||||
| ARO-RAGE | Phase 1/2 | Arrowhead | |||||||||
| ARO-MUC5AC | Phase 1/2a | Arrowhead | |||||||||
| ARO-MMP7 | Phase 1/2 | Arrowhead | |||||||||
| Liver | ARO-HSD | Phase 1/2 | GSK | ||||||||
| ARO-AAT | Phase 2 | Takeda and Arrowhead | |||||||||
| JNJ-3989 | Phase 2 | Janssen | |||||||||
| ARO-XDH | Phase 1 | Horizon | |||||||||
| ARO-C3 | Phase 1/2 | Arrowhead | |||||||||
| JNJ-75220795 | Phase 1 | Janssen | |||||||||
| Muscle | ARO-DUX4 | Pre-Clinical | Arrowhead | ||||||||
The Company operates lab facilities in San Diego, California and Madison, Wisconsin, where its research and development activities, including the development of RNAi therapeutics, take place. The Company’s principal executive offices are located in Pasadena, California.
Consolidation and Basis of Presentation
The Consolidated Financial Statements include the accounts of Arrowhead Pharmaceuticals, Inc. and its subsidiaries (wholly-owned subsidiaries and a variable interest entity that the Company is the primary beneficiary in). Subsidiaries refer to Arrowhead Madison, Inc., Visirna Therapeutics, Inc. (“Visirna”), and Arrowhead Australia Pty Ltd. For subsidiaries in which the Company owns or is exposed to less than 100% of the economics, the Company records net loss attributable to noncontrolling interests in its consolidated statements of operations equal to the percentage of the economic or ownership interests retained in such entity by the respective noncontrolling party.
The Consolidated Financial Statements have been prepared in conformity with U.S. generally accepted accounting principles (“GAAP”). All intercompany transactions and balances have been eliminated. Certain prior period amounts have been reclassified to conform with the current period presentation.
Liquidity
The Company’s primary sources of financing have been through the sale of its securities and revenue from its licensing and collaboration agreements. Research and development activities have required significant capital investment since the Company’s inception and are expected to continue to require significant cash expenditure in the future, particularly as the Company’s pipeline of drug candidates and its headcount have both expanded significantly. Additionally, significant capital investment will be required as the Company’s pipeline matures into later stage clinical trials and as the Company plans to increase its internal manufacturing capabilities.
F-9
At September 30, 2022, the Company had $108.0 million in cash and cash equivalents (including $7.3 million in restricted cash), $268.4 million in short-term investments and $105.9 million in long-term investments to fund operations. During the year ended September 30, 2022, the Company’s cash and cash equivalents and investments balance decreased by $131.1 million which was primarily cash being used to fund the Company’s operations, offset by the $120.0 million upfront payment received from Glaxosmithkline Intellectual Property Limited (Note 2) and $60.0 million cash infusion from the formation of Visirna (Note 2).
In total, the Company is eligible to receive up to $4.9 billion in developmental, regulatory and sales milestones, and may receive various royalties on net sales from its licensing and collaboration agreements, subject to the terms and conditions of those agreements. The revenue recognition for these collaboration agreements is discussed further in Note 2.
Summary of Significant Accounting Policies
Variable Interest Entity (“VIE”)
A VIE is an entity that, by design, either (i) lacks sufficient equity to permit the entity to finance its activities without additional subordinated financial support from other parties; or (ii) has equity investors that do not have the ability to make significant decisions relating to the entity’s operations through voting rights, or do not have the obligation to absorb the expected losses, or do not have the right to receive the residual returns of the entity. The primary beneficiary of a VIE is required to consolidate the assets and liabilities of the VIE. The primary beneficiary is the party that has both (i) the power to direct the activities of the VIE that most significantly impact the VIE’s economic performance, and (ii) the obligation to absorb losses or the right to receive benefits from the VIE that could potentially be significant to the VIE through its interest in the VIE.
On April 25, 2022, the Company entered into a license agreement with Visirna (Note 2) and consolidated Visirna’s financial statements in which the Company has a direct controlling financial interest based on the VIE model.
The Company considers all the facts and circumstances, including its role in establishing Visirna and its ongoing rights and responsibilities to assess whether the Company has the power to direct the activities of Visirna. In general, the parties that make the most significant decisions affecting a VIE and have the right to unilaterally remove those decision-makers are deemed to have the power to direct the activities of a VIE.
The Company also considers all of its economic interests to assess whether the Company has the obligation to absorb losses of Visirna or the right to receive benefits from it that could potentially be significant to Visirna. This assessment requires the Company to apply judgment in determining whether these interests, in the aggregate, are considered potentially significant to Visirna. Factors considered in assessing the significance include: the design of the Visirna, including its capitalization structure, subordination of interests, payment priority, and the reasons why the interests are held by the Company.
Cash, Cash Equivalents and Restricted Cash
All highly liquid interest-bearing investments with short-term are classified as cash equivalents. These investments mainly include commercial paper with maturities of three months or less when purchased. The carrying value of these cash equivalents approximate fair value.
There were $7.3 million and $2.4 million restricted cash at September 30, 2022 and September 30, 2021, respectively, that are primarily held as collateral associated with letters of credit for the Company’s facility leases. The increase in 2022 was mainly due to the Company’s expansion plan in Verona, Wisconsin and San Diego, California.
F-10
Investments
Investment securities are mainly held-to-maturity investments and marketable securities.
These held-to-maturity investments may consist of investment-grade interest bearing instruments, primarily certificates of deposit, money market accounts, government-sponsored enterprise securities, corporate bonds and/or commercial paper, which are stated at amortized cost. The Company does not intend to sell these investment securities and the contractual maturities are not greater than 36 months. Those with maturities less than twelve months are included in short-term investments on the Company’s consolidated balance sheets, while those with remaining maturities in excess of twelve months are included in long-term investments on its consolidated balance sheets. Discounts and premiums to par value of the debt securities are amortized to interest income/expense over the term of the security, and no gains or losses on held-to-maturity investment are realized until they are sold.
The Company’s marketable debt securities consisted of mutual funds that primarily invest in U.S. government bonds, U.S. government agency bonds, and corporate bonds. Dividends from these funds were automatically re-invested. These securities were recorded at fair value, and all unrealized gains/losses were recorded in the Company’s consolidated statement of operations and comprehensive loss. In April 2022, the Company sold all marketable debt securities for $122.3 million.
Property and Equipment
Property and equipment are recorded at cost, which may equal fair market value in the case of property and equipment acquired in conjunction with a business acquisition. Depreciation of property and equipment is recorded using the straight-line method over the respective useful lives of the assets ranging from to seven years . Leasehold improvements are amortized over the lesser of the expected useful life or the remaining lease term.
The Company periodically assesses long-lived assets or asset groups, including property and equipment, for recoverability when events or changes in circumstances indicate that their carrying amounts may not be recoverable. If the Company identifies an indicator of impairment, the Company assesses recoverability by comparing the carrying amount of the asset to the sum of the undiscounted cash flows expected to result from the use and the eventual disposal of the asset. An impairment loss is recognized when the carrying amount is not recoverable and is measured as the excess of carrying value over fair value. There were no impairment charges during 2022, 2021, and 2020.
Intangible Assets Subject to Amortization
Intangible assets subject to amortization include certain patents and license agreements. The Company qualitatively evaluates intangible assets for impairment annually or whenever events or changes in circumstances indicate that it is more likely than not that the carrying amount of intangible assets may exceed their implied fair values. As of September 30, 2022 and 2021, intangible impairment assessments indicated that there was no
Leases
The Company determines whether a contract is, or contains, a lease at inception. All of the Company’s leases are classified as operating leases. Leases with terms greater than one-year are recognized on the Company’s consolidated balance sheets as right-of-use assets that represent the Company’s right to use an underlying asset for the lease term, and lease liabilities that represent its obligation to make lease payments arising from the lease. Lease assets and liabilities are
F-11
Revenue Recognition
On October 1, 2018, the Company adopted Financial Accounting Standards Board (“FASB”) Topic 606 – Revenue for Contracts from Customers which amended revenue recognition principles and provides a single, comprehensive set of criteria for revenue recognition within and across all industries. The Company’s adoption of the revenue standard did not have a material impact on its Consolidated Financial Statements. The Company has not yet achieved commercial sales of its drug candidates to date, however, the new standard is applicable to its ongoing licensing and collaboration agreements. See Note 2.
The revenue standard provides a five-step framework for recognizing revenue as control of promised goods or services is transferred to a customer at an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. To determine revenue recognition for arrangements that it determines are within the scope of the revenue standard, the Company performs the following five steps: (i) identify the contract; (ii) identify the performance obligations; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) the Company satisfies a performance obligation. At contract inception, the Company assesses whether the goods or services promised within each contract are distinct and, therefore, represent a separate performance obligation, or whether they are not distinct and are combined with other goods and services until a distinct bundle is identified. The Company then determines the transaction price, which typically includes upfront payments and any variable consideration that the Company determines is probable to not cause a significant reversal in the amount of cumulative revenue recognized when the uncertainty associated with the variable consideration is resolved. The Company then allocates the transaction price to each performance obligation and recognizes the associated revenue when (or as) each performance obligation is satisfied.
The Company recognizes the transaction price allocated to upfront license payments as revenue upon delivery of the license to the customer and resulting ability of the customer to use and benefit from the license, if the license is determined to be distinct from the other performance obligations identified in the contract. These other performance obligations are typically to perform research and development services for the customer, often times relating to the candidate that the customer is licensing. If the license is not considered to be distinct from other performance obligations, the Company assesses the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied at a point in time or over time. If the performance obligation is satisfied over time, the Company then determines
F-12
the appropriate method of measuring progress for purposes of recognizing revenue from license payments. The Company evaluates the measure of progress each reporting period and, if necessary, adjusts the related revenue recognition.
Typically, the Company’s collaboration agreements entitle it to additional payments upon the achievement of milestones or royalties on sales. The milestones are generally categorized into three types: development milestones, generally based on the initiation of toxicity studies or clinical trials; regulatory milestones, generally based on the submission, filing or approval of regulatory applications such as a Clinical Trial Application (“CTA”) or a New Drug Application (“NDA”) in the United States; and sales-based milestones, generally based on meeting specific thresholds of sales in certain geographic areas. The Company evaluates whether it is probable that the consideration associated with each milestone or royalty will not be subject to a significant reversal in the cumulative amount of revenue recognized. Amounts that meet this threshold are included in the transaction price using the most likely amount method, whereas amounts that do not meet this threshold are excluded from the transaction price until they meet this threshold. At the end of each subsequent reporting period, the Company re-evaluates the probability of a significant reversal of the cumulative revenue recognized for its milestones and royalties, and, if necessary, adjusts its estimate of the overall transaction price. Any such adjustments are recorded on a cumulative catch-up basis, which would affect revenues and net income in the Company’s consolidated statements of operation and comprehensive loss. Typically, milestone payments and royalties are achieved after the Company’s performance obligations associated with the collaboration agreements have been completed and after the customer has assumed responsibility for the respective clinical or preclinical program. Milestones or royalties achieved after the Company’s performance obligations have been completed are recognized as revenue in the period the milestone or royalty was achieved. If a milestone payment is achieved during the performance period, the milestone payment would be recognized as revenue to the extent performance had been completed at that point, and the remaining balance would be recorded as deferred revenue.
The revenue standard requires the Company to assess whether a significant financing component exists in determining the transaction price. The Company performs this assessment at the onset of its licensing or collaboration agreements. Typically, a significant financing component does not exist because the customer is paying for a license or services in advance with an upfront payment. Additionally, future royalty payments are not substantially within the control of the Company or the customer.
The revenue standard requires the Company to allocate the arrangement consideration on a relative standalone selling price basis for each performance obligation after determining the transaction price of the contract and identifying the performance obligations to which that amount should be allocated. The relative standalone selling price is defined in the revenue standard as the price at which an entity would sell a promised good or service separately to a customer. If other observable transactions in which the Company has sold the same performance obligation separately are not available, the Company estimates the standalone selling price of each performance obligation. Key assumptions to determine the standalone selling price may include forecasted revenues, development timelines, reimbursement rates for personnel costs, discount rates and probabilities of technical and regulatory success.
Whenever the Company determines that goods or services promised in a contract should be accounted for as a combined performance obligation over time, the Company determines the period over which the performance obligations will be performed and revenue will be recognized. Revenue is recognized using either the proportional performance method or on a straight-line basis if efforts will be expended evenly over time. Labor hours, costs incurred or patient visits in clinical trials are typically used as the measure of performance. Significant management judgment is required in determining the level of effort required under an arrangement and the period over which the Company is expected to complete its performance obligations. If the Company determines that the performance obligation is satisfied over time, any upfront payment received is initially recorded as deferred revenue on its consolidated balance sheets.
Certain judgments affect the application of the Company’s revenue recognition policy. For example, the Company records short-term (less than one year) and long-term (over one year) deferred revenue based on its best estimate of when such revenue will be recognized. This estimate is based on the Company’s current operating plan and, the Company may recognize a different amount of deferred revenue over the next 12-month period if its plan changes in the future.
Collaborative Arrangements
The Company analyzes its collaborative arrangements to assess whether such arrangements involve joint operating activities performed by parties that are both active participants in the activities and exposed to significant risks and rewards, and therefore are within the scope of FASB Topic 808 - Collaborative Arrangements. For collaborative arrangements that contain multiple elements, the Company determines which units of account are deemed to be within the scope of Topic 808 and which units of account are more reflective of a vendor-customer relationship, and therefore are within the scope of Topic 606. For units of account that are accounted for pursuant to Topic 808, an appropriate recognition method is determined and applied consistently, either by analogy to appropriate accounting literature or by applying a reasonable accounting policy election. For collaborative arrangements that are within the scope of Topic 808,
F-13
Research and Development
Costs and expenses that can be clearly identified as research and development are charged to expense as incurred. Included in research and development costs are operating costs, facilities, supplies, external services, clinical trial and manufacturing costs, overhead directly related to the Company’s research and development operations, and costs to acquire technology licenses.
Earnings per Share
Basic earnings per share is computed using the weighted-average number of common shares outstanding during the period. Diluted earnings per share is computed using the weighted-average number of common shares and dilutive potential common shares outstanding during the period. Dilutive potential common shares primarily consist of stock options and restricted stock units issued to employees.
Income Taxes
Deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to differences between the financial reporting basis and the respective tax basis of the Company’s assets and liabilities, and expected benefits of utilizing net operating loss, capital loss, and tax-credit carryforwards. The Company assesses the likelihood that its deferred tax assets will be realized and, to the extent management does not believe these assets are more likely than not to be realized, a valuation allowance is established. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates or laws is recognized in earnings in the period that includes the enactment date.
Recent Accounting Pronouncements
In December 2019, the FASB issued Accounting Standards Update 2019-12, Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes, which eliminates certain exceptions related to the incremental approach for intra-period allocation, deferred tax recognition requirement for changes in equity method investments and foreign subsidiaries, and methodology for calculating income taxes in an interim period. The guidance also simplifies certain aspects of the accounting for franchise taxes, the accounting for step-up in the tax basis of goodwill, and accounting for change in tax laws or rates. The Company adopted the new standard which became effective for fiscal years and interim periods within those years that begin after December 15, 2020. The adoption of the new standard did not have any material impact on the Company’s Consolidated Financial Statements.
NOTE 2. COLLABORATION AND LICENSE AGREEMENTS
F-14
On November 22, 2021, GSK and the Company entered into an Exclusive License Agreement (the “GSK License Agreement”). Under the GSK License Agreement, GSK has received an exclusive license for ARO-HSD. The exclusive license is worldwide with the exception of greater China, for which the Company retained rights to develop and commercialize ARO-HSD. The Company has completed its Phase 1/2 study of ARO-HSD, and GSK is now wholly responsible for all clinical development and commercialization of ARO-HSD in its territory. Under the terms of the agreement, the Company has received an upfront payment of $120.0 million and is eligible for additional payments of $30.0 million at the start of Phase 2 and $100.0 million upon achieving a successful Phase 2 trial readout and the first patient dosed in a Phase 3 trial. Furthermore, should the Phase 3 trial readout positively, and the potential new medicine receives regulatory approval in major markets, the deal provides for commercial milestone payments to the Company of up to $190.0 million at first commercial sale, and up to $590.0 million in sales-related milestone payments. The Company is further eligible to receive tiered royalties on net product sales in a range of mid-teens to twenty percent.
At the inception of the GSK License Agreement, the Company identified one distinct performance obligation. The Company determined that the key deliverables included the license and certain R&D services, including the Company’s responsibility to complete the Phase 1/2 study, (the “GSK R&D Services”). Due to the specialized and unique nature of the GSK R&D Services and their direct relationship with the license, the Company determined that these deliverables represented one distinct bundle and, thus, one performance obligation. Beyond the GSK R&D Services, which are the responsibility of the Company, GSK will be responsible for managing future clinical development and commercialization in its territory.
The Company determined the initial transaction price totaled $120.0 million, including the upfront payment, which was collected in January 2022. The Company has excluded any future estimated milestones or royalties from this transaction price to date. The Company has allocated the total $120.0 million initial transaction price to its one distinct performance obligation for the ARO-HSD license and the associated GSK R&D Services. As the Company has completed its performance obligation related to this agreement, the upfront payment of $120.0 million was fully recognized as of September 30, 2022. There were $0 in contract assets recorded as accounts receivable and $0 in contract liabilities recorded as deferred revenue as of September 30, 2022.
The Company has also performed certain development and manufacturing activities, including drug substance and drug product manufacture under GMP conditions, for GSK pursuant to the GSK License Agreement, for which the Company has been reimbursed for its costs. The Company recognized $4.8 million in connection with these efforts for the year ended September 30, 2022. There were $4.8 million of contract assets recorded as accounts receivable and $0 of contract liabilities recorded as current deferred revenue as of September 30, 2022.
Horizon Therapeutics Ireland DAC (“Horizon”)
On June 18, 2021, Horizon and the Company entered into a collaboration and license agreement (the “Horizon License Agreement”). Under the terms of the Horizon License Agreement, Horizon received a worldwide exclusive license for ARO-XDH, a previously undisclosed discovery-stage investigational RNAi therapeutic being developed by the Company as a potential treatment for people with uncontrolled gout. The Company conducted all activities through the preclinical stages of development of ARO-XDH, and Horizon is now wholly responsible for clinical development and commercialization of ARO-XDH. In July 2021, the Company received $40.0 million as an upfront payment and is eligible to receive up to $660.0 million in potential development, regulatory and sales milestones. The Company is also eligible to receive royalties in the low- to mid-teens range on net product sales.
At the inception of the Horizon License Agreement, the Company identified one distinct performance obligation. The Company determined that the key deliverables included the license and certain R&D services, including the Company’s responsibilities to conduct all activities through the preclinical stages of development of ARO-XDH (the “Horizon R&D Services”). Due to the specialized and unique nature of these Horizon R&D Services and their direct relationship with the license, the Company determined that these deliverables represented one distinct bundle and, thus, one performance obligation. Beyond the Horizon R&D Services, which are the responsibility of the Company, Horizon will be responsible for managing future clinical development and commercialization of ARO-XDH.
The Company determined the initial transaction price totaled $40.0 million, including the upfront payment. The Company has excluded any future estimated milestones or royalties from this transaction price to date. The Company allocates the total $40.0 million initial transaction price to its one distinct performance obligation for the ARO-XDH license and the associated Horizon R&D Services. Revenue is recognized on a straight-line basis over the estimated timeframe for completing the Horizon R&D Services. The Company determined that the straight-line basis was appropriate as its efforts will be expended evenly over the course of completing its performance obligation. Revenue for the years ended September 30, 2022 and 2021 were $26.7 million and $6.7 million, respectively. There were $0 in contract assets recorded as accounts receivable and $6.7 million in contract liabilities recorded as deferred revenue as of September 30, 2022.
F-15
In addition, the Company has performed certain development and manufacturing activities, including drug substance and drug product manufacture under GMP conditions, for Horizon pursuant to the Horizon License Agreement. The Company recognized $2.5 million and $0 in connection with these efforts for the years ended September 30, 2022 and 2021, respectively. There were $1.3 million of contract assets recorded as accounts receivable and $0 of contract liabilities recorded as current deferred revenue as of September 30, 2022.
Takeda Pharmaceutical Company Limited (“Takeda”)
On October 7, 2020, Takeda and the Company entered into an Exclusive License and Co-Funding Agreement (the “Takeda License Agreement”). Under the Takeda License Agreement, Takeda and the Company will co-develop its ARO-AAT program, the Company’s second-generation subcutaneously administered RNAi therapeutic candidate being developed as a treatment for liver disease associated with alpha-1 antitrypsin deficiency. Within the United States, ARO-AAT, if approved, will be co-commercialized under a 50/50 profit sharing structure. Outside the United States, Takeda will lead the global commercialization strategy and will receive an exclusive license to commercialize ARO-AAT, while the Company will be eligible to receive tiered royalties of 20 % to 25 % on net sales. In January 2021, the Company received $300.0 million as an upfront payment and is eligible to receive potential development, regulatory and commercial milestones of up to $595.0 million.
At the inception of the Takeda License Agreement, the Company identified one distinct performance obligation. The Company determined that the key deliverables included the license and certain R&D services including the Company’s responsibilities to complete the initial portion of the SEQUOIA study, to complete the ongoing Phase 2 AROAAT2002 study and to ensure certain manufacturing of ARO-AAT drug product is completed and delivered to Takeda (the “Takeda R&D Services”). Due to the specialized and unique nature of these Takeda R&D Services and their direct relationship with the license, the Company determined that these deliverables represent one distinct bundle and, thus, one performance obligation. Beyond the Takeda R&D Services, which are the responsibility of the Company, Takeda will be responsible for managing future clinical development and commercialization outside the United States. Within the United States, the Company will also participate in co-development and co-commercialization efforts and will co-fund these efforts with Takeda as part of the 50/50 profit sharing structure within the United States. The Company considers the collaborative activities, including the co-development and co-commercialization, to be a separate unit of account within Topic 808, and as such, these co-funding amounts are recorded as research and development expenses or general and administrative expenses, as appropriate.
The Company determined the initial transaction price totaled $300.0 million, which includes the upfront payment. The Company has excluded any future milestones or royalties from this transaction price to date. The Company has allocated the total $300.0 million initial transaction price to its one distinct performance obligation for the ARO-AAT license and the associated Takeda R&D Services. Revenue is recognized using a proportional performance method (based on actual patient visits completed versus total estimated visits completed for the ongoing SEQUOIA and AROAAT2002 clinical studies). The Company recognized $85.8 million and $90.8 million in connection with these efforts for the years ended September 30, 2022 and 2021, respectively. There were $0 of contract assets recorded as accounts receivable and $123.4 million of contract liabilities recorded as deferred revenue, of which $67.4 million was classified as current deferred revenue, as of September 30, 2022. The Company also recorded $8.6 million as accrued expenses that was primarily driven by co-development and co-commercialization activities.
Janssen Pharmaceuticals, Inc. (“Janssen”)
On October 3, 2018, Janssen, part of the Janssen Pharmaceutical Companies of Johnson & Johnson, and the Company entered into a License Agreement (the “Janssen License Agreement”) and a Research Collaboration and Option Agreement (the “Janssen Collaboration Agreement”). The Company also entered into a stock purchase agreement with JJDC, Inc. (“JJDC”), Johnson & Johnson's venture capital arm (“JJDC Stock Purchase Agreement”). Under the Janssen License Agreement, Janssen has received a worldwide, exclusive license to the Company’s JNJ-3989 (ARO-HBV) program, the Company’s third-generation subcutaneously administered RNAi therapeutic candidate being developed as a potential therapy for patients with chronic hepatitis B virus infection. Beyond the Company’s Phase 1/2 study of JNJ-3989 (ARO-HBV), which the Company was responsible for completing, Janssen is wholly responsible for clinical development and commercialization of JNJ-3989 (ARO-HBV). Under the Janssen Collaboration Agreement, Janssen was able to select three new targets against which the Company would develop clinical candidates. These candidates were subject to certain restrictions and did not include candidates that already were in the Company’s pipeline. The Company was obligated to perform discovery, optimization and preclinical research and development, entirely funded by Janssen, which on its own or in combination with Janssen development work, would have been sufficient to allow the filing of a U.S. Investigational New Drug Application (“IND”) or equivalent, at which time Janssen would have the option to take an exclusive license. If the option was exercised, Janssen would have been wholly responsible for clinical development and commercialization of each optioned candidate. Under the terms of the agreements taken together, the Company has received $175.0 million as an
F-16
upfront payment, $75.0 million in the form of an equity investment by JJDC in the Company’s common stock under the JJDC Stock Purchase Agreement, and milestone and option payments totaling $73.0 million, and the Company may receive up to $1.6 billion in development and sales milestone payments for the Janssen License Agreement, and up to $0.6 billion in development and sales milestone payments for the remaining target covered under the Janssen Collaboration Agreement. The Company is further eligible to receive tiered royalties on product sales up to mid-teens under the Janssen License Agreement and up to low teens under the Janssen Collaboration Agreement. During 2022, Janssen’s option period expired unexercised for two of the three candidates (ARO-JNJ2 and ARO-JNJ3) under the Janssen Collaboration Agreement.
At the inception of Janssen License Agreement and Janssen Collaboration Agreement, the Company identified one distinct performance obligation. Regarding the Janssen License Agreement, the Company determined that the key deliverables included the license and certain R&D services including the Company’s responsibility to complete the Phase 1/2 study of JNJ-3989 (ARO-HBV) and the Company’s responsibility to ensure certain manufacturing of JNJ-3989 (ARO-HBV) drug product is completed and delivered to Janssen (the “Janssen R&D Services”). Due to the specialized and unique nature of these Janssen R&D Services and their direct relationship with the license, the Company determined that these deliverables represent one distinct bundle and, thus, one performance obligation. The Company also determined that Janssen’s option to require the Company to develop up to three new targets is not a material right and, thus, not a performance obligation at the onset of the agreement. The consideration for this option is accounted for separately.
The Company determined the transaction price totaled approximately $252.7 million, which includes the upfront payment, the premium paid by JJDC for its equity investment in the Company, two $25.0 million milestone payments related to JNJ-3989 (ARO-HBV), and estimated payments for reimbursable Janssen R&D Services to be performed. The Company has allocated the total $252.7 million initial transaction price to its one distinct performance obligation for the JNJ-3989 (ARO-HBV) license and the associated Janssen R&D Services. The Company has recognized this transaction price in its entirety as of September 30, 2021, as its performance obligations were substantially completed. Future milestones and royalties achieved will be recognized in their entirety when earned. There were no
The Company has conducted its discovery, optimization and preclinical research and development of JNJ-75220795 (ARO-JNJ1), ARO-JNJ2, and ARO-JNJ3 under the Janssen Collaboration Agreement. All costs and labor hours spent by the Company have been entirely funded by Janssen. Janssen’s option period expired unexercised for two of the three candidates (ARO-JNJ2 and ARO-JNJ3) under the Janssen Collaboration Agreement during 2022. In May 2021, Janssen exercised its option right for JNJ-75220795 (ARO-JNJ1), which resulted in a $10.0 million milestone payment to the Company. This $10.0 million milestone payment was recognized entirely as of September 30, 2021. The Company recognized $3.4 million and $0.5 million of revenue associated with these efforts during September 30, 2022 and 2021, respectively. There were $0.1 million of contract assets recorded as accounts receivable and $0 of contract liabilities recorded as current deferred revenue as of September 30, 2022.
Amgen Inc. (“Amgen”)
On September 28, 2016, Amgen and the Company entered into two collaboration and license agreements and a common stock purchase agreement. Under the Second Collaboration and License Agreement (the “Olpasiran Agreement”), Amgen has received a worldwide, exclusive license to the Company’s novel RNAi Olpasiran (previously referred to as AMG 890 or ARO-LPA) program. These RNAi molecules are designed to reduce elevated lipoprotein(a), which is a genetically validated, independent risk factor for atherosclerotic cardiovascular disease. Under the prior collaboration and license agreement (the “First Collaboration and License Agreement” or the “ARO-AMG1 Agreement”), Amgen received an option to a worldwide, exclusive license to ARO-AMG1, an RNAi therapy for an undisclosed genetically validated cardiovascular target. Under both agreements, Amgen is wholly responsible for clinical development and commercialization.
Under the Olpasiran Agreement and the ARO-AMG1 Agreement, the Company has received $35.0 million in upfront payments, $21.5 million in the form of an equity investment by Amgen in the Company’s common stock, and $30.0 million in milestone payments, and may receive up to an additional $400.0 million in remaining development, regulatory and sales milestone payments. The Company is further eligible to receive up to low double-digit royalties for sales of products under the Olpasiran Agreement. The Company has substantially completed its performance obligations under the Olpasiran Agreement and the ARO-AMG1 Agreement. In July 2019, Amgen informed the Company that it would not be exercising its option for an exclusive license for ARO-AMG1, and as such, there will be no further milestone or royalty payments under the ARO-AMG1 Agreement. In July 2020, Amgen initiated a Phase 2 clinical study of Olpasiran, which resulted in a $20.0 million milestone payment to the Company. There were no no
F-17
Joint Venture and License Agreement with Visirna Therapeutics, Inc. (“Visirna”)
On April 25, 2022, the Company entered into a License Agreement with Visirna (the “Visirna License Agreement”), pursuant to which Visirna received an exclusive license to develop, manufacture and commercialize four of the Company’s RNAi-based investigational cardiometabolic medicines in Greater China (including the People’s Republic of China, Hong Kong, Macau and Taiwan). Pursuant to a Share Purchase Agreement entered into simultaneously with the Visirna License Agreement (the “Visirna SPA”), the Company acquired a majority stake in Visirna (after accounting for shares reserved for Visirna’s employee stock ownership plan) as partial consideration for the Visirna License Agreement. Under the Visirna SPA, entities affiliated with Vivo Capital also acquired a minority stake in Visirna in exchange for $60.0 million in upfront capital to support the operations of Visirna. As further consideration under the Visirna License Agreement, the Company is also eligible to receive potential royalties on commercial sales.
NOTE 3. PROPERTY AND EQUIPMENT
The following table summarizes the Company’s major classes of property and equipment:
| September 30, | |||||||||||
| 2022 | 2021 | ||||||||||
| (in thousands) | |||||||||||
| Computers, software, office equipment and furniture | $ | $ | |||||||||
| Land | |||||||||||
| Research equipment | |||||||||||
| Leasehold improvements | |||||||||||
| Construction in progress | |||||||||||
| Less: Accumulated depreciation and amortization | ( | ( | |||||||||
| Property and equipment, net | $ | $ | |||||||||
Depreciation and amortization expense for property and equipment for the years ended September 30, 2022, 2021, and 2020 was $8.7 million, $6.6 million and $4.2 million respectively.
The increase in the construction in progress during 2022 was mainly due to the developments of manufacturing, laboratory and office facilities in Verona, Wisconsin as well as a new laboratory and office facility in San Diego, California. See Note 7.
F-18
NOTE 4. INVESTMENTS
The Company’s investments consisted of the following:
| As of September 30, 2022 | |||||||||||||||||||||||
| (In thousands) | |||||||||||||||||||||||
| Adjusted Basis | Gross Unrealized Gains | Gross Unrealized Losses | Fair Value | ||||||||||||||||||||
| Short-term investments (due within one year) | |||||||||||||||||||||||
| Held to maturity debt securities | $ | $ | $ | ( | $ | ||||||||||||||||||
| Held to maturity certifiate of deposit | |||||||||||||||||||||||
| Total short-term investments | $ | $ | $ | ( | $ | ||||||||||||||||||
| Long-term investments (Due within one through three years) | |||||||||||||||||||||||
| Held to maturity debt securities | ( | ||||||||||||||||||||||
| Total long-term investments | $ | $ | $ | ( | $ | ||||||||||||||||||
| Marketable debt securities | $ | $ | $ | $ | |||||||||||||||||||
| As of September 30, 2021 | |||||||||||||||||||||||
| (In thousands) | |||||||||||||||||||||||
| Adjusted Basis | Gross Unrealized Gains | Gross Unrealized Losses | Fair Value | ||||||||||||||||||||
| Short-term investments (due within one year) | |||||||||||||||||||||||
| Held to maturity debt securities | $ | $ | $ | $ | |||||||||||||||||||
| Total short-term investments | $ | $ | $ | $ | |||||||||||||||||||
| Long-term investments (Due within one through three years) | |||||||||||||||||||||||
| Held to maturity debt securities | $ | $ | $ | ( | $ | ||||||||||||||||||
| Held to maturity certificate of deposit | |||||||||||||||||||||||
| Total long-term investments | $ | $ | $ | ( | $ | ||||||||||||||||||
| Marketable debt securities | $ | $ | $ | ( | $ | ||||||||||||||||||
F-19
NOTE 5. INTANGIBLE ASSETS
Intangible assets subject to amortization include patents and a license agreement capitalized as part of the Novartis RNAi asset acquisition in March 2015. The following table presents the components of intangible asset:
| Gross Carrying Amount | Accumulated Amortization | Impairment | Net Carrying Amount | Useful Lives | |||||||||||||||||||||||||
| (amounts in thousands) | (in years) | ||||||||||||||||||||||||||||
| As of September 30, 2022 | |||||||||||||||||||||||||||||
| Patents | $ | $ | $ | $ | |||||||||||||||||||||||||
| License | |||||||||||||||||||||||||||||
| Total intangible assets, net | $ | $ | $ | $ | |||||||||||||||||||||||||
| As of September 30, 2021 | |||||||||||||||||||||||||||||
| Patents | $ | $ | $ | $ | |||||||||||||||||||||||||
| License | |||||||||||||||||||||||||||||
| Total intangible assets, net | $ | $ | $ | $ | |||||||||||||||||||||||||
Intangible assets are reviewed annually for impairment and more frequently if potential impairment indicators exist. No impairment indicators were identified during 2022 and 2021.
Intangible assets with definite useful lives are amortized on a straight-line basis over their useful lives. Intangible assets amortization expense in each of 2022, 2021, and 2020 was $1.7
The following table presents the estimated future amortization expense related to intangible assets as of September 30, 2022:
| Amortization Expense | |||||||||||
| Year Ending September 30, | (in thousands) | ||||||||||
| 2023 | $ | ||||||||||
| 2024 | |||||||||||
| 2025 | |||||||||||
| 2026 | |||||||||||
| 2027 | |||||||||||
| Thereafter | |||||||||||
| Total | $ | ||||||||||
NOTE 6. STOCKHOLDERS’ EQUITY
F-20
| Shares | ||||||||||||||
| Par Value | Authorized | Issued | Outstanding | |||||||||||
| (in thousands) | ||||||||||||||
| As of September 30, 2022 | ||||||||||||||
| Common stock | $ | |||||||||||||
| Preferred stock | $ | |||||||||||||
| As of September 30, 2021 | ||||||||||||||
| Common stock | $ | |||||||||||||
| Preferred stock | $ | |||||||||||||
NOTE 7. COMMITMENTS AND CONTINGENCIES
Litigation
From time to time, the Company may be subject to various claims and legal proceedings in the ordinary course of business. If the potential loss from any claim, asserted or unasserted, or legal proceeding is considered probable and the amount is reasonably estimable, the Company will accrue a liability for the estimated loss. There were no contingent liabilities recorded as of the year ended September 30, 2022.
Commitments
On December 20, 2021, the Company completed a purchase of 13 acres of land in the Verona Technology Park in Verona, Wisconsin, which is being developed into an approximately 160,000 square foot drug manufacturing facility and an approximately 140,000 square foot laboratory and office facility which will support the Company’s process development and analytical activities. The Company intends to invest between $200.0 million and $260.0 million into the build out of the facilities. As part of this acquisition, the Company entered into a development agreement with the City of Verona to construct certain infrastructure improvements within the tax incremental district and will be reimbursed up to $16.0 million by the City of Verona by future tax increment revenue generated from the developed property. The total amount of funding that City of Verona will pay under the Tax Incremental Financing program is not guaranteed and will depend on future tax revenues generated from the developed property. The Company will also receive up to $2.5 million of refundable Wisconsin state income tax credits from the Wisconsin Economic Development Corporation (WEDC) as incentives to invest in the local community and create new jobs.
Technology License Commitments
F-21
NOTE 8. LEASES
On November 19, 2021, the Company entered into a new 15 -year lease for approximately 144,000 square feet of office and research and development laboratory space under construction in San Diego, California. This facility will replace the Company’s current office and research facility sublease located in San Diego, California. The increased capacity of this new facility compared to the Company’s current research facility in San Diego will accommodate increased personnel for its expanding pipeline of current and future drug candidates. The estimated rent commencement date for the new lease is in April 2023 after construction and leasehold improvements have been completed. The lease payments, which begin on the rent commencement date, will be approximately $119.0 million over the initial 15 -year term. The Company also estimates payments for operating expenses to be approximately $3.0 million for the first year of the lease, and these payments will continue throughout the initial 15 -year term. The Company expects to pay approximately $31.0 million for leasehold improvements, net of tenant improvement allowances. Pursuant to the lease, within twelve months of the expiration of the initial 15 -year term, the Company has the option to extend the lease for up to one additional ten-year term, with certain annual increases in base rent.
Other Significant Leases
Pasadena, California: The Company leases office space located at 177 Colorado Blvd for its corporate headquarters from 177 Colorado Owner, LLC. The lease began on September 30, 2019 and expires on April 30, 2027. The lease contains an option to renew for one term of five years . On October 23, 2020, the Company entered into a lease expansion to add an additional approximately 24,000 square feet of office with a lease expiration date of April 30, 2027.
San Diego, California: The Company subleases space from Halozyme, Inc. for additional research and development facility in San Diego, California. The term of this sublease commenced on April 1, 2020 and will end on January 14, 2023.
Madison, Wisconsin: The Company leases space for office and laboratory facilities, which had an expiration date of September 30, 2026. The lease was amended in January 2019 and May 2020 to expand the rentable square feet by an additional 40,000 square feet and to extend the lease expiration date to September 30, 2031. The lease contains two options to renew for two terms of five years . In November 2020 and December 2020, the Company entered into amendments to expand the rentable square space by an additional 10,743 square feet for the remainder of the term.
The components of lease assets and liabilities along with their classification on the Company’s consolidated balance sheets were as follows:
| September 30, | |||||||||||||||||
| Lease Assets and Liabilities | Classification | 2022 | 2021 | ||||||||||||||
| (in thousands) | |||||||||||||||||
| Operating lease assets | Right-of-use assets | $ | $ | ||||||||||||||
| Current operating lease liabilities | Lease liabilities | ||||||||||||||||
| Non-current operating lease liabilities | Lease liabilities, net of current portion | ||||||||||||||||
The components of lease cost along with its classification on the Company’s consolidated statements of operations were as follows:
| Year Ended September 30, | |||||||||||||||||||||||
| Lease Cost | Classification | 2022 | 2021 | 2020 | |||||||||||||||||||
| (in thousands) | |||||||||||||||||||||||
| Operating lease cost | Research and development | $ | $ | $ | |||||||||||||||||||
| General and administrative expense | |||||||||||||||||||||||
| Variable lease cost | Research and development | ||||||||||||||||||||||
| General and administrative expense | |||||||||||||||||||||||
| Total | $ | $ | $ | ||||||||||||||||||||
Variable lease cost primarily related to operating expenses associated with the Company’s operating leases. There was $0.3 million and $0 short-term lease cost during the years ended September 30, 2022, and 2021, respectively.
F-22
The following table presents maturities of operating lease liabilities on an undiscounted basis as of September 30, 2022:
| Year | Amounts | ||||
| (in thousands) | |||||
| 2023 | $ | ||||
| 2024 | |||||
| 2025 | |||||
| 2026 | |||||
| 2027 | |||||
| 2028 and thereafter | |||||
| Total | $ | ||||
| Less imputed interest | ( | ||||
| Total operating lease liabilities | $ | ||||
Supplemental cash flow and other information related to leases was as follows:
| Year Ended September 30, | ||||||||||||||
| 2022 | 2021 | |||||||||||||
| Cash paid for amounts included in the measurement of lease liabilities: | ||||||||||||||
| Operating cash flows from operating leases (in thousands) | $ | $ | ||||||||||||
| Weighted-average remaining lease term (in years) | ||||||||||||||
| Weighted-average discount rate | % | % | ||||||||||||
NOTE 9. STOCK-BASED COMPENSATION
The Company has three plans that provide for equity-based compensation. Under the 2004 Equity Incentive Plan (the “2004 Plan”) and 2013 Incentive Plan (the “2013 Plan”), 175,083 and 4,072,137 shares, respectively, of the Company’s common stock are reserved for the grant of stock options, stock appreciation rights, restricted stock awards and performance unit/share awards to employees, consultants and others as of September 30, 2022.
On March 18, 2021, the Company’s Board of Directors approved the Arrowhead Pharmaceuticals, Inc. 2021 Incentive Plan (the “2021 Plan”), which authorizes 8,000,000 shares (subject to certain adjustments) to be awarded for grants of stock options, stock appreciation rights, restricted and unrestricted stock and stock units, performance awards, cash awards and other awards convertible into or otherwise based on shares of the Company’s common stock. The maximum number of shares authorized under the 2021 Plan will be (i) reduced by any shares subject to awards made under the 2013 Plan after January 1, 2021, and (ii) increased by any shares subject to outstanding awards under the 2013 Plan as of January 1, 2021 that, after January 1, 2021, are canceled, expired, forfeited or otherwise not issued under such awards (other than as a result of being tendered or withheld to pay the exercise price or withholding taxes in connection with any such awards) or settled in cash. As of September 30, 2022, the total number of shares reserved for issuance under the 2021 Incentive Plan was 7,190,077 shares, which includes 131,897 shares that were forfeited under the 2013 Plan.
In addition, there were 778,425 shares reserved for options and 838,625 shares reserved for restricted stock units issued as inducement grants to new employees granted outside of the Company’s equity-based compensation plans under Rule 5635(c)(4) of the Nasdaq Listing Rules.
The following table presents a summary of awards outstanding:
| As of September 30, 2022 | |||||||||||||||||||||||
| 2004 Plan | 2013 Plan | 2021 Plan | Total | ||||||||||||||||||||
| Granted and outstanding awards: | |||||||||||||||||||||||
| Options | |||||||||||||||||||||||
| Restricted stock units | |||||||||||||||||||||||
| Total | |||||||||||||||||||||||
F-23
Stock Option Awards
The following table presents a summary of the stock option activity for the year ended September 30, 2022:
| Shares | Weighted- Average Exercise Price Per Share | Weighted- Average Remaining Contractual Term (Years) | Aggregate Intrinsic Value | ||||||||||||||||||||
| Outstanding at September 30, 2021 | $ | ||||||||||||||||||||||
| Granted | |||||||||||||||||||||||
| Cancelled or expired | ( | ||||||||||||||||||||||
| Exercised | ( | ||||||||||||||||||||||
| Outstanding at September 30, 2022 | $ | $ | |||||||||||||||||||||
| Exercisable at September 30, 2022 | $ | $ | |||||||||||||||||||||
The aggregate intrinsic values in the table above represent the total pre-tax intrinsic value (the difference between the Company’s closing stock price and the stock option exercise price) that would have been received by the stock option holders had all stock options been exercised on September 30, 2022. The total intrinsic value of the options exercised during the years ended September 30, 2022, 2021, and 2020 was $27.6 million, $66.9 million and $44.1 million, respectively.
Stock-based compensation expense related to stock options for the years ended September 30, 2022, 2021, and 2020 was $10.8 million, $12.4 million and $9.7 million, respectively.
As of September 30, 2022, the pre-tax compensation expense for all outstanding unvested stock options in the amount of $12.0 million will be recognized in the Company’s results of operations over a weighted average period of 1.5 years.
The fair value of each stock option award is estimated on the date of grant using the Black-Scholes option pricing model. The Black-Scholes option valuation model was developed for use in estimating the fair value of traded options, which do not have vesting restrictions and are fully transferable. The determination of the fair value of each stock option is affected by the Company’s stock price on the date of grant, as well as assumptions regarding a number of highly complex and subjective variables. Because the Company’s employee stock options have characteristics significantly different from those of traded options, and because changes in the subjective input assumptions can materially affect the fair value estimate, the existing models do not necessarily provide a reliable single measure of the fair value of its employee stock options.
The following table provides the assumptions used in the calculation of grant-date fair values of these stock options based on the Black-Scholes option pricing model:
| Year Ended September 30, | |||||||||||||||||
2022(5) | 2021 | 2020 | |||||||||||||||
Expected dividend yield(1) | |||||||||||||||||
Risk-free interest rate(2) | N/A | ||||||||||||||||
Expected volatility(3) | N/A | ||||||||||||||||
Expected term (in years)(4) | N/A | ||||||||||||||||
| Weighted-average grant date fair value per share | N/A | $ | $ | ||||||||||||||
(1) The dividend yield is zero as the Company currently does not pay a dividend.
(2) The risk-free interest rate is based on that of the U.S. Treasury yields with equivalent terms in effect at the time of the grant..
(3) Volatility is estimated based on volatility average of the Company’s common stock price.
(4) The expected term represents the period of time that stock options granted are expected to be outstanding, by using historical exercise patterns and post-vesting termination behavior.
(5) No options were granted during the year ended September 30, 2022.
Restricted Stock Units
Restricted stock units (“RSUs”), including market-based, time-based and performance-based awards, have been granted under the Company’s 2013 and 2021 Plans and as inducements grants granted outside of the Company’s equity-based compensation plans. At vesting, each outstanding RSU will be exchanged for one share of the Company’s common
F-24
stock. RSU awards generally vest subject to the satisfaction of service requirements or the satisfaction of both service requirements and achievement of certain performance targets.
The following table summarizes the activity of the Company’s RSUs:
| Number of RSUs | Weighted- Average Grant Date Fair Value | ||||||||||
| Outstanding as of September 30, 2021 | $ | ||||||||||
| Granted | |||||||||||
| Vested | ( | ||||||||||
| Forfeited | ( | ||||||||||
| Outstanding as of September 30, 2022 | $ | ||||||||||
The fair value of RSUs was determined based on the closing price of the Company’s common stock on the grant date, with consideration given to the probability of achieving service and/or performance conditions for awards.
On July 8, 2022, the Company revised the equity award made to its Chief Executive Officer on January 1, 2022 consisting of 800,000 shares, equal in value of $38.4 million, that was 100% market-based awards. The revised awards consist of 99,521 RSUs and 149,282 performance-based RSUs. No incremental expense resulted from the modification. The fair values of these awards were estimated on the date of grant using a closed-form valuation model (Monte-Carlo).
NOTE 10. FAIR VALUE MEASUREMENTS
The Company employs a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value. The fair value of a financial instrument is the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date using the exit price. Accordingly, when market observable data are not readily available, the Company’s own assumptions are used to reflect those that market participants would be presumed to use in pricing the asset or liability at the measurement date.
Assets and liabilities recorded at fair value on the consolidated balance sheets are categorized based on the level of judgment associated with inputs used to measure their fair values and the level of market price observability, as follows:
Level 1 Unadjusted quoted prices are available in active markets for identical assets or liabilities as of the reporting date.
Level 2 Pricing inputs are other than quoted prices in active markets, which are based on the following:
• Quoted prices for similar assets or liabilities in active markets;
• Quoted prices for identical or similar assets or liabilities in non-active markets; or
• Either directly or indirectly observable inputs as of the reporting date.
Level 3 Pricing inputs are unobservable and significant to the overall fair value measurement, and the determination of fair value requires significant management judgment or estimation.
In certain cases, inputs used to measure fair value may fall into different levels of the fair value hierarchy. In such cases, the level in the fair value hierarchy within which the fair value measurement in its entirety falls has been determined based on the lowest level input that is significant to the fair value measurement in its entirety. Thus, a Level 3 fair value measurement may include inputs that are observable (Level 1 or Level 2) and unobservable (Level 3). The Company’s assessment of the significance of a particular input to the fair value measurement in its entirety requires judgment and consideration of factors specific to the asset or liability.
The Company uses prices and inputs that are current as of the measurement date, including during periods of market disruption. In periods of market disruption, the ability to observe prices and inputs may be reduced for many instruments. This condition could cause an instrument to be reclassified from Level 1 to Level 2, or from Level 2 to Level 3. The Company recognizes transfers between levels at either the actual date of the event or a change in circumstances that caused
F-25
the transfer. At September 30, 2022 and 2021, the Company did not have any financial assets or financial liabilities based on Level 3 measurements
| September 30, 2022 | |||||||||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | ||||||||||||||||||||
| (in thousands) | |||||||||||||||||||||||
| U.S. government bonds | $ | $ | $ | $ | |||||||||||||||||||
| Commercial notes | |||||||||||||||||||||||
| Corporate debt securities | |||||||||||||||||||||||
| Certificate of deposits | |||||||||||||||||||||||
| Money market instruments | |||||||||||||||||||||||
| September 30, 2021 | |||||||||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | ||||||||||||||||||||
| (in thousands) | |||||||||||||||||||||||
| Corporate debt securities | $ | $ | $ | $ | |||||||||||||||||||
| Certificate of deposits | |||||||||||||||||||||||
| Money market instruments | |||||||||||||||||||||||
| Marketable debt securities | |||||||||||||||||||||||
There were no transfers between Levels 1, 2, and 3 of the fair value hierarchy during the years ended September 30, 2022 and 2021.
The carrying amounts of cash and cash equivalents, accounts receivable, accounts payable, and accrued expenses of the Company approximate fair value based on the short maturities of these instruments. At September 30, 2022, the Company did not have any nonrecurring fair value measurements of nonfinancial assets or nonfinancial liabilities.
NOTE 11. INCOME TAXES
Income Tax Provision
F-26
| September 30, | |||||||||||
| 2022 | 2021 | ||||||||||
| (in thousands) | |||||||||||
| Federal: | |||||||||||
| Current | $ | $ | |||||||||
| Deferred | |||||||||||
| State: | |||||||||||
| Current | |||||||||||
| Deferred | |||||||||||
| Foreign: | |||||||||||
| Current | |||||||||||
| Deferred | |||||||||||
| Total: | |||||||||||
| Current | |||||||||||
| Deferred | |||||||||||
| Income tax provision | $ | $ | |||||||||
The following table presents a reconciliation of the tax expense based on the statutory rate to the Company’s actual tax expense in the consolidated statements of operations:
| September 30, | |||||||||||||||||
| 2022 | 2021 | 2020 | |||||||||||||||
| At U.S. federal statutory rate | - | % | - | % | - | % | |||||||||||
| State taxes, net of federal effect | - | % | - | % | - | % | |||||||||||
| Stock compensation | % | - | % | - | % | ||||||||||||
| Valuation allowance | % | % | % | ||||||||||||||
| Other | % | % | - | % | |||||||||||||
| Effective income tax rate | % | % | % | ||||||||||||||
F-27
Deferred Income Taxes
The following table presents the significant components of the Company’s net deferred tax assets and liabilities:
| September 30, | |||||||||||
| 2022 | 2021 | ||||||||||
| (in thousands) | |||||||||||
| Deferred tax assets: | |||||||||||
| Accrued Compensation | $ | $ | |||||||||
| Stock Compensation | |||||||||||
| Capitalized Research & Development | |||||||||||
| California Alternative Minimum Tax | |||||||||||
| Net Operating Losses | |||||||||||
| Intangible Assets | |||||||||||
| Deferred Revenue | |||||||||||
| Right of Use Assets/Lease Liabilities | |||||||||||
| Capital Loss | |||||||||||
| Total gross deferred tax assets | $ | $ | |||||||||
| Valuation allowance | $ | ( | $ | ( | |||||||
| Deferred tax liabilities: | |||||||||||
| Fixed Assets | $ | ( | $ | ( | |||||||
| State taxes | ( | ( | |||||||||
| Total gross deferred tax liability | $ | ( | $ | ( | |||||||
| Net deferred tax assets (liabilities) | $ | $ | |||||||||
The Company has concluded, in accordance with the applicable accounting standards, that it is more-likely-than not that the Company may not realize the benefit of all of its deferred tax assets. Accordingly, management has provided a 100 % valuation allowance against its deferred tax assets until such time as management believes that its projections of future profits as well as expected future tax rates make the realization of these deferred tax assets more-likely-than-not.
Significant judgment is required in the evaluation of deferred tax benefits and differences in future results from the Company’s estimates could result in material differences in the realization of these assets. The Company has performed an assessment of positive and negative evidence regarding the realization of the net deferred tax asset. This assessment included the evaluation of scheduled reversals of deferred tax liabilities, the availability of carry forwards and estimates of projected future taxable income.
As of September 30, 2022, the Company had available gross federal net operating loss (“NOL”) carry forwards of $504.8 million and gross state NOL carry forwards of $626.5 million. The NOLs expire at various dates through 2042.
Uncertainty in Income Taxes
The Company has adopted guidance issued by the FASB that clarifies the accounting for uncertainty in income taxes recognized in an enterprise’s financial statements and prescribes a recognition threshold of more-likely-than not and a measurement process for financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. In making this assessment, a company must determine whether it is more-likely-than not that a tax position will be sustained upon examination, based solely on the technical merits of the position and must assume that the tax position will be examined by taxing authorities.
F-28
| Year Ended September 30, | |||||||||||||||||
| 2022 | 2021 | 2020 | |||||||||||||||
| (in thousands) | |||||||||||||||||
| Beginning balance of unrecognized tax benefits | $ | $ | $ | ||||||||||||||
| Increase for prior period tax positions | |||||||||||||||||
| Ending balance of unrecognized tax benefits | $ | $ | $ | ||||||||||||||
Included in the balance of unrecognized tax benefits at September 30, 2022, 2021 and 2020 were $3.5 million, $0 and $0 respectively, that if the Company recognized, would affect its effective tax rate.
The Company recognizes interest accrued related to unrecognized tax benefits and penalties as income tax expense. During the years ended September 30, 2022 and 2021, the Company recognized $1.4 0
The Company does not foresee any material changes to its gross unrecognized tax benefits within the next twelve months.
The Company and its subsidiaries file income tax returns with the Internal Revenue Service, the state of California and certain other taxing jurisdictions. The Company is subject to income tax examinations by the Internal Revenue Service and by state tax authorities until the net operating losses are settled. The Company is under examination by the state of California for the years 2018 and 2019.
NOTE 12. EMPLOYEE BENEFIT PLANS
The Company sponsors a defined contribution retirement plan which is under Section 401(k) of the Internal Revenue Code and is designed to adhere to ERISA Fiduciary standards. Substantially all of the Company’s employees are eligible to participate this plan. Under the terms of the plan, an eligible employee may elect to contribute a portion of their salary on a pre-tax basis, subject to federal statutory limitations. The plan allows for a discretionary match in an amount up to 100 % of each participant’s first 3 % of compensation contributed plus 50 % of each participant’s next 2 % of compensation contributed.
For the years ended September 30, 2022, 2021, and 2020, the Company recorded expenses for the matching contributions under this plan of $1.7 million, $1.3 million and $0.9 million, respectively.
The Company also provides certain employee benefit plans, including those which provide health and life insurance benefits to employees.
NOTE 13. SUBSEQUENT EVENTS
On November 9, 2022, the Company and Royalty Pharma Investments 2019 ICAV (“Royalty Pharma”) entered into a Royalty Purchase Agreement (the “Royalty Pharma Agreement”), pursuant to which Royalty Pharma agreed to pay up to $410.0 million in cash to the Company in consideration for the Company’s future royalty interest in Olpasiran, a small interfering RNA (siRNA) originally developed by the Company and licensed to Amgen in 2016 under the Olpasiran Agreement.
Pursuant to the Royalty Pharma Agreement, Royalty Pharma paid $250.0 million upfront and agreed to pay up to an additional $160.0 million in aggregate one-time milestone payments due if and when the following milestone events occur: (i) $50.0 million on completion of enrollment in the planned OCEAN Phase 3 clinical trial for Olpasiran, (ii) $50.0 million upon receipt of FDA approval of Olpasiran for an approved indication (reduction in the risk of myocardial infarction, urgent coronary revascularization, or coronary heart disease death in adults with established cardiovascular disease and elevated Lp(a)), and (iii) $60.0 million upon Royalty Pharma’s receipt of at least $70.0 million of royalty payments under the Royalty Pharma Agreement in any single calendar year.
In consideration for the payment of the foregoing amounts under the Royalty Pharma Agreement, Royalty Pharma is entitled to receive all royalties otherwise payable by Amgen to the Company under the Olpasiran Agreement. The Company remains eligible to receive any milestone payments potentially payable by Amgen under the Olpasiran Agreement.
The Royalty Pharma Agreement contains other customary terms and conditions, including representations and warranties, covenants, and indemnification obligations in favor of each party. The above description of the Royalty Pharma Agreement is a summary of the material terms, does not purport to be complete and is qualified in its entirety by reference
F-29
F-30
ATTACHMENTS / EXHIBITS
XBRL TAXONOMY EXTENSION SCHEMA DOCUMENT
XBRL TAXONOMY EXTENSION CALCULATION LINKBASE DOCUMENT
XBRL TAXONOMY EXTENSION DEFINITION LINKBASE DOCUMENT
XBRL TAXONOMY EXTENSION LABEL LINKBASE DOCUMENT
Serious News for Serious Traders! Try StreetInsider.com Premium Free!
You May Also Be Interested In
- Arrowhead Pharmaceuticals Inc. (ARWR) Initiates Phase 1/2a Study of ARO-CFB
- Agents Win Again! Epique Realty Amazes with Incredible Last Benefit Reveal at PowerCON: Delta Dental and Vision
- Australian Oilseeds, Largest APAC Producer of Non-Chemical, Non-GMO “Cold-Processing” Vegetable Oil, Enters into Contract with Woolworths and Costco Australia.
Create E-mail Alert Related Categories
SEC FilingsSign up for StreetInsider Free!
Receive full access to all new and archived articles, unlimited portfolio tracking, e-mail alerts, custom newswires and RSS feeds - and more!
