

Form 6-K BioLineRx Ltd. For: Mar 05
 Tweet
Tweet Share
ShareSECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 6-K
REPORT OF FOREIGN PRIVATE ISSUER
PURSUANT TO RULE 13a-16 OR 15d-16 OF
THE SECURITIES EXCHANGE ACT OF 1934
For the month of March 2015
BioLineRx Ltd.
(Translation of registrant’s name into English)
P.O. Box 45158
19 Hartum Street
Jerusalem 9777518, Israel
(Address of Principal Executive Offices)
Indicate by check mark whether the registrant files or will file annual reports under cover of Form 20-F or Form 40-F:
Form 20-F x Form 40-F o
Indicate by check mark whether the registrant by furnishing the information contained in this form is also thereby furnishing the information to the Commission pursuant to Rule 12g3-2(b) under the Securities Exchange Act of 1934:
Yes o No x
Item 7.01. Regulation FD Disclosure
A copy of the Management Presentation of BioLineRx Ltd. (the “Company”) is furnished as Exhibit 99.1 to this Item 7.01.
The information contained in Item 7.01 of this report and in Exhibit 99.1 shall not be deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), or incorporated by reference in any filing under the Securities Act of 1933, as amended (the “Securities Act”) or the Exchange Act, except as shall be expressly set forth by specific reference in such a filing.
Item 8.01 Other Events.
On March 5, 2015, the Company issued a press release announcing that it has commenced an underwritten offering of American Depositary Shares (“ADSs”), each representing ten of its ordinary shares (“Ordinary Shares”), par value NIS 0.01 per share, pursuant to a prospectus supplement, dated March 5, 2014, to the Company’s prospectus dated August 14, 2012, filed as part of its effective shelf registration statement on Form F-3 (File No. 333-182997) previously filed with, and declared effective by, the Securities and Exchange Commission (SEC).
The Company expects to grant the underwriters an option to purchase up to an additional 15 percent of its ADSs, exercisable for 30 days after the pricing date of the ADSs offering. The offering is subject to market conditions, and there can be no assurance as to whether or when the offering may be completed, or as to the actual size or terms of the offering. A copy of the press release is furnished as Exhibit 99.2.
This Current Report on Form 6-K shall not constitute an offer to sell or the solicitation of an offer to buy nor shall there be any sale of ADSs in any state in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of any such state.
In connection with this offering, the Company has updated certain business and risk factor information. This information and risk factor disclosure is attached as Exhibit 99.3 to this Current Report on Form 6-K and is incorporated herein by reference.
Item 9.01 Financial Statements and Exhibits.
|
(d)
|
Exhibits
|
The following Exhibits are filed as part of this report:
|
Exhibit No.
|
Description of Exhibit
|
|
99.1
|
Company Presentation
|
|
99.2
|
Press Release dated March 5, 2015
|
|
99.3
|
Business and risk factor disclosure
|
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
BioLineRx Ltd.
|
|||
|
By:
|
/s/ Philip Serlin | ||
| Philip Serlin | |||
| Chief Financial and Operating Officer | |||
Dated: March 5, 2015
Exhibit 99.1

Company Presentation
February 2015

|
|
This presentation contains "forward-looking statements." These statements include words like "may," "expects," "believes," “plans,” “scheduled," and "intends," and describe opinions about future events. These forward-looking statements involve known and unknown risks and uncertainties that may cause the actual results, performance or achievements of BioLineRx to be materially different from any future results, performance or achievements expressed or implied by such forward-looking statements.
|
2
Forward Looking Statements

Offering Summary
|
Issuer:
|
BioLineRx Ltd. (NasdaqCM:BLRX / TASE: BLRX)
|
|
Offering Size:
|
~$30 million (100% primary; excluding overallotment)
|
|
Overallotment Option:
|
15.0% (100% primary)
|
|
Offering Type:
|
Confidentially-Marketed Follow-On
|
|
Securities Offered:
|
American Depositary Shares
(1 ADS represents 10 Ordinary Shares)
|
|
Use of Proceeds:
|
• Continued clinical development of BL-8040 platform for AML and other
hematological indications • EU post-marketing and US clinical studies for BL-7010 in celiac disease
• Develop potential product candidates through Novartis collaboration
• Working capital and other general corporate purposes
|
|
Expected Pricing:
|
March 6, 2015 (before market opens)
|
|
Sole Bookrunner:
|
JMP Securities
|
3

BioLineRx Snapshot
• Drug development company focused on oncology & immunology
– Founded in 2003 by Teva and other key players in Israeli Life Sciences industry
• Bridge “development gap” for Israeli assets
– Leverage carefully selected early-stage technology, primarily at academia level,
following proof of concept in animals (at a minimum)
following proof of concept in animals (at a minimum)
• Current pipeline of 10 assets, 6 in clinical development
• Lead clinical programs:
– BL-8040 for AML and other hematological indications
– BL-1040 to prevent ventricular remodeling post AMI
– BL-7010 for celiac disease
• Strategic collaboration with Novartis for co-development of
selected Israeli-sourced programs
selected Israeli-sourced programs
4

Jerusalem
Rehovot
Tel-Aviv
Haifa
Be’er Sheva
Long Term Relationships with Israeli Institutions
Extensive in-licensing track record with majority of
academic & research centers in Israel
Technion
Tel Aviv University
Hebrew University
Weizmann Institute
Ben-Gurion University
Rambam Medical Center
Sheba Medical Center
Hadassah Medical Center
Sourasky Medical Center
5

Main Pipeline Assets
6

Main 2014 Achievements
• R&D Achievements in 2014
– All three lead development programs met their objectives in 2014
• BL-8040 (r/r AML) - phase 2 study partial results
– Clinical evidence for MOA (mobilization and apoptosis)
• BL-7010 (celiac program) - phase 1/2 completion
• BL-1040 (AMI) - completed enrollment for EU pivotal study
• BD Achievements in 2014
– Entered into strategic collaboration with Novartis
– Out-license of BL-5010 (skin lesion program) for EU market
• Deal signed with Omega Pharma, a leader in the OTC market, for Europe and
other selected territories
other selected territories
7

Main 2015 Value Drivers
• BL-8040 stem cell mobilization study
– Top line results by end of Q1 2015
• BL-8040 r/r AML study
– Top line results H2 2015
• BL-1040 CE Mark registration study
– Topline results mid-year 2015
• BL-7010 project
– Final approval of device regulatory path in EU from Notified Body in 2015
– Initiation of EU pivotal study in H2 2015
8

LEAD DEVELOPMENT PROGRAMS
9

10
BL-8040:
BEST-IN-CLASS CXCR4
ANTAGONIST FOR
TREATMENT OF
HEMATOLOGICAL
CANCERS
BEST-IN-CLASS CXCR4
ANTAGONIST FOR
TREATMENT OF
HEMATOLOGICAL
CANCERS

BL-8040 Highlights
• Platform Molecule: Can Address Multiple Cancer Indications
– Acute myeloid leukemia (AML) & other blood cancers
– Received Orphan designation from FDA for lead indications (accelerates development)
• Mode of Action: Inhibits CXCR4 (a cell surface protein)
– Present in high quantities on >70% of tumors; correlates with disease severity
– Inhibition induces cancer cell death
– Exposes cancer cells to treatment by mobilizing from bone marrow to blood circulation
• Status (AML): Phase 2 study ongoing
– Encouraging efficacy and excellent safety results to date
– To conclude in 2H 2015; Interim data in early 2015
• Status (Cell Mobilization): Phase 1 study ongoing
– Results to be reported early 2015

• AML is most common acute leukemia in adults
– Over 60,000 new cases recorded worldwide in 2010, growing to 130,000 by 2020
– 14,000 cases of AML diagnosed in the US in 2012
– Majority of AML patients relapse and require repeated treatment cycles
• AML has poor prognosis - less than 25% five-year survival
– Over 10,000 fatalities from AML in the US in 2012
• AML treatment regimens have changed little in past 30 years
– Treatment of AML is based largely on use of older chemotherapeutic drugs
AML - Treatment and Unmet Medical Need
12

BL-8040 Mechanism Of Action
• Binds CXCR4 with high affinity (1-2 nM)
• Maintains extended inhibition of CXCR4 through long receptor occupancy (>24 hours)
• Works as inverse agonist of CXCR4
• Induces apoptosis of tumor cells dependent on CXCR4 for survival
• Increases sensitivity to anti-cancer agents by mobilizing tumor cells from protective
microenvironment
microenvironment
• Induces terminal differentiation of immature cancer cells
13
BL-8040 directly
induces apoptosis
induces apoptosis
BL-8040
sensitizes tumor cells
to other drugs
BL-8040
Induces terminal
differentiation of
tumor cells
differentiation of
tumor cells
BL-8040
BL-8040
+ SOC
+ SOC
BL-8040
induces tumor
cells mobilization
cells mobilization

BL-8040 Shows Superiority to Current Therapy
14
Time post BL-8040/Mozobil injection
BL-8040 12 mg/kg
Mozobil 3.2 mg/kg
BL-8040 Stimulates Greater Mobilization
of Cells from the Bone Marrow
Compared to Mozobil
of Cells from the Bone Marrow
Compared to Mozobil
BL-8040 Causes Significant Cancer Cell
Death Compared to Mozobil
Death Compared to Mozobil
Drug Concentration
(μm)
(μm)
Mozobil
BL-8040
Beider K., Experimental Hematology 2011;39:282-292
Abraham M., Stem Cells 2007;25:2158 -2166

|
|
BL-8040
|
Mozobil
|
BMS-936564
(MDX1338)
|
|
Affinity for CXCR4
|
1-2 nM
|
84 nM
|
5nM
|
|
Inhibition
|
Inverse agonist
|
Antagonist (partial agonist)
|
Antagonist
|
|
CXCR4 Binding site
|
Extracellular domains in the
CXCR4 receptor |
Trans-membrane regions in the
CXCR4 receptor |
Extracellular domains in the
CXCR4 receptor |
|
Plasma half-life
|
0.3 - 0.7 hr
|
~3-5 hr
|
More than 24hr
|
|
Receptor occupancy
|
More than 24 hr
|
~2 hr
|
Not published
|
|
Cancer Cell Death
|
Induces apoptosis in preclinical
models. Evidence of remarkable apoptosis in samples from patients administrated with 0.75 and 1 mg/kg (phase 2). |
None
|
Demonstrated apoptosis in
preclinical models, modest effect in patients (ASH 2013) |
|
Mobilization
|
6-8 fold increase
(6/8 patients, phase 2a)
|
2.5 fold
(A phase 1/2 study, Blood 2012)
|
2.1-fold increase
(14/24 patients in phase 2 study,
ASH 2013) |
|
Other remarks of BL-8040:
•Synergizes with Rituximab and Bendamustine to stimulate Lymphoma cell death in vitro.
•Synergizes with Bortezomib (Velcade) to stimulate multiple myeloma cell death in vitro.
•Combination of BL-8040 with Imatinib in CML cells overcomes the protective effect of stroma in vitro.
•BL-8040 alone is highly efficient in eliminating lymphoma cells in the bone marrow and combined with Rituximab
significantly reduces tumor load (in vivo). •Synergizes with AC220 to minimize residual disease in FLT3+ AML (in vivo)
|
|||
BL-8040 is Best-in-Class vs. Competitors
15
Superior
Comparative
Inferior

Phase 2a - Treatment of r/r AML patients
Open-label study to evaluate safety and efficacy profile of repeated escalating doses of BL
-8040 in up to 70 adult subjects with relapsed or refractory AML
-8040 in up to 70 adult subjects with relapsed or refractory AML
Study design:
– Dose escalation phase - 3+3 design (last update given re 5th cohort of 1.5 mg/kg)
– Expansion phase: expand safe, efficacious dose group
Treatment:
– 2 consecutive days of BL-8040 monotherapy
– 5 days of BL-8040 + chemotherapy
Endpoints:
– To assess the safety and tolerability of escalating repeated doses of BL-8040 as monotherapy and when
combined with high-dose Ara-C in AML adult subjects with relapsed or refractory disease
combined with high-dose Ara-C in AML adult subjects with relapsed or refractory disease
– To assess the clinical efficacy (response rates) of escalating repeated doses of BL-8040
– To assess the apoptotic effect of BL-8040 on leukemic blasts
– To assess the effect of BL-8040 on mobilization of AML blasts to peripheral blood (PB)
– To assess the single and multiple dose pharmacokinetic profile of BL-8040
Treatment
Follow up
Screening
BM biopsy
BL-8040
Day
Ara-C
1 2 3 4 5 6 7 ------------------------------------------------------------------------------ 30
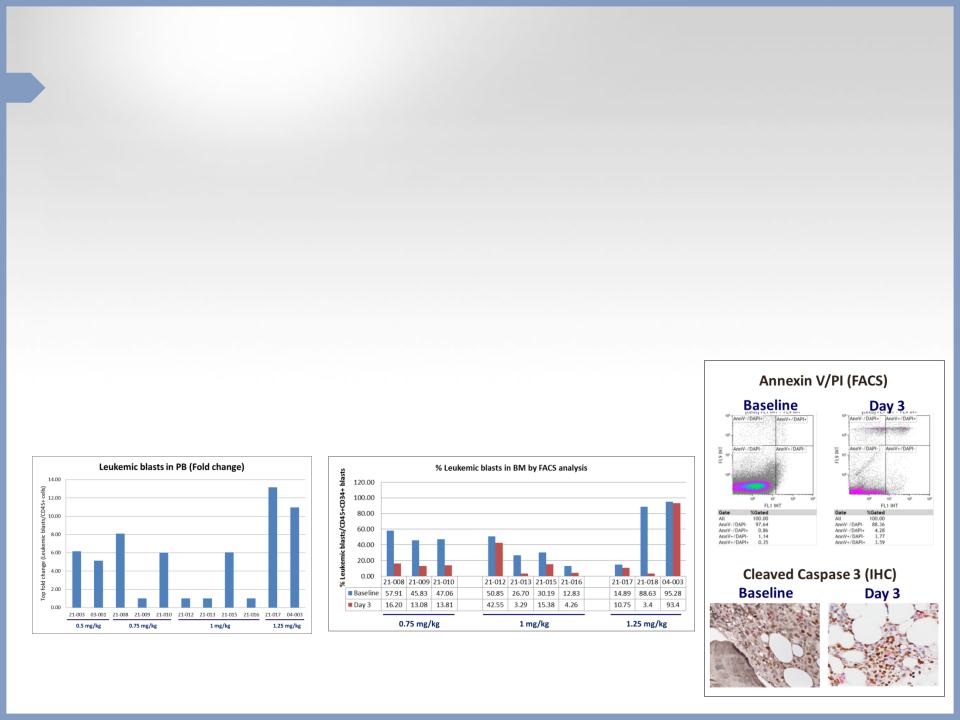
Partial Results in AML Phase 2 Study
• No BL-8040 related SAEs and no AEs were considered DLTs
• Robust leukemic blast mobilization was observed (median of 6-fold
increase)
increase)
• BL-8040 monotherapy decreased amount of leukemic cells in BM by median
of ~70%
of ~70%
• BL-8040 monotherapy achieved 3.5-fold increase in AML cell apoptosis
• Topline results are expected during H2 2015
17

Phase 1 - Single Agent Stem Cell Mobilization
A Phase 1, Two Part Study Exploring the Safety, Tolerability, Pharmacodynamic and
Pharmacokinetic Effect of Ascending Doses of BL-8040 in Healthy Subjects
Pharmacokinetic Effect of Ascending Doses of BL-8040 in Healthy Subjects
Study design:
•Part 1 - Dose escalation, randomized, placebo controlled - up to 4 escalating doses (0.5-1.25 mg/kg) P
•Part 2 - Dose expansion of safe and efficacious dose group
Endpoints:
– Safety and tolerability of escalating repeated doses
– Effect of BL-8040 on mobilization of hematopoietic stem cells (HSC) to peripheral blood (PB)
– Pharmacokinetic profile of BL-8040
– Yields of hematopoietic progenitor cells, immune cells, and other cellular subsets collected by leukapheresis
– Viability, biological activity and repopulating capacity of the cells collected by leukapheresis
Timelines
•Topline results expected by end of Q1/2015
Mobilization
Safety follow up
Safety follow up
Mobilization
Screening
Screening
Part 1
Part 2
BL-8040
1
-28
Day
2
7
3
18

Three New Studies to be Initiated in 2015
|
Study
|
Collaborator
|
Description
|
Results
Œ
|
|
AML
Consolidation Phase 2b
(200 patients)
|
German Study
Alliance Leukemia Group |
Double-blind, placebo-controlled,
repeated-administration, multi treatment cycles |
Topline results
Q4 2017
|
|
AML FLT3-ITD
Phase 1/2
(up to 100 patients)
|
MD Anderson
Cancer Center |
Open-label, 2 parts:
•Dose selection, with Sorafinib and
BL-8040 •Expansion in different FLT3
patients |
Partial results
H2 2016 Final results
Q1 2017
|
|
Myelodysplatic
Syndrome and Aplastic Anemia
Phase 1/2
(up to 25 patients)
|
MD Anderson
Cancer Center |
Open-label, repeated administration,
single-treatment cycle |
Partial results
H2 2016 Final results
H2 2017
|
19

BL-8040 Summary
• CXCR4 is a validated target
• BL-8040 has robust mobilization activity and apoptosis
– Validated in preliminary data from Phase II study in AML, and Phase 1/2 study in multiple
myeloma
myeloma
– BL-8040 has very favorable profile in comparison with leading CXCR4 antagonists
• BL-8040 is an inverse agonist
– Blocks the auto-signaling of CXCR4
• BL-8040 is a platform for a number of hematological indications
– Three new studies planned for 2015
20

BL-1040:
FIRST-IN-CLASS
MYOCARDIAL IMPLANT
FOR PREVENTION OF
VENTRICULAR REMODELING
FOLLOWING AMI
FIRST-IN-CLASS
MYOCARDIAL IMPLANT
FOR PREVENTION OF
VENTRICULAR REMODELING
FOLLOWING AMI
Out-licensed to Bellerophon
(f/k/a Ikaria) and being
developed as Bioabsorbable
Cardiac Matrix (BCM)
(f/k/a Ikaria) and being
developed as Bioabsorbable
Cardiac Matrix (BCM)
21

BL-1040 Highlights
• Indication: Cardiac remodeling post-AMI
• Mode of Action: Provides support to ischemic tissue during healing
• Status: CE Mark registration trial - enrollment completed; top-line results in
mid-2015
mid-2015
• Device designation (including FDA)
• Partnered with Bellerophon BCM (f/k/a Ikaria)
– All program costs funded by Bellerophon BCM
• Market Opportunity: >$1 Billion*
22
*Based on a customized survey and report prepared for BioLineRx by Defined Health

Unmet Medical Need
Vessel occlusion
Tissue damage
23

How Does BL-1040 Work?
Arterial injection deposits
material into infarcted tissue
material into infarcted tissue
Turns from liquid to gel on
contact with infarcted tissue
contact with infarcted tissue
Gel-like scaffold provides
mechanical support to damaged
tissue
mechanical support to damaged
tissue
Transitions to liquid and exits
the body within 6 weeks
the body within 6 weeks
BL-1040
Untreated
Porcine AMI model, day 60
L
V
V
L
V
V
• Dilated
• Dilated
• Thin LV wall
• Thin LV wall
• Normal size
• Normal size
• Normal LV wall
• Normal LV wall
L
V
V
L
V
V
24
Designated as device by regulatory authorities

BL-1040 Clinical studies
Pivotal CE Mark Registration trial progressing at full steam
•Placebo controlled, enrollment of 303 patients P
• 6-month follow-up
•Includes ~90 sites in nine countries (including 16 sites in US)
•Endpoints: end diastolic volume, QLQ, six-minute walk test
•Top-line results expected in mid-2015
US pivotal trial in final discussions with FDA
•Placebo controlled, ~1,000 patients, ~200 sites
•12-month follow-up
•The study expected to start early 2016
25
* Intracoronary Delivery of Injectable Bioabsorbable Scaffold (IK-5001) to Treat
Left Ventricular Remodeling After ST-Elevation Myocardial Infarction - A First-in
-Man Study, Frey N et al., Circ: Cardiovasc Interv. 2014
Left Ventricular Remodeling After ST-Elevation Myocardial Infarction - A First-in
-Man Study, Frey N et al., Circ: Cardiovasc Interv. 2014

BL-1040 Summary
• Huge unmet medical need
– >$1 billion market
• Designated as device in both US and Europe
• Pilot study successfully completed
– No safety or tolerability issues after six months of follow-up
– Promising efficacy in comparison to historical data
• Top-line results of CE mark registration study expected in mid-2015
– Enrollment of 303 patients completed
26

27
BL-7010:
NOVEL GLIADIN
BINDING POLYMER
FOR
NOVEL GLIADIN
BINDING POLYMER
FOR
CELIAC DISEASE

BL-7010: Polymeric Binder for Celiac Disease
• Indication: Celiac disease
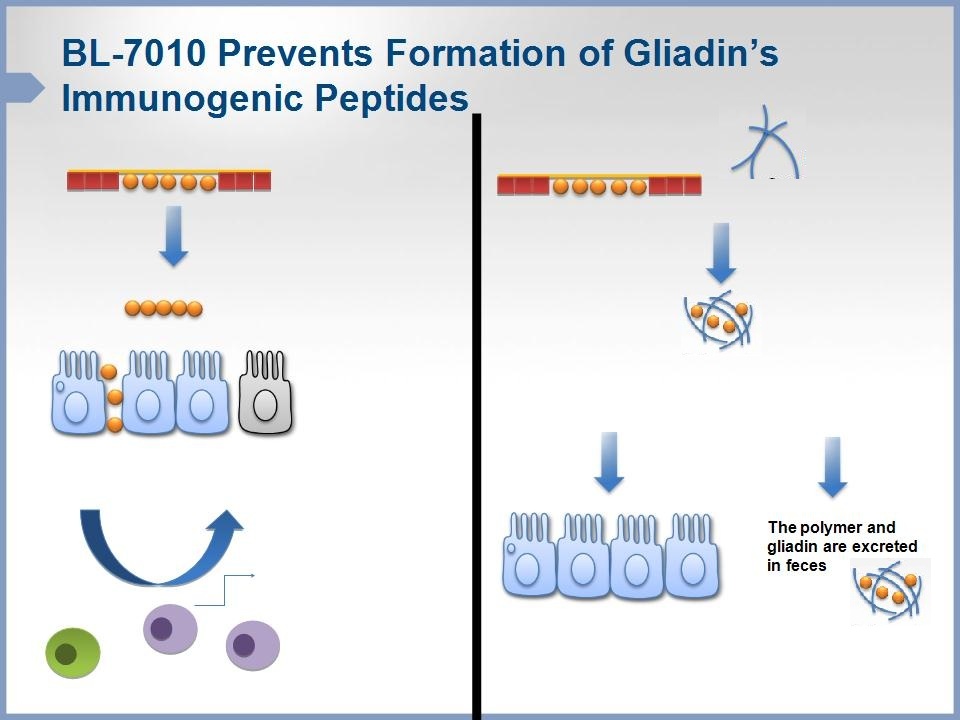
• Mode of Action: Non-absorbable polymer with high affinity to gliadins
(immunogenic peptides contained in gluten)
(immunogenic peptides contained in gluten)
• Status: Phase 1/2 completed
• Product Highlights
– Prevents pathological damage to small intestine
– Non-absorbable
– Non-toxic
28

Celiac Disease - Large Unmet Medical Need
29
• 1% of world’s population suffers from celiac disease
– Number underestimated due to lack of awareness/diagnostic tools
• Market projected to reach $8 billion by 2019
• No current pharmacological agents approved for celiac
– Only treatment option is life-long, strict gluten-free diet (GFD)
– ~20% of celiac patients are symptomatic even with GFD
• Major interest shown by Big Pharma
– AbbVie recently acquired rights to phase 2 asset from Alvine for $70 million upfront
Copolymer of sodium
styrene sulfonate (SS)
and 2-hydroxyethyl
methacrylate (HEMA)
styrene sulfonate (SS)
and 2-hydroxyethyl
methacrylate (HEMA)
Gluten
BL-7010 demonstrates
distinguished specificity
towards gliadin
distinguished specificity
towards gliadin
Prevention of intestinal damage
30
Small intestinal damage with loss of
absorptive villi and hyperplasia of the
crypts, typically leading to malabsorption
APC
BL-7010
Gluten
Gliadin
Enterocytes
Lymphocytes
Inflammatory Cytokines

31
BL-7010 Maintains Normal Structure of GI
non-sensitized
mice
mice
Gluten-sensitized
mice
mice
Gluten-sensitized
mice + BL-7010
mice + BL-7010
5.96 ± 1.23
Villus-to-crypt ratio
2.58 ± 0.43
4.89 ± 1.51
Model: HLA-DQ8/HCD4 transgenic male mice sensitized to gliadin

BL-7010 Clinical Program Overview
• Phase 1/2 study in celiac patients completed
– Positive top-line results presented in early November
• Safe and well tolerated; no serious or dose-limiting side effects
• Optimal dose identified: 1 gram x 3 per day
• Confirmed no systemic absorption; supports medical device classification in Europe
(significantly accelerates potential approval)
(significantly accelerates potential approval)
32
– Single ascending dose
– Safety endpoints
– No efficacy endpoints
– Assessment of systemic exposure
– 14 days repeated administration
– 3 times per day
– Safety w/o efficacy endpoints
– Assessment of systemic exposure

BL-7010 Summary
• Celiac disease is huge unmet medical need
– There are only a handful of clinical-stage programs in development
• BL-7010 has unique and simple MOA
• Successful Phase 1/2 pilot study
– Well tolerated, no systemic exposure
– Will likely be classified as medical device in Europe
• Pivotal study for EU in celiac patients expected to begin in 2015
– 6-week repeated oral administration
– Efficacy endpoints (primary and secondary) and safety endpoints
33

CORPORATE
34

Transformative Collaboration with Novartis
• Novartis selected BioLineRx as its leading partner for identification
and early development of Israeli-sourced drug candidates
and early development of Israeli-sourced drug candidates
– Exclusive first look at all Israeli-based projects scouted by BioLineRx
– Co-develop selected projects through clinical proof-of-concept (POC)
• Provides lasting shareholder value and key insights
– Builds pipeline in conjunction with global leader, gaining Big Pharma perspective
• Financial highlights:
– Upfront $10 million equity investment in BLRX
– Upon selection of project, BioLineRx will receive:
• $5 million option fee (non-dilutive)
• 50% of remaining R&D expenses up to POC (in equity at a premium to market)
– Novartis receives right of first negotiation for full out-license upon clinical POC
35

Financial and Corporate Summary
Cash position
• $29.6 million as of September 30, 2014
– Does not include $10 million received from Novartis in December
• Funds operational capital into 2016
Capital structure
• Traded on NASDAQ and TASE (Symbol: BLRX)
• 39 million shares outstanding; 45 million fully diluted (based on ADSs)
• US shareholders represent ~60% of investor base
– Novartis holds ~13% of Company
Other
• 47 employees, approximately 2/3 with advanced degrees
36

Major Development Milestones - 2015 and 2016
37
BL-7010 (Celiac) CE pivotal study initiation
BL-1040 (AMI) CE mark study completion
BL-8040 (AML) phase 2 completion
BL-8040 (AML) phase 2 partial results*
BL-8040 (SC Mobilization) phase 1 apheresis data
BL-8040 (FLT-3) phase 1/2 initiation
BL-8040 (hMDS & AA) phase 1/2 initiation
BL-8040 (SC Mobilization) phase 1 completion
√
BL-7010 (Celiac ) CE pivotal study completion
BL-8040 (hMDS & AA) phase 1/2 interim results
BL-8040 (SC Mobilization) phase 2 Initiation
BL-8040 (SC Mobilization) phase 2 LPI
BL-8040 (AML) - end of phase 2 meeting
BL-8040 (Consolidation) phase 2b LPI
BL-1040 (AMI) US pivotal study initiation
BL-1040 (AMI) complete CE mark study enrollment
√

38
Exhibit 99.2
For Immediate Release
BioLineRx Announces Underwritten Public Offering
of its American Depositary Shares
Jerusalem, March 5, 2015 - BioLineRx (NASDAQ: BLRX; TASE: BLRX), a clinical-stage biopharmaceutical company dedicated to identifying, in-licensing and developing promising therapeutic candidates, today announced that it has commenced an underwritten public offering of American Depositary Shares (“ADSs”), each representing ten (10) of its Ordinary Shares. All of the ADSs in the offering are to be sold by BioLineRx.
JMP Securities is acting as sole book-running manager for the offering. BioLineRx intends to grant the underwriters a 30-day option to purchase up to an additional 15 percent of the amount sold to cover over-allotments, if any. The offering is subject to market conditions, and there can be no assurance as to whether or when the offering may be completed, or as to the actual size or terms of the offering.
The ADSs will be issued pursuant to a shelf registration statement that was previously filed with, and declared effective by, the Securities and Exchange Commission (SEC). A final prospectus supplement related to the offering will be filed with the SEC and will be available on the SEC's website located at www.sec.gov.
This press release does not constitute an offer to sell or a solicitation of an offer to buy nor shall there be any sale of these securities in any state or jurisdiction in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of any such state or jurisdiction. Any offer, if at all, will be made only by means of a prospectus supplement and accompanying prospectus forming a part of the effective registration statement, copies of which may be obtained, when available, from JMP Securities LLC, 600 Montgomery Street, 10th Floor, San Francisco, California 94111, Attention: Prospectus Department, or by telephone: (415) 835-8985.
About BioLineRx
BioLineRx is a publicly-traded, clinical-stage biopharmaceutical company dedicated to identifying, in-licensing and developing promising therapeutic candidates. The Company in-licenses novel compounds primarily from academic institutions and biotech companies based in Israel, develops them through pre-clinical and/or clinical stages, and then partners with pharmaceutical companies for advanced clinical development and/or commercialization.
BioLineRx’s current portfolio consists of a variety of clinical and pre-clinical projects, including: BL-1040 for prevention of pathological cardiac remodeling following a myocardial infarction, which has been out-licensed to Bellerophon BCM (f/k/a Ikaria) and is in the midst of a pivotal CE-Mark registration trial scheduled for completion in mid-2015; BL-8040, a cancer therapy platform, which is in the midst of a Phase 2 study for acute myeloid leukemia (AML) as well as a Phase 1 study for stem cell mobilization; and BL-7010 for celiac disease, which has successfully completed a Phase 1/2 study.
In December 2014, BioLineRx entered into a strategic collaboration with Novartis for the co-development of selected Israeli-sourced novel drug candidates. The companies intend to co-develop a number of pre-clinical and early clinical therapeutic projects through clinical proof-of-concept for potential future licensing by Novartis.
For more information on BioLineRx, please visit www.biolinerx.com or download the investor relations mobile device app, which allows users access to the Company’s SEC documents, press releases, and events. BioLineRx’s IR app is available on the iTunes App Store as well as the Google Play Store.
Various statements in this release concerning BioLineRx’s future expectations constitute “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. These statements include words such as “may,” “expects,” “anticipates,” “believes,” and “intends,” and describe opinions about future events. These forward-looking statements involve known and unknown risks and uncertainties that may cause the actual results, performance or achievements of BioLineRx to be materially different from any future results, performance or achievements expressed or implied by such forward-looking statements. Some of these risks are: changes in relationships with collaborators; the impact of competitive products and technological changes; risks relating to the development of new products; and the ability to implement technological improvements. These and other factors are more fully discussed in the “Risk Factors” section of BioLineRx’s most recent annual report on Form 20-F filed with the Securities and Exchange Commission on March 17, 2014. In addition, any forward-looking statements represent BioLineRx’s views only as of the date of this release and should not be relied upon as representing its views as of any subsequent date. BioLineRx does not assume any obligation to update any forward-looking statements unless required by law.
Contact:
Tiberend Strategic Advisors, Inc.
Joshua Drumm, Ph.D.
+1-212-375-2664
Andrew Mielach
+1-212-375-2694
or
Tsipi Haitovsky
Public Relations
+972-3-6240871
Exhibit 99.3
Our stockholders or potential investors may be referred to as “you” or “your” in this disclosure and BioLineRx Ltd. is referred to as “the company,” “we,” “our,” or “us”.
|
Our Business
|
We are a clinical stage biopharmaceutical development company dedicated to identifying, in-licensing and developing therapeutic candidates that have advantages over currently available therapies or that address unmet medical needs. Our current development pipeline consists of six clinical-stage therapeutic candidates: BL-1040, a novel polymer solution for use in the prevention of ventricular remodeling following an acute myocardial infarction, or AMI; BL-8040, a novel peptide for the treatment of acute myeloid leukemia (AML), stem cell mobilization and other hematological indications; BL-7010, a novel polymer for the treatment of celiac disease; BL-5010, a customized, proprietary, pen-like applicator containing a novel, acidic, aqueous solution, which is being developed in Europe as a medical device for the non-surgical removal of benign skin lesions; BL-7040, an oligonucleotide for the treatment of inflammatory bowel disease, or IBD; and BL-8020, an orally available treatment for the hepatitis C virus, or HCV, and other viral indications, with a unique mechanism of action involving the inhibition of virus-induced autophagy in host cells. In addition, we have four therapeutic candidates in the preclinical stages of development. We generate our pipeline by systematically identifying, rigorously validating and in-licensing therapeutic candidates that we believe exhibit a relatively high probability of therapeutic and commercial success. None of our therapeutic candidates have been approved for marketing and, to date, there have been no commercial sales of any of our therapeutic candidates. Our strategy includes commercializing our therapeutic candidates through out-licensing arrangements with biotechnology and pharmaceutical companies. We also evaluate, on a case-by-case basis, co-development and similar arrangements and the commercialization of our therapeutic candidates independently.
In December 2014, we entered into a strategic collaboration with Novartis Pharma AG, or Novartis, for the co-development of selected Israeli-sourced novel drug candidates. Under the agreement, we intend, in collaboration with Novartis, to co-develop a number of pre-clinical and early clinical therapeutic projects through clinical proof-of-concept for potential future licensing by Novartis.
Our Product Pipeline
The table below summarizes our current pipeline of therapeutic candidates, including the target indications and status of each candidate and our development partners.
BL-1040
Our first therapeutic candidate, BL-1040 (now called “Bioabsorbable Cardiac Matrix,” or BCM), is a medical device, injected in patients following an AMI, intended for prevention of ventricular remodeling and subsequent congestive heart failure. Ventricular remodeling is the structural alteration of the damaged heart muscle that occurs following an acute heart attack. Once this damage occurs, the weakened heart muscle forces the rest of the heart to compensate. Under this extra workload, the heart muscle dilates, the walls of the heart thin, and the heart further remodels, thereby causing another cycle of dilation and overcompensation. The extra workload to the heart causes further structural damage and can lead to congestive heart failure. BL-1040 is a liquid polymer which is delivered in a bolus injection via the coronary artery during catheterization and flows into the damaged heart muscle, creating a scaffold within injured cardiac muscle designed to enhance cardiac mechanical strength during the healing period and prevent pathological ventricular dilation. BL-1040 remains in the infarct zone for a few months and is excreted through the kidneys. The data from our preclinical trials in various animal models indicate that, by supporting the damaged heart tissue, BL-1040 preserves the normal functioning of the heart, and the data from our clinical trials indicate that BL-1040 should be safe. After consultation by our out-licensing partner, Bellerophon BCM LLC, or Bellerophon, with the FDA, BL-1040 is being developed as a class III medical device under the FDA’s pre-marketing approval, or PMA, regulatory pathway. In December 2011, Bellerophon commenced PRESERVATION 1, a CE Mark registration clinical trial of BL-1040. PRESERVATION 1 aims to evaluate the safety and effectiveness of BL-1040 for prevention of ventricular remodeling when administered following AMI. The trial is a placebo-controlled, randomized, double-blind, multi-country and multi-center trial. BL-1040 is being administered to subjects who had successful percutaneous coronary intervention with stent placement after ST-segment elevation myocardial infarction (STEMI). Enrollment for this trial was completed in December 2014, with 303 AMI patients having been recruited and treated. There are almost 90 sites activated worldwide for this trial, 16 of which are in the United States. Bellerophon expects to report top line results from the study, which includes a six-month follow-up period, in mid-2015.
In 2009, we entered into an out-licensing arrangement with Bellerophon (formerly known as “Ikaria Development Subsidiary One LLC”) with regard to BL-1040, which we amended in January 2015. Under this arrangement, Bellerophon is obligated to use commercially reasonable efforts to complete clinical development of, and to commercialize, BL-1040 or a product related thereto. To date, we have received $17.0 million from Bellerophon, and we are entitled to receive up to an additional $265.5 million from Bellerophon upon achievement of certain development, regulatory, and commercial milestones. In addition, we are entitled to receive from Bellerophon royalties from net sales of any product developed under the arrangement. Pursuant to the January 2015 amendment, a certain milestone and related payments have been adjusted, but the total potential milestone payments to be paid to us under the license agreement remain the same. We believe that Bellerophon has financial resources sufficient to meet its contractual obligations under its agreement with us.
2
We are obligated to pay 28% of all net consideration received under this arrangement to B.G. Negev Technologies and Applications Ltd., or B.G. Negev Technologies, the party from which we in-licensed BL-1040 in 2004. We have agreed to pay Ramot at Tel Aviv University Ltd., or Ramot, a portion of the payments we make to B.G. Negev Technologies in connection with the in-license arrangement to satisfy contractual obligations between B.G. Negev Technologies and Ramot with respect to certain intellectual property rights to the licensed technology. We have also agreed to indemnify Ramot and certain of its related parties in connection with our use of the technology we in-licensed from B.G. Negev Technologies.
BL-8040
Our second clinical-stage therapeutic candidate, BL-8040, is a novel, short peptide that functions as a high-affinity antagonist for CXCR4, which we intend to develop for AML, stem cell mobilization and other hematological indications. CXCR4 is a chemokine receptor that is directly involved in tumor progression, angiogenesis (growth of new blood vessels in the tumor), metastasis (spread of tumor to other organs) and cell survival. CXCR4 is over-expressed in more than 70% of human cancers and its over-expression often correlates with poor prognosis. BL-8040 mobilizes cancer cells from the bone marrow and may therefore sensitize these cells to chemo- and bio-based anti-cancer therapy. In addition, BL-8040 has demonstrated a direct anti-cancer effect by inducing apoptosis (cell death). Multiple pre-clinical studies have shown the safety and efficacy of BL-8040. These studies have shown that BL-8040 is efficient, both alone and in combination with chemotherapy, in reducing bone marrow malignant cells and stimulating their cell death. BL-8040 also mobilizes stem cells from the bone marrow to the peripheral blood, enabling their collection for subsequent autologous or allogeneic transplantation in cancer patients. In September 2013, the FDA granted an Orphan Drug Designation to BL-8040 as a therapeutic for the treatment of AML; and in January 2014, the FDA granted an Orphan Drug Designation to BL-8040 as a treatment for stem cell mobilization.
In June 2013, we commenced a Phase 2 trial for BL-8040 for the treatment of AML. The study is currently being conducted at four sites in the United States, including MD Anderson Cancer Center in Houston, Memorial Sloan-Kettering Cancer Center in New York, Northwestern University Hospital in Chicago, and Mayo Clinic in Jacksonville, as well as at five well-known sites in Israel. The study is a multicenter, open-label study under an Investigational New Drug, or IND, approval from the FDA, designed to evaluate the safety and efficacy profile of repeated escalating doses of BL-8040 in adult subjects with relapsed/refractory AML. As of the date of this prospectus supplement, 19 patients have been enrolled in the study, out of a total expected enrollment of up to 70 patients. Early results of this trial showed that BL-8040, as a stand-alone therapy and in combination with high-dose Cytarabine (Ara-C), is safe at all doses tested to date, and triggers substantial mobilization of cancer cells from the bone marrow to the peripheral blood, thereby increasing the vulnerability of the cells to chemotherapy treatment. In addition, signs of robust apoptosis of cancer cells were observed following administration of higher doses tested.
At the annual ASH conference in December 2014, we presented data from the trial showing that even at the highest dose reached at that time (1.25 mg/kg), there were no dose-limiting toxicity events or serious adverse events, nor early discontinuations attributable to BL-8040. Furthermore, we presented data showing that BL-8040 triggered substantial mobilization of AML cancer cells from the bone marrow to the peripheral blood, with a median 6-fold increase of AML cells in the blood. This mobilization is crucial for exposing a higher ratio of AML cells to accompanying chemotherapy such as Ara-C. Additional results from the trial show that after only two days of BL-8040 monotherapy, there was a median decrease of approximately 70% in the amount of AML cells in the bone marrow, while the levels of normal progenitor cells remained stable. Furthermore, BL-8040 as a monotherapy showed a 3.5-fold increase in cell death (apoptosis) of AML cells, both in the bone marrow and in peripheral blood samples.
3
The dose-escalation stage of the study is expected to be completed in early 2015, while the full study results from both the dose-escalation and dose-expansion stages of the study are expected in the second half of 2015.
Targeting a second AML treatment line, BL-8040 is scheduled to commence a Phase 2b trial, as a consolidation treatment for AML patients who have responded to standard induction treatment, in the first half of 2015. The trial will be conducted in collaboration with the German Study Alliance Leukemia Group. The trial aims to improve the response of AML patients to the second stage of AML treatment, termed consolidation therapy, by eliminating the minimal residual disease left in the bone marrow after the first stage of the standard treatment regimen, called induction therapy. We recently announced the filing of the regulatory submissions required to commence the trial.
In addition, BL-8040 is scheduled to commence a Phase 1/2 trial for the treatment of a third population of AML patients, those with the FLT3-ITD mutation, in the first half of 2015. The Phase 1/2 trial, which will be conducted in collaboration with the MD Anderson Cancer Center, is aimed at improving the response of FLT3-ITD mutated AML patients to treatment with sorafenib (a FLT3 inhibitor). This trial follows the presentation at several conferences during 2014 of positive preclinical results of BL-8040 as a treatment for AML patients with FLT3 mutations.
In September 2014, we announced the dosing of the first patient in a Phase 1 trial for another indication of BL-8040 - as a novel treatment for the mobilization of stem cells from the bone marrow to the peripheral blood circulation, where they can be harvested for transplant supporting the treatment of hematological indications. The study is being conducted at the Hadassah Medical Center in Jerusalem. Part 1 of the study is a randomized, double-blind, placebo-controlled dose escalation study exploring the safety and tolerability of escalating repeated doses of BL-8040 in healthy volunteers. In January 2015, we announced that all healthy volunteers had completed the treatment phase of the study. Following initial analysis of the data, the optimal safe and efficacious dose of BL-8040 was selected to be used as a stand-alone therapy in the second part of the study. Part 2 is an open-label study designed to assess BL-8040’s stem cell mobilization capacity, as well as the yield of cells collected by apheresis. The top line results of both parts of this study are expected by the end of the first quarter of 2015.
We are also planning to conduct a Phase 1/2 trial, again in collaboration with the MD Anderson Cancer Center, for a fifth indication of BL-8040 - as a treatment for hypoplastic myelodysplastic syndrome, or hMDS, and aplastic anemia, or AA. The study will be open label and designed to evaluate the safety, tolerability and efficacy of the combination of BL-8040 with immunosuppressive therapies (hATG, cyclosporine and prednisone). We plan to commence the trial in first half of 2015.
BL-7010
Our third clinical-stage therapeutic candidate, BL-7010, is a novel, non-absorbable, orally available, high-molecular-weight co-polymer intended for the treatment of celiac disease. It has a high affinity for gliadins, the immunogenic proteins present in gluten that cause an immune response in patients with celiac disease. BL-7010 effectively masks gliadins from enzymatic degradation and prevents the formation and absorption of immunogenic peptides that trigger the immune system. BL-7010 is excreted with gliadin from the digestive tract, preventing the absorption of gliadin peptides. This significantly reduces the immune response triggered by gluten. The safety and efficacy of BL-7010 were demonstrated in pre-clinical and clinical studies.
In December 2013, we commenced a Phase 1/2 trial for BL-7010 at Tampere Hospital in Finland. The trial was a two-part (single and repeated administration), double-blind, placebo-controlled, dose escalation study of BL-7010 in up to 40 well-controlled celiac patients. The primary objective of the study was to assess the safety of single and repeated ascending doses of BL-7010. Secondary objectives included an assessment of the systemic exposure, if any, of BL-7010 in the study patients. In November 2014, we reported the final results of the study. Those results confirmed that BL-7010 is safe and well tolerated in both single and repeated-dose administrations. Based on these results, we selected the dosing regimen of one gram, three times per day, of BL-7010 as the optimal repeated dose for the upcoming efficacy study, which is expected to commence in the second half of 2015. In addition, pharmacokinetic analyses revealed no systemic exposure of BL-7010 in plasma and urine samples from all patients at all doses and time points tested, both in the single- and repeated-dose regimens. Based on previous communications with a Notified Body in the European Union, we believe the lack of systemic exposure will likely support a medical-device classification in Europe for BL-7010, which would significantly accelerate its development in Europe.
4
BL-5010
Our fourth clinical-stage therapeutic candidate, BL-5010, is a novel medical device containing a novel, acidic aqueous solution for the non-surgical removal of benign skin lesions. It offers an alternative to painful, invasive and expensive removal treatments including cryotherapy, laser treatment and surgery. Since the treatment is non-invasive, it poses minimal infection risk and eliminates the need for anesthesia, antiseptic precautions and bandaging. The pre-filled device controls and standardizes the volume of solution applied to a lesion, ensuring accurate administration directly on the lesion and preventing both accidental exposure of the healthy surrounding tissue and unintentional dripping. It has an ergonomic design, making it easy to handle, and it will be childproofed. The product has completed a phase 1/2 pilot clinical study for the removal of seborrheic keratosis, or SK, which showed excellent efficacy and cosmetic results, and has received confirmation in Europe for the regulatory pathway classification as a Class 2a medical device.
Our original development plan for BL-5010 consisted of clinical testing for the treatment of SK. However, during discussions in recent years with potential partners for the development and commercialization of BL-5010, we learned that they had more interest in the possibilities of BL-5010 for over-the-counter, or OTC, indications. In December 2014, we entered into an exclusive out-licensing arrangement with a subsidiary of Omega Pharma NV, or Omega Pharma, for the rights to BL-5010 for OTC indications in the territories of Europe, Australia and additional selected countries. We will retain the non-OTC rights to BL-5010 in Omega Pharma’s territories as well as all rights to BL-5010 in the United States and the rest of the world. Under our out-licensing arrangement with Omega Pharma, Omega Pharma is obligated to use commercially reasonable best efforts to obtain regulatory approval in the licensed territory for at least two OTC indications and to commercialize BL-5010 for those two OTC indications. In addition, Omega Pharma will sponsor and manufacture BL-5010 in the relevant regions. Omega Pharma will pay us an agreed amount for each unit sold, and we will be entitled to certain commercial milestone payments. In addition, we will have full access to all clinical and research and development data generated during the performance of the development plan and may use these data in order to develop or license the product in other territories and fields of use where we retain the rights. We expect that the first OTC products will enter the market in 2016. As a result of this out-licensing arrangement, as well as the previous discussions with other potential partners for this product, the development activities for BL-5010 are currently focused on OTC indications. However, we may decide to continue development of BL-5010 for non-OTC indications, including but not limited to, SK.
We are required to pay a portion, within the standard range of sublicense receipt consideration paid to our licensors, of the revenues we receive from our arrangement with Omega Pharma, to Innovative Pharmaceutical Concepts, Inc. or IPC, the party from which we in-licensed BL-5010 in 2007.
BL-7040
Our fifth clinical-stage therapeutic candidate, BL-7040, is an oligonucleotide being developed for the treatment of inflammatory bowel disease (IBD). The compound had already been the subject of phase 1 safety and pharmacokinetics studies and a phase 2a study examining the efficacy of the compound for the treatment of myasthenia gravis, an autoimmune, neurodegenerative disease. BL-7040 showed a high level of safety and efficacy in those trials. The compound was also found to target the innate inflammatory pathway and, therefore, we decided to develop the compound for the treatment of IBD and other inflammatory diseases.
In April 2013, we announced positive results from a phase 2a proof-of-concept study to evaluate the effectiveness of BL-7040 for the treatment of IBD at five sites in Israel. The study showed that BL-7040 is safe and effective in treating ulcerative colitis, a form of IBD. Sixteen of the 22 patients who were enrolled in the clinical trial completed the full five-week course of treatment and two-week follow-up. The primary clinical endpoint in the study – a 3-point and 30% reduction in the Mayo score between baseline and completion of treatment – was achieved. Fifty percent of patients (8 patients) met the primary endpoint, while the remaining 8 patients demonstrated a stable clinical condition or minor improvement.
In November 2013, we announced additional results from this study showing significant improvement of disease measurements in biopsies taken from IBD patients treated with BL-7040. The histological and biochemical analyses of inflammation indicators reinforced the initial positive results of the study described above. During the third quarter of 2014, we conducted a pharmacokinetic study which indicated that BL-7040 reaches the target organ (the colon) and appears to have a local, as opposed to systemic, effect. We are currently discussing this therapeutic candidate with a number of potential co-development partners, as well as planning the next stages of development.
5
BL-8020
Our sixth clinical-stage therapeutic candidate, BL-8020, is an orally available treatment for the hepatitis C virus, or HCV, and other viral indications, with a unique mechanism of action involving the inhibition of virus-induced autophagy in host cells. In April 2013, we commenced a phase 1/2 clinical trial to evaluate the safety, tolerability and effectiveness of BL-8020 at two sites in France. In January 2014, we entered into a collaboration agreement with the licensors of the compound whereby, in consideration for the payment of future royalties to us, we terminated the license agreement, the licensors agreed to take over development of the compound and we agreed to supply, at the licensors’ request and for full payment, the drug product needed for a clinical trial to be administered by the licensors. In August 2014, the licensors decided to terminate the ongoing phase 1/2 trial in HCV due to a very slow recruitment rate, and are now determining the next steps in the clinical development plan of the compound, including an assessment regarding potential additional viral indications for development.
|
Our Product Development Approach
|
As part of our business strategy, we continue to actively source, rigorously evaluate and in-license selected therapeutic candidates. We establish and maintain close relationships with research institutes, academic institutions and biotechnology companies in Israel, including, in some instances, a formal right of first offer for therapeutic compounds in their portfolios. In the last several years, we have extended our sourcing activities to other countries. Before in-licensing, each therapeutic candidate must pass through our thorough screening process. Our Scientific Advisory Board and disease-specific third-party advisors are active in evaluating each therapeutic candidate. Our approach is consistent with our objective of proceeding only with therapeutic candidates that we believe exhibit a relatively high probability of therapeutic and commercial success. To date, we have screened over 2,000 compounds, presented more than 70 candidates to our Scientific Advisory Board for consideration, initiated development of 45 therapeutic candidates and terminated 35 feasibility programs.
Our Strategy
Our objective is to be a leader in developing innovative pharmaceutical and biopharmaceutical products. We continuously identify and in-license therapeutic candidates in order to maximize our potential for commercial success. We repeatedly assess compounds by evaluating their efficacy, safety, total estimated development costs, technological novelty, patent status, market potential and approvability. Our approach to evaluating, in-licensing and developing therapeutic candidates allows us to:
|
|
•
|
continually build our pipeline of therapeutic candidates;
|
|
|
•
|
advance those therapeutic candidates with the greatest potential;
|
|
|
•
|
quickly identify, and terminate the development of, unattractive therapeutic candidates; and
|
|
|
•
|
avoid dependency on a small number of therapeutic candidates.
|
Using this approach, we have successfully advanced six therapeutic candidates into clinical development. Specific elements of our current strategy include the following:
|
|
•
|
Support the successful development and commercialization of therapeutic candidates that have already been partnered. We currently have five programs at various stages of development in our pipeline, which have already been partnered.
|
|
|
•
|
Commercialize additional therapeutic candidates through out-licensing arrangements or, where appropriate, by ourselves. We intend to commercialize many of our other products through out-licensing arrangements with third parties who may perform any or all of the following tasks: completing development, securing regulatory approvals, manufacturing and/or marketing. If appropriate, we may also enter into co-development and similar arrangements with respect to any therapeutic candidate with third parties or commercialize a therapeutic candidate ourselves.
|
6
|
|
•
|
Design development programs that reach critical decisions quickly. At each step of our screening process for therapeutic candidates, a candidate is subjected to rigorous feasibility testing and potential advancement or termination. We believe our feasibility approach reduces costs and increases the probability of commercial success by eliminating less promising candidates quickly before advancing them into more costly preclinical and clinical programs.
|
|
|
•
|
Use our expertise and proprietary screening methodology to evaluate in-licensing opportunities. In order to review and select among various candidates efficiently and effectively, we employ a rigorous screening system we developed. Our Scientific Advisory Board and disease-specific third-party advisors evaluate each candidate. We intend to in-license a sufficient number of therapeutic candidates to allow us to move a new therapeutic candidate into clinical development every 12 to 24 months.
|
|
|
•
|
Leverage and expand our relationships with research institutes, academic institutions and biotechnology companies, including the specific strategic relationships that we have developed with Israeli research and academic institutions, to identify and in-license promising therapeutic candidates. To date, we have successfully in-licensed compounds from major Israeli universities, as well as from Israeli hospitals, technology incubators and biotechnology companies. We continue to maintain close contacts with university technology transfer offices, research and development authorities, university faculty, and many biotechnology companies to actively seek out early stage compounds. In addition, we actively source and evaluate non-Israeli compounds.
|
|
|
•
|
Seek to co-develop certain pre-clinical and early clinical therapeutic projects through clinical proof-of-concept by means of our multi-year strategic collaboration agreement with Novartis. Novartis will evaluate Israeli-sourced projects identified and presented by us for co-development and potential future licensing under the collaboration. Pursuant to an agreement entered into in December 2014, Novartis will evaluate jointly with us both clinical and pre-clinical stage projects presented by us via a Joint Steering Committee, which will determine which projects to advance further in development and on what terms. Projects at or reaching the clinical stage will be eligible for selection by Novartis. Upon selection of a project, Novartis will pay us an option fee of $5 million, as well as fund 50% of the anticipated remaining development costs associated with establishing clinical proof-of-concept, in the form of an additional equity investment in BioLineRx. The companies intend to develop up to three programs pursuant to this collaboration. Under the terms of the agreement, Novartis acquired 5,000,000 ADSs of BioLineRx in a private transaction at a price of $2.00 per ADS for a total equity investment of $10 million and agreed to certain standstill provisions.
|
7
RISK FACTORS
Investing in our Ordinary Shares or ADSs involves a high degree of risk. You should carefully consider the specific risks described below, including, but not limited to, the risks included in our Current Report on Form 6-K filed March 5, 2015, before making an investment decision. Any of the risks we describe below could cause our business, financial condition or operating results to suffer. The market price of our Ordinary Shares and ADSs could decline if one or more of these risks and uncertainties develop into actual events. You could lose all or part of your investment.
Risks Related to Our Financial Condition and Capital Requirements
We are a clinical stage biopharmaceutical development company with a history of operating losses, expect to incur additional losses in the future and may never be profitable.
We are a clinical stage biopharmaceutical development company that was incorporated in 2003. Since our incorporation, we have been focused on research and development. Our most advanced therapeutic candidates are in clinical development. We, or our licensees, as applicable, will be required to conduct significant additional clinical trials before we or they can seek the regulatory approvals necessary to begin commercial sales of our therapeutic candidates. We have incurred losses since inception, principally as a result of research and development and general administrative expenses in support of our operations. We recorded net losses of approximately NIS 24.0 million ($6.5 million) in the nine months ended September 30, 2014, NIS 61.4 million ($17.7 million) in 2013 and NIS 76.3 million ($20.4 million) in 2012. As of September 30, 2014, we had an accumulated deficit of approximately NIS 529.8 million ($143.4 million). We anticipate that we will incur significant additional losses as we continue to focus our resources on prioritizing, selecting and advancing our most promising therapeutic candidates. We may never be profitable and we may never achieve significant sustained revenues.
We cannot ensure investors that our existing cash and investment balances will be sufficient to meet our future capital requirements.
As of September 30, 2014, we held cash and short-term investments of approximately $29.6 million. In December 2014, we received an additional $10.0 million in connection with the strategic collaboration agreement signed with Novartis. We believe that our existing cash and investment balances and other sources of liquidity, not including potential milestone and royalty payments under our out-licensing agreements with Bellerophon and Omega Pharma, will be sufficient to meet our requirements through the end of 2016. We have funded our operations primarily through public and private offerings of our securities and, until 2013, grants from the Office of the Chief Scientist of Israel’s Ministry of the Economy, or the OCS. In addition, we have funded our operations through out-licensing arrangements with respect to our therapeutic candidates. The adequacy of our available funds to meet our operating and capital requirements will depend on many factors including: the number, breadth, progress and results of our research, product development and clinical programs; the costs and timing of obtaining regulatory approvals for any of our therapeutic candidates; the terms and conditions of in-licensing and out-licensing therapeutic candidates; and costs incurred in enforcing and defending our patent claims and other intellectual property rights.
While we will continue to explore alternative financing sources, including the possibility of future securities offerings, government funding, and public and private grants, we cannot be certain that in the future these liquidity sources will be available when needed on commercially reasonable terms or at all, or that our actual cash requirements will not be greater than anticipated. We will also continue to seek to finance our operations through other sources, including out-licensing arrangements or other partnerships or joint ventures. If we are unable to obtain future financing through the methods we describe above or through other means, we may be unable to achieve our business objectives and may be unable to continue operations, which would have a material adverse effect on our business and financial condition.
8
Risks Related to Our Business and Regulatory Matters
If we or our licensees are unable to obtain U.S. and/or foreign regulatory approval for our therapeutic candidates, we will be unable to commercialize our therapeutic candidates.
To date, we have not marketed, distributed or sold an approved product. Currently, we have six clinical-stage therapeutic candidates in development: BL-1040 for the reduction or prevention of ventricular remodeling following an acute myocardial infarction, or AMI; BL-8040 for the treatment of acute myeloid leukemia, or AML, and other hematological indications; BL-7010 for the treatment of celiac disease; BL-5010 for the treatment of benign skin lesions; BL-7040 for the treatment of inflammatory bowel disease, or IBD; and BL-8020 for the treatment of the hepatitis C virus, or HCV, as well as other viral indications. Our therapeutic candidates are subject to extensive governmental regulations relating to development, clinical trials, manufacturing and commercialization of drugs and devices. We may not obtain marketing approval for any of our therapeutic candidates in a timely manner or at all. In connection with the trials for our clinical products and other therapeutic candidates that we are currently developing or may seek to develop in the future, either on our own or through out-licensing arrangements, we face the risk that:
|
|
·
|
a therapeutic candidate or medical device may not prove safe or efficacious;
|
|
|
·
|
the results with respect to any therapeutic candidate may not confirm the positive results from earlier preclinical studies or clinical trials;
|
|
|
·
|
the results may not meet the level of statistical significance required by the U.S. Food and Drug Administration, or FDA, or other regulatory authorities; and
|
|
|
·
|
the results will justify only limited and/or restrictive uses, including the inclusion of warnings and contraindications, which could significantly limit the marketability and profitability of the therapeutic candidate.
|
Any delay in obtaining, or the failure to obtain, required regulatory approvals will materially and adversely affect our ability to generate future revenues from a particular therapeutic candidate. Any regulatory approval to market a product may be subject to limitations on the indicated uses for which we may market the product or may impose restrictive conditions of use, including cautionary information, thereby limiting the size of the market for the product. We and our licensees, as applicable, also are, and will be, subject to numerous foreign regulatory requirements that govern the conduct of clinical trials, manufacturing and marketing authorization, pricing and third-party reimbursement. The foreign regulatory approval process includes all of the risks associated with the FDA approval process that we describe above, as well as risks attributable to the satisfaction of foreign requirements. Approval by the FDA does not ensure approval by regulatory authorities outside the United States. Foreign jurisdictions may have different approval processes than those required by the FDA and may impose additional testing requirements for our therapeutic candidates.
Clinical trials involve a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results.
We have limited experience in conducting and managing the clinical trials necessary to obtain regulatory approvals, including FDA approval. Clinical trials are expensive and complex, can take many years and have uncertain outcomes. We cannot predict whether we or our licensees will encounter problems with any of the completed, ongoing or planned clinical trials that will cause us, our licensees or regulatory authorities to delay or suspend clinical trials, or delay the analysis of data from completed or ongoing clinical trials. We estimate that clinical trials of our most advanced therapeutic candidates will continue for several years, but they may take significantly longer to complete. Failure can occur at any stage of the testing and we may experience numerous unforeseen events during, or as a result of, the clinical trial process that could delay or prevent commercialization of our current or future therapeutic candidates, including but not limited to:
|
|
·
|
delays in securing clinical investigators or trial sites for the clinical trials;
|
|
|
·
|
delays in obtaining institutional review board and other regulatory approvals to commence a clinical trial;
|
|
|
·
|
slower than anticipated patient recruitment and enrollment;
|
|
|
·
|
negative or inconclusive results from clinical trials;
|
9
|
|
·
|
unforeseen safety issues;
|
|
|
·
|
uncertain dosing issues;
|
|
|
·
|
an inability to monitor patients adequately during or after treatment; and
|
|
|
·
|
problems with investigator or patient compliance with the trial protocols.
|
A number of companies in the pharmaceutical, medical device and biotechnology industries, including those with greater resources and experience than us, have suffered significant setbacks in advanced clinical trials, even after seeing promising results in earlier clinical trials. Despite the results reported in earlier clinical trials for our therapeutic candidates, we do not know whether any phase 3 or other clinical trials we or our licensees may conduct will demonstrate adequate efficacy and safety to result in regulatory approval to market our therapeutic candidates. If later-stage clinical trials of any therapeutic candidate do not produce favorable results, our ability to obtain regulatory approval for the therapeutic candidate may be adversely impacted, which will have a material adverse effect on our business, financial condition and results of operations.
Even if we obtain regulatory approvals, our therapeutic candidates will be subject to ongoing regulatory review and if we fail to comply with continuing U.S. and applicable foreign regulations, we could lose those approvals and our business would be seriously harmed.
Even if products we or our licensees develop receive regulatory approval or clearance, we or our licensees, as applicable, will be subject to ongoing reporting obligations and the products and the manufacturing operations will be subject to continuing regulatory review, including FDA inspections. The results of this ongoing review may result in the withdrawal of a product from the market, the interruption of the manufacturing operations and/or the imposition of labeling and/or marketing limitations. Since many more patients are exposed to drugs and medical devices following their marketing approval, serious but infrequent adverse reactions that were not observed in clinical trials may be observed during the commercial marketing of the product. In addition, the manufacturer and the manufacturing facilities we or our licensees, as applicable, will use to produce any therapeutic candidate will be subject to periodic review and inspection by the FDA and other, similar foreign regulators. Later discovery of previously unknown problems with any product, manufacturer or manufacturing process, or failure to comply with regulatory requirements, may result in actions such as:
|
|
·
|
restrictions on such product, manufacturer or manufacturing process;
|
|
|
·
|
warning letters from the FDA or other regulatory authorities;
|
|
|
·
|
withdrawal of the product from the market;
|
|
|
·
|
suspension or withdrawal of regulatory approvals;
|
|
|
·
|
refusal to approve pending applications or supplements to approved applications that we or our licensees submit;
|
|
|
·
|
voluntary or mandatory recall;
|
|
|
·
|
fines;
|
|
|
·
|
refusal to permit the import or export of our products;
|
|
|
·
|
product seizure or detentions;
|
|
|
·
|
injunctions or the imposition of civil or criminal penalties; or
|
|
|
·
|
adverse publicity.
|
10
If we, or our licensees, suppliers, third party contractors, partners or clinical investigators are slow to adapt, or are unable to adapt, to changes in existing regulatory requirements or the adoption of new regulatory requirements or policies, we or our licensees may lose marketing approval for any of our products, if any of our therapeutic products are approved, resulting in decreased or lost revenue from milestones, product sales or royalties.
We rely on third parties to conduct our clinical trials and provide other services, and those third parties may not perform satisfactorily, including by failing to meet established deadlines for the completion of such services.
We do not have the ability to conduct certain preclinical studies and clinical trials independently for our therapeutic candidates, and we rely on third parties, such as contract laboratories, contract research organizations, medical institutions and clinical investigators to conduct these studies and our clinical trials. Our reliance on these third parties limits our control over these activities. The third-party contractors may not assign as great a priority to our clinical development programs or pursue them as diligently as we would if we were undertaking such programs directly. Accordingly, these third-party contractors may not complete activities on schedule, or may not conduct the studies or our clinical trials in accordance with regulatory requirements or with our trial design. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, or if their performance is substandard, we may be required to replace them. Although we believe that there are a number of other third-party contractors that we could engage to continue these activities, replacement of these third parties will result in delays. As a result, our efforts to obtain regulatory approvals for, and to commercialize, our therapeutic candidates may be delayed. The third-party contractors may also have relationships with other commercial entities, some of whom may compete with us. If the third-party contractors assist our competitors, our competitive position may be harmed.
In addition, our ability to bring future products to market depends on the quality and integrity of data that we present to regulatory authorities in order to obtain marketing authorizations. Although we attempt to audit and control the quality of third-party data, we cannot guarantee the authenticity or accuracy of such data, nor can we be certain that such data has not been fraudulently generated. The failure of these third parties to carry out their obligations would materially adversely affect our ability to develop and market new products and implement our strategies.
We depend on out-licensing arrangements to develop, market and commercialize our therapeutic candidates.
We depend on out-licensing arrangements to develop, market and commercialize our therapeutic candidates. We have limited experience in developing, marketing and commercializing therapeutic candidates. Dependence on out-licensing arrangements subjects us to a number of risks, including the risk that:
|
|
·
|
we may not be able to control the amount and timing of resources that our licensees devote to our therapeutic candidates;
|
|
|
·
|
our licensees may experience financial difficulties;
|
|
|
·
|
our licensees may fail to secure adequate commercial supplies of our therapeutic candidates upon marketing approval, if at all;
|
|
|
·
|
our future revenues depend heavily on the efforts of our licensees;
|
|
|
·
|
business combinations or significant changes in a licensee’s business strategy may adversely affect the licensee’s willingness or ability to complete its obligations under any arrangement with us;
|
|
|
·
|
a licensee could move forward with a competing therapeutic candidate developed either independently or in collaboration with others, including our competitors; and
|
|
|
·
|
out-licensing arrangements are often terminated or allowed to expire, which would delay the development and may increase the development costs of our therapeutic candidates.
|
In 2009, we entered into an exclusive, royalty-bearing worldwide out-licensing arrangement with Bellerophon with respect to BL-1040, which we amended in 2015. Under the arrangement, Bellerophon is obligated to use commercially reasonable efforts to complete clinical development of, and to commercialize, BL-1040 or a product related thereto. In December 2014, we entered into an exclusive out-licensing arrangement with Omega Pharma for the rights to BL-5010 for over-the-counter, or OTC, indications in the territories of Europe, Australia and additional selected countries. Under the arrangement, Omega Pharma is obligated to use commercially reasonable best efforts to obtain regulatory approval in the licensed territory for at least two OTC indications and to commercialize BL-5010 for those two OTC indications. In addition, we have co-development collaborations with partners for BL-8020, BL-8030 and BL-9020 whereby such partners have development and commercialization rights in certain territories.
11
If we or any of our licensees, including Bellerophon, Omega Pharma and our co-development partners, breach or terminate their agreements with us, or if any of our licensees otherwise fail to conduct their development and commercialization activities in a timely manner or there is a dispute about their obligations, we may need to seek other licensees, or we may have to develop our own internal sales and marketing capability for our therapeutic candidates. Our dependence on our licensees’ experience and the rights of our licensees will limit our flexibility in considering alternative out-licensing arrangements for our therapeutic candidates. Any failure to successfully develop these arrangements or failure by our licensees to successfully develop or commercialize any of our therapeutic candidates in a competitive and timely manner, will have a material adverse effect on the commercialization of our therapeutic candidates.
We depend on our ability to identify and in-license technologies and therapeutic candidates.
We employ a number of methods to identify therapeutic candidates that we believe are likely to achieve commercial success. In addition to our internal research and business developments efforts, we employ a rigorous screening system we developed. In addition, our Scientific Advisory Board and disease-specific third-party advisors evaluate each therapeutic candidate. However, there can be no assurance that our internal research efforts or our screening system will accurately or consistently select among various therapeutic candidates those that have the highest likelihood to achieve, and which ultimately achieve, commercial success. As a result, we may spend substantial resources developing therapeutic candidates that will not achieve commercial success and we may not advance those therapeutic candidates with the greatest potential for commercial success.
An important element of our strategy is maintaining relationships with universities, medical institutions and biotechnology companies in order to in-license potential therapeutic candidates. We may not be able to maintain relationships with these entities and they may elect not to enter into in-licensing agreements with us or to terminate existing agreements. Recently, a number of global pharmaceutical companies have set up operations in Israel, both with and without Israeli government funding, in order to identify and in-license new technologies. The presence of these global companies with significantly greater resources than we have may increase the competition with respect to the in-licensing of promising therapeutic candidates. We may not be able to acquire licenses on commercially reasonable terms, or at all. Failure to license or otherwise acquire necessary technologies could materially and adversely affect our business, financial condition and results of operations.
If we cannot meet requirements under our in-license agreements, we could lose the rights to our therapeutic candidates, which could have a material adverse effect on our business.
We depend on in-licensing agreements with third parties to maintain the intellectual property rights to our therapeutic candidates. Regarding the therapeutic candidates in clinical trials, we have in-licensed rights from B.G. Negev Technologies, the technology transfer company of Ben Gurion University, with respect to our BL-1040 therapeutic candidate; from Biokine Therapeutics Ltd., or Biokine, with respect to our BL-8040 therapeutic candidate; from Gestion Univalor, Limited Partnership, or Univalor, for our BL-7010 therapeutic candidate; from Innovative Pharmaceutical Concepts, Inc., or IPC, with respect to our BL-5010 therapeutic candidate; and from the Yissum Research Development Company of the Hebrew University of Jerusalem Ltd., or Yissum, with respect to our BL-7040 therapeutic candidate. See “Summary — Our Product Pipeline.” Our in-license agreements require us to make payments and satisfy performance obligations in order to maintain our rights under these agreements. The royalty rates and revenue sharing payments vary from case to case but generally range from 22% to 29.5% of the consideration we receive from sublicensing the applicable therapeutic candidate. In some instances, we are required to pay a substantially lower percentage (generally less than 5%) if we elect to commercialize the subject therapeutic candidate independently. Due to the relatively advanced stage of development of the compound licensed from Biokine, our license agreement with Biokine provides for royalty payments of between 40-60% of the consideration we receive from sublicensing and between 10-12% of net sales, subject to certain limitations, should we independently sell products. The amount of the royalty for either direct sales or sublicensing is dependent on the aggregate amount of our investment in connection with the Biokine agreement, decreasing as the amount of our investment in the project increases. These in-license agreements last either throughout the life of the patents that are the subject of the agreements, or with respect to other licensed technology, for a number of years after the first commercial sale of the relevant product.
12
In addition, we are responsible for the cost of filing and prosecuting certain patent applications and maintaining certain issued patents licensed to us. If we do not meet our obligations under our in-license agreements in a timely manner, we could lose the rights to our proprietary technology which could have a material adverse effect on our business, financial condition and results of operations.
Modifications to our therapeutic candidates, or to any other therapeutic candidates that we may develop in the future, may require new regulatory clearances or approvals or may require us or our licensees, as applicable, to recall or cease marketing these therapeutic candidates until clearances are obtained.
Modifications to our therapeutic candidates, after they have been approved for marketing, if at all, or to any other pharmaceutical product or medical device that we may develop in the future, may require new regulatory clearance, or approvals, and, if necessitated by a problem with a marketed product, may result in the recall or suspension of marketing of the previously approved and marketed product until clearances or approvals of the modified product are obtained. The FDA requires pharmaceutical products and device manufacturers to initially make and document a determination of whether or not a modification requires a new approval, supplement or clearance. A manufacturer may determine in conformity with applicable regulations and guidelines that a modification may be implemented without pre-clearance by the FDA; however, the FDA can review a manufacturer’s decision and may disagree. The FDA may also on its own initiative determine that a new clearance or approval is required. If the FDA requires new clearances or approvals of any pharmaceutical product or medical device for which we or our licensees receive marketing approval, if any, we or our licensees may be required to recall such product and to stop marketing the product as modified, which could require us or our licensees to redesign the product and will have a material adverse effect on our business, financial condition and results of operations. In these circumstances, we may be subject to significant enforcement actions.
If a manufacturer determines that a modification to an FDA-cleared device could significantly affect the safety or efficacy of the device, would constitute a major change in its intended use, or otherwise requires pre-clearance, the modification may not be implemented without the requisite clearance. We or our licensees may not be able to obtain those additional clearances or approvals for the modifications or additional indications in a timely manner, or at all. For those products sold in the European Union, or EU, we, or our licensees, as applicable, must notify the applicable EU Notified Body, an organization appointed by a member State of the EU either for the approval and monitoring of a manufacturer’s quality assurance system or for direct product inspection, if significant changes are made to the product or if there are substantial changes to the quality assurance systems affecting the product. Delays in obtaining required future clearances or approvals would materially and adversely affect our ability to introduce new or enhanced products in a timely manner, which in turn would have a material adverse effect on our business, financial condition and results of operations.
If our competitors develop and market products that are more effective, safer or less expensive than our current or future therapeutic candidates, our future prospects will be negatively impacted.
The life sciences industry is highly competitive, and we face significant competition from many pharmaceutical, biopharmaceutical and biotechnology companies that are researching and marketing products designed to address the indications for which we are currently developing therapeutic candidates or for which we may develop therapeutic candidates in the future. Specifically, we are aware of several other companies who currently market and/or are in the process of developing products that address AMI, AML, celiac disease, skin lesions, IBD, and HCV and other viral indications.
There are no generally accepted products approved for structural support to prevent cardiac remodeling following an AMI. One group of product candidates that are currently in clinical development includes stem cell therapies to restore heart muscle cells following an AMI, with large Phase 3 trials expected to be completed in 2018 or 2019. We do not expect BL-1040 to compete with, or replace, current treatments for congestive heart failure following AMI, but instead believe it will become part of the treatment regimen used in conjunction with other therapies. In addition, because BL-1040 can be delivered by a minimally invasive percutaneous coronary intervention procedure, we do not believe it will directly compete with devices that are used to treat congestive heart failure, which are designed for administration during open heart surgery or by intra-cardiac injection involving a thoracotomy procedure. These include mesh restraining devices, for example HeartNet™; injectable biopolymers, for example Algisyl LVR™; and implantable electro stimulation devices, for example, CardioFit™. In addition, volume reduction surgery or cardiac assist devices, or pumps, are sometimes used to treat patients with congestive heart failure.
13
Approved treatments for AML currently include chemotherapy (Doxorubicin, Cyclophosphamide, Vincristine), radiation therapy and stem cell transplantation. In addition there are a number of potentially competitive compounds under development that act as CXCR4 inhibitors, including, among others, AMD 3100 (Mozobil), which is being developed by Genzyme and Sanofi; LY-2510924, which is being developed by Lilly; Ulocuplumab (MDX-1338; BMS-936564) developed by Medarex and Bristol Myaers Squibb; F-50067 developed by Pierre Fabre; burixafor developed by TaiGen Biotechnology Co; and POL-6326 developed by Polyphor Ltd; PTX-9908 developed by Chemokine Therapeutics Corp. In addition there are a number of potentially competitive compounds under development to treat AML including, among others, Dacogen (decitabine), which is being developed by Eisai and Johnson & Johnson; Vidaza (azacitidine), which is being developed by Celgene; Vosaroxin, which is being developed by Sunesis Pharmaceuticals; Midostaurin, which is being developed by Novartis; Quizartinib, which is being developed by Ambit; Volasertib, which is being developed by Boehringer Ingelheim; fludarabine, which is being developed by Sanofi; nintedanib and BI-836858, both of which are under development by Boehringer Ingelheim; dasatinib (Sprycel) developed under BMS; RG-6016 under development by Roche; OCV-501, under development by Otsuka Pharmaceutical; ibrutinib developed by Pharmacyclics, under license from Celera, and in collaboration with Janssen Biotech; CPI-613 developed by Cornerstone Pharmaceuticals; F-14512 developed by Pierre Fabre; SL-401 developed by Stemline Therapeutics; pacritinib developed by CTI BioPharma Corp; sonidegib developed by Novartis; venetoclax developed by AbbVie; lirilumab developed by Innate Pharma in collaboration with BMS; selinexor developed by Karyopharm Therapeutics; Ganetespib developed by Synta Pharmaceuticals; crenolanib, which is being developed by Arog Pharmaceuticals, under license from Pfizer; BVD-ERK developed by BioMed Valley Discoveries; tosedostat developed by CTI BioPharma; pidilizumab developed by Medivation, under license from CureTech; sorafenib (Nexavar) developed by Bayer; Bortezomib developed by Janssen and Takeda; Uprosertib developed by GSK; PLX-3397 developed by Plexxikon Inc.; Lenalidomide developed by Celgene; erlotinib developed by Roche Astellas and Chugai; Trametinib developed by GSK; Vorinostat developed by Merck and Co.; Selumetinib developed by Astra Zeneca; SGI-110 developed by Astex Pharmaceuticals;; OCV-501 developed by Otsuka Pharmaceuticals; Birinapant developed by Tetralogic Pharmaceuticals; Alvocidib developed by Tolero Pharmaceuticals Inc; Pracinostat developed by MEI Pharma; Rigosertib developed by Onconova Therapeutics , Baxter International and Symbio; Sapacitabine developed by Cyclacel Pharmaceuticals; and RP-323 under development by Rich Pharmaceuticals. Some of these treatments are currently developed for specific AML patient populations and lines of treatment (e.g., AC220 developed by Ambit Biosciences) and not for the entire AML population. Some of these treatments can be developed for administration to AML patients in combination with BL-8040.
Several compounds are currently under development for celiac disease including larazotide acetate (Alba Therapeutic Corp.), which inhibits the activity of Zonulin; and latiglutenase (Alvine Pharmaceuticals Inc.), which is a combination of gluten targeting proteases and endopeptidases. Celiac patients are prescribed a gluten-free diet to relieve their disease symptoms. Nevertheless the symptoms persist in most cases despite the patient’s following a gluten-free diet. BL-7010, as well as the treatments specified above, is envisioned to be prescribed to patients who are on a gluten-free diet but still suffer from disease symptoms.
Skin lesions are generally removed using cryotherapy (liquid nitrogen), laser therapy, photodynamic therapy, electrodessication and curettage and several cream-based treatments. Picato (Leo Pharma) and Metvix® (Galderma Pharma) are cream-based treatments for skin lesions which have been approved in many countries.
IBD is often treated with currently marketed steroids, immunomodulators and immunomodulatory antibodies. Approved treatments for IBD currently include anti-TNFs, such as Remicade (infliximab, Janssen Biotech, Inc., a Johnson & Johnson company, Merck & Co. and Mitsubishi Tanabe Pharma), Humira (adalimumab, Abbott Laboratories and Eisai Co.), Cimzia (certolizumab, UCB, Inc.) and Simponi (golimumab, Janssen Biotech, Inc., Merck & Co. and Mitsubishi Tanabe Pharma), as well as antibodies inhibiting immune cell migration such as Tysabri (natalizumab, Biogen and Elan) and Vedolizumab (Takeda). In addition, there are generic brands of mesalazine, a 5-aminosalicylate, and the recently launched Budesonide MMX (Cosmo Pharmaceuticals, Ferring Pharmaceuticals and Santarus). The first biosimilar version of infliximab was approved for use in Europe in 2013. We are also aware of a number of potentially competitive compounds under development, including Xeljanz (tofacitinib, Pfizer Inc.), a Jak 1 inhibitor; Vedolizumab (Takeda, Millenium Pharmaceuticals), a MAdCAM inhibitor /integrin alpha-4/beta-7 antagonist; Ustekinomab (Johnson & Johnson), an anti-IL-12/IL23 mAb; JM-300 (Ajinomoto), an Integrin alpha-4/beta-7 antagonist; Etrolizumab a beta 7 targeting mAb developed by Roche; LP-02 developed by Lipid Therapeutics; and DIMS-0150 (Kappaproct) a TLR9-targeting oligo developed by InDex Pharmaceuticals.
14
HCV treatment consists of either a combination of interferon and ribavirin alone or together with a combination of direct anti-viral agents (DAAs) of several classes including NS3/4 protease inhibitors, NS5A inhibitors and NS5B inhibitors. Recently, treatment regimens that do not include interferon have been approved, and treatment regimens without ribavirin are at advanced stages of development. Approved anti-HCV treatments include Sovaldi (sofosbuvir) and Harvoni (a fixed combination of sofosbuvir and ledipasvir), both developed by Gilead Sciences; Viekira Pak (a fixed combination of paritaprevir/r + ombitasvir + dasabuvir ) developed by AbbVie; Olysio (simeprevir, Janssen Therapeutics and Medivir); Victrelis (boceprevir, Merck and Co); vaniprevir (developed by Merck and Co ); Incivek (telaprevir, Janssen Pharmaceuticals and Vertex Pharmaceuticals); asunaprevir and daclatasvir (developed by Bristol Myers Squibb); Compounds under development include: elbasvir (Merck and Co.) and ACH-3102 (developed by Achillion). BL-8020’s mechanism of action suggests that it could potentially be suitable for treatment of other viral infections, each of which has numerous competing treatments approved or in advanced stages of development.
An important element of our strategy for identifying future products is maintaining relationships with universities, medical institutions and biotechnology companies in order to in-license potential therapeutic candidates, and we compete with respect to this in-licensing with a number of global pharmaceutical companies, both with and without Israeli government funding. The presence of these global companies with significantly greater resources than we have may increase the competition with respect to the in-licensing of promising therapeutic candidates. Our failure to license or otherwise acquire necessary technologies could materially and adversely affect our business, financial condition and results of operations.
We and our contract manufacturers are, and will be, subject to FDA and other comparable agency regulations.
We and our contract manufacturers are, and will be, required to adhere to FDA regulations setting forth cGMP for drugs and Quality System Regulations, or QSR, for devices. These regulations cover all aspects of the manufacturing, testing, quality control and recordkeeping relating to our therapeutic candidates. We and our manufacturers may not be able to comply with applicable regulations. We and our manufacturers are and will be subject to unannounced inspections by the FDA, state regulators and similar regulators outside the United States. Our failure, or the failure of our third party manufacturers, to comply with applicable regulations could result in the imposition of sanctions on us, including fines, injunctions, civil penalties, failure of regulatory authorities to grant marketing approval of our therapeutic candidates, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of our candidates or products, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect regulatory approval and supplies of our therapeutic candidates, and materially and adversely affect our business, financial condition and results of operations.
We have no experience selling, marketing or distributing products and no internal capability to do so.
We currently have no sales, marketing or distribution capabilities and no experience in building a sales force or distribution capabilities. To be able to commercialize any of our therapeutic candidates upon approval, if at all, we must either develop internal sales, marketing and distribution capabilities, which will be expensive and time consuming, or enter into out-licensing arrangements with third parties to perform these services.
If we decide to market any of our other therapeutic candidates on our own, we must commit significant financial and managerial resources to develop a marketing and sales force with technical expertise and with supporting distribution capabilities. Factors that may inhibit our efforts to commercialize our products directly and without strategic partners include:
|
|
·
|
our inability to recruit and retain adequate numbers of effective sales and marketing personnel;
|
|
|
·
|
the inability of sales personnel to obtain access to or persuade adequate numbers of physicians to prescribe our therapeutic candidates;
|
15
|
|
·
|
the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines; and
|
|
|
·
|
unforeseen costs and expenses associated with creating and sustaining an independent sales and marketing organization.
|
We may not be successful in recruiting the sales and marketing personnel necessary to sell any of our therapeutic candidates upon approval, if at all, and even if we do build a sales force, it may not be successful in marketing our therapeutic candidates, which would have a material adverse effect on our business, financial condition and results of operations.
Our business could suffer if we are unable to attract and retain key employees.
Our success depends upon the continued service and performance of our senior management and other key personnel. The loss of the services of these personnel could delay or prevent the successful completion of our planned clinical trials or the commercialization of our therapeutic candidates or otherwise affect our ability to manage our company effectively and to carry out our business plan. We do not maintain key-man life insurance. Although we have entered into employment agreements with all of the members of our senior management team, members of our senior management team may resign at any time. High demand exists for senior management and other key personnel in the pharmaceutical industry. There can be no assurance that we will be able to continue to retain and attract such personnel.
Our growth and success also depend on our ability to attract and retain additional highly qualified scientific, technical, sales, managerial and finance personnel. We experience intense competition for qualified personnel, and the existence of non-competition agreements between prospective employees and their former employers may prevent us from hiring those individuals or subject us to suit from their former employers. In addition, if we elect to independently commercialize any therapeutic candidate, we will need to expand our marketing and sales capabilities. While we attempt to provide competitive compensation packages to attract and retain key personnel, many of our competitors are likely to have greater resources and more experience than we have, making it difficult for us to compete successfully for key personnel. If we cannot attract and retain sufficiently qualified technical employees on acceptable terms, we may not be able to develop and commercialize competitive products. Further, any failure to effectively integrate new personnel could prevent us from successfully growing our company.
We expect to rely upon third-party manufacturers to produce therapeutic supplies for phase 3 clinical trials, and commercialization, of our therapeutic candidates. If we manufacture any of our therapeutic candidates in the future, we will be required to incur significant costs and devote significant efforts to establish and maintain manufacturing capabilities.
We currently have laboratories that are compliant with both current good manufacturing practices, or cGMP, and Good Laboratory Practices, or GLP, and allow us to manufacture drug products for our current clinical trials. If we decide to perform any phase 3 clinical trial, or commercialize, any therapeutic candidate on our own, we anticipate that we will rely on third parties to produce the therapeutic supplies. We have limited personnel with experience in drug or medical device manufacturing and we lack the resources and capabilities to manufacture any of our therapeutic candidates on a commercial scale. The manufacture of pharmaceutical products and medical devices requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of pharmaceutical products and medical devices often encounter difficulties in production, particularly in scaling up initial production. These problems include difficulties with production costs and yields and quality control, including stability of the therapeutic candidate.
We do not currently have any long-term agreements with third party manufacturers for the supply of any of our therapeutic candidates. We believe that our current supply of therapeutic candidates is sufficient to complete our current clinical trials. However, if we require additional supplies of our therapeutic candidates to complete our clinical trials or if we elect to commercialize our products independently, we may be unable to enter into agreements for clinical or commercial supply, as applicable, with third party manufacturers, or may be unable to do so on acceptable terms. Even if we enter into these agreements, it is likely that the manufacturers of each therapeutic candidate will be single source suppliers to us for a significant period of time.
16
Reliance on third party manufacturers entails risks to which we would not be subject if we manufactured therapeutic candidates ourselves, including:
|
|
·
|
reliance on the third party for regulatory compliance and quality assurance;
|
|
|
·
|
limitations on supply availability resulting from capacity and scheduling constraints of the third parties;
|
|
|
·
|
impact on our reputation in the marketplace if manufacturers of our products, once commercialized, fail to meet customer demands;
|
|
|
·
|
the possible breach of the manufacturing agreement by the third party because of factors beyond our control; and
|
|
|
·
|
the possible termination or nonrenewal of the agreement by the third party, based on its own business priorities, at a time that is costly or inconvenient for us.
|
The failure of any of our contract manufacturers to maintain high manufacturing standards could result in injury or death of clinical trial participants or patients being treated with our products. Such failure could also result in product liability claims, product recalls, product seizures or withdrawals, delays or failures in testing or delivery, cost overruns or other problems, which would have a material adverse effect on our business, financial condition and results of operations.
Risks Related to Our Industry
Even if our therapeutic candidates receive regulatory approval or do not require regulatory approval, they may not become commercially viable products.
Even if our therapeutic candidates are approved for commercialization, they may not become commercially viable products. For example, if we or our licensees receive regulatory approval to market a product, approval may be subject to limitations on the indicated uses or subject to labeling or marketing restrictions which could materially and adversely affect the marketability and profitability of the product. In addition, a new product may appear promising at an early stage of development or after clinical trials but never reach the market, or it may reach the market but not result in sufficient product sales, if any. A therapeutic candidate may not result in commercial success for various reasons, including:
|
|
·
|
difficulty in large-scale manufacturing;
|
|
|
·
|
low market acceptance by physicians, healthcare payors, patients and the medical community as a result of lower demonstrated clinical safety or efficacy compared to other products, prevalence and severity of adverse side effects, or other potential disadvantages relative to alternative treatment methods;
|
|
|
·
|
insufficient or unfavorable levels of reimbursement from government or third-party payors;
|
|
|
·
|
infringement on proprietary rights of others for which we or our licensees have not received licenses;
|
|
|
·
|
incompatibility with other therapeutic products;
|
|
|
·
|
other potential advantages of alternative treatment methods;
|
|
|
·
|
ineffective marketing and distribution support;
|
|
|
·
|
lack of cost-effectiveness; or
|
|
|
·
|
timing of market introduction of competitive products.
|
17
If we are unable to develop commercially viable products, either on our own or through licensees, our business, results of operations and financial condition will be materially and adversely affected.
We could be adversely affected if healthcare reform measures substantially change the market for medical care or healthcare coverage in the United States.
In 2010, the U.S. Congress adopted the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act (collectively, the PPACA), important legislation regarding health insurance which may have far-reaching consequences for most health care companies, including biopharmaceutical companies like us. Under the new legislation, substantial changes are going to be made to the current system for paying for healthcare in the United States, including changes made in order to extend medical benefits to those who currently lack insurance coverage.
Extending coverage to a large population could substantially change the structure of the health insurance system and the methodology for reimbursing medical services, drugs and devices. These structural changes could entail modifications to the existing system of private payors and government programs (Medicare, Medicaid and State Children’s Health Insurance Program), creation of a government-sponsored healthcare insurance source, or some combination of both, as well as other changes. Restructuring the coverage of medical care in the United States could impact the reimbursement for prescribed drugs and biopharmaceuticals, such as those we and our licensees are currently developing. If reimbursement for our approved products, if any, is substantially reduced in the future, or rebate obligations associated with them are substantially increased, our business could be materially and adversely impacted.
Extending medical benefits to those who currently lack coverage will likely result in substantial cost to the U.S. federal government, which may force significant changes to the healthcare system in the United States. Much of the funding for expanded healthcare coverage may be sought through cost savings. While some of these savings may come from realizing greater efficiencies in delivering care, improving the effectiveness of preventive care and enhancing the overall quality of care, much of the cost savings may come from reducing the cost of care.
Cost of care could be reduced by decreasing the level of reimbursement for medical services or products (including those biopharmaceuticals currently being developed by us or our licensees), or by restricting coverage (and, thereby, utilization) of medical services or products. In either case, a reduction in the utilization of, or reimbursement for, any product for which we receive marketing approval in the future could have a materially adverse effect on our financial performance.
The PPACA also requires the medical device industry to subsidize healthcare reform in the form of a 2.3% excise tax on U.S. sales of certain medical devices beginning January 1, 2013 and also includes new regulatory mandates and other measures designed to constrain medical costs, as well as stringent new reporting requirements of financial relationships between device manufacturers and physicians and hospitals.
If third-party payors do not adequately reimburse customers for any of our therapeutic candidates that are approved for marketing, they might not be purchased or used, and our revenues and profits will not develop or increase.
Our revenues and profits will depend heavily upon the availability of adequate reimbursement for the use of our approved candidates, if any, from governmental or other third-party payors, both in the United States and in foreign markets. Reimbursement by a third-party payor may depend upon a number of factors, including the third-party payor’s determination that the use of an approved product is:
|
|
·
|
a covered benefit under its health plan;
|
|
|
·
|
safe, effective and medically necessary;
|
|
|
·
|
appropriate for the specific patient;
|
|
|
·
|
cost-effective; and
|
|
|
·
|
neither experimental nor investigational.
|
18
Obtaining reimbursement approval for a product from each government or other third-party payor is a time-consuming and costly process that could require us or our licensees to provide supporting scientific, clinical and cost-effectiveness data for the use of our products to each payor. Even when a payor determines that a product is eligible for reimbursement, the payor may impose coverage limitations that preclude payment for some uses that are approved by the FDA or comparable foreign regulatory authorities. Reimbursement rates may vary according to the use of the product and the clinical setting in which it used, may be based on payments allowed for lower-cost products that are already reimbursed, may be incorporated into existing payments for other products or services, and may reflect budgetary constraints and/or imperfections in Medicare, Medicaid or other data used to calculate these rates.
Regardless of the impact of the PPACA on us, the U.S. government, other governments and commercial payors have shown significant interest in pursuing healthcare reform and reducing healthcare costs. Any government-adopted reform measures could cause significant pressure on the pricing of healthcare products and services, including those biopharmaceuticals currently being developed by us or our licensees, in the United States and internationally, as well as the amount of reimbursement available from governmental agencies or other third party payors. The continuing efforts of the U.S. and foreign governments, insurance companies, managed care organizations and other payors to contain or reduce healthcare costs may compromise our ability to set prices at commercially attractive levels for our products that we may develop, which in turn could adversely impact how much or under what circumstances healthcare providers will prescribe or administer our products, if approved. Changes in healthcare policy, such as the creation of broad limits for diagnostic products, could substantially diminish the sale of or inhibit the utilization of diagnostic tests, increase costs, divert management’s attention and adversely affect our ability to generate revenues and achieve consistent profitability. This could materially and adversely impact our business by reducing our ability to generate revenue, raise capital, obtain additional collaborators and market our products, if approved.
Further, the Centers for Medicare and Medicaid Services, or CMS, frequently change product descriptors, coverage policies, product and service codes, payment methodologies and reimbursement values. Third-party payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates, and both CMS and other third-party payors may have sufficient market power to demand significant price reductions.
Our business has a substantial risk of clinical trial and product liability claims. If we are unable to obtain and maintain appropriate levels of insurance, a claim could adversely affect our business.
Our business exposes us to significant potential clinical trial and product liability risks that are inherent in the development, manufacturing and sales and marketing of human therapeutic products. Although we do not currently commercialize any products, claims could be made against us based on the use of our therapeutic candidates in clinical trials. We currently carry life science liability insurance covering general liability with a coverage amount of $10.0 million per occurrence, products liability with an annual coverage amount of $10.0 million in the aggregate, and clinical trial insurance with a coverage amount of $10.0 million in the aggregate. The maximum indemnity for a single occurrence, claim or circumstance under this insurance is $10.0 million. In addition to this policy, we carry excess liability insurance with a coverage amount of $10.0 million which increases the coverage limit provided by our life science insurance package. However, our insurance may not provide adequate coverage against potential liabilities. Furthermore, clinical trial and product liability insurance is becoming increasingly expensive. As a result, we may be unable to maintain current amounts of insurance coverage or obtain additional or sufficient insurance at a reasonable cost to protect against losses that could have a material adverse effect on us. If a claim is brought against us, we might be required to pay legal and other expenses to defend the claim, as well as damages awards beyond the coverage of our insurance policies resulting from a claim brought successfully against us. Furthermore, whether or not we are ultimately successful in defending any claims, we might be required to direct significant financial and managerial resources to such defense, and adverse publicity is likely to result.
19
We deal with hazardous materials and must comply with environmental, health and safety laws and regulations, which can be expensive and restrict how we do business.
Our activities and those of our third-party manufacturers on our behalf involve the controlled storage, use and disposal of hazardous materials, including microbial agents, corrosive, explosive and flammable chemicals and other hazardous compounds. We and our manufacturers are subject to U.S. federal, state, local, Israeli and other foreign laws and regulations governing the use, manufacture, storage, handling and disposal of these hazardous materials. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards prescribed by these laws and regulations, we cannot eliminate the risk of accidental contamination or injury from these materials. In addition, if we develop a manufacturing capacity, we may incur substantial costs to comply with environmental regulations and would be subject to the risk of accidental contamination or injury from the use of hazardous materials in our manufacturing process.
In the event of an accident, government authorities may curtail our use of these materials and interrupt our business operations. In addition, we could be liable for any civil damages that result, which may exceed our financial resources and may seriously harm our business. Although our Israeli insurance program covers certain unforeseen sudden pollutions, we do not maintain a separate insurance policy for any of the foregoing types of risks. In addition, although the general liability section of our life sciences policy covers certain unforeseen, sudden environmental issues, pollution in the United States and Canada is excluded from the policy. In the event of environmental discharge or contamination or an accident, we may be held liable for any resulting damages, and any liability could exceed our resources. In addition, we may be subject to liability and may be required to comply with new or existing environmental laws regulating pharmaceuticals or other medical products in the environment.
Risks Related to Intellectual Property
Our access to most of the intellectual property associated with our therapeutic candidates results from in-license agreements with universities, research institutions and biotechnology companies, the termination of which would prevent us from commercializing the associated therapeutic candidates.
We do not conduct our own initial research with respect to the identification of our therapeutic candidates. Instead, we rely upon research and development work conducted by third parties as the primary source of our therapeutic candidates. As such, we have obtained our rights to our therapeutic candidates through in-license agreements entered into with universities, research institutions and biotechnology companies that invent and own the intellectual property underlying our candidates. There is no assurance that such in-licenses or rights will not be terminated or expire due to a material breach of the agreements, such as a failure on our part to achieve certain progress milestones set forth in the terms of the in-licenses or due to the loss of the rights to the underlying intellectual property by any of our licensors. There is no assurance that we will be able to renew or renegotiate an in-licensing agreement on acceptable terms if and when the agreement terminates. We cannot guarantee that any in-license is enforceable or will not be terminated or converted into a non-exclusive license in the future. The termination of any in-license or our inability to enforce our rights under any in-license would materially and adversely affect our ability to commercialize certain of our therapeutic candidates.
We currently have in-licensing agreements relating to our lead therapeutic candidates under clinical development. In January 2005, we in-licensed the rights to BL-1040 under a license agreement with B.G. Negev Technologies. Under the BL-1040 license agreement, we are obligated to use commercially reasonable efforts to develop the licensed technology in accordance with a specified development plan, including meeting certain specified diligence goals. In September 2012, we in-licensed the rights to BL-8040 under a license agreement from Biokine. Under the BL-8040 license agreement, we are obligated to make commercially reasonable, good faith efforts to sublicense or commercialize BL-8040 for fair consideration. In February 2011, we in-licensed the rights to BL-7010 from Univalor. Under the BL-7010 license agreement, we are obligated to use commercially reasonable efforts to develop the licensed technology in accordance with a specified development plan, including meeting certain specified diligence goals. In November 2007, we in-licensed the rights to BL-5010 under a license agreement with IPC. Under the BL-5010 license agreement, we are obligated to use commercially reasonable efforts to develop the licensed technology in accordance with a specified development plan, including meeting certain specified diligence goals. In June 2011, we in-licensed the rights to BL-7040 under a license agreement from Yissum. Under the BL-7040 license agreement, we are responsible for, and are required to exert, reasonable commercial efforts to carry out the development, regulatory, manufacturing, and marketing work necessary to develop and commercialize products under the agreement in accordance with a specified development plan. In January 2012, we in-licensed the rights to BL-8020 under a license agreement from Panmed and Genoscience. Under the BL-8020 license agreement, we were obligated to use commercially reasonable efforts to develop and commercialize the licensed technology in accordance with a specified development plan. Due to a number of considerations, including a re-prioritization of our pipeline, we agreed with the licensors that as of April 1, 2014, the license agreement would be terminated and that we would enter into a collaboration agreement whereby, among other things, the licensors agreed to take over development of the drug in consideration for 28% of future sublicense receipts by the licensors, and we agreed to supply, at the licensors’ request and in consideration for full payment, the drug needed for a clinical trial to be administered by the licensors.
20
Each of the foregoing in-licensing agreements, or the obligation to pay royalties thereunder, will generally remain in effect until the expiration, under the applicable agreement, of all of the licensing, royalty and sublicense revenue obligations to the applicable licensors, determined on a product-by-product and country-by-country basis. We may terminate the BL-1040 in-licensing agreement by providing 60 days’ prior written notice to B.G. Negev Technologies. We may terminate the BL-8040 in-licensing agreement upon 90 days’ prior written notice to Biokine. We may terminate the BL-7010 in-licensing agreement, the BL-5010 in-licensing agreement or the BL-7040 in-licensing agreement upon 30 days’ prior written notice to the respective licensor.
Any party to any of the foregoing in-licensing agreements may terminate the respective agreement for material breach by the other party if the breaching party is unable to cure the breach within an agreed upon period, generally 30 days to 90 days, after receiving written notice of the breach from the non-breaching party. Each of the foregoing in-licensing agreements provide that with respect to any termination for material breach, if the breach is not susceptible to cure within the stated period and the breaching party uses diligent, good faith efforts to cure such breach, the stated period will be extended by an additional 30 days. In addition, either party to one of the foregoing in-licensing agreements may terminate the agreement upon notice to the other upon the occurrence of certain bankruptcy events.
Patent protection for our products is important and uncertain.
Our success depends, in part, on our ability, and the ability of our licensees and licensors to obtain patent protection for our therapeutic candidates, maintain the confidentiality of our trade secrets and know how, operate without infringing on the proprietary rights of others and prevent others from infringing our proprietary rights.
We try to protect our proprietary position by, among other things, filing U.S., European, Israeli and other patent applications related to our proprietary products, technologies, inventions and improvements that may be important to the continuing development of our therapeutic candidates. As of December 31, 2014 we owned or exclusively licensed for uses within our field of business 18 patent families that, collectively, contain 54 issued patents, three allowed patent applications and over 40 pending patent applications relating to our clinical candidates. We are also pursuing patent protection for other drug candidates in our pipeline.
Because the patent position of biopharmaceutical companies involves complex legal and factual questions, we cannot predict the validity and enforceability of patents with certainty. Our issued patents and the issued patents of our licensees or licensors may not provide us with any competitive advantages, or may be held invalid or unenforceable as a result of legal challenges by third parties. Thus, any patents that we own or license from others may not provide any protection against competitors. Our pending patent applications, those we may file in the future or those we may license from third parties may not result in patents being issued. If these patents are issued, they may not provide us with proprietary protection or competitive advantages against competitors with similar technology. The degree of future protection to be afforded by our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage.
Patent rights are territorial; thus, the patent protection we do have will only extend to those countries in which we have issued patents. Even so, the laws of certain countries do not protect our intellectual property rights to the same extent as do the laws of the United States and Israel. For example, the patent laws of China and India are relatively new and are not as developed as are older, more established patent laws of other countries. Competitors may successfully challenge our patents, produce similar drugs or products that do not infringe our patents, or produce drugs in countries where we have not applied for patent protection or that do not respect our patents. Furthermore, it is not possible to know the scope of claims that will be allowed in published applications and it is also not possible to know which claims of granted patents, if any, will be deemed enforceable in a court of law.
21
Our technology may infringe the rights of third parties. The nature of claims contained in unpublished patent filings around the world is unknown to us and it is not possible to know which countries patent holders may choose for the extension of their filings under the Patent Cooperation Treaty, or other mechanisms. Any infringement by us of the proprietary rights of third parties may have a material adverse effect on our business, financial condition and results of operations.
If we are unable to protect the confidentiality of our trade secrets or know-how, such proprietary information may be used by others to compete against us.
We rely on a combination of patents, trade secrets, know-how, technology, trademarks and regulatory exclusivity to maintain our competitive position. We generally try to protect trade secrets, know-how and technology by entering into confidentiality or non-disclosure agreements with parties that have access to it, such as our licensees, employees, contractors and consultants. We also enter into agreements that purport to require the disclosure and assignment to us of the rights to the ideas, developments, discoveries and inventions of our employees, advisors, research collaborators, contractors and consultants while we employ or engage them. However, these agreements can be difficult and costly to enforce or may not provide adequate remedies. Any of these parties may breach the confidentiality agreements and willfully or unintentionally disclose our confidential information, or our competitors might learn of the information in some other way. The disclosure to, or independent development by, a competitor of any trade secret, know-how or other technology not protected by a patent could materially adversely affect any competitive advantage we may have over any such competitor.
To the extent that any of our employees, advisors, research collaborators, contractors or consultants independently develop, or use independently developed, intellectual property in connection with any of our projects, disputes may arise as to the proprietary rights to this type of information. If a dispute arises with respect to any proprietary right, enforcement of our rights can be costly and unpredictable and a court may determine that the right belongs to a third party.
Legal proceedings or third-party claims of intellectual property infringement may require us to spend substantial time and money and could prevent us from developing or commercializing products.
The development, manufacture, use, offer for sale, sale or importation of our therapeutic candidates may infringe on the claims of third-party patents. A party might file an infringement action against us. The cost to us of any patent litigation or other proceeding, even if resolved in our favor, could be substantial. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively because of their substantially greater financial resources. Uncertainties resulting from the initiation and continuation or defense of a patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace. Patent litigation and other proceedings may also absorb significant management time. Consequently, we are unable to guarantee that we will be able to manufacture, use, offer for sale, sell or import our therapeutic candidates in the event of an infringement action. At present, we are not aware of pending or threatened patent infringement actions against us.
In the event of patent infringement claims, or to avoid potential claims, we may choose or be required to seek a license from a third party and would most likely be required to pay license fees or royalties or both. These licenses may not be available on acceptable terms, or at all. Even if we were able to obtain a license, the rights may be non-exclusive, which could potentially limit our competitive advantage. Ultimately, we could be prevented from commercializing a therapeutic candidate or be forced to cease some aspect of our business operations if, as a result of actual or threatened patent infringement claims, we are unable to enter into licenses on acceptable terms. This inability to enter into licenses could harm our business significantly. At present, we have not received any written demands from third parties that we take a license under their patents nor have we received any notice form a third party accusing us of patent infringement.
Our license agreements with our licensees, including Bellerophon, Omega Pharma and our other co-development partners, contain, and any contract that we enter into with licensees in the future will likely contain, indemnity provisions that obligate us to indemnify the licensee against any losses arising from infringement of third party intellectual property rights. In addition, our in-license agreements contain provisions that obligate us to indemnify the licensors against any damages arising from the development, manufacture and use of products developed on the basis of the in-licensed intellectual property.
22
We may be subject to other patent-related litigation or proceedings that could be costly to defend and uncertain in their outcome.
In addition to infringement claims against us, we may in the future become a party to other patent litigation or proceedings, including interference or re- examination proceedings filed with the U.S. Patent and Trademark Office or opposition proceedings in other foreign patent offices regarding intellectual property rights with respect to our products and technology, as well as other disputes regarding intellectual property rights with licensees, licensors or others with whom we have contractual or other business relationships. Post-issuance oppositions are not uncommon and we, our licensee or our licensor will be required to defend these opposition procedures as a matter of course. Opposition procedures may be costly, and there is a risk that we may not prevail.
In July 2014 and October 2014, Bellerophon was notified by the European Patent Office in July 2014 and October 2014 that Notices of Opposition to two European patents that Bellerophon licensed from us, one of which covers the BCM intended commercial product described above, have been filed with the European Patent Office. A Notice of Opposition initiates a process during which the European Patent Office can decide to reconsider an issued patent and modify or revoke some or all of the patent claims. As our licensee, Bellerophon has the right to respond to the Notices of Opposition before the European Patent Office makes a decision whether or not any or all of the patent claims are to be modified or revoked. Bellerophon filed a response to the first patent opposition in December 2014 and intends to file a response in the near future for the second patent opposition as Bellerophon and BioLineRx believe the two issued patents were properly examined and appropriately granted by the European Patent Office. Furthermore, Bellerophon and BioLineRx believe the arguments made in the Notices of Opposition misstate the facts and lack scientific merit.
We may be subject to damages resulting from claims that we or our employees or contractors have wrongfully used or disclosed alleged trade secrets of their former employers.
Many of our employees and contractors were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that we or any employee or contractor has inadvertently or otherwise used or disclosed trade secrets or other proprietary information of his or her former employers. Litigation may be necessary to defend against these claims. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. A loss of key research personnel or their work product could hamper or prevent our ability to commercialize certain therapeutic candidates, which could severely harm our business, financial condition and results of operations. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
Risks Related to our Ordinary Shares and ADSs
We may be a passive foreign investment company, or PFIC, for U.S. federal income tax purposes in 2015 or in any subsequent year. There may be negative tax consequences for U.S. taxpayers that are holders of our Ordinary Shares or our ADSs.
We will be treated as a PFIC for U.S. federal income tax purposes in any taxable year in which either (i) at least 75% of our gross income is “passive income” or (ii) on average at least 50% of our assets by value produce passive income or are held for the production of passive income. Passive income for this purpose generally includes, among other things, certain dividends, interest, royalties, rents and gains from commodities and securities transactions and from the sale or exchange of property that gives rise to passive income. Passive income also includes amounts derived by reason of the temporary investment of funds, including those raised in a public offering. In determining whether a non-U.S. corporation is a PFIC, a proportionate share of the income and assets of each corporation in which it owns, directly or indirectly, at least a 25% interest (by value) is taken into account. We believe that we were a PFIC during certain prior years and, although we have not determined whether we will be a PFIC in 2015, or in any subsequent year, our operating results for any such years may cause us to be a PFIC. If we are a PFIC in 2015, or any subsequent year, and a U.S. shareholder does not make an election to treat us as a “qualified electing fund,” or QEF, or make a “mark-to-market” election, then “excess distributions” to a U.S. shareholder, and any gain realized on the sale or other disposition of our Ordinary Shares or ADSs will be subject to special rules. Under these rules: (i) the excess distribution or gain would be allocated ratably over the U.S. shareholder’s holding period for the Ordinary Shares (or ADSs, as the case may be); (ii) the amount allocated to the current taxable year and any period prior to the first day of the first taxable year in which we were a PFIC would be taxed as ordinary income; and (iii) the amount allocated to each of the other taxable years would be subject to tax at the highest rate of tax in effect for the applicable class of taxpayer for that year, and an interest charge for the deemed deferral benefit would be imposed with respect to the resulting tax attributable to each such other taxable year. In addition, if the U.S. Internal Revenue Service, or the IRS, determines that we are a PFIC for a year with respect to which we have determined that we were not a PFIC, it may be too late for a U.S. shareholder to make a timely QEF or mark-to-market election. U.S. shareholders who hold our Ordinary Shares or ADSs during a period when we are a PFIC will be subject to the foregoing rules, even if we cease to be a PFIC in subsequent years, subject to exceptions for U.S. shareholders who made a timely QEF or mark-to-market election. A U.S. shareholder can make a QEF election by completing the relevant portions of and filing IRS Form 8621 in accordance with the instructions thereto. A QEF election generally may not be revoked without the consent of the IRS. Upon request, we will annually furnish U.S. shareholders with information needed in order to complete IRS Form 8621 (which form would be required to be filed with the IRS on an annual basis by the U.S. shareholder) and to make and maintain a valid QEF election for any year in which we or any of our subsidiaries are a PFIC.
23
The market prices of our Ordinary Shares and ADSs are subject to fluctuation, which could result in substantial losses by our investors.
The stock market in general and the market prices of our Ordinary Shares on the TASE and ADSs on the Nasdaq, in particular, are subject to fluctuation, and changes in these prices may be unrelated to our operating performance. We expect that the market prices of our Ordinary Shares and ADSs will continue to be subject to wide fluctuations. The market price of our Ordinary Shares and ADSs are and will be subject to a number of factors, including:
|
|
·
|
announcements of technological innovations or new products by us or others;
|
|
|
·
|
announcements by us of significant acquisitions, strategic partnerships, in-licensing, out-licensing, joint ventures or capital commitments;
|
|
|
·
|
expiration or terminations of licenses, research contracts or other collaboration agreements;
|
|
|
·
|
public concern as to the safety of drugs we, our licensees or others develop;
|
|
|
·
|
general market conditions;
|
|
|
·
|
the volatility of market prices for shares of biotechnology companies generally;
|
|
|
·
|
success of research and development projects;
|
|
|
·
|
departure of key personnel;
|
|
|
·
|
developments concerning intellectual property rights or regulatory approvals;
|
|
|
·
|
variations in our and our competitors’ results of operations;
|
|
|
·
|
changes in earnings estimates or recommendations by securities analysts, if our Ordinary Shares or ADSs are covered by analysts;
|
|
|
·
|
statements about the Company made in the financial media or by bloggers on the Internet;
|
|
|
·
|
changes in government regulations or patent decisions;
|
|
|
·
|
developments by our licensees; and
|
|
|
·
|
general market conditions and other factors, including factors unrelated to our operating performance.
|
24
These factors and any corresponding price fluctuations may materially and adversely affect the market price of our Ordinary Shares and result in substantial losses by our investors.
Additionally, market prices for securities of biotechnology and pharmaceutical companies historically have been very volatile. The market for these securities has from time to time experienced significant price and volume fluctuations for reasons unrelated to the operating performance of any one company. In the past, following periods of market volatility, shareholders have often instituted securities class action litigation. If we were involved in securities litigation, it could have a substantial cost and divert resources and attention of management from our business, even if we are successful.
Our Ordinary Shares are traded on the TASE and our ADSs are listed on the Nasdaq Capital Market. Trading in our securities on these markets takes place in different currencies (dollars on the Nasdaq Capital Market and NIS on the TASE), and at different times (resulting from different time zones, different trading days and different public holidays in the United States and Israel). The trading prices of our securities on these two markets may differ due to these factors, the factors listed above, or other factors. Any decrease in the price of our securities on one of these markets could cause a decrease in the trading price of our securities on the other market.
Future sales of our Ordinary Shares or ADSs could reduce the market price of our Ordinary Shares and ADSs.
Substantial sales of our Ordinary Shares or ADSs, either on the TASE or on the Nasdaq, may cause the market price of our Ordinary Shares or ADSs to decline. Sales by us or our securityholders of substantial amounts of our Ordinary Shares or ADSs, or the perception that these sales may occur in the future, could cause a reduction in the market price of our Ordinary Shares or ADSs.
As a result of previous financings, we have warrants outstanding for the purchase of approximately 4,200,000 ADSs at an average exercise price of $3.71 per ADS. In addition we have stock options granted to directors, employees and consultants for the purchase of approximately 32,500,000 Ordinary Shares with an average exercise price of $0.25 per Ordinary Share (equivalent to 3,250,000 ADSs with an average exercise price of approximately $2.45 per ADS).
In May 2014, we entered into a purchase agreement with Lincoln Park Capital Fund, LLC, or LPC, for the sale, from time to time, of up to $20 million of our ADSs to LPC. During the 36-month term of this purchase agreement, we control the timing and amount of any sales to LPC, if and when we decide, in accordance with the agreement. LPC has no right to require us to sell any ADSs to LPC, but LPC is obligated to make purchases as we direct, subject to certain conditions. The purchase price related to any sales to LPC is based on the prevailing market prices of our ADSs immediately preceding the notice of sale to LPC, without any fixed discount. The agreement may be terminated by us at any time, at our sole discretion, without any cost or penalty. As of the date of this annual report, we have not yet sold any ADSs to LPC under the purchase agreement.
The issuance of any additional Ordinary Shares, any additional ADSs, or any securities that are exercisable for or convertible into our Ordinary Shares or ADSs, may have an adverse effect on the market price of our Ordinary Shares and ADSs and will have a dilutive effect on our shareholders.
Raising additional capital by issuing securities may cause dilution to existing shareholders.
We may need to raise substantial future capital to continue to complete clinical development and commercialize our products and therapeutic candidates and to conduct the research and development and clinical and regulatory activities necessary to bring our therapeutic candidates to market. Our future capital requirements will depend on many factors, including:
|
|
·
|
the failure to obtain regulatory approval or achieve commercial success of our therapeutic candidates, including BL-1040, BL-8040, BL-7010, BL-5010, BL-7040 and BL-8020;
|
25
|
|
·
|
our success in effecting out-licensing arrangements with third-parties;
|
|
|
·
|
our success in establishing other out-licensing arrangements;
|
|
|
·
|
the success of our licensees in selling products that utilize our technologies;
|
|
|
·
|
the results of our preclinical studies and clinical trials for our earlier stage therapeutic candidates, and any decisions to initiate clinical trials if supported by the preclinical results;
|
|
|
·
|
the costs, timing and outcome of regulatory review of our therapeutic candidates that progress to clinical trials;
|
|
|
·
|
the costs of establishing or acquiring specialty sales, marketing and distribution capabilities, if any of our therapeutic candidates are approved, and we decide to commercialize them ourselves;
|
|
|
·
|
the costs of preparing, filing and prosecuting patent applications, maintaining and enforcing our issued patents and defending intellectual property-related claims;
|
|
|
·
|
the extent to which we acquire or invest in businesses, products or technologies and other strategic relationships; and
|
|
|
·
|
the costs of financing unanticipated working capital requirements and responding to competitive pressures.
|
If we raise additional funds through licensing arrangements with third parties, we may have to relinquish valuable rights to our therapeutic candidates, or grant licenses on terms that are not favorable to us. If we raise additional funds by issuing equity or convertible debt securities, we will reduce the percentage ownership of our then-existing shareholders, and these securities may have rights, preferences or privileges senior to those of our existing shareholders. Following this offering, we will have a limited amount of authorized ordinary shares available under our Articles of Association. Therefore, in order to issue additional ordinary shares or ADSs in the future, we would be required to seek the approval of our shareholders to increase the amount of authorized ordinary shares. If our shareholders vote against such a proposal, it would limit our ability to raise equity capital in the future, and could have an adverse effect on our business and operations. See also “— Future sales of our Ordinary Shares or ADSs could reduce the market price of our Ordinary Shares and ADSs.”
As a foreign private issuer, we are permitted to follow certain home country corporate governance practices instead of applicable SEC and Nasdaq requirements, which may result in less protection than is accorded to investors under rules applicable to domestic issuers.
As a foreign private issuer, we are permitted to follow certain home country corporate governance practices instead of those otherwise required under the Marketplace Rules of the Nasdaq for domestic issuers. For instance, we may follow home country practice in Israel with regard to, among other things, composition of the Board of Directors, director nomination procedure, composition of the compensation committee, approval of compensation of officers, and quorum at shareholders’ meetings. In addition, we will follow our home country law, instead of the Marketplace Rules of the Nasdaq, which require that we obtain shareholder approval for certain dilutive events, such as for the establishment or amendment of certain equity based compensation plans, an issuance that will result in a change of control of the company, certain transactions other than a public offering involving issuances of a 20% or more interest in the company and certain acquisitions of the stock or assets of another company. Following our home country governance practices as opposed to the requirements that would otherwise apply to a United States company listed on the Nasdaq may provide less protection than is accorded to investors under the Marketplace Rules of the Nasdaq applicable to domestic issuers.
In addition, as a foreign private issuer, we are exempt from the rules and regulations under the U.S. Securities Exchange Act of 1934, as amended (the “Exchange Act”), related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we are not required under the Exchange Act to file annual, quarterly and current reports and financial statements with the SEC as frequently or as promptly as domestic companies whose securities are registered under the Exchange Act.
26
If we are unable to satisfy the requirements of Section 404 of the Sarbanes-Oxley Act as they apply to a foreign private issuer that is listed on a U.S. exchange, or our internal controls over financial reporting are not effective, the reliability of our financial statements may be questioned and our stock price and ADS price may suffer.
Section 404 of the Sarbanes-Oxley Act requires companies subject to the reporting requirements of the U.S. securities laws to do a comprehensive evaluation of its and its subsidiaries’ internal controls over financial reporting. To comply with this statute, we are required to document and test our internal control procedures, and our management is required to assess and issue a report concerning our internal controls over financial reporting. In addition, our independent registered public accounting firm may be required to issue an opinion on management’s assessment of those matters.
The continuous process of strengthening our internal controls and complying with Section 404 is complicated and time-consuming. Furthermore, as our business continues to grow both domestically and internationally, our internal controls will become more complex and will require significantly more resources and attention to ensure our internal controls remain effective overall. During the course of its testing, our management may identify material weaknesses or significant deficiencies, which may not be remedied in a timely manner to meet the deadline imposed by the Sarbanes-Oxley Act. If our management cannot favorably assess the effectiveness of our internal controls over financial reporting, or our independent registered public accounting firm identifies material weaknesses in our internal controls, investor confidence in our financial results may weaken, and the market price of our securities may suffer.
Risks Related to our Operations in Israel
We conduct our operations in Israel and therefore our results may be adversely affected by political, economic and military instability in Israel and its region.
Our headquarters, all of our operations and some of our suppliers and third party contractors are located in central Israel and our key employees, officers and most of our directors are residents of Israel. Accordingly, political, economic and military conditions in Israel and the surrounding region may directly affect our business. Since the establishment of the State of Israel in 1948, a number of armed conflicts have taken place between Israel and its Arab neighbors. Any hostilities involving Israel or the interruption or curtailment of trade within Israel or between Israel and its trading partners could adversely affect our operations and results of operations and could make it more difficult for us to raise capital. During the summer of 2006, Israel was engaged in an armed conflict with Hezbollah, a Lebanese Islamist Shiite militia group and political party; during the winter of 2008-2009 and the autumn of 2012, Israel was engaged in armed conflicts with Hamas, a militia group and political party operating in the Gaza Strip; and during the summer of 2014, another escalation in violence among Israel, Hamas and other groups took place. This escalation became known as “Operation Protective Edge.” These conflicts involved missile strikes against civilian targets in various parts of Israel, as well as civil insurrection of Palestinians in the West Bank in some cases, and negatively affected business conditions in Israel. In addition, Israel faces threats from more distant neighbors, in particular Iran. Iran is also believed to have a strong influence among extremist groups in the region, such as Hamas in Gaza, Hezbollah in Lebanon, and various rebel militia groups in Syria. Recent political uprisings and social unrest in various countries in the Middle East and North Africa are affecting the political stability of those countries. The year 2014 saw the rise of an Islamic fundamentalist group known as ISIS. Following swift operations, ISIS gained control of large areas in the Middle East, including in Iraq and Syria, which have contributed to the turmoil experienced in these areas. As a result, the United States armed forces have recently engaged in limited operations against ISIS. This instability may lead to deterioration of the political relationships that exist between Israel and these countries, and has raised concerns regarding security in the region and the potential for armed conflict. These situations may escalate in the future to more violent events which may affect Israel and us. Among other things, this instability may affect the global economy and marketplace through changes in oil and gas prices. Any armed conflicts, terrorist activities or political instability in the region could adversely affect business conditions and could harm our results of operations. For example, any major escalation in hostilities in the region could result in a portion of our employees being called up to perform military duty for an extended period of time. Parties with whom we do business have sometimes declined to travel to Israel during periods of heightened unrest or tension, forcing us to make alternative arrangements when necessary. In addition, the political and security situation in Israel may result in parties with whom we have agreements involving performance in Israel claiming that they are not obligated to perform their commitments under those agreements pursuant to force majeure provisions in the agreements.
27
Our commercial insurance does not cover losses that may occur as a result of events associated with the security situation in the Middle East. Although the Israeli government currently covers the reinstatement value of direct damages that are caused by terrorist attacks or acts of war, we cannot assure you that this government coverage will be maintained. Any losses or damages incurred by us could have a material adverse effect on our business. Any armed conflicts or political instability in the region would likely negatively affect business conditions and could harm our results of operations.
Further, in the past, the State of Israel and Israeli companies have been subjected to an economic boycott. Several countries still restrict business with the State of Israel and with Israeli companies. These restrictive laws and policies may have an adverse impact on our operating results, financial condition or the expansion of our business.
Our operations may be disrupted as a result of the obligation of management or key personnel to perform military service.
Many of our male employees in Israel, including members of our senior management, are obligated to perform one month, and in some cases more, of annual military reserve duty until they reach the age of 40 (or older, for officers or reservists with certain occupations) and, in the event of a military conflict, may be called to active duty. In response to increases in terrorist activity, there have been periods of significant call-ups of military reservists, and recently some of our employees have been called up in connection with armed conflicts. It is possible that there will be military reserve duty call-ups in the future. Our operations could be disrupted by the absence of a significant number of our employees or of one or more of our key employees. Such disruption could materially adversely affect our business, financial condition and results of operations.
Because a certain portion of our expenses is incurred in currencies other than the NIS, our results of operations may be harmed by currency fluctuations and inflation.
Our reporting and functional currency is the NIS, and we pay a substantial portion of our expenses in NIS. The revenues from our out-licensing and co-development arrangements are payable in U.S. dollars and we expect our revenues from future licensing arrangements to be denominated in U.S. dollars or in euros. As a result, we are exposed to the currency fluctuation risks relating to the recording of our revenues in NIS. For example, if the NIS strengthens against either the U.S. dollar or the euro, our reported revenues in NIS may be lower than anticipated. The Israeli rate of inflation has generally not offset or compounded the effects caused by fluctuations between the NIS and the U.S. dollar or the euro. From time to time, we engage in hedging transactions. Although the Israeli rate of inflation has not had a material adverse effect on our financial condition during 2012, 2013 or 2014, we may, in the future, decide to enter into currency hedging transactions to decrease the risk of financial exposure from fluctuations in the exchange rates of the currencies mentioned above in relation to the NIS. These measures, however, may not adequately protect us from material adverse effects. Effective January 1, 2015, we intend to change our reporting and functional currency to the dollar, which we expect will reduce, to some extent, our exposure to the currency fluctuation risks mentioned above.
We have received Israeli government grants and loans for the operation of a biotechnology incubator and for certain research and development expenditures. The terms of these grants and loans may require us to satisfy specified conditions in order to manufacture products and transfer technologies outside of Israel. We may be required to pay penalties in addition to repayment of the grants and loans. Such grants and loans may be terminated or reduced in the future, which would increase our costs.
Our research and development efforts, including the operation of a biotechnology incubator which we terminated at the end of 2013, have been financed, in part, through grants and loans that we have received from the OCS. Of our 10 current development projects, two were approved for funding by the OCS: BL-1040 and BL-7040. In addition, before we in-licensed BL-8040, Biokine had received funding for the project from the OCS, and as a condition to OCS consent to our in-licensing of BL-8040, we were required to agree to abide by any obligations resulting from such funding. We therefore must comply with the requirements of the Israeli Law for the Encouragement of Industrial Research and Development, 1984, and related regulations, or the Research Law, with respect to these projects. As of September 30, 2014, we received approximately NIS 76.1 million ($20.6 million) in funding from the OCS, of which approximately NIS 53.7 million ($14.5 million) was funding provided to our biotechnology incubator, which we undertook to repay from proceeds received from the sale of products developed under the incubator project. Under the terms of the biotechnology incubator, we had recorded a first-degree floating lien on all assets of the incubator against the repayment of funding received by the incubator for the projects developed under it. As previously described, we terminated the incubator at the end of 2013, and recorded the projects as having been terminated for repayment purposes. As of September 30, 2014, we have paid the OCS approximately NIS 24.3 million ($6.6 million) in royalties under our approved programs. As of September 30, 2014, we have a contingent obligation to the OCS in the total amount of NIS 22.4 million (approximately $6.0 million) under all of our approved programs, of which NIS 20.4 million ($5.5 million) are attributed to projects recorded by the Company as terminated for repayment purposes but which still require a formal termination process with the OCS. When know-how, technology or products are developed using OCS grants, the terms of these grants and the Research Law restrict the transfer of that know-how (as well as know-how that is derived from funded know-how) and the development or manufacture of those products out of Israel without the prior approval of the OCS. Therefore, the discretionary approval of an OCS committee will be required for any transfer to third parties of our therapeutic candidates developed with OCS funding, for the purpose of the commercialization of our product candidates. We received approval in 2009 for the out-licensing of BL-1040 to Bellerophon; however the out-licensing of BL-7040 and BL-8040 to any party outside of Israel will be subject to the prior approval of the OCS. There is no assurance that we will receive the required approvals should we wish to transfer this technology or development out of Israel in the future. Furthermore, the OCS committee may impose certain conditions on any arrangement under which we transfer technology or development out of Israel. Transfers of know-how from OCS funded programs, including our biotechnology incubator, even if approved by the OCS, may be subject to restrictions set forth in the Research Law, and may include payments to the OCS.
28
The transfer abroad of the manufacturing of any OCS-supported product or technology is also subject to various conditions, including the payment of increased royalties equal to, in the aggregate, up to 300% of the total grant amounts received in connection with the product or technology, plus interest, depending on the portion of total manufacturing that is performed outside of Israel. Payment of the increased royalties would constitute the repayment amount required with respect to the OCS grants received for the development of the products or technology for which the manufacturing is performed outside of Israel. In addition, any decrease in the percentage of manufacture performed in Israel of any product or technology, as originally declared in the application to the OCS with respect to the product or technology, may require us to notify, or to obtain the approval of, the OCS, and may result in increased royalty payments to the OCS of up to 300% of the total grant amounts received in connection with the product or technology, plus interest, depending on the portion of total manufacturing that is performed outside of Israel. In addition, the OCS has the discretion to permit overseas manufacture in excess of the declared percentage (deviations of up to 10% do not require consent, but the OCS must be notified). Consent is contingent upon payment of additional royalties, at rates and subject to ceilings set out in the relevant regulations, up to three times the amount of the grants. Furthermore, the transfer of OCS-supported know-how, and the transfer of OCS-supported manufacturing or manufacturing rights of products, technologies or know-how inside or outside of Israel is subject to payment of transfer fees. Maximal transfer fees with respect to the transfer of know-how are as follows: up to three times the original grant received plus accrued interest as of the date of transfer, when the OCS Research Committee is satisfied that the core research and development activity will remain in Israel, and up to six times the value of the original grant in the case of liquidation of activities in Israel. Therefore, if aspects of our technologies are deemed to have been developed with OCS funding, the discretionary approval of an OCS committee would be required for any transfer to third parties inside or outside of Israel of know how or manufacturing or manufacturing rights related to those aspects of such technologies. These restrictions may impair our ability to sell our technology assets or to outsource or transfer development or manufacturing activities with respect to any product or technology. These restrictions continue to apply even after we have repaid any grants, in whole or in part, unless otherwise agreed by the designated OCS committee.
We cannot be certain that any approval of the OCS will be obtained on terms that are acceptable to us, or at all. Furthermore, if we undertake a transaction involving the transfer to a non-Israeli entity of technology developed with OCS funding pursuant to a merger or similar transaction, the consideration available to our shareholders may be reduced by the amounts we are required to pay to the OCS. If we fail to comply with the conditions imposed by the OCS, including the payment of royalties with respect to grants received, we may be required to refund any payments previously received, together with interest and penalties, and may be subject to criminal penalties.
29
Provisions of Israeli law may delay, prevent or otherwise impede a merger with, or an acquisition of, our company, which could prevent a change of control, even when the terms of such a transaction are favorable to us and our shareholders.
Israeli corporate law regulates mergers, requires tender offers for acquisitions of shares above specified thresholds, requires special approvals for transactions involving directors, officers or significant shareholders and regulates other matters that may be relevant to these types of transactions. For example, a merger may not be consummated unless at least 50 days have passed from the date that a merger proposal was filed by each merging company with the Israel Registrar of Companies and at least 30 days from the date that the shareholders of both merging companies approved the merger. In addition, a majority of each class of securities of the target company must approve a merger. Moreover, a full tender offer can only be completed if the acquirer receives the approval of at least 95% of the issued share capital (provided that a majority of the offerees that do not have a personal interest in such tender offer shall have approved the tender offer, except that if the total votes to reject the tender offer represent less than 2% of the company’s issued and outstanding share capital, in the aggregate, approval by a majority of the offerees that do not have a personal interest in such tender offer is not required to complete the tender offer), and the shareholders, including those who indicated their acceptance of the tender offer, may, at any time within six months following the completion of the tender offer, claim that the consideration for the acquisition of the shares did not reflect their fair market value and petition the court to alter the consideration for the acquisition accordingly (unless the acquirer stipulated in the tender offer that a shareholder that accepts the offer may not seek appraisal rights, and the acquirer or the company published all required information with respect to the tender offer prior to the date indicated for response to the tender offer).
Furthermore, Israeli tax considerations may make potential transactions unappealing to us or to our shareholders whose country of residence does not have a tax treaty with Israel exempting such shareholders from Israeli tax. For example, Israeli tax law does not recognize tax-free share exchanges to the same extent as U.S. tax law. With respect to mergers, Israeli tax law allows for tax deferral in certain circumstances but makes the deferral contingent on the fulfillment of numerous conditions, including a holding period of two years from the date of the transaction during which sales and dispositions of shares of the participating companies are restricted. Moreover, with respect to certain share swap transactions, the tax deferral is limited in time, and when such time expires, the tax becomes payable even if no actual disposition of the shares has occurred.
These and other similar provisions could delay, prevent or impede an acquisition of us or our merger with another company, even if such an acquisition or merger would be beneficial to us or to our shareholders.
We have received Israeli government grants and loans for the operation of a biotechnology incubator and for certain research and development expenditures. The terms of these grants and loans may require us to satisfy specified conditions in order to manufacture products and transfer technologies outside of Israel. We may be required to pay penalties in addition to repayment of the grants and loans. Such grants and loans may be terminated or reduced in the future, which would increase our costs.
It may be difficult to enforce a U.S. judgment against us and our officers and directors named in this prospectus in Israel or the United States, or to serve process on our officers and directors.
We are incorporated in Israel. All of our executive officers and the majority of our directors reside outside of the United States, and all of our assets and most of the assets of our executive officers and directors are located outside of the United States. Therefore, a judgment obtained against us or any of our executive officers and directors in the United States, including one based on the civil liability provisions of the U.S. federal securities laws, may not be collectible in the United States and may not be enforced by an Israeli court. It also may be difficult for you to effect service of process on these persons in the United States or to assert U.S. securities law claims in original actions instituted in Israel.
30
Your rights and responsibilities as a shareholder will be governed by Israeli law which may differ in some respects from the rights and responsibilities of shareholders of U.S. companies.
We are incorporated under Israeli law. The rights and responsibilities of the holders of our Ordinary Shares are governed by our Articles of Association and Israeli law. These rights and responsibilities differ in some respects from the rights and responsibilities of shareholders in typical U.S.-based corporations. In particular, a shareholder of an Israeli company has a duty to act in good faith toward the company and other shareholders and to refrain from abusing its power in the company, including, among other things, in voting at the general meeting of shareholders on matters such as amendments to a company’s articles of association, increases in a company’s authorized share capital, mergers and acquisitions and interested party transactions requiring shareholder approval. In addition, a shareholder who knows that it possesses the power to determine the outcome of a shareholder vote or to appoint or prevent the appointment of a director or executive officer in the company has a duty of fairness toward the company. There is limited case law available to assist us in understanding the implications of these provisions that govern shareholders’ actions. These provisions may be interpreted to impose additional obligations and liabilities on holders of our Ordinary Shares that are not typically imposed on shareholders of U.S. corporations.
31
Serious News for Serious Traders! Try StreetInsider.com Premium Free!
You May Also Be Interested In
- BioLineRx Announces Poster Presentation on Apheresis Center Efficiency and CXCR4 Antagonists including APHEXDA® (motixafortide) in Patients with Multiple Myeloma at the ASFA 2024 Annual Meeting
- Arthrex Wins Both Gold and Silver Medals at 2024 Edison Awards
- Illinois Speaker Chris Welch Recommended by Proviso Township Democratic Organization to Serve as Committeeman
Create E-mail Alert Related Categories
SEC FilingsSign up for StreetInsider Free!
Receive full access to all new and archived articles, unlimited portfolio tracking, e-mail alerts, custom newswires and RSS feeds - and more!
